

Abstract
Deficiency of interleukin-1 receptor antagonist is a rare autoinflammatory disease that affects infants early in life. It often presents with systemic inflammation, skin and bone involvement. We present a 5-month-old boy who was hospitalized due to generalized erythematous pustular eruption with secondary impetigo, cellulitis, bronchiolitis, and elevated inflammatory markers. The patient was unresponsive to multiple courses of intravenous antibiotics, systemic, and topical steroid medications. The patient was evaluated by dermatology and rheumatology services among other subspecialities. Skin biopsy showed changes consistent with psoriasiform dermatitis, while bone scans showed multifocal osteomyelitis. The patient was started empirically on anakinra with improvement at 72 hours upon administration. This is one of the youngest reported case in the literature to be started on anakinra empirically prior to genetic confirmation of the mutation. A comprehensive literature review revealed that approximately 20 genetically confirmed patients, including our patient, have been reported with this genetic disease. It is imperative to recognize this disease early to achieve adequate response and remission. Therefore, clinical symptoms and the associated differential diagnosis for this disease should be constantly reassessed and reviewed by pediatricians and subspecialists to detect the disease as early as possible and reduce the high morbidity and mortality associated with delayed diagnosis and treatment.
Keywords: deficiency of interleukin-1 receptor antagonist, autoinflammatory disease, pediatrics
Introduction
Deficiency of interleukin-1 receptor antagonist (DIRA) is a rare, autosomal recessive disorder in which loss-of-function mutations in the gene IL1RN lead to unopposed action of interleukin-1 (IL-1) with resultant life-threatening systemic inflammation with prominent skin and bone involvement. 1 Since first reported in 2009, a handful of cases have been described worldwide, 1 though its rarity makes its true global incidence unknown.
Background
DIRA is a rare, autosomal recessive inflammatory disease in which loss-of-function mutations in the gene IL1RN lead to the absence of IL-1 receptor antagonist (IL-1 Ra), allowing unopposed IL-1 activation and an increased response to proinflammatory cytokines IL-1α and IL-1β stimulation. 1 2 The IL-1 Ra competitively binds to the IL-1 receptor, preventing the IL-1 receptor/IL-1α/IL-1β from forming a signaling complex, 3 4 which leads to unopposed action of the IL-1 with resultant life-threatening systemic inflammation with prominent skin and bone involvement. 1
The interleukin-1 family is a group of cytokines that play a central role in the regulation of immune and inflammatory responses to infections or sterile insults. 4 IL-1 has a wide range of biological functions, some of which include acting as a leukocytic pyrogen, a mediator of fever and a leukocytic endogenous mediator, and an inducer of acute-phase reactants and lymphocyte-activating factor (LAF). 5 6 The activation of IL-1 has been associated with autoinflammatory diseases, metabolic syndromes, acute inflammatory processes, chronic inflammation, and malignancy. 5 6
Clinical Manifestation
DIRA may present at or within days of birth and is characterized by skin and bone involvement in the setting of systemic inflammation, and in the absence of fever unless superinfection is present. Although the disease is autosomal recessive with 100% penetrance, the extent of DIRA's presenting features are variable and dependent on the domain affected by the described mutation. This explains why some patients presents primarily skin pathology with minimal bone involvement, whereas others may present mild skin changes with widespread bone pathology. Affected patients may develop pustular rash, in which there may be discrete crops of pustules or a severe generalized pustular eruption that may resemble generalized pustular psoriasis. Other variable features include failure to thrive, lung disease, generalized ichthyosis-like changes, nail changes, stomatitis, mouth ulcers, conjunctivitis, lung involvement, vasculitis, hepatosplenomegaly, joint swelling, periostitis, and osteomyelitis. 7 8 The myriad dermatologic conditions that may be considered as part of DIRA's differential diagnosis include acrodermatitis enteropathica, acute generalized exanthematous pustulosis, benign neonatal pustulosis, bullous impetigo, dermatitis herpetiformis, eosinophilic pustular folliculitis, folliculitis, IgA pemphigus, infantile acne, infantile acropustulosis, infantile atopic dermatitis (with or without superinfection), infantile seborrheic dermatitis, Langerhans cell histiocytosis, pustular psoriasis, scabies, and toxic epidermal necrolysis.
Diagnosis
Diagnostic tests are directed toward the identification of etiology ( Table 1 ). Nevertheless, DIRA can only be diagnosed by genetic analysis. 8
Table 1. Diagnostic tests and possible findings consistent with deficiency of interleukin-1 receptor antagonist.
| Blood tests | • Increased erythrocyte sedimentation rate • Increased C-reactive protein • Leukocytosis, thrombocytosis, and anemia can present with chronic inflammation |
| Skin biopsy | • Epidermal neutrophilic pustules • Pustule formation along hair follicles • Epidermal acanthosis (skin thickening) • Hyperkeratosis (scaling) • Heavy neutrophil infiltrate of dermis • Thrombosis in blood vessels |
| Radiographs | • Balloon-like swelling of the ends of the ribs • Periosteal elevation along multiple long bones • Heterotopic ossification around the hip • Multifocal osteolytic lesions |
| Bone biopsy | • Aseptic purulent osteomyelitis • Fibrosis • Sclerosis • Vasculitis in adjacent fat and connective tissue |
| Brain MRI | • Evidence of vasculitis |
| DNA analysis | • Point mutations or large genomic deletion involving the IL1RN gene resulting in the absence of functional interleukin-1 receptor antagonist |
Abbreviation: MRI, magnetic resonance imaging.
Management
Acute management of symptoms associated with DIRA includes supportive care. High doses of corticosteroids can reduce symptoms; however, definite treatment involves life-long administration of a recombinant IL-1 Ra, also known as anakinra. Daily subcutaneous administration of anakinra replaces the missing protein. Skin changes may respond within days and bone changes may resolve within months.
Prognosis
Long-term prognosis is good if diagnosed and treated early in life, allowing the patient to possibly live a normal life. If untreated, DIRA results in multiorgan failure and death in early childhood. 9 10
Case Presentation
We describe the clinical presentation of a 5-month-old infant with DIRA whose early and rare clinical presentation made it a diagnostic challenge for pediatricians and subspecialists. The patient initially developed erythematous skin patches that were treated with oral antibiotics, with subsequent bronchiolitis approximately 1 month prior to admission. Despite medical treatment, the patient continued with worsening skin findings that included erythema, edema, and suppurative lesions, associated with cough, nasal congestion, and difficulty breathing.
Past medical history was remarkable for a preterm vaginal birth at 36 weeks, birth weight 3.4 kg, length 21 inches, and born to a nonconsanguineous primiparous couple, whose families live in the northwest region of Puerto Rico. The pregnancy was complicated by hyperemesis gravidarum. At 1 week, the patient presented with unspecified dermatitis that was treated with a topical steroid cream with mild improvement. At 3 weeks, he was diagnosed with a right femur fracture of unknown etiology, for which he was hospitalized for intravenous (IV) antibiotic therapy and leg immobilization. At 4 months, he was hospitalized due to persistent emesis, clinical sepsis was suspected, and IV antibiotics were given. Due to reflux, the patient underwent multiple formula changes, including goat milk for 2 weeks. Developmental milestones were delayed and consistent with that of 2 months old. In total, 1 month prior to admission, the patient was evaluated by a pediatric geneticist who ordered a karyotype and metabolic workup; results were found XY and within normal limits, respectively. Family history was unremarkable.
Physical examination showed an acutely ill-appearing male with severe whole-body edema, with diffuse erythematous plaques, desquamation, pustules, and yellow crusted crops. He also had distal interphalangeal joint contracture of left third phalange, nail changes, mouth ulcers ( Fig. 1 ), yellow nasal discharge, bilateral wheezes, and rhonchi. The rest of his physical findings were normal. Laboratory workup showed leukocytosis (25.5 × 10 3 /µL), microcytic hypochromic anemia (Hgb 8.8 g/dL, MCV 66.8), and thrombocytosis (728 × 10 3 /µL); with an elevated erythrocyte sedimentation rate 59 mm/h and c-reactive protein 5.72 mg/dL. The patient was admitted to the inpatient floor for further management. Workup was directed toward the evaluation of the dermatological findings ( Table 1 ).
Fig. 1.
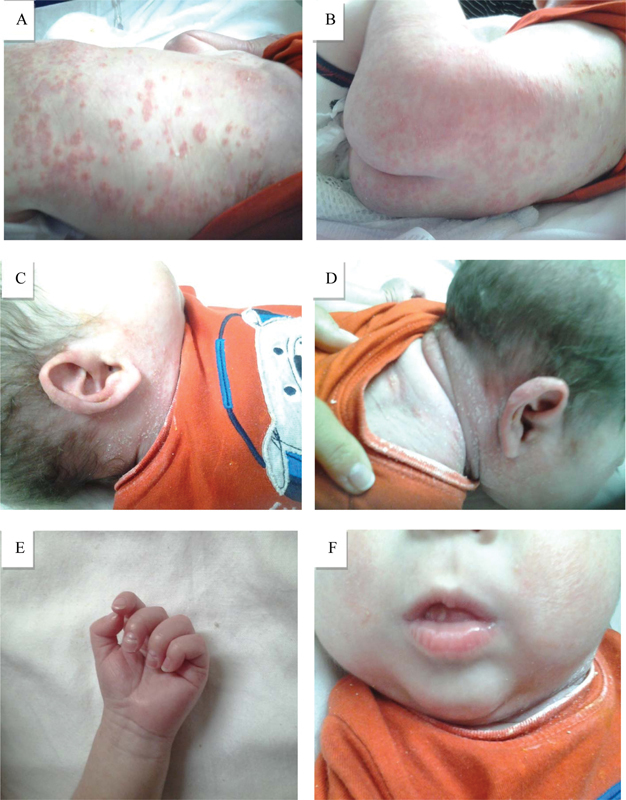
Skin manifestations of the disease.
An interdisciplinary approach was utilized with consultation to multiple subspecialists, including allergy and immunology, dermatology, genetics, gastroenterology, infectious disease and rheumatology to rule out immunologic deficiencies, metabolic diseases, malnutrition, resistant infectious processes, and autoimmune diseases, respectively. Metabolic and immunologic workup was unremarkable in the absence of an infectious process. Despite adequate nutrition, multiple courses of IV antibiotic therapy, systemic, and topical steroid therapy; the patient maintained persistent leukocytosis and elevated inflammatory markers, while his skin condition worsened. Microscopic examination of the dermatologic lesions revealed psoriasiform dermatitis with parakeratosis and subcorneal neutrophils. Skin bacterial cultures showed normal skin flora. Due to the combination of physical and histologic findings, DIRA was suspected.
A bone scan with and without gallium showed increased radionucleotide localization in the upper thoracic spine, anterior aspect of one of the mid left ribs, and left femur consistent with an inflammatory process and osteomyelitis in proximal left femur. As the patient continued to deteriorate, he became unresponsive to steroid therapy to which he had been partially responsive, developed intermittent fever, facial ichthyosis, conjunctivitis, and denudation of the skin involving 98% of his body surface area ( Fig. 2 ). He was then transferred to the intensive care unit due to high risk of sepsis for IV antibiotic therapy, IV hydration, skin care, and empiric administration of subcutaneous anakinra. Blood sample for genetic sequencing of IL1RN gene for the diagnosis of DIRA was drawn and sent for genetic analysis. After 72 hours of daily anakinra administration at 0.5 mg/kg/day, the patient's condition improved. Follow-up bone scan showed increased radionucleotide localization on the left femur, upper thoracic spine, anterior aspect of multiple right ribs, two of mid left ribs in the anterior aspect, right acetabulum, proximal metaphysis of the right tibia and distal metaphysis, consistent with multifocal osteomyelitis and osteitis ( Fig. 3 ). Anakinra was increased to 1 mg/kg/day, after which the patient's skin continued to improve ( Fig. 4 ). A femoral central line was placed and thrombosed, for which Enoxaparin sodium was administered.
Fig. 2.
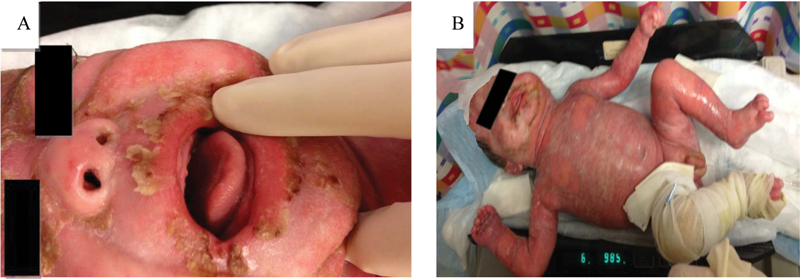
Clinical progression.
Fig. 3.
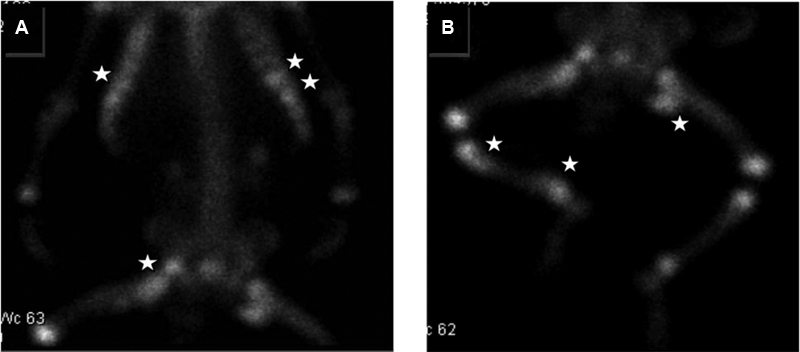
Bone scan showing orthopedic anomalies.
Fig. 4.
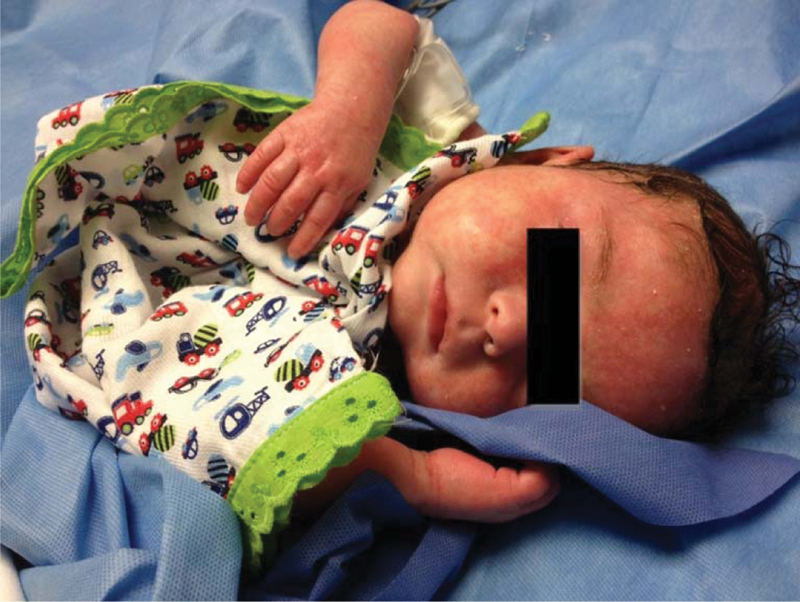
Clinical progression of skin manifestation after anakinra administration.
Anakinra was slowly increased until reaching an administration dose of 2 mg/kg/day. A follow-up bone scan performed 1 month after treatment with anakinra showed persistent changes in proximal left femur with improvement as compared with previous study, and increased activity noted in proximal and distal metaphysis of right tibia; consistent with improved multifocal osteomyelitis. Inflammatory markers and leukocytosis remained elevated. The patient remained hemodynamically stable and afebrile, and due to negative blood cultures and lack of clinical deterioration after 76 days, the patient was discharged home on anakinra (2 mg/kg/day) at 13 mg subcutaneously daily with rheumatology follow-up.
In total, 4 weeks after discharge, the diagnosis of DIRA was confirmed by genetic Sanger sequencing of IL1RN gene, as homozygous for a 175-kb deletion on chromosome 2q (OMIM 612852).
Discussion
DIRA is a genetic inflammatory disease, first reported in 2009, 1 9 10 in which a germline mutation in IL1RN causes the absence of IL-1 Ra, subsequently producing an unopposed cascade of proinflammatory cytokines that causes a life-threatening systemic inflammation. 1 9 Cases originating from Puerto Rico, United States, Canada, Germany, Brazil, Netherlands, India, Türkiye, and Lebanon have been reported. 1 A 175-kb deletion on chromosome 2q13, a founder mutation in five Puerto Rican patients with DIRA was reported with an estimated allele frequency of 1.3% and an incidence of 1 in 6,300 births in Puerto Rico. 1 2 9 Its clinical manifestations resemble those of inflammatory diseases with multiorgan involvement. The prominent role of interleukin-1 cytokines in the development of skin and bone manifestations in affected patients suggests that interleukin-1 plays a role in the pathophysiology of autoinflammatory bone disorders such as chronic recurrent multifocal osteomyelitis and the syndrome with synovitis, acne, pustulosis, hyperostosis, and osteitis. Elevated levels of IL-1β have been associated with preterm labor; deficiency of interleukin-1 receptor antagonist may therefore explain the premature birth of some infants with the disease. 10
In the case reported, the patient presented neonatal onset pustulosis with erythematous scaly plaques, failure to thrive, joint contractures, conjunctivitis, radiologic findings of multifocal osteomyelitis and osteitis, and Doppler ultrasound notable for right femoral vein thrombosis. Pulmonary involvement has also been reported in the literature, with respiratory problems in infants shortly after birth; ranging from respiratory distress, apnea, or aspiration pneumonia to interstitial lung disease, pulmonary fibrosis, and ultimately respiratory failure. 11 12 The patient in this case was diagnosed with bronchiolitis at 5 months. This may have been a confounder, and potentially part of DIRA's early presentation as part of his clinical course. Due to high suspicion of deficiency of interleukin-1 receptor antagonist and the patient's marked clinical deterioration despite high steroid therapy, anakinra was empirically initiated while awaiting genetic analysis for the IL1RN gene mutation. Reported acute anakinra-related adverse effects include transient injection-site reactions (the most common characterized by mild erythema, ecchymosis, inflammation, and pain lasting 2 to 4 weeks), catarrhal symptoms, headache, nausea, diarrhea, and abdominal pain. Late onset toxicity includes weight gain, infection, neutropenia, thrombocytopenia, liver toxicity, and hypersensitivity reactions. None of which were reported in our patient. Anti-IL1 immunosuppressive medications, such as anakinra, rilonacept and canakinumab, are utilized as the therapy of choice in inflammasomopathies. However, anakinra is not currently approved by the U.S. Food and Drug Administration for the use in children less than 2 years of age, although the literature establishes the successful use of the recombinant medication in select patients within this age group. 1 The patient's mother was oriented about anakinra, side effects and complications, and therapeutic indications. Due to its off-label use, future administration in similar cases may require literature review to ascertain the optimal therapeutic dose.
The patient showed remarkable improvement while on anakinra therapy. At the time of discharge, the patient's inflammatory markers and complete blood cell counts improved but remained elevated. Patients with DIRA who carry the chromosomal deletion have undergone clinical remission and normalization of acute-phase reactant levels and complete blood cell counts. However, like our patient, a Puerto Rican patient homozygous for the founder mutation, presented a rapid clinical response after anakinra administration and dose increase, but remained with elevated inflammatory markers. 1
Conclusion
To our knowledge, our patient is the sixth Puerto Rican patient with clinical and genetically confirmed findings for the homozygous mutation consistent with DIRA. 12 He is one of the youngest cases in the literature to be started on anakinra empirically prior to genetic confirmation. 12 DIRA must be included in the differential diagnosis of a pediatric patient who presents with neonatal generalized pustulosis and treat accordingly on the basis of suspicion to reduce the high morbidity and mortality associated with delayed diagnosis and treatment. Screening of newborns may be warranted in high-risk populations.
Footnotes
Conflict of Interest None declared.
References
- 1.Aksentijevich I, Masters S L, Ferguson P J et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360(23):2426–2437. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CeRéMAI, French reference center for autoinflammatory diseases . Touitou I, Galeotti C, Rossi-Semerano L, Hentgen V, Piram M, Koné-Paut I. The expanding spectrum of rare monogenic autoinflammatory diseases. Orphanet J Rare Dis. 2013;8(01):162. doi: 10.1186/1750-1172-8-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cowen E W, Goldbach-Mansky R. DIRA, DITRA, and new insights into pathways of skin inflammation: what's in a name? Arch Dermatol. 2012;148(03):381–384. doi: 10.1001/archdermatol.2011.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldbach-Mansky R. Immunology in clinic review series; focus on autoinflammatory diseases: update on monogenic autoinflammatory diseases: the role of interleukin (IL)-1 and an emerging role for cytokines beyond IL-1. Clin Exp Immunol. 2012;167(03):391–404. doi: 10.1111/j.1365-2249.2011.04533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gery I, Gershon R K, Waksman B H. Potentiation of the T-lymphocyte response to mitogens. I. The responding cell. J Exp Med. 1972;136(01):128–142. doi: 10.1084/jem.136.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenstreich D L, Vogel S N, Jacques A R, Wahl L M, Oppenheim J J. Macrophage sensitivity to endotoxin: genetic control by a single codominant gene. J Immunol. 1978;121(05):1664–1670. [PubMed] [Google Scholar]
- 7.Verbsky J W. Monogenic causes of inflammatory disease in rheumatology. Curr Opin Rheumatol. 2012;24(05):506–514. doi: 10.1097/BOR.0b013e32835689b9. [DOI] [PubMed] [Google Scholar]
- 8.Minkis K, Aksentijevich I, Goldbach-Mansky R et al. Interleukin 1 receptor antagonist deficiency presenting as infantile pustulosis mimicking infantile pustular psoriasis. Arch Dermatol. 2012;148(06):747–752. doi: 10.1001/archdermatol.2011.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brau-Javier C N, Gonzales-Chavez J, Toro J R. Chronic cutaneous pustulosis due to a 175-kb deletion on chromosome 2q13: excellent response to anakinra. Arch Dermatol. 2012;148(03):301–304. doi: 10.1001/archdermatol.2011.2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reddy S, Jia S, Geoffrey R et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360(23):2438–2444. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassidy J T, Petty R E, Laxer R M, Lindsley C B. Elsevier Health Sciences; 2010. Textbook of Pediatric Rheumatology E-Book. [Google Scholar]
- 12.Diaz A.2019Deficiency of the Interleukin-1 Receptor Antagonist (DIRA)In Auto-Inflammatory Syndromes.Springer; Cham, Switzerland: 69–83. [Google Scholar]