

Abstract
The growing discovery and development of novel radiopharmaceuticals and radiolabeling methods requires an increasing capacity for radiochemistry experiments. However, such studies typically rely on radiosynthesizers designed for clinical batch production rather than research, greatly limiting throughput. Two general solutions are being pursued to address this: developing new synthesis optimization algorithms to minimize how many experiments are needed, and developing apparatus with enhanced experiment throughput. We describe here a novel high-throughput system based on performing arrays of droplet-based reactions at 10 μL volume scale in parallel. The automatic robotic platform can perform a set of 64 experiments in ~3 h (from isotope loading to crude product, plus sampling onto TLC plates), plus ~1 h for off-line radio-TLC analysis and radioactivity measurements, rather than the weeks or months that would be needed using a conventional system. We show the high repeatability and low crosstalk of the platform and demonstrate optimization studies for two 18F-labeled tracers. This novel automated platform greatly increases the practicality of performing arrays of droplet reactions by eliminating human error, vastly reducing tedium and fatigue, minimizing radiation exposure, and freeing up radiochemist time for other intellectually valuable pursuits.
Keywords: Microreactor, Automation, High-throughput Radiochemistry, Droplet Radiochemistry, Synthesis Optimization
Graphical Abstract
Introduction
Positron emission tomography (PET) is a non-invasive imaging modality that uses trace amounts of radiolabeled compounds to image specific biochemical processes within living subjects with high sensitivity and specificity1,2. To investigate different biological processes, thousands of different radiotracers have been developed3,4, and as new biological targets are discovered, there is ongoing need to develop tracers to image these targets. Currently, it can take many years between new target discovery and the development of a useful imaging tracer, and even longer for translation into the clinic. Though there are several factors in this timeline, the difficulty in synthesizing novel radiolabeled compounds with sufficient reliability and yield at each stage of development is a significant bottleneck.
The majority of available radiosynthesizers are designed to safely and automatically perform routine large-scale batch production of radiotracers5,6. Several characteristics of these systems are poorly suited to early stage radiotracer development and synthesis optimization. For example, the need to wait for decay of residual radioactivity within the system between experiments (e.g. overnight for F-18) severely limits experimental throughput. Each experiment may provide a limited amount of data, unless manual interventions are performed to make measurements of radioactivity or radiochemical composition at multiple stages throughout the synthesis process. Additionally, the typical reactor size requires relatively large quantities of expensive precursors (e.g., 1-10 mg for 1 mL reaction volume) per experiment. Finally the high value of these systems in service of clinical production often limits their availability for research purposes.
A variety of approaches are being developed to overcome these limitations. One strategy is to try to reduce the number of experiments needed to achieve the desired optimization goals. Using a design of experiments (DoE) approach, which helps to identify the most critical reaction parameters (factors), Bowden et al. showed more than 2x improvement in experimental efficiency for optimization of copper-mediated radiofluorination of arylstannanes7. Machine learning approaches are also being developed to enable synthesis optimization through reduced numbers of experiments8,9.
Another strategy is to modify radiochemistry hardware or experimental approaches in order to increase throughput and decrease experimental costs. For example, Zhang et al. leveraged the high sensitivity of LC-MS/MS to detect ultra-low amounts of product when performing reactions in conventional radiosynthesizers using nanomolar concentrations of non-radioactive isotopes (e.g. [19F]fluoride instead of [18F]fluoride)10. The low concentrations simulate the typical concentrations encountered in 18F-radiochemistry, and the authors observed a good correlation between the synthesis performance of MDL100907 when using F-19 or F-18, enabling increased throughput by avoiding the waiting time for radioactive decay, and finding conditions that could be directly translated to a conventional synthesizer. However, each data point still consumes a full batch quantity of precursor and other reagents.
In another approach, Laube et al. reported performing >50 vial-based reactions per day (25-50 μL scale) from a single batch of [18F]fluoride by using multi-vial heating blocks to carry out groups of simultaneous reactions to investigate the syntheses of [18F]FDG and a celecoxib analog11. This approach both increases throughput and decreases reagent usage, but required extensive manual handling, and there is a chance that some degree of re-optimization would be needed when translating the optimal conditions to a larger sized vial in a conventional radiosynthesizer12.
Flow-chemistry methods, where the precursor and radioactive material are mixed and flowed through a heated reactor in a continuous fashion, have been used to perform reagent-efficient optimization (generally 10-40 μL range) but in a more automated fashion. Using the Advion Nanotek capillary-based synthesizer, investigators have shown the possibility to sequentially perform dozens of optimization reactions per day from a single batch of radioisotope to conveniently explore the influence of temperature or flow rate (which affects reagent ratios, residence time, or concentration)13. Scale-up of optimal conditions is then achieved by scaling up the volumes (i.e. running the flow system for a longer time). However, some reaction parameters (e.g. reaction solvent, or the conditions for azeotropic drying of [18F]fluoride, which are done outside of the flow system) cannot be investigated in a high-throughput manner. In a similar approach, our lab has previously experimented with polydimethylsiloxane (PDMS) microfluidic chips for generating mixtures of reagents (~120 nL each) with programmable composition and pH for optimization of labeling of antibody fragments with the prosthetic group N-succinimidyl-4-[18F]fluorobenzoate ([18F]SFB)14,15, but these devices and studies were limited to room temperature aqueous conditions.
We have recently reported an approach for reagent-efficient high-throughput reaction optimization in which up to 64 reactions could be performed simultaneously in the form of ~10 μL droplets trapped on arrays of hydrophilic sites patterned on Teflon-coated silicon “chips”16,17. Because all steps are performed at each reaction site, any conditions of the overall synthesis process (drying conditions, base, precursor, and other reagents amounts, reaction volume, solvent, temperature, and duration) can be studied in a high-throughput manner. Once the optimal conditions are found, the conditions can be directly transferred to production via the use of an automated droplet radiosynthesizer18. Droplet-based radiochemistry systems have successfully been used to synthesize a wide range of 18F-labeled tracers including [18F]FDG19, [18F]Fallypride16,17, [18F]FET20, [18F]FDOPA21, [18F]FBB20, [18F]Flumazenil17, [18F]PBR0617, [18F]FEPPA17, [18F]FPEB22, and [18F]AMBF3-TATE23, with several demonstrated at the scale of one or more clinical doses20,24.
While the high-throughput droplet radiochemistry technique has been used to perform hundreds of experiments per week17, it requires a large amount of manual pipetting operations to add reagents and collect and analyze crude reaction products. Experiments are thus very tedious and prone to human error. To address these factors, and to minimize radiation exposure, we developed a fully-automated robotic platform for optimization. It automatically performs all of the liquid transfer operations and system control, including delivering isotope and reagents to reaction sites, performing evaporations or reactions, collecting products into individual reservoirs, and spotting crude samples onto thin-layer chromatography (TLC) plates (e.g. for rapid multi-lane radio-TLC analysis25). This new platform has the potential to increase the accessibility and throughput of high-throughput radiochemistry. In this paper, we describe the design, characterization, and proof-of-concept demonstrations of the robotic platform.
Methods
Robotic System
The overall system design is shown in Figure 1. With a size of 63.5 x 40.6 x 55.9 cm3 (W x H x D), it can fit within a small mini-cell (68 x 50 x 61 cm3 interior; Von Gahlen, Zevenaar, Netherlands). The system consists of three main elements: a work area (where microfluidic multi-reaction chips are operated, where reagents and collected products are stored, and where TLC plates are spotted), a fluidics head with multiple piezoelectric dispensers for reagent dispensing and a pipetting system for liquid transfers, and an XYZ motion gantry to move the fluidics head around the work area.
Figure 1:

(A) 3D rendering of the optimization platform showing the geometry and major components. (B) Photograph of the system inside a minicell. The fluidics head is shown in the inset to illustrate the piezoelectric reagent dispensers and pipette system.
Work Area
The work area (Figure 2A) consists of a multi-heater platform for operating four multi-reaction chips simultaneously with independent temperature control16,17 (Figure 2B), four plate nests that hold standard microwell plates, a pipette tip remover, and a priming sensor. Chips were aligned to the heaters with the aid of Delrin walls on two sides of each heater. Typical plate nest configuration was: (i) 384-position pipette tip rack (epT.I.P.S. 384, Eppendorf, Hamburg, Germany), (ii) a 96-well plate (Costar 3363, Corning Inc. Corning, NY, USA) containing reagents (e.g. precursors, dilution series), (iii) a 96-position strip-well plate (TRC9601 plate, TLS0801 stripwells, Bio-Rad Laboratories, Inc. Hercules, CA, USA) for collection of crude products, and (iv) a custom TLC plate holder with a laddered design to accommodate eight 50 mm wide TLC plates for parallel separation of 8 samples (0.5 μL spotted at 4.5 mm pitch)25 (Figure 2C). The infrared (IR) liquid priming sensor (OCB350L250Z, Optek-Danulat GmbH, Essen, Germany) was used to ensure the piezoelectric dispensers and pipetting system (and associated tubing) are fully filled with reagent or water, respectively. Pipette tips were removed with a forked tool and collected in a waste container (Supporting Information Section S1.1).
Figure 2:

(A) Overview of the system work area showing major components of the system. (B) Close-up photograph of the heaters with chips installed for 64 parallel reactions. In this photograph the pipette system is retracted so that the piezoelectric dispensers can be used to deliver reagents to the chip. (C) Detail of “stacked” structure of the TLC plate holder that allows up to 64 samples to be spotted on multi-lane TLC plates.
Fluidics head
The fluidics head (Supporting Information Section S1.2 comprises a set of seven non-contact piezoelectric dispensers (INKX0514300A and INKX0514100A, The Lee Company, Westbrook CT, USA) and a custom pipette cone, designed to mate with the disposable tips for aspirating and dispensing liquids. The piezoelectric dispensers were used for the dispensing of reagents shared across many reaction sites, such as collection solutions and [18F]fluoride solutions. Dispensers were each connected via 0.03” ID or 0.01” ID PTFE tubing dip tubes to septum capped reagent vials, comprising either 20 mL scintillation vials (03-340-25Q, Thermo Fischer Scientific, Waltham, MA, USA), 5 mL V-vials (NextGen V Vial, Wheaton Industries, New Jersey, USA), or 1.5mL V-vials (μVial 09-1400, Microliter Analytical Supplies Inc., Suwanee, Georgia, USA) based on the total volume needed of the corresponding reagent. Each vial was connected to the output of a pneumatic valve (S070B-5DG, SMC Corporation, Tokyo, Japan) allowing the vial to be either pressurized (inert nitrogen) or vented. The pipetting system was used for delivery of varied reagents (e.g. precursor prepared in different concentrations or solvents for optimization) and for collecting crude products from reaction sites. The pipette cone was mounted on vertical slide (8381K2, McMaster-Carr) with its position (extended or retracted) controlled using a dual-acting pneumatic actuator (6498K003, McMaster-Carr, Elhurst IL, USA). When retracted, attached pipette tips would be out of the way of the dispensers, allowing reagent dispensing without removing the tip. To attach a tip, the pipette cone was extended and pressed into the tip with a pressure of 20 psig (~13N force). The pipette cone was also fitted with an electrical microswitch that could be used to precisely determine the Z-axis position of the top of the multi-reaction chips. The pipette cone was connected to a syringe pump (Microlab PSD/4, Hamilton Company, Reno NV, USA) equipped with 250 μL syringe (with ~100 nL volume accuracy) mounted at the side of the workspace, via 0.03” ID PTFE tubing (~1 m long). The syringe pump could switch between the pipette cone and a DI water reservoir, allowing filling of the tubing with DI water to improve responsiveness and accuracy compared to air. Details of priming of the fluidics systems is described in the Supporting Information Section S1.3.
XYZ Gantry
Fast motion actuators were selected to minimize radioactive decay during movement operations. The X and Y axes consist of belt-driven slides (LCR30, Parker Hannifin Corporation, Irwin PA, USA), arranged in an H-formation, providing 40 cm travel in the X-direction and 21 cm in Y, with a maximum speed of 57 cm/s and positioning repeatability of ±100μm. For the Z-axis, a 12 mm/rev pitch lead-screw slide (MLC028, PBC Linear, Roscoe IL, USA) was used, with a repeatability of ±20 μm and maximum speed of 12 cm/s. All three axes were powered by stepper motors (eCLM-S233F, Parker Hannifin) using hall-effect sensors to define a home position in the top back-left-most position. A full explanation of the coordinate system and component positioning is provided in the Supporting Information Section S2.
Control system and software
Front-end control was implemented using a LabView program (National Instruments, Riverside, CA, USA) which controlled communications with external devices (microcontroller, data acquisition modules (DAQs), syringe pump), initialize all equipment, load configuration files and populate global variables, read method file, and perform all listed method steps. The system configuration and calibration information are described in XML files (Supporting Information, Section S3) while the method file, written by the user in a custom scripting language (Supporting Information, Section S4), is used to define the optimization study. The system was operated with a Windows computer, but any computer / operating system that can run LabView can be used.
The control system (Figure 3) comprises multiple subsystems driven by the front-end computer. The temperature control system for the heater platform has been previously described17. Briefly, signals from integrated heater thermocouples were amplified (MAX31856, Adafruit Industries, New York New York, USA) and measured via a DAQ (USB-202, Measurement Computing Corporation, Norton MA, USA) which also digitally controlled a dedicated relay and fan per heater, allowing closed-loop on-off software temperature control. The syringe pump was controlled via RS485 serial commands from LabView via a USB to RS485 adapter (USB-485B, Sima S. Enterprises, Los Angeles, CA, USA). A separate DAQ (USB-201, Measurement Computing Corporation) monitored the analog output of the priming sensor to detect liquid and controlled the reset signal for priming sensor recalibration.
Figure 3:

Block diagram of the control system. Blue lines represent gas pathways, solid red represents analog signals, dashed red represents digital signals, and black represents serial communication.
All other systems were interfaced to a microcontroller (Arduino Mega, Arduino AG, Sommerville MA, USA) in communication with the front-end computer via USB. Custom firmware was written in C++ and compiled using the GNU C++ compiler. The stepper motors (and built-in encoders) were connected to closed-loop stepper drivers (CL57T, OMC Corporation, Nanjing City, China) which were in turn controlled via step and direction signals from the microcontroller for each axis. A stepping algorithm was implemented in the microcontroller to allow smooth acceleration and deceleration (Supporting Information, Section S5). The pneumatic system contained two electronic pressure regulators (ITV0050-3UMS, SMC Corporation) – one for the reagent driving pressure for the dispensers, and one for the pipette cone pneumatic actuator. Pressure setpoints were controlled by converting digital outputs from the microcontroller to analog signal signals via digital potentiometer voltage dividers (10 kΩ, AD5220, Analog Devices, Norwood, MA, USA) in conjunction with op-amp follower circuits (1x amplification, AD8012, Analog Devices), and analog signals from the regulators representing current pressure were monitored via the microcontroller. The valves controlling pressure to each dispenser reagent reservoir, and the two valves to actuate the pneumatic cylinder for the pipette cone, were interfaced via a Darlington transistor array (ULN2803A, Texas Instruments Inc., Dallas, Texas, USA) to digital outputs of the microcontroller. The reagent dispensers were powered by dedicated spike-and-hold drivers (IECX0501350A, Lee Company) triggered by the microcontroller to cause the desired open duration.
System Calibrations
System positions were determined as described in Supporting Information Section S2, while heater temperatures, pressure regulators, automated TLC spotting protocol, and piezoelectric dispensers were calibrated as described in the Supporting Information, Section S6.
Reagents
[18F]fluoride in [18O]H2O was obtained from the UCLA Crump Cyclotron and Radiochemistry Center. Anhydrous methanol (MeOH, 99.8%), anhydrous acetonitrile (MeCN, 99.8%), acetone (99.9%), ammonium formate (NH4HCO2, 97%), dichloromethane (DCM, ≥99.8%), 2-3-dimethyl-2-butanol (thexyl alcohol, 98%), tetrahydrofuran (THF, ≥99.9%), hexanes (HPLC grade), pyridine (99.8%), triethylamine (TEA, 99%), trifluoroacetic acid (TFA, 99%), ethanol (EtOH, >99.5%) N,N-dimethylacetamide (DMA, 99.8%), 1,3-dimethyl-2-imidazolidinone (DMI, ≥99%), and N,N-dimethylformamide (DMF, 99.8%) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Tetrabutylammounium bicarbonate (TBAHCO3, 75mM in ethanol), tetrakis(pyridine)copper(II) triflate (Cu(py)4OTf2, 95%), cesium carbonate (Cs2CO3, >99%), and 3-[3,4-dimethoxy-5-[[(2S)-1-prop-2-enylpyrrolidin-2-yl]methylcarbamoyl]phenyl]propyl 4-methylbenzenesulfonate (precursor for [18F]fallypride, >95%) were purchased from ABX Advanced Biochemical Compounds (Radeberg, Germany). Deionized (DI) water was obtained from a Milli-Q water purification system (IQ 7000, EMD Millipore Corporation, Berlin, Germany). N-methyl-2-pyrrolidone (NMP, >99%) and tetraethylammonium trifluoromethanesulfonate (TEAOTf, >99%) were purchased from Tokyo Chemical Industry (Tokyo, Japan). FBnTP precursor ((4-methylphenylboronic acid pinacol ester)triphenylphosphonium triflate) and reference standard (4-fluorobenzyl-triphenylphosphonium) were generously provided by Dr. Kuo-shyan Lin (University of British Columbia, Canada).
Teflon-coated silicon chips (containing 4x4 arrays of 3 mm diameter surface tension traps) for performing parallel reactions were fabricated as described previously16.
Analytical methods
Measurements of radioactivity were made in a calibrated dose calibrator (CRC-25PET, Capintec Inc., Florham Park, NJ, USA) or a gamma counter (Wizard 3” 1480, PerkinElmer, Waltham, MA, USA). To ensure repeatable dose calibrator measurements, a custom acrylic holder was machined to hold Eppendorf tubes, strip wells, and chips in consistent positions within the dose calibrator chamber. Gamma counting of samples was performed for 45 s, and an empty well was measured every 8 samples for background subtraction.
Fluorination efficiency was determined via multi-lane radio-TLC methods25. TLC plates (silica gel 60 F254 , 200 mm x 50 mm, Merck KgaA, Darmstadt, Germany) were cut into 50 mm x 50 mm pieces before use. Samples (0.5 μL) were spotted 10 mm from the bottom edge of the TLC plates. Up to 8 samples were spotted per plate at 4.5 mm spacing between “lanes”. Development distance was 35 mm. TLC plates containing crude [18F]Fallypride samples were developed in a mobile phase of 30.0% TEA, 14.7% acetone, 18.8% THF, and 36.5% hexanes (v/v)22. For samples of crude [18F]FBnTP, the mobile phase was 9:2 DCM:MeOH (v/v)26. Readout of plates was performed by covering with a glass microscope slide (75 x 50 x 1 mm3, Fisher Scientific, Hampton, NH, USA) using Cerenkov luminescence imaging with 5 min exposure time as previously described25, and fluorination efficiency for each sample (lane) was computed from the resulting images using (manual) region of interest analysis as previously described25. Collection efficiency for a reaction was computed by dividing the activity of the collected crude product by the initial activity, correcting for decay. The initial activity was estimated via measurement of an Eppendorf tube (“aliquot vial”) loaded with the same volume as dispensed to all reaction sites. The crude RCY was determined by multiplying the fluorination efficiency by the collection efficiency. To determine the activity of a single reaction site on a chip (e.g. residual activity after collecting the crude product), the total chip activity was first measured in a dose calibrator, then multiplied by the fraction of the total activity corresponding to that reaction site. This fraction was determined by obtaining a CLI image of the chip (covered with 1 mm glass microscope slide, 5 min exposure time unless otherwise noted) and then dividing the integrated pixel intensity within the desired reaction site by the total integrated pixel intensity of all reaction sites.
Robotic system characterization
Characterization of the performance of the ceramic heater system and pipetting system, the piezoelectric reagent dispenser repeatability, assessment of cross-contamination during synthesis operations, and repeatability of parallel synthesis, are described in detail in the Supporting Information, Section S7.
Optimization of [18F]Fallypride synthesis
To validate the overall system, we performed a study of the synthesis of [18F]Fallypride, including conditions for which we have previously reported the performance using manually-performed droplet reactions16,17. Figure 4A shows the experimental layout. One set of 32 reactions was performed with constant amount of precursor (230 nmol) but different amounts of TBAHCO3 (8 values, 120-480 nmol, n=4 replicates each). Another set of 32 reactions was performed with a fixed amount of TBAHCO3 (240 nmol) but different amounts of the precursor (8 values, 3.65 – 468 nmol, n=4 replicates each).
Figure 4:

(A) Map of reaction conditions for [18F]Fallypride optimization experiments with varied amount of TBAHCO3 and precursor. Reactions were all performed in 1:1 v/v thexyl alcohol:MeCN at 110°C for 7 min. (B) Map of conditions for [18F]FBnTP optimization experiments. All reactions were performed for 5 min with 10 nmol Cs2CO3, 300 nmol TEAOTf, 450 nmol precursor, and 680 nmol Cu(py)4OTf2.
To prepare the experiment, a stock precursor solution (154 mM) in 1:1 v/v thexyl alcohol:MeCN was prepared and additional concentrations (77 – 0.6 mM) were prepared by dilution with the same solvent mixture. 250 μL of each stock precursor concentration were loaded into wells of a 96 well plate (Costar 3363, Corning Inc., Glendale, AZ, USA) for the reactions with varied precursor amount, and an additional 250 μL of 38.5 mM solution was loaded into another well for the remaining reactions. A stock collection solution was prepared by mixing 20 mL 9:1 MeOH:DI water and connecting to a reagent dispensers. A 1.5 mL activity stock solution was prepared from [18F]fluoride, DI water, and TBAHCO3 (final concentrations ~740 MBq/mL, 24 mM TBAHCO3) and then loaded into a second dispenser, and a 1.5 mL stock solution of 30 mM TBAHCO3 in DI water was loaded into a third dispenser.
Synthesis of [18F]Fallypride at each reaction site was similar to our previous report16. First, 5 μL of the [18F]fluoride/TBAHCO3 stock solution was dispensed to each reaction site. Additional amounts of TBAHCO3 solution (0-7 μL) were then dispensed to achieve the desired total amount of TBAHCO3 for each reaction. The droplets were then dried by heating all chips at 92°C for 30s, 98°C for 30 s, and 105°C for 60s. Next, 6 μL of precursor solution was loaded to each reaction site, with concentration chosen to achieve the desired precursor amount for each reaction site. The fluorination reaction was performed by heating all chips at 110°C for 7 min. Individual reactions were collected using 4 collection cycles (i.e. adding 10 μL of collection solution to the reaction site, mixing via pipette, and transferring via pipette to a dedicated location in a strip-well plate). A 0.5 μL sample of each crude product was spotted to a location on a TLC plate. At the end of the experiment, activity of the collected products and aliquot vial was measured, and TLC plates were developed and imaged. Additional activity measurements and CLI imaging of chips were performed after [18F]fluoride drying, fluorination, and collection steps.
Optimization of [18F]FBnTP synthesis
Next we performed an optimization study of a synthesis for which we had not previously studied, namely the copper-mediated synthesis of [18F]FBnTP27. Based on previous study of the droplet-based synthesis of [18F]FDOPA via a similar copper-mediated route, we found significant impact of reaction solvent and temperature, and thus implemented the experimental layout in Figure 4B to explore the influence of solvent (DMF, DMI, NMP, and DMA, each with 3.8% v/v pyridine) and temperature (100, 110, 120, and 130°C) in the synthesis of of [18F]FBnTP.
To prepare the experiment, stock solutions of the FBnTP precursor (90 mM) were prepared in each of the 4 solvent mixtures, and, similarly, stock solutions of Cu(py)4OTf2 (136 mM) were prepared in each solvent mixture. 20 mL of collection stock solution was prepared by mixing 2:3 v/v MeCN:DI water and loading into a reagent dispenser. A 1.5 mL stock solution of [18F]fluoride was prepared by mixing aqueous [18F]fluoride with DI water and adding Cs2CO3 and TEAOTf (~740 MBq/mL, 1 mM Cs2CO3, 30 mM TEAOTf,), corresponding to 10 nmol of Cs2CO3, 300 nmol TEAOTf, and ~7.4 MBq per reaction site.
To perform the droplet reactions, 10 μL of the [18F]fluoride stock solution was first dispensed to each reaction site. Droplets were dried by heating all chips to 92°C for 15 s, 98°C for 15 s, and 105°C for 60 s. Immediately before fluorination, for each solvent mixture, 125 μL of the corresponding precursor and Cu(py)4OTf2 stock solutions were mixed together to create 4 new precursor stock solutions (each containing 45 mM FBnTP precursor and 68 mM Cu(py)4OTf2). The system then deposited 10 μL to each reaction site according to the experimental plan, and fluorination was performed by heating chips for 5 min to the temperatures in the experimental plan. Crude products and measurements were collected in the same manner as for [18F]Fallypride.
Some batches of [18F]FBnTP were purified via radio-HPLC using an analytical column (ZORBAX RR Eclipse Plus C18, 4.6 x 100 mm, 3.5 μm particle size, Agilent, Santa Clara, CA, USA) using a mobile phase of DI water and MeCN (66:34 v/v) with 0.1%TFA (v/v) and flow rate of 1.2 mL/min. The radio-HPLC system setup comprised a Smartline HPLC system (Knauer, Berlin, Germany) equipped with a degasser (Model 5050), pump (Model 1000), UV detector (254 nm; Eckert & Ziegler, Berlin, Germany), gamma-radiation detector (BFC-4100, Bioscan, Inc., Poway, CA, USA), and counter (BFC-1000; Bioscan, Inc., Poway, CA, USA). The purified product was then formulated via solid-phase extraction using a C18 cartridge (Sep-Pak Plus Short, WAT020515, Waters Corporation, Milford, MA, USA) pre-conditioned before use with 3 mL of EtOH followed by 20 mL of DI water. The purified product was diluted with 20 mL of DI water and then slowly loaded onto the cartridge followed by rinsing with 20 mL of DI water. The final product was eluted with 1 mL of EtOH and diluted with DI water to 10 mL. This final diluted product was analyzed on the same HPLC system to confirm radiochemical purity using mobile phase of water and MeCN (60:40 v/v) with 0.1%TFA (v/v) and flow rate of 1.2 mL/min. Co-injection of the final diluted [18F]FBnTP and reference standard was performed to confirm product identity.
Results
System characterization
Several experiments were performed to establish the ability of the system to accurately assess reaction performance. First, one of the piezoelectric reagent dispensers was loaded with 1.5 mL [18F]fluoride/TBAHCO3 stock solution (~3.7 MBq/mL, 25 mM) and 10 μL was dispensed sequentially into each of 96 strip-wells. Gamma counting (with decay correction) of the individual separated (and capped) strip wells revealed high repeatability of dispensed amount, with a standard deviation of 1.9% (n=96) (Figure 5A). Based on this excellent repeatability, we were able to estimate the initial activity on each reaction site during a high-throughput experiment by dispensing an equal portion into an “aliquot vial” that could be measured via dose calibrator or gamma counter.
Figure 5:

(A) Reagent dispensing uniformity. Graph shows gamma counter measurements (decay-corrected) for 96 individual dispenses of [18F]fluoride into wells of a strip-well plate. (B) Reaction uniformity. Crude RCY of [18F]Fallypride synthesized under identical conditions at 16 reaction sites. (Conditions: TBAHCO3 amount: 240 nmol, precursor amount: 230 nmol, reaction temperature: 110°C, reaction time: 5 min).
We also assessed uniformity of [18F]fluoride dispensing and drying on chip reaction sites (n=64) and found excellent site-to-site uniformity and chip-to-chip uniformity (see Supporting Information Section S7.1).We further assessed uniformity by performing replicate [18F]Fallypride syntheses under identical conditions (n=16). Performance was highly consistent across reactions with fluorination efficiency of 90.4 ± 0.7 % (n=16), collection efficiency of 91 ± 2 % (n=16) and crude RCY of 83 ± 1 % (n=16) (Figure 5B). Full details can be found in Supporting Information Sections S7.2 and S7.3. A cross-contamination test was performed by dispensing a [18F]fluoride/TBAHCO3 solution to alternate reaction sites on a chip and then performing a drying step (see Supporting Information Section S7.4), and the unused reaction sites contained negligible radioactivity.
Optimization of [18F]Fallypride synthesis
Results of the 64-reaction [18F]Fallypride study (exploring the influence of precursor amount and TBAHCO3 amount) are summarized in Figure 6, with more details available in the Supporting Information Section S7.5. Excellent agreement was observed between the reaction performance in this automated study and results of a previous manual study16. The current study found the optimal condition (180 nmol TBAHCO3, 230 nmol precursor) had a crude RCY of 92.5 ± 0.5 % (n=4), while in the prior study, the optimum condition (240 nmol TBAHCO3, 230 nmol precursor) had a crude RCY of 92 ± 1% (n=2)16. While in the current study, the crude RCY for 240 nmol TBAHCO3 (89 ± 5%, n=8) was not significantly different from the prior study, and the inclusion of intermediate base amounts allowed us to find the more optimal 180 nmol value, which also provides more robustness (lower sensitivity to deviations in amount of TBAHCO3).
Figure 6:

(A) Crude RCY for the droplet synthesis of [18F]Fallypride as a function of the amount of TBAHCO3 with constant precursor amount of 230 nmol. (B) Crude RCY as a function of the amount of precursor with amount of TBAHCO3 fixed at 240 nmol. Other conditions were fixed: reaction temperature: 110°C, reaction time: 5min. Results of the current study (black symbols) are compared to results of manually-performed experiments previously reported16 (red symbols).
Optimization of [18F]FBnTP synthesis
During initial [18F]FBnTP experiments, we discovered that there can be significant evaporation of the collection solution from the microwells during the 1.34 h collection process. For the collection solution used in this case (3:2 v/v MeCN:H2O), this evaporation could change the composition, and we suspect that this affected the solubility of the product in the collection solution, resulting in sampling error when the TLC spotting occurred. This is consistent with our observations of unexpectedly low product signal (and low overall signal) when developing and imaging the TLC plates. To overcome this issue, we modified the control software to immediately transfer a sample of the crude product to TLC right after the collection step, rather than first collecting all reaction droplets into microwells, and then subsequently transferring samples of all microwells onto TLC plates. This change appeared to eliminate the sampling error, and we confirmed that samples spotted onto TLC plates were stable, with no difference in the resulting TLC separation regardless of the time the sample was on the plate before the developing step.
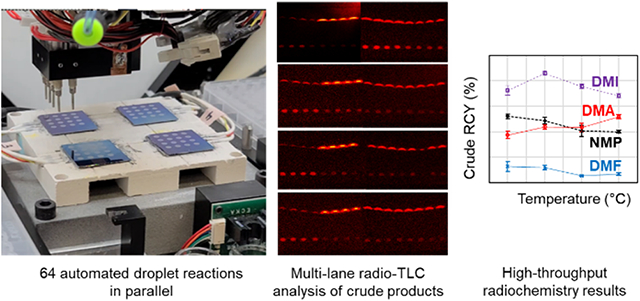
Results of the improved [18F]FBnTP study (exploring impact of reaction solvent and temperature) are summarized in Figure 7, with additional details in Supporting Information Section S7.6. The solvent had a particularly large impact on reaction performance, and the optimal condition was found to be 110°C in DMI (with 3.8% v/v pyridine), providing a fluorination efficiency of 89±1% (n=4), collection efficiency of 97±2% (n=4) and overall crude RCY of 86±2% (n=4). These results compare favorably to the reported macroscale reaction with a fluorination conversion of ~60% (isolated RCY not reported)27. The full set of 64 reactions was automatically performed in 180 min, with an additional ~60 min needed to perform manual radioactivity measurements and TLC analysis. Of the 180 min, the majority of the time (120 min) was spent collecting crude reaction products from the reaction sites into the microwell plate. A detailed analysis of the timing for high-throughput experiments is given in the Supporting Information Section S7.8.
Figure 7:

Effect of temperature and reaction solvent on the performance of the synthesis of [18F]FBnTP. (A) Fluorination efficiency, (B) collection efficiency, and (C) crude RCY. Each of the indicated solvents contains 3.8% v/v pyridine. Fixed conditions: 5 min reaction time, 10 nmol Cs2CO3, 300 nmol TEAOTf, 450 nmol precursor, 680 nmol Cu(py)4OTf2.
Using the optimal condition, the droplet radiosynthesis of [18F]FBnTP was slightly scaled up, by using higher initial [18F]fluoride activity, to an amount sufficient for preclinical imaging studies (~120 MBq). After purification via radio-HPLC and cartridge formulation, the RCY was 66 ± 6% (n=3), with excellent radiochemical purity of 100% (n=3). The overall preparation time including on-chip fluorination, HPLC purification and cartridge formulation was only 42 ± 1 min (n=3). Taking advantage of this rapid and efficient production format, the activity yield was 49 ± 3% (n=3).
Discussion
We previously showed that arrays of droplet reactions provide an efficient means to perform optimization studies by enabling 64 simultaneous reactions while consuming a total amount of precursor equivalent to a single conventional synthesis17. After optimization, the synthesis can then be automated via a miniature droplet radiosynthesizer18, and, if needed, scaled to higher activity levels by increasing the starting activity20,24. In the present work we have developed and demonstrated a novel platform to implement this technique in a highly automated fashion to increase throughput as well as safety for the radiochemist. The high uniformity of operations of the automated platform (exemplified by the consistent performance of n=16 replicate syntheses of [18F]Fallypride), combined with the observation of negligible crosstalk, confirms that the automated platform can be relied upon to perform large sets of independent reactions with high reliability. The close agreement of the results of our 64-reaction exploration of the synthesis of [18F]Fallypride with prior manually-performed experiments16 further validates the current platform as an optimization tool. Due to the wide compatibility of droplet reactions with a variety of different 18F-labeled tracers and labeling methods28, and the successful optimization of a reaction not previously studied in droplet format (i.e. copper-mediated radiofluorination route to produce [18F]FBnTP), we expect this platform to have wide applicability to other 18F-radiotracers and likely other isotopes.
The robotic system software provides a high degree of customizability without a steep learning curve. Experiments are programmed by stringing together a series of parameterizable unit operations using a flexible scripting language. In practice, a new optimization experiment can often be defined by editing a previously-created program in <15 min by a user familiar with the scripting language. Beyond the reaction parameters studied here (base amount, precursor amount, solvent, reaction temperature), the platform can also be used to explore a variety of different parameters for every stage of the synthesis (see Table 1).
Table 1:
Examples of parameters that can be optimized in an 18F-radiosynthesis
| Stage of radiosynthesis process | Parameter(s) |
|---|---|
| [18F]fluoride loading and activation |
|
| Reactions (fluorination, deprotection, etc.) |
|
| Collection |
|
Compared to previously reported manual optimization studies16,17, the automated platform presented here provides enormous practical and safety advantages by eliminating tedious manual pipetting steps and minimizing radiation exposure. For example, in the [18F]Fallypride experiment described here, there were a total of 768 pipetting operations (including all reagent loading and liquid transfer steps, but not counting complex manipulations such as “mixing”) and 138 pipette tip changes. It is not difficult to imagine that when performing such an experiment manually, there is a high likelihood of human error, such as forgetting a pipetting step, pipetting the wrong reagent, or dispensing or aspirating to/from the wrong reaction site17; these errors are eliminated with the automated system. Although, in principle, manual operation could be simplified using multi-channel pipettes, the increased complexity of reagent preparation and the need to continually adjust tip spacing to match either a microwell plate (4.5 mm or 9 mm spacing) or chips (5 mm spacing), makes this impractical in the current chip and heater design.
From choice of actuators to relative positioning of components within the work area, the hardware system was designed for rapid operation and high throughput. The described experiments each took ~3 h of automated system operation, plus an additional ~1 h of manual effort to perform radioactivity measurements and Cerenkov imaging of chips (i.e. at intermediate steps and after the collection step), measure radioactivity of collected crude products, and to develop and image the TLC plates. Further system developments are underway to enable these radioactivity measurements to be performed in situ, which will increase safety and reduce overall experiment time. Even including the manual interventions, the effective time per data point (i.e., 3 h / 64 reactions = 2.8 min/reaction) is extremely short when compared with conventional radiochemistry apparatus, in which experiments (each taking up to several hours) are performed sequentially. Leveraging this throughput allows a study that would normally require many months of experiments to be completed in just days. We contemplate that the current throughput can potentially be even further enhanced by operating the system multiple times per day, increasing the number of parallel reaction sites, or parallelizing the pipetting operations. Additionally, in principle, the robotic system could assist with reagent preparation (e.g. prepare dilution series) to further reduce the experimental setup time.
Performing high-throughput radiochemistry studies requires a high-throughput method to analyze the radiochemical composition of crude reaction products. While typical analysis is performed via radio-HPLC, the high time requirement per sample (>15-40 min for cleaning, equilibration, injection and separation) makes this infeasible for 64 sequential samples before they decay to unusable levels. In this work we used multi-lane TLC techniques25, which are particularly convenient due to the capability for simultaneous multi-lane separation and high-resolution Cerenkov luminescence imaging-based readout of 8 samples per plate. We recently reported a systematic approach for determining the optimal mobile phase for TLC separation that, for analysis of [18F]Fallypride enabled comparable resolution to HPLC, and that will enable extension of high-resolution TLC methods to additional radiopharmaceutical compounds22. We are also investigating the use of other high-throughput analysis techniques such as ultra-performance liquid chromatography (UPLC), which can be optimized to reduce the time per sample to the order of ~1 min29.
There are a few cases in which the open nature of the droplet reactions in our platform could introduce some limitations. (i) Currently the platform would not be able to handle reactions where the radioisotope, intermediate species, or product is volatile. (ii) Atmospheric exposure could be an issue for certain reactions involving reagents sensitive to oxygen or moisture (e.g. copper-mediated radiofluorination). However, in this work, the observed yield of the open-droplet copper-mediated radiosynthesis of [18F]FBnTP exceeded that reported for (closed) vial-based reactions, and thus atmospheric exposure does not appear to have a significant adverse impact. (iii) Special measures must be taken for reactions involving volatile solvents as the solvent can rapidly evaporate at elevated temperature, which can limit the duration of reactions. We have found that the issue can be addressed by replenishing the solvent at regular intervals, using short reaction times (e.g. 30 s), or by switching to higher boiling point solvents17.
In addition to rapid synthesis optimization, the platform described here could potentially also assist with labeling of libraries of radiopharmaceutical compounds to screen in vitro or in vivo properties30, or to generate training data for novel machine learning approaches in radiochemistry8.
Conclusions
We developed a robotic, high-throughput radiochemistry platform that fits inside most commercially-available mini-cells and hot cells and can perform a set of 64 droplet-based reactions on patterned Teflon-coated silicon chips nearly simultaneously. The process is highly automated, only requiring manual intervention for intermediate radioactivity measurements and analysis of final products. The system automates all aspects of the synthesis including isotope dispensing, isotope drying by evaporation, reagent loading, heating to activate the radiolabeling reaction, cooling, collecting crude product into microwell plates or tubes, and transferring crude samples to TLC plates for analysis. In characterization experiments, performance of replicate reactions was highly repeatable and negligible crosstalk among different reaction sites was observed. We performed a 64-reaction study to explore the effects of amount of TBAHCO3 and precursor in the synthesis of [18F]Fallypride and found performance closely matched a similar prior study in which experiments were conducted manually. As a proof-of-concept of novel radiosynthesis optimization, we investigated the impact of reaction temperature and solvent on the copper-mediated radiosynthesis of [18F]FBnTP27. As a result of the 64-reaction study, we found high-performing conditions for the synthesis and demonstrated that the conditions could be combined with purification and formulation to achieve a high RCY (66%) in a 42 min synthesis time. The platform will enable routinely performing droplet-array-based radiochemistry studies17 without the tedious pipetting, chance of human error, and radiation exposure of manual techniques.
Supplementary Material
Synopsis:
To overcome throughput limits on conventional radiosynthesizers, we developed an automated robotic system to rapidly and safely increase radiochemistry throughput by orders of magnitude via arrays of parallel droplet reactions.
Acknowledgments
We thank Jeffery Collins for providing [18F]fluoride ion for these studies. We thank Dr. Kuo-shyan Lin for providing FBnTP precursor and reference standard. Microfluidic reaction chips were produced in the UCLA Nanofabrication Laboratory (NanoLab), and we thank the staff for technical support. We also thank Jeffery Collins, Alejandra Rios, and Travis S. Laferriere-Holloway for valuable discussions about experimental setup and findings. This work was supported in part by the National Cancer Institute (R21 CA212718, R33 CA240201), National Institute of Mental Health (R44 MH097271), the National Institute of Biomedical Imaging and Bioengineering (R01 EB032264, T32 EB002101), and the National Institutes of Health (S10 OD026942).
Footnotes
Disclosures
Dr. van Dam is a co-founder of Sofie, Inc. (Dulles, VA). Drs. van Dam and Jones are co-founders of DropletPharm, Inc. (Sherman Oaks, CA).
Additional Supporting Information
Further technical details (e.g. CAD files, LabVIEW files, firmware source code) are available upon reasonable request.
References
- (1).Phelps ME PET: Molecular Imaging and Its Biological Applications, 1st ed.; Springer Science & Business Media: Berlin, Germany, 2004. [Google Scholar]
- (2).Ametamey SM; Honer M; Schubiger PA Molecular Imaging with PET. Chem. Rev 2008, 108 (5), 1501–1516. 10.1021/cr0782426. [DOI] [PubMed] [Google Scholar]
- (3).Radiosynthesis Database of PET Probes (RaDaP). http://www.nirs.qst.go.jp/research/division/mic/db2/ (accessed 2017–05-08). [Google Scholar]
- (4).Molecular Imaging and Contrast Agent Database (MICAD); National Center for Biotechnology Information (US): Bethesda (MD), 2004. [PubMed] [Google Scholar]
- (5).Collins J; Waldmann CM; Drake C; Slavik R; Ha NS; Sergeev M; Lazari M; Shen B; Chin FT; Moore M; Sadeghi S; Phelps ME; Murphy JM; Dam R. M. van. Production of Diverse PET Probes with Limited Resources: 24 18F-Labeled Compounds Prepared with a Single Radiosynthesizer. PNAS 2017, 114 (43), 11309–11314. 10.1073/pnas.1710466114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Bruton L; Scott PJH Automated Synthesis Modules for PET Radiochemistry. In Handbook of Radiopharmaceuticals; John Wiley & Sons, Ltd, 2020; pp 437–456. 10.1002/9781119500575.ch13. [DOI] [Google Scholar]
- (7).Bowden GD; Pichler BJ; Maurer A A Design of Experiments (DoE) Approach Accelerates the Optimization of Copper-Mediated 18 F-Fluorination Reactions of Arylstannanes. Scientific Reports 2019, 9 (1), 11370. 10.1038/s41598-019-47846-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Webb EW; Scott PJH Potential Applications of Artificial Intelligence and Machine Learning in Radiochemistry and Radiochemical Engineering. PET Clinics 2021, 16 (4), 525–532. 10.1016/j.cpet.2021.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ahmadi F; Simchi M; Perry JM; Frenette S; Benali H; Soucy J-P; Massarweh G; Shih SCC Integrating Machine Learning and Digital Microfluidics for Screening Experimental Conditions. Lab Chip 2022. 10.1039/D2LC00764A. [DOI] [PubMed] [Google Scholar]
- (10).Zhang X; Dunlow R; Blackman BN; Swenson RE Optimization of 18F-Syntheses Using 19F-Reagents at Tracer-Level Concentrations and Liquid Chromatography/Tandem Mass Spectrometry Analysis: Improved Synthesis of [18F]MDL100907. Journal of Labelled Compounds and Radiopharmaceuticals 2018, 61 (5), 427–437. 10.1002/jlcr.3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Laube M; Wodtke R; Kopka K; Kniess T; Pietzsch J 18F-Chemistry in HPLC Vials - a Microliter Scale Radiofluorination Approach. Nuclear Medicine and Biology 2021, 96-97, S61. 10.1016/S0969-8051(21)00367-X. [DOI] [Google Scholar]
- (12).Lazari M; Irribarren J; Zhang S; van Dam RM Understanding Temperatures and Pressures during Short Radiochemical Reactions. Applied Radiation and Isotopes 2016, 108, 82–91. 10.1016/j.apradiso.2015.12.037. [DOI] [PubMed] [Google Scholar]
- (13).Pascali G; Matesic L; Collier TL; Wyatt N; Fraser BH; Pham TQ; Salvadori PA; Greguric I Optimization of Nucleophilic 18F Radiofluorinations Using a Microfluidic Reaction Approach. Nat. Protocols 2014, 9 (9), 2017–2029. 10.1038/nprot.2014.137. [DOI] [PubMed] [Google Scholar]
- (14).Chen Y-C; Liu K; Shen CK-F; van Dam RM On-Demand Generation and Mixing of Liquid-in-Gas Slugs with Digitally Programmable Composition and Size. J. Micromech. Microeng 2015, 25 (8), 084006. 10.1088/0960-1317/25/8/084006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Liu K; Lepin EJ; Wang M-W; Guo F; Lin W-Y; Chen Y-C; Sirk SJ; Olma S; Phelps ME; Zhao X-Z; Tseng H-R; van Dam RM; Wu AM; Shen CK-F Microfluidic-Based 18F-Labeling of Biomolecules for Immuno–Positron Emission Tomography. Mol Imaging 2011, 10 (3), 168–176. 10.2310/7290.2010.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Rios A; Wang J; Chao PH; Dam R. M. van. A Novel Multi-Reaction Microdroplet Platform for Rapid Radiochemistry Optimization. RSC Adv. 2019, 9 (35), 20370–20374. 10.1039/C9RA03639C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rios A; Holloway TS; Chao PH; De Caro C; Okoro CC; van Dam RM Microliter-Scale Reaction Arrays for Economical High-Throughput Experimentation in Radiochemistry. Sci Rep 2022, 12 (1), 10263. 10.1038/s41598-022-14022-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang J; Chao PH; van Dam RM Ultra-Compact, Automated Microdroplet Radiosynthesizer. Lab Chip 2019, No. 19, 2415–2424. 10.1039/C9LC00438F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wang J; Chao PH; Hanet S; Dam R. M. van. Performing Multi-Step Chemical Reactions in Microliter-Sized Droplets by Leveraging a Simple Passive Transport Mechanism. Lab Chip 2017, 17 (24), 4342–4355. 10.1039/C7LC01009E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lisova K; Wang J; Hajagos TJ; Lu Y; Hsiao A; Elizarov A; van Dam RM Economical Droplet-Based Microfluidic Production of [18F]FET and [18F]Florbetaben Suitable for Human Use. Sci Rep 2021, 11 (1), 20636. 10.1038/s41598-021-99111-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wang J; Holloway T; Lisova K; van Dam RM Green and Efficient Synthesis of the Radiopharmaceutical [18F]FDOPA Using a Microdroplet Reactor. React. Chem. Eng 2020, 5 (2), 320–329. 10.1039/C9RE00354A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Laferriere-Holloway TS; Rios A; Lu Y; Okoro CC; van Dam RM A Rapid and Systematic Approach for the Optimization of Radio Thin-Layer Chromatography Resolution. J Chromatogr A 2023, 1687, 463656. 10.1016/j.chroma.2022.463656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lisova K; Sergeev M; Evans-Axelsson S; Stuparu AD; Beykan S; Collins J; Jones J; Lassmann M; Herrmann K; Perrin D; Lee JT; Slavik R; van Dam RM Microscale Radiosynthesis, Preclinical Imaging and Dosimetry Study of [18F]AMBF3-TATE: A Potential PET Tracer for Clinical Imaging of Somatostatin Receptors. Nuclear Medicine and Biology 2018, 61, 36–44. 10.1016/j.nucmedbio.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang J; Chao PH; Slavik R; van Dam RM Multi-GBq Production of the Radiotracer [18F]Fallypride in a Droplet Microreactor. RSC Adv. 2020, 10 (13), 7828–7838. 10.1039/D0RA01212B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wang J; Rios A; Lisova K; Slavik R; Chatziioannou AF; van Dam RM High-Throughput Radio-TLC Analysis. Nuclear Medicine and Biology 2020, 82–83, 41–48. 10.1016/j.nucmedbio.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kim D-Y; Kim H-J; Yu K-H; Min J-J Synthesis of [18F]-Labeled (2-(2-Fluoroethoxy)Ethyl)Tris(4-Methoxyphenyl)Phosphonium Cation as a Potential Agent for Positron Emission Tomography Myocardial Imaging. Nuclear Medicine and Biology 2012, 39 (7), 1093–1098. 10.1016/j.nucmedbio.2012.03.008. [DOI] [PubMed] [Google Scholar]
- (27).Zhang Z; Zhang C; Lau J; Colpo N; Bénard F; Lin K-S One-Step Synthesis of 4-[18F]Fluorobenzyltriphenylphosphonium Cation for Imaging with Positron Emission Tomography. J Labelled Compd Rad 2016, 59 (11), 467–471. 10.1002/jlcr.3436. [DOI] [PubMed] [Google Scholar]
- (28).Wang J; van Dam RM High-Efficiency Production of Radiopharmaceuticals via Droplet Radiochemistry: A Review of Recent Progress. Mol Imaging 2020, 19, 1–21. 10.1177/1536012120973099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Franck D; Nann H; Davi P; Schubiger PA; Ametamey SM Faster Analysis of Radiopharmaceuticals Using Ultra Performance Liquid Chromatography (UPLC®) in Combination with Low Volume Radio Flow Cell. Applied Radiation and Isotopes 2009, 67 (6), 1068–1070. 10.1016/j.apradiso.2009.02.077. [DOI] [PubMed] [Google Scholar]
- (30).Gagnon MKJ; Hausner SH; Marik J; Abbey CK; Marshall JF; Sutcliffe JL High-Throughput in Vivo Screening of Targeted Molecular Imaging Agents. Proc Natl Acad Sci U S A 2009, 106 (42), 17904–17909. 10.1073/pnas.0906925106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.