

Abstract
Growing evidence has proved that RNA editing enzyme ADAR1, responsible for detecting endogenous RNA species, was significantly associated with poor response or resistance to immune checkpoint blockade (ICB) therapy. Here, a genetically engineered nanovesicle (siAdar1-LNP@mPD1) was developed as an RNA interference nano-tool to overcome tumor resistance to ICB therapies. Small interfering RNA against ADAR1 (siAdar1) was packaged into a lipid nanoparticle (LNP), which was further coated with plasma membrane extracted from the genetically engineered cells overexpressing PD1. siAdar1-LNP@mPD1 could block the PD1/PDL1 immune inhibitory axis by presenting the PD1 protein on the coating membranes. Furthermore, siAdar1 could be effectively delivered into cancer cells by the designed nanovesicle to silence ADAR1 expression, resulting in an increased type I/II interferon (IFN-β/γ) production and making the cancer cells more sensitive to secreted effector cytokines such as IFN-γ with significant cell growth arrest. These integrated functions confer siAdar1-LNP@mPD1 with robust and comprehensive antitumor immunity, as evidenced by significant tumor growth regression, abscopal tumor prevention, and effective suppression of lung metastasis, through a global remodeling of the tumor immune microenvironment. Overall, we provided a promising translatable strategy to simultaneously silence ADAR1 and block PDL1 immune checkpoint to boost robust antitumor immunity.
Keywords: genetically engineered cell membrane, lipid nanoparticle, ADAR1 silence, PDL1 blocking, cancer immunotherapy
Graphical abstract
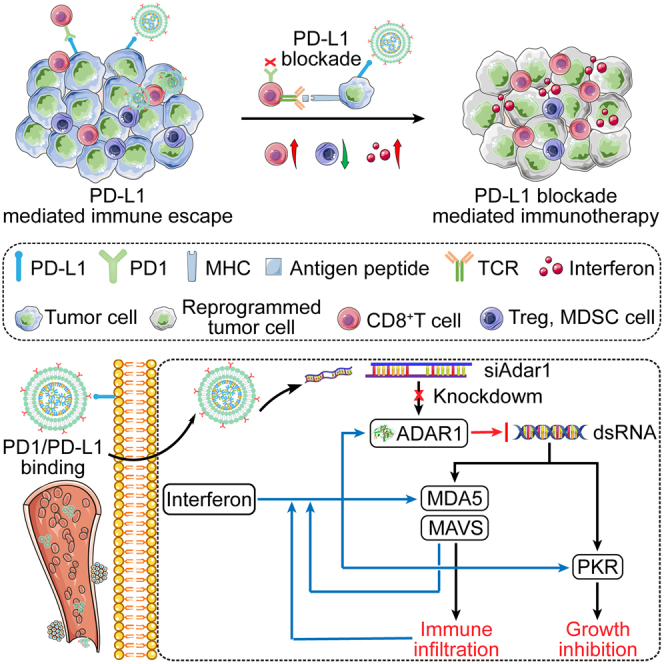
Liu and colleagues reported a biomimetic nanovesicle by cloaking the genetically engineered cell membrane on the surface of lipid nanoparticle loaded with siRNA. These nanovesicles can effectively knock down ADAR1 and block the PD1/PDL1 immunosuppressive pathway to enhance immune checkpoint blockade efficiency.
Introduction
Immune checkpoint blockade (ICB) therapy, such as antibody blockade of programmed death 1/programmed death-ligand 1 (PD1/PDL1), has shown remarkable clinical successes.1 PD1 is mainly expressed on activated T lymphocytes, whereas PDL1 is its ligand and expressed on various cells. The interaction of PD1 with PDL1 leads to T cell exhaustion. Blockade of their interaction with antibodies or other inhibitors can therefore boost the immune response against cancer cells. Nevertheless, only a small proportion of patients (around 20%) can respond well to ICB.2,3 The poor response may be due to the low infiltration of tumor-killing T cells, the tumor immunosuppressive microenvironment, or inadequate antigen presentation.4 Therefore, it is highly pursued to identify new therapeutic targets and develop alternative approaches for elevating the therapeutic efficiency of immunotherapy. Recently, ADAR1 involved in adenosine-to-inosine (A-to-I) RNA editing was found to be one of the intrinsic immunosuppressors for ICB resistance.5 By analyzing The Cancer Genome Atlas database, Liang’s group and Levanon’s group revealed that ADAR1 expression and A-to-I editing levels were significantly elevated in tumor compared with their matched normal tissues, with the highest expression in breast, thyroid, head and neck, and lung cancers.6,7 The hyper-editing and neutralizing of immunogenic dsRNA by ADAR1 lead to the inactivation of the MDA5-MAVS-IFN signaling pathway, which finally assists cancer cells to suppress inflammatory responses for escaping from host immunosurveillance.8 Ishizuka et al. found that disabling ADAR1 in tumor cells via CRISPR-Cas9 editing can enhance tumor sensitivity to type I and type II interferons (IFNs), helping to overcome ICB resistance by reshaping the tumor microenvironment (TME).9 Thus, ADAR1 can be used as a key target for developing novel strategies to improve responses to ICB therapy. However, there is no drug that specifically inhibits ADAR1 to decrease the hyper-editing events in tumor progression up to now.
RNA interference represented by siRNA holds great promise in cancer therapy.10,11 In fact, siRNA drugs in the form of lipid nanoparticles (LNPs) have been approved to treat several diseases in the clinic.12,13,14 However, the complicated interactions between LNPs and the physiological environment severely compromise the in vivo performance, such as surface dissolution, protein adsorption, nonspecific nanoparticle uptake, and clearance by the reticuloendothelial system.15 Cell membrane decoration on the nanoparticle surface is an effective strategy to enhance circulation durations and specific targeting capabilities during in vivo applications.16 For example, our group has developed a fusion nanovesicle assembly from the genetically engineered cell membranes (GECMs), which are modified to overexpress PD1 for tumor immunotherapy.17,18 In addition to disrupting the PD1/PDL1 immune inhibitory axis, PD1-overexpressing genetically engineered cell membrane vesicles (GECVs) can also act as tumor-targeting vehicles by exploiting the binding between the PD1 receptor on GECVs and PDL1 on cancer cells.17,19,20
Here, we report a GECM-cloaked LNP as the siRNA delivery vehicle for in vivo silencing ADAR1 and blocking PD1/PDL1 immunosuppressive pathway to amplify immune response to ICB therapy (Figure 1A). Specifically, the synthetic ADAR1-siRNA was loaded into the LNP (siAdar1-LNP) with high encapsulation efficiency. Subsequently, the siAdar1-loaded LNP was coated with a layer of the GECM with PD1 overexpression (siAdar1-LNP@mPD1). We validated the function and performance of our designed nanovesicles in 4T1 tumor models with different tumorigenic types, which showed great potential in cancer immunotherapy for future clinical translation.
Figure 1.

Preparation and characterization of siAdar1-LNP@mPD1 nanovesicles
(A) Schematic illustration of siAdar1-LNP@mPD1 nanovesicle preparation and the principle of antitumor immune response. (B–D) CLSM images (B), flow cytometry analysis (C), and western blot (WB) analysis (D) of stably expressing PD1 on engineered CHO cell membrane. Scale bar, 5 μm. (E) The interaction between mPD1 NVs and anti-PD1 antibody detected by co-immunoprecipitation (coIP). For immunoprecipitation, anti-PD1 antibody was used to pull down mPD1 NVs, while IgG was used as the control. (F) CLSM images of mPD1 NVs binding to PDL1-EGFP that is intentionally overexpressed on 4T1 cells, the co-localization of mPD1 NVs and PDL1 proteins are represented by the yellow colored dots as indicated by white arrows. Scale bar, 20 μm). mPD1 NVs (40 μg/mL) were incubated with 4T1 cells at 4°C for 1 h. (G) CLSM images of siAdar1-LNP with Cy5-labeled siAdar1 (red fluorescence) and LNP loaded with DiO (green fluorescence). Scale bars, 20 μm. (H) Gel fluorescence imaging of free siAdar1 and siAdar1-LNP. (I and J) Transmission electron microscopy images (I) and dynamic light scattering (DLS) results (J) of siAdar1-LNP and siAdar1-LNP@mPD1. Scale bar, 100 nm. (K and L) SDS-PAGE electrophoresis patterns (K) and WB analysis (L) of siAdar1-LNP (I), mPD1 NVs (II), and siAdar1-LNP@mPD1 (III).
Results
Characterization of siAdar1-LNP@mPD1
Different from our and other previous studies using HEK293 cells or even cancer cells as the parent cells with undesirable immunogenicity or the risk of oncogenicity,17,20 we used Chinese hamster ovary (CHO) cells as the host cells for the production of PD1-expressing cell membranes. As the most common workhorse of newly approved recombinant biotherapeutic products, CHO cells can be stably integrated with exogenous genes with low immunogenicity.21 We first constructed a stably transfected CHO line (PD1-CHO) expressing PD1 protein with mCherry tag (PD1-mCherry) on the cell membrane by using a lentiviral vector through our established method (Figures S1 and S2).17 Note that the mCherry tag was used only as a fluorescent indicator for PD1, and it has been removed in other experiments to rule out its influence on PD1 function. Notably, the PD1 proteins were obviously expressed on the cell membrane, as verified by confocal laser scanning microscopy (CLSM) images that showed the red fluorescence of PD1-mCherry co-localized with the green fluorescence of membrane-staining dye DiO (Figure 1B). Furthermore, flow cytometry (FCM) analysis revealed that approximately 99% of the genetically modified CHO cells successfully expressed PD1 protein (Figure 1C). The presence of recombinant PD1 protein on CHO cell membranes was further verified by western blot (WB) analysis (Figures 1D and S3A). In addition, the PD1 contents in CHO cell membranes were measured by enzyme-linked immunosorbent assay (ELISA) (Figure S4), and the ratio of PD1 to total proteins was calculated to be 3.65%. These results confirmed that the PD1 proteins overexpressing CHO cells were successfully established.
Next, the PD1-anchored cell membrane (mPD1) was extracted and assembled into nanovesicles (mPD1 NVs) by sonication. As shown in Figures 1E and S3B, correct outside-out orientation of PD1 protein on the mPD1 NVs surface was proved by co-immunoprecipitation (coIP) assay, as evidenced by the fact that the anti-PD1 antibody could pull down the majority of mPD1 NVs. PDL1 proteins, which are highly expressed on the surface of tumor cells, are well known to interact with PD1 receptors on activated cytotoxic T cells to accomplish tumor immune escape.22 Therefore, inhibiting the PD1/PDL1 connection can significantly boost the immune response against tumor cells. From the coIP assay, the correct orientation of PD1 proteins on the mPD1 NVs allowed the vesicles to bind with PDL1. To further investigate this interaction, 4T1 cells were transfected with the PDL1-EGFP plasmid to generate PDL1-EGFP-expressing cells, which were further incubated with the mPD1 NVs. As expected, the co-localization signal of EGFP and PD1 verified the effective binding of mPD1 NVs to PDL1 on cancer cells. The presence of unmerged red fluorescent dots suggested that mPD1 NVs might also bind to endogenous PDL1 in 4T1 cells (Figure 1F). Accordingly, FCM analysis showed that mPD1 NVs but not the control vesicles (without PD1 presentation, m NVs) could effectively bind to 4T1 cells. However, such binding could be blocked by PDL1 antibody pretreatment (Figure S5). Next, coIP assays were also utilized to confirm whether mPD1 NVs can bind PDL1. For this purpose, mPD1 NVs or m NVs were co-incubated with recombinant mouse PDL1 protein at 4°C for 1 h, then the precipitates were collected by centrifugation and subjected to WB analysis. Figure S6 revealed a distinct band corresponding to the PDL1 protein in the group treated with mPD1 NVs. These results suggested that PD1-anchored NVs could specifically bind to tumor cells through the interaction between PD1 and PDL1, which might serve as a PDL1 blockade for ICB therapy.
Encouraged by the specific binding ability of mPD1 NVs, we investigated the effects of blocking the PD1/PDL1 immune checkpoint with mPD1 NVs on immune cells. To this end, CFSE-labeled CD8+ T cells were incubated with 4T1 cells pretreated with either PBS, m NVs, or mPD1 NVs. After incubation for 5 days, FCM analysis revealed that CD8+ T cells in the mPD1 NVs group proliferated more obviously than those cells incubated with non-treated or m NV-treated 4T1 cells (Figure S7). CD69, a well-established hallmark of T cell activation,23 was also significantly elevated on CD8+ T cells in the mPD1 NV-treated group (Figures S8A and S8B). Consistent with these findings, CD25 as an IL-2 receptor alpha chain (IL-2Rα) that promotes clonal expansion of T cells,24 was markedly increased in this group (Figures S8C and S8D). These results suggest that mPD1 NVs possess the potential to augment the immune response by disrupting PD1/PDL1 interactions between T cells and cancer cells.
Then, the siAdar1-LNP was prepared according to the previously reported method by an ethanol-injection process.25 To demonstrate the effective encapsulation, we fabricated the double fluorescence-labeled siAdar1-LNP with a larger size under mild stirring (Cy5-labeled siAdar1; DiO-labeled LNP), which was photographed by CLSM. As shown in Figure 1G, the CLSM images exhibited merged (yellow) fluorescence signals of Cy5 and DiO dye, indicating that the siRNA was successfully encapsulated inside the LNP. Meanwhile, the prepared siAdar1-LNP showed no electrophoretic shift in 3% agarose gel compared with free siAdar1, further validating the effective packaging of siAdar1 into LNPs (Figure 1H). The superiority of the siAdar1-LNP delivery system was demonstrated by an encapsulation efficiency of 94%, as determined by RiboGreen quantification assays,26 compared with only 37% for direct loading of siAdar1 into nanovesicles that only self-assembled from cell membranes (Figure S9). The high encapsulation efficiency of siAdar1-LNP is because LNP can provide a highly positive and dense core for trapping negatively charged siRNA.
Then, the mPD1 was used to coat siAdar1-LNP (siAdar1-LNP@mPD1) by repeated sonication and co-extrusion through a series of different pore-sized polycarbonate membranes with a mini extruder. The size and morphology of siAdar1-LNP and siAdar1-LNP@mPD1 were observed by transmission electron microscopy (TEM). Compared with siAdar1-LNP, siAdar1-LNP@mPD1 exhibited a distinct core-shell structure (Figure 1I). The hydrodynamic diameter of siAdar1-LNP was about 141 nm (polydispersity index [PDI], 0.12), while that of siAdar1-LNP@mPD1 was increased to 181 nm (PDI, 0.21) according to dynamic light scattering (DLS) measurement (Figures 1J; Table S1). Due to the mild aggregation nature of nanoparticles in solution, the size of the nanoparticles in wet measurement condition (DLS) was somewhat larger than that detected by TEM (dry conditions).27 Compared with the positive charge of siAdar1-LNP, the zeta potential of the membrane-coated siAdar1-LNP@mPD1 decreased to −6.9 ± 0.9 mV (Figure S10A; Table S1). The siAdar1-LNP@mPD1 showed good colloidal stability when suspended in the culture medium containing 10% of FBS at 4°C during 10-day observation period (Figure S10B). By comparison, the siAdar1-LNP showed an increase in size and PDI under identical conditions over time (Figure S10C). To further assess functional stability, the siAdar1-LNP@mPD1 stored at different time points were co-cultured with 4T1 cells for 48 h each. The quantitative reverse-transcriptase PCR (qRT-PCR) results showed that siAdar1-LNP@mPD1 stored for 0, 5, and 10 days were all able to effectively reduce ADAR1 mRNA levels, without any significant differences (Figure S10D). Coomassie blue staining revealed that the siAdar1-LNP@mPD1 had nearly the same electrophoresis patterns as mPD1 NVs (Figure 1K). Accordingly, the PD1 protein was also retained after coating of the cell membrane as demonstrated by WB analysis (Figures 1L and S11). Furthermore, we performed coIP assay to confirm whether the siAdar1-LNP@mPD1 maintain PD1 function. Accordingly, siAdar1-LNP@m or siAdar1-LNP@mPD1 were co-incubated with recombinant mouse PDL1 protein at 4°C for 1 h, and the precipitates were collected by centrifugation and subjected to WB analysis. Figure S12 showed a much stronger band of PDL1 protein in the siAdar1-LNP@mPD1 precipitation group than the siAdar1-LNP@m precipitation group without PD1 overexpression, confirming the reserve of the PD1 protein function of cell membrane after squeezing with LNP to construct siAdar1-LNP@mPD1. In addition, the fluorescence emission spectrum of siAdar1-LNP@mPD1 was also very similar to PD1 NVs with mCherry tag (λem at 545 nm, Figure S13). Overall, the above results demonstrated that PD1-anchored cell membrane was successfully coated on siAdar1-LNP.
siRNA delivery by nanovesicles for gene expression silence in vitro
ADAR1 expression is a common phenomenon in almost all types of cancers, as revealed by The Cancer Genome Atlas database7 and supported by our exploration. As shown in Figures S14 and S15, the expression of ADAR1 (p150 and p110 isoforms) could be obviously detected in cancer cells with different degrees, such as mouse breast cancer 4T1 cells, mouse colon cancer CT26 cells, mouse liver cancer Hepa 1-6 and H22 cells, mouse colon cancer MC38 cells, human hepatocellular carcinoma SMMC-7721 cells, human hepatocellular carcinoma HepG2 cells, and human cervical cancer HeLa cells, but not in normal cells of mouse hepatocyte BNL CL.2 and human immortalized oral epithelial human immortalized oral epithelial cells (HIOECs). To more favorably demonstrate the potential value of ADAR1 as a therapeutic target, we selected 4T1 cancer cells with the highest ADAR1 level as the model for following investigations. Firstly, we investigated the gene silencing efficiency of three different siRNA sequences for targeting ADAR1 of 4T1 cells by a commercial transfection reagent (Lipofectamine 3000). The qRT-PCR and WB assay results showed that siAdar1-mus-3302 (si3302) could effectively reduce the level of ADAR1 mRNA and downregulate the expression of ADAR1 protein (Figure S16). Thereafter, si3302 was used for preparing the siAdar1-LNP nanovesicles. Before exploring their gene silencing effect, we first evaluated the biocompatibility by the standard Cell Counting Kit-8 (CCK-8) test. Negligible toxicity was observed in both normal mouse fibroblast NIH3T3 cells or mouse 4T1 cells when incubated with even as high as 200 nM siAdar1-LNP (based on the concentration of siAdar1) (Figure 2A). Then, the cellular internalization of siRNA within LNPs was investigated on CLSM. Remarkably, the green fluorescence signal of 5-FAM-labeled siAdar1 was observed in 4T1 cells after 4 h of incubation, and the signal enhanced along with the increase in incubation time (Figure 2B). In contrast, almost no fluorescence of 5-FAM appeared in 4T1 cells even incubated with free siAdar1 for more than 48 h. Therefore, LNPs could significantly promote the cellular uptake of siRNA payloads. To investigate the biological activity of siAdar1 after being delivered by LNPs, the mRNA level and protein expression of ADAR1 were then evaluated. As shown in Figure 2C, the mRNA of ADAR1 was nearly unchanged in scrambled siRNA-loaded LNPs (siNC-LNP) and the free siAdar1 group. However, the expression of ADAR1 mRNA was apparently downregulated in the siAdar1-LNP-treated group, which was comparable with the positive control-treated group transfected by Lipofectamine 3000. Correspondingly, the WB analysis for protein expression (Figures 2D, 2E, and S17) also presented the same trend, indicating the ADAR1 silencing effect of siAdar1-LNP.
Figure 2.

In vitro immune checkpoint inhibition and ADAR1 silence of siAdar1-LNP@mPD1 nanovesicle
(A) Relative viability of 4T1 cells and NIH3T3cells after incubation with siAdar1-LNP, as determined by CCK-8 assays. Error bar: mean ± SD (n = 5). (B) CLSM images of 4T1 cells treated with 5-FAM-labeled siAdar1-LNP for 4, 24, and 48 h, or free siAdar1 (for 48 h). Scale bar, 20 μm. (C–E) The qRT-PCR analysis of the relative expression of ADAR1 mRNA (C), WB analysis (D), and the relative expression (E) of ADAR1 protein from 4T1 cells after different treatments (G1 to G5). G1, blank control without any treatment; G2, siNC-LNP; G3, free siAdar1; G4, siAdar1-LNP; G5, positive control with siAdar1 transfected by Lipofectamine 3000 (mean ± SD; ∗∗∗∗p < 0.0001; n = 3). (F) CLSM images of the nanoparticle internalization behaviors in 4T1 cells when treated with siAdar1-LNP@m, siAdar1-LNP@mPD1, and siAdar1-LNP@mPD1 + anti-PDL1. The siAdar1 was labeled with 5-FAM for fluorescence imaging. Scale bar, 20 μm. (G) Schematic diagram of IFN-γ response (left) and the relative viability of 4T1 cells treated by INF-γ with different concentrations (0.1–0.5 μg/mL) (right). 4T1 cells were pretreated with PBS or siAdar1-LNP@mPD1 before INF-γ incubation (mean ± SD; ∗∗∗∗p < 0.0001; n = 5).
In view of the fact that PDL1 blocking by antibody is able to induce its subsequent internalization,28 we anticipated that the endocytosis of siAdar1-LNP@mPD1 could be mediated by the specific interaction of mPD1 and PDL1. Compared with control nanocarriers (siAdar1-LNP coated with native cell membrane, siAdar1-LNP@m), siAdar1-LNP@mPD1 increased the delivery of siAdar1 to 4T1 cells after 24 h of incubation. However, such enhanced uptake could be abolished when the cancer cells were pretreated with PDL1 antibody (Figure 2F). These results demonstrate that the PD1 membrane cloaking is in favor of LNPS to deliver siRNA into cancer cells. To provide more experimental evidence that the binding of mPD1 to PDL1 can promote endocytosis of siAdar1-LNP@mPD1, we further performed the following studies. First, 4T1 cells were incubated with Cy5-labeled siAdar1-LNP@m or siAdar1-LNP@mPD1 at low temperature (on ice) to eliminate energy-dependent endocytosis but allow ligand-receptor interaction.29 After 1 h of incubation, the supernatant was removed and the cells were incubated at 37°C for another 4 h to allow the internalization of the remaining nanoparticles pre-adherent to cell membrane in the previous step. CLSM results demonstrated significantly stronger Cy5 fluorescence in the cells treated with siAdar1-LNP@mPD1 than that in cells treated with siAdar1-LNP@m, with some co-localization with green fluorescence of LysoTracker DND-26 (Figure S18). This result is consistent with the previous findings reported by Zhai et al. that PD1-overexpressing cell membrane-decorated nanoparticles can be internalized in tumor cells through the binding between PD1 and PDL1.19
According to previous reports, the loss or downregulation of ADAR1 makes cancer cells more sensitive to IFN-γ.30 We tested this possibility by stimulating ADAR1-deficient 4T1 cells (treated by siAdar1-LNP@mPD1 for 48 h) and control cells (no treatments) with IFN-γ and comparing their cell viability. In contrast with the control cells that were highly resistant to IFN-γ, the cell viability of the siAdar1-LNP@mPD1-treated cells gradually decreases with increasing concentrations of IFN-γ (Figure 2G). Compared with cells treated with siAdar1-LNP@m, cells treated with siAdar1-LNP@mPD1 exhibited a higher susceptibility to INF-γ due to the enhanced cellular uptake (Figure S19). In addition, under the same concentration of INF-γ (0.5 μg/mL), cells treated with siAdar1-LNP showed comparable survival rate to siAdar1-LNP@mPD1, possibly due to the positive charge on the surface of siAdar1-LNP, which facilitates cellular uptake. This result suggests that the silence of ADAR1 by our nanovesicles can sensitize cancer cells to IFN-γ, which has a prospective potential for tumor therapy.
In vivo antitumor effect of siAdar1-LNP@mPD1
Before investigating the antitumor effect of siAdar1-LNP@mPD1, we firstly evaluated whether the as-prepared nanovesicles could effectively deliver siAdar1 into the tumor site. To this end, the subcutaneous (s.c.) 4T1 tumor models were established,31 and Cy5-labeled siAdar1-LNP@mPD1 were intravenously injected into the tumor-bearing mice (Figure 3A). After 6 h of injection, the fluorescence signal in the tumor area became obviously stronger in the siAdar1-LNP@mPD1-treated mice in comparison with free siAdar1 with limited signals (Figure 3B), because the nanoscaled formulations can passively enter into tumor tissue via the enhanced permeability and retention (EPR) effect.32 Impressively, the fluorescence signal could still be observed after 48 h, which was also brighter than the siAdar1-LNP@m group (Figure 3B) due to the binding effect between the PD1 receptor on siAdar1-LNP@mPD1 and PDL1 on cancer cells to enhance tumor accumulation.17,19,20 For quantitative analysis, the mice were sacrificed at 24 h post-injection and the major organs including tumors were excised for ex vivo imaging. As shown in Figures 3C and 3D, the fluorescence signal mainly appeared in liver and kidney in addition to tumor tissue, which is consistent with the previous report describing that the nanoscaled particles were cleared by the liver and then followed by degradation into small species in kidney for kidney-dependent clearance.33 Even so, tumor tissue of the mice receiving siAdar1-LNP@mPD1 still exhibited the highest siRNA-related fluorescence signal among these groups. In addition, the biodistribution of siAdar1-LNP without cell membrane cloaking was also examined. As shown in Figure S20, siAdar1-LNP showed a noticeable Cy5-siAdar1 signal in the lung compared with siAdar1-LNP@m or siAdar1-LNP@mPD1 with cell membrane decoration (Figure 3C), but with negligible accumulation at tumor sites. This result is consistent with the previous report describing that positively charged nanoparticles always led to severe aggregation under physiological conditions and is prone to deposit in pulmonary capillaries after intravenous injection, thus limiting their in vivo circulation for tumor accumulation via the EPR effect.34,35 Collectively, these results suggested that LNP as a vehicle could significantly improve the accumulation of siRNA in tumor site, which could be further strengthened by the PD1-engineered cell membrane decoration.
Figure 3.

Therapeutic effect on the subcutaneous 4T1 tumor model
(A) Schematic representation of the therapeutic timeline of siAdar1-LNP@mPD1 on the subcutaneous 4T1 tumor model. (B) In vivo fluorescence imaging of the 4T1 tumor-bearing mouse at different time points after intravenous (i.v.) injection of siAdar1-LNP@mPD1, siAdar1-LNP@m, or free siAdar1. (C) Ex vivo fluorescence imaging of excised major organs and tumors from the mice after 24 h i.v. injection of different formulation. (D) Analysis of the mean fluorescence intensity (MFI) of major organs and tumors by ImageJ software. (E) Average and individual tumor growth kinetics of mice after i.v. injection with PBS, siAdar1-LNP@m, siNC-LNP@mPD1, or siAdar1-LNP@mPD1 (mean ± SD; n = 10; ∗∗p < 0.01, ∗∗∗∗p < 0.0001). (F and G) Photographs (F) and weights (G) of tumors excised from all treated mice on day 20 (mean ± SD; n = 10; ∗∗p < 0.01, ∗∗∗∗p < 0.0001). (H) H&E, Ki67, and TUNEL staining of tumor slices after different treatments. Scale bars, 100 μm. (I–K) The qRT-PCR analysis of the relative expression of ADAR1 mRNA (I), WB analysis (J), and the relative expression (K) of ADAR1 protein from excised 4T1 tumor cells after different treatments (mean ± SD; ∗∗∗∗p < 0.0001; n = 3).
Based on the above results, we next assessed the therapeutic effect of siAdar1-LNP@mPD1 on mice after intravenous injection (repeated once every 3 days for a total of 4 times), and the body weight and tumor size of mice were measured every second day (Figure 3A). Tumor growth was suppressed considerably by siAdar1-LNP@m and siNC-LNP@mPD1, which enabled ADAR1 silencing or PDL1 blockade-based therapy, respectively, but siAdar1-LNP@mPD1 with the combined functions was much more potent in suppressing tumor growth (Figure 3E). On day 20 post-treatment, all excised tumors were photographed (Figures 3F and S21) and weighed (Figure 3G). Correspondingly, the tumor weights of the siAdar1-LNP@mPD1-treated group were much less than that of the PBS-treated group. To further provide evidence of the improved efficacy, histological analysis was performed. siAdar1-LNP@mPD1 treatment was found to result in the highest levels of proliferation inhibition (Ki67 staining), apoptosis (TUNEL staining), and necrosis (H&E staining) in tumor tissues compared with any other groups (Figure 3H). qRT-PCR (Figure 3I) and WB (Figures 3J and S22) revealed that the ADAR1 in tumor tissues was apparently downregulated after treatment with siAdar1-LNP@mPD1 and siAdar1-LNP@m, while siNC-LNP@mPD1 treatment had no effect on ADAR1 expression compared with the control group. There were no notable weight changes of mice for all groups throughout the course of treatment (Figure S23). Therefore, treatment of siAdar1-LNP@mPD1 successfully slowed down tumor growth through a self-sensitization strategy by the co-delivery of siRNA for ADAR1 silence and GECM with PDL1 blocking ability for ICB.
To elucidate the underlying mechanisms of tumor suppression, we analyzed the immune response in tumor tissues. As depicted in Figure 4A, loss of ADAR1 not only could sensitize tumor to IFN as demonstrated previously, but also could promote the secretion of IFN and increase tumor inflammation.9 We thus investigated the effect of nanovesicles on IFN-β/IFN-γ secretion in tumors by ELISA. As shown in Figures 4B and S24, siAdar1-loaded LNP (siAdar1-LNP) to silence ADAR1 in cancer cells could significantly promote intra-tumoral levels of IFN-β and INF-γ in comparison with the levels in the PBS-treated group. Most significantly, their levels in the siAdar1-LNP@mPD1-treated group were highest among all groups. This is due to the additive effect of mPD1 to reactivate T cells to secrete more antineoplastic cytokines for antitumor immunity. In addition, siAdar1-LNP@mPD1 treatment also led to a higher level of other pro-inflammation cytokines (IFN-β, IFN-γ, TNF-α, and IL-12) while inhibiting the secretion of anti-inflammatory cytokines (IL-10 and TGF-β1) (Figure 4B). Flow cytometry of immune populations confirmed increased frequency of effector T cells (CD3+CD8+ and CD3+CD4+) in tumor tissues receiving siAdar1-LNP@mPD1 treatment, while immunosuppressive cells such as Treg (CD4+Foxp3+) and MDSC (CD11b+Ly-6G+) exhibited a marked decline relative to the control group (Figures 4C–4F, S25, S26, and S27). Consistently, immunofluorescence staining showed the significantly increased intra-tumoral infiltration of CD8+ and CD4+ T cells in siAdar1-LNP@mPD1-treated mice compared with the PBS-treated mice (Figure 4G). All these results suggest that our developed nanovesicles integrated dual functions by blocking innate and adaptive immune checkpoints (i.e., ADAR1 and PDL1) can intensively reshape the immunosuppressive TME and boost antitumor immunity. These functions result in significant enhancement of the therapeutic efficacy against primary tumors.
Figure 4.

Immune response in the tumor site
(A) Schematic diagram of IFN production after treatment. (B) Cytokine levels (IFN-β, IFN-γ, TNF-α, IL-12, IL-10, and TGF-β1) in excised tumors after different treated mice by ELISA analysis. G1, PBS; G2, siAdar1-LNP@m; G3, siNC-LNP@mPD1; G4, siAdar1-LNP@mPD1 (mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n = 3). (C–F) Typical FCM plots and quantification results of CD3+CD8+ T cells (C), CD3+CD4+ T cells (D), Tregs (E), and MDSCs (F) in tumors of four different treatment groups (mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n = 3). (G) Multiple immunofluorescence images displayed the infiltrated CD4+ T cells (red) and CD8+ T cells (green) within tumors tissue. Scale bar, 100 μm. An enlarged image of the upper panel is shown in the lower panel. Scale bar, 25 μm. (H) Volcano plot of disregulated genes in tumor tissues with siAdar1-LNP@mPD1 treatment versus PBS treatment. (I) Heatmap showing disregulated genes in mice tumors after siAdar1-LNP@mPD1 and PBS treatments (fold change ≥ ± 10, false discovery rate < 0.05; n = 3). (J) KEGG enrichment analysis of the disregulated genes between siAdar1-LNP@mPD1 and PBS treatment.
To further explain the overall molecular mechanisms of the immune response in tumor tissues, we performed RNA sequencing analysis of tumor tissues after different treatments. Volcano plot revealed that, following siAdar1-LNP@mPD1 treatment, 116 transcripts were significantly disregulated when compared with those that received PBS treatment (Figure 4H). Among these disregulated genes, 55 were downregulated and 61 were upregulated. The heatmap showed the increased expression of IFN receptors (Ifnar1, Ifnar2), pro-inflammatory chemokines (Cxcl9, Cxcl10), and CD8+ T cell infiltration markers (Cd8a, Cd8b1) after the treatment with siAdar1-LNP@mPD1, indicating an effective immunological response in tumors (Figure 4I). KEGG enrichment analysis further identified that the disregulated genes after siAdar1-LNP@mPD1 treatment were enriched in immune-associated signaling pathways, such as those involved in cytokine-cytokine receptor interaction, chemokine signaling pathway, TNF-β signaling pathway, Th1 and Th2 cell differentiation, T cell receptor signaling pathway, natural killer cell-mediated cytotoxicity, etc. (Figure 4J). These transcriptome profiles revealed that blocking PDL1 exposure and knocking down of ADAR1 levels by siAdar1-LNP@mPD1 can inflame the TME and activate the immune system at the transcriptional level.
Finally, we examined blood routine and biochemistry analysis in serum to determine the biocompatibility of siAdar1-LNP@mPD1. The biochemical indexes of siAdar1-LNP@mPD1-treated mice did not show any obvious variations (Figure S28). Similarly, following siAdar1-LNP@mPD1 therapy, histological investigation of major organs such as the heart, liver, spleen, lung, and kidney maintained normal tissue morphology (Figure S29). These findings indicated that our siAdar1-LNP@mPD1 was biocompatible for in vivo applications.
To verify the universality of the siAdar1-LNP@mPD1 treatment strategy in other tumor models, we established a CT26 s.c. colon tumor model, which also showed a high expression of ADAR1, as shown in Figure S14. As depicted in Figure S30A, CT26 tumor-bearing mice were randomly divided into six groups and treated with PBS, anti-PDL1, siNC-LNP@m, siAdar1-LNP@m, siNC-LNP@mPD1, or siAdar1-LNP@mPD1, respectively, for a total of 3 times every 4 days. Throughout the treatment period, no significant alterations in the body weight of the mice were observed in any of the experimental groups (Figure S30B). Among these groups, siAdar1-LNP@m and siNC-LNP@mPD1 could moderately delay tumor growth due to the single function of ADAR1 silencing or PDL1 blockade (Figures S30C, and S30D). Notably, siAdar1-LNP@mPD1 substantially inhibited tumor progression, and even outperformed the anti-PDL1 antibody to overcome the low response rate and drug resistance of the standard checkpoint inhibitor (Figures S30C and S30D). After the 12th day of treatment, the excised tumors were photographed (Figure S30E) and weighed (Figure S30F). Obviously, the tumor weights of the siAdar1-LNP@mPD1 group were significantly lower than those of the other groups. qRT-PCR (Figure S30G) and WB (Figures S30H and S30I) demonstrated that siAdar1-LNP@mPD1 and siAdar1-LNP@m treatment significantly downregulated the expression of ADAR1 in tumor tissues compared with the group without siAdar1. Therefore, siAdar1-LNP@mPD1, which inhibits tumor growth by blocking the PD1/PDL1 interaction on tumor cells and downregulating ADAR1 expression in cells, achieved great success not only in the 4T1 tumor model but also in the CT26 tumor model.
siAdar1-LNP@mPD1 suppresses lung metastasis of 4T1 tumors
Immune-memory effects, which are distinct characteristics induced by immunotherapy, are crucial for durable protection against tumor recurrence/metastasis. Therefore, mice that underwent the above treatments were rechallenged with 4T1 cells by intravenous injection on day 27, while the residual primary tumor was surgically resected from each mouse 1 week in advance to avoid ethical violations (Figure 5A). Two weeks later, photographs of the lungs revealed that the PBS-treated mice had extensive lung metastasis, while siAdar1-LNP@m and siNC-LNP@mPD1 with single function showed some degree of lung metastasis inhibition (Figures 5B and S31). In sharp contrast, tumor metastasis to lung was dramatically suppressed by siAdar1-LNP@mPD1 with combination effects of ADAR1 silencing and PDL1 blocking, with the number of metastasis nodules reducing from 27 ± 5 to 3 ± 2 per lung (Figure 5C). Correspondingly, H&E staining also revealed that siAdar1-LNP@mPD1 significantly reduced the formation of metastatic foci in lung (Figure 5B).
Figure 5.

Therapeutic effect on a lung metastasis 4T1 tumor model
(A) Schematic representation of the timeline of a lung metastasis inhibition model of 4T1 tumor-bearing mice after different treatments. (B) Representative photographs (upper two panels) and H&E staining (lower panel) of isolated lungs on the 40th day post-treatment. Scale bar, 5 mm. (C) Summary data analysis of lung metastasis nodules in (B) (mean ± SD; ∗∗p < 0.01, ∗∗∗∗p < 0.0001; n = 5). (D–F) Typical FCM plots of CD44+ cells (gated on CD4+ or CD8+ T cells) (D), effector memory T cells (TEM) (gated on CD4+CD44+ or CD8+CD44+ T cells) (E), and relevant quantification results (F) in the spleens after various treatments (mean ± SD; ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; n = 3).
To further investigate the underlying immunological memory responses mediated by siAdar1-LNP@mPD1-based immunotherapy, we detected memory T cells in the spleen of mice by FCM after different treatments (Figures 5D–5F and S32). In comparison with other groups, siAdar1-LNP@mPD1 treatment had the highest proportion of memory T cells (CD8+CD44+ and CD4+CD44+). In-depth analysis further validated that these upregulated memory T cells were more inclined to differentiate toward the effector phenotype (TEM, CD44+CD62L−). This phenotype can respond quickly to secondary antigenic attack.36 Collectively, these results indicate that siAdar1-LNP@mPD1-mediated dual blockade therapy generates more powerful and effective antitumor immune memory responses to inhibit tumor recurrence and metastasis.
Abscopal effect of siAdar1-LNP@mPD1
In the following study, we investigated whether the local administration of siAdar1-LNP@mPD1 also could generate systemic immune responses to inhibit distant tumors as well. To this end, bilateral 4T1 tumor models were constructed by injecting 4T1 tumor cells s.c. into the left (1 × 106) and right (5 × 105) flanks of mice as primary and abscopal tumors, respectively. The primary tumors were treated with PBS, siAdar1-LNP@m, siNC-LNP@mPD1, and siAdar1-LNP@mPD1, whereas the abscopal tumors were shielded from any treatments (Figure 6A). We measured the tumor volume over time to determine the immunotherapeutic efficiency and the degree of the abscopal effect of various therapies (Figures 6B–6D). In comparison with the PBS group, we found that siAdar1-LNP@mPD1 treatment undoubtedly elicited powerful therapeutic effectiveness in fighting the progression of primary tumors. More importantly, siAdar1-LNP@mPD1 treatment also greatly hindered the growth of distant tumors (without treatments) compared with the PBS group. In comparison, siAdar1-LNP@m and siNC-LNP@mPD1 inhibited the growth of tumors better than PBS but were less effective than siAdar1-LNP@mPD1. These results substantiated a systemic antitumor immune response induced by siAdar1-LNP@mPD1, which could simultaneously inhibit the growth of primary and distant tumors. Notably, the body weight of mice with siAdar1-LNP@mPD1 treatment was not significantly diminished, suggesting no severe systemic toxicity (Figure S33). In addition, 50% of mice in the siAdar1-LNP@mPD1 group survived over 60 days, whereas all the mice in other groups survived no longer than 42 days (Figure 6E), which can be due to the enhanced immunotherapy efficiency and promoted abscopal effect.
Figure 6.

Therapeutic effect on a bilateral 4T1 tumor model
(A) Schematic representation of the therapeutic timeline of siAdar1-LNP@mPD1 on a bilateral 4T1 tumor model. The primary tumors were treated by intratumoral injection of different formulations, and the abscopal tumors were not treated. (B and C) Growth curves and the average volume of primary tumors (B) and abscopal tumors (C) in different groups were recorded every other day, including: G1, PBS; G2, siAdar1-LNP@m; G3, siNC-LNP@mPD1; G4, siAdar1-LNP@mPD1 (mean ± SD; n = 10). (D) Quantification results of average tumor volume on the day 20 post-treatment (mean ± SD; ∗∗p < 0.01, ∗∗∗∗p < 0.0001; n = 3). (E) Survival curves of the mice after different treatments as indicated (∗∗∗∗p < 0.0001; n = 10). (F) Schematic diagram of tumor microenvironment reshaped by siAdar1-LNP@mPD1. (G) Multiple immunofluorescence images displayed the infiltrated CD4+ T cells (red) and CD8+ T cells (green) within abscopal tumors. Scale bar, 100 μm. Enlarged images of the upper panel are shown in the lower panel. Scale bar, 25 μm. (H) Quantification results of different tumor-infiltrating immune cells in abscopal tumors after different treatments as indicated (mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; n = 3). (I) Cytokine levels in excised tumors after different treatments examined by ELISA analysis (mean ± SD; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n = 3). (J) Typical FCM plots of CD3+CD8+ T cells and quantification results of CD3+CD8+ and CD3+CD4+ T cells in the blood after various treatments. CD8+ and CD4+ values (right panel) are calculated by multiplying the proportion of positive cells, respectively, indicated in the left panel and in Figure S37B (gated on P1 region) by a normalization factor. The normalization factor is the percentage of P1 region in ALL EVENTS (FSC/SSC) as shown in Figure S37A (mean ± SD; ∗p < 0.05, ∗∗p < 0.01; n = 3).
To elucidate the mechanism of the abscopal effect of siAdar1-LNP@mPD1, we analyzed the infiltration of tumor-infiltrating immune cells in abscopal tumors of the treated mice. As shown in the immunofluorescent staining results (Figure 6G), the siAdar1-LNP@mPD1-treated mice demonstrated dramatically increased CD8+ and CD4+ T cell infiltration in the tumor, which was consistent with the FCM analysis (Figures 6H and S34). Furthermore, the frequency of MDSCs and Tregs in distal tumor tissues of siAdar1-LNP@mPD1-treated mice was the lowest among all treatment groups (Figures 6H, S35, and S36). The ELISA analysis was like the result of s.c. treatment model in Figure 4B, which showed elevated secretion of cytokines associated with immune cell activation (IFN-β, IFN-γ, TNF-α, and IL-12) and also declined secretion of immunosuppressive cytokines (IL-10 and TGF-β1) in the abscopal tumors after siAdar1-LNP@mPD1 treatment (Figure 6I).
To further evaluate the systemic immune response, the populations of CD8+ and CD4+ T cells in peripheral blood were analyzed, which were significantly increased in all effective groups including the siAdar1-LNP@mPD1-treated group (CD8+, 1.88% ± 0.25%; CD4+, 6.19% ± 1.43%), siAdar1-LNP@m-treated group (CD8+, 1.19% ± 0.24%; CD4+, 3.31% ± 0.52%), and siNC-LNP@mPD1-treated group (CD8+, 0.91% ± 0.14%; CD4+, 2.72% ± 0.55%); but, still, the siAdar1-LNP@mPD1-treated group showed the highest percentage of CD8+ and CD4+ T cells (Figures 6J and S37). These findings supported the ability of siAdar1-LNP@mPD1-mediated local treatment to elicit systemic antitumor immunity with obvious abscopal effect.
Discussion
This study is inspired from the recent findings that ADAR1 (as an emerging innate immune checkpoint) frequently impedes sensing of the immunogenic dsRNA, and thus can mediate resistance to ICB therapy.9 Although ADAR1 has been demonstrated to be one of potential targets to improve ICB efficacy, there are no clinically viable ADAR1 inhibitors up to now.37 Of course, it is not hard to imagine how to exploit ADAR1-dependent immune regulating strategy by considering the great potential of LNPs in siRNA therapeutics.38 In this work, we used (2,3-dioleoyloxy-propyl)-trimethylammonium-chloride (DOTAP), 1,2-dipalmitoyl-sn-glycerol-3-phosphocholine (DPPC), cholesterol, N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-sn-glycerol-3-phosphoethanolamine, and sodium salt (DSPE-mPEG2000) to construct LNPs to deliver ADAR1 targeted siRNA (siAdar1-LNP). However, without the decoration of any specific targeting ligands, most of the reported LNP formulations including clinically approved LNPs (mainly for liver diseases) are likely to accumulate in the liver if injected intravenously39; how to turn LNP delivery from intrinsic liver tropism to other organs/tissues (tumors in this case) is an important prerequisite for ADAR1 silence by siRNA. Another important issue is how to improve the therapeutic efficiency based on dual blockade of ADAR1 and PD1/PDL1, given that simply combing siAdar1-LNP and PD1/PDL1 inhibitors may be suboptimal due to their nonsynchronous in vivo behaviors and concurrent increase in undesired side effects.
Alternatively, we employ a biomimetic strategy to simultaneously silence ADAR1 and block PDL1 checkpoint using an immune nano-regulator (siAdar1-LNP@mPD1) by decorating the LNP with a shell of cell membrane. The cell membrane is originated from genetically engineered PD1-overexpressing CHO cells. Our group and others previously verified that these engineered cellular vesicles are competent in PD1/PDL1 blocking with merits of highly efficacy and limited toxicity.17,19,20,40 Compared with these reported vesicles isolated from immortalized cell lines that might have carcinogenic risk, we specially selected CHO cells (one of the most common cell factories) to production of PD1-expressing cell membranes from the perspective of biological safety. Although inevitably trapped in liver due to normal hepatic clearance, a phenomenon that is also found in other PD1-overexpression membrane-decorated nanoparticles,19 our membrane camouflaged LNP is still capable of delivering sufficient siRNA into the tumor site (Figure 3C), taking advantage of the specific interaction of PD1 on siAdar1-LNP@mPD1 and PDL1 on cancer cells.
Looking into the future, more critical issues need to be resolved. We notice that our developed nano-regulator can only delay the growth but not completely remove the established tumor, probably because the chosen tumor model (4T1) belongs to the triple-negative breast cancer with a severe immune “cold” environment,41 which is further specifically reflected by a strong ADAR1 expression (Figures S14 and S15). That is, the therapeutic effect might be strengthened if we could replace siRNA with a more robust gene regulation tool such as CRISPR machinery. In addition, despite our preliminary biocompatibility assay that verified limited toxicity in vivo, tremendous efforts are still strongly needed to optimize the LNP compositions, administration routes/schedules, and targeting approaches to further increase therapeutic efficacy and limit short- and long-term toxicity of these membrane cloaked LNPs. In the future, more work is needed to comprehensively assess the risk/benefit of siAdar1 delivery to off-target tissues such as the liver or kidney, particularly in terms of immune-related adverse events. Moreover, another critical issue is how to obtain this platform with the upgraded demands of high-throughput, good manufacturing practice, and excellent quality control in view of clinical translation. Given the tremendous advances in the production of engineered autologous cells, the selection of patient-derived cells as donor cells may be feasible to provide PD1-overexpressing cell membranes in clinical quality and quantity.
In summary, we have constructed a biomimetic nanovesicle by cloaking the GECM on the surface of LNPs. These nanovesicles can effectively knock down ADAR1 and bind to PDL1 of cancer cells, which induces diverse functions such as increasing the production of IFNs, making cancer cells more sensitive to IFNs, and specifically recognizing and blocking PDL1 to overcome ADAR1-regulated resistance to ICB. The application of our nanovesicles on the 4T1 and CT26 tumor models with different types of occurrences confirm their excellent antitumor immunity from multiple aspects, including the local tumor suppression effect, immunological memory effect, and abscopal effect, which is found to be associated with the significant enhancement and activation of effector immune cells and elevation of antineoplastic cytokine secretion to reshape the TME. We envision that this novel strategy can be generalized to treat different tumor types with high expression of ADAR1, and thus provides an effective solution to enhance the therapeutic efficacy of ICB.
Materials and methods
Materials
DOTAP, cholesterol, DPPC, and DSPE-mPEG2000 were purchased from AVT Pharmaceutical (Shanghai, China). Cell culture products (Gibco) were obtained from Thermo Fisher Scientific. RIPA lysis buffer and the Cytosol Protein Extraction Kit were purchased from Beyotime Biotechnology (Shanghai, China). The protease inhibitor cocktail was purchased from MedChemExpress (Shanghai, China). The Lipofectamine 3000 transfection kit was purchased from Invitrogen (Carlsbad). TransZol Up and Easy II Protein Quantitative Kits were obtained from TransGen Biotech (Beijing, China). Hieff qPCR SYBR Green Master Mix was purchased from Shanghai Yeasen BioTechnologies (Shanghai, China). CCK-8 was purchased from Dojindo Laboratories (Kumamoto, Japan). 4′,6-Diamidino-2-phenylindole (DAPI) was purchased from Sigma-Aldrich. DiO and DiI were purchased from Us Everbright (Suzhou, China). All other chemicals with analytical reagent grade were provided by Sinopharm Chemical Reagent (Shanghai, China). Anti-GAPDH, anti-β-actin (ab8226), anti-PD1 (ab214421), anti-PDL1 (ab213480), and anti-Ki67 (ab231172) antibodies were purchased from ABclonal (Wuhan, China). Anti-ADAR1 antibody and polybrene were purchased from Santa Cruz Biotechnology (Shanghai, China). Anti-CD3-APC (17-0032-82), anti-CD4-FITC (11-0042-85), anti-CD8-PE (12-0081-82), anti-CD25-PerCP-Cy5.5 (46-0251-82), anti-CD69-FITC (11-0691-82), anti-Foxp3-PE-Cy7 (25-5773-80), anti-CD11b-APC (17-0112-82), anti-Ly-6G/Ly-6C-PE (12-5931-82), anti-CD44-PE-Cy7 (250441-82), and anti-CD62L-PerCP-Cy5.5 (45-0621-82) antibodies were purchased from Thermo Fisher Scientific (eBioscience). Mouse ELISA Kits (IFN-β, IFN-γ, TNF-α, IL-12p70, IL-10, and TGF-β1) were purchased from Boster Biological Technology. Recombinant mouse PDL1 protein was purchased from Acmec Biochemical (Shanghai, China). The siRNA (siAdar1) targeting ADAR1 mRNA was synthesized by GenePharma (Shanghai, China), and the sequences are summarized in Table 1.
Table 1.
The sequences of siRNA used in this study
| siRNA (siAdar1) | Sequence |
|---|---|
| siAdar1-mus-3302 | sense: 5′-UUGACGCUUGUUUCCUUGGTT-3′ |
| antisense strand: 5′-CCAAGGAAACAAGCGUCAATT-3′ | |
| siAdar1-mus-1809 | sense: 5′- UUUGUGCAUGCACUCAAGCTT-3′ |
| antisense strand: 5′-GCUUGAGUGCAUGCACAAATT-3′ | |
| siAdar1-mus-829 | sense: 5′- UUUCAGCCAUGUCAAGAGGTT-3′ |
| antisense strand: 5′-CCUCUUGACAUGGCUGAAATT-5′ |
Cell culture
Mouse breast cancer (4T1) cells, CHO cells, normal mouse hepatocyte (BNL CL.2) cells, mouse colon cancer (CT26) cells, mouse liver cancer (Hepa 1-6) cells, human hepatocellular carcinoma (SMMC-7721) cells, human hepatocellular carcinoma (HepG2) cells, and human cervical cancer (HeLa) cells were cultured with DMEM medium (10% FBS, 1% penicillin and streptomycin) in standard incubators (37°C, humidified atmosphere of 5% CO2). Mouse colon cancer (MC38) cells and mouse liver cancer (H22) cells were cultured at 37°C in the incubator with RPMI-1640 medium (10% FBS, 1% penicillin and streptomycin). HIOECs were cultured with KGM-Gold medium (1% penicillin and streptomycin). Except for the CHO cell line derived from GMP (Jena, Germany), all other cell lines were derived from ATCC (Manassas, VA).
Establishment and characterization of genetically engineered stable cell lines
First, the PD1 plasmid encoding mouse PD1 with mCherry-tag at the C-terminal region (pCDH-CMV-PD1-mCherry-Puro) was designed and cloned into pCDH-CMV mammalian expression vector. The PD1 plasmid was confirmed by the DNA sequencing. Lentiviruses were produced from 293FT cells that were transiently transfected with the above plasmids with the Lipofectamine 3000 Transfection Kit. Then, the CHO cells were infected with the lentivirus and incubated with 5 μg/mL polybrene. Two days after infection, the engineered CHO cells were cultured in DMEM with 10% FBS and screened with puromycin (5 μg/mL) or blasticidin (10 μg/mL) to select cells stably expressing murine PD1 for a week. Finally, the PD1-mCherry stable expressing CHO cells were cultured and expanded in DMEM medium with 10% FBS.
The cells were then fixed with 4% paraformaldehyde and stained with the DAPI and the membrane dye (DiO) for 10 min. After washing with PBS 3 times, the fluorescence was observed by CLSM (ZEISS), with that the excitation wavelengths for mCherry, DAPI, and DiO were 561, 405, and 488 nm, respectively. Flow cytometry was also used to verify the expression of PD1 on cells. Finally, the stable expression of PD1-mCherry in the engineered CHO cells was further characterized by the WB assay list below, where the PD1 cell membrane fractions were used as samples, and anti-PD1 antibody (1 μg/mL) or anti-GAPDH antibody (1:5,000) was the corresponding primary antibody.
Cell membrane extraction
To extract cell membrane, PD1-mCherry stable expressing CHO cells (CHO-PD1) were cultured in a DMEM medium containing 10% FBS. The cells were harvested with trypsin and washed with cold PBS 3 times by centrifugation with 800 × g for 10 min. Then, the Membrane and Cytosol Protein Extraction Kits (Beyotime, P0033) were used to extract the cell membrane. Then, 5 × 107 cells were resuspended with 1 mL membrane protein extraction reagent A containing 1 mM PMSF for 30 min on ice. Subsequently, the cell suspensions were freeze-thawed 3 times in liquid nitrogen and at room temperature, respectively. Then, the entire solution was spun down and the supernatant was collected after centrifugation with 500 × g for 10 min. Thereafter, the above supernatant was centrifuged at 14,000 × g for 30 min, and then the precipitates of PD1-mCherry CHO cell membrane (mPD1) were obtained. Cell membranes were re-dispersed in PBS and stored separately at −80°C for further usage. Membrane protein concentration was quantified using the Easy II Protein Quantitative Kit (BCA assay).
Western Blot (WB)
Cell lysates were prepared in RIPA lysis buffer containing PMSF and protease inhibitor cocktail for 30 min on ice, and the protein concentration of the generated cell lysates was quantified by BCA protein assay. Afterward, each sample was added with loading buffer and heated to 100°C for 10 min before being transferred into 10% SDS-PAGE gel. The gels were run at 110 V for 2 h and then transferred onto the nitrocellulose (NC) membrane. After being blocked with 5% BSA for 1 h, the NC membranes were incubated with specific primary antibodies overnight at 4°C. Next, NC membranes were washed with 1× TBST and then incubated with the HRP-conjugated corresponding secondary antibodies (1:5,000) for 1 h at room temperature. Finally, they were tracked by SuperSignal West Pico PLUS Chemiluminescent Substrate and recorded by the ChemiDoc MP imaging system. Primary antibodies were as follows: PD1, ADAR1, GAPDH, and β-actin antibodies.
Co-immunoprecipitation (coIP) assay
To prepare nanovesicles, 0.8 mg/mL of mPD1 (based on total protein concentration) was sonicated in an ice bath for 10 min. In this experiment, immunoprecipitation was used to verify the correct orientation of PD1 protein on nanovesicles. In brief, the nanovesicles with surface presenting PD1 (NVs/PD1) were dispersed in 1 mL ice-cold PBS and then incubated with 100 μL of 50% protein A/G agarose beads for 2 h at 4°C with gentle rotation. The above mixture was centrifuged at 10,000 rpm for 15 min at 4°C to precipitate the protein A/G agarose beads that were nonspecifically bonded with NVs/PD1. The collected supernatant was then co-incubated with PD1 primary antibody (10 μg/mL) and protein A/G agarose beads in the rotation condition overnight at 4°C. Afterward, the beads were centrifuged at 4°C for 15 min (10,000 rpm) and washed gently with ice-cold PBS 3 times. Finally, the samples were analyzed by the above-mentioned WB assay using the relevant antibodies. Meanwhile, the group that used IgG to substitute PD1 primary antibody was used as the control.
Cell binding assay
pCDH-CMV-PDL1-EGFP-Puro plasmid (constructed from previous work of our group17) was transiently transfected into 4T1 cells using the Lipofectamine 3000 kit. After 24 h, the medium was changed to a fresh medium containing PD1 NVs (40 μg/mL based on total protein content) and incubated with 4T1 cells at 4°C for 1 h. Subsequently, cells were fixed with 4% paraformaldehyde solution, washed with cold PBS, and stained with the membrane dye (DiO) and DAPI. Then, the cells were imaged by CLSM (the excitation wavelengths for mCherry, DAPI, and EGFP were 561, 405, and 488 nm, respectively). Similarly, DiI-stained mPD1 NVs or m NVs were co-cultured with 4T1 cells and analyzed by flow cytometry.
ICB stimulates T cell proliferation and activation
The following protocol was used to isolate CD8+ T cells from mouse spleen. First, cells were aspirated from the spleen using a syringe. The resulting cell suspension was passed through a 40-μm cell strainer to remove large debris. The cells were collected by centrifugation at 800 × g for 5 min and then suspended in red blood cell lysis buffer for 10 min at room temperature to eliminate red blood cells. CD8+ T cells were then isolated using CD8 (TIL) MicroBeads (Miltenyi Biotec, 130-116-478) according to the manufacturer’s protocol.
4T1 cells were seeded at a density of 3 × 104 cells for activation assays or 6 × 104 cells for proliferation assays in 24-well plates and allowed to incubate for 24 h. The cells were then treated with either mPD1 NVs or m NVs at a concentration of 40 μg/mL based on total protein content. After 48 h of incubation, CD8+ T cells (2 × 105) were added to the wells containing KBM 581 medium supplemented with IL-2 (10 ng/mL, R&D Systems, MX2918061) and anti-CD3ε antibody (5 μg/mL, BioLegend, 100331). After 36 h of incubation, T cells were harvested and stained with anti-CD69-FITC and anti-CD25-PerCP-Cy5.5 for activation analysis by flow cytometry. For proliferation assays, CD8+ T cells were pre-stained with 5 μM CFSE working solution (Selleck, S8269). After incubation with 4T1 cells for 5 days, CFSE fluorescence of T cells was detected by flow cytometer.
Preparation of siAdar1-LNP@mPD1
siAdar1-LNP was prepared using a modified method as described previously for siRNA delivery.25 In brief, DOTAP, DPPC, cholesterol, and DSPE-mPEG2000 (with molar ratios of 50:10:38.5:1.5) were dissolved in ethanol to obtain a lipid mixture. On the other hand, siAdar1 was dissolved in 20 mM citrate buffer (pH 4.0). The lipid mixture (10 mg/mL) was mixed with 20 μM siRNA at a ratio of 1:2 (v/v) through ice bath ultrasound sonication for 5 min. The vesicles were collected and concentrated by ultrafiltration through a 100-kDa molecular weight cutoff membrane, and further washed twice with PBS to remove free siRNA. The encapsulation efficiency of siRNA in LNP was determined by measuring the fluorescence of the crude product of siAdar1-LNP before ultrafiltration using the RiboGreen kit (MKBio, Shanghai). Encapsulation efficiency = (F2 – F1)/F2 × 100%, where F2 and F1 are the fluorescence intensity in the presence and absence of 1% of Triton X-100, respectively.
siAdar1-LNP@mPD1 was prepared by an ultrasound method. First, mPD1 (0.8 mg/mL) was entirely broken by ice bath ultrasound sonication for 5 min. Then, siAdar1-LNP (1 mg/mL) were mixed with 0.8 mg of mPD1 (total protein) at a volume ratio of 1:1 under sonication for 5 min, followed by being successively passed through 0.4-, 0.2-, and 0.1-μm pore-sized polycarbonate membrane filters another 11 times.
The characterization of nanovesicles
The morphology of siAdar1-LNP and siAdar1-LNP@mPD1 were imaged by TEM (JEOL, Japan). The DLS, PDI, and zeta potential of nanovesicles were characterized using Zeta Sizer (NanoZS, Malvern, UK). The PD1 protein on siAdar1-LNP@mPD1 was characterized by Coomassie brilliant blue and WB experiments, where the PD1 cell membrane fractions or siAdar1-LNP@mPD1 were used as samples, and the anti-PD1 antibody (1 μg/mL) was used as the corresponding primary antibody.
qRT-PCR
Differently treated cells were collected and analyzed for gene expression by qRT-PCR assay.42 In brief, total RNA was isolated from homogenized cells in 1 mL of TransZol Up. The homogenate was mixed with chloroform for complete phase separation and then centrifuged at 12,000 × g for 15 min at 4°C. The upper aqueous phase of RNA was transferred to a new tube with an equal buffer volume for precipitation. The RNA was washed with different buffers in a microcentrifuge column and centrifuged. Finally, the total RNA was eluted with RNase-free water, and the RNA concentration was quantified by NanoDrop 2000.43 In addition, cDNA synthesis was performed on 1 μg total RNA using a synthesis kit according to the company’s protocol. qRT-PCR was performed by a one-step qRT-PCR system (Applied Biosystems, Foster City, CA) using Hieff qPCR SYBR Green Master Mix. Gene expression primers: mouse ADAR1 and 18S rRNA were synthesized by Biosune Biotechnology (Shanghai, China), and the sequences are summarized in Table 2. 18S rRNA was used as a housekeeping gene, and data were analyzed using the 2–ΔΔCT method.44
Table 2.
The primers sequences are given below
| Genes | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| ADAR1 | TGCTAGCCAGAGAGCTCAGA | GGATTCACAACTCCAGGGG |
| 18S rRNA | AGAAACGGCTACCACATCCA | CACCAGACTTGCCCTCCA |
Cellular uptake of siAdar1-LNP@mPD1
Cells (1 × 105) were seeded in a confocal dish and cultured for 24 h to allow cell adhere. After that, 5-FAM-labeled siAdar1-LNP (100 nM based on siAdar1 concentration) was added and incubated for 4, 24, or 48 h. Afterward, the cells were fixed with 4% paraformaldehyde and stained with DAPI for 10 min and then imaged under CLSM with excitation wavelengths of DAPI at 405 nm and FAM at 488 nm, respectively. Meanwhile, to verify the targeting ability of PD1 protein, 4T1 cells pretreated with anti-PDL1 antibody (10 μg/mL) for 2 h before addition of siAdar1-LNP@mPD1 were selected as the competition control group.
In vitro gene silencing and protein knockdown with siAdar1-LNP
4T1 cells (2 × 105) were seeded into 6-well plates and allowed to grow at 50%–60% density, followed by the treatment with siAdar1-LNP, siNC-LNP, or free siAdar1 (100 nM based on siRNA concentration). Lipofectamine 3000 carrying the same concentration of siAdar1 was used as the positive control. After 48 h of incubation, cells were digested with trypsin and collected by centrifugation at 300 × g for 3 min. The ADAR1 silencing effect was evaluated by qRT-PCR and WB assays, respectively.
IFN sensitivity assay
4T1 cells were plated in a 96-well plate at a density of 1 × 104 cells per well and incubated for 12 h. The cells were treated with siAdar1-LNP@mPD1 (100 nM based on siAdar1 concentration) for 48 h and then replaced by the fresh culture medium containing different concentrations of IFN-γ (0.1, 0.2, 0.3, 0.4, or 0.5 μg/mL) for further incubation for 24 h. After washing with PBS thrice, 10 μL of CCK-8 solution mixed with 90 μL of culture medium was added to the plate and incubated for 30 min at 37°C. The absorbance at 450 nm was measured at a microplate reader (Spectra 206 Max M5, Molecular Devices). To compare different siRNA formulations, the cells were exposed to siAdar1-LNP, siAdar1-LNP@m, siNC-LNP@mPD1, and siAdar1-LNP@mPD1 at an equivalent siRNA concentration of 100 nM. After 48 h of incubation, the culture medium was replaced with fresh medium containing IFN-γ (0.5 μg/mL) and further incubated for another 24 h. The relative cell viability (%) was calculated and expressed as the mean ± standard deviation (SD) of five samples as: cell viability (%) = (ODsample − ODblank)/(ODcontrol − ODblank) × 100%. ODsample is the absorbance of treated cells by nanovesicles, while ODblank and ODcontrol are the absorbances of the CCK-8 solution itself and control cells without nanovesicle treatment.
In vivo distribution of siAdar1-LNP@mPD1
All animal experiments were strictly conducted following the guidelines approved by the Animal Ethics Committee of Mengchao Hepatobiliary Hospital of Fujian Medical University. The Balb/c mice were brought from Wushi Laboratory Animal (Shanghai, China) for in vivo experiments. The 4T1 cells (1 × 106, 100 μL) were mixed with Matrigel (Corning, Corning, NY) (v/v = 1:1) and then injected s.c. in the right back hind leg of mice. When 4T1 tumor volume reached about 50–60 mm3, the tumor-bearing mice were divided into three groups and intravenously injected with 100 μL of Cy5-labeled siAdar1-LNP@mPD1, siAdar1-LNP@m, siAdar1-LNP, or free siAdar1 (containing 25 μg of siRNA). Then, at different times post-injection (0, 2, 4, 6, 8, 10, 12, or 24 h), the mice were observed on the small animal imaging system (PerkinElmer IVIS Spectrum, MA). After 48 h of injection, the mice were sacrificed, and their major organs (heart, liver, spleen, lung, and kidney) and tumors were excised and imaged. The fluorescence intensity was further analyzed using ImageJ software to assess the tissue distribution of nanovesicles.
Antitumor efficiency on the s.c. 4T1 tumor model
4T1 tumor-bearing mice (tumor volume reached about 50–60 mm3) were randomly divided into four groups for the following treatments. PBS (100 μL), siAdar1-LNP@m, siNC-LNP@mPD1, and siAdar1-LNP@mPD1 (containing 25 μg of siRNA) were administered into tumor-bearing mice by tail vein injection for a total of 4 times once every 4 days.
To check the histological changes in tumor after treatment, one mouse from each group was sacrificed 3 days after the last treatment. The excised tumors were prepared in paraffin sections for further H&E staining, Ki67 immunohistochemical, and TUNEL immunofluorescence staining analysis. The body weight and tumor size of the mice were recorded every 2 days. The tumor volume (V) was calculated to be V = a × b2/2, where a is the longest and b is the shortest diameter of the tumor, respectively. After 20 days of treatments, all excised tumors were weighed and photographed. Furthermore, the expression of ADAR1 in tumors was analyzed by qRT-PCR and WB assays, respectively.
On the other hand, four healthy mice were given the above treatments. After 20 days, major organs were removed and sectioned by paraffin embedding for H&E staining to analyze the systemic toxicity of nanovesicles. In addition, the biological safety of siAdar1-LNP@mPD1 was evaluated by blood routine and serum biochemical analysis. Blood biochemistry and blood routine were detected by the CX5 automatic blood cell analyzer (CX5, Beckman Coulter).
Antitumor efficiency on the s.c. CT26 tumor model
CT26 tumor-bearing mice with a tumor volume of approximately 50–60 mm3 were randomly divided into to six groups to receive the following treatments: 100 μL of PBS, anti-PDL1, siNC-LNP@m, siAdar1-LNP@m, siNC-LNP@mPD1, and siAdar1-LNP@mPD1. Except for anti-PDL1 (BioLegend, 124328), which was administered by intraperitoneal injection at a dose of 5 mg/kg, other formulations were injected intravenously through the tail vein as indicated above. The administration schedule is a total of 3 times once every 4 days. Body weight and tumor size of the mice were recorded every 2 days, and tumor volume (V) was calculated using the formula V = a × b2/2, where a and b represent the longest and shortest diameters of the tumor, respectively. After 12 days of treatment, tumors were excised, weighed, and photographed. In addition, the expression of ADAR1 in the tumors was analyzed by qRT-PCR and WB assays.
Evaluation of the lung metastasis
The s.c. 4T1 tumor-bearing mice were treated as described above. On day 20, the residual tumors were surgically removed from the mice, and then the wound was sutured and disinfected. After 1 week, the mice were injected with 5 × 105 4T1 cells through the tail vein. On the 40th day, the lungs were isolated from treated mice to photograph and count the metastasis nodules. The lung tissue was then fixed, embedded in paraffin, sectioned, stained with H&E, and placed under a microscope to observe the pulmonary metastasis.
Antitumor evaluation on the bilateral 4T1 tumor model
To construct a bilateral mouse breast cancer model, one side of each mouse was s.c. injected with 1 × 106 4T1 cells, and the other side was s.c. injected with 5 × 105 4T1 cells to simulate primary tumors and distant metastatic tumors, respectively. When the tumor volume of one side reached about 100 mm3 while the other side’s tumor volume reached approximately 50 mm3, the tumor-bearing mice were randomly divided into four groups for follow-up experiments. Each tumor-bearing mouse was intratumorally (i.t.) injected into the primary tumor site with 50 μL of PBS, siAdar1-LNP@m (5 μg of siAdar1), siNC-LNP@mPD1 (5 μg of siNC), and siAdar1-LNP@mPD1 (5 μg of siAdar1) for a total of 4 times every 4 days. The survival of tumor-bearing mice was assessed by monitoring tumor volume. Mice were considered as dead when they showed signs of impaired health or the tumor volume exceeded 1,500 cm3. Meanwhile, to evaluate potential toxicity, the changes of mouse body weight were monitored during treatment.
Flow cytometry analysis in vivo
To study the infiltration of immune cells in the tumors, the 4T1 tumor-bearing mice were treated with indicated formulations. Then, the tumors were isolated on the 20th day. Afterward, the tumors were treated in RPMI-1640 with collagenase type IV (1 mg/mL), hyaluronidase (0.2 mg/mL), and DNase I (0.02 mg/mL) for 3 h at 37°C. Then, the cells were isolated through a 40-μm cell strainer and by Ficoll-Paque density gradient centrifugation at 800 × g for 30 min at 4°C. Afterward, the cells were collected and then analyzed by flow cytometry. The list of antibodies used for FCM is as follows: anti-CD3-APC, anti-CD4-FITC, and anti-CD8-PE antibodies for effector T cells, anti-CD11b-APC and anti-Ly-6G-PE antibodies for MDSCs, and anti-CD4-FITC and anti-Foxp3-PE-Cy7 antibodies for Tregs.
To examine immune memory effects, the memory T cells were isolated from the spleen according to the above method. The cells were harvested and then analyzed by FCM after staining with anti-CD4-FITC, anti-CD8-PE, anti-CD44-PE-Cy7, and anti-CD62L-PerCP-Cy5.5 antibodies.
Cytokine analysis for tumor lysate
To examine the cytokine secretion, the tumors were isolated from the mice receiving different treatments on the 20th day. The tumors were weighed and lysed in RIPA lysis buffer containing PMSF and protease inhibitor cocktail. The tumor tissues were homogenized with 5-mm magnetic beads at 60 Hz for 6 min and centrifuged at 10,000 rpm for 10 min. Subsequently, the collected supernatant was used for cytokine detection using a Mouse ELISA Kit (IFN-β, IFN-γ, TNF-α, IL-12p70, IL-10, TGF-β1) according to the manufacturer’s protocol.
RNA sequencing and bioinformatic analysis
The tumors were isolated from the mice receiving the aforementioned treatments, which were then quickly frozen in liquid N2. Afterward, total RNA was extracted by using a Trizol reagent kit following the manufacturer’s instructions. Then, mRNA was further enriched and fragmented to prepare cDNA by reverse transcription. Finally, transcriptome libraries were established and sequenced by the Illumina novaseq6000 platform (paired-end, 150 bp).
All qualified raw reads were aligned to the mouse genome (mm10, https://www.gencodegenes.org) using STAR, then expression levels of genes were further quantified using FPKM (transcripts per million) value and differential expression analysis between different groups was conducted by Cufflinks v.2.2.1, and KEGG pathway enrichment analysis was conducted with the clusterProfiler package using differentially expressed genes (|log2 fold change| > 10, p < 0.05). The infiltration level of each immune cell type was inferred by CIBERSORT.
Statistical analysis
All results are presented as the mean ± SD or min to max with all points as indicated. Statistical analysis of data was performed through one-way variance for comparison among multiple groups or the two-tail paired Student’s t test for comparison between two groups. The survival rate was compared using the log rank test. All statistical analyses were carried out by GraphPad Prism 8.0.2. ∗p < 0.05 was represented as statistically significant. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Pearson’s correlation coefficient was used to evaluate the correlation matrices.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant nos. 62175031 and 21904021), the Scientific Foundation of Fujian Province (grant nos. 2022J011282 and 2021J011281), the Scientific Foundation of Fuzhou City (grant nos. 2022-Y-007, 2021-R-105, and 2021-S-wt3), the Medical Innovation Project of Fujian Province (grant no. 2021CXA043), the Fuzhou “14th Five-Year” Clinical Key Specialty (grant no. 20220203), the Major Research Projects for Young and Middle-aged Talent of Fujian Provincial Health Commission (2021ZQNZD013).
Author contributions
X.Z., M.W., and X.L. conceived the idea, designed the study, and provided the conceptual framework for the study. L.D., X.Z., P.Y., Y.W., and H.L. performed the experiments. L.D., X.Z., F.P., and Y.Z. analyzed the data. L.D., X.Z., and M.W. wrote the manuscript. X.L. supervised the project and revised the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.04.011.
Contributor Information
Ming Wu, Email: wmmj0419@163.com.
Xiaolong Liu, Email: xiaoloong.liu@gmail.com.
Supplemental information
Data availability
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information.
References
- 1.Petitprez F., Meylan M., de Reyniès A., Sautès-Fridman C., Fridman W.H. The tumor microenvironment in the response to immune checkpoint blockade therapies. Front. Immunol. 2020;11:784. doi: 10.3389/fimmu.2020.00784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian S.L., Drake C.G., Pardoll D.M. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park Y.-J., Kuen D.-S., Chung Y. Future prospects of immune checkpoint blockade in cancer: from response prediction to overcoming resistance. Exp. Mol. Med. 2018;50:1–13. doi: 10.1038/s12276-018-0130-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shiromoto Y., Sakurai M., Minakuchi M., Ariyoshi K., Nishikura K. ADAR1 RNA editing enzyme regulates R-loop formation and genome stability at telomeres in cancer cells. Nat. Commun. 2021;12:1654. doi: 10.1038/s41467-021-21921-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han L., Diao L., Yu S., Xu X., Li J., Zhang R., Yang Y., Werner H.M.J., Eterovic A.K., Yuan Y., et al. The genomic landscape and clinical relevance of A-to-I RNA editing in human cancers. Cancer Cell. 2015;28:515–528. doi: 10.1016/j.ccell.2015.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paz-Yaacov N., Bazak L., Buchumenski I., Porath H.T., Danan-Gotthold M., Knisbacher B.A., Eisenberg E., Levanon E.Y. Elevated RNA editing activity is a major contributor to transcriptomic diversity in tumors. Cell Rep. 2015;13:267–276. doi: 10.1016/j.celrep.2015.08.080. [DOI] [PubMed] [Google Scholar]
- 8.Xu L.-D., Öhman M. ADAR1 editing and its role in cancer. Genes (Basel) 2018;10:12. doi: 10.3390/genes10010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishizuka J.J., Manguso R.T., Cheruiyot C.K., Bi K., Panda A., Iracheta-Vellve A., Miller B.C., Du P.P., Yates K.B., Dubrot J., et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature. 2019;565:43–48. doi: 10.1038/s41586-018-0768-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 11.Deng Y., Wang C.C., Choy K.W., Du Q., Chen J., Wang Q., Li L., Chung T.K.H., Tang T. Therapeutic potentials of gene silencing by RNA interference: principles, challenges, and new strategies. Gene. 2014;538:217–227. doi: 10.1016/j.gene.2013.12.019. [DOI] [PubMed] [Google Scholar]
- 12.Coelho T., Adams D., Silva A., Lozeron P., Hawkins P.N., Mant T., Perez J., Chiesa J., Warrington S., Tranter E., et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013;369:819–829. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 13.Beloqui A., Solinís M.Á., Rodríguez-Gascón A., Almeida A.J., Préat V. Nanostructured lipid carriers: promising drug delivery systems for future clinics. Nanomedicine. 2016;12:143–161. doi: 10.1016/j.nano.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Yonezawa S., Koide H., Asai T. Recent advances in siRNA delivery mediated by lipid-based nanoparticles. Adv. Drug Deliv. Rev. 2020;154–155:64–78. doi: 10.1016/j.addr.2020.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu G., Zhao X., Zhang Y., Xu J., Xu J., Li Y., Min H., Shi J., Zhao Y., Wei J., et al. Engineering biomimetic platesomes for pH-responsive drug delivery and enhanced antitumor activity. Adv. Mater. 2019;31:1900795. doi: 10.1002/adma.201900795. [DOI] [PubMed] [Google Scholar]
- 16.Hu C.-M.J., Fang R.H., Wang K.-C., Luk B.T., Thamphiwatana S., Dehaini D., Nguyen P., Angsantikul P., Wen C.H., Kroll A.V., et al. Nanoparticle biointerfacing by platelet membrane cloaking. Nature. 2015;526:118–121. doi: 10.1038/nature15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu M., Zheng D., Zhang D., Yu P., Peng L., Chen F., Lin Z., Cai Z., Li J., Wei Z., et al. Converting immune cold into hot by biosynthetic functional vesicles to boost systematic antitumor immunity. iScience. 2020;23:101341. doi: 10.1016/j.isci.2020.101341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu P., Zheng D., Zhang C., Wu M., Liu X. Protocol to prepare functional cellular nanovesicles with PD1 and TRAIL to boost antitumor response. STAR Protoc. 2021;2:100324. doi: 10.1016/j.xpro.2021.100324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhai Y., Wang J., Lang T., Kong Y., Rong R., Cai Y., Ran W., Xiong F., Zheng C., Wang Y., et al. T lymphocyte membrane-decorated epigenetic nanoinducer of interferons for cancer immunotherapy. Nat. Nanotechnol. 2021;16:1271–1280. doi: 10.1038/s41565-021-00972-7. [DOI] [PubMed] [Google Scholar]
- 20.Zhang X., Wang C., Wang J., Hu Q., Langworthy B., Ye Y., Sun W., Lin J., Wang T., Fine J., et al. PD-1 blockade cellular vesicles for cancer immunotherapy. Adv. Mater. 2018;30:e1707112. doi: 10.1002/adma.201707112. [DOI] [PubMed] [Google Scholar]
- 21.Omasa T., Onitsuka M., Kim W.-D. Cell engineering and cultivation of Chinese hamster ovary (CHO) Cells. Curr. Pharm. Biotechnol. 2010;11:233–240. doi: 10.2174/138920110791111960. [DOI] [PubMed] [Google Scholar]
- 22.Riley J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009;229:114–125. doi: 10.1111/j.1600-065X.2009.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayes P.A., Hance K.W., Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018;17:509–527. doi: 10.1038/nrd.2018.75. [DOI] [PubMed] [Google Scholar]
- 24.Valenzuela J., Schmidt C., Mescher M. The roles of IL-12 in providing a third signal for clonal expansion of naive CD8 T Cells. J. Immunol. 2002;169:6842–6849. doi: 10.4049/jimmunol.169.12.6842. [DOI] [PubMed] [Google Scholar]
- 25.Ickenstein L.M., Garidel P. Lipid-based nanoparticle formulations for small molecules and RNA drugs. Expert Opin. Drug Deliv. 2019;16:1205–1226. doi: 10.1080/17425247.2019.1669558. [DOI] [PubMed] [Google Scholar]
- 26.Leung A.K.K., Hafez I.M., Baoukina S., Belliveau N.M., Zhigaltsev I.V., Afshinmanesh E., Tieleman D.P., Hansen C.L., Hope M.J., Cullis P.R. Lipid nanoparticles containing siRNA synthesized by microfluidic mixing exhibit an electron-dense nanostructured core. J. Phys. Chem. C. 2012;116:18440–18450. doi: 10.1021/jp303267y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding L., Lin X., Lin Z., Wu Y., Liu X., Liu J., Wu M., Zhang X., Zeng Y. Cancer cell-targeted photosensitizer and therapeutic protein co-delivery nanoplatform based on a metal-organic framework for enhanced synergistic photodynamic and protein therapy. ACS Appl. Mater. Inter. 2020;12:36906–36916. doi: 10.1021/acsami.0c09657. [DOI] [PubMed] [Google Scholar]
- 28.Li C.-W., Lim S.-O., Chung E.M., Kim Y.-S., Park A.H., Yao J., Cha J.-H., Xia W., Chan L.-C., Kim T., et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33:187–201.e10. doi: 10.1016/j.ccell.2018.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Summers H.D., Rees P., Holton M.D., Brown M.R., Chappell S.C., Smith P.J., Errington R.J. Statistical analysis of nanoparticle dosing in a dynamic cellular system. Nat. Nanotechnol. 2011;6:170–174. doi: 10.1038/nnano.2010.277. [DOI] [PubMed] [Google Scholar]
- 30.Liu H., Golji J., Brodeur L.K., Chung F.S., Chen J.T., DeBeaumont R.S., Bullock C.P., Jones M.D., Kerr G., Li L., et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat. Med. 2019;25:95–102. doi: 10.1038/s41591-018-0302-5. [DOI] [PubMed] [Google Scholar]
- 31.Xiong W., Yeung N., Wang S., Liao H., Wang L., Luo J. Breast cancer induced bone osteolysis prediction using temporal variational autoencoders. BME Front. 2022;2022:9763284. doi: 10.34133/2022/9763284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Y., van der Meel R., Chen X., Lammers T. The EPR effect and beyond: strategies to improve tumor targeting and cancer nanomedicine treatment efficacy. Theranostics. 2020;10:7921–7924. doi: 10.7150/thno.49577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Min H., Wang J., Qi Y., Zhang Y., Han X., Xu Y., Xu J., Li Y., Chen L., Cheng K., et al. Biomimetic metal–organic framework nanoparticles for cooperative combination of antiangiogenesis and photodynamic therapy for enhanced efficacy. Adv. Mater. 2019;31:e1808200. doi: 10.1002/adma.201808200. [DOI] [PubMed] [Google Scholar]
- 34.Oupicky D., Ogris M., Howard K.A., Dash P.R., Ulbrich K., Seymour L.W. Importance of lateral and steric stabilization of polyelectrolyte gene delivery vectors for extended systemic circulation. Mol. Ther. 2002;5:463–472. doi: 10.1006/mthe.2002.0568. [DOI] [PubMed] [Google Scholar]
- 35.Du J.Z., Sun T.M., Song W.J., Wu J., Wang J. A tumor-acidity-activated charge-conversional nanogel as an intelligent vehicle for promoted tumoral-cell uptake and drug delivery. Angew. Chem. Int. Ed. Engl. 2010;49:3621–3626. doi: 10.1002/anie.200907210. [DOI] [PubMed] [Google Scholar]
- 36.McGranahan N., Furness A.J.S., Rosenthal R., Ramskov S., Lyngaa R., Saini S.K., Jamal-Hanjani M., Wilson G.A., Birkbak N.J., Hiley C.T., et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–1469. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang T., Yin C., Fedorov A., Qiao L., Bao H., Beknazarov N., Wang S., Gautam A., Williams R.M., Crawford J.C., et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature. 2022;606:594–602. doi: 10.1038/s41586-022-04753-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Akinc A., Maier M.A., Manoharan M., Fitzgerald K., Jayaraman M., Barros S., Ansell S., Du X., Hope M.J., Madden T.D., et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019;14:1084–1087. doi: 10.1038/s41565-019-0591-y. [DOI] [PubMed] [Google Scholar]
- 39.Gillmore J.D., Gane E., Taubel J., Kao J., Fontana M., Maitland M.L., Seitzer J., O’Connell D., Walsh K.R., Wood K., et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021;385:493–502. doi: 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X., Wang J., Chen Z., Hu Q., Wang C., Yan J., Dotti G., Huang P., Gu Z. Engineering PD-1-presenting platelets for cancer immunotherapy. Nano Lett. 2018;18:5716–5725. doi: 10.1021/acs.nanolett.8b02321. [DOI] [PubMed] [Google Scholar]
- 41.Sagiv-Barfi I., Kohrt H.E.K., Czerwinski D.K., Ng P.P., Chang B.Y., Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA. 2015;112:E966–E972. doi: 10.1073/pnas.1500712112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shrestha S., Jang S.R., Shrestha B.K., Park C.H., Kim C.S. Engineering 2D approaches fibrous platform incorporating turmeric and polyaniline nanoparticles to predict the expression of βIII-Tubulin and TREK-1 through qRT-PCR to detect neuronal differentiation of PC12 cells. Mater. Sci. Eng. C. 2021;127:112176. doi: 10.1016/j.msec.2021.112176. [DOI] [PubMed] [Google Scholar]
- 43.Shrestha S., Shrestha B.K., Joong O.K., Park C.H., Kim C.S. Para-substituted sulfonic acid-doped protonated emeraldine salt nanobuds: a potent neural interface targeting PC12 cell interactions and promotes neuronal cell differentiation. Biomater. Sci. 2021;9:1691–1704. doi: 10.1039/d0bm01034k. [DOI] [PubMed] [Google Scholar]
- 44.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available on reasonable request. All data relevant to the study are included in the article or uploaded as supplemental information.