

Abstract
Bacteria first develop tolerance after antibiotic exposure; later genetic resistance emerges through the population of tolerant bacteria. Bacterial persister cells are the multidrug-tolerant subpopulation within an isogenic bacteria culture that maintains genetic susceptibility to antibiotics. Because of this link between antibiotic tolerance and resistance and the rise of antibiotic resistance, there is a pressing need to develop treatments to eradicate persister cells. Current anti persister cell strategies are based on the paradigm of “awakening” them from their low metabolic state before attempting eradication with traditional antibiotics. Herein, we demonstrate that the low metabolic activity of persister cells can be exploited for eradication over their metabolically active counterparts. We engineered gold nanoclusters coated with adenosine triphosphate (AuNC@ATP) as a benchmark nanocluster that kills persister cells over exponential growth bacterial cells and prove the feasibility of this new concept. Finally, using AuNC@ATP as a new research tool, we demonstrated that it is possible to prevent the emergence of antibiotic-resistant superbugs with an anti-persister compound. Eradicating persister cells with AuNC@ATP in an isogenic culture of bacteria stops the emergence of superbug bacteria mediated by the sub-lethal dose of conventional antibiotics. Our findings lay the groundwork for developing novel nano-antibiotics targeting persister cells, which promise to prevent the emergence of superbugs and prolong the lifespan of currently available antibiotics.
Keywords: Antimicrobial Gold nanoclusters, Persister cells, antibiotic resistance, outer membrane proteins, lipid homeostasis
Graphical Abstract

INTRODUCTION
The rise of antibiotic resistance and the declining discovery of new antibiotics has created a global health crisis [1–3]. In addition, we knew that bacteria could survive lethal doses of antimicrobial agents, not through genetic mutations that confer the resistance but instead through reducing their metabolism, resulting in the depletion of intracellular levels of adenosine triphosphate (ATP) [4]. In doing so, they enter a low metabolic state called persister cells, which exhibit multidrug tolerance [5, 6]. Persister cells do not grow in the presence of antimicrobial agents but can resuscitate after the treatment [7, 8]. Therefore, recurrent and chronic infections are generally associated with persister cells [8–12].
Evidence has now accumulated, demonstrating that persister cells promote antibiotic resistance rates [13, 14]. In addition, we now know that bacteria that encounter antibiotics first become tolerant, and then genetic resistance emerges through this tolerance [15, 16]. Because of this link between antibiotic tolerance and resistance and the rise of antibiotic resistance, there is a pressing need to develop treatments to eradicate persister cells. However, killing persister cells is an unmet clinical need because no Food and Drug Administration (FDA) approved antibiotics are effective against persister cells, and there are no potential prospects in the global preclinical bacterial pipeline [17]. Ongoing research into the application of ultrasmall gold nanoclusters (AuNCs have a total diameter of < 4 nm) that exhibit antibacterial activity for bacterial infection treatment and diagnosis has demonstrated its advantages as a potential alternative solution for conventional antibiotics in the fight against multidrug-resistant gram-negative bacteria [18–24].
Most antimicrobial AuNCs were discovered in experiments that tested the ability of compounds to inhibit bacterial growth [25–28]. However, they are often ineffective for treating non-growing that are less metabolically active. For instance, previous attempts to use antimicrobial AuNCs to kill bacterial cells with low metabolic activity have failed, with a 97% reduction in antimicrobial efficacy compared to that achieved in bacteria with an active metabolism [28]. This finding demonstrates that existing antimicrobial AuNCs are inadequate to address persister cells with intracellular ATP depletion. Another key translational hurdle is the production of intracellular reactive oxygen species (ROS), the primary mechanism of action leading to the death of bacteria triggered by existing AuNC strategies, which the risk of turning metabolically active cells into persister cells [29, 30].
In support of this idea, numerous independent studies have shown that compounds inducing ROS production in bacteria promote the formation of persister cells. For example, paraquat, a potent inducer of ROS and a widely used herbicide, pushes the metabolically active bacterial cells to become persister cells [31]. Another example is salicylate-induced ROS also causes an increase in persister cell formation [32]. It has also been shown that sublethal antibiotic treatment leads to multidrug resistance through ROS-induced mutagenesis [33]. These results question the appropriateness of the development of antimicrobial AuNCs, relying on ROS production as the primary mechanism of action to combat antibiotic resistance [23, 28, 34, 35]. Therefore, for antimicrobial AuNCs to stay relevant in the clinical battlegrounds to combat antibiotic resistance, it is imperative to eradicate persister cells, one root cause of antibiotic resistance. In addition, designing a new class of AuNCs with new antimicrobial mechanisms of action that are not dependent on ROS is needed.
Current anti persister cell strategies are based on the paradigm of “awakening” them from their low metabolic state before attempting eradication with traditional antibiotics [36]. Therefore, we created a nontraditional antimicrobial chemotherapy strategy based on AuNCs as an adjuvant to eradicate persister cells by conventional antibiotics [37]. However, persister cells formed by multidrug-resistant bacteria can easily survive this therapeutic approach because when the bacteria become resistant to the partner antibiotic, the AuNC/antibiotic combination fails to eradicate persister cells. Herein, we now show that the low metabolic activity of persister cells can be exploited to target them over their metabolically active counterpart with the outcome of complete eradication.
RESULTS AND DISCUSSION
Characterization of gold nanoclusters coated with adenosine triphosphate (AuNC@ATP).
Figure 1 displays the characterization data for the AuNC@ATP. Transmission electron microscopy (TEM) confirms that AuNC@ATP are highly uniform with average core sizes around 2.45 ± 0.43 nm. In addition, the UV-visible spectra show a lack of a prominent surface plasmon peak at around 500 nm, consistent with the small size of the particles [38]. The AuNC@ATP are negatively charged, with a zeta potential in phosphate buffer of − 30 ± 2 mV, respectively. The molecular weight (g/mol) of AuNC was calculated as follows: (i) Weight of one AuNC (g) = Volume (nm3) * density of gold (19.32 g/cm3) * 10−21 cm3/nm3 = 1.487 * 10−19 g. (ii) Molecular weight (g/mol) = Weight of AuNC (g) * 6.022 × 1023 mol−1 = 8.95 * 104 g/mol. The ATP molecular weight is 507 g/mol, meaning that AuNC (with 2.45 nm in diameter) is 176 times heavier than ATP. Therefore, 99.4 % of the weight of AuNC@ATP is due to AuNC. Moreover, the amount of ATP in AuNC@ATP (1 µg/ml or 11.2 nM) was estimated to be 1079 nM based on the ATP bioluminescent assay Kit (Supplementary Figure S1). Based on this calculation, we estimate that one AuNC@ATP contains 96 molecules of ATP.
Figure 1. Characterization of gold nanoclusters coated with adenosine triphosphate (AuNC@ATP).

(A) Schematics of AuNC@ATP, a picture of the solution of as-synthesized AuNC@ATP, was analyzed using UV-Vis spectroscopy to demonstrate the absence of the plasmon resonance band at 520 nm. (B) Transmission electron microscopy (TEM) image of AuNC@ATP. Magnification and scale bar (5 nm) in the pictures. (C) The particle distribution of AuNC@ATP was measured by TEM.
AuNC@ATP disrupts the membrane integrity by increasing permeability without causing bacterial cell lysis.
The outer membrane of gram-negative bacteria (OM) is asymmetric, with phospholipids (PLs) in the inner leaflet and lipopolysaccharides (LPS) in the outer leaflet [39]. The biosynthesis of PLs is completed at the cytoplasmic (CM), also known as the inner membrane (IM), which makes the translocation from the IM to the OM (anterograde transport) essential to fill the OM [40]. The translocation of PLs from the OM to the IM (retrograde transport) is believed to maintain the asymmetric LPS/PL structure of OM [40]. In the stationary phase, cells can no longer replace those PLs lost from the IM through new synthesis or stimulating retrograde transport. Thus, the balance between anterograde and retrograde transport is essential for the gram-negative bacteria in the persister cell state to ensure the permeability barrier of OM and integrity of the cytoplasmic membrane required for cell viability [41, 42]. It has previously been observed that anterograde transfer is ATP-independent but could be abolished by ATP hydrolysis [43].
Furthermore, contrary to anterograde transport, ATP drives retrograde PL transport [43]. We resonate that AuNC@ATP-mediated cell death of the stationary phase occurs through the disruption of lipid homeostasis, which has been shown to activate a novel cell death pathway [44]. We first investigated whether AuNC@ATP-induced alterations of OM permeability using 8-Anilino-1-naphthalene sulfonic acid (ANS), a fluorescent lipophilic dye which shows enhanced fluorescence when in a hydrophobic environment [45–49]. As a positive control, we chose Colistin (polymyxin E) which disrupts the OM [50, 51]. The stationary-phase culture of multidrug-resistant gram-negative bacteria, including Pseudomonas aeruginosa (P. aeruginosa), Escherichia coli (E. coli) and Klebsiella pneumoniae (K. pneumoniae), were treated with either AuNC@ATP or Colistin. We found that the interaction between gram-negative bacteria and AuNC@ATP carries a negative charge on the surface, leading to the impairment of OM permeability, as evidenced by the increased ANS fluorescence (Figure 2a). For P. aeruginosa, the impact of AuNC@ATP on OM permeability was comparable to the one induced by Colistin. While for E. coli and K. pneumoniae, AuNC@ATP induced more damage than Colistin, which is positively charged (Figure 2a). Next, we examine whether AuNC@ATP triggers cytoplasmic membrane (CM) disruption. Propidium iodide (PI) is a membrane-impermeant stain which only labels bacteria with a compromised IM [52]. We found that after AuNC@ATP treatment, the PI can penetrate the CM, suggesting damage to CM (Figure 2b). For P. aeruginosa and K. pneumoniae, the impact of AuNC@ATP on OM permeability was comparable to the one induced by Colistin. While for E. coli, AuNC@ATP induced more damage than Colistin (Figure 2a).
Figure 2. Exposure to AuNC@ATP leads to the activation of stress that disrupts the outer membrane (OM) and cytoplasmic membrane permeability (CM).

First, the stationary phase culture of gram-negative bacteria was exposed to AuNC@ATP and Colistin (positive control). Then the permeability of the OM and IM was assessed by measuring the fluorescence of (A) 8-Anilino-1-naphthalene sulfonic acid (ANS) and (B) propidium iodide (PI), respectively. ANS is a compound that changes fluorescence depending on the polarity of its surrounding environment. In the presence of intact gram-negative bacterial cells in an aqueous environment, ANS is weakly fluorescent. Still, if the OM is disturbed, the ANS can penetrate the nonpolar phospholipid bilayer, resulting in a measurable increase in fluorescence. PI is a membrane-impermeable DNA stain; it can only label bacteria with a compromised CM. Still, if the CM is disturbed, the PI can penetrate the CM and binds to DNA, resulting in a measurable increase in fluorescence.
To demonstrate that AuNC@ATP is active against bacteria cells in the growth-arrested (persister cell) phase, we compared the bactericidal activity of AuNC@ATP and Ofloxacin against the stationary-phase gram-negative bacteria. We found that AuNC@ATP sterilizes the stationary-phase culture of P. aeruginosa, E. coli and K. pneumoniae (Figure 3a). Conversely, Ofloxacin fails to eradicate the stationary-phase culture (Figure 3a), which broadly supports the work of other studies in this area linking the bacterial metabolic state with antibiotic lethality [4, 53]. Furthermore, these results suggest that exposure to AuNC@ATP triggest a gram-negative cell envelope stress that activates cell death.
Figure 3. AuNC@ATP kills gram-negative bacteria in the growth-arrested state without causing bacterial cell lysis.

(A) the stationary phase culture of gram-negative bacteria resuspended in phosphate-buffered saline (PBS) and exposed to either AuNC@ATP or Ofloxacin for 4h. After the treatment, the drugs were removed, and the number of surviving bacteria was assessed by measuring the colony-forming unit per millilitre (CFU/mL). (B) AuNC@ATP-mediated no-lytic cell death of the stationary phase culture of gram-negative bacteria. After the treatment of the stationary phase culture of gram-negative bacteria with PBS, AuNC@ATP (16.8 µM), Ofloxacin (8.3 µM) and Colistin (1.3 mM), the presence of proteins in collected supernatant of each treatment was assessed by using a Pierce BCA Protein Assay kit.
Given that one of the most prominent features of cell death triggered by disruption of PL homeostasis is that cell death does not occur by cell lysis [44]; therefore we analyzed whether AuNC@ATP causes cell lysis using the presence of protein in the supernatant of gram-negative bacteria treated with AuNC@ATP as a proxy of bacterial cell membrane lysis. Colistin and Ofloxacin cause bacteria cell lysis by forming membrane pores and peptidoglycan composition [54–56]. However, in contrast to these conventional antibiotics, no cell lysis was observed after exposure of gram-negative bacteria to AuNC@ATP, as seen by the absence of proteins in the supernatant (Figure 3b). Moreover, to prove that the entire protein pool was still in bacteria following AuNC@ATP treatment, we lysed the AuNC@ATP-treated bacterial cells with Colistin. As expected, the protein levels were comparable to that obtained from the supernatant of cells treated with Colistin (Supplementary Figure S2).
Furthermore, AuNC@ATP mediated no lytic cell death, contrary to the mechanism of action of existing antimicrobial AuNCs that results in lysis [28, 57]. The scanning electron microscopy (SEM) analysis of morphological features of cells shows that PA14 treated with AuNC@ATP (16. 8 µM) exhibited no bacteria lysis but blebbing surfaces and OM vesicles (OMVs) were also visible (Supplementary Figure S3). OMVs are membrane-enclosed spherical entities released by gram-negative bacteria and are essential for bacterial survival under stress conditions [58]. In our case, the membrane blebbing results from cell envelope disturbances can be caused by the unbalanced biosynthesis of cell wall components, such as outer membrane proteins (OMPs). This may partly explain why the AuNC@ATP disrupts the membrane integrity by increasing permeability without causing bacterial cell lysis. To explore further the changes in the OM during cell death, we stained AuNC@ATP (16. 8 µM) treated PAO1-GPF (10145GFP) cells with the red fluorescent dye FM 4–64 to visualize OM of dead cells. Our FM 4–64 data demonstrates OM is not ruptured after treatment with AuNC@ATP (Supplementary Figure S4). This effort showed that AuNC@ATP functions through a mechanism distinct from known classes of bactericidal antibiotics, including cationic antimicrobial polypeptides and existing antimicrobial nanoparticles.
To further support that the mechanism of action of AuNC@ATP is distinct from existing antimicrobial AuNCs, where ROS production is essential to achieve bacterial killing [28, 35], we confirm that the internalization of AuNC@ATP in bacterial cells does not increase intracellular ROS using the fluorescent probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA). As expected, no change in the DCFH-DA fluorescence intensity was observed in the stationary phase culture of P. aeruginosa (PA14) treated with AuNC@ATP compared to Ofloxacin and untreated PA14 cells (Supplementary Figure S5). Cumulatively, the data in this section support that after transiting through OM, AuNC@ATP leads to the fusion of the inner leaflet of OM with the outer leaflet of the cytoplasmic membrane, which leads to bacteria death without cell lysis.
The accumulation of unfolded outer membrane proteins (OMPs) causes AuNC@ATP lethality.
We next assessed that ability of AuNC@ATP to induce a stress response that causes a lethal accumulation of unfolded OMPs. Bacteria respond to misfolded or unfolded β-barrel OMPs in the periplasm by inducing sigma factor (σE)-dependent transcription of stress genes in the cytoplasm. The β-barrel structure of OMPs is not found in the cytoplasmic membrane because they represent open pores that enable the diffusion of water, ions and hydrophilic molecules up to 600 Da [59]. To activate the σE response, unfolded OMPs in the periplasm trigger a proteolytic cascade involving ClpXP, an ATP-dependent cytoplasmic protease that destroys a transmembrane protein (RseA) that usually binds to and inhibits σE [59, 60]. When the σE system is activated, genes are transcribed from σE-dependent promoters, leading to the upregulation of OMP folding pathways to prevent an accumulation of the highly toxic unfolded OMPs [59]. However, if the σE response is triggered in cells lacking ClpXP, the upregulation of OMP folding pathways cannot be activated, causing cell death due to the accumulation of the unfolded OMPs in the periplasm. Therefore, to test whether exposure to AuNC@ATP building-up of unfolded OMPs in the periplasm and causing envelope damage, we compared the antimicrobial activity of AuNC@ATP against P. aeruginosa (PA14) and PA14 (ΔClpXP). We found that PA14 (ΔClpXP) is eight times more susceptible to AuNC@ATP than PA14 (ClpXP). The minimum inhibitory concentration (MIC) of PA14 (ClpXP) and PA14 (ΔClpXP) were 2.23 µM and 0.28 µM, respectively (Figure 4a and b).
Figure 4. The accumulation of unfolded outer membrane proteins (OMPs) causes AuNC@ATP lethality.

(A and B) Suppression on the growth of P. aeruginosa (PA14) and its genetic mutant harbouring a genetic deletion of ClpXP protease (ΔClpXP) incubated with AuNC@ATP at different concentrations. The growth of P. aeruginosa in lysogeny broth (LB) was assessed by measuring the optical density at 600 nm (OD600 nm) (N = 3). (C) Schematic showing that AuNC@ATP exert their antibacterial activities mainly by inducing stress that triggers multiple perturbations causing accumulation of toxic unfolded OMPs in the periplasmic space.
Contrary to AuNc@ATP, the MIC of ATP against PA14 (ΔClpXP) and PA14 were similar above 10 mM. These data strongly imply that the enhanced antimicrobial effect of AuNc@ATP against PA14 (ΔClpXP) was not determined by the ATP ligand identity or its density on the surface but by the overall AuNc@ATP as an entire entity and compound (Supplementary Figure S6). Cumulatively, this supports that the internalization of AuNC@ATP in bacterial cells induces a lethal accumulation of unfolded OMPs. In line with the fact that cell death mediated by AuNC@ATP occurs without releasing periplasmic proteins and cytosolic components into the supernatant, the hypersusceptibility of PA14 (ΔClpXP) to AuNC@ATP indicates that cell death occurs through the lethal accumulation of unfolded OMPs in the periplasmic space (Figure 4c and Figure S2). We conclude that AuNC@ATP has antimicrobial activity attributable to stress responses building up of unfolded OMP accumulation in the periplasm, disrupting bacterial membranes by altering lipid homeostasis and asymmetry.
Although AuNC@ATP-mediated cell death through a lethal accumulation of unfolded OMPs, the molecular mechanism of how it impairs the σE response remains unanswered. We, therefore, examined the effect of AuNC@ATP on the OMP biogenesis in E. coli (MDR). The porins OmpF and OmpC are highly abundant, and both restrict the passage to compounds with a size-exclusion limit of about 600 Da [61]. Furthermore, like most OMPs, OmpC and OmpF are synthesized as unfolded precursors in the cytoplasm and translocated across the cytoplasmic membrane via the Sec translocase. Once in the periplasm, the OMPs are kept unfolded by the periplasmic chaperones survival protein A (SurA) and seventeen kilodalton protein (Skp), which prevent them from forming misfolded aggregates and are guided to the ß-barrel assembly machinery (BAM). With its core protein BamA, the BAM complex finally inserts the OMPs into the OM [62].
To demonstrate that AuNC@ATP could trigger the accumulation of OMPs in the periplasm after treatment, we carried out the western blot analysis with anti-OmpC. OMPs of Gram-negative bacteria are heat-modifiable proteins, i.e., proteins altering their molecular forms (trimers or monomers) and, accordingly, their electrophoretic mobilities on SDS-PAGE gels depending upon the folded state [63, 64]. Thus, an SDS-PAGE gel-shift assay can follow folding since the native state of OMPs resists denaturation in SDS at room temperature, and the folded OMP migrates faster than the unfolded OMP in the full-length form. As expected, OmpC was present in the periplasm of the periplasmic fraction of untreated E. coli (MDR), and the band observed on the SDS-PAGE gel corresponded to the folded OmpC monomer (36 kDa). Furthermore, the position of this band in the absence and presence of heat (95°C) was similar, confirming that it is the folded OmpC monomer. Our result supports that some β-barrel proteins begin to fold before interacting with the Bam complex [65], suggesting that the folding of OmpC is initiated in the periplasm.
In contrast, after treatment with AuNC@ATP (4.5 µM), OmpC migrated as a folded trimer (72 kDa) on SDS-PAGE gels (Supplementary Figure S7). Our thermal stability of the OmpC trimer may be due to the AuNC@ATP binding, as previous research already demonstrates that ATP stabilizes the protein against thermal denaturation [66]. Furthermore, the unfolded trimer was observed with similar thermal stability (Supplementary Figure S7). Cumulatively, this supports that AuNC@ATP treatment induces the accumulation and trimerization of OmpC in the periplasm. This mechanism may contribute to the impairment of OMPs biogenesis and homeostasis in Gram-negative bacteria and partly explain why the AuNC@ATP disrupts the membrane integrity by increasing permeability without causing bacterial cell lysis.
A remarkable feature of the periplasmic space of gram-negative bacteria is that it contains more than 300 proteins and provides a unique protein folding and stabilization environment because it has no ATP [67, 68]. ATP was found to keep the proteins in their soluble form and prevent them from aggregation [69]. Furthermore, ATP regulates the solubility of a significantly large set of proteins [70]. Finally, the computational simulation demonstrates that ATP can unfold a single chain of hydrophobic macromolecules [71]. ATP tends to bind to the surface regions with high flexibility and a high degree of hydration, which are also vulnerable to thermal perturbations [66]. In this case, a model for oligomer OmpC formation would likely be an accumulation of AuNC@ATP around the unfolded and folded monomers aggregate. The thermal stability of the OmpC trimer supports this scenario. Thus, these findings partly explain why AuNC@ATP triggers the trimerization of OMPs in the periplasm.
Persister cells are more susceptible to AuNC@ATP than metabolically active bacterial cells.
Recent studies have shown a reduction in ATP levels in bacterial persister cells [10, 72, 73]. The decrease in ATP levels correlates with reduced proteolysis of functional proteins by different ATP-dependent proteases [74]. Since AuNC@ATP induces a lethal accumulation of unfolded OMPs, the lethality of AuNC@ATP should increase as bacteria transition from a metabolically active state to a metabolically repressed state (i.e., low ATP levels) due to reduced proteolysis of unfolded OMPs. Moreover, persister cells can no longer synthesize new PLs to replace those lost from OM upon stimulating retrograde transport. We reasoned that these features could be exploited to eradicate persister cells with low metabolic activity. To test this hypothesis, we compared the bactericidal activity of AuNC@ATP against exponential and persister cells. Passage through the stationary phase is associated with the formation of persister cells, which usually represent about 1% of the total bacterial population [9, 12]. Therefore, the 48-h-old-stationary phase was treated with Ofloxacin for 24h to eradicate non-persister cells (Figure 5a). Ofloxacin was used at 415 μM, corresponding to 30-fold the MIC for the PAO1 wild-type strain. The surviving persister cells were concentrated and redispersed in phosphate-buffered saline (PBS) to prevent persister cells from awakening from their metabolically repressed state. We confirmed that persister cells have a reduction in ATP levels compared to bacterial cells in the exponential growth phase (Figure 5b). We found that AuNC@ATP (2.2 µM) leads to a 7-log reduction in bacterial numbers (CFU/ml) compared to less than 1-log reduction when bacteria are in the exponential growth phase (Figure 5c). Additionally, we sterilized the inoculum of persister cells (108 CFU/ml) with 4.45 µM of AuNC@ATP. However, only a 3-log reduction was observed when bacteria were in the exponential growth phase (from 108 to 105 CFU/ml). However, no eradication was observed in persister cells after exposure to the ATP up to 10 mM, proving that the entire entity of AuNC@ATP was required to eradicate persister cells (Supplementary Figure S8).
Figure 5. Persister cells are more susceptible to AuNC@ATP than metabolically active bacterial cells.

(A) Schematic showing the isolation of persister cells from the stationary phase culture of P. aeruginosa (PA14) using Ofloxacin. (B) ATP levels were measured in isolated persister cells and exponentially growing PA14. (C) Persister cells and exponentially growing PA14 were redispersed in phosphate-buffered saline (PBS) with AuNC@ATP (without carbon sources). After the AuNC@ATP treatment, the number of surviving bacteria was assessed by measuring the colony-forming unit per millilitre (CFU/mL). Cells during the AuNC@ATP treatments at different concentrations were plated to generate the dose-response curves (N = 3).
We also examined the ability of AuNC@ATP (4.5 µM) to eradicate exponential-phase cells in the presence of 200 µg/mL carbonyl cyanide m-chlorophenyl hydrazone (i.e., CCCP-induced persister cells). Our results indicate that the AuNC@ATP eradicates persister cells prepared from exponential-phase cells (Supplementary Figure S9). However, no eradication was observed in exponential-phase cells after exposure to the AuNC@ATP (4.5 µM), proving that persister cells are more susceptible to AuNC@ATP than their metabolically active counterpart (Supplementary Figure S9). Although the Gram-positive persister cells are behind the cope of this manuscript, the ofloxacin-inducing persister cell of Staphylococcus aureus (MDR) could be eradicated by AuNC@ATP and death also occur without cells lysis as evidenced by the absence of protein in the supernatant after treatment with AuNC@ATP (Supplementary Figure S10). This result suggests that AuNC@ATP is a compound that acts on multiple targets. Therefore, the molecular mechanism of AuNC@ATP against persistent Gram-positive cells will be described in the next manuscript.
Contrary to the previous works reporting a 97% reduction in the antimicrobial efficacy of AuNCs when bacteria exhibit low metabolic activity [28], our findings demonstrate that AuNC@ATP-mediated cell death increases as bacteria transition from a metabolically active state to a metabolically repressed state (i.e., low ATP levels). This support the conclusion that AuNC@ATP is a new class of antimicrobial nanoclusters with a unique and novel mechanism of action. This AuNC@ATP-mediated persister cell death is contrary to the bactericidal activity of conventional antibiotics, where lethality decreases as the bacteria transition from a metabolically active state to a metabolically repressed state [4]. This effort showed that contrary to current anti-persister cell strategies are based on the paradigm of “awakening” them from their low metabolic state before attempting eradication with traditional antibiotics, the low metabolic activity of persister cells can be exploited for eradication over their metabolically active counterparts. AuNC@ATP is presented as a benchmark nanocluster that proves the feasibility of this concept.
P. aeruginosa fails to produce pyocyanin in the presence of a sub-lethal dose of AuNC@ATP.
During infections, P. aeruginosa is often in contact with other pathogens [75]. It has been shown that pyocyanin (PYO), a small molecule produced by P. aeruginosa, increases the persister cell population of other pathogens in contact with P. aeruginosa. The gram-negative coccobacillus Acinetobacter baumannii (A. baumannii) currently leads the WHO list of pathogens in critical need of new therapeutic development. A. baumannii formed 0.07 and 0.02% persister cells in the presence of amikacin and carbenicillin [76]. However, this increased 4- and 3-fold in the presence of PYO [76]. Given that PYO promotes persister cell formation from neighbouring bacteria in coinfection with P. aeruginosa, our next goal was to elucidate whether AuNC@ATP could act as an inhibitor of PYO production. The Chloroform–hydrochloric acid method was used to assess the PYO production. As a result, we found that PYO production by P. aeruginosa (PA14) was 1.15 ± 0.20 μg/mL and 0.23 ± 0.19 μg/mL in the presence of 0.42 and 0.56 µM of AuNC@ATP, respectively, compared to 5.92 ± 0.25 µg/mL in the absence of AuNC@ATP (Figure 6). We conclude that AuNC@ATP is a multifunctional platform. Therefore, apart from being used to eradicate or prevent the growth of bacteria, AuNC@ATP may act as an anti-virulence agent that can attenuate P. aeruginosa PYO production. Several infections associated with the cytotoxic effects of PYO have been reported [77]. For instance, PYO increases interleukin-8 expression by human airway epithelial cells [78] and mediates tissue damage leading to necrosis during lung infection [79]. The FDA has recently approved five anti-virulence drugs, including two immunoglobulins (BabyBIG and BAT for Clostridium botulinum) and three monoclonal antibodies (raxibacumab and obiltoxaximab for Bacillus anthracis and bezlotoxumab for C. difficile) [80]. Along this line, our data show that although AuNC@ATP is an antimicrobial nanocluster, it could also be used as an anti-virulence drug for P. aeruginosa. In future investigations, it might be possible to use AuNC@ATP for anti-virulence therapy. The rationale is that when virulence traits are suppressed, the bacteria are rendered benign and are more easily cleared by the host’s immune system [81].
Figure 6. P. aeruginosa fails to produce pyocyanin in the presence of a sub-lethal dose of AuNC@ATP.

(A) Pyocyanin production by P. aeruginosa (PA14) in lysogeny broth (LB) with AuNC@ATP (N = 3). The inset shows the chemical structure of pyocyanin. After centrifugation, the pyocyanin was collected, and the optical density was measured at 520 nm (OD520 nm). The pyocyanin concentration was determined by multiplying the OD520 values by 17.072, and the results were expressed in µg/mL. (B) Picture of extracted pyocyanin converted colour to red with HCl.
Bacteria do not develop resistance to AuNC@ATP and could prevent sub-lethal antibiotic treatment from inducing multidrug resistance.
An ideal antibiotic would show low potential for resistance development alongside an ability to eradicate persister cells. To address resistance rates more quantitatively, we serial-passaged two biologically independent cultures of P. aeruginosa from (PAO1) in sub-lethal concentrations of AuNC@ATP, as well as two control antibiotics: Tobramycin (targeting protein synthesis) and Ofloxacin (targeting DNA gyrase). During 21 passages, we successfully isolated mutants resistant to all the control antibiotics, showing no AuNC@ATP-resistant mutants emerged (Figure 7a). For Tobramycin and Ofloxacin, the resistance gradually increased throughout the experiment with more than 30-fold and 1500-fold increase in MIC, respectively. While the resistance level remained constant for AuNC@ATP, indicating that P. aeruginosa did not acquire partial resistance to AuNC@ATP. Of note, PAO1 resistance gradually increased after 20 passages in cultivation media containing subinhibitory concentrations of silver nanoparticles [82]. This further supports the conclusion that AuNC@ATP is a new class of antimicrobial nanoclusters with a distinct mechanism of action.
Figure 7. Bacteria do not develop resistance to AuNC@ATP and prevent sub-lethal antibiotic treatment from inducing resistance.
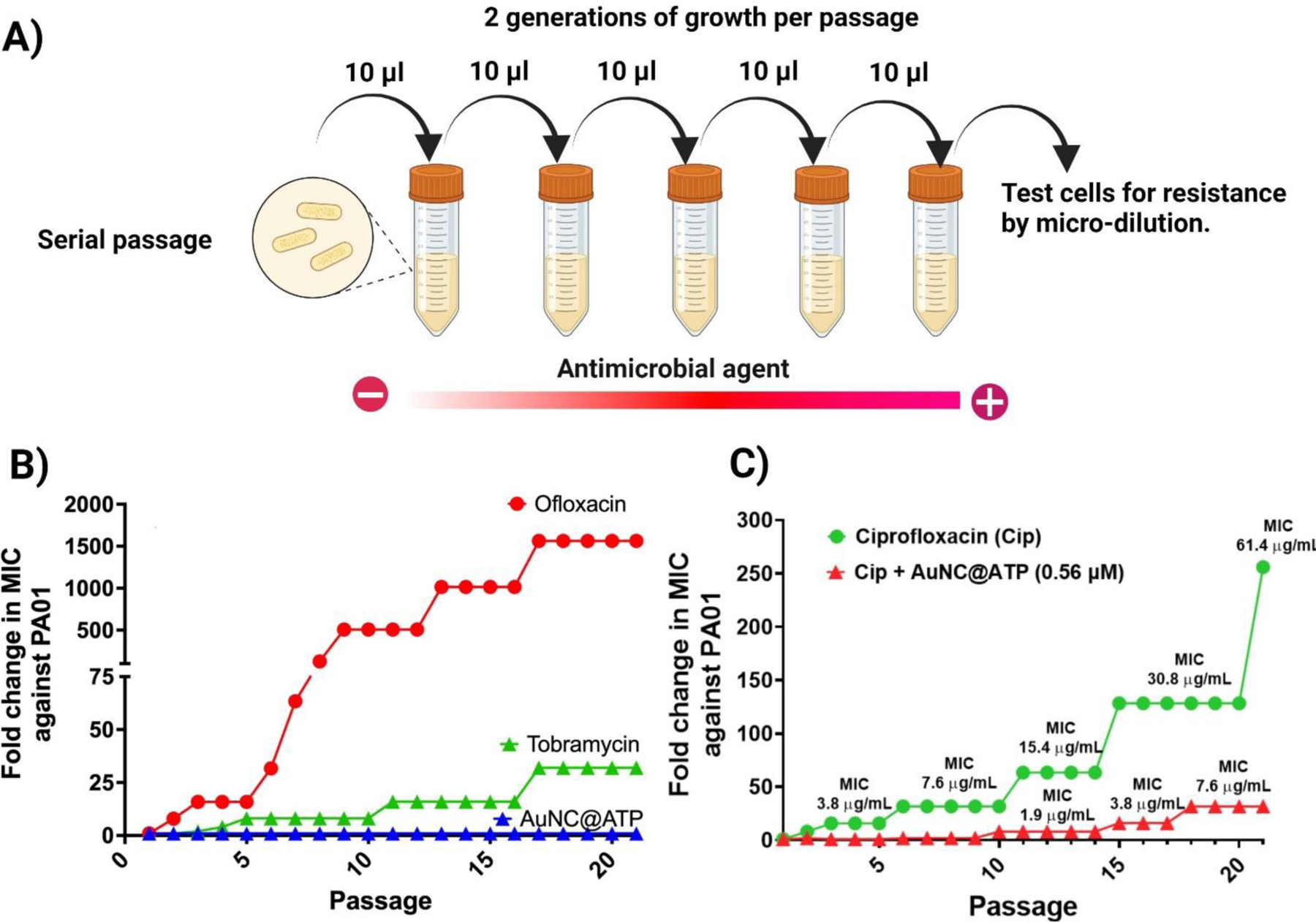
(A) Schematic showing the serial passage experiment. The fold change in minimum inhibitory concentration (MIC) was measured as the ratio between the MIC at passage n/ initial MIC. (B) Resistance development of susceptible PAO1 during serial passaging in sub-MIC dosing of Ofloxacin, Tobramacy and AuNC@ATP following 21 passages (1 passage per 24 h). (C) Resistance development of susceptible PAO1 during serial passaging in sub-MIC dosing of Ciprofloxacin in the absence or presence of AuNC@ATP (0.56 μM).
It is suggested that persister cells are the leading cause of the emergence of genetic antimicrobial resistance [14]. Given that AuNC@ATP targets persister cells over their metabolically active counterpart, it could serve as a valuable research tool to kill the multidrug-tolerant subpopulation within an isogenic culture of bacteria genetically susceptible to antibiotics in order to demonstrate that resistance emerges from a multidrug-tolerant subpopulation of bacterial cells. To test this idea, we repeated our serial passaging study with two biologically independent cultures of PAO1. Throughout 21 passages, PAO1 quickly acquires resistance to Ciprofloxacin with a more than 250-fold increase in MIC (Figure 7b). However, PAO1 resistance to Ciprofloxacin drastically decreases in the presence of AuNC@ATP (1.12 µM), which has been demonstrated earlier in this work to selectively kills persister cells (i.e., > 2-log reduction in CFU/ml vs over their metabolically active counterpart) (Figure 7b). This result supports our conclusion that AuNC@ATP may be a new research tool to demonstrate the association between persister cells and antibiotic resistance development. In addition, this study supports evidence from previous observations that persister cells promote antibiotic resistance rates [14, 73].
AuNC@ATP prevents the cross-resistance that triggers the emergence of superbugs upon exposure to the sub-lethal dose of antibiotics.
Superbugs are strains of bacteria that are resistant to several types of antibiotics. Cross-resistance refers to the situation where one antibiotic confers resistance to other drugs within an antibiotic class or to unrelated drugs with different mechanisms of action [83]. Cross-resistance to β-lactam antibiotics is observed in bacterial populations that evolve during exposure of P. aeruginosa to sublethal concentrations of Ciprofloxacin [84]. We demonstrate that a strategy addressing the persister cells is a promising approach to fight against the emergence of multidrug-resistant superbugs. Thanks to the AuNC@ATP, we can now prove that the presence of persister cells within the isogenic culture of bacteria genetically susceptible to Ciprofloxacin is the leading cause of cross-resistance that triggers the emergence of superbugs and their metabolically active counterpart play a minor role. We demonstrated this by examining the antibiotic resistance profile of PAO1 isolate after 21 passages in media containing subinhibitory concentrations of Ciprofloxacin without (i.e., PAO1Cip21) and with AuNC@ATP (i.e., PAO1Cip21-AuNC@ATP).
We found that PAO1Cip21 is multidrug-resistant to carbapenems, a class of atypical β-lactam antibiotics (Meropenem and Imipenem), aminoglycosides (Tobramycin and Amikacin), polymyxins (Colistin and Polymyxin-B), aztreonam and cephalosporins (Cefepime and Ceftazidime) as evidenced by the decrease in inhibition zone diameter test for antimicrobial activity compared the ancestor PAO1 (Figure 8). Therefore, it can be assumed that the P. aeruginosa superbug strain (PAO1Cip21) emerges during exposure to sublethal concentrations of Ciprofloxacin. In contrast, PAO1Cip21-AuNC@ATP show an inhibition zone diameter similar to that observed with the ancestor PA01 (Figure 8). Thus, it can be suggested that persister cells are the leading cause of the emergence of P. aeruginosa superbug strain during exposure to sublethal concentrations of Ciprofloxacin. Furthermore, this study confirms that persister cells are associated with enhanced antibiotic resistance development from fluoroquinolone [85]. Cumulatively, the data in this section lays the groundwork for developing novel nano-antibiotic adjuvants such as AuNC@ATP as it would stop the development of superbugs with the benefit of prolonging the lifespan of current antibiotics.
Figure 8. AuNC@ATP prevents the cross-resistance triggers by the sub-lethal fluoroquinolones.

(A) Schematic showing the disk diffusion test used to determine the antimicrobial susceptibility profile of PAO1 isolate after 21 passages in media containing subinhibitory concentrations of Ciprofloxacin without (PAO1Cip21) and with AuNC@ATP (PAO1Cip21-AuNC@ATP). (B) Cross-resistance of PAO1Cip21 and PAO1Cip21-AuNC@ATP against different antipseudomonal antibiotics. The vertical axis labels indicate the antibiotic tested for cross-tolerance, and the horizontal axis labels indicate the fold change in the inhibition zone compared to the susceptible P. aeruginosa (PAO1 ancestor).
Nonclinical safety and toxicology of AuNC@ATP after intravenous and intraperitoneal injection with multiple doses.
The quantitative parameters of toxicity of AuNC@ATP in mice are presented in Table 1. The median lethal dose (LD50) for a single injection of AuNC@ATP administered intravenously (IV) and intraperitoneally (IP) to mice was 205.12 and 346 mg/kg, respectively. For comparison, The LD50 for a single injection of tobramycin sulphate administered by IV and IP to mice was 77 and 262 mg/kg, respectively [86]. Furthermore, the highest tolerated dose of AuNC@ATP that can be administered intravenously to mice without causing any signs of toxicity (i.e., IV-MTD) was 38.19 mg/kg. The IP-MTD was estimated to be between 95mg/kg (no death) and 195 mg/kg (1/10 death mice) for intraperitoneal injection. No clinical laboratory signs of toxicity were found after IP injection of AuNC@ATP 3 times/day for 14 days at a dose equivalent to the IV-MTD. As shown in Figure 9, we observe that AuNC@ATP did not affect any measured hematology and clinical chemistry parameters. Since AuNCs are cleared from the body through the liver and kidneys [87, 88], one particular interest has been focused on liver and kidney toxicity. Changes in alanine transferase (ALT), aspartate transferase (AST), and total bilirubin (TBIL) levels typically indicate liver injury. The changes in creatinine (CR) and blood urea nitrogen (BUN) levels are associated with kidney injury. No abnormal liver and kidney function-related parameters were observed compared to the control group (PBS) (Figure 9). Cumulatively, the data in this section demonstrates the lack of toxicity of AuNC@ATP. It should be pointed out that AuNC@ATP causes hemolysis in the test tube, but IV-administered AuNC@ATP at an MTD dose (38 gm/kg) does not support this in vitro hemolysis study. The mice have sacrificed 24 hrs after the AuNC@ATP injection. Red blood cell levels show no difference between the PBS-treated group (control) and the AuNC@ATP-treated group (Supplementary Figure S11).
Table 1.
Values of the median lethal dose and highest tolerated dose for a single administration of AuNC@ATP to adult mice.
| Route of administration | Single dose injection of AuNC@ATP | |
|---|---|---|
| Median lethal dose (LD50) | Highest tolerated dose (MTD) | |
| Intravenous injection (IV) | 205.12 mg/kg | 38.19 mg/kg |
| Intraperitoneal injection (IP) | 345.67 mg/kg | 95 to 195 mg/kg |
Figure 9. Multiple doses administration of AuNC@ATP is not toxic to mice.
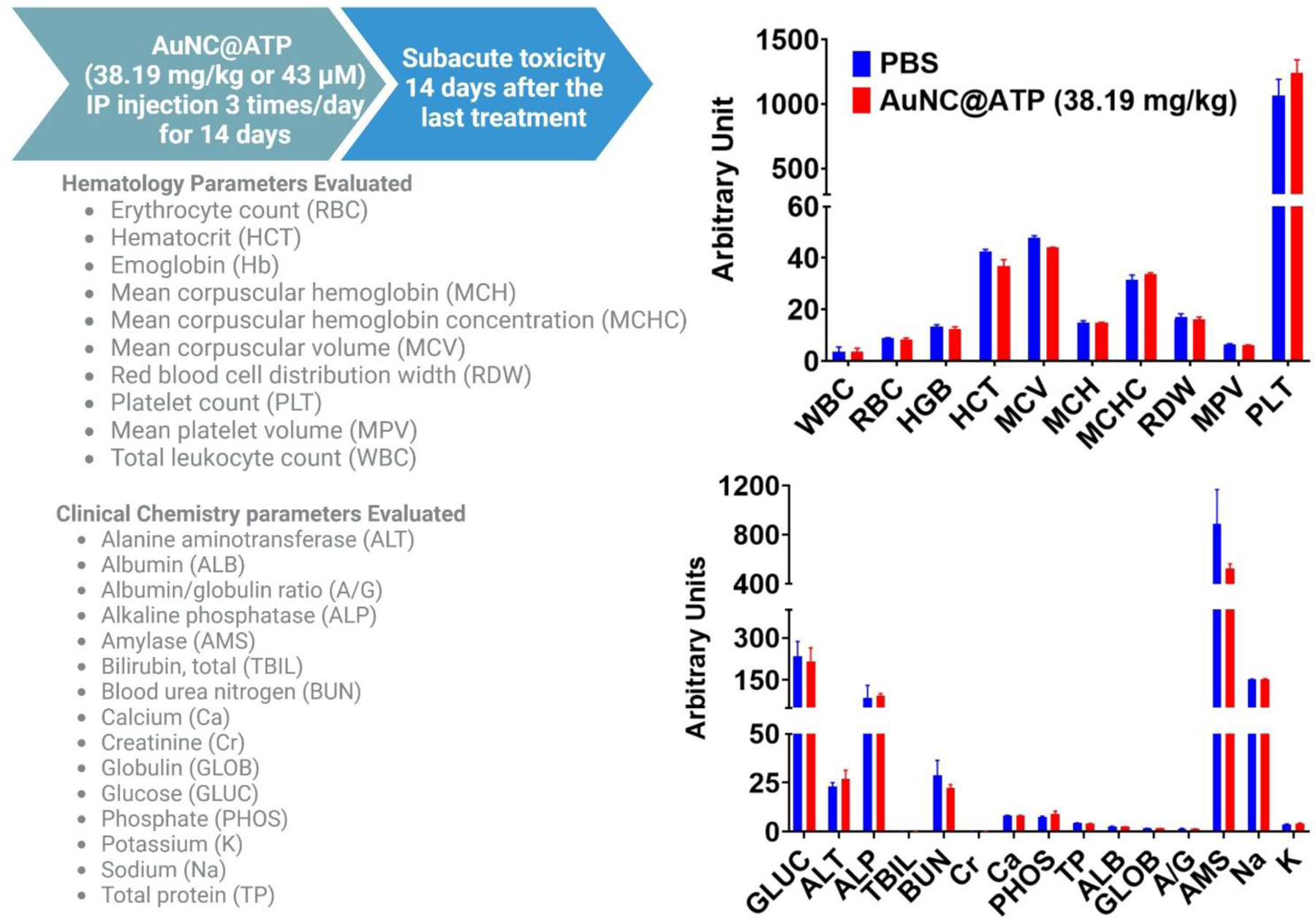
Effect of AuNC@ATP on hematology and clinical chemistry parameters at 14 days post-treatment at a dose of 38.19 mg/kg administered intraperitoneally (IP) three times a day for 14 days. The parameters evaluated are listed in the figure. Group ten mice (five female and five male) were used. Phosphate-buffered saline (PBS) was used as vehicle control.
CONCLUSIONS
To summarize, we engineered gold nanoclusters coated with adenosine triphosphate (AuNC@ATP) as a benchmark nanocluster that eradicates persister cells over exponential growth bacterial cells and prove that the low metabolic activity of persister cells can be leveraged for eradication and modulation of pseudomonal virulence. We also demonstrate that AuNC@ATP could serve as a valuable research tool to probe the association between persister cells and the development of antibiotic resistance. Finally, the repeated dose toxicity studies in mice demonstrate its potential safety. Cumulatively, these findings show the promise of AuNC@ATP to eradicate persister cell-driven infectious diseases.
EXPERIMENTAL SECTION
Synthesis of AuNC@ATP
Freshly prepared aqueous solutions of HAuCl4 (20 mM, 5 mL) and ATP (50 g, 50 mL) were mixed in water (790 mL). After that, an aqueous NaOH solution (1 M, 60 mL) was added to the mixture. Then the mixture was boiled for 5 minutes, and the AuNC@ATP solution was cooled to room temperature. After synthesis, the AuNC@ATP was collected by centrifugation (4000 g for 60 min) of AuNC@ATP in Pall Macrosep Advance centrifugal devices (membrane, MWCO = 3000). Finally, the AuNC@ATP was washed three times with deionized water by centrifugation. The resulting AuNC@CPP were lyophilized and dried entirely before further use.
AuNC@ATP characterization
Absorption spectra of AuNC@ATP were recorded in the visible domain of the electromagnetic spectrum (400–800 nm) using an absorption spectrophotometer (spectramMax M2, Molecular Devices, Downington, PA). Furthermore, transmission electron microscopy (TEM) images of AuNC@ATP were acquired to analyze the morphology and measure the core size. In addition, the zeta potential of the AuNC@ATP was measured using a Malvern Zetasizer Nano ZS at nanoComposix, San Diago, CA 92111.
Bacterial strains, growth media, and conditions
We used multidrug-resistant bacteria from the CDC & FDA Antibiotic Resistance Isolate Bank. In addition, the P. aeruginosa PA01 strain was obtained from ATCC, and the PA14 and PA14 (ΔClpXP) were obtained from Newman’s lab at the California Institute of Technology. In all experiments, bacterial cells were cultured in 10 mL of lysogeny broth (LB) at 37 °C and were aerated at 200 rpm in 50 mL plastic polypropylene tubes. Exponential phase cultures were prepared as follows: a stationary overnight culture was diluted 1 : 1000 in LB and incubated at 37 °C with aeration at 200 rpm until the optical density at 600 nm (OD600) = 0.3 was reached. The optical density at 600 nm (OD600) was read every 15 minutes in a microplate reader (spectramMax M2, Molecular Devices, Downington, PA) to generate growth curves.
Resistance development
Serial passage MICs were performed in 96-well microtiter panels. First, an aliquot of the well with the highest concentration permitting growth was taken and back diluted (1/100) in new media from inoculated microtiter panels. After overnight incubation at 37 °C, this suspension was diluted to a 0.5 McFarland standard turbidity and used to inoculate a new MIC panel, resulting in a final concentration of 1.5 × 106 CFU/ml. Panels were incubated according to CLSI guidelines, MICs were recorded, and the next inoculum was prepared from the well containing the highest drug concentration that identically allowed growth as described above. Twenty-one repeat passages were performed.
Susceptibility of bacteria using disc diffusion assay
Strains of PA01 (ancestor), PAO1 isolate after 21 passages in media containing subinhibitory concentrations of Ciprofloxacin without (i.e., PAO1Cip21) and with AuNC@ATP (i.e., PAO1Cip21-AuNC@ATP) were tested for susceptibility against a panel of antipseudomonal drugs using the disc diffusion method according to the Clinical Laboratory Standard Institute (CLSI) guidelines. In brief, glycerol stocks of each bacteria were streaked on LB plates and grown overnight at 37 °C. An inoculum was prepared by diluting several individual colonies in PBS with a 0.5 McFarland Standard turbidity. The inoculum was then spread with sterile cotton swabs on Mueller Hinton agar plates supplemented with 5% sheep blood. Discs containing 5 μg of antipseudomonal drugs were dispensed on the surface of the plate. After 24 h incubation at 37 °C, the inhibition zone was measured using a digital calliper.
Persister cells generation
Persister cells of the P. aeruginosa (PA14) were isolated by treating 250 ml of stationary phase cultures with Ofloxacin (415 µM final concentration). After 24 h treatment, the samples were washed with PBS, and then persister cells were concentred in 10 ml of PBS. The number of persister cells was estimated by serially diluting to determine the colony-forming unit per millilitre (CFU/ml). CCCP-induced persister cells were generated by pretreating P. aeruginosa (PA14) exponential cultures to CCCP (200 μg/mL) for 24 h at 37°C in LB.
Assessment of intracellular ATP level and total protein
The ATP levels of exponential and Ofloxacin-induced persisters cells were measured using a BacTiter Glo kit (Promega) according to the manufacturer’s instructions. According to the manufacturer’s instructions, the protein levels were determined using the bicinchoninic acid (BCA) assay (Thermo Scientific, Pierce).
Bactericidal activity against persister cells
All the bactericidal tests were performed on PBS without a carbon source to prevent persister cells from waking up from their low metabolic activity. Ofloxacin-induced persister cells were challenged with antimicrobial agents at the concentrations listed in the text. After the survival bacteria were washed 3× with PBS, the pellet was resuspended in 100 μL of PBS and then spread on an LB plate. The plate was incubated for 72 h at 37 °C before assessing growth. Next, AuNC@ATP was added to the solution of CCCP-induced persister cells and incubated for 24 h at 37°C. After the treatment, survival bacteria cells were assessed as described above.
In vivo cytotoxicity
Animal Treatment and Sample Collection. Stanford University’s Administrative Panel approved all animal work in Laboratory Animal Care. The 10−12 week-old BALB/c mice were purchased from Jackson Laboratories (Sacramento, CA) and housed in Stanford University’s animal resource facility according to standard guidelines in which food and water were provided ad libitum in a room maintained at 12 h dark/light cycles. Mice (Male = 5 each group and Female =5 each group) BALB/c mice were divided into two groups, including control (PBS) and AuNC@ATP. The treatments were administered intravenously (IV) or intraperitoneally (IP) at doses listed in the text. The AuNC@ATP was administered by IP 3 times daily for 14 consecutive days for the sub-acute toxicity study. Then mice were sacrificed 14 days after the last injection. Blood was collected for further investigation of serum chemistry and hematology. Blood samples were subjected to toxicity analysis. An inferior vena cava blood collection was performed at the sacrifice. Blood (150 μL) was placed in a K2 EDTA tube for hematological analysis, and the left blood sample was placed in a 1.5 mL Eppendorf tube for serum extraction. The serum was separated by centrifuging the blood to remove the cellular fraction for liver and renal function testing. It should be noted that mice were sacrificed 24 hrs after AuNC@ATP infection for the single dose IV acute toxicity.
Hemolysis assay in vitro
To examine for hemolysis, we mixed 0.3 mL of mice red blood cells (RBCs) suspension with 0.7 mL of AuNC@ATP in a 1.5 mL centrifuge tube by gentle pipetting to give the final desired concentrations of 4.5 µM in PBS. An equal volume of PBS and 0.1 % Triton X-100 were used as negative and positive controls of hemolysis, respectively. The mixtures were incubated at 37 °C for 24 h with mixing to minimize sedimentation before centrifugation at 12 000 rpm for 30 min unlysed RBCs. The absorbance of the lysed supernatant was then measured for its hemoglobin content.
Generation of intracellular reactive oxygen species (ROS)
DCFH-DA (2’,7’-dichlorofluorescein diacetate) dye was applied to test the intracellular ROS concentration, which could be cleaved by the intracellular non-specific esterase into the DCFH. DCFH, in the presence of ROS, would be further oxidized into fluorescent DCF (2’,7’-dichlorofluorescein). DCFH-DA in DMSO (10 µM) was added to the treated bacterial solution and further incubated at 37 °C, 200 rpm for 15 min. After that, the bacterial cells were centrifuged at 8000 g for 5 min, washed three times with PBS, and resuspended in ultrapure water to the original volume (1 mL). The microplate reader was used to measure the concentration of the produced DCF at the excitation/emission wavelength of 488/525 nm. In this experiment, the fluorescence intensity of DCF directly reflects the amount of ROS generation. The ROS amount was then normalized to the total cell number, which was reflected by the optical density at 600 nm (OD600). Finally, the relative ROS production level was calculated by normalizing the ROS level from the treated groups with the production level of the PBS-treated group.
Outer membrane permeability assay
After treatment with antimicrobial agents, the stationary phase culture of gram-negative bacteria was diluted with PBS to form a suspension with the optical density at 600 nm (OD600) = 0.5 and add 50 μl of 3 mg/ml 8-Anilino-1-naphthalene sulfonic acid (ANS). After equilibration for 30 min at 37 °C, the cells were washed with PBS by centrifugation (8000g for 5 min) and resuspended in fresh PBS solution (1 ml). Then the fluorescence intensity between 450–650 nm was measured with excitation at 380 nm.
Cytoplasmic membrane permeability assay
After treatment with antimicrobial agents, the stationary phase culture of gram-negative bacteria was diluted with PBS to form a suspension with the optical density at 600 nm (OD600) = 0.5 and add 5 μl of 1 mg/ml propidium iodide (PI dissolved in sterile deionized H2O). After equilibration for 30 min at 37 °C, the cells were washed with PBS by centrifugation (8000g for 5 min) and resuspended in fresh PBS solution (1 ml). Then the fluorescence intensity between 550–800 nm was measured with excitation at 530 nm.
Pyocyanin quantitation assay
Pyocyanin concentration was determined as described by Essar et al. [89]. First, an aliquot of the exponential growth culture of PA14 at the optical density at 600 nm (OD600) = 0.5 was taken and back diluted (1/100) in 50 mL of lysogeny broth (LB) in the absence and presence of sub-lethal doses of AuNC@ATP, incubated at 37 °C and were aerated at 200 rpm to maximize pyocyanin production. After 24h, the bacterial cells were removed from the cultures by centrifugation (8000g for 5 min). Then, pyocyanin was extracted from the supernatant by adding Chloroform to a total of 50% total volume. The mixture was vortexed vigorously for 30 seconds, and let the sample settled for 10 minutes to allow for the aqueous phase to separate. Next, the blueish Chloroform fraction was carefully transferred to the new tube. Then 0.1 N HCl was added to 20% total volume and vortexed vigorously for 30 seconds. After this step, the chloroform fraction turns from blue to clear, and the small aqueous fraction of added acid turns from clear to pink. Again, the sample settled for 10 minutes to allow for the aqueous phase to separate. Finally, the aqueous fraction was removed, and the absorbance at 520 nm (OD520) was measured. Concentrations, expressed as micrograms of pyocyanin produced per millilitre of culture supernatant, were determined by multiplying the OD520 by 17.072. Next, the concentration values were normalized to the cell density of each sample (OD600).
Periplasmic protein extraction and Western blotting
Periplasmic protein preparation from stationary phase E. Colie was done using the Spheroplasting by lysozyme and sucrose method [90]. Briefly, The 400 mL stationary phase culture bacterial pellet was treated with AuNC@ATP (4.5 µM finale concentration). After 24h, the bacterial cells were removed from the treatment by centrifugation (8000g for 5 min). Then, 0.5 mL of the bacterial pellet was resuspended in 1 mL of 30 mM Tris-HCl, pH 8, 20%sucrose, 4 mM EDTA, 0.5 mg/mL lysozyme and 1 mM PMSF. The mixture was incubated for 60 min in a 30ºC water bath with gentle shaking. After 2 min, MgCl2 was added at 10 mM final concentration. Finally, the suspension was centrifuged (11 000 x g for 15 min at 4ºC) to collect the periplasmic protein in the supernatant. The periplasmic proteins from the untreated control were extracted using the same procedure.
For Western blot analysis, the periplasmic proteins samples were run on Mini-PROTEAN TGX Stain-Free precast gels. Periplasmic proteins were loaded in 2 µg/ml with and without prior boiling at 95 ºC for 5 min. The reducing agent DTT was not used in the experiment. Following SDS-PAGE, proteins were transferred to trans blot turbo mini-size PVDF membrane (BioRad). Membranes were blocked by incubation in TBS-T (TBS with 0.1% (w/v) Tween 20) containing 3% BSA for 30 h at room temperature and incubated with the primary antibody (anti-OmpC) in TBS-T + 5% skimmed milk for 2 hrs at room temperature. After washing with TBS-T, blots were incubated with a secondary antibody (goat anti-rabbit) in TBS-T for 1 h at room temperature. Protein signals were visualized using Bio-Rad Universal Hood II Imaging System.
Statistical analysis
Data are represented as the mean ± s.d. Statistical analyses (details in figure legends) were calculated with GraphPad Prism Ver. 9 (GraphPad, San Diego, CA). A p-value of <0.05 was considered statistically significant.
Supplementary Material
Highlights.
The low metabolic activity of persister cells can be exploited for eradication over their metabolically active counterparts.
AuNC@ATP kills persister cells over exponential growth bacterial cells
Bacteria become more sensitive to AuNC@ATP when they enter a dormant state which would typically allow them to survive exposure to traditional antibiotics
Eradicating persister cells with AuNC@ATP in an isogenic culture of bacteria stops the emergence of superbug bacteria mediated by the sub-lethal dose of conventional antibiotics.
AuNC@ATP has antimicrobial activity attributable to the stress response that builds up from unfolded outer membrane proteins accumulation in the periplasm leading to the disruption of bacterial membranes by altering lipid homeostasis and asymmetry.
Acknowledgements
We acknowledge the support from the National Institute of Health, National Institute of Allergy and Infectious Diseases (R21-AI154097-01), Stanford’s Maternal and Child Health Research Institute, the Stanford SPARK translational medicine program, the Department of Otolaryngology, Head and Neck Surgery at Stanford University and the Stanford Initiative to Cure Hearing Loss including generous gifts from the Bill and Susan Oberndorf Foundation. In addition, this research was jointly funded by RNID (RNID T10), and the Fondation pour l’Audition (FPA RD 2020–12). Finally, we thank Professor Dianne k. Newman from the Biology and Bioengineering Division of the California Institute of Technology for sharing their PA14 and PA14 (ΔClpXP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHORSHIP STATEMENT
All persons who meet authorship criteria are listed as authors, and all authors certify that they have participated sufficiently in the work to take public responsibility for the content, including participation in the concept, design, analysis, writing, or revision of the manuscript. Furthermore, each author certifies that this material or similar material has not been and will not be submitted to or published in any other publication before its appearance in the Nanotoday.
Declaration of interests
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests:
We declare that L. A. B. and P. L. S. M. are listed inventors on a provisional patent regarding the antimicrobial activity described in this paper.
References
- [1].Murray CJ, Ikuta KS, Sharara F, Swetschinski L, Aguilar GR, Gray A, Han C, Bisignano C, Rao P, Wool E, The Lancet, 399 (2022) 629–655. [Google Scholar]
- [2].Larsson D, Flach C-F, Nature Reviews Microbiology, 20 (2022) 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bombaywala S, Mandpe A, Paliya S, Kumar S, Environmental Science and Pollution Research, 28 (2021) 24889–24916. [DOI] [PubMed] [Google Scholar]
- [4].Lopatkin AJ, Stokes JM, Zheng EJ, Yang JH, Takahashi MK, You L, Collins JJ, Nature microbiology, 4 (2019) 2109–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cohen NR, Lobritz MA, Collins JJ, Cell host & microbe, 13 (2013) 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brauner A, Fridman O, Gefen O, Balaban NQ, Nature Reviews Microbiology, 14 (2016) 320–330. [DOI] [PubMed] [Google Scholar]
- [7].Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K, FEMS microbiology letters, 230 (2004) 13–18. [DOI] [PubMed] [Google Scholar]
- [8].Fisher RA, Gollan B, Helaine S, Nature Reviews Microbiology, 15 (2017) 453–464. [DOI] [PubMed] [Google Scholar]
- [9].Lewis K, Nature Reviews Microbiology, 5 (2007) 48–56. [DOI] [PubMed] [Google Scholar]
- [10].Lewis K, Annual review of microbiology, 64 (2010) 357–372. [DOI] [PubMed] [Google Scholar]
- [11].Santa Maria PL, Kaufman AC, Bacacao B, Thai A, Chen X, Xia A, Cao Z, Fouad A, Bekale LA, Otology & Neurotology, 42 (2021) e1263–e1272. [DOI] [PubMed] [Google Scholar]
- [12].Lewis K, Bacterial biofilms, (2008) 107–131.
- [13].Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S, Science, 305 (2004) 1622–1625. [DOI] [PubMed] [Google Scholar]
- [14].Windels EM, Michiels JE, Fauvart M, Wenseleers T, Van den Bergh B, Michiels J, The ISME journal, 13 (2019) 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lewis K, Shan Y, Science, 355 (2017) 796-796. [DOI] [PubMed] [Google Scholar]
- [16].Levin-Reisman I, Ronin I, Gefen O, Braniss I, Shoresh N, Balaban NQ, Science, 355 (2017) 826–830. [DOI] [PubMed] [Google Scholar]
- [17].Theuretzbacher U, Outterson K, Engel A, Karlén A, Nature Reviews Microbiology, 18 (2020) 275–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zheng K, Xie J, ACS nano, 14 (2020) 11533–11541. [DOI] [PubMed] [Google Scholar]
- [19].Xie Y, Zhang Q, Zheng W, Jiang X, ACS Applied Materials & Interfaces, 13 (2021) 35306–35314. [DOI] [PubMed] [Google Scholar]
- [20].Zhao X, Tang H, Jiang X, ACS nano, 16 (2022) 10066–10087. [DOI] [PubMed] [Google Scholar]
- [21].Zheng W, Jia Y, Zhao Y, Zhang J, Xie Y, Wang L, Zhao X, Liu X, Tang R, Chen W, Nano Letters, 21 (2021) 1992–2000. [DOI] [PubMed] [Google Scholar]
- [22].Li J, Cha R, Zhao X, Guo H, Luo H, Wang M, Zhou F, Jiang X, Acs Nano, 13 (2019) 5002–5014. [DOI] [PubMed] [Google Scholar]
- [23].Zheng K, Setyawati MI, Leong DT, Xie J, Bioactive materials, 6 (2021) 941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zheng Y, Liu W, Qin Z, Chen Y, Jiang H, Wang X, Bioconjugate Chemistry, 29 (2018) 3094–3103. [DOI] [PubMed] [Google Scholar]
- [25].Tang M, Zhang J, Yang C, Zheng Y, Jiang H, Frontiers in Chemistry, 8 (2020) 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yougbare S, Chang T-K, Tan S-H, Kuo J-C, Hsu P-H, Su C-Y, Kuo T-R, International journal of molecular sciences, 20 (2019) 2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pang Z, Li Q, Jia Y, Yan W, Qi J, Guo Y, Hu F, Zhou D, Jiang X, Chemical science, 12 (2021) 14871–14882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zheng K, Setyawati MI, Leong DT, Xie J, ACS nano, 11 (2017) 6904–6910. [DOI] [PubMed] [Google Scholar]
- [29].Zheng Y, Wei M, Wu H, Li F, Ling D, Journal of Nanobiotechnology, 20 (2022) 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zheng K, Xie J, Accounts of Materials Research, 2 (2021) 1104–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wu Y, Vulić M, Keren I, Lewis K, Antimicrobial agents and chemotherapy, 56 (2012) 4922–4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang T, El Meouche I, Dunlop MJ, Scientific reports, 7 (2017) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kohanski MA, DePristo MA, Collins JJ, Molecular cell, 37 (2010) 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Meng J, Gao Y, Li W, Wang J, Chen X, Talanta, 234 (2021) 122618. [DOI] [PubMed] [Google Scholar]
- [35].Zheng K, Setyawati MI, Leong DT, Xie J, Chemistry of Materials, 30 (2018) 2800–2808. [Google Scholar]
- [36].Allison KR, Brynildsen MP, Collins JJ, Nature, 473 (2011) 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cao Z, Chen X, Chen J, Xia A, Bacacao B, Tran J, Sharma D, Bekale LA, Santa Maria PL, Nanoscale, 14 (2022) 10016–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Amendola V, Pilot R, Frasconi M, Maragò OM, Iatì MA, Journal of Physics: Condensed Matter, 29 (2017) 203002. [DOI] [PubMed] [Google Scholar]
- [39].Nikaido H, Microbiology and molecular biology reviews, 67 (2003) 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Lundstedt E, Kahne D, Ruiz N, Chemical Reviews, 121 (2020) 5098–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Guest RL, Rutherford ST, Silhavy TJ, Trends in Microbiology, 29 (2021) 334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Malinverni JC, Silhavy TJ, Proceedings of the National Academy of Sciences, 106 (2009) 8009–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Low W-Y, Thong S, Chng S-S, Proceedings of the National Academy of Sciences, 118 (2021) e2110055118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sutterlin HA, Shi H, May KL, Miguel A, Khare S, Huang KC, Silhavy TJ, Proceedings of the National Academy of Sciences, 113 (2016) E1565–E1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Stryer L, Journal of molecular biology, 13 (1965) 482–495. [DOI] [PubMed] [Google Scholar]
- [46].Haynes DH, Staerk H, The Journal of Membrane Biology, 17 (1974) 313–340. [DOI] [PubMed] [Google Scholar]
- [47].Young K, Silver LL, Journal of Bacteriology, 173 (1991) 3609–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Falchi FA, Maccagni EA, Puccio S, Peano C, De Castro C, Palmigiano A, Garozzo D, Martorana AM, Polissi A, Deho G, Journal of bacteriology, 200 (2018) e00487–00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Malojčić G, Andres D, Grabowicz M, George AH, Ruiz N, Silhavy TJ, Kahne D, Proceedings of the National Academy of Sciences, 111 (2014) 9467–9472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sabnis A, Hagart KL, Klöckner A, Becce M, Evans LE, Furniss RCD, Mavridou DA, Murphy R, Stevens MM, Davies JC, Elife, 10 (2021) e65836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Manioglu S, Modaresi SM, Ritzmann N, Thoma J, Overall SA, Harms A, Upert G, Luther A, Barnes AB, Obrecht D, Nature communications, 13 (2022) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ma B, Fang C, Zhang J, Wang M, Luo X, Hou Z, Bio-protocol, 10 (2020) e3548-e3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Völzing KG, Brynildsen MP, MBio, 6 (2015) e00731–00715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Khondker A, Rheinstädter MC, Communications Biology, 3 (2020) 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Vincent S, Glauner B, Gutmann L, Antimicrobial agents and chemotherapy, 35 (1991) 1381–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kohanski MA, Dwyer DJ, Collins JJ, Nature Reviews Microbiology, 8 (2010) 423–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang L, Li S, Yin J, Yang J, Li Q, Zheng W, Liu S, Jiang X, Nano Letters, 20 (2020) 5036–5042. [DOI] [PubMed] [Google Scholar]
- [58].Kulp A, Kuehn MJ, Annual review of microbiology, 64 (2010) 163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Mitchell AM, Silhavy TJ, Nature Reviews Microbiology, 17 (2019) 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Konovalova A, Grabowicz M, Balibar CJ, Malinverni JC, Painter RE, Riley D, Mann PA, Wang H, Garlisi CG, Sherborne B, Proceedings of the National Academy of Sciences, 115 (2018) E6614–E6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Prajapati JD, Kleinekathöfer U, Winterhalter M, Chemical reviews, 121 (2021) 5158–5192. [DOI] [PubMed] [Google Scholar]
- [62].Rigel NW, Silhavy TJ, Current opinion in microbiology, 15 (2012) 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hussain S, Peterson JH, Bernstein HD, Mbio, 12 (2021) e01696–01621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rocque WJ, McGroarty EJ, Biochemistry, 28 (1989) 3738–3743. [DOI] [PubMed] [Google Scholar]
- [65].Sikdar R, Peterson J, Anderson D, Bernstein H, 2017.
- [66].Ou X, Lao Y, Xu J, Wutthinitikornkit Y, Shi R, Chen X, Li J, JACS Au, 1 (2021) 1766–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Allen WJ, Phan G, Waksman G, Advances in Protein Chemistry and Structural Biology, 78 (2009) 51–97. [DOI] [PubMed] [Google Scholar]
- [68].Weiner JH, Li L, Biochimica et Biophysica Acta (BBA)-Biomembranes, 1778 (2008) 1698–1713. [DOI] [PubMed] [Google Scholar]
- [69].Patel A, Malinovska L, Saha S, Wang J, Alberti S, Krishnan Y, Hyman AA, Science, 356 (2017) 753–756. [DOI] [PubMed] [Google Scholar]
- [70].Sridharan S, Kurzawa N, Werner T, Günthner I, Helm D, Huber W, Bantscheff M, Savitski MM, Nature communications, 10 (2019) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Sarkar S, Mondal J, The Journal of Physical Chemistry B, 125 (2021) 7717–7731. [DOI] [PubMed] [Google Scholar]
- [72].Wainwright J, Hobbs G, Nakouti I, Archives of Microbiology, 203 (2021) 5899–5906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Manuse S, Shan Y, Canas-Duarte SJ, Bakshi S, Sun W-S, Mori H, Paulsson J, Lewis K, PLoS biology, 19 (2021) e3001194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Yeom J, Groisman EA, Science Signaling, 14 (2021) eabc4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Rezzoagli C, Granato ET, Kümmerli R, Journal of medical microbiology, 69 (2020) 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bhargava N, Sharma P, Capalash N, Infection and immunity, 82 (2014) 3417–3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sultan M, Arya R, Kim KK, International Journal of Molecular Sciences, 22 (2021) 12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Denning GM, Wollenweber LA, Railsback MA, Cox CD, Stoll LL, Britigan BE, Infection and immunity, 66 (1998) 5777–5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Lau GW, Ran H, Kong F, Hassett DJ, Mavrodi D, Infection and immunity, 72 (2004) 4275–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ogawara H, The Journal of Antibiotics, 74 (2021) 24–41. [DOI] [PubMed] [Google Scholar]
- [81].Allen RC, Popat R, Diggle SP, Brown SP, Nature Reviews Microbiology, 12 (2014) 300–308. [DOI] [PubMed] [Google Scholar]
- [82].Panáček A, Kvítek L, Smékalová M, Večeřová R, Kolář M, Röderová M, Dyčka F, Šebela M, Prucek R, Tomanec O, Nature nanotechnology, 13 (2018) 65–71. [DOI] [PubMed] [Google Scholar]
- [83].El Kary N, El Rassy E, Azar N, Choucair J, American Journal of Infection Control, 44 (2016) 1736–1737. [DOI] [PubMed] [Google Scholar]
- [84].Jørgensen KM, Wassermann T, Jensen PØ, Hengzuang W, Molin S, Høiby N, Ciofu O, Antimicrobial agents and chemotherapy, 57 (2013) 4215–4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Barrett TC, Mok WW, Murawski AM, Brynildsen MP, Nature communications, 10 (2019) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Gol’dberg L, Filippos’ iants S, Belova I, Vertogradova T, Shepelevtseva N, Antibiotiki, 24 (1979) 60–67. [PubMed] [Google Scholar]
- [87].Tsoi KM, MacParland SA, Ma X-Z, Spetzler VN, Echeverri J, Ouyang B, Fadel SM, Sykes EA, Goldaracena N, Kaths JM, Nature materials, 15 (2016) 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Loynachan CN, Soleimany AP, Dudani JS, Lin Y, Najer A, Bekdemir A, Chen Q, Bhatia SN, Stevens MM, Nature nanotechnology, 14 (2019) 883–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Essar DW, Eberly L, Hadero A, Crawford I, Journal of bacteriology, 172 (1990) 884–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Imperi F, Ciccosanti F, Perdomo AB, Tiburzi F, Mancone C, Alonzi T, Ascenzi P, Piacentini M, Visca P, Fimia GM, Proteomics, 9 (2009) 1901–1915. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.