

Abstract
Background
GJB2 mutations are among the most important causes of deafness, and their prevalence varies greatly among different countries and ethnic groups. This study aimed to determine the pathogenic mutation spectrum of GJB2 in patients with nonsyndromic hearing loss (NSHL) in Western Guangdong and to explore the pathogenic characteristics of the c.109G>A locus.
Methods
In total, 97 NSHL patients and 212 normal controls (NC) were included in this study. Genetic sequencing analyses were performed on GJB2.
Results
In the NSHL group, the main pathogenic mutations in GJB2 were as follows: c.109G>A, c.235delC, and c.299_300delAT with allele frequencies of 9.28%, 4.12%, and 2.06%, respectively. c.109G>A was the most frequently detected pathogenic mutation in this region. In the NC group, the allele frequency of c.109G>A among 30–50 years old subjects was markedly lower than that among 0–30 years old subjects (5.31% vs. 11.11%, p < 0.05).
Conclusion
We found the pathogenic mutation spectrum of GJB2 in this region and showed that c.109G>A was the most common GJB2 mutation with unique characteristics, such as clinical phenotypic heterogeneity and delayed onset. Therefore, the c.109G>A mutation should be considered as an essential marker for routine genetic assessment of deafness, which can also be beneficial for preventing deafness.
Keywords: c.109G>A (p.Val37Ile), delayed onset, hearing loss, nonsyndromic hearing loss (NSHL), phenotypic heterogeneity
In this study, hotspot mutations in the GJB2 gene of all subjects were sequenced and comprehensively analyzed to investigate the mutation spectrum of this region and the pathogenic characteristics of the c.109G>A mutation. It was speculated that the pathogenicity of the c.109G>A mutation was delayed, and some adolescents with c.109G>A mutation did not have true normal hearing. These findings provide a better understanding of the genetic basis of NSHL, facilitating earlier diagnosis and genetic counseling for the disease.

1. INTRODUCTION
According to World Health Organization (WHO) data, there were 430 million patients with hearing loss globally, accounting for 5% of the world population (WHO, 2021). The Asia‐Pacific region is a high‐risk area for deafness. Congenital hearing loss is one of the most common forms of congenital disabilities. Approximately 20,000–30,000 cases of congenital deafness have been reported annually in China. In addition, a considerable proportion of infants suffer from delayed hearing loss, and new deafness occurs in approximately 60,000–80,000 children each year (Dai et al., 2007).
To date, it is reported that more than 120 gene and over 1000 variants are connected to hearing loss. In China, nonsyndromic hearing loss (NSHL) is mainly related to mutations in GJB2 (OMIM:121011), SLC26A4 (OMIM:274600), 12S rRNA (mitochondrial, OMIM:561000), and GJB3 (OMIM:612644), with the mutation frequencies of 27.13%, 20.85%, 2.26%, and 2.01%, respectively (Liu et al., 2020). GJB2 mutation is the most common genetic factor associated with hearing loss. Approximately 81.6% of patients with NSHL carrying GJB2 mutations have severe or profound hearing loss, whereas the remaining patients have mild‐to‐profound hearing loss. These cases are mainly distributed across two levels centered at 70 dB or higher than 95 dB. The severity and onset of the disease are associated with different mutations in the GJB2 gene (Liu et al., 2005). However, its prevalence varies greatly among different countries or ethnic groups. Epidemiological analyses have shown that the detection rates of GJB2 mutations are 43% in Israel, 17% in Tunisia, 14% in Australia, and 5% in Korea (Kenneson et al., 2002). Dai et al. (2009) reported that the frequency of GJB2 mutations varied from 4% to 30.4% among 23 regions of China. Duan et al. (2021) reported that in patients with NSHL from Qinghai, China, the mutation rates of GJB2 were 20.88% (48 of 230) and 9.5% (20 of 210) in the Han and minority ethnic groups, respectively. Epidemiological analyses have shown that the most common GJB2 pathogenic variants are c.35delG (OMIM:121011.0005) in Caucasians, c.167delT (OMIM:121011.0010) in German Ashkenazi Jews, and c.235delC (OMIM:121011.0014) in Asian populations (Dai et al., 2019; Kenneson et al., 2002). Numerous studies have suggested that c.35delG, c.176_191del16, c.235delC, and c.299_300delAT are the most common GJB2 mutations with certain pathogenicity in China (Dai et al., 2019; Liu et al., 2020), and the four most prevalent variants accounting for 88.0% of the detected mutations (Dai et al., 2009). Recently, it has been reported that the c.109G>A (p.Val37Ile, OMIM:121011.0023) mutation is frequently detected in patients with deafness, with a mutation rate of approximately 1.5–18.2% (Huang, Yang et al., 2015; Wu et al., 2017; Kim et al., 2013). Nevertheless, the pathogenicity of the c.109G>A mutation is controversial, and it is sometimes considered nonpathogenic. Lin et al. (2022) identified a high prevalence and pathogenic role of c.109G>A. There are few studies on the pathogenic characteristics of the c.109G>A mutation. The mutation spectrum of deafness in Western Guangdong has remained unclear to date.
In this study, hotspot mutations in the GJB2 gene of all subjects were sequenced and comprehensively analyzed to investigate the mutation spectrum of this region and the pathogenic characteristics of the c.109G>A mutation. It was speculated that the pathogenicity of the c.109G>A mutation was delayed, and some adolescents with c.109G>A mutation did not have true normal hearing. These findings provide a better understanding of the genetic basis of NSHL, facilitating earlier diagnosis and genetic counseling for the disease.
2. MATERIALS AND METHODS
2.1. Ethical compliance
The study protocol was approved by the Ethics Committee of the Zhaoqing No.2 People's Hospital. After obtaining written informed consent from all participants or their guardians, demographic and clinical data were collected.
2.2. Subject
A total of 97 patients with NSHL without other illnesses were recruited from Zhaoqing City and Yunfu City, located in the Western region of Guangdong Province. The severity of deafness was estimated by auditory brainstem response (ABR) or pure tone audiometry (PTA) as mild (<40 dB), moderate (41–70 dB), severe (71–95 dB), and profound (>95 dB). Peripheral venous blood samples (2 mL) were collected from each participant for genetic testing. In addition, 212 subjects with normal hearing were randomly selected, and their GJB2 sequences were analyzed simultaneously.
2.3. Genetic testing and analysis
Genomic DNA was extracted using a QIAamp DNA Micro Kit. The concentration and purity of DNA were tested using a SimpliNano™ spectrophotometer (GE Healthcare, USA). PCR amplification was conducted on exon 2 of GJB2 using the forward primer 5’‐GTGTAAGAGTTGGTGTTTGCT‐3′ and the reverse primer 5’‐GAGCAGTCCACAGTGTTGG‐3′. Amplified products were directly sequenced in both directions at the Beijing Genomics Institute (Guangzhou, China). The sequencing results were aligned with the reference sequence, NM_004004.6. Sequence analysis was performed using Chromas, DNASTAR 7.10, and DNAMAN 8.0. The allele frequencies and mutation rates of each locus or group were analyzed using the SPSS 26 software (IBM Corp., USA). Statistical significance was set at p < 0.05.
3. RESULTS
In this study, 97 NSHL patients were enrolled, including 62 male and 35 female participants. All participants were Han Chinese and came from different families. Their average age was 9.92 years, ranging from 2 to 20 years. Their sensorineural hearing loss was more than moderate (>40 dB) in most participants. While 212 subjects with normal hearing, including 109 male and 103 female participants, aged from 0 to 50 years, were recruited. There were 31, 22, 46, 67, and 46 persons in 0–10, 10–20, 20–30, 30–40, and 40–50 years old group, respectively. Han accounted for 99.53% (211 of 212), which was consistent with the proportion of that in this region (99.27%) (Table 1).
TABLE 1.
Demographics data of studied subjects.
| Groups | Number (cases) | Number of male | Number of female | Number of Han ethnic |
|---|---|---|---|---|
| NSHL | 97 | 62 (63.91%) | 35 (36.08%) | 97 (100.00%) |
| NC | 212 | 109 (51.42%) | 103 (48.58%) | 211 (99.53%) |
3.1. Mutation spectrum of GJB2 genes in NSHL patients
GJB2 mutations were found in 39 of 97 patients with NSHL, including three pathogenic mutations (c.109G>A, c.235delC, and c.299_300delAT) and two polymorphic mutations (c.79G>A and c.341A>G). There were 23 cases (23.71%) with pathogenic mutations. As shown in Table 2, the allele frequency of c.109G>A was 9.28% (18 of 194), and its mutation rate was as high as 39.13% (18 of 46). The second most common mutation was c.235delC, with an allele frequency of 4.12% (eight of 194). Only three cases of c.299_300delAT mutations were detected, with an allele frequency of 2.06% (four of 194). There were 16 cases of other GJB2 benign mutations, among which c.79G>A and c.341A>G were predominant.
TABLE 2.
Allele frequencies of GJB2 mutations in patients with NSHL.
| Nucleotide change | Amino acid change | Mutation type | Number of alleles | Allele frequency a (%) | ACMG/AMP classification | Pathogenicity score of CADD | Damage prediction by MSC 99% confidence interval | ||
|---|---|---|---|---|---|---|---|---|---|
| CADD score | MSC‐CADD score | MSC‐CADD‐ impact‐Pred | |||||||
| NM_004004.6: c.235del | NP_003995.2: p.(Leu79Cysfs*3) | Frame‐shift | 8 | 4.12 | Pathogenic | – | 32.000 | 0.001 | High |
| NM_004004.6: c.299_300del | NP_003995.2: p.(His100Argfs*14) | Frame‐shift | 4 | 2.06 | Pathogenic | – | 32.000 | 0.001 | High |
| NM_004004.6: c.109G>A | NP_003995.2: p.(Val37Ile) | Missense | 18 | 9.28 | Pathogenic | 22.5 | 10.550 | 0.001 | High |
| NM_004004.6: c.79G>A | NP_003995.2: p.(Val27Ile) | Missense | 21 | 10.82 | Benign | 22.9 | 16.990 | 0.001 | High |
| NM_004004.6: c.341A>G | NP_003995.2: p.(Glu114Gly) | Missense | 13 | 6.70 | Benign | 14.72 | 2.639 | 0.001 | High |
Note: CADD pathogenicity score and MSC‐CADD for the GJB2.
Abbreviations: ACMG/AMP, American College of Medical Genetics and Genomics/Association for Molecular Pathology; CADD, combined annotation‐dependent depletion (https://cadd.gs.washington.edu/). MSC, mutation significance cutoff (http://pec630.rockefeller.edu:8080/MSC/).
Percentage of alleles (97 × 2 = 194). GJB2(NM_004004.6).
Four of the 23 patients with pathogenic mutations were compound heterozygous for c.109G>A mutation and six carried homozygous mutations. Of the six homozygotes, c.109G>A, c.235delC, and c.299_300delAT (Figure 1a) were found in three, two, and one patient with NSHL, respectively (Figure 1d). The remaining 13 patients had a single heterozygous mutation. Eight patients had c.109G>A (Figure 1b), three patients had c.235delC (Figure 1c), and two patients had c.299_300delAT mutation. The c.109G>A mutation exhibited the highest detection rate (p < 0.05), indicating that it might be a hotspot mutation in this region.
FIGURE 1.

Sequencing results and the frequencies of pathogenic mutations in GJB2. (a) The location of c.299_300delAT homozygous mutation. (b) The location of c.109G>A heterozygous mutation. (d) The location of c.235delC heterozygous mutation. The black arrows indicate single nucleotide mutations. (d) Frequencies of GJB2 mutations in 23 cases with hearing loss. GJB2 (NM_004004.6).
Moreover, patients with c.109G>A mutation showed some clinical phenotypic heterogeneities. Three patients with the c.109G>A homozygous mutation exhibited profound (n = 1, Figure 2b), severe (n = 1), and moderate (n = 1), respectively (Table 3). Four patients with c.109G>A and compound other GJB2 pathogenic mutations also showed moderate deafness (n = 2), or more than severe deafness (n = 2). Furthermore, the clinical phenotypes of eight patients with c.109G>A heterozygous mutations varied from mild to profound hearing loss. Of these patients, three children had delayed hearing loss (Table 3).
FIGURE 2.
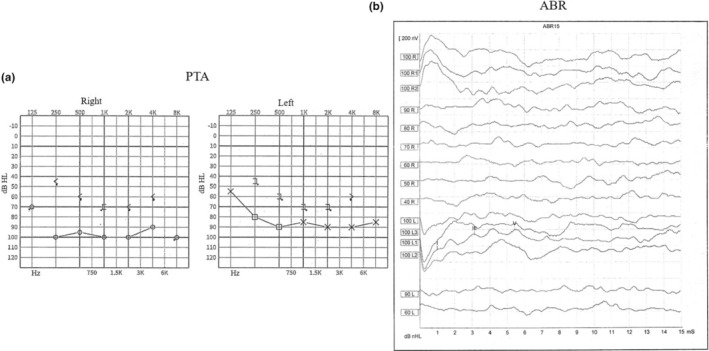
Representative audiograms results. (a) Pure tone audiometry (PTA) result of a severe subject. (b) Auditory brainstem response (ABR) result of a profound hearing loss patient.
TABLE 3.
Correlation between GJB2 c.109G>A mutation spectrum and clinical phenotypes of hearing loss.
| Genotype | Allele 1 | Allele 2 | Hearing level | Case | Morbidity age |
|---|---|---|---|---|---|
| Homozygous | c.109G>A | c.109G>A | Profound | 1 | 0 |
| Homozygous | c.109G>A | c.109G>A | Severe | 1 | 0 |
| Homozygous | c.109G>A | c.109G>A | Moderate | 1 | 0 |
| Compound heterozygous | c.109G>A | c.235delC | Moderate | 1 | 0 |
| Compound heterozygous | c.109G>A | c.79G>A | Profound | 1 | 2 |
| Compound heterozygous | c.109G>A | c.79G>A | Severe | 1 | 2 |
| Compound heterozygous | c.109G>A | c.79G>A/c.341A>G | Moderate | 1 | 0 |
| Single heterozygous | c.109G>A | Nv | Profound | 2 a | 0 |
| Single heterozygous | c.109G>A | Nv | Severe | 2 | 0 |
| Single heterozygous | c.109G>A | Nv | Moderate | 3 | 0 |
| Single heterozygous | c.109G>A | Nv | Mild | 1 | 2 |
Note: The severity of hearing loss was described as: mild, 21–40 dB; moderate, 41–70 dB; severe, 71–95 dB; profound, >95 dB.
Abbreviation: Nv, no variant.
Besides single heterozygous of c.109G>A in GJB2, one patient also carried compound heterozygous of c.919‐2A>G and c.1229C>T in SLC26A4. No other mutations were found in the four common genes (GJB2, SLC26A4, GJB3, and 12S rRNA) among other patients in Table 3. GJB2 (NM_004004.6), SLC26A4 (NM_000441.1), GJB3 (NM_001005752.1), 12S rRNA (NC_012920.1).
3.2. Characteristics of c.109G>A mutation in subjects with normal hearing
Except for one case of heterozygous c.299_300delAT and one case of compound heterozygous c.109G>A and c.235delC, only c.109G>A mutation was detected in the normal hearing group. c.109G>A was the most common mutation, with an allelic frequency of 8.02% (34 of 424), which was not significantly different from that in the deafness group (9.28%, 18 of 194) (p > 0.05). In the normal‐hearing population, the allele frequency of c.109G>A in all subjects with less than 30 years of age was approximately 10%, with an average of 11.11% (22 of 198), whereas the allele frequency of c.109G>A was 5.31% (12 of 226) in subjects aged 30 to 50. There was a significant difference in the allele frequency of c.109G>A between 0 and 30 years old and between 30 and 50 years old subjects (p < 0.05). The mutation rates of c.109G>A homozygous or compound heterozygous also showed a similar trend in different age groups: 6.25% (two of 32), 4.55% (one of 22), 2.17% (one of 46), 1.49% (one of 67), and 0% (zero of 46) in 0–10, 10–20, 20–30, 30–40 and 40–50 years old subjects, respectively. A high proportion of c.109G>A homozygous or compound heterozygous mutations was found at earlier ages, and its mutation rate declined approximately 30% every 10 years of age (Table 4).
TABLE 4.
GJB2 c.109G>A mutation spectrum in the general hearing group.
| Mutations | Age groups | ||||
|---|---|---|---|---|---|
| 0–10 years (n = 31) | 10–20 years (n = 22) | 20–30 years (n = 46) | 30–40 years (n = 67) | 40–50 years (n = 46) | |
| Homozygous c.109G>A | 2 | 1 | 0 | 1 | 0 |
| Compound heterozygous c.109G>A and c.235delC | 0 | 0 | 1 | 0 | 0 |
| Heterozygous c.109G>A | 4 | 3 | 8 | 5 | 5 |
| Other mutations | 0 | 0 | 0 | 0 | 1 a |
| Carrier rate of homozygous or compound heterozygous c.109G>A (%) | 6.45 | 4.5 | 2.17 | 1.49 | 0 |
| Allele frequency of c.109G>A (%) | 12.90 | 11.4 | 9.78 | 5.22 | 5.43 |
Heterozygous c.299_300delAT mutation. GJB2 (NM_004004.6).
4. DISCUSSION
The mutation spectrum of hearing loss in Zhaoqing City and Yufu City locating in the Western of Guangdong has remained unclear. In our study, we found that c.109G>A, c.235delC, and c.299_300delAT were the most common pathogenic mutations in GJB2, with allele frequencies of 9.28% (18 of 194), 4.12% (eight of 194), and 2.06% (four of 194), respectively in 97 NSHL patients. The c.109G>A mutation accounted for 39.13% (18 of 46) of all the pathogenic GJB2 alleles, indicating that it is the most frequently detected hotspot mutation in this region. A previous study reported that c.235delC is the most common mutation in GJB2, with the highest carrier frequency. The study revealed that c.176_191del16 and c.35delG are also common mutations, with allele frequencies of 0.75% (31 of 4126) and 0.29% (12 of 4126) among Chinese NSHL patients, respectively (Dai et al., 2009). In contrast, the last two mutations were not detected in our study. These results confirmed racial and regional differences in the GJB2 mutation spectrum.
Additionally, the overall GJB2 mutation rate was 23.71% (23 of 97) in this study, which was higher than that of patients with sensorineural hearing loss in Southwestern China (17.27%) (Qing et al., 2015). This was because the genetic tests performed in Southwestern China were based on the Deafness Gene Variant Detection Array Kit (CapitalBio) without the involvement of c.109G>A mutation (Qing et al., 2015). However, our study revealed that the allele frequency of c.109G>A in the GJB2 gene was 9.28% (18 of 194), which was not only higher than that of c.235delC (4.12%) in our study, but also higher than that reported by Dai et al. (6.7%, 185 of 2744) (Dai et al., 2009), but lower than that (15.05%, 90 of 598) reported by Huang et al. (15.05%, 90 of 598) (Huang et al., 2018). This may be due to racial and regional differences in the mutation rates of the GJB2 gene. Additionally, the c.109G>A mutation has been reported to have high carrier rates in patients with hearing loss in Taiwan (11.6%) and Korea (8.87%) (Huang, Yang et al., 2015; Kim et al., 2013), whereas the allele frequency of this mutation was 16.5% in Japan (Tsukada et al., 2010). Taken together, the c.109G>A mutation has a high frequency among patients with deafness in many regions of East Asia. Therefore, more attention should be paid to c.109G>A mutation.
Up to now, the pathogenicity of c.109G>A remains controversial (Dai et al., 2009). It may be related to the initial identification history of this mutation. It was first detected in subjects with normal hearing, and thus classified as a polymorphic site (Kelley et al., 1998). However, it was considered to be a pathogenic mutation soon (Abe et al., 2000). With further in‐depth studies, the pathogenicity of this mutation has been recognized by many otologists worldwide (Huang, Huang et al., 2015; Tsukada et al., 2010; Yu et al., 2020). Huang, Huang et al. (2015) studied 3864 patients with deafness and demonstrated that the frequency of this mutation was much higher in patients than in normal controls. Another study reported that no homozygous or compound heterozygous c.109G>A mutations were detected in 484 infants with normal hearing (Huang, Yang et al., 2015). Several in vitro and in vivo experiments have suggested that the c.109G>A homozygous mutation can affect the adhesion between connexins and leads to deafness due to the inability to form gap junction channels (Chai et al., 2015; Lin et al., 2019). Recently, the ClinGen Hearing Loss Expert Panel confirmed the pathogenic role of c.109G>A in hearing loss based on allelic, case–control, and segregation evidences (Shen et al., 2019). In our study, we performed PCR and sequencing for GJB2, SLC26A4, 12S rRNA, and GJB3 in these participants. And only one patient with heterozygous c.109G>A carried compound heterozygous c.919‐2A>G and c.1229C>T of SLC26A4 (Table 3). Otherwise, no other variations of the three common genes were found among other patients in Table 3. It can be speculated that these symptoms of deafness may be caused by c.109G>A. Meanwhile, the possible effects of the mutations on GJB2 gene were analyzed with online bioinformatics software Combined Annotation Dependent Depletion (CADD) and the Mutation Significance Cutoff (MSC). The c.109G>A mutation was predicted to be pathogenicity by CADD with a score of 22.5 and by MSC with Impact‐Pred “high” (Table 2). Moreover, in this study, three cases with the c.109G>A homozygous mutation and four cases with the c.109G>A compound heterozygous mutation displayed moderate‐to‐profound hearing loss, which indicated the pathogenicity of c.109G>A.
In addition, we found that the pathogenicity of c.109G>A has some special characteristics, such as clinical heterogeneity and late‐onset characteristics, which may also be the reasons why its pathogenicity is controversial. In our study, patients with different genotype of c.109G>A mutations (homozygous, compound heterozygous, or single heterozygous) displayed moderate‐to‐profound deafness (Table 3), which was consistent with the results of Huang, Yang et al. (2015). Moreover, Chai et al. (2015) reported that the rates of homozygous c.109G>A in severe and mild‐to‐moderate deafness cases were 1.63% (14/857) and 12.5% (11/88), respectively, supporting the clinical heterogeneity of c.109G>A. We speculated that the larger Isoleucine side chain produced by c.109G>A mutation may affect the formation of hydrogen bonds of peptide chains, resulting in unstable gap connection channels or even inability to form gap connection channels, thus leading to mild‐to‐profound deafness. But the further research is needed.
In this cohort, three children in the NSHL group with normal hearing screening developed hearing loss after 2 years of age (Table 3), which suggested the pathogenicity of the c.109G>A mutation may be attributed to delayed hearing loss. On the contrary, we found some interesting phenomenon that in our NC group, the allele frequency of c.109G>A in 30–50 years old subjects (5.31%, 12 of 226) decreased by half compared with its frequency in 0–30 years old participants (11.11%, 22/198) (p < 0.05; Table 4). It was speculated that the pathogenicity of the c.109G>A mutation may be delayed, and perhaps some adolescents did not have true normal hearing. These temporary hearing loss‐negative individuals might develop hearing abnormalities with increasing age and were removed from the NC group. Therefore, the mutation rate of the c.109G>A homozygous or compound heterozygous mutation was gradually diluted by true hearing loss‐negative individuals. This hypothesis can be further verified by regular changes in the mutation rate of c.109G>A. A high proportion of c.109G>A homozygous or compound heterozygous mutations was in the lower age groups of the general hearing population. c.109G>A mutation rate declined slowly over time at a rate of approximately 30% every 10 years. Therefore, hearing impairment may not develop in some children carrying the c.109G>A mutation, but they likely have a high risk of delayed hearing impairment, which can be masked by their temporary normal hearing. Perhaps the allele frequency and regular changes in the c.109G>A mutation in NC group also further explain the clinical heterogeneity and late onset of deafness in c.109G>A. Similarly, Wu et al. (2017) found that children with GJB2 c.109G>A/c.109G>A or c.109G>A/c.235delC mutations may develop hearing impairment at an annual rate of 1 dB during long‐term follow‐up. Chai et al. (2015) revealed that 65% (11 of 17) and 35% (six of 17) of patients with homozygous c.109G>A presented with congenital and delayed deafness, respectively. These findings indicated that this mutation can cause delayed deafness. Furthermore, recently, it was (Shen et al., 2017) reported that the c.109G>A mutation was associated with sudden deafness. Therefore, the c.109G>A mutation in GJB2 is related to autosomal recessive nonsyndromic hearing loss and is correlated with age, which might provide a reasonable explanation for the detection of the c.109G>A homozygous mutation in subjects with normal hearing. However, most studies have neglected the long‐term follow‐up of populations carrying the c.109G>A homozygous mutation and/or have unintentionally included patients with mild hearing loss as healthy controls, which leads to a high frequency of this mutation in the normal population.
5. CONCLUSIONS
In summary, c.109G>A, c.235delC, and c.299_300delAT were the main pathogenic mutations in the GJB2 gene in the Western of Guangdong Province. c.109G>A was the most common GJB2 mutation in patients with NSHL. Our results suggest that c.109G>A is a pathogenic variant associated with delayed onset hearing impairment. Therefore, more attention should be paid to the genetic testing of the c.109G>A mutation in patients with NSHL.
AUTHOR CONTRIBUTIONS
Shaoming Liang, Zhao Wang, Weihong Li, and Zhichao Chen designed the study and performed the experiments. Zhao Wang and Zhichao Chen analyzed the data. Shaoming Liang and Shimin Yuan drafted and edited the manuscript.
FUNDING INFORMATION
This study was supported by the Natural Science Foundation Project of Guangdong Province (2018A030313960) and the Medical Research Fund Project of Guangdong (A2018561).
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests at the time of publication.
ETHICS STATEMENT
The study protocol was approved by the Ethics Committee of the Zhaoqing No.2 People's Hospital.
ACKNOWLEDGMENTS
The authors thank all participants and their parents, as well as the faculty staff of the Zhaoqing Rehabilitation Center and Yunfu Special Education School, for their support and assistance in this study. The authors also would like to express their gratitude to Professor Yueqiu Tan for providing professional genetic analysis proposals.
Liang, S. , Li, W. , Chen, Z. , Yuan, S. , & Wang, Z. (2023). Analysis of GJB2 gene mutations spectrum and the characteristics of individuals with c.109G>A in Western Guangdong. Molecular Genetics & Genomic Medicine, 11, e2185. 10.1002/mgg3.2185
Contributor Information
Shaoming Liang, Email: lshaoming3@163.com.
Zhao Wang, Email: luckbird100@163.com.
DATA AVAILABILITY STATEMENT
The datasets used and/or analyzed during the present study are available from the corresponding author upon a reasonable request.
REFERENCES
- Abe, S. , Usami, S. , Shinkawa, H. , Kelley, P. M. , & Kimberling, W. J. (2000). Prevalent connexin 26 gene (GJB2) mutations in Japanese. Journal of Medical Genetics, 37(1), 41–43. 10.1136/jmg.37.1.41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai, Y. , Chen, D. , Sun, L. , Li, L. , Chen, Y. , Pang, X. , Zhang, L. , Wu, H. , & Yang, T. (2015). The homozygous p.V37I variant of GJB2 is associated with diverse hearing phenotypes. Clinical Genetics, 87(4), 350–355. 10.1111/cge.12387 [DOI] [PubMed] [Google Scholar]
- Dai, P. , Huang, L. H. , Wang, G. J. , Gao, X. , Qu, C. Y. , Chen, X. W. , Ma, F. R. , Zhang, J. , Xing, W. L. , Xi, S. Y. , Ma, B. R. , Pan, Y. , Cheng, X. H. , Duan, H. , Yuan, Y. Y. , Zhao, L. P. , Chang, L. , Gao, R. Z. , Liu, H. H. , … Han, D. M. (2019). Concurrent hearing and genetic screening of 180,469 neonates with follow‐up in Beijing, China. American Journal of Human Genetics, 105(4), 803–812. 10.1016/j.ajhg.2019.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, P. , Yu, F. , Han, B. , Liu, X. Z. , Wang, G. J. , Li, Q. , Yuan, Y. Y. , Liu, X. , Huang, D. L. , Kang, D. Y. , Zhang, X. , Yuan, H. J. , Yao, K. , Hao, J. S. , He, J. , He, Y. , Wang, Y. Q. , Ye, Q. , Yu, Y. J. , … Wong, L. J. (2009). GJB2 mutation spectrum in 2,063 Chinese patients with nonsyndromic hearing impairment. Journal of Translational Medicine, 7, 26. 10.1186/1479-5876-7-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, P. , Yu, F. , Han, B. , Yuan, Y. Y. , Li, Q. , Wang, G. J. , Liu, X. , He, J. , Huang, D. L. , Kang, D. Y. , Zhang, X. , Yuan, H. J. , Schmitt, E. , Han, D. Y. , & Wong, L. J. (2007). The prevalence of the 235delC GJB2 mutation in a Chinese deaf population. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 9(5), 283–289. 10.1097/gim.0b013e31804d2371 [DOI] [PubMed] [Google Scholar]
- Duan, S. H. , Guo, Y. F. , Chen, X. J. , & Li, Y. (2021). Genetic mutations in patients with nonsyndromic hearing impairment of minority and Han Chinese ethnicities in Qinghai, China. The Journal of International Medical Research, 49(4), 3000605211000892. 10.1177/03000605211000892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, B. , Han, M. , Wang, G. , Huang, S. , Zeng, J. , Yuan, Y. , & Dai, P. (2018). Genetic mutations in non‐syndromic deafness patients in Hainan Province have a different mutational spectrum compared to patients from mainland China. International Journal of Pediatric Otorhinolaryngology, 108, 49–54. 10.1016/j.ijporl.2018.02.015 [DOI] [PubMed] [Google Scholar]
- Huang, S. S. , Huang, B. Q. , Wang, G. J. , Yuan, Y. Y. , & Dai, P. (2015). The relationship between the p.V37I mutation in GJB2 and hearing phenotypes in Chinese individuals. PLoS ONE, 10(6), e0129662. 10.1371/journal.pone.0129662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. , Yang, X. L. , Chen, W. X. , Duan, B. , Lu, P. , Wang, Y. , & Xu, Z. M. (2015). Prevalence of p.V37I variant of GJB2 among Chinese infants with mild or moderate hearing loss. International Journal of Clinical and Experimental Medicine, 8(11), 21674–21678. [PMC free article] [PubMed] [Google Scholar]
- Kelley, P. M. , Harris, D. J. , Comer, B. C. , Askew, J. W. , Fowler, T. , Smith, S. D. , & Kimberling, W. J. (1998). Novel mutations in the connexin 26 gene (GJB2) that cause autosomal recessive (DFNB1) hearing loss. American Journal of Human Genetics, 62(4), 792–799. 10.1086/301807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenneson, A. , Van Naarden Braun, K. , & Boyle, C. (2002). GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 4(4), 258–274. 10.1097/00125817-200207000-00004 [DOI] [PubMed] [Google Scholar]
- Kim, S. Y. , Park, G. , Han, K. H. , Kim, A. , Koo, J. W. , Chang, S. O. , Oh, S. H. , Park, W. Y. , & Choi, B. Y. (2013). Prevalence of p.V37I variant of GJB2 in mild or moderate hearing loss in a pediatric population and the interpretation of its pathogenicity. PLoS One, 8(4), e61592. 10.1371/journal.pone.0061592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, X. , Li, G. , Zhang, Y. , Zhao, J. J. , Lu, J. W. , Gao, Y. G. , Liu, H. H. , Li, G. L. , Yang, T. , Song, L. , & Wu, H. (2019). Hearing consequences in Gjb2 knock‐in mice: Implications for human p.V37I mutation. Aging, 11(18), 7416–7441. 10.18632/aging.102246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y. F. , Lin, H. C. , Tsai, C. L. , & Hsu, Y. C. (2022). GJB2 mutation spectrum in the Taiwanese population and genotype‐phenotype comparisons in patients with hearing loss carrying GJB2 c.109G>a and c.235delC mutations. Hearing Research, 413, 108135. 10.1016/j.heares.2020.108135 [DOI] [PubMed] [Google Scholar]
- Liu, X. W. , Wang, J. C. , Wang, S. Y. , Li, S. J. , Zhu, Y. M. , Ding, W. J. , Xu, C. Y. , Duan, L. , Xu, B. C. , & Guo, Y. F. (2020). The mutation frequencies of GJB2, GJB3, SLC26A4 and MT‐RNR1 of patients with severe to profound sensorineural hearing loss in Northwest China. International Journal of Pediatric Otorhinolaryngology, 136, 110143. 10.1016/j.ijporl.2020.110143 [DOI] [PubMed] [Google Scholar]
- Liu, X. Z. , Pandya, A. , Angeli, S. , Telischi, F. F. , Arnos, K. S. , Nance, W. E. , & Balkany, T. (2005). Audiological features of GJB2 (connexin 26) deafness. Ear and Hearing, 26(3), 361–369. 10.1097/00003446-200506000-00011 [DOI] [PubMed] [Google Scholar]
- Qing, J. , Zhou, Y. , Lai, R. S. , Hu, P. , Ding, Y. , Wu, W. J. , Xiao, Z. , Ho, P. T. , Liu, Y. Y. , Liu, J. , Du, L. L. , Yan, D. , Goldstein, B. J. , Liu, X. Z. , & Xie, D. H. (2015). Prevalence of mutations in GJB2, SLC26A4, and mtDNA in children with severe or profound sensorineural hearing loss in southwestern China. Genetic Testing and Molecular Biomarkers, 19(1), 52–58. 10.1089/gtmb.2014.0241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, J. , Oza, A. M. , Del Castillo, I. , Duzkale, H. , Matsunaga, T. , Pandya, A. , Kang, H. P. , Mar‐Heyming, R. , Guha, S. , Moyer, K. , Lo, C. , Kenna, M. , Alexander, J. J. , Zhang, Y. , Hirsch, Y. , Luo, M. , Cao, Y. , Wai Choy, K. , Cheng, Y. F. , … ClinGen Hearing Loss Working Group . (2019). Consensus interpretation of the p.Met34Thr and p.Val37Ile variants in GJB2 by the ClinGen Hearing Loss Expert Panel. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 21(11), 2442–2452. 10.1038/s41436-019-0535-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, N. , Peng, J. , Wang, X. , Zhu, Y. , Liu, W. , Liu, A. , & Lu, Y. (2017). Association between the p.V37I variant of GJB2 and hearing loss: A pedigree and meta‐analysis. Oncotarget, 8(28), 46681–46690. 10.18632/oncotarget.17325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada, K. , Nishio, S. , Usami, S. , & Deafness Gene Study Consortium . (2010). A large cohort study of GJB2 mutations in Japanese hearing loss patients. Clinical Genetics, 78(5), 464–470. 10.1111/j.1399-0004.2010.01407.x [DOI] [PubMed] [Google Scholar]
- World Health Organization . (2021). Deafness and hearing loss. WHO. https://www.who.int/health‐topics/hearing‐loss [Google Scholar]
- Wu, C. C. , Tsai, C. H. , Hung, C. C. , Lin, Y. H. , Lin, Y. H. , Huang, F. L. , Tsao, P. N. , Su, Y. N. , Lee, Y. L. , Hsieh, W. S. , & Hsu, C. J. (2017). Newborn genetic screening for hearing impairment: A population‐based longitudinal study. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 19(1), 6–12. 10.1038/gim.2016.66 [DOI] [PubMed] [Google Scholar]
- Yu, X. Y. , Lin, Y. , Xu, J. , Che, T. J. , Li, L. , Yang, T. , & Wu, H. (2020). Molecular epidemiology of Chinese Han deaf patients with bi‐allelic and mono‐allelic GJB2 mutations. Orphanet Journal of Rare Diseases, 15(1), 29. 10.1186/s13023-020-1311-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author upon a reasonable request.