

Abstract
Age-related and chemotherapy-induced bone loss depends on cellular senescence and the cell secretory phenotype. However, the factors secreted in the senescent microenvironment that contribute to bone loss remain elusive. Here, we report a central role for the inflammatory alternative complement system in skeletal bone loss. Through transcriptomic analysis of bone samples, we identified complement factor D, a rate-limiting factor of the alternative pathway of complement, which is among the most responsive factors to chemotherapy or estrogen deficiency. We show that osteoblasts and osteocytes are major inducers of complement activation, while monocytes and osteoclasts are their primary targets. Genetic deletion of C5ar1, the receptor of the anaphylatoxin C5a, or treatment with a C5AR1 inhibitor reduced monocyte chemotaxis and osteoclast differentiation. Moreover, genetic deficiency or inhibition of C5AR1 partially prevented bone loss and osteoclastogenesis upon chemotherapy or ovariectomy. Altogether, these lines of evidence support the idea that inhibition of alternative complement pathways may have some therapeutic benefit in osteopenic disorders.
Keywords: C5aR1, chemotherapy, C5aR inhibitors, osteoporosis, complement system, senescence, bone remodeling
Graphical abstract
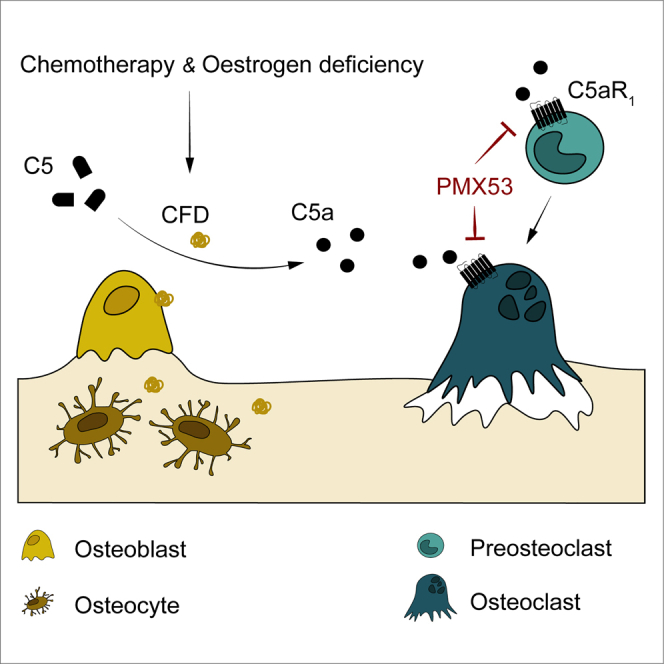
Ventura and colleagues identified a central role for the inflammatory alternative complement system in pathological skeletal bone loss. Genetic or pharmacological inhibition of C5AR1 prevented doxorubicin-induced bone loss. Therefore, repurposing C5AR1 inhibitors could become therapeutic for osteopenic pathologies.
Introduction
Bone is remodeled constantly throughout life depending on a delicate balance between osteoclastic resorption and osteoblastic new bone formation. Deregulation of this process causes deterioration of bone composition, structure, and function, leading to pathological states such as osteoporosis. The increasing human lifespan is having an important social, medical, and economic impact related to late-in-life chronic diseases, such as age-related bone loss and osteoporosis.1 Osteoporosis is triggered by various types of internal and external cellular stressors such as aging, hormone deficiency, inflammation, and exposure to radiation and genotoxic drugs. Most, if not all, converge to cause bone cells to enter the senescence program.2,3
Cells become senescent as a consequence of low-grade accumulated damage, which causes stable cell-cycle arrest.4 Cellular senescence is defined as one of the hallmarks of aging, since the number of senescent cells increases with age.5 Moreover, transplantation of small numbers of senescent cells has been proven sufficient to induce physical dysfunction in young mice, whereas senolytics improve physical function and increase lifespan in old mice.6 Osteocytes are the longest-lived cells in bone, living more than 20 years; hence they are more likely to accumulate molecular damage over time.7,8 In a seminal study, Farr et al. reported that eliminating senescent cells in 22-month-old mice improved their bone mass and strength and resulted in better bone microarchitecture.9 Recently, it has been shown that intrinsic aging of skeletal stem cells promotes bone loss by establishing a local pro-inflammatory niche.10 Interestingly, exposure to youthful circulation through parabiosis did not improve bone loss in an aging model.10 In addition, growing evidence suggests that conditioned medium (CM) from local senescent cells impairs osteoblast mineralization and increases osteoclast progenitor survival, promoting osteoclastogenesis.5,11 Therefore, senescent cells are now thought to mediate important aspects of age-related and chemotherapy-induced bone loss.11
Senescent cells produce inflammatory cytokines, chemokines, and matrix-degrading molecules, collectively known as the senescence-associated secretory phenotype (SASP), which have important detrimental effects on the tissue microenvironment and establish a state of chronic low-grade inflammation.12,13 Accordingly, a major analysis of whole-organism gene expression dynamics during aging identified a consistent increase in inflammatory and immune response genes.14 A number of cytokines, including CSF1 and RANKL, have been implicated in senescence-induced bone loss.10,15 However, the exact mechanisms and molecules involved by which senescent cells and SASP alter bone remodeling remains elusive. On this basis, we hypothesize that the generation of senescent cells, and their associated SASP, impacts on the function of bone cells through paracrine and cell-autonomous mechanisms, leading to loss of bone homeostasis.
In this study, we identified increased expression of complement factor D (Cfd), a rate-limiting factor in the alternative pathway of the complement system, as being one of the most responsive factors to osteopenic triggers such as chemotherapy or estrogen deficiency. Although the complement system is essential for innate immune responses, locally activated complement can contribute to disease development and progression.16 We present both genetic and pharmacologic evidence showing the causal role of activation of the alternative complement pathway in the recruitment and activation of osteoclasts and consequent impairment of skeletal health.
Results
Chemotherapy-induced senescence leads to bone loss
To study the effects of senescence in chemotherapy-induced bone loss, we established a mouse model by administration of a single intraperitoneal dose of 10 mg/kg of doxorubicin (DoxoR). DoxoR is an anthracycline agent used to treat several types of cancers that induces DNA damage, promotes senescence, and has adverse effects on bone density in patients.11,17,18 This dose is similar to the one that human patients receive, and induces an antitumor response but is far below the maximally tolerated dose in mice.18 We combined this treatment with the senolytic compound ABT263 by daily oral administration of 50 mg/kg in 2 alternate weeks (Figure 1A). ABT263 is a specific inhibitor of BCL-2 and BCL-xL that selectively induces apoptosis of senescent cells.19 DoxoR induced a reduction in the weight of mice after 30 days, whereas ABT263 treatment had no significant influence on mouse weight (Figure S1A). Bone samples from the femur and tibia, enriched in osteoblasts and osteocytes by removal of the bone marrow, were collected 30 days after DoxoR treatment. Consistent with the induction of senescence in vivo, bone samples exhibited increased mRNA levels of p21 (Cdkn1a), p16 (Cdkn2a), and the SASP component Csf2 at 30 days after DoxoR treatment. Moreover, although it did not modify basal expression, ABT263 partially prevented the increase in mRNA of Cdkn1a, Cdkn2a, and Csf2, suggesting that ABT263 can reduce the number of DoxoR-induced senescent bone cells (Figure 1B).
Figure 1.

Chemotherapy-induced senescence leads to bone loss
(A) Schematic representation of the DoxoR-induced senescence model. At day 0, 15-week-old mice received either vehicle or DoxoR (10 mg/kg, intraperitoneal DoxoR). For an entire week, starting at days 7 and 21, mice received ABT263 (50 mg/kg, daily oral gavage) or vehicle 2. (B) qRT-PCR of senescence markers Cdkn1a, Cdkn2a, Il1α, Il6, and Csf2. RNA isolated from the tibiae and femurs of vehicle-, DoxoR-, ABT263-, and DoxoR + ABT263-treated mice. n = 4–6. (C) Quantitative parameters and 3D representation of cortical bone of tibiae measured by μ-CT. n = 6–10. (D) Quantitative parameters and 3D representation of the trabecular compartment of tibiae measured by μ-CT. n = 6–10. Data shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. One-way ANOVA.
We analyzed the bone microarchitecture at the tibial metaphysis and mid-diaphysis by micro-computed tomography scanning (μ-CT). DoxoR-induced senescence led to impaired trabecular and cortical bone architecture. DoxoR-treated mice presented lower cortical bone volume associated with reduced cortical thickness, whereas the bone perimeter around the midshaft was not affected (Figure 1C). Moreover, tibiae also presented lower trabecular bone volumes, resulting from a significantly lower trabecular number (Figure 1D). Furthermore, ABT263 prevented all these osteopenic effects on bone parameters to about the same extent that it prevented the expression of senescent cell markers in bone samples (Figures 1B–1D). Altogether, these data show that even a single DoxoR insult leads to increased expression of senescence gene markers and permanent bone loss.
Chemotherapy-induced senescence promotes osteoclast function
We performed histomorphometric analysis of tibiae of mice treated with DoxoR and/or ABT263. Osteoblast and osteocyte numbers were not modified in any of the conditions. By contrast, tartrate-resistant acid phosphatase (TRAP) staining showed that DoxoR almost doubled the number of osteoclasts covering the bone surface. Again, treatment with ABT263 did not modify osteoclast numbers in control mice but reduced the increase in the number of osteoclasts in DoxoR-treated mice (Figures 2A and S1B). qRT-PCR analysis of bones further confirmed these data. The expression of the osteoblastic genes Runx2, Osx/Sp7, and Col1a1 was not modified in any condition, whereas the expression of the osteoclastic markers Mmp9, Ctsk (Cathepsin K), and Acp5 (TRAP) was increased in bones from DoxoR-treated mice and restored to control levels upon treatment with ABT263 (Figure 2B). An increase in bone marrow adipose tissue, due to disruption of the equilibrium between adipogenesis and osteogenesis, has been observed in pathological bone conditions, including aging, estrogen deficiency, obesity, and diabetes.20,21 Therefore, we analyzed the number of adipocytes in the bone marrow and the expression of adipocyte marker genes in the bone marrow of our treated mice. The number of adipocytes and the mRNA expression of Pparg, Cebpb, Fasn, Lipe, and Pnpla2 in the bone marrow were not modified under any condition, suggesting that, at least in adult mice (15 weeks old), enhanced adipogenesis does not occur 30 days after DoxoR treatment (Figures 2C and S1C).
Figure 2.

Chemotherapy-induced senescence promotes osteoclast function
(A) Quantification of the number of osteoblasts per bone surface (osteoblasts/mm) and osteocytes per cortical bone area (osteocytes/mm2) from H&E-stained sections of tibiae. The number of osteoclasts per trabecular bone surface (osteoclasts/mm) was quantified from TRAP-staining of tibiae. n = 4–6. (B) Gene expression of osteoblastic (Runx2, Sp7, and Col1a1) and osteoclastic (Nfatc1, Mmp9, Ctsk, and Trap) markers quantified through qRT-PCR from RNA isolated from bone (tibiae and femur) devoid of bone marrow. n = 4–8. (C) Quantification of mRNA of adipogenic markers (Pparg, Cebpb, Fasn, Lipe, and Pnpla2) from bone marrow. n = 4–6. (D and E) qRT-PCR relative quantification of osteoblastic (Runx2, Osx, and Col1a1) osteocytic (Dmp1 and Sost) and adipogenic (Cebpa, Cebpb, and Pparg) markers from control and senescent cells. Bone marrow mesenchymal stromal cells (BMMSCs) and osteocytes were treated for 24 h with 100 nM DoxoR and left 7 days to become fully senescent. n = 6–10. (F) Gene expression of osteoblastic and adipogenic markers. RNA was isolated from BMMSCs cultured with conditioned medium from control or senescent BMMSCs, respectively. n = 8–10. (G) Gene expression of osteoclastic markers. RNA was isolated from RAW264.7 cultured with conditioned medium from control or senescent BMMSCs, respectively. n = 5–6. Data shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (A–C) One-way ANOVA. (D–G) Student’s t test.
We also analyzed the effects of DoxoR-induced senescence on differentiation and SASP induction in primary cultures of osteogenic cells. DoxoR-induced senescence was characterized by complete proliferative cell arrest, increased cell size, and complexity, SA-β-galactosidase staining and induction of SASP in primary bone marrow-derived mesenchymal stem cells (BMMSCs) and osteocytes (Figures S2A–S2F). The expression of the osteogenic markers Runx2 and Col1a1 and the adipogenic markers Cebpa and Pparg was lower in senescent BMMSCs (Figure 2D). Similarly, DoxoR-induced senescent primary osteocytes showed lower expression of Runx2, Osx/Sp7, and Col1a1 (Figure 2E). The trilineage differentiation potential of BMMSCs was also assessed through colorimetric analysis by staining with alizarin red (osteoblastic), oil red O (adipogenic), and Alcian blue (chondrogenic). In agreement with the gene expression results, induction of senescence by DoxoR resulted in a lower differentiation potential of BMMSCs to the three lineages (Figure S3A). These data suggest an intrinsic cell-autonomous defect in the differentiation potential of cultured senescent osteogenic cells.
Senescence-induced decoupling of bone formation and resorption in vivo relies on SASP, since pharmacological blockade of SASP strongly reduces bone loss induced by aging or DoxoR.9,11 To test this further, primary murine BMMSCs and RAW264.7 murine macrophage cells were exposed to CM collected from control or senescent BMMSCs. We confirmed that exposure to CM from donor senescent BMMSCs also promoted senescence in the recipient cells (a senescence bystander effect22) (Figure S3B). In addition, senescent-cell CM reduced the expression of the osteogenic genes Runx2, Osx/Sp7, and Col1a1 as well as the adipogenic genes Cebpa and Pparg in recipient BMMSCs (Figure 2F). RAW264.7 macrophage cells cultured in senescent-cell CM displayed a much lower bystander effect than BMMSCs (Figure S3C). More importantly, instead of impaired differentiation, these cells expressed the osteoclastic genes Mmp9, Ctsk, and Acp5 (Figure 2G). Altogether, the evidence suggests that secreted factors from senescent cells promote osteoclastic specification of precursors.
Expression of complement factor D is induced by senescent bone cells upon chemotherapy
To identify factors that are differentially secreted by DoxoR-induced senescent bone cells, we performed transcriptome analysis of bone samples. Tibiae and femur samples, enriched in osteocytes and osteoblasts by removal of bone marrow, were obtained 30 days following administration of a single DoxoR dose. RNA sequencing (RNA-seq) analysis showed 31 protein-coding genes differentially expressed (FDR < 0.05) between control and DoxoR-treated mice (Figure 3A; Table S1). We found strong enrichment of biological processes related to responses to chemical and external stimuli, immune responses, and interferon-responsive genes (Figure 3B). Our results agree with previous data from models related to senescence and aging.12,14,23 Gene set enrichment analysis revealed that senescent bones had significantly increased innate immune responses and bone resorption than control (Figure 3C). Altogether, we focused on Cfd as the best candidate gene as it (1) is upregulated in senescent bones, (2) encodes for a secreted protein, and (3) is directly related to the innate immune system. Cfd is a secreted serine protease essential for the initiation and amplification of the alternative complement pathway.16 This is consistent with previous findings showing that Cfd is highly expressed in osteocytes from the tibia and femur.24 Moreover, a search of public databases indicated that Cfd expression is increased in several senescent conditions, such as osteoblast replicative senescence, MSCs from aged humans, and natural aging of bone in mice (E-MTAB-1391)25 (Gene Expression Omnibus [GEO] accession no. GSE97311).26 In addition, Cfd was found to be overexpressed in human skeletal pathological conditions such as non-union fractures and osteoarthritis (GEO accession no. GSE494).27 The complement system consists of three distinct pathways that converge in the activation of proteases that finally generate the anaphylatoxins C3a and C5a.28 Activation of the alternative pathway, the oldest evolutionary pathway of the system, is finely tuned by the action of complement factor B (CFB) and the rate-limiting protease CFD.16,29 Using qRT-PCR, we confirmed that Cfd and Cfb expression was increased in the bone marrow-depleted bones of mice treated with DoxoR (Figure 3D). These increases were completely reverted in mice treated with the senolytic ABT263. Expression of C3ar1 and C5ar1, the receptors of anaphylatoxins C3a and C5a, displayed only minor, non-significant, increases in DoxoR-treated mice. Since Cfb and Cfd might also be expressed in other cell types of the bone microenvironment, we analyzed their expression in the bone marrow of the same animals. The expression of Cfb, Cfd, C3ar1, and C5ar1 remained unchanged upon DoxoR administration (Figure 3E). In addition, we analyzed the expression of Cfd, Cfb, C3ar1, and C5ar1 in a different murine model of osteoporosis. Sixty days after surgical ovariectomy (OVX), there were also increased levels of Cfd in the bones of these mice (Figure 3F). Altogether, these data show higher expression of Cfd in bone cells under different osteopenic conditions.
Figure 3.

Expression of complement factor D is induced in bone cells upon chemotherapy
(A) Heatmap analysis of the genes differentially expressed between control and DoxoR-treated mice (n = 4; FDR < 0.05). (B) Gene ontology (GO) enrichment analysis performed by g.profiler of upregulated genes in DoxoR mice. Most enriched biological processes are shown. (C) Genes set enrichment analysis (GSEA) was performed starting from the ranked gene list. Significant enrichment score curves, the normalized enrichment score (NES), and FDR are shown. (D) Expression of complement genes (Cfd, Cfb, C3ar1, and C5ar1) in mRNA isolated from the tibiae and femurs of control, ABT263-, DoxoR-, or DoxoR + ABT263-treated mice. n = 4–8. (E) qRT-PCR of complement genes in RNA isolated from bone marrow of control, ABT263-, DoxoR-, and DoxoR +ABT263-treated mice. n = 3–4. (F) mRNA relative expression of complement genes from RNA isolated from bones of sham or ovariectomized (OVX) mice, 60 days after surgery. n = 5–7. Data are shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (D) One-way ANOVA.
The complement system promotes monocyte migration and osteoclastogenesis
Cells from the osteogenic lineage, mainly osteoblasts and osteocytes, express higher levels of Cfd and Cfb than hematopoietic stem progenitor cells (HSPCs), BMMSCs, macrophages, osteoclasts, and endothelial cells. Conversely, the C3a and C5a receptors C3ar1 and C5ar1 are highly enriched in cultured macrophages and osteoclasts (Figure 4A). The C5ar1 transcripts are almost 20-fold (in murine bone samples) and 80-fold (in the bone marrow) more abundant than the C3ar1 transcripts, suggesting a higher dependency of monocyte/macrophage function on C5AR1 signaling (Figure 4B). Overall, the results reveal that osteoblasts and osteocytes might be major modulators of the complement pathway, while monocytes/macrophages and osteoclasts would be the primary target cells.
Figure 4.

The complement system promotes monocyte migration and osteoclastogenesis
(A) Normalized expression against Tbp expression (2−ΔCt) of complement genes (Cfd, Cfb, C3ar1, and C5ar1) from primary bone cells: BMMSCs, osteoblasts (OB), osteocytes (OCy), hematopoietic stem progenitor cells (HSPCs), bone marrow-derived macrophages (BMM), osteoclasts (OC), and endothelial cells (ECs). n = 6–10. (B) Normalized relative expression against Tbp (2−ΔCt) of complement receptor genes (C3ar1 and C5ar1) from bone (tibiae and femur) and bone marrow (BM) cells. n = 4–6. (C) Chemotaxis of THP1 cells toward medium without FBS (control), medium without FBS + 0.1 μg/mL of C5a (C5a), and THP1 cells pre-treated 30 min with 0.9 μg/mL PMX53 toward medium without FBS + 0.1 μg/mL C5a (PMX53 + C5a) was evaluated. After 2 h of migration, RFU (Ex/Em = 485/520 nm) was measured. n = 5–8. (D) Western blotting of phosphoS6, phosphoHSP27, phosphoERK, and the loading control β-actin of THP1 cells untreated or treated with PMX53 and/or C5a for 30 min. The experiment was performed three times with similar results. (E) Number of BM-Monocytes untreated or treated with 0.1 μg/mL C5a for 0, 24, or 48 h. n = 5–7. (F) Osteoclastic relative gene expression of primary BM-Monocytes control or treated with 0.1 μg/mL C5a for 5 days. n = 4. Data are shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. One-way ANOVA.
Next, we determined the functional responses of complement activation in cells of the bone microenvironment. Treatment of primary HSPCs of the bone marrow with DoxoR, ABT263, or both, reduced the number of colonies obtained from these cells (Figure S4A). However, the analysis of their differentiation capacity using colony formation unit assays in vitro showed no major changes in the ratios of their progeny upon treatment with ABT263 and/or DoxoR (Figure S4B). Expression of the genes of the alternative complement system after addition of DoxoR showed increased mRNA levels of Cfb, C3ar1, and C5ar1 upon DoxoR addition that were not reverted by ABT263 (Figure S4C). Treatment of BMMSCs with the anaphylatoxins C3a and C5a led to a decrease in the expression of the osteogenic markers Runx2, Osx/Sp7, and Col1a1 when treated with C3a. Similar experiments did not detect changes in the adipogenic expression markers Pparg, Cebpb, or Fasn by either C3a or C5a (Figures S5A and S5B). Moreover, treatment of primary osteocytes with C3a or C5a did not modify their transcriptional program (Figure S5C). We also investigated whether monocyte migration is affected by the anaphylatoxin C5a. Transwell assays showed that C5a efficiently acted as a chemoattractant for THP1 monocytes, whereas a specific C5AR1 antagonist, PMX53,30 blocked their chemoattractive effect (Figure 4C). In monocytes, C5a signals through the mTOR/S6K and ERK pathways and leads to increased phosphorylation of HSP27 (Figures 4D and S5D), which has been shown to be required for monocyte migration and involved in osteoporosis.31,32 We also analyzed the effect of C5a on primary monocytes isolated from bone marrow. The addition of C5a did not significantly modify monocyte proliferation or survival (Figure 4E) but increased the expression levels of macrophage and osteoclast markers such as Csf1r, Rank, Nfatc1, Mmp9, Ctsk, and Acp5 (Figure 4F). Similar experiments in RAW264.7 macrophage cells demonstrated that anaphylatoxins did not alter proliferation in the presence or absence of osteoclastic differentiation upon treatment with RANKL for 1 week (Figures S5E and S5G). More importantly, C5a increased the expression of osteoclastic markers when cells were cultured in the absence or presence of RANKL (Figures S5F and S5H). Altogether, our data support the idea that anaphylatoxin C5a acts as a chemoattractant for monocytes and a promoter of osteoclastogenesis.
C5ar1 deficiency prevents bone loss induced by chemotherapy or OVX
In view of our results, we hypothesized that blocking C5a signaling could prevent bone loss induced by either DoxoR or estrogen deficiency. To this end, we used C5ar1 knockout mice (C5ar1-KO) in a DoxoR-induced osteopenic model. As shown previously, DoxoR caused weight loss in control mice over a 30-day period. C5ar1-deficient mice had lower basal weight than controls and developed further weight reduction after DoxoR treatment (Figure S6A). As shown above, DoxoR-treated control mice presented a lower cortical bone volume (Figure S6B). Moreover, tibiae also presented a lower trabecular bone volume resulting from a lower trabecular number and thickness (Figure 5A). Deficiency in C5AR1 function partially prevented the lower trabecular bone phenotype by decreasing the deleterious effects of DoxoR on bone volume, trabecular number, and number of osteoclasts bound to bone surfaces (Figures 5A, 5B, and S6C). Gene expression analysis indicated that, after 30 days, DoxoR treatment led to increased levels of Ctsk and Acp5 in bone samples. However, although baseline levels of these genes were similar between C5ar1-KO and control mice, DoxoR treatment was unable to increase the expression of Ctsk and Acp5 mRNAs in C5ar1-KO mice (Figure 5C). We also isolated monocytes from the bone marrow of these mice and performed chemotactic assays upon C5a stimulation. As expected, addition of C5a was chemoattractive for wild-type (WT) monocytes. However, WT monocytes exposed to C5a upon addition of a C5AR1 antagonist (PMX53) or monocytes from C5ar1-KO mice were refractory to C5a-induced migration (Figure 5D). Moreover, primary bone marrow monocytes from C5ar1-KO mice showed diminished osteoclastogenic potential, as demonstrated by reduced expression of the Mmp9, Ctsk, and Acp5 genes (Figure 5E). BMMSCs isolated from C5ar1-KO mice showed similar differentiation potential to the osteoblastic lineage as the WT BMMSCs, and lower differentiation potential to the adipogenic or chondrogenic lineages than WT BMMSCs (Figure S7A).
Figure 5.

C5ar1 deficiency partially prevented bone loss induced by chemotherapy
(A) Quantitative parameters and 3D representation of the trabecular compartment of tibiae measured by μ-CT from wild-type (WT), WT/DoxoR, C5aR1−/−, and C5aR1−/−/DoxoR experimental groups. n = 9–13. (B) Quantification of the number of osteoblasts per bone surface (osteoblasts/mm) and osteocytes per cortical bone area (osteocytes/mm2) from H&E-stained sections of tibiae. The number of osteoclasts per trabecular bone surface (osteoclasts/mm) was quantified from TRAP staining of tibiae. n = 4–6. (C) qRT-PCR of osteoblastic (Runx2, Osx, and Col1a1) and osteoclastic (Nfatc1, Mmp9, Ctsk, and Trap) markers. RNA isolated from the tibiae and femurs of WT, WT DoxoR, C5aR1−/−, and C5aR1−/−/DoxoR experimental groups. n = 5–9. (D) Chemotaxis of primary monocytes isolated from WT and C5aR1−/− mice toward medium without FBS, medium without FBS + 0.1 μg/mL of C5a (C5a). Migration of WT monocytes pre-treated for 30 min with 0.9 μg/mL PMX53 toward medium without FBS + 0.1 μg/mL C5a was also evaluated n = 4–6. (E) qRT-PCR of osteoclastic gene expression (Csf1r, Rank, Nfatc1, Mmp9, Ctsk, and Trap) from primary WT and C5aR1−/− of primary WT and C5aR1−/− monocytes. n = 8–11. Data are shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. (A–D) One-way ANOVA. (E) Student’s t test.
We also analyzed the effects of C5ar1 deficiency in osteopenia induced by OVX. OVX strongly induced weight gain in control mice over a 60-day period. C5ar1-deficient mice had a lower mean weight than controls and reduced weight gain after OVX (Figure S7B). μ-CT analysis indicated that cortical bone volume and thickness were reduced by OVX to the same extent in control and C5ar1-KO mice (Figure 6A). Ovariectomized mice also displayed decreased trabecular bone mass due to a reduced number and thickness of trabeculae. Interestingly, C5ar1 deficiency significantly prevented the loss of trabecular bone mass and increased the number of trabeculae and their thickness, compared with those in ovariectomized control mice (Figure 6B). In female ovariectomized control mice there was a slight, non-significant increase in osteoclast numbers compared with control mice. This increase induced by OVX was absent in ovariectomized C5ar1-KO mice (Figures 6C and S7C). Similar to the results obtained from DoxoR-treated male KO mice, estrogen deficiency was unable to increase the expression of Ctsk and Acp5/TRAP mRNAs in C5ar1-KO female mice (Figure 6D).
Figure 6.

C5ar1 deficiency prevented bone loss induced by loss of estrogens
(A) μ-CT quantitative parameters and 3D representation of cortical bone tibiae from an estrogen deficiency model by performing sham or ovariectomy (OVX) surgery on WT or C5aR1−/− female mice. n = 9–13. (B) Quantitative parameters and 3D representation of the trabecular compartment of tibiae. n = 9–13. (C) Quantification of the number of osteoblasts per bone surface (osteoblasts/mm) and osteocytes per cortical bone area (osteocytes/mm2) from H&E-stained sections of tibiae. The number of osteoclasts per trabecular bone surface (osteoclasts/mm) was quantified from TRAP staining of tibiae. n = 4–5. (D) Expression of osteoblastic (Runx2, Osx, and Col1a1) and osteoclastic (Nfatc1, Mmp9, Ctsk, and Trap) markers. RNA isolated from the tibiae and femurs of sham or OVX WT and C5aR1−/− mice. n = 6–9. Data shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. One-way ANOVA.
Using pharmacological treatment of DoxoR-treated mice with the C5AR1 antagonist PMX53, we also corroborated the findings observed in C5ar1-KO mice. PMX53 is rapidly distributed in the plasma but also has a fast elimination rate, which can be somewhat improved by daily subcutaneous administration33 (Figure 7A). Daily treatment with PMX53 did not produce significant changes in the weight of either control or DoxoR-treated mice (Figure S8A). The lower cortical bone volume and cortical thickness induced by DoxoR were not improved by PMX53 treatment (Figure 7B). However, PMX53 treatment partially prevented the reduction in trabecular bone volume and trabecular number upon DoxoR administration (Figure 7C). The remaining bone loss could arise from the involvement of additional complement pathways (i.e., C3aR), additional inflammatory signals activated by DoxoR (as shown by enhanced expression of genes involved in inflammatory responses in the RNA-seq analysis [Figure 3B]), or from reduced activity of PMX53 due to its rapid pharmacokinetics in vivo.34 In any case, the increases in the number of osteoclasts induced by DoxoR were completely abrogated by PMX53 treatment (Figures 7D and S8B). This reduction in the number of osteoclasts was confirmed through qRT-PCR analysis of bone samples. The increased expression of the osteoclastic markers Mmp9 and Acp5/TRAP in DoxoR-treated mice was recovered upon their treatment with PMX53 (Figure 7E). Altogether, these data show that deficiency or inhibition of C5AR1 signaling prevents bone loss by reducing monocyte recruitment and osteoclastogenesis upon DoxoR treatment or estrogen deficiency in mice.
Figure 7.

Pharmacological inhibition of C5AR1 prevented bone loss induced by chemotherapy
(A) Schematic representation of the pharmacological inhibition of C5aR1 in the DoxoR-induced senescence model. At day 0, 15-week-old mice received either vehicle or DoxoR (10 mg/kg, i.p.). From day 1 to day 30, mice received PMX53 (1 mg/kg, daily subcutaneous) or vehicle 2. (B) Quantitative parameters and 3D representation of the cortical bone of tibiae measured by μ-CT. n = 8–14. (C) Quantitative parameters and 3D representation of the trabecular compartment of tibiae measured by μ-CT. n = 8–14. (D) Quantification of the number of osteoblasts per bone surface (osteoblasts/mm) and osteocytes per cortical bone area (osteocytes/mm2) from H&E-stained sections of tibiae. The number of osteoclasts per trabecular bone surface (osteoclasts/mm) was quantified from TRAP staining of tibiae. n = 4–6. (E) qRT-PCR of osteoblastic (Runx2, Osx, and Col1a1) and osteoclastic (Nfatc1, Mmp9, Ctsk, and Trap) markers. RNA isolated from the tibiae and femurs of vehicle-, PMX53-, DoxoR-, and DoxoR + PMX53-treated mice. n = 6–10. Data are shown as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. One-way ANOVA.
Discussion
One in three women and at least one in six men will suffer an osteoporotic fracture in their lifetime. It is estimated that, in the European Union alone, more than 23 million men and women are at high risk of osteoporotic fractures. Osteoporosis, and the 4.3 million fragility fractures that it causes, costs the health care systems of Europe more than €56 billion each year.35 Therefore, there is a need for new drugs that lack side effects such as osteonecrosis of the jaw and atypical femur fractures and capitalize on the emerging knowledge of bone biology. Herein, we provide evidence that complement activation is a critical mediator of monocyte recruitment and osteoclast specification, which results in bone loss upon chemotherapy or estrogen deficiency.
A causal role of senescence in age-dependent bone loss has been established through genetic and pharmacological approaches.9 Our data broaden this concept to chemotherapy-induced bone loss, since we showed that pharmacological depletion of DoxoR-induced senescent cells improves bone health. In our model, all senescent cells of the body are targeted by ABT263.19 However, although we cannot rule out the involvement of additional cell types from the bone microenvironment, several lines of evidence point to senescent cells of mesenchymal origin as the most likely culprits in the bone microenvironment. First, exposure to a youthful circulation through parabiosis or reconstitution with young hematopoietic stem cells did not improve bone loss in a murine aging model.10 Second, eliminating senescent osteoclast progenitors while sparing senescent osteocytes has no effect on age-associated bone loss.36 Third, osteocytes are the longest living cells in bone and are thus more susceptible to accumulating damage over time, which affects about 11% of senescent osteocytes with aging.37
The deleterious effects of senescent cells on bone rely on induction of the SASP. Treatment of mice with pharmacological agents that suppress the SASP rescues bone health in a manner virtually identical to senescent cell clearance.9,11,38 The SASP promotes an inflammatory state, as demonstrated in our transcriptomic studies, where interferon-responsive genes were highlighted among those highly induced after 30 days of DoxoR administration. Increased expression of the pro-inflammatory SASP acts on osteoblasts to impair their function, reduces the number of MSCs, and shifts their differentiation toward adipogenesis.10,20,39,40 However, in our model of DoxoR-induced bone loss, we could not detect a decrease in the number of osteoblasts or an increase in bone marrow adiposity, at least after 30 days of DoxoR administration. As in most osteopenic conditions, we found in our chemotherapy-induced senescence, as well as in estrogen deficiency models, a major increase in osteoclast numbers on bone surfaces. Although evidence regarding the role of senescence in bone loss by estrogen deficiency is conflicting, estrogen deficiency is clearly associated with a pro-inflammatory state in the bone microenvironment.3,41 Therefore, the pro-inflammatory state secondary to estrogen deficiency may not necessarily reflect a canonical SASP, but might involve increased complement pathway activity as a shared inflammatory mechanism. Attraction of osteoclast precursors to remodeling sites and their maturation are chemotactically controlled by factors secreted from osteoblasts and osteocytes. Among these, CSF1 and the RANKL/OPG ratio have been identified as triggers of osteopenic conditions.10,15,42 However, we did not find alterations in the expression levels of Csf1, RANKL (Tnfsf11), or OPG (Tnfrsf11b) when we sequenced the RNA from the bones of DoxoR-treated mice. Instead, we found increases in Cfd, the rate-limiting protease of the alternative complement pathway, in DoxoR-induced and OVX-induced bone loss. The innate complement system senses danger signals and is activated during tissue injury to promote healing and organ regeneration.28 However, aberrant activation of the complement system contributes to the pathogenesis of several inflammatory diseases. The underlying mechanisms include (1) increased and persistent activation, as in systemic lupus erythematosus, anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis, periodontitis, and osteoarthritis16,43,44 and (2) altered expression or function of various complement regulators as in paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, and C3 glomerulopathies.45 Our findings provide a rationale to include activation of the alternative complement pathway as a new inflammatory mechanism triggering skeletal bone loss.
The majority of complement proteins are generated in the liver and released into the bloodstream. However, the final inflammatory products, anaphylatoxins C3a and C5a, are generated by the action of CFD at specific locations by tissue-resident cells. For instance, CFD from bone marrow has been shown to prime mesenchymal cells toward the adipogenic lineage and increase bone marrow adiposity. Mice deficient in Cfd, although lacking the basal bone phenotype, are resistant to bone loss induced by caloric restriction.46 We demonstrate that CFB and CFD expression is highly enriched in osteoblasts and osteocytes, whereas the anaphylatoxin receptors C3AR1 and C5AR1 are mostly expressed in hematopoietic osteoclast precursors. These data support the notion that alternative complement works as an additional osteocyte-osteoclast coupling factor that might direct homing of osteoclast precursors and promote their osteoclastogenesis. Consistent with this hypothesis, it is known that optimal homing and engraftment of hematopoietic stem cells also requires the alternative complement pathway.47 In vitro evidence suggests that activation of the complement pathway was able to promote osteoclast differentiation.48 Moreover, C5a, the most potent inflammatory complement fragment through C5aR1 activation, has been shown to mediate the release of a number of additional inflammatory cytokines, such as IL6, IL8, RANKL, and CXCL10.49 Our relative cellular expression and functional data suggest that the C5a/C5aR1 axis is the major branch involved in bone homeostasis, although additional complement pathways could also contribute. Mouse genetics showed that, although C5-deficient mice did not have any severe bone phenotype, C5ar1 and C5ar2 global KO mice showed higher bone mass.50 The effect on bone was more pronounced in C5ar1 than C5ar2, implying the existence of different mechanisms.50 In fact, the evidence suggests that C5aR2 is not pro-inflammatory and that its function is to dampen C5aR1 signaling.51
Given the major role of C5a/C5aR1 in a number of human pathologies, pharmacological agents have been developed to inhibit C5aR1 and thus keep inflammation in check.52 Small-molecule inhibitors such as PMX53 or PMX205 have shown some potential therapeutic effects in neutropenic inflammatory diseases and amyotrophic lateral sclerosis.53 However, these drugs are only in the preclinical stage and show a short half-life in blood and very rapid secretion.33 The most advanced drug to date is the competitive C5aR1 inhibitor Avacopan, which is specific for the human receptor and potently blocks C5a-induced responses. As it is orally bioavailable and has a good safety profile, the drug has been shown to be effective for the treatment of ANCA-associated vasculitis.54 Very recently, Avacopan (Tavneos) was approved for medical use for ANCA-associated vasculitis in Japan, the United States, and Europe in 2021. Therefore, repurposing Avacopan for the treatment of osteopenic pathologies should pose few contraindications. However, further studies should be performed to optimize the dosing and timing of administration or to evaluate combinatorial approaches with other anti-resorptive therapies.
Materials and methods
Cell cultures
Primary osteocytes were isolated using a protocol from Sánchez-de-Diego et al.55 In brief, tibiae, femurs, and calvariae from five P1-P3 mice were pooled and digested on a rotating shaker at 37°C. After several alternate digestions with trypsin (0.025%)/collagenase II (1 mg/mL) in α-MEM and EDTA solution (5 mM EDTA in PBS containing 1% BSA [pH 7.4]), digestions 7–10 were pooled by centrifugation. The cell pellet was resuspended in α-MEM and plated on type I collagen-coated culture plates. Osteocytes were cultured in α-MEM supplemented with heat-inactivated 5% FBS, 5% calf serum, 2 mM L-glutamine, 1 mM sodium pyruvate, and 100 U/mL penicillin and 0.1 mg/mL streptomycin (P/S) in a humidified incubator for up to 7 days.
For bone marrow-derived cell isolation, hindlimbs from 6- to 8-week-old C57BL6/J mice were harvested. The distal and proximal epiphyses of the tibiae and femurs were excised, and bone marrow was removed by centrifugation at >20,000 × g for 15 s at 4°C. Murine BMMSCs were isolated as described previously.56 In brief, after marrow isolation, cells were cultured in complete DMEM (supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate and P/S). Non-adherent cells were discarded after 3 h. Media was replaced every 12 h until 70% confluence was reached. At this stage, cells were lifted by incubation with 0.25% trypsin/0.02% EDTA for 5 min at room temperature. Lifted cells were expanded for further experiments.
Bone marrow-derived macrophages (BMMs) were isolated following the protocol described by Guo et al.57 The isolated bone marrow cells from one mouse were incubated for 8 min on ice with 3 mL of red blood cell lysis buffer. Lysis was stopped by adding 6 mL of complete α-MEM medium (supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, and P/S), and cells were centrifuged at 250 × g and 4°C for 10 min. Cells were resuspended in 4 mL PBS and gently added to 2 mL of Ficoll Biocoll isotonic separating solution (Biochrom). After 20 min at 250 × g (centrifuge break <2), the ring containing mononucleated cells was collected. Cells were rinsed with PBS and centrifuged at 1,200 rpm for 5 min. Finally, BMMs were resuspended in complete α-MEM medium supplemented with 30 ng/mL M-CSF (PeproTech) for 5 days. Medium and M-CSF1 supplementation were changed every 48 h.
Bone marrow-derived monocytes were isolated using the Monocyte Isolation Kit (BM) (Miltenyi Biotec). CD11b+ monocytes were isolated from total isolated bone marrow cells using microbeads for magnetic separation according to the manufacturer’s protocol. Monocytes were collected in RPMI-1640 medium supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, and P/S and used directly for the chemotactic migration assay.
HSPCs were isolated from total bone-marrow using the EasySep Mouse Hematopoietic Progenitor Cell Isolation Kit (STEMCELL Technologies). Isolated cells were maintained for 7 days in Ham’s F12 Nutrient Mix medium supplemented with 10 mM HEPES, 1× P/S-glutamine, 1× insulin-transferrin-selenium-ethanolamine (all from Gibco), 1 mg/mL polyvinyl alcohol (Sigma), 100 ng/mL thrombopoietin (PeproTech), and 10 ng/mL stem cell factor (PeproTech).
Endothelial cells were isolated from mouse lungs, following the protocol described by Wang et al.58 In brief, minced lungs were digested at 37°C for 45 min using 2 mg/mL type I collagenase. Digested tissue was filtered through a 70-μm strainer and the cell suspension was incubated with CD31-conjugated Dynabeads for 15 min at room temperature with vigorous shaking (rat anti-mouse CD31 antibody from BD Pharmingen and sheep anti-rat IgG Dynabeads from Thermo Fisher Scientific). When seeded cells reached confluence a second sorting was performed using CD31-conjugated Dynabeads. Endothelial cells were seeded in 0.1% gelatin-coated dishes. The human monocytic cell line THP1 was cultured in RPMI-1640 supplemented with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, P/S, 0.1 mM non-essential amino acid solution, 50 μM 2-mercaptoethanol, and 10 mM HEPES. The murine macrophage-like cell line RAW264.7 was maintained as macrophages in complete α-MEM or differentiated into osteoclasts by supplementing the medium with 15 ng/mL RANKL (PreproTech) for 5 days.
Cellular senescence was induced by treating cells with 100 nM DoxoR (Cell Signaling Technology) for 24 h. Cells became completely senescent after 7 days. In vitro modulation of the complement pathway was performed by treating cells with 1 μg/mL C3a, 0.1 μg/mL C5a (both from R&D), or 0.9 μg/mL PMX53 (MedChemExpress). All complement experiments were performed with complete medium supplemented with heat-inactivated FBS. Treatment times were as follows: in cell signaling experiments, cells were exposed to ligands and inhibitor for 30 min; in chemotaxis assays, cells were pre-exposed for 30 min with the inhibitor and for the 2 h of migration to C5a; in differentiation-modulation experiments, cells were treated daily with C3a or C5a, for 5–7 days.
CM experiments
After 7 days of DoxoR treatment, 48-h CM was collected from control and senescent BMMSCs. CM was filtered through a 0.2-μm filter. Receptor cells were treated with 50% CM and 50% fresh medium for 5–7 days.
Mouse treatment
To define the chemotherapy-induced senescence model, 15-week-old WT C57BL/6 male mice were randomly assigned to one of the four experimental groups: vehicle, ABT263, DoxoR, and DoxoR + ABT263. At day 0, mice were injected intraperitoneally with vehicle (saline solution) or 10 mg/kg DoxoR (Cell Signaling Technology). At days 7 and 21, mice were dosed daily for 7 days via oral gavage with vehicle (10% ethanol, 30% polyethylene glycol 400 and 60% Phosal 50 PG) or 50 mg/kg of ABT263 (Chemgood). Mice were weighed every 3 days.
To study the impact of complement pathway inhibition in vivo, both genetic and pharmacological models were used. C5aR-KO (C5ar1tm1Cge) mice were purchased from The Jackson Laboratory. Fifteen-week-old male WT and C5aR-KO mice were randomly assigned to a vehicle and a DoxoR group. Eight-week-old female WT and C5aR-KO mice underwent a bilateral OVX, and an equal number of mice underwent sham surgery (Sham). Female mice were weighed every 3–5 days and euthanized at day 60 after surgery. Pharmacological modulation was performed on the DoxoR-induced senescence model. The day after DoxoR injection, mice were treated daily with vehicle (saline solution) or 1 mg/kg PMX53 by subcutaneous administration (MedChemExpress).
Mice were housed at 23°C ± 1°C under a 12-h light/12-h dark cycle with access to food and water ad libitum. All procedures were approved by the Ethics Committee for Animal Experimentation of the Generalitat of Catalunya.
Bone histology
The right tibiae, cleaned of soft tissue, were fixed in 4% PFA for 24 h at 4°C and decalcified in 14% EDTA (pH 7.4) (in PBS) for 6 weeks. Bones were embedded in paraffin and 5-μm sections were cut and stained with hematoxylin and eosin for osteoblast and osteocyte quantification. Osteoclasts were quantified by staining for TRAP. For TRAP staining, deparaffinized and rehydrated slides were incubated with naphthyl AS-BI phosphate (Sigma-Aldrich) for 1 h at 37°C and osteoclasts were developed with pararosaniline hydrochloride (Sigma-Aldrich) for 3 min. The tissue was counterstained with 0.02% fast green solution (Sigma-Aldrich) for 45 s. Cells were quantified from images captured using an Axio Imager M2 microscope (Zeiss).
μ-CT
High-resolution images from fixed tibiae (4% PFA, 24 h at 4°C), were acquired using a computerized microtomography imaging system (Skyscan 1272, Bruker microCT) in accordance with the recommendations of the American Society of Bone and Mineral Research. Samples were scanned in air at 70 kV and a power of 10 W (143 μA) with an exposure time of 2,700 ms, using a 1-mm aluminum filter and an isotropic voxel size of 11 μm. Two-dimensional (2D) images were obtained every 1° of a 180° rotation. Images were reconstructed using the NRecon software (version 1.7.1.0, Bruker) and analyzed with a CT Analyzer (version 1.17.7.2, SkyScan).59,60 For trabecular measurements, manual ROIs were selected, starting 0.6 mm from the distal growth plate of the tibiae and extending to the diaphysis for 2 mm. For cortical parameters, a 3-mm section starting from the bifurcation of the fibula was selected. Trabecular and cortical ROIs were 3D represented using CT-Vol (version 2.3.2.0, SkyScan).
Chemotactic migration assay
The chemotactic migration of THP1 and primary monocytes was analyzed using the InnoCyte Monocyte Cell Migration Assay (Merck Millipore). Cells were serum starved for 3 h and, when needed, exposed to 0.9 μg/mL of PMX53 for 30 min before the start of the assay. Upper chambers were pre-humidified with 20 μL of medium without FBS for 30 min. A total of 1 × 105 cells/100 μL were added to the upper chamber of the assay plate. The lower chambers were filled with medium with FBS (positive control) medium without FBS (negative control) or 0.1 μg/mL C5a (rmC5a, R&D) as a chemotactic factor. After 2 h, the cells in the lower compartment were stained with calcein-AM. Fluorescence was measured using a plate reader at an excitation wavelength of 485 nm and an emission wavelength of 520 nm.
Cell number quantification
Living cells were labeled with 5 μg/mL Hoechst 33342 (Sigma-Aldrich) in PBS for 10 min. Labeling was performed at 0, 24, and 48 h in independent wells. Several fields were analyzed using a Leica DM-IRB inverted microscope. The total number of cells and percentage of multinucleated cells were counted using ImageJ.
Gene expression
Total RNA was isolated from cell cultures or murine tibia and femur cells using the TRIsure reagent (BioLine). Purified RNA was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Quantitative PCR was carried out using the QuantStudio 7 Pro Real-Time PCR System using TaqMan primers (Thermo Fischer Scientific) with SensiFAST Probe Hi-ROX Mix (Bioline). Fold changes in gene expression were calculated using the ΔΔCt method, where all transcripts were normalized to TATA binding protein (Tbp) expression.
RNA isolation and sequencing
Within 5 min of the animal’s sacrifice, long bones of the hindlimbs were prepared for high-quality RNA isolation. In brief, bones were cleaned of soft tissue, and the distal and proximal epiphyses were excised. Bone marrow was removed by centrifugation at >20,000 × g for 15 s at 4°C. TRIsure was added to marrow-free bones, which were then flash frozen in liquid nitrogen and stored at −80°C. For RNA isolation, both tibiae and femurs from each mouse were subjected to 1 cycle of 45 s at full speed using a Polytron (Kinematic PT). Total RNA was separated from bone minerals, DNA, and protein by phenol-chloroform extraction. The RNA-containing fraction was then purified using the RNeasy Mini Kit (QIAGEN) and treated with DNase I (RNase-Free DNase Set, QIAGEN ).
DNA-free RNA samples (n = 4 for each condition) were quantified using the Qubit RNA BR Assay kit (Thermo Fisher Scientific), and RNA integrity was estimated with the Agilent RNA 6000 Pico Bioanalyzer 2100 Assay (Agilent). The RNA-seq libraries were prepared with the KAPA Stranded mRNA-Seq Illumina Platforms Kit (Roche) following the manufacturer's recommendations. In brief, 500 ng of total RNA was used for enrichment of the poly(A) fraction with oligo-dT magnetic beads following mRNA fragmentation. Strand specificity was achieved during the second-strand synthesis performed in the presence of dUTP instead of dTTP. The blunt-ended double-stranded cDNA was 3′ adenylated, and Illumina platform-compatible adaptors with unique dual indexes and unique molecular identifiers (Integrated DNA Technologies) were ligated. The ligation product was enriched with 15 PCR cycles. The size and quality of the libraries were assessed in a High-Sensitivity DNA Bioanalyzer assay (Agilent). The libraries were sequenced using the HiSeq 4000 (Illumina) platform with a read length of 2 × 76 bp + 8 bp + 8 bp using the HiSeq 4000 SBS kit (Illumina).
Reads were mapped against the mouse reference genome (GRCh38) with STAR/2.5.3a using the ENCODE parameters for long RNA. Genes were quantified with RSEM/1.3.0 using the gencode annotation M23. Quality control of the mapping and quantification steps was performed using “gtfstats” from GEM-Tools 1.7.1. Differential expression analysis was performed with DESeq2/1.18 using the default parameters. Unwanted sources of variation were removed with surrogate variable analysis using the sva R package.59 Genes with FDR < 5% were considered significant. Heatmaps of differentially expressed genes were generated using the DESeq2 “rlog” transformation of the counts. Top categories were selected according to the adjusted p values.
Western blot assay
Cell cultures were lysed in 1% (w/v) SDS, 10 mM Tris, 1 mM EDTA (pH 8.0). Quantification of the protein content was performed using the BCA protein assay kit (Thermo Scientific). Equal amounts of protein from cell lysates were resolved by PAGE and transferred to Immobilon-P membranes. Primary antibodies against phospho-S6 (Cell Signaling Technology, 2211), phospho-Hsp27 (Cell Signaling Technology, 2401), and phospho-ERK1/2 (Cell Signaling Technology, 91011) were used at a 1:1,000 dilution. Antibody against β-actin (Abcam, 6276) was used at 1:4,000 dilution. Immunoreactive bands were detected with horseradish-peroxidase-conjugated secondary antibodies and an EZ-ECL kit (Biological Industries). Quantification of the band intensities was performed with Fujifilm Multi Gauge software using β-actin for normalization.
Statistical analysis
The Shapiro-Wilk test was performed to analyze deviations from a normal distribution of the measures. Statistical analyses were performed by applying a two-tailed unpaired Student’s t test or one-way ANOVA with a Tukey multiple comparisons post hoc test (GraphPad Prism 8). Quantitative data are presented as mean ± SEM. Differences were considered significant at ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Data availability
RNA-seq data was deposited in the GEO repository (GEO accession no. GSE205237).
Acknowledgments
We thank B. Torrejón, E. Castaño, and M. Ortega from the CCiT-UB-Bellvitge and L. Mulero, J. Llamas from IDIBELL S&T Services for technical assistance. C.P.-L. is a recipient of an F.P.U. fellowship from the Spanish Ministry of Education. A.D. is a recipient of an F.P.I. fellowship from the Spanish Ministry of Science and Innovation (MCIN/AEI/10.13039/501100011033). This research was supported by grants PDC2021-121776-I00 and PID2020-117278GB-I00 from MCIN/AEI/10.13039/501100011033 and ISCIII /MINECO (PT17/0009/0019) and co-funded by FEDER “Una manera de hacer Europa” or “NextGenerationEU”/PRTR and grant 202038-30 from La Marató de TV3.
Author contributions
C.P.-L., J.L.R., and F.V. conceived and designed the experiments. C.P.-L., C.S.-d.-D., A.D., A.E.-C., J.A.V., and A.M.-L. performed the experiments. C.P.-L., C.S.-d.-D., A.D., A.A., A.E.-C., J.L.R., and F.V. analyzed the data. C.P.-L. and F.V. wrote the paper.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.04.022.
Supplemental information
References
- 1.Compston J.E., McClung M.R., Leslie W.D. Osteoporosis. Lancet. 2019;393:364–376. doi: 10.1016/S0140-6736(18)32112-3. [DOI] [PubMed] [Google Scholar]
- 2.Farr J.N., Khosla S. Cellular senescence in bone. Bone. 2019;121:121–133. doi: 10.1016/j.bone.2019.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khosla S., Farr J.N., Monroe D.G. Cellular senescence and the skeleton: pathophysiology and therapeutic implications. J. Clin. Invest. 2022;132 doi: 10.1172/JCI154888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wan M., Gray-Gaillard E.F., Elisseeff J.H. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res. 2021;9:41. doi: 10.1038/S41413-021-00164-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goltzman D. The aging skeleton. Adv. Exp. Med. Biol. 2019;1164:153–160. doi: 10.1007/978-3-030-22254-3_12. [DOI] [PubMed] [Google Scholar]
- 6.Xu M., Pirtskhalava T., Farr J.N., Weigand B.M., Palmer A.K., Weivoda M.M., Inman C.L., Ogrodnik M.B., Hachfeld C.M., Fraser D.G., et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018;24:1246–1256. doi: 10.1038/s41591-018-0092-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farr J.N., Kaur J., Doolittle M.L., Khosla S. Osteocyte cellular senescence. Curr. Osteoporos. Rep. 2020;18:559–567. doi: 10.1007/S11914-020-00619-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonewald L.F. The amazing osteocyte. J. Bone Miner. Res. 2011;26:229–238. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farr J.N., Xu M., Weivoda M.M., Monroe D.G., Fraser D.G., Onken J.L., Negley B.A., Sfeir J.G., Ogrodnik M.B., Hachfeld C.M., et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med. 2017;23:1072–1079. doi: 10.1038/nm.4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ambrosi T.H., Marecic O., McArdle A., Sinha R., Gulati G.S., Tong X., Wang Y., Steininger H.M., Hoover M.Y., Koepke L.S., et al. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature. 2021;597:256–262. doi: 10.1038/S41586-021-03795-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao Z., Murali B., Ren Q., Luo X., Faget D.V., Cole T., Ricci B., Thotala D., Monahan J., Van Deursen J.M., et al. Therapy-induced senescence drives bone loss. Cancer Res. 2020;80:1171–1182. doi: 10.1158/0008-5472.CAN-19-2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu Q., Katlinskaya Y.V., Carbone C.J., Zhao B., Katlinski K.V., Zheng H., Guha M., Li N., Chen Q., Yang T., et al. DNA-Damage-Induced type I interferon promotes senescence and inhibits stem cell function. Cell Rep. 2015;11:785–797. doi: 10.1016/J.CELREP.2015.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tchkonia T., Zhu Y., Van Deursen J., Campisi J., Kirkland J.L. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Invest. 2013;123:966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schaum N., Lehallier B., Hahn O., Pálovics R., Hosseinzadeh S., Lee S.E., Sit R., Lee D.P., Losada P.M., Zardeneta M.E., et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature. 2020;583:596–602. doi: 10.1038/S41586-020-2499-Y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim H.N., Xiong J., MacLeod R.S., Iyer S., Fujiwara Y., Cawley K.M., Han L., He Y., Thostenson J.D., Ferreira E., et al. Osteocyte RANKL is required for cortical bone loss with age and is induced by senescence. JCI Insight. 2020;5 doi: 10.1172/JCI.INSIGHT.138815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barratt J., Weitz I. Complement factor D as a strategic target for regulating the alternative complement pathway. Front. Immunol. 2021;12:712572. doi: 10.3389/fimmu.2021.712572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hadji P., Ziller M., Maskow C., Albert U., Kalder M. The influence of chemotherapy on bone mineral density, quantitative ultrasonometry and bone turnover in pre-menopausal women with breast cancer. Eur. J. Cancer. 2009;45:3205–3212. doi: 10.1016/J.EJCA.2009.09.026. [DOI] [PubMed] [Google Scholar]
- 18.Demaria M., O’Leary M.N., Chang J., Shao L., Liu S., Alimirah F., Koenig K., Le C., Mitin N., Deal A.M., et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7:165–176. doi: 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang J., Wang Y., Shao L., Laberge R.-M., Demaria M., Campisi J., Janakiraman K., Sharpless N.E., Ding S., Feng W., et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016;22:78–83. doi: 10.1038/nm.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ambrosi T.H., Scialdone A., Graja A., Sch€ A., Jank A.M., Bocian C., Woelk L., Fan H., Logan D.W., Schürmann A., et al. Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration cell stem cell article adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell. 2017;20:771–784.e6. doi: 10.1016/j.stem.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J., Lu L., Liu Y., Yu X. Bone marrow adiposity during pathologic bone loss: molecular mechanisms underlying the cellular events. J. Mol. Med. 2022;100:167–183. doi: 10.1007/s00109-021-02164-1. [DOI] [PubMed] [Google Scholar]
- 22.Nelson G., Wordsworth J., Wang C., Jurk D., Lawless C., Martin-Ruiz C., von Zglinicki T. A senescent cell bystander effect: senescence-induced senescence. Aging Cell. 2012;11:345–349. doi: 10.1111/j.1474-9726.2012.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Cecco M., Ito T., Petrashen A.P., Elias A.E., Skvir N.J., Criscione S.W., Caligiana A., Brocculi G., Adney E.M., Boeke J.D., et al. L1 drives IFN in senescent cells and promotes age-associated inflammation Activation of L1 and IFN-I in cellular senescence. Nature. 2019;566:73–78. doi: 10.1038/s41586-018-0784-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Youlten S.E., Kemp J.P., Logan J.G., Ghirardello E.J., Sergio C.M., Dack M.R.G., Guilfoyle S.E., Leitch V.D., Butterfield N.C., Komla-Ebri D., et al. Osteocyte transcriptome mapping identifies a molecular landscape controlling skeletal homeostasis and susceptibility to skeletal disease. Nat. Commun. 2021;12:2444. doi: 10.1038/S41467-021-22517-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papatheodorou I., Moreno P., Manning J., Mu A.M., George N., Fexova S., Fonseca N.A., Füllgrabe A., Ullgrabe A.F., Huang N., et al. Expression Atlas update: from tissues to single cells. Nucleic Acids Res. 2020;48:77–83. doi: 10.1093/nar/gkz947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodríguez S.A., Grochová D., Mckenna T., Borate B., Trivedi N.S., Erdos M.R., Eriksson M. Global genome splicing analysis reveals an increased number of alternatively spliced genes with aging. Aging Cell. 2016;15:267–278. doi: 10.1111/acel.12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brophy R.H., Zhang B., Cai L., Wright R.W., Sandell L.J., Rai M.F. Transcriptome comparison of meniscus from patients with and without osteoarthritis. Osteoarthritis Cartilage. 2018;26:422–432. doi: 10.1016/j.joca.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reis E.S., Mastellos D.C., Hajishengallis G., Lambris J.D. New insights into the immune functions of complement. Nat. Rev. Immunol. 2019;19:503–516. doi: 10.1038/s41577-019-0168-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katschke K.J., Wu P., Ganesan R., Kelley R.F., Mathieu M.A., Hass P.E., Murray J., Kirchhofer D., Wiesmann C., Van Lookeren Campagne M. Inhibiting alternative pathway complement activation by targeting the factor D exosite. J. Biol. Chem. 2012;287:12886–12892. doi: 10.1074/jbc.M112.345082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu H., Kim H.R., Deepak R.V.K., Wang L., Chung K.Y., Fan H., Wei Z., Zhang C. Orthosteric and allosteric action of the C5a receptor antagonists. Nat. Struct. Mol. Biol. 2018;25:472–481. doi: 10.1038/s41594-018-0067-z. [DOI] [PubMed] [Google Scholar]
- 31.Rousseau S., Dolado I., Beardmore V., Shpiro N., Marquez R., Nebreda A.R., Arthur J.S.C., Case L.M., Tessier-Lavigne M., Gaestel M., et al. CXCL12 and C5a trigger cell migration via a PAK1/2-p38α MAPK-MAPKAP-K2-HSP27 pathway. Cell. Signal. 2006;18:1897–1905. doi: 10.1016/j.cellsig.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Daswani B., Gupta M.K., Gavali S., Desai M., Sathe G.J., Patil A., Parte P., Sirdeshmukh R., Khatkhatay M.I. Monocyte proteomics reveals involvement of phosphorylated HSP27 in the pathogenesis of osteoporosis. Dis. Markers. 2015;2015:196589. doi: 10.1155/2015/196589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar V., Lee J.D., Clark R.J., Noakes P.G., Taylor S.M., Woodruff T.M. Preclinical pharmacokinetics of complement C5a receptor antagonists PMX53 and PMX205 in mice. ACS Omega. 2020;5:2345–2354. doi: 10.1021/acsomega.9b03735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar V., Lee J.D., Clark R.J., Noakes P.G., Taylor S.M., Woodruff T.M. Preclinical pharmacokinetics of complement C5a receptor antagonists PMX53 and PMX205 in mice. ACS Omega. 2020;5:2345–2354. doi: 10.1021/ACSOMEGA.9B03735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanis J.A., Norton N., Harvey N.C., Jacobson T., Johansson H., Lorentzon M., McCloskey E.V., Willers C., Borgström F. Scope 2021: a new scorecard for osteoporosis in Europe. Arch. Osteoporos. 2021;16:82. doi: 10.1007/s11657-020-00871-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim H.N., Chang J., Iyer S., Han L., Campisi J., Manolagas S.C., Zhou D., Almeida M. Elimination of senescent osteoclast progenitors has no effect on the age-associated loss of bone mass in mice. Aging Cell. 2019;18:e12923. doi: 10.1111/acel.12923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Farr J.N., Fraser D.G., Wang H., Jaehn K., Ogrodnik M.B., Weivoda M.M., Drake M.T., Tchkonia T., Lebrasseur N.K., Kirkland J.L., et al. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res. 2016;31:1920–1929. doi: 10.1002/jbmr.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu M., Tchkonia T., Ding H., Ogrodnik M., Lubbers E.R., Pirtskhalava T., White T.A., Johnson K.O., Stout M.B., Mezera V., et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA. 2015;112:E6301–E6310. doi: 10.1073/pnas.1515386112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sui B., Hu C., Liao L., Chen Y., Zhang X., Fu X., Zheng C., Li M., Wu L., Zhao X., Jin Y. Mesenchymal progenitors in osteopenias of diverse pathologies: differential characteristics in the common shift from osteoblastogenesis to adipogenesis. Sci. Rep. 2016;6 doi: 10.1038/srep30186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim H.N., Chang J., Shao L., Han L., Iyer S., Manolagas S.C., O’Brien C.A., Jilka R.L., Zhou D., Almeida M. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell. 2017;16:693–703. doi: 10.1111/acel.12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu D., Cline-Smith A., Shashkova E., Perla A., Katyal A., Aurora R. T-cell mediated inflammation in postmenopausal osteoporosis. Front. Immunol. 2021;12:687551. doi: 10.3389/fimmu.2021.687551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu Y., Li X., Zhi X., Cong W., Huang B., Chen H., Wang Y., Li Y., Wang L., Fang C., et al. RANKL from bone marrow adipose lineage cells promotes osteoclast formation and bone loss. EMBO Rep. 2021;22:e52481. doi: 10.15252/embr.202152481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mödinger Y., Löffler B., Huber-Lang M., Ignatius A. Complement involvement in bone homeostasis and bone disorders. Semin. Immunol. 2018;37:53–65. doi: 10.1016/j.smim.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Wang Q., Rozelle A.L., Lepus C.M., Scanzello C.R., Song J.J., Meegan Larsen D., Crish J.F., Bebek G., Ritter S.Y., Lindstrom T.M., et al. Identification of a central role for complement in osteoarthritis. Nat. Med. 2011;17:1674–1679. doi: 10.1038/nm.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohebnasab M., Eriksson O., Persson B., Sandholm K., Mohlin C., Huber-Lang M., Keating B.J., Ekdahl K.N., Nilsson B. Current and future approaches for monitoring responses to anti-complement therapeutics. Front. Immunol. 2019;10:2539. doi: 10.3389/fimmu.2019.02539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aaron N., Kraakman M.J., Zhou Q., Liu Q., Costa S., Yang J., Liu L., Yu L., Wang L., He Y., et al. Adipsin promotes bone marrow adiposity by priming mesenchymal stem cells. Elife. 2021;10:e69209. doi: 10.7554/eLife.69209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adamiak M., Ciechanowicz A., Chumak V., Bujko K., Ratajczak J., Brzezniakiewicz-Janus K., Kucia M., Ratajczak M.Z. Novel evidence that alternative pathway of complement cascade activation is required for optimal homing and engraftment of hematopoietic stem/progenitor cells. Stem Cell Rev. Rep. 2022;18:1355–1365. doi: 10.1007/s12015-021-10318-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tu Z., Bu H., Dennis J.E., Lin F. Efficient osteoclast differentiation requires local complement activation. Blood. 2010;116:4456–4463. doi: 10.1182/blood-2010-01-263590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mödinger Y., Rapp A., Pazmandi J., Vikman A., Holzmann K., Haffner-Luntzer M., Huber-Lang M., Ignatius A. C5aR1 interacts with TLR2 in osteoblasts and stimulates the osteoclast-inducing chemokine CXCL10. J. Cell Mol. Med. 2018;22:6002–6014. doi: 10.1111/jcmm.13873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kovtun A., Bergdolt S., Hägele Y., Matthes R., Lambris J.D., Huber-Lang M., Ignatius A. Complement receptors C5aR1 and C5aR2 act differentially during the early immune response after bone fracture but are similarly involved in bone repair. Sci. Rep. 2017;7:1–12. doi: 10.1038/s41598-017-14444-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X.X., Clark R.J., Woodruff T.M. C5aR2 activation broadly modulates the signaling and function of primary human macrophages. J. Immunol. 2020;205:1102–1112. doi: 10.4049/jimmunol.2000407. [DOI] [PubMed] [Google Scholar]
- 52.Pandey S., Maharana J., Li X.X., Woodruff T.M., Shukla A.K. Emerging insights into the structure and function of complement C5a receptors. Trends Biochem. Sci. 2020;45:693–705. doi: 10.1016/j.tibs.2020.04.004. [DOI] [PubMed] [Google Scholar]
- 53.Li X.X., Lee J.D., Massey N.L., Guan C., Robertson A.A.B., Clark R.J., Woodruff T.M. Pharmacological characterisation of small molecule C5aR1 inhibitors in human cells reveals biased activities for signalling and function. Biochem. Pharmacol. 2020;180:114156. doi: 10.1016/j.bcp.2020.114156. [DOI] [PubMed] [Google Scholar]
- 54.Jayne D.R.W., Merkel P.A., Schall T.J., Bekker P., ADVOCATE Study Group Avacopan for the treatment of ANCA-associated vasculitis. N. Engl. J. Med. 2021;384:599–609. doi: 10.1056/nejmoa2023386. [DOI] [PubMed] [Google Scholar]
- 55.Sánchez-de-Diego C., Pedrazza L., Pimenta-Lopes C., Martinez-Martinez A., Dahdah N., Valer J.A., Garcia-Roves P., Rosa J.L., Ventura F. NRF2 function in osteocytes is required for bone homeostasis and drives osteocytic gene expression. Redox Biol. 2021;40:101845. doi: 10.1016/j.redox.2020.101845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Valer J.A., Sánchez-de-Diego C., Gámez B., Mishina Y., Rosa J.L., Ventura F. Inhibition of phosphatidylinositol 3-kinase α ( PI 3Kα) prevents heterotopic ossification. EMBO Mol. Med. 2019;11:e10567. doi: 10.15252/emmm.201910567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo Y., Xie C., Li X., Yang J., Yu T., Zhang R., Zhang T., Saxena D., Snyder M., Wu Y., Li X. Succinate and its G-protein-coupled receptor stimulates osteoclastogenesis. Nat. Commun. 2017;8:15621. doi: 10.1038/NCOMMS15621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang J., Niu N., Xu S., Jin Z.G. A simple protocol for isolating mouse lung endothelial cells. Sci. Rep. 2019;9:1458. doi: 10.1038/s41598-018-37130-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reimand J., Arak T., Adler P., Kolberg L., Reisberg S., Peterson H., Vilo J. g:Profiler-a web server for functional interpretation of gene lists (2016 update) Nucleic Acids Res. 2016;44:W83–W89. doi: 10.1093/NAR/GKW199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leek J.T., Johnson W.E., Parker H.S., Jaffe A.E., Storey J.D. The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data was deposited in the GEO repository (GEO accession no. GSE205237).