

Abstract
The gut microbiota produce hundreds of small molecules, many of which modulate host physiology. Although efforts have been made to identify biosynthetic genes for secondary metabolites, the chemical output of the gut microbiome consists predominantly of primary metabolites. Here, we introduce the gutSMASH algorithm for identification of primary metabolic gene clusters and use it to systematically profile gut microbiome metabolism, identifying 19,890 gene clusters in 4,240 high-quality microbial genomes. We find marked differences in pathway distribution among phyla, reflecting distinct strategies for energy capture. These data explain taxonomic differences in short-chain fatty acid production and suggest a characteristic metabolic niche for each taxon. Analysis of 1,135 subjects from a Dutch population-based cohort shows that the level of microbiome-derived metabolites in plasma and faeces is almost completely uncorrelated with the metagenomic abundance of corresponding metabolic genes, indicating a crucial role for pathway-specific gene regulation and metabolite flux. This work is a starting point for understanding differences in how bacterial taxa contribute to the chemistry of the microbiome.
Keywords: specialized primary metabolism, metabolic gene clusters, gut microbiome, gutSMASH
Introduction
The pathways encoding the production of microbial metabolites are often physically clustered in the genome, in regions known as metabolic gene clusters (MGCs). Current tools for computational prediction of metabolic pathways focus on gene clusters for natural product biosynthesis 1 or generic primary metabolism2,3. Here, we introduce an algorithm, gutSMASH, to profile known and predicted novel specialized primary metabolic gene clusters from the gut microbiome, which we define as gene clusters encoding primary metabolic pathways that are taxon-specific, niche-defining and important for (host-)microbiome interactions. We use this tool to perform a systematic analysis of primary metabolic gene clusters in bacterial strains from the gut microbiome, and identify the prevalence and abundance of each of these pathways across a large population-based cohort as well as a clinical cohort. Although gutSMASH has been built to specifically predict MGCs from anaerobic human gut bacteria, this tool can also be applied to microbial communities that inhabit other (animal) body sites.
Algorithms that identify physically clustered genes have become a mainstay of bacterial pathway identification4-6; taking into account the conserved physical clustering of genes prevents false positive hits based on sequence similarity alone. This principle has been widely applied in the field of natural product biosynthesis, e.g. in antiSMASH1 which predicts biosynthetic gene clusters (BGCs) by detecting physically clustered protein domains using profile hidden Markov Models (pHMMs). Here, we tailored this gene cluster detection framework to detect MGCs involved in primary metabolism and bioenergetics.
Results
As a starting point, we constructed a dataset of 51 primary metabolic pathways from the gut microbiome with biochemical or genetic literature support (including MGCs as well as pathways encoded by a single genes) and identified core enzymes (i.e., required for pathway function) to serve as a signature for the detection rules (Figure 1, Table S1; see Methods for details). To more accurately predict MGCs of interest, we performed three computational procedures. First, for core enzymes belonging to 12 of the protein superfamilies that are known to catalyze diverse types of reactions and were most commonly found across a wide range of pathways, we constructed phylogenies and used them to create clade-specific pHMMs to detect specific subfamilies (see SI results Phylogenetic analysis of protein superfamilies to identify pathway-specific clades). Second, we designed pathway-specific rules for each MGC type in our dataset (see Methods). These rules were validated and optimized by detailed manual visual inspection and analysis of MGC sequence similarity networks made using BiG-SCAPE7, generated from gutSMASH results on a set of 1,621 microbial genomes (Online Data: https://gutsmash.bioinformatics.nl/help.html#Validation); see SI results Validation of gutSMASH detection rules by evaluating their predictive performance) (Table S2 & S3). Third, despite the fact that most specialized primary metabolic pathways are encoded in MGCs, there are also single-protein pathways that are in charge of the secretion of key specialized primary metabolites in the gut microbial ecosystem, such as serine dehydratase, which produces ammonia and pyruvate from serine8. For this reason, we also built 10 clade-specific pHMMs to detect these (see Methods section Assessing single-protein pathway abundance within representative human gut bacteria). The above procedures led to the design of a set of detection rules included in the gutSMASH framework to identify both known and putative MGCs that are potentially relevant for metabolite-mediated microbiome-associated phenotypes and also assess the presence/absence patterns of single-protein pathways across microbial genera by using custom pHMMs (not included in gutSMASH detection rule set). While obtaining a precise estimate of precision and recall of the gutSMASH algorithm is infeasible due to the absence of large-scale experimentally verified MGCs from diverse taxa, additional manual validation on a dataset of 18 experimentally verified homologues of gutSMASH-detected MGCs, as well as on a dataset of 42 MGCs from five model organisms from different phyla, showed no false negatives or false positives (see SI Results Validation of gutSMASH detection rules by evaluating their predictive performance for details).
Figure 1: Development and design of detection rules for gutSMASH.
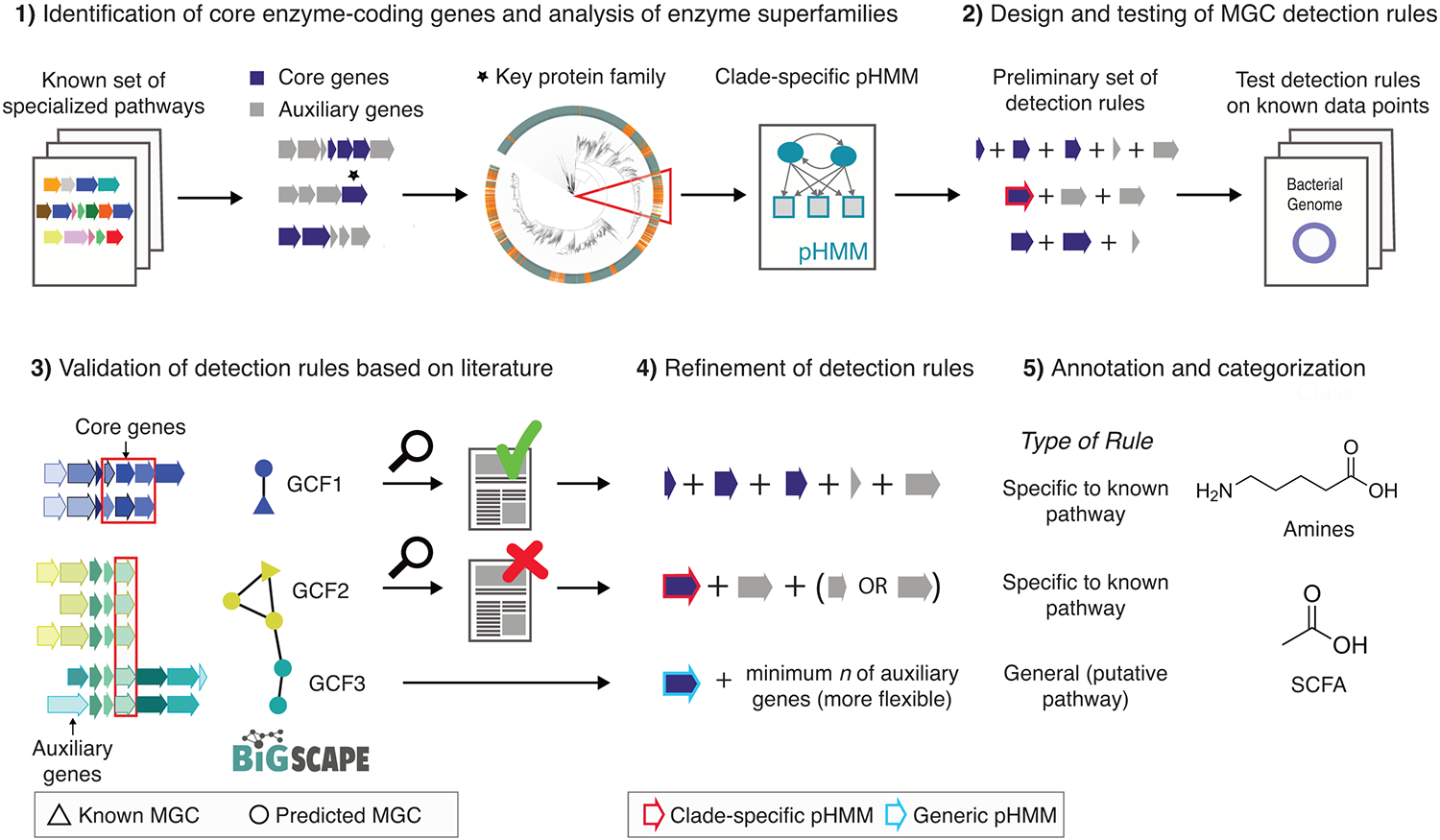
(1) A set of known and characterized MGC-encoded pathways were curated from the literature. Protein domains were identified across all MGCs and core enzymatic domains were manually identified. For enzymatic domains belonging to broad multifunctional enzyme families, protein superfamily phylogenies were built to create clade-specific pHMMs. (2) These domains were incorporated in the initial detection rules. The detection rules were run on a test set, and all the MGC predicted by the same rule were grouped together and (3) run through BiG-SCAPE, which grouped the MGCs into gene cluster families (GCFs). (4) Based on literature analysis of GCF members, detection rules were manually fine-tuned to either include or exclude MGC architectures that were either related to specialized primary metabolism or not. (5) Finally, fine-tuned detection rules were annotated and categorized into different MGC classes based on their metabolic end products.
To profile the metabolic capacity of strains from the human gut microbiome, we selected a set of 4,240 unique high-quality reference genomes consisting of 1,520 genomes from the Culturable Genome Reference (CGR) collection9, 2,308 genomes from the Microbial Reference Genomes collection of the Human Microbiome Project (HMP) consortium10 and 414 additional genomes from the class Clostridia to account for their metabolic versatility11 (Table S4). We refrained from including metagenome-assembled genomes in this analysis, as they often lack the taxon-specific genomic islands12 on which many specialistic metabolic functions are encoded. In total, gutSMASH predicted 19,890 MGCs across these genomes that are clear homologues of MGCs for our set of known pathway types (See Methods: Evaluating the functional potential of the human microbiome using gutSMASH).
The combined results of the gutSMASH MGC scanning and the single-protein pHMM detection across the three reference collections provide unique insights into the metabolic traits encoded by the genomes of human gut bacteria. While some genera harbor a small set of highly conserved pathways, (e.g., Akkermansia, Faecalibacterium), other genera contain much larger interspecies differences (Figure 2A). The genus Clostridium displays remarkable metabolic versatility, with 43 distinct MGC-encoded metabolic pathways present across members of this genus (Figure 2A); this corroborates earlier results by Viera-Silva et al., who showed high dissimilarity of metabolic module repertoires in Clostridia13. Clostridial strains that are indistinguishable by 16S sequencing often harbor distinct gene cluster ensembles (Suppl. Figure 1), suggesting that specialization in primary metabolism leads to functional differentiation even among closely related strains. Clostridium is a clear outlier: by comparison, the next most numerous set of metabolic pathways are found within the Enterobacteriaceae (e.g., Salmonella, Escherichia, Enterobacter, and Klebsiella) with 22-25 metabolic pathways. Intriguingly, many of the metabolic pathways encoded by Clostridium and members of the Enterobacteriaceae are non-overlapping (with 23/43 Clostridium pathways not being identified among Enterobacteriaceae), highlighting the distinct metabolic strategies these microbes employ within the gut (Figure 2A). The Bacteroides, Actinobacteria (Eggerthella and Collinsella) and Verrucomicrobia (Akkermansia) harbor a more restricted set of primary metabolic pathways, likely reflecting versatility in upstream components of their metabolism (i.e., glycan foraging and other forms of substrate utilization).
Figure 2: Distribution of known pathways across most representative genera in the human gut.
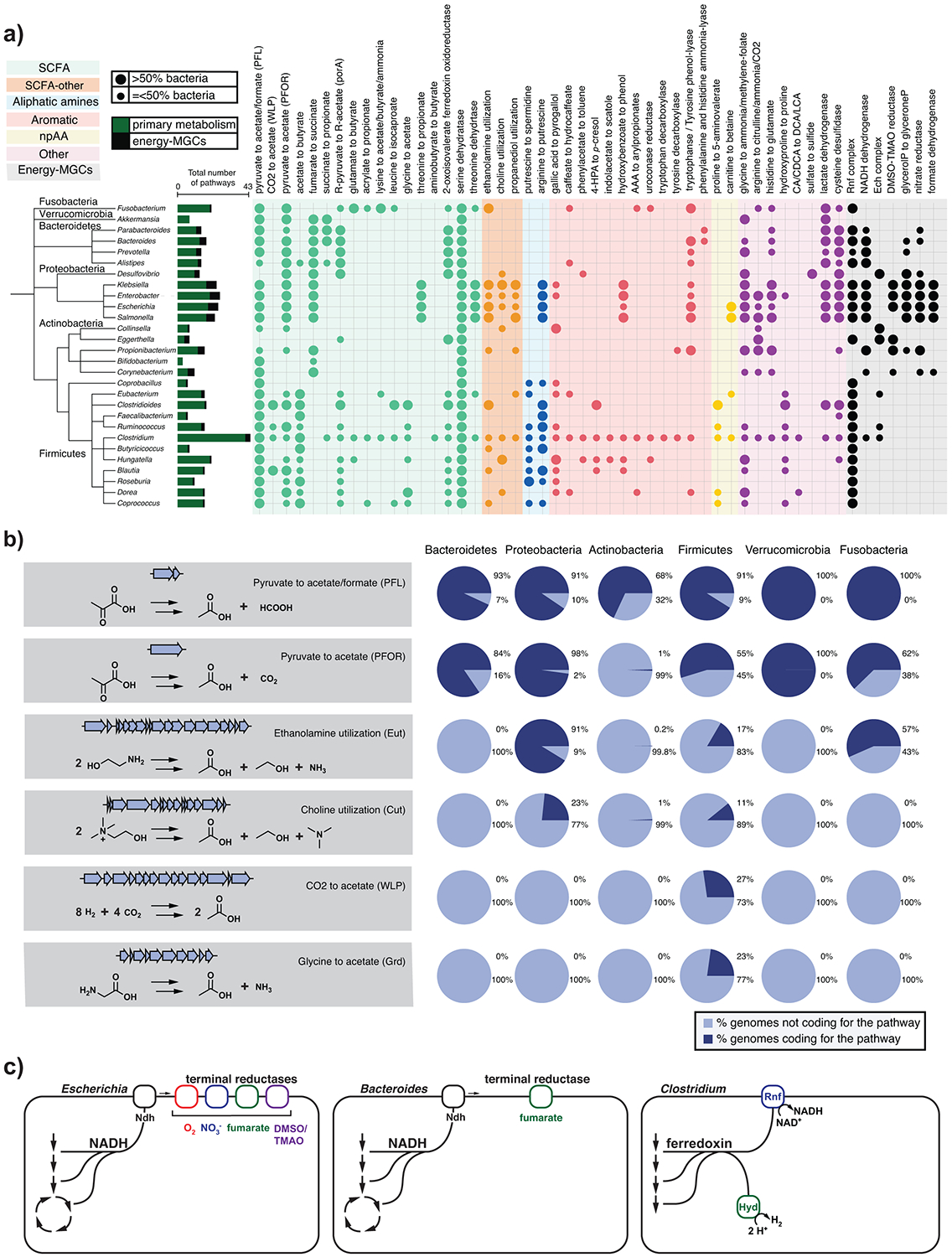
(a) Circles represent the absence/presence of known pathways in each genus. Larger circles indicate cases in which more than 50% of the genomes for a genus encode the pathway, while smaller circles indicate cases in which 50% or fewer of the genomes encode it. Colored ranges indicate a categorization of MGCs by chemical class of their product, in which npAA represents nonproteinogenic amino acids and SCFA represents short-chain fatty acids. Taxonomic assignments were applied using the Genome Taxonomy Database release 95 (GTDB)14. The tree was generated using phyloT (https://phylot.biobyte.de/) and visualized using iTOL15. Raw data are available in Table S5. (b) Distribution of the main acetate synthesis pathways at phylum level. Some of the pathways are ubiquitous across the five major phyla (e.g. pyruvate to acetate/formate [PFL]), while others are only found in Firmicutes (CO2 to acetate [WLP]). Raw data for the pie charts is available in Table S6. Genes and gene clusters depicted are representatives from Bacteroides thetaiotaomicron (PFL & PFOR), Salmonella enterica (Eut), Clostridium sporogenes (Cut), Clostridium difficile (WLP) and Clostridium sticklandii (Grd). (c) Bioenergetic strategies in Escherichia that has a variety of alternate electron acceptors to choose from compared to Bacteroides and Clostridium. Abbreviations: PFL, pyruvate formate-lyase; PFOR, pyruvate:ferredoxin oxidoreductase; Eut, ethanolamine utilization; Cut, choline utilization; WLP, Wood-Ljungdahl Pathway; Grd, glycine reductase; CA, cholic acid; CDCA, chenodeoxycholic acid; DCA, deoxycholic acid; LCA, lithocholic acid; TMAO, trimethylamine N-oxide; DMSO, dimethylsulfoxide; SCFA, short-chain fatty acid; Ndh, NADH dehydrogenase, Rnf, Rhodobacter nitrogen fixation like complex; Hyd, hydrogenase.
Our results provide insights into the metabolic strategies that microbes use to produce short chain fatty acids (SCFAs). As expected, butyrate production is found mainly in certain Firmicutes and Fusobacteria, however some Alistipes sp. within the Bacteroidetes phylum have genes for the acetate to butyrate pathway (Figure 2A). This is consistent with previous reports that Alistipes sp. produce small amounts of this compound16. On the other hand, propionate production is largely confined to (and conserved in) the Bacteroidetes. However, the phylogenetic distribution of pathways that generate acetate -- the most concentrated molecule produced in the gut17 -- has not yet been described. Two pathways for the conversion of pyruvate to acetate -- pyruvate formate-lyase (pyruvate to acetate/formate) and pyruvate:ferredoxin oxidoreductase (PFOR) -- are widely distributed across microbial strains from diverse phyla (Figure 2B). Two observations suggest that these two pathways are the most prolific source of acetate in the gut. First, some strains known to produce large quantities of acetate rely entirely on one or both of the pathways. Second, each one uses pyruvate as a substrate, consistent with a model in which these pathways are the primary conduit through which carbohydrate-derived carbon is converted to acetate. Additional taxon-specific pathways for acetate include the CO2 to acetate pathway and the glycine to acetate pathway (each specific to a subset of Firmicutes), as well as the choline and ethanolamine utilization pathways (widespread among Enterobacteriaceae and each found in different clades of Firmicutes) (Figure 2A).
Our results demonstrate a striking difference in mechanisms for energy capture by three of the major bacterial genera in the gut: Bacteroides, Escherichia, and Clostridium. When growing aerobically with glucose, E. coli generates most of its energy by channelling electrons through membrane bound cytochromes using oxygen as the terminal electron acceptor (Figure 2C). However, oxygen is limiting in the gut. Under anaerobic conditions, bacteria from the genus Escherichia employ alternate terminal electron acceptors such as nitrate, dimethyl sulfoxide (DMSO), trimethylamine N-oxide (TMAO), and fumarate by substituting alternate terminal reductases into their electron transport system (Figure 2C). However, in the healthy gut these alternate electron acceptors are either absent or available in limited amounts, likely explaining why these facultative anaerobes represent a small proportion of the healthy microbiome18. In contrast to the diversity of terminal reductases used by the Escherichia, Bacteroides genomes encode only fumarate reductase (Figure 2C). They use a unique pathway, carboxylating phosphoenolpyruvate (PEP) to form fumarate, which they use as a terminal electron acceptor to run an anaerobic electron transport chain involving NADH dehydrogenase and fumarate reductase, ultimately forming propionate. Thus, the metabolic strategy employed by Bacteroides ensures a steady stream of electron acceptor to fuel their metabolism. Clostridia do not utilize similar mechanisms for energy capture as members of the genera Escherichia and Bacteroides. Recent analyses suggest that they use the Rnf complex for generating a proton motive force19,20. Several pathways encoded by the genomes of Clostridium (e.g., acetate to butyrate, aromatic amino acids [AAA] to arylpropionates, leucine to isocaproate) (Figure 2A) consist of an electron bifurcating acyl-CoA dehydrogenase enzyme. This complex bifurcates electrons from NADH to the low potential electron carrier ferredoxin which can then donate electrons to the Rnf complex which functions as a proton or sodium pump, generating an ion motive force. Although much still is to be learned about Clostridial metabolism, our findings suggest that their metabolism operates at a different scale of the redox tower compared to Bacteroides and Enterobacteriaceae, using low potential electron carriers to fuel their metabolism.
Next, we set out to determine the prevalence and abundance of each pathway in a cohort of human samples. We used BiG-MAP21 to profile the relative abundance of each MGC class across 1,135 metagenomes from the population-based LifeLines DEEP cohort22, by mapping metagenomic reads against a collection of 5,655 non-redundant MGCs detected in our set of reference genomes (Figure 3A,B; Extended Data Figure 1; Suppl. Figure 2). Some pathways, such as CO2 to acetate (acetogenesis) and butyrate production from acetate or glutamate, as well as polyamine-forming pathways, were found in >99% of microbiomes. Others, such as 1,2-propanediol utilization and p-cresol production, both associated with negative effects on gut health23,24, were observed at detectable levels in only 75% and 53% of the samples, respectively. In terms of abundance, it is striking that for example the bile acid-induced (bai) operon for the formation of the secondary bile acids deoxycholic acid and lithocholic acid, which has been characterized from very low-abundance Clostridium scindens strains25, was still shown to be present in relatively high abundance across a subset of subjects. Analysis of the mapped reads showed that the vast majority of these mapped to a homologous MGC from the genus Dorea instead (Suppl. Figure 2), for which the physiological relevance remains to be established. While two of the three acetate-forming pathways (PFL and PFOR) were consistently found at high abundance levels, the abundance of all butyrate-forming pathways is highly variable across subjects, with a ~13-fold difference between lower and upper quartiles in the abundance distribution of the glutamate-to-butyrate pathway, and a >130-fold difference between the 10th percentile and the 90th percentile.
Figure 3. Prevalence and abundance of specialized primary metabolic pathways across 1,135 human microbiome samples.
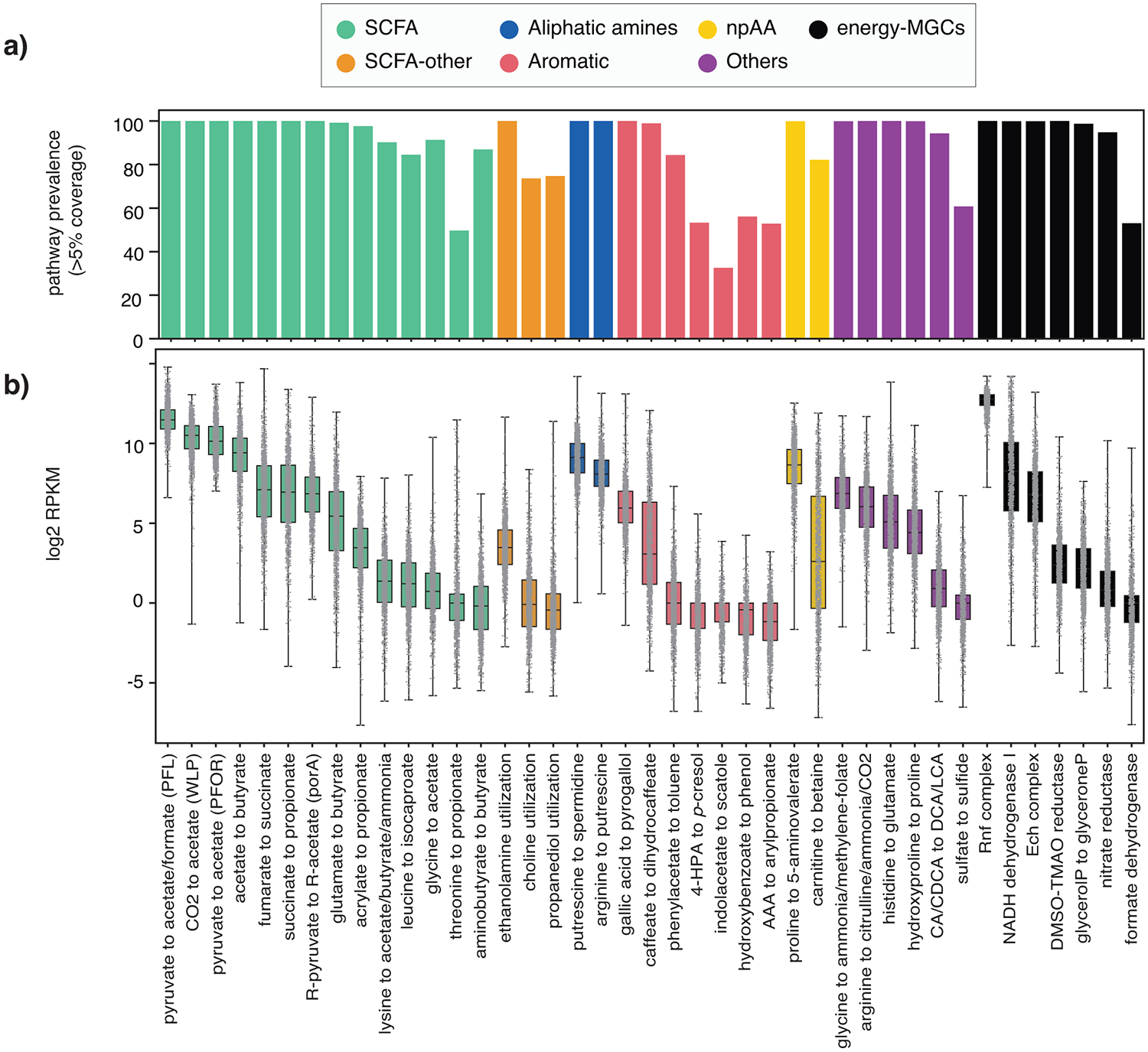
(a) Prevalence of each of the 41 known MGC-encoded pathway classes across all microbiomes, measured as the percentage of samples in which core enzyme-coding genes of at least one reference MGC belonging to a given class were covered by metagenomic reads across >5% of their sequence length. This cutoff was kept low to avoid false negatives due to limited sequencing depth for low-abundance taxa (raw data available at Table S7). (b) Distributions of log2 RPKM relative abundance values of all 41 known pathway classes, categorized by product class, across all LifeLines DEEP metagenomes (n=1,135; raw count data available at Table S8). All samples are represented by a dot in the box plot, representing the log2 RPKM value for a given sample. The box limits indicate the quartiles of the dataset while the whiskers extend to 1.5x the interquartile range; center line denotes the median.
The wide variability in the metagenome abundance of each pathway raises the question of whether metagenomic abundance of a pathway correlates with the level of its small molecule product in the host. To address this question, we systematically compared the level of each pathway with the quantity of the corresponding metabolite as determined by plasma metabolomics. We find a striking lack of correlation between pathway and metabolite levels (r ranging from −0.04 to 0.24, Figure 4A & Extended Data Figure 2); also when abundances of multiple MGC types with the same end products were summed, correlations remained low (Table S9). These data indicate that gene abundances in metagenomes are not (on their own) a useful predictor of plasma metabolic outputs. This lack of correlation may be due to several factors, such as dietary differences, transcriptional regulation linked to substrate availability, varying dynamics of diffusion and import of the metabolites out of the lumen into the host, secondary fermenters that degrade the end product of some of the gutSMASH-predicted pathways, and the existence of pathways with similar substrate/product profiles that are yet unknown. In order to assess the effect of nutrient import, we also quantified metagenomic pathway abundance correlation with available faecal metabolomic data for SCFA from the LifeLines DEEP cohort, and this showed similarly low correlations (ranging from −0.16 to 0.11, see Table S10). We also mapped reads from 81 samples from the integrative human microbiome project (iHMP), including 41 Crohn’s disease patients, 17 ulcerative colitis patients and 23 healthy subjects (SI Table 11), to the same set of gutSMASH MGCs (Figure 4B & C). Correlating pathway abundance levels on these samples further confirmed this pattern, with overall correlations ranging from −0.32 to 0.34 (see Figure 4B). Correlations did increase when splitting samples by disease status ranging from −0.50 to 0.52 for healthy samples, −0.51 to 0.53 for Ulcerative Colitis (UC) patients and −0.37 to 0.42 for Crohn Disease (CD) patients, see Table S11), suggesting that large-scale physiological differences (e.g., differences in absolute microbial abundance) among human subjects are prominent confounding factors. Overall, our findings have important implications for analyses that make metabolic inferences from gene abundances26 or the abundances of individual strains27. We speculate that a more detailed understanding of the influence of diet, differences in gene regulation, characteristic pathway flux (turnovers per unit time per protein copy), which may also be affected by secondary fermenters, and pharmacokinetic characteristics (e.g., absorption, distribution, metabolism, and excretion) could ultimately enable the prediction of metabolite abundance from metagenome abundance. Indeed, when we compared mapping of metatranscriptomic reads for the 81 iHMP samples (mixed phenotypes) for which paired metagenomic/metatranscriptomic/metabolomic data were available, we already observed slightly higher correlations (ranging from −0.22 to 0.37, Figure 4B), although the difference with the metagenomic data from the same samples was not statistically significant (cochran’s Q test coefficient ranging from 0.0013 to 0.983). At an FDR of <0.1, while we observed 5 significant associations between gutSMASH pathways and their corresponding metabolites for samples with paired metagenome/metabolome data, we observed 6 significant associations for samples with paired metatranscriptome/metabolome data (Table S11). The correlations from a larger set of 271 iHMP metatranscriptomic/metabolomic samples, from which complete metadata was available (Table S12), also seemed to show slightly stronger signals compared to the metagenome/metabolome data, with overall correlations ranging from −0.17 to 0.34 (see Figure 4B), although no direct comparison could be made in the absence of metagenome data. When split out across the three phenotypes, correlations ranged from −0.28-0.38 for healthy, −0.27-0.27 for UC and −0.24-0.42 for CD and yielded 10 significant associations between pathway expression values and their metabolites. The correlations across these datasets varied quite a lot depending on the pathway (see Figure 4A), suggesting that the expression of some pathways is more specifically predictive for metabolite abundances. For instance, strong correlations were found between the CA/CDCA to DCA/LCA (bai operon) pathway with deoxycholic acid, possibly due to the fact that it is a taxonomically restricted pathway without known alternative pathways leading to the same products. In contrast, some pathways showed low or even slightly negative correlations, which may be explained by, e.g., pathway competition, diffusion/transport differences or consumption by other bacteria. Overall, systematic detection of the relevant genes and gene clusters by gutSMASH provides a technological foundation for future studies to study how various factors influence microbial metabolite production and accumulation in the lumen as well as in plasma, by allowing mapping of metatranscriptomic data to these accurately defined and categorized sets of genomic loci across a wide range of conditions. Measuring absolute microbial abundance across samples will likely greatly help in this as well28.
Figure 4. Pathway correlations with metabolomic data.
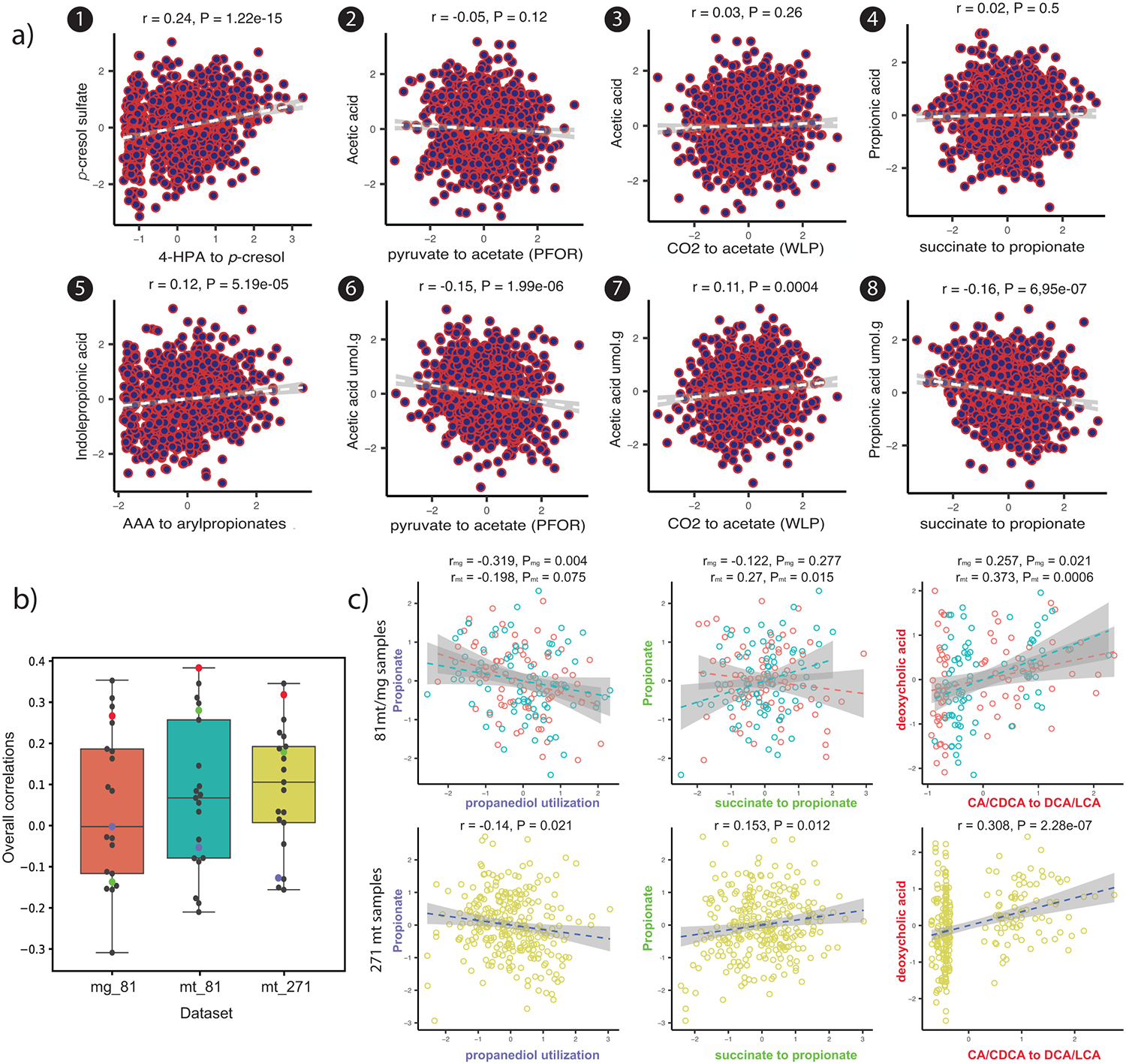
(a) Limited correlation of genetic pathway abundance with abundance of metabolites in blood plasma (correlation plots 1-5) and faeces (correlation plots 6-8) from the LifeLines DEEP cohort (n=1,055). The correlation plots 2-4 and 6-8 correspond to pathway association with plasma and faecal levels of the same short-chain fatty acids, respectively. The x-axis indicates abundance of pathways and the y-axis indicates abundance levels of metabolites in plasma or faces. The grey line shows the best linear fit, with 95% confidence interval. Spearman correlation (two sided) is used to check the relationship between pathway abundances and metabolite levels after adjusting for age, sex and read depth. The rank-based Spearman correlation coefficient and empirical p-value are also shown. Spearman correlation (two sided) is used to check the relationship between pathway abundances and metabolite levels after adjusting for age, sex and read depth. (b) Overall correlation boxplots between gutSMASH-predicted pathways and the iHMP data considering the 81 samples with paired metagenome/metabolome/metatranscriptome data when considering the metagenome/metabolome correlations (mg_81, red), and the metatranscriptome/metabolome correlations (mt_81, turquoise), as well as correlations for the 271 samples with metatranscriptome/metabolome data (mt_271, yellow). Individual data points are shown in the dot plot. The box limits indicate the quartiles of the dataset while the whiskers extend to 1.5x the interquartile range; center line denotes the median. (c) Correlation (Spearman, two sided) plots for three specific pathways within each dataset, with the mg_81 and mt_81 datasets being shown above in red/turquoise and the mt_271 dataset being shown below in yellow. For each pathway, a different colour has been used for the axis labels: purple for propanediol utilization, green for succinate to propionate and red for CA/CDCA to DCA/LCA; the corresponding data point in the box plot in (b) has been colored accordingly.
Discussion
The gutSMASH software constitutes a comprehensive automated tool designed to identify niche-defining primary metabolic pathways from genome sequences or metagenomic contigs—even a full-fledged metabolic network reconstruction software like PathwayTools29 (which uses the extensive MetaCyc database30) lacks detection capabilities for 2 out of the 41 MGC-encoded pathways detected by gutSMASH (Table S13). We also assessed the overlap of pathways between gutSMASH and GenomeProperties31 and only 5 out of the 41 MGC-encoded pathways can be systematically annotated using the latter (Table S13). Moreover, the identification of MGCs provides considerably increased confidence that detected homologues for a given pathway are truly working together. Downstream, detected MGCs can be used as input for read-based tools such as HUMAnN32 or BiG-MAP21 to measure abundance or expression levels of the encoded pathways. On top of these functionalities, the gutSMASH framework also facilitates identifying new (i.e., uncharacterized) pathways in the microbiome. To this end, we designed an additional set of rules, referred as general rules in Figure 1, to detect primary metabolic gene clusters of unknown function that harbor at least one of the following key enzymes: Fe-S flavoenzymes33, glycyl-radical enzymes, 2-hydroxyglutaryl-CoA-dehydratase-related enzymes, and/or enzymes involved in oxidative decarboxylation. After running gutSMASH on the 4,240 microbial genomes and pulling out the putative MGCs (see SI methods Analysis of distant homologues and putative MGCs from CGR, HMP and Clostridioides dataset), we found 12,256 putative MGCs from 760 different species, that, after redundancy filtering at 90% sequence similarity, were classified into 932 GCFs. Within these, we manually prioritized a range of gene clusters with unprecedented enzyme-coding gene content highlighted in Extended Data Figure 3 & 4 (see SI Results Analysis of putative clusters and distant homologues: relevant candidates to study further). These putative MGCs can be a potential source to discover new pathways and metabolites. Thus, gutSMASH can be a valuable tool in the field of enzyme/pathway discovery, to link metabolites to gene clusters and to identify genes responsible for microbiome-associated phenotypes
Methods
gutSMASH is a Python-based pipeline that has been built from antiSMASH version 5.0 source code. The latest command line version is freely available and can be downloaded and installed from here: https://github.com/victoriapascal/gutsmash/tree/gutsmash
Finding pathway signatures for known and characterized MGCs
To create a new set of detection rules, 41 known and characterized MGCs were gathered from literature and used as positive controls. The protein sequences of these MGCs were searched using hmmscan (HMMER suit version 3.1b2, February 2015; http://hmmer.org/). From the resulting pHMM profile hits, auxiliary and core domains were manually identified for each pathway, to ultimately determine the pathway signature and specify it in the corresponding detection rule. To discern and more precisely identify key enzymes of interest sharing a keystone domain, we used custom-made pHMMs following a procedure described in the SI Methods section: Towards a more robust MGC identification by building new HMM profiles. Altogether, the knowledge on the core enzyme coding-genes and the newly-built pHMMs helped to construct a preliminary set of detection rules to predict known pathways.
New HMM profiles for robust MGC identification
Certain core domains are shared across diverse pathways, including the PFL-like domain and the HGD-D domain. In total, 13 keystone domains were found to be ubiquitous in multiple pathways (see Table S14). Hence, to increase gutSMASH precision and discern between enzyme subfamilies of interest, 12 protein superfamily phylogenies were constructed by aligning the protein sequences harbouring the domain of interest from the MGC collection (described in Methods section Exploring the yet unknown metabolic diversity by creating general detection rules; for an example, see Suppl. Figure 3), the respective reference proteome34 at a 15% or 35% co-membership threshold (the latter only for the domains Gly_radical and Acyl-CoA_dh_1) and any experimentally characterized UniProt representatives. After aligning the sequences with hmmalign35, approximately-maximum-likelihood phylogenetic trees using FastTree 2.136 were inferred to further annotate the tree with iTOL15. Thus, from the desired and functionally relevant clades, specific pHMMs were built by extracting the amino acid sequence of the clade-specific proteins, aligning them with Clustal Omega, trimming the edges of the multiple sequence alignment using Jalview37, re-aligning all the sequences with Clustal Omega and finally building a pHMM using hmmbuild (HMMER suite version 3.1b2, February 2015; http://hmmer.org/). Subsequently, for all the newly created pHMMs, sensitivity was assessed using 10-fold jackknife cross-validation. Each clade was divided randomly into training and testing sets. The protein sequences from the training set were aligned using Clustal Omega and used to create a pHMM. Next, the protein sequences of the test set were hmmscanned (HMMER suit version 3.1b2, February 2015; http://hmmer.org/) against the newly built testing pHMMs. When a sequence scored positively for multiple domains in the same region, only the domain with a higher bit score was picked out. Sensitivity then accounted for the number of sequences positively associated with the correct pHMM out of the total number of sequences in the testing set. The same procedure was repeated 10 times. The pHMMs with a true positive rate higher than 0.85 across the 10 rounds were included in the detection rules. In total, 43 newly built pHMMs were included in the corresponding detection rules (Table S15). Moreover, a pHMM to capture succinate dehydrogenase/fumarate reductase was built by aligning 10 protein sequences of such enzymes and building the model from this alignment using a hmmbuild. To also competitively score similar Pfam domains, Hhsearch pre-computed results obtained from the Pfam FTP (http://ftp.ebi.ac.uk/pub/databases/Pfam/current_release/database_files/) were parsed and included in the gutSMASH code.
Testing and validating detection rules for known pathways
To evaluate the performance of the preliminary set of detection rules, a total of 1,621 bacterial genomes, including 1,520 genomes from the CGR collection9 and 101 manually selected genomes from the most representative bacterial genera in the human gut, were used as input for gutSMASH (see Table S3). The predicted MGCs were classified based on the detection rule they were predicted from, to later run BiG-SCAPE on each sub-collection. The resulting networks were screened individually to evaluate the taxonomic and architectural diversity, to assess if any architectural variant or taxon (based on literature) was missing from the MGC pool or was incorrectly predicted by the detection rule. Hence, this procedure ultimately helped to tweak the detection rules to predict true homologues of the known pathways (see Supplementary Results and Table S16 for more details). After two iterations of fine-tuning and testing, all detection rules were performing as intended and constituted the new set of detection rules of gutSMASH version 1.0.
gutSMASH customized databases and output visualization
The antiSMASH version 5.0 source code was further tailored to meet gutSMASH functionality. The 32,144 predicted MGCs obtained from running gutSMASH on the CGR, HMP and Clostridiales collections (see Methods section Evaluating functional potential of gut bacteria using gutSMASH for more insights), were used to create the ClusterBlast database. In a similar way, 59 positive controls carrying the known pathways (from which we created the specific-to-known-pathway detection rules) were used to create the KnownClusterBlast database. These databases facilitate comparative gene cluster analysis using BLAST38. Thus, they allow assessing how broadly distributed an MGC is across bacteria (in the case of ClusterBlast) or evaluating the similarity between the predicted MGC and a known and functionally characterized MGC (when using KnownClusterBlast).
Another functionality of antiSMASH is to classify coding genes based on the domains into five major functional categories: core biosynthetic, additional biosynthetic, transport-related, regulatory, resistance and other, using the pmCOG (primary metabolism Clusters of Orthologous Groups) tool, which is embedded in antiSMASH (there originally named smCOG for ‘secondary’ metabolism Clusters of Orthologous Groups). Thus, the pHMM library pmCOG uses was updated to include relevant domains found in specialized primary metabolism. Also, two other important functional categories were added: electron transport-related genes and encapsulation genes.
Exploring unknown metabolic diversity using general rules
With the objective of creating general detection rules to predict putative MGCs, a similar approach used to screen the surrounding genes around a Fe-S flavoenzyme coding gene was used33. Some of the representative known pathways share proteins with biochemically similar functions; these include, for instance, pyruvate formate lyase-like enzymes that are found in the threonine-to-propionate pathway, the choline utilization pathway and the pyruvate-to-acetate pathways. In order to cover a large amount of sequence diversity, we created a database that included 11,000 complete genomes and 98,886 draft genomes available in Genbank (in February 2017) in order to use clusterTools39, a software to find remote homologues of known MGCs. As input, a subset of the known pathways used to design the detection rules for known pathways were used as input (see Table S17). The output of several iterated clusterTools searches were grouped to acquire a collection of over 29,000 clusters. For visualization and manual scoring purposes, MultiGeneBlast40 was run using the clusterTools output as input. Thus, MGCs harbouring at least half of the genes from the query gene list and with a cumulative BLAST score higher than 1,000 were included in the MGC collection. In order to filter out redundant sequences, we used Mmseqs241 at a 95% similarity cut-off. From the resulting network of 1,599 groups, a maximum of 1 random representative plus singletons were picked creating a ‘non-redundant’ set of almost 3,200 clusters. This collection was screened for gene clusters harbouring the baiCD or baiH coding gene (Oxidored_FMN and Pyr_redox_2), pyruvate-formate lyase (PFL-like or Gly_radical), pyruvate ferredoxin (POR, POR_N or PFOR_II), thiamine pyrophosphate enzyme (TPP_enzyme_C) and 2-hydroxyglutaryl-CoA dehydratase (HGD-D), each of which are keystone domains in charge of important anaerobic reactions. This helped create general detection rules, by identifying which other enzyme-coding Pfam domains are found around these ‘anchor’ domains in flanking regions; this was systematically analyzed per gene cluster family to make sure that the general rules captured all major families of homologous MGCs of interest. Also, when validating the specific-to-known pathway detection rules, whenever a specific rule predicted interesting MGCs that were variants of the representative pathway with likely differing functions, a general rule was created out of the specific one by loosening up the Pfam requirements. The full list of general rules can be found in Table S18.
Assessing single-protein pathway abundance
To include single-protein pathways in our analysis to assess the overall abundance of specialized primary metabolic pathways, 10 enzyme families were selected for downstream analysis. Following the same procedure as described in the Methods section: Towards a more robust MGC identification by building new HMM profiles, protein phylogenies were built for each protein superfamily. Similarly, from the pathway-specific monophyletic clades, we built new pHMMs. A bitscore threshold for each newly built pHMM was calibrated in order to identify with high confidence proteins belonging to the same functional clades. To this end, the protein sequences that composed the superfamily phylogeny were subjected to an hmmsearch run with the new pHMM. The bitscore reported by hmmsearch for the most distantly related protein within the pathway-specific clade was chosen as the threshold for that specific pHMM. Next, the protein sequences from the CGR, HMP and Clostridiales collections (further information in Methods section: Evaluating the functional potential of the human microbiome using gutSMASH) were scanned using the newly built pHMMs. Finally, the hmmsearch output tables for each pHMM were parsed so that the proteins with a bitscore equal or higher to the chosen threshold were deemed hits. In those cases in which the single-protein sequence codes for two Pfam domains, as for instance the serine dehydratase (SDH_alpha and SDH_beta), one of the Pfam domains was selected to create a protein phylogeny to further build a clade-specific pHMM, in this case SDH_alpha. Then, the protein sequences from the three collections were subjected to hmmsearch runs with both the clade-specific pHMM and the other co-occurring Pfam domain (in this case SDH_beta). The sequences that harbour both the specific pHMM at the chosen threshold and the co-occurring domain with an e-value ≤ 10−05 were deemed hits.
Evaluating the functional potential of the human microbiome
To evaluate the metabolic potential of the human microbiome, gutSMASH was run on three different genome collections: (1) the CGR collection, with 1,520 CGR genomes deposited under the PRJNA482748, (2) the HMP reference genomes, with 2,146 HMP bacterial genomes downloaded in September 2019 from here: https://www.hmpdacc.org/hmp/catalog/grid.php?dataset=genomic and (3) 414 Clostridiales complete genomes under the taxid 186802. The genomic FASTA sequence of these genomes was used as input for gutSMASH, which used Prodigal42 to annotate genes across all of them in a consistent way. Moreover, in order to assess which MGC belonged to known pathways, the KnownClusterBlast (see SI Methods gutSMASH customized databases and output visualization) option was enabled. Thus, from the KnownClusterBlast output, the predicted regions were classified as known when the following two requirements were met: (1) an overall pathway similarity of at least 50% and at least half of the genes with a minimum protein sequence similarity of 40% or (2) an overall similarity of 60% and half of the genes with protein sequence similarity higher than 30%. However, in order not to penalize MGCs with similar domain profiles but substantially larger sizes, the requirements to be considered “known” slightly changed for the KnownClusterBlast MGCs longer than 17 coding genes. In this case, the same requirements as described above were used but instead of considering candidates with at least half of the coding genes having either 30 or 40% minimum sequence identity, one third of the genes were required to be present with the same minimum sequence identity. This was the case for the ethanolamine utilization operon, the bai operon characterized from C. scindens ATCC35704 (CA/CDCA to DCA/LCA pathway), the acetyl-CoA pathway (CO2 to acetate (WLP)), the tetrathionate to thiosulfate pathway and the NADH dehydrogenase I complex. Thus, all the MGCs that did not satisfy these conditions were classified as putative MGCs. The phylogenetic tree In Figure 2 was generated using phyloT v2 (https://phylot.biobyte.de/). The GDTB database14 was used to assign the taxonomy to the genomes of the three collections (when present) and those taxonomic identifiers were the ones used for the subsequent pathway absence/presence analysis. Finally, the tree was annotated using iTOL15.
Analysis of distant homologues and putative MGCs
The putative MGCs predicted from the CGR, HMP and Clostridiales genome collections were selected following the definition of “known” and “putative” gene clusters stated in the Evaluating the functional potential of the human microbiome using gutSMASH Methods section. To account for redundant MGCs, protein sequences extracted from all gene clusters were subjected to a redundancy filtering of 90% sequence similarity using mMseqs2. From the resulting clustering, two random representatives were chosen from each group, including the singletons. The resulting non-redundant collection of 3,040 putative MGCs was used as input for BiG-SCAPE using the default thresholds. The network in Extended Data Figure 3 was constructed and annotated using Cytoscape v3.043.
Mapping metagenomics reads from cohort samples to MGCs
The HMP, CGR and Clostridiales-predicted MGCs were used as input for BiG-MAP21, a tool that assesses gene cluster abundance or expression across metagenomics or metatranscriptomics data, respectively, by mapping the genomic reads onto the gene cluster sequences. The BiG-MAP family module grouped the 32,146 MGCs into 5,655 GCFs. Next, the reads of 1,135 participants of the population-based cohort LifeLines-DEEP44 (quality-filtered using KneadData version 0.7.2) were mapped onto the resulting 5,655 Mash45 representative MGCs by using BiG-MAP.map module. (All Lifelines participants signed an informed consent form before sample collection. The ethics review board of the University Medical Center Groningen has approved the study with reference number M12.113965.) To assess the abundance of known pathways, the RPKM values from the known MGCs (following the definition of “known” stated in Methods section Evaluating the functional potential of the human microbiome using gutSMASH) were pulled out. The RPKM values of all the MGCs predicted by the same detection rule were merged. The pathway abundance (RPKM) was computed by dividing the gene clusters in 2kb-sized bins, and assessing the lower quartile number of reads mapping the 2kb bins for each gene cluster and sample. In contrast, a pathway was annotated as present in a sample when reads from that sample were found to be mapping to at least 5% of the core region of that MGC. This threshold was kept low to enable detection of MGCs from low-abundant microbes and avoid false negatives due to limited sequencing depth. The lowest percentage identity of reads mapped to MGCs was 78% at the nucleotide level, which instilled confidence that finding multiple reads mapping to different locations within a MGC provides sufficient evidence for its presence in a sample. The pathway prevalence was also computed using 10%, 20%, 30%, 40%, 50%, 60%, 70% and 80% core coverage thresholds (Extended Data Figure 1), and results for increasing thresholds were consistent with gradual loss of detection capability for pathways known to be associated with low-abundance bacteria, such as the AAA to arylpropionate pathway (aromatic amino acid reductive branch). To also take into account25etatranscriptomicsc and faecal metabolome data, the 81 paired metagenomes, metatranscriptomes and metabolomes from the Inflammatory Bowel Disease Multi’omics Database (IBDMDB) study46 were similarly analysed using BiG-MAP. In this case, to speed up calculation, only the bacterial genomes whose gutSMASH run predicted at least one “known” (following the definition as described above) MGC were used as input for the BiG-MAP.family module. The same “known” MGC collection as described above was used. In total, for the BiG-MAP.family module, 1764 gutSMASH runs were used, which included 8,109 gene clusters that were downsized to 6,301. This reduced MGC reference collection was then used by the BiG-MAP.map module that aligned the metagenomic and25etatranscriptomicsc to the reference collection independently. The same procedure was followed for the 271 samples with paired metatranscriptome and metabolome data.
Correlating pathway abundance with metabolite concentrations
To evaluate the correlation between the gene cluster abundance and metabolite concentrations, the masses of 7 metabolites derived from several gutSMASH predicted gene clusters could be found in the Mass Spectrometry (MS) data of the plasma measured in LifeLines DEEP22,44. Untargeted metabolomics profiling was done using flow-injection time-of-flight mass spectrometry (FI-MS) as described by Chen et al47. These metabolites included acetic acid, indolepropionic acid, isovaleric acid, p-cresol, p-cresol sulfate, phenylacetic acid and propionic acid (see Figure 4 and Extended Data Figure 2). Both metabolite and pathway abundance (RPKM counts) were inverse-rank-transformed and the linear regression was applied to adjust covariates including age, sex and metagenomic sequencing depth (only for pathway abundance). Metabolite and pathway abundance residuals from the linear regression model were then used to perform Spearman correlation test. Finally, the Benjamini Hochberg method was applied to control for false discovery rate (FDR). The RPKM counts of the gutSMASH-predicted pathways involved in the synthesis of Short Chain Fatty Acids (SCFA) were correlated in the same manner with the faecal SCFA MS data also collected from the LifeLines DEEP cohort. Specifically, the SCFAs measured in the faecal metabolomes were acetate, propionate, butyrate and caproate.
In order to further assess the relationship between MGC abundance/expression with faecal metabolite concentration, the data derived from analysing the IBDMDB data with BiG-MAP was used similarly as the Lifelines DEEP data. In this case, gene cluster abundance and expression values were correlated with the following faecal metabolites: betaine, butyrate, deoxycholate, glutamate, hydrocinnamate, indole-3-propionate, lithocholate, p-hydroxyphenylacetate, phenylacetate, proline, propionate, putrescine, spermidine, succinate and trimethylamine-N-oxide. Correlations were made for each individual subgroup in the dataset that included Croh’'s disease (CD), Ulcerative Colitis (UC) and healthy samples.
Extended Data
Extended Data Figure 1: Pathway prevalence using different core coverage thresholds.

Pathway prevalence was computed by assessing the number of reads (per sample) mapping to known gene clusters at a certain core coverage cut-off. The figure illustrates how the pathway prevalence gradually changes when increasing the core coverage cut-off from 10 to 80%.
Extended Data Figure 2: Limited correlation of genetic pathway abundance with metabolites abundance in blood plasma.

This figure shows correlation plots for additional metabolites not shown in Figure 4a. Spearman correlation (two sided with rho and empirical p-value are reported) is used to check the relationship between pathway abundances and metabolite levels after adjusting for age, sex and read depth. n= 1054 biologically independent samples.
Extended Data Figure 3: Network of putative non-redundant MGCs predicted by gutSMASH.

From all the unknown predicted MGCs, a redundancy filtering of 0.9 sequence similarity was applied using MMseqs2. From each cluster, two representatives were picked, and all representatives were used as input for BiG-SCAPE using the default cut-offs. The network contains 2,921 nodes and 7,474 edges. The MGCs have been classified into four different categories based on the key enzyme classes they code for. The GR (glycyl-radical) category is composed of MGCs that include pyruvate formate-lyase (PFL-like) and/or glycyl radical (Gly_radical), OD (oxidative decarboxylation) involves MGCs with at least one of the following Pfam domains: Pyruvate ferredoxin/flavodoxin oxidoreductase (POR), Pyruvate flavodoxin/ferredoxin oxidoreductase, thiamine diP-bdg (POR_N), Pyruvate:ferredoxin oxidoreductase core domain II (PFOR_II) and Thiamine pyrophosphate enzyme, C-terminal TPP binding domain (TPP_enzyme_C). The Flavoenzymes category is a combination of MGCs harbouring at least one of the custom-made BaiCD and BaiH pHMMs. HGD-D-related MGCs, as the name states, include enzymes matching any of the 2-hydroxyglutaryl-CoA dehydratase, D-component (HGD-D)-related pHMM domains.
Extended Data Figure 4: Subset of unknown MGCs predicted by gutSMASH manually picked.

The network/nodes present in the left side of the figure represent the subnetwork extracted from the complete network in Extended Data Figure 3. The arrows have been coloured-coded based on the Pfam domains found in the protein-coding sequences and the functional annotations of these proteins.
Supplementary Material
Acknowledgements
This work was supported by the Chan-Zuckerberg Biohub (M.A.F.), DARPA awards HR0011-15-C-0084 and HR0112020030 (M.A.F.), NIH awards R01 DK101674, DP1 DK113598, and P01 HL147823 (to M.A.F.); the Leducq Foundation, and an ERC Starting Grant [948770-DECIPHER to M.H.M.]. A.Z. is supported by the ERC Starting Grant 715772, Netherlands Organization for Scientific Research NWO-VIDI grant 016.178.056, the Netherlands Heart Foundation CVON grant 2018-27, and the NWO Gravitation grant ExposomeNL 024.004.017. J.F. is supported by the ERC Consolidator grant (grant agreement No. 101001678), NWO-VICI grant VI.C.202.022, the Dutch Heart Foundation IN-CONTROL (CVON2018-27), the Netherlands Organ-on-Chip Initiative, and an NWO Gravitation project (024.003.001) funded by the Ministry of Education, Culture and Science of the government of The Netherlands. L.C. is supported by the Foundation de Cock-Hadders grant (20:20-13) and a joint fellowship from the University Medical Centre Groningen and China Scholarship Council (CSC201708320268). D.D. was supported by NIH awards K08 DK110335, R35 GM142873, and R01 AT011396.
Footnotes
Competing Interests Statement
M.A.F. is a co-founder and director of Federation Bio, a co-founder of Revolution Medicines, and a member of the scientific advisory board of NGM Biopharmaceuticals. D.D. is a co-founder of Federation Bio. M.H.M. is a co-founder of Design Pharmaceuticals and a member of the scientific advisory board of Hexagon Bio. The remaining authors declare no competing interests.
Data Availability
The LifeLines DEEP cohort raw metagenomic sequencing data, metabolome data and human phenotypes (i.e. age and sex) used for the analysis presented in this study are available at the European Genome-phenome Archive under accession EGAS00001001704. Taxonomic assignments of bacteria were performed according to the Genome Taxonomy Database release 95 (https://gtdb.ecogenomic.org/). Lists of accessions of genome assemblies used are available in Tables S3 and S4. iHMP multi-omics data were downloaded from https://ibdmdb.org. Raw sequence data of the iHMP are also available from the NCBI sequence read archive (SRA) via BioProject PRJNA398089, metatranscriptome data through GEO Series accession number GSE111889, and metabolomics data at the Metabolomics Workbench (http://www.metabolomicsworkbench.org; Project ID PR000639).
Code Availability
The gutSMASH source code is available freely under an open-source AGPL-3.0 license from https://github.com/victoriapascal/gutsmash/ .
References
- 1.Blin K et al. antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karp PD et al. The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinform 20, 1085–1093 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abubucker S et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Computational Biology 8, e1002358 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Medema MH & Fischbach MA Computational approaches to natural product discovery. Nat. Chem. Biol 11, 639–648 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ziemert N, Alanjary M & Weber T The evolution of genome mining in microbes — a review. Nat. Prod. Rep 33, 988–1005 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Medema MH, de Rond T & Moore BS Mining genomes to illuminate the specialized chemistry of life. Nat. Rev. Genet 22, 553–571 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navarro-Muñoz JC et al. A computational framework to explore large-scale biosynthetic diversity. Nat. Chem. Biol 16, 60–68 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitamoto S et al. Dietary L-serine confers a competitive fitness advantage to Enterobacteriaceae in the inflamed gut. Nat Microbiol 5, 116–125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zou Y et al. 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol 37, 179–185 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lloyd-Price J et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature 550, 61–66 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tracy BP, Jones SW, Fast AG, Indurthi DC & Papoutsakis ET Clostridia: the importance of their exceptional substrate and metabolite diversity for biofuel and biorefinery applications. Curr. Opin. Biotechnol 23, 364–381 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Maguire F et al. Metagenome-assembled genome binning methods with short reads disproportionately fail for plasmids and genomic Islands. Microb Genom 6, mgen000436 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vieira-Silva S et al. Species-function relationships shape ecological properties of the human gut microbiome. Nat Microbiol 1, 16088 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Parks DH et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol 36, 996–1004 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Letunic I & Bork P Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reclassification of Bacteroides putredinis (Weinberg et al., 1937) in a new genus Alistipes gen. nov., as Alistipes putredinis comb. Nov., and description of Alistipes finegoldii sp. Nov., from human sources. Syst. Appl. Microbiol 26, 182–188 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Cummings JH, Pomare EW, Branch WJ, Naylor CP & Macfarlane GT Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28, 1221–1227 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones SA et al. Anaerobic respiration of Escherichia coli in the mouse intestine. Infect. Immun 79, 4218–4226 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tremblay PL, Zhang T, Dar SA, Leang C & Lovley DR The Rnf complex of Clostridium ljungdahlii is a proton-translocating ferredoxin:NAD+ oxidoreductase essential for autotrophic growth. Mbio 4, e00406–12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y et al. Clostridium sporogenes uses reductive Stickland metabolism in the gut to generate ATP and produce circulating metabolites. Nat Microbiol 7, 695–706 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andreu VP et al. BiG-MAP: an automated pipeline to profile metabolic gene cluster abundance and expression in microbiomes. mSystems 6, e0093721. (2021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tigchelaar EF et al. Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: study design and baseline characteristics. BMJ Open 5, e006772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faber F et al. Respiration of microbiota-derived 1,2-propanediol drives Salmonella expansion during colitis. PloS Pathog. 13, e1006129 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andriamihaja M et al. The deleterious metabolic and genotoxic effects of the bacterial metabolite p-cresol on colonic epithelial cells. Free Radic. Biol. Med 85, 219–227 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Funabashi M et al. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 582, 566–570 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mallick H et al. Predictive metabolomic profiling of microbial communities using amplicon or metagenomic sequences. Nat. Commun 10, 3136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Douglas GM et al. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol 38, 685–688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vandeputte D et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 551, 507–511 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Karp PD et al. Pathway Tools version 23.0 update: software for pathway/genome informatics and systems biology. Brief. Bioinform 22, 109–126 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caspi R et al. The MetaCyc database of metabolic pathways and enzymes — a 2019 update. Nucleic Acids Res. 48, D445–D453 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richardson LJ et al. Genome properties in 2019: a new companion database to InterPro for the inference of complete functional attributes. Nucleic Acids Res. 47, D564–D572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Franzosa EA et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962–968 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pascal Andreu V, Fischbach MA & Medema MH Computational genomic discovery of diverse gene clusters harbouring Fe-S flavoenzymes in anaerobic gut microbiota. Microb Genom 6, e000373 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 34.Chen C et al. Representative proteomes: a stable, scalable and unbiased proteome set for sequence analysis and functional annotation. PloS One 6, e18910 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eddy SR A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211 (2009). [PubMed] [Google Scholar]
- 36.Price MN, Dehal PS & Arkin AP FastTree 2 – approximately maximum-likelihood trees for large alignments. PloS ONE 5, e9490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waterhouse AM, Procter JB, Martin DMA, Clamp M & Barton GJ Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Camacho C et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de los Santos ELC & Challis GL clusterTools: proximity searches for functional elements to identify putative biosynthetic gene clusters. BioRxiv (2017), doi: 10.1101/119214. [DOI] [Google Scholar]
- 40.Medema MH, Takano E & Breitling R Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol. Biol. Evol 30, 1218–1223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinegger M & Söding J Mmseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol 35, 1026–1028 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Hyatt D et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhernakova A et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ondov BD et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 17, 132 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lloyd-Price J et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 569, 655–662 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen L et al. Influence of the microbiome, diet and genetics on inter-individual variation in the human plasma metabolome. Nat. Med 28, 2333–2343 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The LifeLines DEEP cohort raw metagenomic sequencing data, metabolome data and human phenotypes (i.e. age and sex) used for the analysis presented in this study are available at the European Genome-phenome Archive under accession EGAS00001001704. Taxonomic assignments of bacteria were performed according to the Genome Taxonomy Database release 95 (https://gtdb.ecogenomic.org/). Lists of accessions of genome assemblies used are available in Tables S3 and S4. iHMP multi-omics data were downloaded from https://ibdmdb.org. Raw sequence data of the iHMP are also available from the NCBI sequence read archive (SRA) via BioProject PRJNA398089, metatranscriptome data through GEO Series accession number GSE111889, and metabolomics data at the Metabolomics Workbench (http://www.metabolomicsworkbench.org; Project ID PR000639).
The gutSMASH source code is available freely under an open-source AGPL-3.0 license from https://github.com/victoriapascal/gutsmash/ .