

Abstract
The Plasmodium falciparum aspartic protease plasmepsin X (PMX) is essential for the egress of invasive merozoite forms of the parasite. PMX has therefore emerged as a new potential antimalarial target. Building on peptidic amino alcohols originating from a phenotypic screening hit, we have here developed a series of macrocyclic analogues as PMX inhibitors. Incorporation of an extended linker between the S1 phenyl group and S3 amide led to a lead compound that displayed a 10-fold improved PMX inhibitory potency and a 3-fold improved half-life in microsomal stability assays compared to the acyclic analogue. The lead compound was also the most potent of the new macrocyclic compounds in in vitro parasite growth inhibition. Inhibitor 7k cleared blood-stage P. falciparum in a dose-dependent manner when administered orally to infected humanized mice. Consequently, lead compound 7k represents a promising orally bioavailable molecule for further development as a PMX-targeting antimalarial drug.
1. Introduction
The mosquito-borne disease malaria impacts half of the world′s population in tropical and subtropical areas causing ∼600 thousand deaths annually, with infants and pregnant women representing particularly high-risk groups.1 Malaria is also a significant cause of poverty in endemic countries.2 Malaria morbidity and mortality has been significantly reduced over recent decades through a combination of approaches, including vector control, insecticide-impregnated bed nets, and chemoprophylactic and chemotherapeutic means.3 In its 2015 Global Technical Strategy, the World Health Organization set a target of 90% reduction in clinical cases and deaths due to malaria by 2030. However, efforts to reach this target are challenged by the spread of drug-resistant Plasmodium parasites, the causative agent of malaria. Plasmodium strains resistant to nearly all clinically used drugs, including artemisinins, the current front-line antimalarial drug class,4−6 have been reported. New drugs for the treatment of acute uncomplicated malaria (Medicines for Malaria Target Product Profile-1, TPP-1) and chemoprotection (TPP-2) are therefore urgently needed to replenish the arsenal of antimalarial drugs.7 While recently approved antimalarial drug products have been dominated by combination therapies based on existing drugs,8 a current focus is on the discovery of new drugs that act through novel modes of action.
Plasmodium aspartic proteases, termed plasmepsins (PMs), have been explored as potential antimalarial drug targets for nearly three decades,9−12 but to date, no PM inhibitor has advanced to the clinic. Ten PM isoforms are encoded in the genome of Plasmodium falciparum (Pf), the deadliest species affecting humans. Seven P. falciparum PMs (I–V, IX, and X) are expressed during the asexual blood stages of the parasite lifecycle that are responsible for all clinical manifestations of the disease, so these enzymes have been considered as potential drug targets. Extensive work on PMs I–IV, which play roles in the proteolytic breakdown of hemoglobin in the parasite digestive vacuole, has shown this group of enzymes to be highly redundant, rendering them unfit as drug targets.13 More recent efforts have focused on the non-degradative PMs, all of which appear essential for parasite viability. PMV is expressed in the parasite endoplasmic reticulum, where it functions to modify a subclass of parasite proteins that are exported into the host red blood cell (RBC).14 PMIX and PMX are maturases essential for the egress (escape) of invasive forms of the parasite, called merozoites, from the host RBC and for merozoite invasion of fresh RBCs.15,16 Several amino alcohol-based peptidomimetic inhibitors of PMIX and/or PMX have been described that potently inhibit parasite proliferation in vitro. Phenotypic screening hit 1 was the first identified compound of this type, proving to be a potent inhibitor of PMX, while another amino alcohol-based compound 2, was shown to inhibit both PMIX and PMX (Figure 1).15,16 Aminohydantoins have also been developed as non-peptidic PMIX and PMX inhibitors. A potent PMX inhibitor, compound 3, resulted from the optimization of an HTS hit and was further optimized to generate analogues with in vivo activity in murine malaria models.17,18 Aminohydantoin 4 was shown to be a dual PMIX and PMX inhibitor and also displayed high antimalarial potency in vitro and in vivo.19
Figure 1.
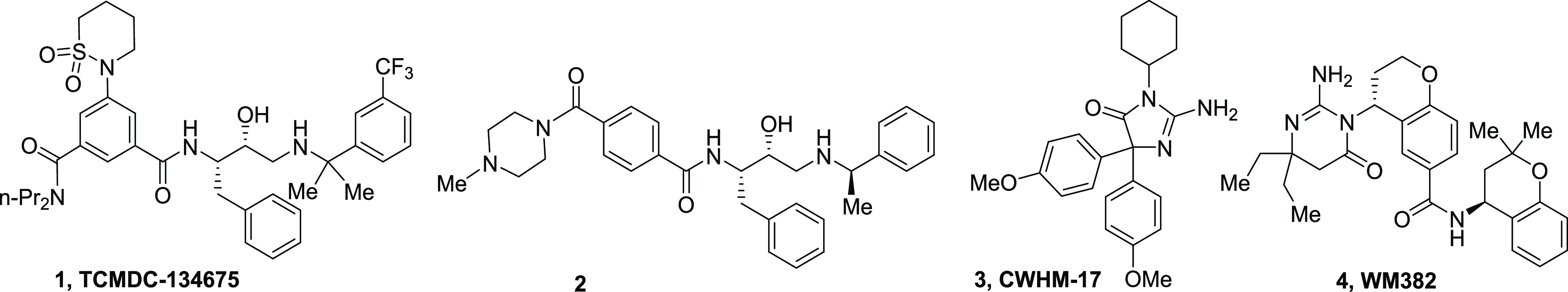
Representative PMIX and/or PMX inhibitors.
Our efforts in PM inhibitor antimalarial drug discovery started with peptidomimetic hydroxyethylamine compound 5, which was identified as a potent inhibitor of P. falciparum growth in a phenotypic HTS screen performed by GSK (Figure 2).20 Early analysis showed that hit 5 was a PM inhibitor, using PMI, PMII, and PMIV as accessible model proteins.21 Work aimed at determining crucial substructures for PM inhibitory potency and reducing inhibition of the human aspartic protease cathepsin D (CatD) resulted in analogue series 6.22 The structurally simplest analogue from this series, compound 6a, was one of the most potent inhibitors of parasite growth. Further examination showed that compound 6a inhibits not only degradative PMs but also PMX, as indicated by the capacity of 6a to inhibit maturation of the P. falciparum PMX substrate subtilisin-like protease-1 (SUB1), a key effector in merozoite egress.23 However, in vitro absorption, distribution, metabolism, and excretion (ADME) profiling of inhibitor 6a (vide infra) revealed that it has low microsomal stability; thus, it was not reasonable to further investigate efficacy in vivo. Since macrocyclization of peptidic compounds can often improve ADME properties,24−27 we were prompted to use 6a as a basis to design macrocyclic analogues 7. Here, we describe the generation of potent, subnanomolar macrocyclic PMX inhibitors that hold potential for development as a new class of antimalarial drugs.
Figure 2.
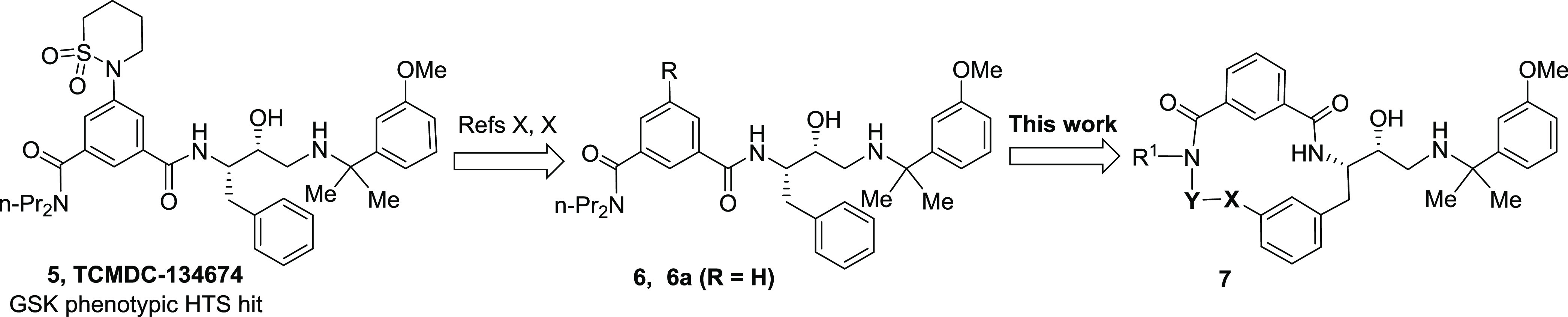
Development of phenotypic screening hit 5 to macrocyclic peptidic inhibitor series 7.
2. Results and Discussion
2.1. Synthesis of Macrocyclic PM Inhibitors
Principal building blocks 16, 18, and 22 for the synthesis of macrocyclic amides 7a–k were prepared according to Scheme 1. Enantioenriched protected glyceraldehyde 8 was transformed into sulfinimine 10 in the reaction with (S)-tert-butanesulfinamide (9),28 which was subjected to the diastereoselective Grignard reaction to give sulfonamide 11.29,30
Scheme 1. Synthesis of Building Blocks 16, 18, and 22 for the Preparation of Inhibitors 7a–k.
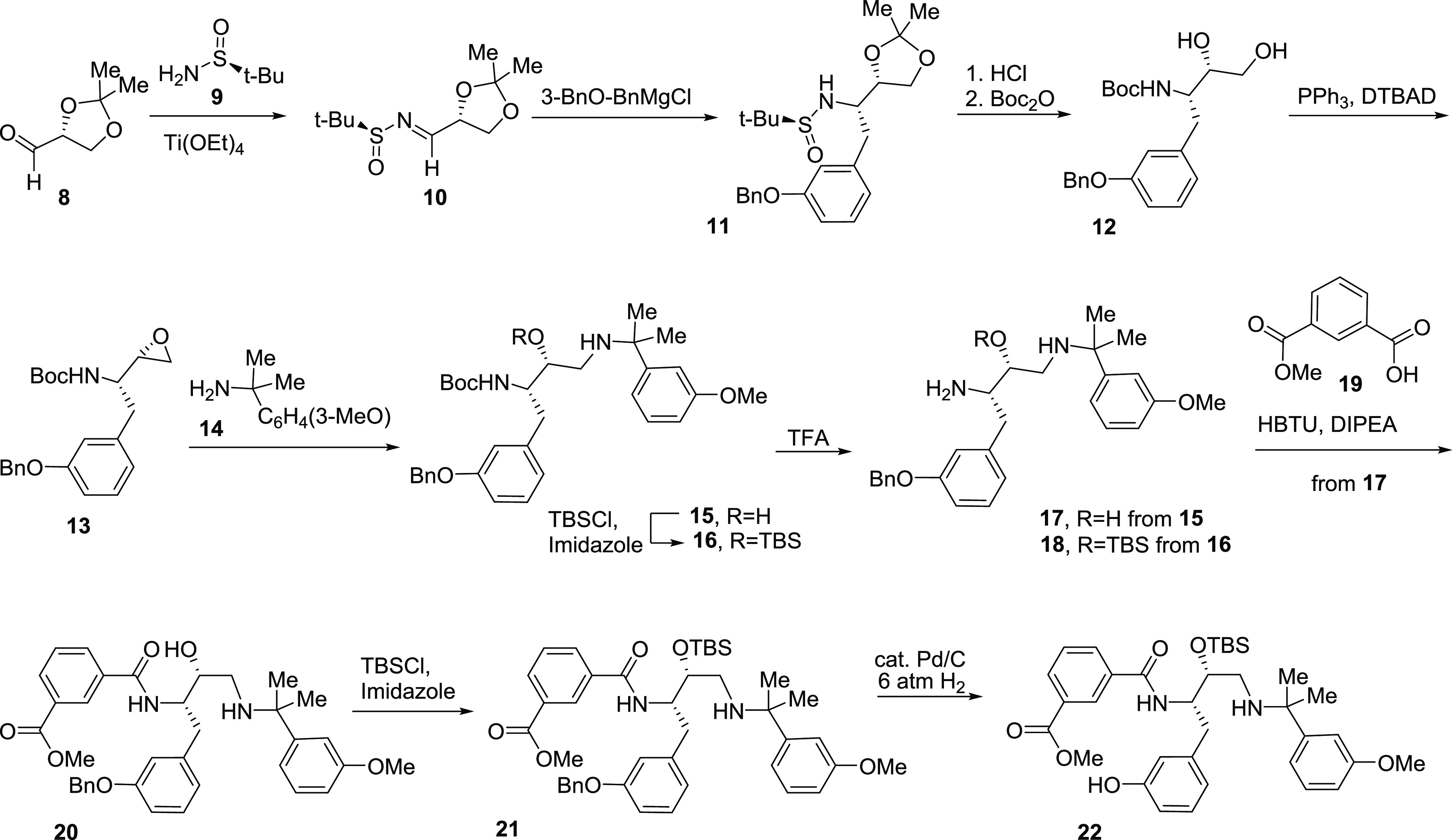
Acetonide and sulfinyl groups in intermediate 11 were removed by treatment with acid, and amine was N-Boc protected to give amino alcohol derivative 12. The Mitsunobu reaction provided aminoepoxide 13, which was ring-opened with benzylamine derivative 14. The resulting diaminoalcohol 15 was O-TBS protected to give an intermediate 16. Both compounds 15 and 16 were subjected to Boc-deprotection to give amines 17 and 18, respectively. Amine 17 was N-coupled with isophthalic acid monoester 19 to produce amide 20. O-TBS protection provided intermediate 21, which was O-debenzylated under hydrogenolysis conditions to provide the building block 22.
Building block 22 was used to prepare the target compounds 7a,e,g,h (Scheme 2). O-Alkylation with alcohols 23a–d in Mitsunobu reaction conditions provided intermediates 24a–d. These were subjected to cleavage of the N-nosyl group leading to amines 25a–d. Hydrolysis of the ester group in intermediates 25a–d followed by macrolactamization and removal of the O-TBS group resulted in target compounds 7a,e,g,h.
Scheme 2. Synthesis of Inhibitors 7a,e,g,h.
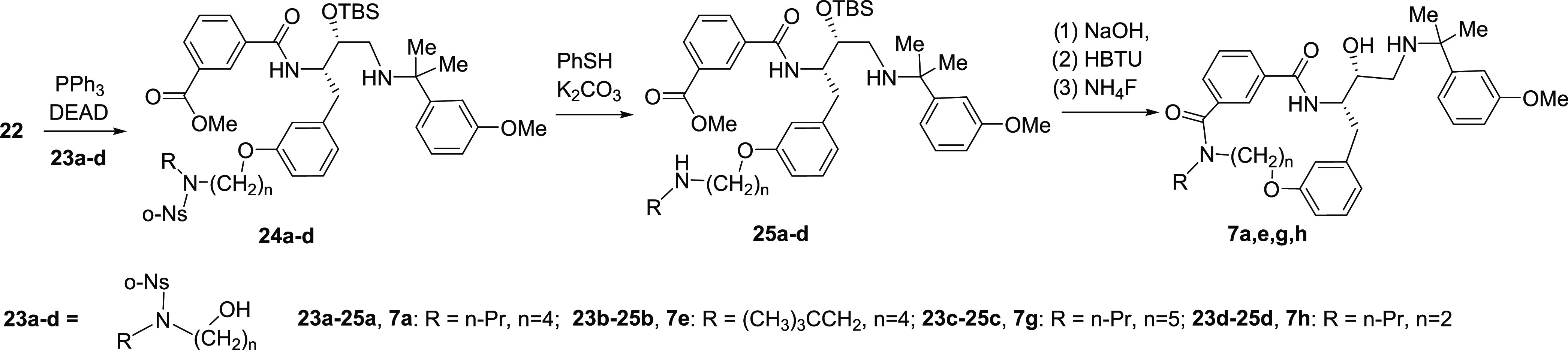
Inhibitors 7b–d,f were prepared starting from amines 26a–d (Scheme 3). These were acylated with isophthalic acid monoester 19 to give isophthalic monoamides 27a–d. Hydrolysis and subsequent coupling with the building block 18 (Scheme 1) provided intermediates 28a–d. O-Benzyl deprotection in compounds 28a–d provided alcohols 29a–d, which were subjected to macrocyclization and O-desilylation, leading to target compounds 7b–d,f.
Scheme 3. Synthesis of Inhibitors 7b–d,f.
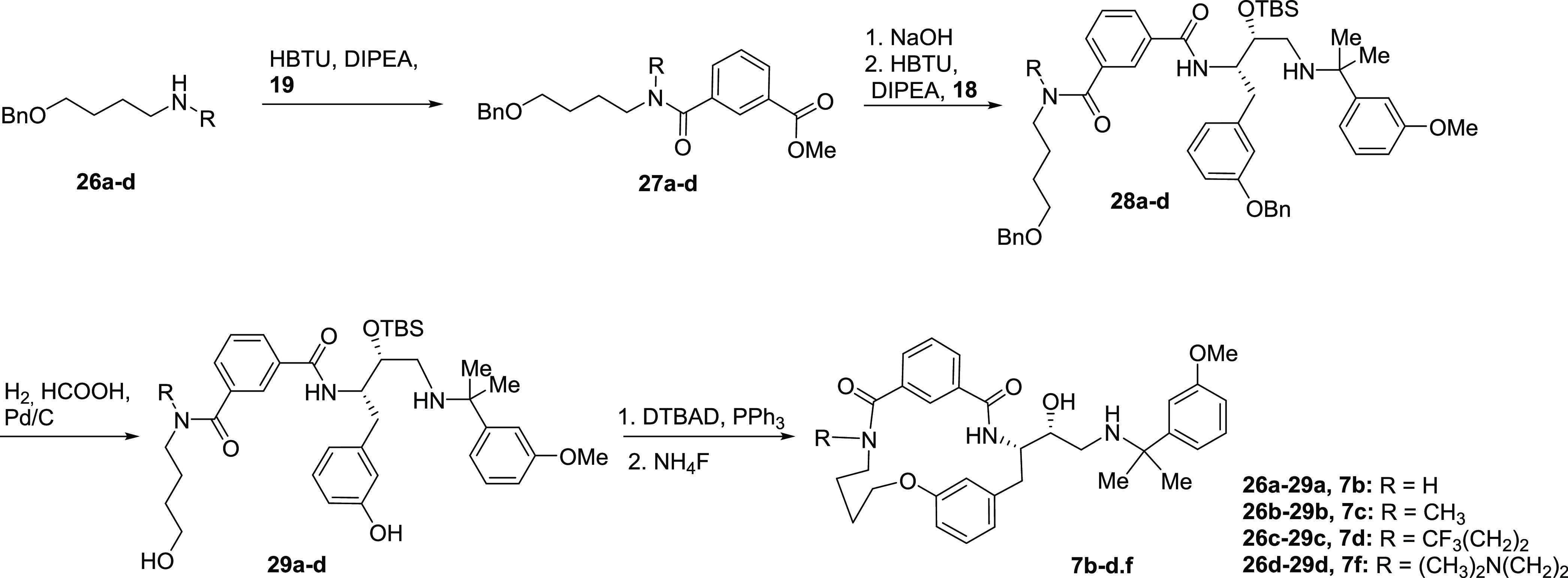
Target compound 7i was prepared from building block 16 (Scheme 4). This was O-debenzylated by hydrogenolysis to give an intermediate 30. The phenolic hydroxyl group in compound 30 was triflated, and the triflate 31 was subjected to coupling with a vinyltin reagent to give intermediate 32. Hydroboration–oxidation of the double bond in intermediate 32 provided alcohol 33, which was O-alkylated with alfa-bromo amide 34. The resulting amide 35 was reduced with borane to an intermediate amine, which was coupled to isophthalic acid monoester 19 to give amide 36. Hydrolysis of the ester group and the cleavage of the N-Boc in intermediate 36 was followed by macrocyclization to give target compound 7i.
Scheme 4. Synthesis of Inhibitor 7i.
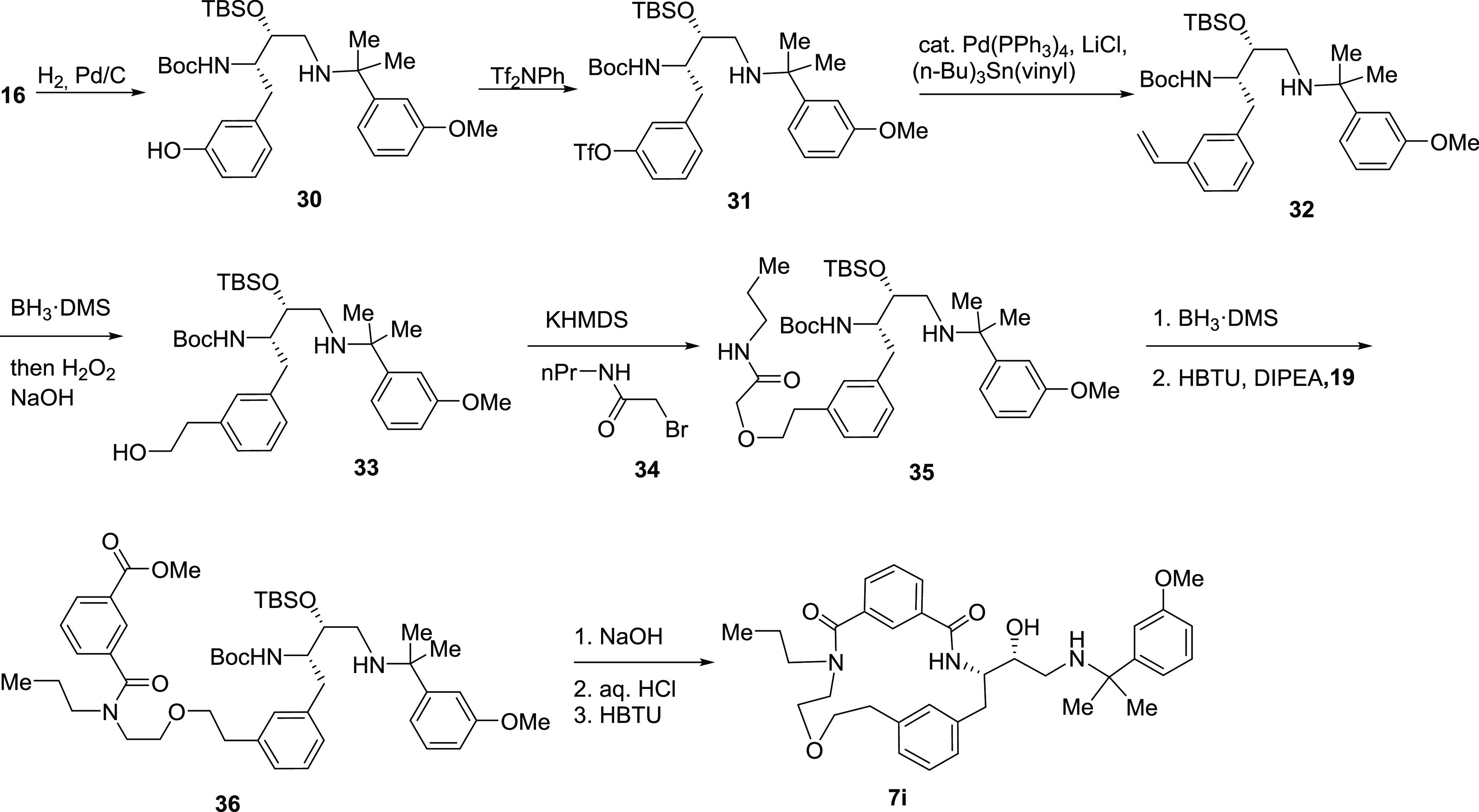
Target compounds 7j,k were prepared according to Scheme 5. Intermediate 32 was subjected to Boc-deprotection, and the resulting amine 37 was coupled to amines 38a,b. Ring-closing metathesis of intermediates 39a,b resulted in macrocyclic amides 40a,b. Hydrogenation of a double bond and desilylation provided target compounds 7j,k.
Scheme 5. Synthesis of Inhibitors 7j,k.
2.2. Plasmepsin Inhibitory Potency and Structure–Activity Relationship (SAR)
Macrocycles 7a–k were tested for their ability to inhibit recombinant P. falciparum PMX as well as the hemoglobinase PMIV in order to determine selectivity for these enzymes and to examine the association between parasite growth-inhibitory activity and inhibition of the enzymatic activity of the PM isoforms. Human aspartic proteases CatD and BACE1 were used for selectivity counter screens (Tables 1 and 2). Acyclic inhibitor 6a was previously shown to be a PMX inhibitor by its capacity to inhibit intracellular maturation of P. falciparum SUB1. Using recombinant PMX, the PMX inhibitory potency of compound 6a was confirmed in enzymatic assays to be in the low nanomolar range (Table 1, entry 1). Macrocyclization via the phenolic oxygen and carboxamide resulted in inhibitor 7a with very similar PMX and PMIV inhibitory potency but somewhat reduced selectivity against CatD. Exploration of substitution of the carboxamide nitrogen showed that hydrogen, methyl, and trifluoropropyl had little impact on PMX inhibitory potency, while PMIV inhibition was significantly reduced (compounds 7b–d) (Table 1, entries 2–4). Compound 7e bearing a more lipophilic pivaloyl substituent showed slightly improved PMX inhibitory potency and good selectivity against CatD and BACE1. Installation of a dimethylaminoethyl substituent as a solubilizing group at the carboxamide resulted in compound 7f with 4-fold reduced PMX inhibitory potency compared to analogue 7a but increased selectivity against PMIV, CatD, and BACE1.
Table 1. PMX, PMIV, CatD, and BACE1 Inhibition by Macrocycles 6a and 7a–f.

| # | cmpd. | R | PMX, nM | PMIV, nM | CatD, nM | BACE1, nM |
|---|---|---|---|---|---|---|
| 1 | 6a | - | 6.2 ± 0.3 | 58 ± 3a | 1150 ± 50a | n.d. |
| 2 | 7a | n-Pr | 4.1 ± 0.3 | 60 ± 3 | 270 ± 20 | 380 ± 20 |
| 3 | 7b | H | 11.2 ± 0.7 | 510 ± 30 | 650 ± 30 | 910 ± 40 |
| 4 | 7c | Me | 5.3 ± 3.0 | 170 ± 20 | 320 ± 20 | 1030 ± 50 |
| 5 | 7d | CF3(CH2)2 | 8.6 ± 2.4 | 61 ± 4 | 230 ± 20 | 2100 ± 100 |
| 6 | 7e | (CH3)3CCH2 | 2.5 ± 0.2 | 40 | 630 | 1900 ± 100 |
| 7 | 7f | (CH3)2NCH2CH2 | 16.7 ± 1.5 | 1730 ± 90 | 6800 ± 340 | 52400 ± 1600 |
Data from ref (22).
Table 2. PMX, PMIV, CatD, and BACE1 Inhibition by Macrocycles 7g–h.

| # | cmpd. | Y | X | PMX, nM | PMIV, nM | CatD, nM | BACE1, nM |
|---|---|---|---|---|---|---|---|
| 1 | 7g | –(CH2)5– | O | 6.9 ± 0.49 | 190 | 1200 | 6000 ± 300 |
| 2 | 7h | –(CH2)2– | O | 6.8 ± 0.59 | 200 | 600 | 1850 ± 90 |
| 3 | 7i | –(CH2)2–O–CH2– | CH2 | 4.6 ± 0.07 | 81±4 | 1650±80 | 22 700 ± 1100 |
| 4 | 7j | –(CH2)3– | CH2 | 3.8 ± 0.69 | 74±4 | 1370±70 | 3020 ± 150 |
| 5 | 7k | –(CH2)4– | CH2 | 0.4 ± 0.03 | 17±2 | 330±20 | 2030 ± 100 |
Installation of a longer or a shorter linker between amide and phenyl groups (compounds 7g and 7h; Table 2, entries 1,2) did not significantly alter PMX inhibitory potency but reduced the capability to inhibit PMIV, CatD, and BACE1. Shifting an oxygen atom position in the linker (compound 7i; Table 2, entry 3) or use of a shorter linker comprising only CH2 groups (compound 7j; Table 2, entry 4) also maintained PMX and PMIV inhibitory potency similar to the parent compound, but reduced potency against CatD and BACE1.
Surprisingly an analogue of macrocycle 7a containing only CH2 groups within the linker, compound 7k (Table 2, entry 5), showed 10-fold improved PMX inhibitory potency and 3-fold improved PMIV inhibitory potency, while inhibition of CatD and BACE1 was similar to the parent compound 7a. Collectively, these results showed that potent and selective macrocyclic PMX inhibitors could be derived through appropriate cyclization of compounds based on the linear peptidic hydroxyethylamine compounds 6 (Figure 2).
To gain insights into the structural features of 7a and 7k that might confer their differential PMX inhibitory potency, the crystal structure of P. falciparum PMX bound to the small inhibitor WM382 (compound 4) (PDB: 7TBC) was used for in silico docking of 7a and 7k in ICM-Pro (Molsoft31).
As a control for this operation, WM382 was first docked into PMX, providing an ICM-Pro score of −16 and a root-mean-square deviation (RMSD) value of 0.97 Å from the experimentally determined WM382 X-ray coordinates. Macrocycles 7a and 7k were found to dock in a similar manner in the active site of PMX (Figure 3) with a slightly better ICM-Pro score for 7k (−12) as compared to 7a (−8), consistent with the greater potency of 7k. Predicted hydrogen bonds and hydrophobic interactions appeared to stabilize both inhibitors in the PMX active site, with each of the macrocycle hydroxyl extensions in the vicinity of Asp457, one of the two catalytic aspartyl residues.
Figure 3.

Macrocycles 7a and 7k docked in the PMX active site. (A) Chemical structures of 7a and 7k. (B) The molecular surface of PMX (PDB: 7TBC) shown as a semi-transparent envelope, illustrating the size and shape of the enzyme active site pocket containing both 7a and 7k docked inhibitors. The inhibitors are shown as sticks, yellow for 7a (C: yellow, N: blue, O: red) and green for 7k (C: green, N: blue, O: red). The ICM-pro docking scores (dimensionless) are −8 for 7a and −12 for 7k, with an RMSD value of 3.3 Å between the poses of the two inhibitors. (C) Vicinity of the PMX (PDB: 7TBC) active site in cartoon representation with the inhibitors and side chain residues shown as sticks, colored and labeled according to their properties. The catalytic aspartic acid dyad (Asp266 and Asp457) is labeled in red. Docked inhibitors are shown as sticks, 7a in yellow (C: yellow, N: blue, O: red) and 7k in green (C: green, N: blue, O: red). Hydrogen bonds are represented by dashed lines, yellow for 7a and green for 7k.
2.3. Macrocyclic PM Inhibitors Display P. falciparum Growth-Inhibitory Activity In Vitro
The capacity of compounds 7a–k to inhibit P. falciparum asexual blood-stage proliferation in vitro was assessed using two different P. falciparum strains: the chloroquine-sensitive strain 3D7 and the chloroquine-resistant strain Dd2. As shown in Table 3, all of the compounds 7a–k displayed high growth-inhibitory potency against both P. falciparum strains. The correlation between in vitro parasite growth-inhibitory activity and PMX enzyme inhibitory activity was not perfect, suggesting some cell-permeability or stability issues. However, the most potent macrocyclic PMX inhibitor, 7k (Table 2, entry 5), also displayed the most potent growth-inhibitory activity. Notably, there was a generally poor correlation between parasite growth-inhibitory activity and inhibition of the hemoglobin degradative enzyme PMIV. This is consistent with previous evidence that inhibition of PMIV cannot be exploited for antimalarial drug discovery due to significant redundancy in the hemoglobin degradation pathway.13 Based on these results, it was tentatively concluded that parasite growth inhibition was likely primarily through the inhibition of PMX.
Table 3. P. falciparum Growth Inhibition In Vitro and Association with PM Inhibition.
| # | cmpd | flow cytometry 3D7a EC50 (nM) | flow cytometry Dd2b EC50 (nM) | PMX, nM | PMIV, nM |
|---|---|---|---|---|---|
| 1 | 7a | 1.0 ± 0.3 | 4.7 ± 0.5 | 6.2 ± 0.3 | 58 ± 3c |
| 2 | 7b | 6.4 ± 0.9 | 13.1 ± 3.1 | 4.1 ± 0.3 | 60 ± 3 |
| 3 | 7c | 2.6 ± 0.1 | 4.9 ± 0.9 | 11.2 ± 0.7 | 510 ± 30 |
| 4 | 7d | 6.2 ± 1.0 | 9.5 ± 1.4 | 5.3 ± 3.0 | 170 ± 20 |
| 5 | 7e | 2.2 ± 0.6 | 3.5 ± 0.4 | 8.6 ± 2.4 | 61 ± 4 |
| 6 | 7f | 6.5 ± 1.2 | 9.6 ± 0.7 | 2.5 ± 0.2 | 40 |
| 7 | 7g | 6.7 ± 2.3 | 9.6 ± 1.0 | 6.9 ± 0.49 | 190 |
| 8 | 7h | 0.8 ± 0.4 | 6.7 ± 1.0 | 6.8 ± 0.59 | 200 |
| 9 | 7i | 2.0 ± 0.3 | 2.0 ± 0.3 | 4.6 ± 0.07 | 81 ± 4 |
| 10 | 7j | 2.7 ± 0.5 | 4.4 ± 0.5 | 3.8 ± 0.69 | 74 ± 4 |
| 11 | 7k | 0.6 ± 0.1 | 0.9 ± 0.2 | 0.4 ± 0.03 | 17 ± 2 |
3D7: chloroquine-sensitive P. falciparum strain.
Dd2: chloroquine-resistant P. falciparum strain.
Data from ref (22).
2.4. P. falciparum Growth Inhibition by Macrocycles is through the Inhibition of PMX
To examine the mode of action of selected parasite growth-inhibitory macrocyclic inhibitors, highly synchronized P. falciparum cultures containing newly invaded ring-stage parasites were treated with 7a and 7k at fully growth-inhibitory concentrations (20 nM; at least 20 × EC50).
Microscopic monitoring of the treated cultures showed that even at these high compound concentrations, the parasites developed normally from rings to the multinucleated schizont stage in the presence of the compounds but then failed to rupture to allow merozoite egress and RBC invasion. This phenotype is characteristic of the inhibition of PMX. Furthermore, the lack of any effects on parasite development suggested the absence of significant off-target activity against other parasite pathways required for intraerythrocytic development. Consistent with this, Western blot analysis of schizonts isolated from cultures treated for 44 h as in Figure 4A with compounds 7a, 7j, and 7k showed a clear defect in maturation of the PMX substrate SUB1, with an accumulation of the SUB1 p54 intermediate species rather than the mature SUB1 p47 form that predominates in control parasites as a result of PMX activity (Figure 4B and C). At the concentration used, inhibitor 7k appeared to be more potent than compounds 7a and 7j in preventing SUB1 p54-to-p47 maturation, consistent with the lower IC50 and EC50 values for 7k in the PMX enzyme inhibition and parasite growth assays, respectively.
Figure 4.

Macrocycle PMX inhibitors have no effect on intraerythrocytic parasite maturation but prevent egress and maturation of SUB1. (A) Light microscopic images of Giemsa-stained P. falciparum 3D7 parasites allowed to mature for 44 h in the presence of vehicle only (dimethyl sulfoxide (DMSO), control) or macrocycle compounds 7a and 7k (20 nM). Development of ring-stage parasites to the multinucleated schizont stage occurred similarly in all cultures. However, while new ring-stage parasites arising from successful egress and invasion were beginning to become evident by this time point in control cultures (DMSO panel, white arrows), schizont rupture and appearance of new rings did not occur in the cultures containing the PMX inhibitors, even following extended further incubation. Scale bar, 20 μm. (B) Simplified schematic of proteolytic maturation of the egress effector SUB1. Conversion of the precursor form to p54 is through autocatalytic removal32 of the prodomain region (PD), whereas conversion of p54 to p47 is mediated by PMX.15,16,33 (C) Left, Western blot analysis of P. falciparum 3D7 schizonts allowed to mature in the presence of the indicated macrocycle compounds (20 nM), showing defective SUB1 maturation in parasites treated with the indicated macrocycle compounds. Right, Western blot of the same extracts probed with a monoclonal antibody (mAb) specific to the rhoptry protein RAP1, which is a substrate for cleavage by the related P. falciparum protease PMIX. The compounds had no discernible effect on the maturation of RAP1. The results shown are typical of 3 independent experiments.
We were unable to directly measure the effects of the compounds on PMIX activity due to recombinant enzymes not being available to us. However, none of the 3 macrocycles 7a, 7j, and 7k had any impact on the profile of maturation of the parasite protein RAP, which is a substrate for the related plasmepsin PMIX, suggesting that the compounds do not affect PMIX activity at these concentrations.
To glean further insights into the mode and duration of action of the most potent macrocycle compounds, we used washout experiments to examine the reversibility of the egress and PMX inhibition mediated by the compounds. To do this, parasites treated with compounds 7a, 7j, and 7k for the entire ∼44 h period of intraerythrocytic maturation, as in Figure 4A, were washed extensively to remove the drugs and then either immediately analyzed by Western blot or returned to culture, with or without the presence of added RBCs. The schizont cultures lacking added RBCs were supplemented with compound C2, which blocks the discharge of exonemes, the parasite secretory organelles in which PMX and SUB1 are stored before egress. The schizonts without added RBCs were sampled for Western blot analysis, while the schizont cultures containing RBCs were examined by microscopy and flow cytometry after a further 4 h in culture to quantify schizont rupture and new ring formation. As shown in Figure 5, the washout of compounds 7a and 7j in the presence of C2 resulted in subsequent efficient conversion of the accumulated SUB1 p54 intermediate to the p47 form, indicating reversal of the PMX inhibition mediated by these compounds. This presumably occurred in the exonemes. In contrast, no conversion of SUB1 p54 to p47 occurred following the washout of the most potent macrocycle 7k. Remarkably, new ring formation from the washed schizonts was completely consistent with this pattern; while the washout of schizonts pretreated with 7a and 7j allowed an efficient generation of new rings, minimal rings were produced from schizonts following the washout of 7k. These findings indicate effectively irreversible inhibition of SUB1 maturation and egress by compound 7k over the time period examined, presumably due to continued blockade of PMX activity within the exonemes following washout. This could be due to a relatively long residence time of engagement of 7k with its intracellular PMX target and/or inefficient washout of 7k due to intracellular “trapping” of the compound.
Figure 5.

Washout experiments indicate low reversibility of PMX and egress inhibition mediated by macrocycle 7k. (A) Washout of the macrocycle inhibitors allows restoration of SUB1 maturation in the case of compounds 7a and 7j but not in the case of 7k. (B) Washout of the macrocycle inhibitors allows restoration of parasite egress (schizont rupture) and new ring formation in the case of compounds 7a and 7j but not in the case of 7k. The plot shows both new ring formation and levels of residual unruptured schizonts after 3.5 h of further culture following washout.
2.5. Macrocyclization Leads to Improved Metabolic Stability
The primary motivation underlying our development of macrocyclic analogues of parent PMX inhibitor 6a was to improve its drug-like properties. The metabolic stability was the most vulnerable point as inhibitor 6a displayed a short half-life in the mice microsomal assay (Table 4, entry 1). Macrocyclic analogue 7a showed slightly improved metabolic stability (Table 4, entry 2). N-Substitution at the amide group in compounds 7b–f produced notable effects on metabolic stability in vitro. Removal of the n-propyl group improved stability (compound 7b, Table 4, entry 3). Substitution with N-methyl or N-trifluoropropyl (compounds 7c,d, Table 4, entries 4,5) increased the stability compared to compounds 6a and 7a, while the N-pivaloyl-substituted compound 7e was only slightly better (Table 4, entry 6). A dimethylaminoethyl substituent (compound 7f, Table 4, entry 7) dramatically improved the microsomal stability, which could be attributed to the charged nature of compound 7f preventing its entry into microsomes. The length of the linker, shifting the oxygen in the linker by one position, or replacement of the oxygen with a methylene group had little effect on microsomal stability (compounds 7g–k, Table 4, entries 8–12). Compared to parent compound 7a, the macrocycle with a linker comprising only methylene groups (compound 7k) showed increased stability (Table 4, entry 12).
Table 4. Microsomal Stability and Plasma Protein Binding.
| microsomal
stabilitya |
plasma protein binding, % | |||
|---|---|---|---|---|
| # | cmpd. | half-life, min | CL(int), μL/min/mg | |
| 1 | 6a | 6.40 | 216.40 | 99.1 |
| 2 | 7a | 8.67 | 159.91 | 97.1 |
| 3 | 7b | 31.27 | 44.32 | 97.26 |
| 4 | 7c | 16.93 | 81.87 | 96.49 |
| 5 | 7d | 20.41 | 67.91 | 98.75 |
| 6 | 7e | 13.73 | 100.93 | 99.65 |
| 7 | 7f | >120 | ND | 93.05 |
| 8 | 7g | 7.64 | 181.47 | 99.21 |
| 9 | 7h | 13.89 | 99.78 | 96.16 |
| 10 | 7i | 8.39 | 165.20 | 97.13 |
| 11 | 7j | 14.37 | 96.43 | 99.33 |
| 12 | 7k | 16.92 | 81.90 | 99.73 |
Fluconazole (negative control) and propranolol (positive control).
Plasma protein binding by the parent compound 6a was very high (Table 4, entry 1). Macrocyclic analogue 7a showed reduced plasma protein binding (Table 4, entry 2), and modifications of the inhibitors leading to compounds 7c, 7f, and 7h further reduced plasma protein binding (Table 4, entries 4,7,9). The most potent PMX and parasite growth-inhibitory inhibitor 7k showed very high plasma protein binding, comparable to the parent compound 6a.
2.6. In Vivo PK-Tox and In Vivo Antimalarial Potency of Inhibitor 7k
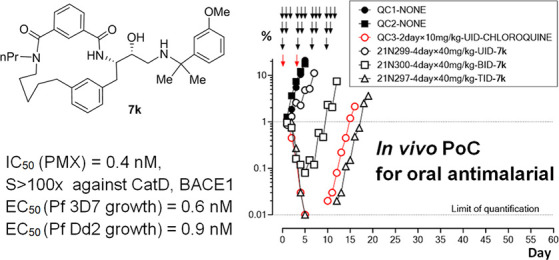
Compound 7k was well tolerated following oral administration of a dose of 50 mg/kg. No adverse effects were observed during a 24 h observation period (Table 5). The same dose was used in the pharmacokinetics (PK) experiment. Relative to the administered dose, low exposure was observed with a maximal concentration of compound 7k (Cmax) of just 0.064 μg/mL (Figure 6). However, since this concentration exceeds concentrations that showed activity in vitro, antiparasitic activity in vivo was expected. Compound 7k was rapidly cleared from the circulation (t1/2 = 53 min); thus, it was considered likely that multiple administrations per day might be required during efficacy testing in vivo.
Table 5. In Vivo PK-Tox Properties of Inhibitor 7k.
| assay | result |
|---|---|
| tolerability p.os 50 mg/kg up to 24 h | no toxic effect (score 0) |
| PK p.os 50 mg/kg, data points 30, 60, 120, 240, 360 minb NMRI female mice 10 weeks old ENVIGO, formulation: suspension, stabilized with HMC, 0.5% | t1/2 = 53 min |
| Tmax = 60 min | |
| Cmax = 0.064 μg/mL | |
| AUC0–t = 13.1 μg/mL*min | |
| AUC0-inf_obs = 13.3 μg/mL*min |
Figure 6.

Plasma concentrations (±SEM) of inhibitor 7k after p.o. administration in mice at a dose of 50 mg/kg.
Inhibitor 7k was selected to execute a proof-of-concept (PoC) of the efficacy of the chemical family against asexual blood stages of P. falciparumin vivo in a standardized P. falciparum humanized mouse model (the PfalcHuMouse model). Given the low exposure expected for compound 7k in PfalcHuMice, three dose regimens (QD, BID, and TID) were tested at a well-tolerated dose to maximize the time above a threshold efficacious concentration in vivo. As shown in Figure 7, inhibitor 7k cleared parasitemia from the peripheral blood in a dose-dependent manner when administered orally. Importantly, the data showed that inhibitor 7k mediated parasite clearance rates comparable to those produced by chloroquine at 10 mg/kg for 2 days in the same experimental system (Figure 7A). Flow cytometry and microscopy analysis showed a relative accumulation of circulating mature schizonts compared to untreated controls on day 3 of the assay (Figure 7B) after two cycles of drug exposure. This suggested that the schizont rupture and reinfection of RBCs were the most sensitive stages of the parasite to inhibitor 7k. Pharmacokinetic modeling of the concentrations of inhibitor 7k over the entire period of administration based on measurements of drug plasma concentration at 71.0, 72.5, 73.0, and 78.0 hours post-treatment suggested a rapid absorption with no accumulation (Figure 7C). Interestingly, inhibitor 7k was shown to be as potent as chloroquine killing P. falciparumin vivo in the PfalchHuMouse model in comparable treatment regimens (ref Demarta-Gatsi et al. AAC. DOI: 10.1128/aac.01574-22) (Figure 7D).34 The total Cmin in the individuals treated with 7k were 51, 32, and 85 ng/mL for the individuals treated with UID, BID, and TID, respectively. A preliminary first estimate of the minimal parasiticidal concentration (MPC) of 85.3 ng/mL could be calculated since the individual treated BID showed the maximum parasite clearance rate for the first 48 h.
Figure 7.

Therapeutic efficacy of inhibitor 7k against P. falciparumin vivo. (a) Parasitemia in peripheral blood of PfalcHuMice: untreated (QC1 and QC2), treated with chloroquine, or treated with inhibitor 7k at 40 mg/kg, p.o., UID, BID, or TID. (b) Distribution of stages of P. falciparum in peripheral blood of untreated and 7k-treated PfalcHuMice after one parasite cycle of drug exposure (day 3 of the assay). The figure shows flow cytometry plots of human erythrocytes (not stained with the anti-mouse erythrocyte mAb TER-119 conjugated with phycoerythrine) stained with the nucleic acid dye SYTO-16. The red rectangles inside flow cytometry plots indicate the area of viable parasites. The photographs show Giemsa-stained blood smears of the very same cytometry plots representative of the parasite cells found in the corresponding regions of viability. (c) Pharmacokinetic modeling of the concentrations of inhibitor 7k during the in vivo testing. Experimental data are represented by open symbols. The red lines indicate the preliminary estimate of minimal parasiticidal concentration (MPC) calculated from the experimental results. (d) PK/PD analysis of parasite killing in vivo induced by inhibitor 7k in comparison with chloroquine as a standard reference antimalarial drug. The data shown in the plot are the day of recrudescence (DoR) versus the total exposure in the blood of 7k of each individual mouse normalized by their respective individual parasite burdens as described.34 Data for Chloroquine are from historical data available at TAD for a set of PfalcHuMice treated p.o. UID, for 4 days, with different dose levels of the drug. In this plot, mapping to similar areas of the plot indicates equipotency for parasite killing in the PfalcHuMouse model.
The PoC study allowed a preliminary PK/PD assessment of the antimalarial effect shown by 7k in the PfalcHuMouse model (Figure 7D). We addressed this assessment by analyzing the day of recrudescence (DoR) as a function of the total exposure in the blood of the drugs tested, as recently described.34 DoR is defined as the the day at which parasitemia reaches again the parasitemia at treatment inception after a transient decline in parasitemia and is used as a measurement of parasite killing induced by the drugs. The amount of parasite killing (i.e., DoR) in one individual was correlated to the total exposure of the drug (7k and chloroquine as a comparator) in blood in the very same individual. In this study, the data of mice for which a DoR could be defined (mice treated with BID and TID 7k at 40 mg/kg) were compared to historical data of Chloroquine, evaluated in the same experimental system and with the same methodology, available at The Art of Discovery.
Clearance of parasitemia in vivo by compound 7k necessitated repeated administration of the compound due to its relatively short plasma lifetime (t1/2 = 53 min, Table 5), which could be attributed to low microsomal stability (Table 4, entry 12). For comparison, compound 7f, which displays relatively high microsomal stability (Table 4, entry 7), was also investigated for antimalarial efficacy in vivo in the mouse model. However, treatment with compound 7f three times a day for 4 days at 40 mg/kg (Supporting information, Figure S1) had only a weak effect on parasite burden.
Conclusions
A series of potent, subnanomolar macrocyclic peptidic PMX inhibitors 7 has been developed that hold potential for development as a new class of antimalarial drugs that interrupt the malaria life cycle through a hitherto unexploited mode of action. Macrocyclic compounds were designed based on their acyclic analogues originating from a phenotypic screening hit by linking the S1 subpocket occupying the phenyl group with an amide residing in the S3 subpocket. Modification of the linker type and length led to compound 7k with a 10-fold improved PMX potency. Compound 7k showed low nanomolar potency also as an inhibitor of the hemoglobin degradative enzyme PMIV, but >100× selectivity against the human aspartic proteases CatD and BACE1. Compounds 7a,j,k did not have an impact on the profile of maturation of the parasite protein RAP1, which is a substrate for the related plasmepsin PMIX, suggesting that the compounds do not affect PMIX activity. All of the compounds 7a–k inhibited in vitroP. falciparum growth (3D7 and Dd2 strains), with compound 7k being the most potent, consistent with the PMX inhibition data. Notably, there was a poor correlation between parasite growth-inhibitory activity and inhibition of the hemoglobin degradative enzyme PMIV. Macrocyclic inhibitor 7k showed a 3-fold improved half-life in the microsomal stability test compared to the acyclic analogue 6a. Another way to improve the microsomal stability was the installation of a dimethylaminoethyl group at the S3 amide substructure (compound 7f). Both compounds 7f and 7k were investigated for their ability to eliminate P. falciparum from an infected humanized mice model. The less potent PMX inhibitor 7f produced a measurable reduction in parasitemia when orally administered 3 times a day (40 mg/kg) for 4 days (see the Supporting Information). However, compound 7k, when administered 3 times a day (40 mg/kg) for 4 days, eliminated P. falciparum from peripheral blood at a rate comparable to chloroquine single-dose administration for 2 days (10 mg/kg). The need for repeated administration was attributed to the relatively rapid clearance of compound 7k (t1/2 = 53 min) as determined by PK assay in mice. In summary, we have developed an antimalarial lead compound 7k acting as a PMX inhibitor, which demonstrates proof of principle for an orally administered drug in a P. falciparum infection model in vivo. Further development of analogues with a simplified route to synthesis and improved PK properties is now needed in order to achieve a next-generation lead suitable for a single-dose antimalarial treatment regimen.
Experimental Section
Antibodies and Reagents
The generation of polyclonal rabbit antibodies to P. falciparum SUB1 has been described previously.35 The RAP1-specific mouse mAb 2.2936 was a kind gift from the European Malaria Reagent Repository (www.malariaresearch.eu). The P. falciparum cGMP-dependent protein kinase (PKG) inhibitor C237,38 was a kind gift from Simon Osborne, LifeArc.
Maintenance of P. falciparumIn Vitro and Growth Assays
Asexual blood-stage forms of P. falciparum strains 3D7 and Dd2 were maintained at 37 °C in an atmosphere of 1% O2, 5% CO2, and 94% N2 in human RBCs in RPMI 1640 medium containing AlbuMAX II (Thermo Fisher Scientific) and supplemented with 2 mM l-glutamine. The synchronization of parasite cultures was as described previously.39 Briefly, this involved isolating mature schizont forms by centrifugation over cushions of 70% (v/v) isotonic Percoll (GE Healthcare, Life Sciences) and then allowing the parasites to undergo rupture in the presence of fresh RBCs for 2 h under gentle shaking conditions (100 rpm). Residual schizonts were then removed through a second Percoll separation, followed by treatment with 5% (w/v) sorbitol to finally obtain a highly synchronized preparation of newly invaded ring-stage parasites.
Growth assays were used to assess the capacity of the macrocycles to interfere with parasite proliferation in vitro. For this, synchronous cultures of ring-stage parasites (0.1% parasitemia, 2% hematocrit) were dispensed in duplicate into flat-bottomed 96-well plates (100 μL per well) containing 1 μL of test compounds serially diluted into DMSO such that final concentrations of the compounds ranged from 5 μM to 0.01 nM. Following culture for 96 h (a total of 2 erythrocytic cycles), samples from each well were fixed with an equal volume of 0.2% glutaraldehyde in phosphate-buffered saline (PBS) and stored at 4 °C until analysis by flow cytometry. Fixed parasites were stained with SYBR Green (Thermo Fisher Scientific, 1:10,000 dilution) for 20 min at 37 °C and then analyzed by flow cytometry on a BD FACSVerse using BD FACSuite software. Parasitemia was quantified by recording 10,000 events (cells) per well and filtering with appropriate forward and side scatter parameters and gating for SYBR Green stain-positive (infected RBCs) and negative RBCs using a 527/32 detector configuration. All data were analyzed using FlowJo software, and EC50 values were calculated in GraphPad Prism 8.0 using nonlinear regression, variable slope (four parameters). Growth stage progression and parasite morphology were also monitored by microscopic examination at selected time points using Giemsa-stained thin blood films.
Washout Assays
Synchronous parasite cultures were cultured from ring to schizont stage (∼44 h) in the presence of DMSO alone (0.04% v/v; control) or macrocycle compounds (20 nM final concentration). Schizonts were isolated by Percoll enrichment (still in the presence of the test compound), then either immediately frozen or further incubated in the absence of the drug but in the presence of C2 (1 μM) for 1 h at 37°C in the absence of added RBCs or for 3.5 h in the presence of added RBCs. The schizonts without added RBCs were then frozen too, and all schizont samples were then thawed into CHAPS–urea extraction buffer, subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and analyzed by Western blot, probing with antibodies against SUB1 or RAP1. For schizonts cultured in the presence of added RBCs, ring formation was assessed in two ways: cultures were examined microscopically by Giemsa stain of thin films and were also fixed with glutaraldehyde and processed for quantification of ring formation by flow cytometry as described above.
Enzyme Bioassays
A fluorescence resonance energy transfer (FRET)-based assay was performed to assess the ability of the compounds to inhibit PMIV, CatD, and BACE1. Serial dilutions of test compounds were dispensed in a white 96-well plate. Enzymes were then added: PMIV or CatD in 0.1 M NaOAc buffer, pH 4.5, 10% glycerol, 0.01% Tween 20, at final concentrations of 0.5 and 1.5 nM, respectively, or BACE1 was added to 0.1 M NaOAc buffer, pH 4.5, 0.01% Triton X-100 at a final concentration of 10 nM. The mixture was incubated for 20 min at 37 °C. PMIV and CatD substrate: DABCYL-ERNleFLSFP-EDANS (AnaSpec Inc.), a final concentration of 5 μM or BACE1 substrate: RE-EDANS-EVNLDAEFK-DABCYL-R (Calbiochem), a final concentration of 10 μM were then added. Substrate hydrolysis was detected as an increase in fluorescence at Em 490 nm, Ex 336 nm, 37 °C for 20 min and 60 min with BACE1. IC50 values were calculated with GraphPad Prism 8.0 using the nonlinear regression, variable slope (four parameters). Compounds were tested in triplicate.
Recombinant P. falciparum PMX Inhibition Assays: Kinetics and IC50 Calculations
The proteolytic activity of PMX was quantified at room temperature by continuously monitoring the cleavage of the fluorogenic peptide substrate Rh2N (DABCYL-HSFIQEGKEE-EDANS) (Ex. 340, Em. 492). The fluorescence of this peptide is quenched by the spatial proximity of the DABCYL/EDANS groups in the uncleaved peptide and is increased upon cleavage at the internal F–I bond. The enzymatic reaction was performed in white Nunc 96-well microtiter plates by adding the following to each well: 50 μL of rPMX at a 0.72 nM intermediate dilution in 25 mM sodium acetate (pH 5.5), 0.005% Tween 20 digestion buffer (DB), and 1 μL of the inhibitor from a serial dilution ranging from 5 to 0.005 μM in 100% anhydrous DMSO. After an incubation time of 5 min to allow interaction of the inhibitors with PMX, 50 μL of the fluorogenic substrate Rh2N (3.2 μM intermediate dilution in DB) was added to the wells. Control wells contained no inhibitor, and the plate was blanked against the substrate only in DB. Subsequent fluorescence increase was monitored using a SpectraMax M5e plate reader and SoftMax Pro 7.1.0 software, with readings taken every 3 min for 60 min and using excitation and emission values of 340 and 492 nm, respectively. Initial rates were calculated over the first 28 min of the assay, during which period progress curves were linear, and IC50 values were calculated with GraphPad Prism 9.5.0 using the nonlinear regression, [inhibitor] vs response, variable slope (four parameters). All experiments were performed in duplicate.
Docking of 7a and 7k into the PMX Crystallographic Structure
Flexible noncovalent docking of the macrocyclic compounds 7a and 7k was performed using ICM-Pro software (version 3.9-2e/MacOSX, Molsoft LLC). The inhibitors were drawn within the ICM molecule editor, converted to 3D, and appended to a chemical table for docking in batch. The WM382-inhibited X-ray structure (PDB ID: 7TBC) was converted to an ICM object for the docking procedure. In this process, hydrogen atoms were added to the PMX model, side chains of His, Pro, Asn, Gln, and Cys residues were optimized in an energetically favorable protonation state, and the WM382 inhibitor was used to define the PMX active site pocket. The WM382 inhibitor was then moved away from the receptor along with all water molecules. The potential energy maps of the PMX active site pocket and docking preferences were set up using the program default parameters. Energy terms were based on the all-atom vacuum force-field ECEPP/3, and conformational sampling was based on the biased probability Monte Carlo (BPMC) procedure.40 Two independent docking runs were performed per ligand, with a length of simulation (thoroughness) varying from 3 to 5 and the selection of 2 docking poses. Ligands were ranked according to their ICM energetics (ICM score, unitless), which weighs the internal force-field energy of the ligand combined with other ligand–receptor energy parameters.
Liver Microsomal Metabolic Stability
Microsomal stability assays are performed as described elsewhere.41 Test compounds and pooled mouse liver microsomes are incubated in potassium phosphate buffer in the presence of the cofactor NADPH at 37 °C. Reaction mixtures (700 μL) contain a final concentration of 5 μM test compound, 0.05% DMSO, 0.5 mg/mL microsomes, and ∼0.7–0.9 mM NADPH in 100 mM potassium phosphate buffer at pH 7.4. Reactions are terminated by the addition of 100 μL of the reaction mixture to 900 μL of acetonitrile after 0, 5, 15, 30, 45, and 60 min of incubation. Samples were centrifuged, and the resultant supernatant was analyzed by ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS). The amount of remaining compound is calculated and compared to the initial amount of test compound (zero time point). Verapamil is used as a positive control, and samples prepared without cofactor NADPH are used as a negative control (0 and 60 min). The in vitro metabolic stability parameters (half-life value (t1/2) and intrinsic clearance value (Clint)) are calculated from the slope of the Ln of concentration remaining vs the time curve.
Plasma Protein Binding
Plasma protein binding was performed with rapid equilibrium dialysis (RED) device inserts containing a dialysis membrane with a molecular weight cut-off of 8000 Daltons. Inserts were placed in the single-use RED plate without prior preparation.
The test compound at a concentration of 5 μM was added to mouse plasma and dialyzed against phosphate-buffered saline (PBS, pH 7.4) for 2 h at 37 °C. Propranolol hydrochloride was used as a positive control (high PPB), and fluconazole was used as negative control (low PPB). After dialysis, the drug concentration in the buffer and plasma compartments was quantified by liquid chromatography and tandem mass spectrometry (LC-MS/MS) analysis.
Tolerability and Pharmacokinetics Testing
NMRI female mice (10–12 weeks old, 29–32 g) were obtained from ENVIGO (The Netherlands) and housed prior to treatment under standard conditions (acclimatization period of 1 week, 21–23 °C, 12-h light/dark cycle, relative humidity 45–65%) with unlimited access to food (R70 diet from Lantmännen) and water. The experimental procedures were performed in accordance with the guidelines of the European Community and local laws and policies (Directive 2010/63/EU), and all of the procedures were approved by Food and Veterinary Service, Riga, Latvia. Animals were weighed on the day of the treatment (before treatment) to calculate the required amount of compound for the corresponding dose. For tolerability and PK studies, the mice were dosed via a peroral (PO) bolus administration (volume 10 mL/kg). Toxicity signs were scored 0–2, 5, 10, 15, 30, 60, 120, 240 min, and 24 h after administration using a widely used 0–6 score system.
For the pharmacokinetics experiment, blood samples were collected in tubes containing heparin. Blood was sampled from the tail vein 30, 60, 120, 240, and 360 min after the first and last administrations of the compounds. Tubes were centrifuged at +4 °C 10,000g for 3 min. Plasma samples were collected and stored at −20 °C until analysis.
Quantitative Determination of VAD259 in Mouse Blood Plasma by UPLC/MS/MS
Concentrations of VAD259 in mouse plasma were measured by ultra-performance liquid chromatography–tandem mass spectrometry using Waters Acquity UPLC H-class chromatograph coupled to the Waters Xevo TQ-S micro mass spectrometer using the Waters Acquity UPLC BEH-C18 column (2.1 mm × 50 mm, 1.7 μm). Mobile phase A was 0.1% aqueous formic acid, and mobile phase B was acetonitrile. The solvent was pumped in gradient mode from 5% B to 98% B, the flow rate was 0.4 mL/min, and the method total run time was 5 min.
The mass spectrometer was operated in positive electrospray ionization mode (ESI+), and data acquisition was performed in multiple reaction monitoring (MRM) modes with ionization parameters as follows: capillary energy of 3.0 kV, cone voltage of 25 V, and collision energy of 20 eV. Specific MRM transition m/z 586.5 to m/z 438.3 was used for VAD259 quantification.
Blood plasma samples were processed using plasma protein precipitation. Then, 20 μL of plasma was mixed with 300 μL of acetonitrile/methanol (3:1/v:v) mixture, and the sample was centrifuged at 1000g for 10 min. The supernatant was further diluted 3 times with 0.1% aqueous formic acid and subjected to UPLC/MS/MS analysis.
In Vivo Efficacy against P. falciparum in the TAD PfalcHuMouse Model
Therapeutic efficacy studies were performed at TAD using a standardized humanized mouse model of P. falciparum malaria31 with modifications.34 Briefly, 22–28 g female NOD-SCID IL-2Rγnull mice (NSG) (Charles River, France) were engrafted with human erythrocytes (hE) (Basque Center of Transfusion and Human Tissues, Galdakao, Spain, Centro de Transfusiones de la Comunidad de Castilla y León, Valladolid, Spain and Bank of Blood and Tissues, Barcelona, Spain) by intraperitoneal (i.p.) and/or intravenous (i.v., via tail lateral vein) daily inoculation of 50–75% hematocrit cell suspensions in RPMI 1640 medium, 25% (vol/vol) decomplemented human serum, and 3.1 mM hypoxanthine. The volume of injections was 1 and 0.7 mL for i.p. or i.v. inoculation, respectively.
When engrafted mice (i.e., HuMice) had more than 40% of circulating hE in peripheral blood, HuMice were infected with P. falciparum Pf3D70087/N9 by inoculation of 0.3 mL of an erythrocyte suspension containing 1.17 × 108 erythrocytes parasitized per mL via the lateral vein. The inoculum was obtained from peripheral blood of CO2-euthanized donor mice harboring 5–10% parasitemia.
Before drug administration, each infected mouse was randomly assigned to its corresponding treatment. Drug treatment started at ∼1.3% patent parasitemia (Day 1). The treatment was administered by oral gavage with 20G straight reusable feeding needles (Fine Science Tools GmbH) at 10 mL·kgbodyweight–1 unless otherwise stated.
Parasitemia was measured in serial 2 μL of samples of tail blood by flow cytometry and expressed as the % of parasitized erythrocytes with respect to the total erythrocytes in circulation. A qualitative analysis of the effect of treatment on P. falciparum Pf3D70087/N9 was assessed by microscopy with Giemsa-stained blood smears and flow cytometry by staining with TER-119-Phycoerythrine (a marker of murine erythrocytes) and SYTO-16 (nucleic acid dye) and acquisition in an Attune NxT Acoustic Focusing Flow Cytometer (Invitrogen), as previously described.42
The concentrations of drugs were measured in samples of peripheral blood (25 μL) taken at different times after the first dosing, mixed with 25 μL of Milli-Q H2O, and immediately frozen on a thermal block at −80 °C. The frozen samples were stored at −80 °C until analysis. Blood from control humanized mice was used for the preparation of standard curves, calibration, and quality control purposes. The drugs were extracted from 10 μL of lysates obtained by protein precipitation of blood samples using standard liquid–liquid extraction methods. The samples were analyzed by LC-MS/MS for quantification in a Waters Micromass UPLC-TQD (Waters, Manchester, U.K.). Blood concentration vs time was analyzed by non-compartmental analysis (NCA) using Phoenix WinNonlin vers.9.2 (Certara) or R or Excel (Microsoft), from which exposure-related values (tmax, Cmax, and AUC0–t) were estimated.
The clearance of parasitized erythrocytes from the peripheral blood of mice was assessed by measuring the parasite reduction ratio (PRR) calculated as the ratio between parasitemia at Day n + 2 divided by parasitemia at Day n for each individual of the study.
The parasiticidal effect of drugs in vivo was assessed by measuring the day of recrudescence (DoR) as described.34 In short, DoR was defined as the day at which parasitemia after drug treatment reached the % parasitemia at treatment inception. Drug-treated mice were deemed cured if there was no detectable parasitemia 60 days after treatment.
Ethical approvals
The studies were approved by The Art of Discovery Institutional Animal Care and Use Committee (TAD-IACUC), certified by the Biscay County Government (Bizkaiko Foru Aldundia, Basque Country, Spain) to evaluate animal research projects from Spanish institutions according to point 43.3 from Royal Decree 53/2013, from the 1st of February (BOE-A-2013–1337). All experiments were carried out in accordance with European Directive 2010/63/EU. The results from the animal experiments were reported following ARRIVE guidelines (https://www.nc3rs.org.uk/arrive-guidelines), except for disclosure of business trade confidential information. The human biological samples were sourced ethically, and their research use was in accord with the terms of the informed consent.
Chemistry
General
All chemicals were used as obtained from commercial sources, and all reactions were performed under an argon atmosphere in oven-dried (120 °C) glassware unless noted otherwise. Anhydrous toluene, Et2O, tetrahydrofuran (THF), and CH2Cl2 were obtained by passing commercially available anhydrous solvents through activated alumina columns.
NMR spectra were recorded on 300 and 400 MHz spectrometers with chemical shift values (δ) in ppm using the residual solvent signal as an internal standard. UPLC/MS analysis was performed on a Waters Acquity column: Acquity UPLC BEH-C18 (2.1 mm × 50 mm, 1.7 μm, (30.0 ± 5.0) °C); gradient, 0.01% TFA in water/CH3CN 90/10% to 10/90%; flow rate, 0.5 mL/min; run time, 8 min; detector, PDA (photodiode matrix), 220–320 nm, SQ detector with an electrospray ion source (APCI). Analytical thin-layer chromatography (TLC) was performed on precoated silica gel F-254 plates. High-resolution mass spectra (HRMS) were recorded on a Waters Synapt G2-Si TOF MS instrument using the ESI technique. Specific rotation was recorded on a Kruss P3000 instrument. Purification by preparative reverse phase chromatography was performed using Waters Atlantis Prep OBD T3 Column 30 mm × 100 mm, 5 μm. The purity of all target inhibitors was confirmed to be ≥95% by the reversed-phase high-performance liquid chromatography (HPLC) assay.
General Procedure A for Boc-Deprotection of Amines
To a solution of Boc-protected amine (1 equiv) in anhydrous dichloromethane (DCM) (1.5 mL/1 mmol of the substrate), trifluoroacetic acid (20 equiv) was added, and the reaction mixture was stirred at room temperature for 1 h. All volatiles were removed under reduced pressure. Then, 1 M aqueous NaOH solution (20 mL) was added to the residue. The mixture was extracted with diethyl ether (3 × 20 mL). The combined organic phase was washed with brine, dried over anhydrous Na2SO4, and evaporated under reduced pressure.
General Procedure B for Silylation of the Secondary Hydroxyl Group
To a stirred solution of alcohol (1 equiv) in N,N-dimethylformamide (DMF) (8.5 mL/1 mmol of alcohol), imidazole (5–10 equiv) and TBSCl (5–10 equiv) were added. The reaction mixture was stirred at 80 °C overnight. DMF was evaporated under reduced pressure. The residue was dissolved in EtOAc (20 mL) and water (20 mL). The aqueous phase was separated and washed with EtOAc (2 × 20 mL). The combined organic phase was washed with brine (20 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure.
General Procedure C for the Intermolecular Mitsunobu Reaction and Denosylation of the Amino Group
To a solution of phenol (1 equiv) and aliphatic alcohol (3–5 equiv) in anhydrous DCM (3.7 mL/0.1 mmol of the phenol), PPh3 (3–5 equiv) was added, followed by either diethyl azodicarboxylate (DEAD) or di-tert-butyl azodicarboxylate (DTBAD) (3–4 equiv). The reaction mixture was stirred at room temperature for 2 h. The reaction mixture was evaporated, and the residue was purified by silica gel column chromatography using diethyl ether. Fractions with product were combined and evaporated under reduced pressure. The residue was used in the next step without further purification.
To a solution of protected amine from the previous step in MeCN (3 mL/0.1 mmol of the amine), K2CO3 (4–10 equiv) was added, followed by thiophenol (2–5 equiv). The reaction mixture was stirred at room temperature for 2 h. The reaction mixture was diluted with DCM (20 mL) and washed with water (20 mL) and 0.1 M NaOH solution (20 mL). The aqueous phase was extracted with DCM (2 × 25 mL). The combined organic phase was washed with brine, dried over anhydrous Na2SO4, and evaporated. A pure material was obtained by column chromatography using gradient elution from DCM to 5% MeOH in DCM.
General Procedure D for Hydrolysis of Methyl Benzoates
One molar aqueous NaOH solution (2–6 equiv) was added to a solution of benzoate (1.0 equiv) in THF (2.5 mL/0.1 mmol of the benzoate). The resulting solution was stirred until the conversion of the starting material was complete. The solvent was evaporated under reduced pressure.
General Procedure E for Macrolactamization
To a solution of deprotected amino acid (1 equiv) in anhydrous DMF (1 mL/0.01 mmol of the amino acid), HBTU (1.5 equiv) was added. The reaction mixture was stirred at room temperature for 1 h. DMF was evaporated under reduced pressure. The residue was suspended in DCM and filtered through a small silica column, using gradient elution from DCM to 10% MeOH in DCM.
General Procedure F for the Desilylation of Secondary Alcohols
To a solution of silylated macrocycle (1 equiv) in MeOH (3 mL/0.01 mmol of the macrocycle), NH4F (20–30 equiv) was added. The reaction mixture was stirred until complete conversion of the starting material was observed in HPLC. The reaction mixture was evaporated under reduced pressure.
General Procedure G for the Synthesis of Amides
A solution of carboxylic acid (1–2 equiv) in anhydrous DCM (1 mL/0.1 mmol of the acid) was cooled to 0 °C. N,N-diisopropylethylamine (DIPEA, 1–4 equiv) was added, followed by 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU, 1–2 equiv). The reaction mixture was stirred at 0 °C for 10 min. A solution of amine (1–2 equiv) in DCM (1 mL/0.2 mmol) was added, and the reaction mixture was allowed to warm up to room temperature and stirred for 30 min. The solvent was evaporated under reduced pressure. The residue was dissolved in EtOAc (40 mL), and the solution was washed with 1 M HCl (40 mL), 1 M NaOH (40 mL), and brine (40 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure.
General Procedure H for the Debenzylation of Alcohols
To a solution of benzyl ether (1 equiv) in MeOH (9 mL/0.1 mmol of the benzyl ether), 10% Pd on carbon (0.1–0.2 equiv) was added, followed by formic acid (2.2 mL/0.1 mmol of the benzyl ether). The reaction mixture was stirred under a hydrogen atmosphere (1–6 atm). After the reaction was complete, the reaction mixture was filtered through a syringe filter (Teflon; 0.45 μm, washed with EtOAc). The filtrate was washed with 12% aqueous ammonia solution (18 mL/0.1 mmol of the benzyl ether). EtOAc was added until two layers were visible. Phases were separated, and the aqueous phase was extracted with EtOAc (2 × 30 mL). The combined organic phase was washed with brine, dried over anhydrous Na2SO4, and evaporated. The full conversion was achieved after stirring under a hydrogen atmosphere (1 atm) for 3 h. A pure material was obtained by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM.
General Procedure I for the Intramolecular Mitsunobu Reaction (Cyclization)
To a solution of diol (1 equiv) in anhydrous DCM (8 mL/0.1 mmol of the diol), PPh3 (2–3 equiv) was added, followed by DTBAD (2–3 equiv). The reaction mixture was stirred at room temperature for 15 min. The reaction mixture was evaporated under reduced pressure, and the residue was purified by silica gel column chromatography using diethyl ether. Fractions with product were combined and evaporated under reduced pressure. The residue was used in the next step without further purification.
General Procedure J for the Ring-Closing Metathesis and the Hydrogenation of Double Bond
To a solution of bis-alkene (1 equiv) in anhydrous toluene (4.4 mL/0.01 mmol of the bis-alkene), Zhan Catalyst-1B (0.1 equiv) was added. The reaction mixture was stirred at 55 °C overnight. The reaction mixture was evaporated under reduced pressure. The residue was filtered through silica using 30% EtOAc in hexanes, evaporated under reduced pressure, and the residue was used in the next step without further purification.
The residue from the previous step was dissolved in MeOH (8.7 mL/0.1 mmol of the bis-alkene). Then, 10% Pd on carbon (0.1 equiv) was added, and the reaction mixture was stirred under a hydrogen atmosphere (1 atm). After the reaction was complete, the reaction mixture was filtered through a syringe filter (Teflon; 0.45 μm, washed with MeOH).
(S)-N-((E)-((S)-2,2-Dimethyl-1,3-dioxolan-4-yl)methylene)-2-methylpropane-2-sulfinamide (10)
To a stirred solution of tert-butylsulfinamide (9) (3.49 g, 28.8 mmol, 1.5 equiv) in anhydrous DCM (200 mL) was added glyceraldehyde 8 (5.00 g, 19.2 mmol, 1 equiv). Ti(OEt)4 (20.1 mL, 96.0 mmol, 5 equiv) was added, and the reaction mixture was stirred at room temperature for 1 h. The mixture was cooled in an ice bath for 5 min. Deionized water (30 mL) was added. After stirring for 5 min, the mixture was filtered through Celite, washing with EtOAc. The filtrate was concentrated under reduced pressure, washed with brine (50 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification by column chromatography on silica gel using 10% EtOAc in hexanes afforded product 10 as a yellow oil (3.37 g, 75% yield). 1H NMR (300 MHz, chloroform-d): δ 8.07 (d, J = 4.1 Hz, 1H), 4.84 (ddd, J = 6.8, 5.1, 4.1 Hz, 1H), 4.22 (dd, J = 8.5, 6.8 Hz, 1H), 4.04 (dd, J = 8.5, 5.1 Hz, 1H), 1.45 (s, 3H), 1.42 (s, 3H), 1.20 (s, 9H). Corresponds with the literature.43
(S)-N-((S)-2-(3-(Benzyloxy)phenyl)-1-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)ethyl)-2-methylpropane-2-sulfinamide (11)
A solution of imine 10 (1.70 g, 7.30 mmol, 1 equiv) in THF (7.3 mL) was cooled to −78 °C in a dry ice/acetone bath. Freshly prepared 0.7 M 3-(benzyloxy)benzylmagnesium chloride solution in THF (10.4 mL, 7.30 mmol, 1 equiv) was added dropwise. The reaction mixture was allowed to slowly warm up to room temperature and stirred for 1 h. The reaction mixture was quenched with sat. aq. NH4Cl solution (25 mL) and extracted with EtOAc (3 × 50 mL). The combined organic phase was washed with brine (25 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification by column chromatography on silica gel using 90% DCM/9% EtOAc/1% MeOH afforded product 11 as a yellow oil (2.07 g, 66% yield). 1H NMR (400 MHz, chloroform-d): δ 7.45–7.35 (m, 4H), 7.35–7.29 (m, 1H), 7.20 (dd, J = 9.0, 7.5 Hz, 1H), 6.86–6.76 (m, 3H), 5.05 (s, 2H), 4.15 (td, J = 6.3, 5.1 Hz, 1H), 4.05 (dd, J = 8.5, 6.6 Hz, 1H), 3.95 (dd, J = 8.5, 6.1 Hz, 1H), 3.66 (dtd, J = 7.3, 6.2, 5.1 Hz, 1H), 3.55 (d, J = 6.1 Hz, 1H), 2.99 (dd, J = 13.9, 7.3 Hz, 1H), 2.72 (dd, J = 13.9, 6.3 Hz, 1H), 1.47 (s, 3H), 1.32 (s, 3H), 1.11 (s, 9H). 13C NMR (101 MHz, Chloroform-d): δ 159.0, 139.1, 137.1, 129.6, 128.7, 128.1, 127.5, 122.3, 116.3, 113.1, 109.5, 77.2, 70.0, 65.9, 58.5, 56.2, 37.8, 26.6, 24.93, 22.6. HRMS-ESI (m/z) calcd for C24H34NO4S [M + H]+ 432.2209. Found 432.2202. [α]D20 = 30.4 (c 1.0, CHCl3).
tert-Butyl ((2S,3S)-1-(3-(Benzyloxy)phenyl)-3,4-dihydroxybutan-2-yl)carbamate (12)
To a stirred solution of compound 11 (2.25 g, 5.21 mmol, 1 equiv) in diethyl ether (50 mL), 4 M HCl solution in dioxane (2.61 mL, 10.4 mmol, 2 equiv) was added, and the reaction mixture was stirred at room temperature for 20 min. A precipitate was filtered, and the filtered solid was dissolved in MeOH (50 mL). Then, 4 M HCl solution in dioxane (2.61 mL, 10.4 mmol, 2 equiv) was added, and the reaction mixture was stirred at room temperature overnight. All volatiles were removed under reduced pressure. The residue was dissolved in DCM (50 mL). DIPEA (3.61 mL, 20.9 mmol, 4 equiv) was added, followed by a solution of (Boc)2O (1.82 g, 8.34 mmol, 1.6 equiv) in DCM (10 mL). The reaction mixture was stirred at room temperature for 1 h. Imidazole (2.13 g, 31.3 mmol, 6 equiv) was added. The reaction mixture was stirred at room temperature for 20 min. The mixture was washed with 1% HCl solution (3 × 50 mL) and brine (30 mL) and dried over anhydrous Na2SO4. Evaporation under reduced pressure afforded product 12 as a beige solid (1.80 g, 89% yield). 1H NMR (400 MHz, chloroform-d): δ 7.48–7.29 (m, 5H), 7.25–7.21 (m, 1H), 6.85 (ddt, J = 12.5, 7.5, 1.3 Hz, 3H), 5.06 (s, 2H), 4.58 (d, J = 8.5 Hz, 1H), 3.83 (qd, J = 8.5, 4.4 Hz, 1H), 3.73–3.54 (m, 2H), 3.32 (d, J = 8.5 Hz, 1H), 3.05 (dd, J = 14.2, 4.4 Hz, 1H), 2.90 (dd, J = 14.3, 7.5 Hz, 1H), 2.68 (br s, 2H), 1.39 (s, 9H). 13C NMR (101 MHz, chloroform-d): δ 159.2, 157.2, 139.0, 137.1, 129.8, 128.7, 128.1, 127.7, 122.2, 116.2, 113.2, 80.6, 73.1, 70.1, 63.0, 52.2, 36.6, 28.4. HRMS-ESI (m/z) calcd for C22H29NO5Na [M+Na]+ 410.1943. Found 410.1942. [α]D20 = 10.5 (c 1.4, CHCl3).
tert-Butyl ((2S,3R)-1-(3-(Benzyloxy)phenyl)-3-hydroxy-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)carbamate (15)
The diol 12 (2.10 g, 5.42 mmol, 1 equiv) was dissolved in chloroform (50 mL) in a pressure tube. Triphenylphosphine (2.13 g, 8.13 mmol, 1.5 equiv) was added, followed by di-tert-butyl azodicarboxylate (1.87 g, 8.13 mmol, 1.5 equiv). The tube was sealed, and the reaction mixture was stirred at 80 °C overnight. Chloroform was evaporated under reduced pressure, and the residue was diluted with dry i-PrOH (5 mL). Amine 14 (2.69 g, 16.3 mmol, 3 equiv) was added, and the reaction mixture was stirred at 80 °C for 5 h. i-PrOH was evaporated under reduced pressure. Purification by column chromatography on silica gel using diethyl ether afforded product 15 as a yellow oil (1.80 g, 62% yield). Excess of amine 14 was recovered by the washing column with EtOAc. 1H NMR (400 MHz, chloroform-d): δ 7.45–7.41 (m, 2H), 7.40–7.35 (m, 2H), 7.34–7.28 (m, 1H), 7.23 (t, J = 8.1 Hz, 1H), 7.19 (t, J = 7.8 Hz, 1H), 7.03–6.95 (m, 2H), 6.86–6.72 (m, 4H), 5.04 (s, 2H), 4.57 (d, J = 9.2 Hz, 1H), 3.80 (s, 3H), 3.79–3.73 (m, 2H), 3.28 (dt, J = 7.6, 4.5 Hz, 1H), 2.92 (dd, J = 14.1, 4.7 Hz, 1H), 2.81 (dd, J = 14.2, 7.6 Hz, 1H), 2.45 (dd, J = 11.6, 4.3 Hz, 1H), 2.37 (dd, J = 12.5, 3.4 Hz, 1H), 1.45 (s, 6H), 1.37 (s, 9H). 13C NMR (101 MHz, chloroform-d): δ 159.7, 159.0, 156.2, 149.3, 139.6, 137.2, 129.5, 129.3, 128.7, 128.0, 127.7, 122.3, 118.5, 116.1, 112.9, 112.6, 111.2, 79.6, 71.2, 70.0, 55.7, 55.3, 53.6, 44.4, 36.8, 29.7, 29.6, 28.5. HRMS-ESI (m/z) calcd for C32H43N2O5 [M + H]+ 535.3172. Found 535.3179. [α]D20 = −10.5 (c 1.2, CHCl3).
tert-Butyl ((2S,3R)-1-(3-(Benzyloxy)phenyl)-3-((tert-butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)carbamate (16)
A solution of 15 (4.63 g, 8.66 mmol, 1 equiv), imidazole (5.90 g, 86.8 mmol, 10 equiv), and TBSCl (13.1 g, 86.8 mmol, 10 equiv) in DMF (60 mL) was stirred at 50 °C overnight. DMF was evaporated under reduced pressure. The residue was dissolved in EtOAc (30 mL) and water (30 mL). The aqueous phase was separated and extracted with EtOAc (2 × 20 mL). The combined organic phase was washed with brine (20 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification by column chromatography on silica gel using gradient elution from 5 to 30% EtOAc in hexanes afforded product 16 as a colorless oil (4.63 g, 82%). 1H NMR (400 MHz, chloroform-d): δ 7.45–7.41 (m, 2H), 7.38 (ddd, J = 7.5, 6.6, 1.3 Hz, 2H), 7.34–7.29 (m, 1H), 7.29–7.23 (m, 1H), 7.20–7.13 (m, 1H), 7.06–6.96 (m, 2H), 6.78 (ddd, J = 11.1, 5.9, 2.1 Hz, 4H), 6.52–6.24 (m, 1H), 5.03 (s, 2H), 4.07–3.92 (m, 1H), 3.80 (s, 3H), 3.65 (d, J = 20.3 Hz, 1H), 2.83 (dd, J = 14.0, 7.7 Hz, 1H), 2.54 (dd, J = 15.0, 8.0 Hz, 1H), 2.49 (br s, 1H), 1.69–1.57 (m, 2H), 1.46 (s, 3H), 1.44 (s, 3H), 1.42 (s, 9H), 0.87 (s, 9H), −0.02 (s, 3H), −0.07 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 159.8, 159.0, 156.0, 149.3, 140.5, 137.3, 129.4, 129.3, 128.7, 128.0, 127.6, 121.9, 118.5, 115.6, 112.8, 112.5, 111.3, 78.6, 71.3, 70.0, 56.5, 55.6, 55.3, 44.8, 37.6, 29.7, 29.3, 28.6, 26.0, 18.2, −4.7, −4.8. HRMS-ESI (m/z) calcd for C38H57N2O5Si [M + H]+ 649.4037. Found 649.4049. [α]D20 = −26.3 (c 1.5, CHCl3).
(2R,3S)-3-Amino-4-(3-(benzyloxy)phenyl)-1-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-ol (17)
The title compound was obtained as a yellow oil (420 mg, 94% yield) from carbamate 15 (550 mg, 1.03 mmol, 1 equiv) and trifluoroacetic acid (1.58 mL, 20.6 mmol, 20 equiv), following general procedure A. A pure material was obtained by column chromatography using gradient elution from DCM to 5% MeOH in DCM. 1H NMR (400 MHz, methanol-d4): δ 7.44–7.37 (m, 2H), 7.37–7.31 (m, 2H), 7.31–7.26 (m, 1H), 7.26–7.20 (m, 1H), 7.20–7.13 (m, 1H), 7.03–6.97 (m, 2H), 6.84–6.79 (m, 2H), 6.78–6.71 (m, 2H), 5.04 (s, 2H), 3.77 (s, 3H), 3.51 (ddd, J = 8.6, 5.2, 3.4 Hz, 1H), 2.95 (dt, J = 9.0, 4.9 Hz, 1H), 2.79 (dd, J = 13.5, 4.8 Hz, 1H), 2.49 (dd, J = 11.7, 3.4 Hz, 1H), 2.38 (ddd, J = 17.0, 12.6, 8.8 Hz, 2H), 1.45 (s, 3H), 1.45 (s, 3H). 13C NMR (101 MHz, methanol-d4): δ 161.2, 160.3, 150.1, 142.1, 138.7, 130.6, 130.4, 129.5, 128.9, 128.6, 123.0, 119.4, 116.9, 113.9, 113.2, 112.6, 74.5, 70.9, 57.3, 56.9, 55.8, 46.2, 40.1, 30.2, 29.0. HRMS-ESI (m/z) calcd for C27H35N2O3 [M + H]+ 435.2648. Found 435.2644. [α]D20 = 2.9 (c 0.7, CHCl3).
(2R,3S)-4-(3-(Benzyloxy)phenyl)-2-((tert-butyldimethylsilyl)oxy)-N1-(2-(3-methoxyphenyl)propan-2-yl)butane-1,3-diamine (18)
The title compound was obtained as a white solid foam (2.25 g, 92% yield) from carbamate 16 (2.90 g, 4.47 mmol, 1 equiv) and trifluoroacetic acid (6.9 mL, 89.4 mmol, 20 equiv) by following general procedure A. A pure material was obtained by column chromatography using gradient elution from DCM to 5% MeOH in DCM. 1H NMR (400 MHz, methanol-d4): δ 7.45–7.39 (m, 2H), 7.39–7.32 (m, 2H), 7.32–7.26 (m, 1H), 7.26–7.22 (m, 1H), 7.22–7.17 (m, 1H), 7.03–6.97 (m, 2H), 6.85 (ddd, J = 8.2, 2.6, 0.9 Hz, 1H), 6.81 (t, J = 2.0 Hz, 1H), 6.79–6.76 (m, 1H), 6.74 (dt, J = 7.6, 1.2 Hz, 1H), 5.07 (s, 2H), 3.76 (s, 3H), 3.64 (td, J = 5.7, 4.2 Hz, 1H), 3.14 (ddd, J = 8.4, 6.3, 4.2 Hz, 1H), 2.68 (dd, J = 13.6, 6.3 Hz, 1H), 2.49–2.41 (m, 3H), 1.45 (s, 3H), 1.42 (s, 3H), 0.92 (s, 9H), 0.05 (s, 3H), 0.03 (s, 3H). 13C NMR (101 MHz, methanol-d4): δ 161.3, 160.4, 150.1, 141.9, 138.8, 130.7, 130.3, 129.5, 128.9, 128.5, 122.7, 119.3, 116.8, 114.0, 113.4, 112.3, 75.8, 70.9, 57.6, 56.8, 55.7, 45.0, 40.1, 30.5, 28.6, 26.4, 19.0, −4.1, −4.4. HRMS-ESI (m/z) calcd for C33H49N2O3Si [M + H]+ 549.3512. Found 549.3539. [α]D20 = −3.4 (c 1.2, CHCl3).
Methyl 3-(((2S,3R)-1-(3-(Benzyloxy)phenyl)-3-hydroxy-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)carbamoyl)benzoate (20)
A solution of monomethyl isophthalate 19 (111 mg, 0.617 mmol, 1.2 equiv) in DCM (10 mL) was cooled to 0 °C. HOBT·H2O (118 mg, 0.617 mmol, 1.2 equiv) was added, followed by EDCl (109 μL, 0.617 mmol, 1.2 equiv) 20 min later. After stirring for 1 h, a solution of amine 17 (261 mg, 0.514 mmol, 1 equiv) and DIPEA (445 μL, 2.57 mmol, 5 equiv) in DCM (5 mL) was added. The reaction mixture was stirred at room temperature for 5 h. The solvent was evaporated under reduced pressure, and the residue was dissolved in EtOAc (30 mL) and water (30 mL). The aqueous phase was separated and extracted with EtOAc (2 × 15 mL). The combined organic phase was washed with brine (15 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification by column chromatography on silica gel using 50% EtOAc in hexanes afforded product 20 as a colorless oil (195 mg, 64% yield). 1H NMR (400 MHz, methanol-d4): δ 8.27 (td, J = 1.8, 0.6 Hz, 1H), 8.12 (dt, J = 7.8, 1.3 Hz, 1H), 7.81 (ddd, J = 7.8, 1.8, 1.2 Hz, 1H), 7.51 (td, J = 7.8, 0.6 Hz, 1H), 7.35–7.23 (m, 5H), 7.19–7.10 (m, 3H), 7.02–6.96 (m, 2H), 6.87 (dd, J = 2.6, 1.6 Hz, 1H), 6.81 (dt, J = 7.5, 1.2 Hz, 1H), 6.77 (ddd, J = 8.3, 2.6, 0.9 Hz, 1H), 6.71 (ddd, J = 8.2, 2.4, 1.1 Hz, 1H), 5.01–4.90 (m, 2H), 4.24 (ddd, J = 10.0, 7.4, 4.5 Hz, 1H), 3.89 (s, 3H), 3.71 (s, 3H), 3.69–3.63 (m, 1H), 3.10 (dd, J = 13.9, 4.5 Hz, 1H), 2.75 (dd, J = 13.9, 10.0 Hz, 1H), 2.53 (dd, J = 12.0, 3.4 Hz, 1H), 2.44 (dd, J = 12.1, 6.9 Hz, 1H), 1.47 (s, 6H). 13C NMR (101 MHz, methanol-d4): δ 169.2, 167.6, 161.2, 160.2, 149.5, 141.6, 138.8, 136.5, 133.2, 132.8, 131.7, 130.30, 130.27, 129.9, 129.4, 129.2, 128.8, 128.5, 123.0, 119.3, 116.8, 114.0, 113.2, 112.6, 73.4, 70.9, 57.0, 56.5, 55.6, 52.8, 46.9, 37.7, 30.0, 28.7. HRMS-ESI (m/z) calcd for C36H41N2O6 [M + H]+ 597.2965. Found 597.2980. [α]D20 = −41.3 (c 1.1, CHCl3).
Methyl 3-(((2S,3R)-1-(3-(Benzyloxy)phenyl)-3-((tert-butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)carbamoyl)benzoate (21)
The title compound was obtained as a colorless oil (1.05 g, 84% yield) from compound 20 (1.05 g, 1.76 mmol, 1 equiv), TBSCl (1.33 g, 8.80 mmol, 5 equiv), and imidazole (599 mg, 8.80 mmol, 5 equiv) by following general procedure B. A pure material was obtained by column chromatography using gradient elution from hexanes to 30% EtOAc in hexanes. 1H NMR (400 MHz, chloroform-d): δ 9.65 (d, J = 6.9 Hz, 1H), 8.51 (td, J = 1.8, 0.6 Hz, 1H), 8.18 (dt, J = 7.7, 1.4 Hz, 1H), 8.13 (ddd, J = 7.8, 1.8, 1.2 Hz, 1H), 7.53 (t, J = 7.8 Hz, 1H), 7.43–7.39 (m, 2H), 7.38–7.33 (m, 2H), 7.32–7.23 (m, 2H), 7.21–7.13 (m, 1H), 7.01–6.94 (m, 2H), 6.84–6.75 (m, 4H), 5.02 (s, 2H), 4.52–4.42 (m, 1H), 3.89 (s, 3H), 3.74 (s, 3H), 3.64 (ddd, J = 4.6, 3.1, 1.3 Hz, 1H), 3.20 (dd, J = 13.7, 5.2 Hz, 1H), 2.69 (d, J = 12.1 Hz, 1H), 2.56 (ddd, J = 11.8, 4.7, 1.2 Hz, 1H), 2.40 (dd, J = 13.7, 10.3 Hz, 1H), 1.56 (s, 3H), 1.52 (s, 3H), 0.80 (s, 9H), −0.12 (s, 3H), −0.16 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 166.5, 166.3, 159.9, 159.0, 148.3, 139.9, 137.2, 135.9, 132.3, 132.0, 130.6, 129.7, 129.5, 128.9, 128.7, 128.0, 127.7, 127.6, 121.8, 118.3, 115.5, 113.2, 112.6, 111.5, 70.0, 68.7, 58.0, 55.7, 55.2, 52.3, 44.1, 38.6, 29.7, 29.3, 25.9, 18.1, −4.8, −5.1. HRMS-ESI (m/z) calcd for C42H55N2O6Si [M + H]+ 711.3829. Found 711.3833. [α]D20 = −39.3 (c 1.8, CHCl3).
Methyl 3-(((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-1-(3-hydroxyphenyl)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)carbamoyl)benzoate (22)
To a solution of compound 21 (500 mg, 0.703 mmol, 1 equiv) in i-PrOH (20 mL), 10% Pd on carbon (150 mg, 0.141 mmol, 0.2 equiv) was added, and the reaction mixture was stirred under a hydrogen atmosphere (6 atm) for 16 h. The reaction mixture was filtered through a syringe filter (Teflon; 0.45 μm; washed with MeOH). The filtrate was evaporated under reduced pressure. Purification by column chromatography on silica gel using gradient elution from DCM to 2% MeOH in DCM afforded product 22 as a colorless oil (390 mg, 89% yield). 1H NMR (400 MHz, chloroform-d): δ 9.60 (d, J = 7.0 Hz, 1H), 8.50–8.47 (m, 1H), 8.18 (dt, J = 7.8, 1.4 Hz, 1H), 8.10 (ddd, J = 7.8, 1.8, 1.2 Hz, 1H), 7.53 (td, J = 7.8, 0.6 Hz, 1H), 7.29–7.22 (m, 1H), 7.09 (t, J = 7.8 Hz, 1H), 7.02–6.93 (m, 2H), 6.81–6.75 (m, 2H), 6.72–6.63 (m, 2H), 4.53–4.44 (m, 1H), 3.89 (s, 3H), 3.75 (s, 3H), 3.70 (ddd, J = 4.8, 3.2, 1.4 Hz, 1H), 3.12 (dd, J = 13.8, 5.7 Hz, 1H), 2.71 (dd, J = 11.9, 1.6 Hz, 1H), 2.60 (ddd, J = 11.9, 4.6, 1.1 Hz, 1H), 2.41 (dd, J = 13.8, 9.8 Hz, 1H), 1.56 (s, 3H), 1.51 (s, 3H), 0.80 (s, 9H), −0.09 (s, 3H), −0.13 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 166.7, 166.5, 159.8, 156.6, 148.2, 139.8, 135.7, 132.4, 132.1, 130.6, 129.7, 129.5, 128.9, 127.7, 121.2, 118.3, 116.0, 113.8, 112.6, 111.5, 68.9, 57.9, 55.8, 55.3, 52.3, 44.2, 38.3, 29.6, 29.2, 25.9, 18.1, −4.8, −5.1. HRMS-ESI (m/z) calcd for C35H49N2O6Si [M + H]+ 621.3360. Found 621.3367. [α]D20 = −40.4 (c 1.4, CHCl3).
Methyl 3-(((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)-1-(3-(4-((2-nitro-N-propylphenyl)sulfonamido)butoxy)phenyl)butan-2-yl)carbamoyl)benzoate (25a)
The title compound was obtained as a colorless oil (30 mg, 71% yield) from compound 22 (36 mg, 0.058 mmol, 1 equiv), alcohol 23a (91 mg, 0.29 mmol, 5 equiv), DEAD (30 μL, 0.17 mmol, 3 equiv), PPh3 (46 mg, 0.17 mmol, 3 equiv), thiophenol (12 μL, 0.12 mmol, 2 equiv), and K2CO3 (32 mg, 0.23 mmol, 4 equiv), following general procedure C. 1H NMR (400 MHz, chloroform-d): δ 9.65 (d, J = 6.9 Hz, 1H), 8.50 (td, J = 1.8, 0.5 Hz, 1H), 8.18 (dt, J = 7.8, 1.4 Hz, 1H), 8.12 (ddd, J = 7.8, 1.9, 1.2 Hz, 1H), 7.53 (td, J = 7.7, 0.5 Hz, 1H), 7.30–7.21 (m, 1H), 7.14 (t, J = 8.0 Hz, 1H), 6.98 (ddd, J = 7.8, 1.8, 0.9 Hz, 1H), 6.95 (dd, J = 2.5, 1.7 Hz, 1H), 6.77 (ddd, J = 8.2, 2.5, 0.9 Hz, 1H), 6.75–6.69 (m, 3H), 4.47 (dtt, J = 9.3, 5.7, 3.1 Hz, 1H), 3.93 (t, J = 6.3 Hz, 2H), 3.89 (s, 3H), 3.74 (s, 3H), 3.68–3.64 (m, 1H), 3.18 (dd, J = 13.7, 5.2 Hz, 1H), 2.77–2.65 (m, 3H), 2.63–2.55 (m, 3H), 2.40 (dd, J = 13.7, 10.3 Hz, 1H), 1.87–1.73 (m, 2H), 1.67 (dtd, J = 9.4, 7.4, 5.3 Hz, 2H), 1.59–1.46 (m, 8H), 0.92 (t, J = 7.4 Hz, 3H), 0.79 (s, 9H), −0.12 (s, 3H), −0.16 (s, 3H). 13C NMR (101 MHz, CDCl3): δ 166.5, 166.3, 159.9, 159.3, 148.3, 139.8, 135.9, 132.3, 132.0, 130.5, 129.6, 129.5, 128.9, 127.7, 121.5, 118.3, 115.1, 112.9, 112.5, 111.5, 68.7, 67.8, 58.0, 55.7, 55.2, 52.3, 51.9, 49.6, 44.1, 38.6, 29.7, 29.3, 27.3, 26.6, 25.8, 23.1, 18.1, 11.9, −4.9, −5.18. HRMS-ESI (m/z) calcd for C42H64N3O6Si [M + H]+ 734.4564. Found 734.4561. [α]D20 = −36.0 (c 1.3, CHCl3).
Methyl 3-(((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)-1-(3-(4-((N-neopentyl-2-nitrophenyl)sulfonamido)butoxy)phenyl)butan-2-yl)carbamoyl)benzoate (25b)
The title compound was obtained as a yellowish oil (50 mg, 82% yield) from compound 22 (50 mg, 0.081 mmol, 1 equiv), alcohol 23b (111 mg, 0.322 mmol, 4 equiv), DTBAD (56 mg, 0.24 mmol, 3 equiv), PPh3 (84 mg, 0.322 mmol, 4 equiv), thiophenol (33 μL, 0.321 mmol, 4 equiv), and K2CO3 (67 mg, 0.48 mmol, 6 equiv), following general procedure C. 1H NMR (400 MHz, chloroform-d): δ 9.57 (d, J = 6.9 Hz, 1H), 8.47 (t, J = 1.7 Hz, 1H), 8.16 (dt, J = 7.8, 1.4 Hz, 1H), 8.09 (dt, J = 7.9, 1.5 Hz, 1H), 7.51 (t, J = 7.8 Hz, 1H), 7.24 (t, J = 7.9 Hz, 1H), 7.12 (t, J = 8.0 Hz, 1H), 6.99–6.94 (m, 2H), 6.78–6.71 (m, 3H), 6.70–6.66 (m, 1H), 4.44 (dtd, J = 9.6, 6.0, 3.1 Hz, 1H), 3.92 (t, J = 6.1 Hz, 2H), 3.88 (s, 3H), 3.73 (s, 3H), 3.70 (d, J = 4.2 Hz, 1H), 3.14 (dd, J = 13.7, 5.6 Hz, 1H), 3.04–2.95 (m, 2H), 2.71 (dd, J = 11.9, 1.5 Hz, 1H), 2.66 (s, 2H), 2.59 (dd, J = 11.8, 4.4 Hz, 1H), 2.42 (dd, J = 13.7, 9.7 Hz, 1H), 2.03–1.91 (m, 2H), 1.79 (p, J = 6.4 Hz, 2H), 1.56 (s, 3H), 1.52 (s, 3H), 1.07 (s, 9H), 0.78 (s, 9H), −0.12 (s, 3H), −0.16 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 166.5, 166.3, 159.8, 159.0, 148.0, 139.8, 135.7, 132.3, 131.9, 130.5, 129.54, 129.46, 128.8, 127.7, 121.7, 118.3, 115.3, 112.7, 112.5, 111.5, 68.7, 67.3, 59.9, 57.8, 55.9, 55.2, 52.3, 49.5, 44.2, 38.4, 31.1, 29.5, 29.1, 27.9, 26.9, 25.8, 23.3, 18.0, −4.9, −5.1. HRMS-ESI (m/z) calcd for C44H68N3O6Si [M + H]+ 762.4877. Found 762.4885. [α]D20 = −35.9 (c 1.1, CHCl3).
Methyl 3-(((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)-1-(3-((5-(propylamino)pentyl)oxy)phenyl)butan-2-yl)carbamoyl)benzoate (25c)
The title compound was obtained as a yellowish oil (50 mg, 83% yield) from compound 22 (50 mg, 0.081 mmol, 1 equiv), alcohol 23c (80 mg, 0.24 mmol, 3 equiv), DEAD (51 μL, 0.32 mmol, 4 equiv), PPh3 (106 mg, 0.403 mmol, 5 equiv), thiophenol (17 μL, 0.16 mmol, 2 equiv), and K2CO3 (44 mg, 0.32 mmol, 4 equiv), following general procedure C. 1H NMR (400 MHz, chloroform-d): δ 9.65 (d, J = 6.9 Hz, 1H), 8.50 (td, J = 1.8, 0.6 Hz, 1H), 8.17 (dt, J = 7.8, 1.4 Hz, 1H), 8.12 (ddd, J = 7.8, 1.9, 1.2 Hz, 1H), 7.53 (td, J = 7.8, 0.5 Hz, 1H), 7.25 (t, J = 8.0 Hz, 1H), 7.14 (t, J = 8.0 Hz, 1H), 6.97 (ddd, J = 7.7, 1.9, 0.9 Hz, 1H), 6.95 (dd, J = 2.5, 1.6 Hz, 1H), 6.77 (ddd, J = 8.1, 2.5, 0.9 Hz, 1H), 6.75–6.67 (m, 3H), 4.51–4.42 (m, 1H), 3.91 (t, J = 6.3 Hz, 2H), 3.89 (s, 3H), 3.74 (s, 3H), 3.66 (ddd, J = 4.6, 3.1, 1.4 Hz, 1H), 3.18 (dd, J = 13.7, 5.2 Hz, 1H), 2.77–2.67 (m, 1H), 2.63 (dd, J = 7.7, 6.6 Hz, 2H), 2.61–2.55 (m, 3H), 2.39 (dd, J = 13.7, 10.3 Hz, 1H), 1.83–1.71 (m, 2H), 1.61–1.42 (m, 12H), 0.91 (t, J = 7.4 Hz, 3H), 0.79 (s, 9H), −0.12 (s, 3H), −0.16 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 166.5, 166.3, 159.8, 159.4, 148.37, 139.8, 135.8, 132.3, 132.0, 130.5, 129.53, 129.45, 128.8, 127.7, 121.4, 118.3, 115.1, 112.8, 112.5, 111.5, 68.7, 67.9, 58.0, 55.7, 55.2, 52.3, 52.1, 50.0, 44.1, 38.6, 30.0, 29.6, 29.4, 29.3, 25.8, 24.0, 23.3, 18.1, 11.9, −4.9, −5.1. HRMS-ESI (m/z) calcd for C43H66N3O6Si [M + H]+ 748.4721. Found 748.4718. [α]D20 = −30.2 (c 1.0, CHCl3).
Methyl 3-(((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)-1-(3-(2-((2-nitro-N-propylphenyl)sulfonamido)ethoxy)phenyl)butan-2-yl)carbamoyl)benzoate (25d)
The title compound was obtained as a yellowish oil (31 mg, 50% yield) from compound 22 (55 mg, 0.089 mmol, 1 equiv), alcohol 23d (77 mg, 0.27 mmol, 3 equiv), DEAD (28 μL, 0.18 mmol, 2 equiv), PPh3 (70 mg, 0.27 mmol, 3 equiv), thiophenol (18 μL, 0.18 mmol, 2 equiv), and K2CO3 (49 mg, 0.36 mmol, 4 equiv), following general procedure C. 1H NMR (400 MHz, chloroform-d): δ 9.64 (d, J = 6.9 Hz, 1H), 8.50 (dt, J = 1.8, 0.9 Hz, 1H), 8.18 (dt, J = 7.8, 1.4 Hz, 1H), 8.12 (ddd, J = 7.8, 1.8, 1.2 Hz, 1H), 7.53 (t, J = 7.8 Hz, 1H), 7.26 (t, J = 7.9 Hz, 1H), 7.15 (t, J = 7.7 Hz, 1H), 6.98 (ddd, J = 7.8, 1.8, 0.9 Hz, 1H), 6.96 (t, J = 2.1 Hz, 1H), 6.81–6.70 (m, 4H), 4.51–4.42 (m, 1H), 4.04 (t, J = 5.2 Hz, 2H), 3.89 (s, 3H), 3.74 (s, 3H), 3.69–3.63 (m, 1H), 3.18 (dd, J = 13.7, 5.3 Hz, 1H), 2.99 (dd, J = 5.8, 4.7 Hz, 2H), 2.72 (dd, J = 11.7, 1.4 Hz, 1H), 2.64 (dd, J = 7.8, 6.8 Hz, 2H), 2.59 (ddd, J = 11.8, 4.7, 1.2 Hz, 1H), 2.40 (dd, J = 13.7, 10.2 Hz, 1H), 1.58–1.51 (m, 8H), 0.93 (t, J = 7.4 Hz, 3H), 0.79 (s, 9H), −0.12 (s, 3H), −0.16 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 166.5, 166.3, 159.9, 159.2, 148.3, 139.9, 135.9, 132.3, 132.0, 130.6, 129.6, 129.5, 128.9, 127.7, 121.7, 118.3, 115.3, 112.7, 112.5, 111.5, 68.7, 67.3, 58.0, 55.7, 55.2, 52.3, 51.8, 48.9, 44.2, 38.6, 29.6, 29.3, 25.8, 23.3, 18.1, 11.9, −4.8, −5.1. HRMS-ESI (m/z) calcd for C40H60N3O6Si [M + H]+ 706.4251. Found 706.4254. [α]D20 = −40.8 (c 1.2, CHCl3).
(S)-4-((R)-1-Hydroxy-2-((2-(3-methoxyphenyl)propan-2-yl)amino)ethyl)-12-propyl-7-oxa-3,12-diaza-1,6(1,3)-dibenzenacyclotridecaphane-2,13-dione (7a)
The hydrolysis of methyl benzoate 25a (32 mg 0.044 mmol, 1 equiv) was performed using 1 M aqueous NaOH solution (89 μL, 0.89 mmol, 2 equiv), following procedure D. The crude product was used in the next step without purification. The macrolactamization was performed using HBTU (25 mg, 0.65 mmol, 1.5 equiv), following procedure E. The crude product was used in the next step without purification. The desilylation was performed using NH4F (47 mg, 1.3 mmol, 30 equiv), following procedure F. Full conversion was achieved after stirring at 40 °C overnight. Purification by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM afforded product 7a as an amorphous solid (17 mg, 68% yield). 1H NMR (400 MHz, methanol-d4) for a mixture of rotamers (0.72:0.20:0.08): δ 7.75–7.35 (m, 4H), 7.26–6.92 (m, 4H), 6.91–3.63 (m, 4H), 4.50–4.07 (m, 2H), 3.91–3.64 (m, 5H), 3.63–3.36 (m, 2H), 3.27–2.96 (m, 3H), 2.91–2.37 (m, 3H), 2.21–1.39 (m, 12H), 1.01 (t, J = 7.4 Hz, 2.15H), 0.73 (t, J = 7.3 Hz, 0.25H), 0.60 (t, J = 7.4 Hz, 0.6H). 13C NMR (101 MHz, methanol-d4) for a major rotamer: δ 173.0, 168.8, 161.3, 159.1, 149.2, 141.8, 138.4, 135.5, 130.8, 130.5, 130.34, 130.31, 129.6, 124.7, 122.7, 119.3, 118.1, 113.7, 113.2, 112.7, 74.0, 67.8, 57.3, 56.3, 55.6, 50.1, 47.9, 47.3, 37.4, 29.8, 28.6, 26.7, 26.6, 21.8, 11.7. HRMS-ESI (m/z) calcd for C35H46N3O5 [M + H]+ 588.3437. Found 588.3453. [α]D20 = 15.4 (c 1.7, CHCl3).
(S)-4-((R)-1-Hydroxy-2-((2-(3-methoxyphenyl)propan-2-yl)amino)ethyl)-12-propyl-7-oxa-3,12-diaza-1,6(1,3)-dibenzenacyclotridecaphane-2,13-dione (7e)
The hydrolysis of methyl benzoate 25b (50 mg 0.066 mmol, 1 equiv) was performed using 1 M aqueous NaOH solution (131 μL, 0.131 mmol, 2 equiv), following procedure D. The crude product was used in the next step without purification. The macrolactamization was performed using HBTU (37 mg, 0.98 mmol, 1.5 equiv), following procedure E. The crude product was used in the next step without purification. The desilylation was performed using NH4F (73 mg, 2.0 mmol, 30 equiv), following procedure F. Full conversion was achieved after stirring at room temperature for 64 h. The crude product was purified by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM. Additional purification by preparative reverse phase HPLC using gradient elution from 10 to 95% MeCN in 0.1% aqueous HCOOH solution afforded product 7e as an amorphous solid (15 mg, 37% yield). 1H NMR (400 MHz, methanol-d4) for a major rotamer: δ 7.68 (dt, J = 7.5, 1.7 Hz, 1H), 7.60 (t, J = 1.6 Hz, 1H), 7.55–7.50 (m, 1H), 7.50–7.46 (m, 1H), 7.17 (t, J = 8.2 Hz, 1H), 7.11 (t, J = 7.8 Hz, 1H), 7.01–6.96 (m, 2H), 6.82 (d, J = 7.7 Hz, 1H), 6.74–6.70 (m, 1H), 6.64–6.60 (m, 1H), 6.60–6.57 (m, 1H), 4.10 (ddd, J = 11.5, 8.1, 3.6 Hz, 1H), 3.77–3.69 (m, 6H), 3.54–3.45 (m, 1H), 3.40–3.32 (m, 3H), 3.19 (dd, J = 13.5, 3.6 Hz, 1H), 2.63 (dd, J = 13.4, 11.2 Hz, 1H), 2.55 (dd, J = 11.9, 3.3 Hz, 1H), 2.41 (dd, J = 11.9, 7.5 Hz, 1H), 1.64–1.48 (m, 4H), 1.46 (s, 3H), 1.45 (s, 3H), 1.06 (s, 9H). 13C NMR (101 MHz, methanol-d4) for a major rotamer: δ 174.4, 168.8, 161.2, 159.2, 149.9, 141.8, 138.7, 136.0, 130.7, 130.5, 130.3, 130.2, 129.4, 125.4, 122.7, 119.4, 117.9, 113.3, 113.2, 112.5, 74.1, 67.8, 56.9, 56.8, 56.1, 55.7, 52.2, 47.5, 37.7, 35.4, 30.2, 29.0, 28.9, 26.8, 26.7. HRMS-ESI (m/z) calcd for C37H50N3O5 [M + H]+ 616.3750. Found 616.3769. [α]D20 = −2.1 (c 0.9, CHCl3).
(S)-4-((R)-1-Hydroxy-2-((2-(3-methoxyphenyl)propan-2-yl)amino)ethyl)-13-propyl-7-oxa-3,13-diaza-1,6(1,3)-dibenzenacyclotetradecaphane-2,14-dione (7g)
The hydrolysis of methyl benzoate 25c (50 mg 0.067 mmol, 1 equiv) was performed using 1 M aqueous NaOH solution (134 μL, 0.134 mmol, 2 equiv), following procedure D. The crude product was used in the next step without purification. The macrolactamization was performed using HBTU (38 mg, 0.10 mmol, 1.5 equiv), following procedure E. The crude product was used in the next step without purification. The desilylation was performed using NH4F (50 mg, 1.3 mmol, 20 equiv), following procedure F. Full conversion was achieved after stirring at room temperature for 120 h. Purification by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM afforded product 7g as an amorphous solid (9 mg, 22% yield). 1H NMR (400 MHz, methanol-d4) for a mixture of rotamers (0.8:0.2): δ 7.60 (d, J = 6.2 Hz, 1H), 7.49–7.37 (m, 3H), 7.19 (t, J = 7.9 Hz, 1H), 7.07 (t, J = 7.8 Hz, 1H), 7.03–6.96 (m, 3H), 6.80 (d, J = 7.6 Hz, 1H), 6.77–6.69 (m, 1H), 6.65 (d, J = 8.0 Hz, 1H), 4.53–4.37 (m, 2H), 4.17–4.08 (m, 1H), 3.75 (s, 3H), 3.68–3.52 (m, 3H), 3.52–3.40 (m, 1H), 3.29–3.22 (m, 1H), 2.58 (t, J = 12.5 Hz, 1H), 2.51 (dd, J = 11.9, 3.3 Hz, 1H), 2.42 (dd, J = 12.0, 7.3 Hz, 1H), 1.80 (ddd, J = 29.3, 14.1, 6.6 Hz, 2H), 1.47 (s, 6H), 1.04 (t, J = 7.4 Hz, 2.4H), 0.80 (br s, 0.6H). 13C NMR (101 MHz, methanol-d4) for a major rotamer: δ 173.6, 169.5, 161.2, 158.4, 149.8, 142.3, 137.3, 135.5, 131.2, 130.9, 130.4, 130.3, 125.4, 122.8, 120.5, 119.3, 113.3, 112.4, 74.7, 63.9, 56.9, 56.2, 55.6, 49.0, 47.6, 47.4, 38.5, 30.3, 28.5, 21.7, 11.7. HRMS-ESI (m/z) calcd for C33H42N3O5 [M + H]+ 560.3124. Found 560.3136. [α]D20 = −10.1 (c 1.0, CHCl3).
(S)-4-((R)-1-Hydroxy-2-((2-(3-methoxyphenyl)propan-2-yl)amino)ethyl)-13-propyl-7-oxa-3,13-diaza-1,6(1,3)-dibenzenacyclotetradecaphane-2,14-dione (7h)
The hydrolysis of methyl benzoate 25d (57 mg 0.081 mmol, 1 equiv) was performed using 1 M aqueous NaOH solution (162 μL, 0.162 mmol, 2 equiv), following procedure D. The crude product was used in the next step without purification. The macrolactamization was performed using HBTU (46 mg, 0.121 mmol, 1.5 equiv), following procedure E. The crude product was used in the next step without purification. The desilylation was performed using NH4F (76 mg, 2.0 mmol, 25 equiv), by following procedure F. Full conversion was achieved after stirring at room temperature for 100 h. The crude product was purified by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM. Additional purification by preparative reverse phase HPLC using gradient elution from 10 to 95% MeCN in 0.1% aqueous HCOOH solution afforded product 7h as an amorphous solid (9 mg, 19% yield). 1H NMR (400 MHz, methanol-d4) for a mixture of rotamers (0.87:0.13): δ 7.78–7.25 (m, 5H), 7.25–6.95 (m, 4H), 6.93–6.85 (m, 1H), 6.84–6.76 (m, 1H), 6.76–6.52 (m, 1H), 4.64–4.10 (m, 2H), 4.10–3.89 (m, 1H), 3.88–3.69 (m, 4H), 3.69–3.53 (m, 2H), 3.53–3.36 (m, 2H), 3.28–3.20 (m, 1H), 2.96–2.66 (m, 2H), 2.58 (t, J = 12.5 Hz, 1H), 1.90–1.51 (m, 8H), 1.04 (t, J = 7.4 Hz, 2.6H), 0.80 (br s, 0.4H). 13C NMR (101 MHz, methanol-d4) for a major rotamer: δ 173.6, 169.5, 161.2, 158.4, 149.8, 142.3, 137.3, 135.5, 131.2, 130.9, 130.4, 130.3, 125.4, 122.8, 120.5, 119.3, 113.3, 112.4, 74.7, 63.9, 56.9, 56.3, 55.6, 49.0, 47.6, 47.4, 38.5, 30.3, 28.5, 21.7, 11.7. HRMS-ESI (m/z) calcd for C33H42N3O5 [M + H]+ 560.3124. Found 560.3136. [α]D20 = 3.4 (c 0.8, CHCl3).
Methyl 3-((4-(Benzyloxy)butyl)carbamoyl)benzoate (27a)
The title compound was obtained as a colorless oil (117 mg, 68% yield) from acid 19 (90 mg, 0.50 mmol, 1 equiv), HBTU (190 mg, 0.50 mmol, 1 equiv), DIPEA (87 μL, 0.50 mmol, 1 equiv), and amine 26a (90 mg, 0.50 mmol, 1 equiv), following general procedure G. A pure material was obtained by column chromatography on silica gel using gradient elution from 10% EtOAc to 30% EtOAc in hexanes. 1H NMR (400 MHz, chloroform-d): δ 8.36 (td, J = 1.8, 0.6 Hz, 1H), 8.13 (dt, J = 7.8, 1.4 Hz, 1H), 7.97 (ddd, J = 7.8, 1.9, 1.2 Hz, 1H), 7.47 (td, J = 7.8, 0.6 Hz, 1H), 7.35–7.22 (m, 5H), 6.71–6.62 (m, 1H), 4.53 (s, 2H), 3.92 (s, 3H), 3.57–3.46 (m, 4H), 1.80–1.70 (m, 4H). 13C NMR (101 MHz, chloroform-d) δ 166.6, 166.5, 138.3, 135.2, 132.3, 131.9, 130.5, 128.9, 128.5, 127.8, 127.6, 73.2, 70.1, 52.5, 40.1, 27.3, 26.6. HRMS-ESI (m/z) calcd for C20H24NO4 [M + H]+ 342.1705. Found 342.1721.
Methyl 3-((4-(Benzyloxy)butyl)(methyl)carbamoyl)benzoate (27b)
The title compound was obtained as a colorless oil (120 mg, 45% yield) from acid 19 (149 mg, 0.825 mmol, 1.1 equiv), HBTU (313 mg, 0.825 mmol, 1.1 equiv), DIPEA (143 μL, 0.825 mmol, 1.1 equiv), and crude amine 26b (145 mg, 0.750 mmol, 1 equiv), following general procedure G. A pure material was obtained by column chromatography on silica gel using gradient elution from 10% EtOAc to 30% EtOAc in hexanes. 1H NMR (400 MHz, chloroform-d) for 2 rotamers: δ 8.11–8.02 (m, 2H), 7.58 (t, J = 6.6 Hz, 1H), 7.47 (q, J = 8.1 Hz, 1H), 7.39–7.23 (m, 5H), 4.53 (s, 1H), 4.40 (s, 1H), 3.92 (s, 3H), 3.56 (q, J = 6.0, 5.6 Hz, 2H), 3.35 (t, J = 6.1 Hz, 1H), 3.25 (t, J = 7.5 Hz, 1H), 3.08 (s, 1.5H), 2.93 (s, 1.5H), 1.83–1.64 (m, 3H), 1.49–1.40 (m, 1H). 13C NMR (101 MHz, chloroform-d) for 2 rotamers: δ 170.9, 170.4, 166.5, 138.7, 138.4, 137.2, 131.5, 131.3, 130.5, 130.4, 128.8, 128.5, 128.1, 128.0, 127.8, 127.7, 73.1, 70.1, 69.6, 52.4, 51.3, 47.5, 37.6, 32.9, 27.6, 27.2, 26.8, 25.4, 24.0. HRMS-ESI (m/z) calcd for C21H26NO4 [M + H]+ 356.1862. Found 356.1871.
Methyl 3-((4-(Benzyloxy)butyl)(3,3,3-trifluoropropyl)carbamoyl)benzoate (27c)
The title compound was obtained as a yellow oil (253 mg, 95% yield) from acid 19 (110 mg, 0.611 mmol, 1 equiv), HBTU (232 mg, 0.611 mmol, 1 equiv), DIPEA (317 μL, 1.83 mmol, 3 equiv), and amine 26c (168 mg, 0.611 mmol, 1 equiv), following general procedure G. A pure material was obtained by column chromatography on silica gel using 2% MeOH in DCM. 1H NMR (400 MHz, chloroform-d) for 2 rotamers: δ 8.09 (dt, J = 7.7, 1.5 Hz, 1H), 8.03 (t, J = 1.7 Hz, 1H), 7.55 (dt, J = 7.6, 1.5 Hz, 1H), 7.52–7.42 (m, 1H), 7.37–7.31 (m, 2H), 7.30–7.21 (m, 3H), 4.60–4.33 (m, 2H), 3.92 (s, 3H), 3.78–3.42 (m, 3H), 3.39–3.18 (m, 3H), 2.66–2.16 (m, 2H), 1.75–1.34 (m, 4H). 13C NMR (101 MHz, chloroform-d) for a major rotamer: δ 171.0, 166.3, 138.4, 136.7, 131.1, 130.8, 130.6, 128.9, 128.5, 127.82, 127.77, 127.71, 73.1, 69.5, 52.5, 50.2, 39.7, 31.9 (q, J = 29.2 Hz), 26.78, 26.02. HRMS-ESI (m/z) calcd for C23H27NO4F3 [M + H]+ 438.1906. Found 438.1901.
Methyl 3-((4-(Benzyloxy)butyl)(2-(dimethylamino)ethyl)carbamoyl)benzoate (27d)
The title compound was obtained as a yellow oil (2.1 g, 83% yield) from acid 19 (1.10 g, 6.11 mmol, 1 equiv), HBTU (2.32 mg, 6.11 mmol, 1 equiv), DIPEA (1.06 mL, 6.11 mmol, 1 equiv), and crude amine 26d (1.8 g, 7.3 mmol, 1.2 equiv), following general procedure G. A pure material was obtained by column chromatography on silica gel using 2% MeOH in DCM. 1H NMR (400 MHz, methanol-d4) for 2 rotamers: δ 8.14–7.95 (m, 2H), 7.68–7.39 (m, 2H), 7.38–7.16 (m, 5H), 4.57–4.30 (m, 2H), 3.70–3.51 (m, 3H), 3.41–3.21 (m, 6H), 2.68–2.38 (m, 2H), 2.36–1.96 (m, 6H), 1.87–1.32 (m, 4H). 13C NMR (101 MHz, methanol-d4) for 2 rotamers: δ 173.0, 167.7, 139.81, 139.70, 139.59, 138.3, 132.2, 131.8, 131.5, 131.4, 130.2, 129.38, 129.26, 128.89, 128.71, 128.65, 128.58, 73.96, 73.85, 71.0, 58.3, 57.2, 53.9, 50.7, 48.1, 46.6, 45.74, 45.58, 44.0, 28.1, 27.6, 26.7, 25.5. HRMS-ESI (m/z) calcd for C24H33N2O4 [M + H]+ 413.2440. Found 413.2445.
N1-(4-(Benzyloxy)butyl)-N3-((2S,3R)-1-(3-(benzyloxy)phenyl)-3-((tert-butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)isophthalamide (28a)
The hydrolysis of methyl benzoate was performed using compound 27a (205 mg, 0.601 mmol, 1 equiv) and 1 M NaOH solution (1.80 mL, 1.80 mmol, 3 equiv), following general procedure D. Full conversion was achieved after stirring at 50 °C for 4 h. Crude acid was used in the next step without further purification. The title compound was obtained as a colorless oil (190 mg, 37% yield over 2 steps) from amine 18 (330 mg, 0.601 mmol, 1 equiv), HBTU (342 mg, 0.902 mmol, 1.5 equiv), DIPEA (208 μL, 1.20 mmol, 2 equiv), and crude acid from the previous step, following general procedure G. A pure material was obtained by column chromatography on silica gel using gradient elution from 10% EtOAc in hexanes to 100% EtOAc. 1H NMR (400 MHz, methanol-d4): δ 8.22 (td, J = 1.9, 0.6 Hz, 1H), 7.93 (ddd, J = 7.8, 1.8, 1.1 Hz, 1H), 7.87 (ddd, J = 7.8, 1.9, 1.1 Hz, 1H), 7.53 (td, J = 7.7, 0.5 Hz, 1H), 7.39–7.33 (m, 2H), 7.33–7.29 (m, 5H), 7.29–7.27 (m, 1H), 7.27–7.24 (m, 1H), 7.24–7.18 (m, 2H), 7.16 (t, J = 7.8 Hz, 1H), 7.01–6.95 (m, 2H), 6.84–6.82 (m, 1H), 6.82–6.80 (m, 1H), 6.80–6.75 (m, 2H), 5.01 (s, 2H), 4.48 (s, 2H), 4.43 (td, J = 7.6, 4.4 Hz, 1H), 3.72 (td, J = 4.4, 2.9 Hz, 1H), 3.70 (s, 3H), 3.55–3.50 (m, 2H), 3.43–3.37 (m, 2H), 2.86 (dd, J = 13.7, 7.7 Hz, 1H), 2.68 (dd, J = 13.7, 7.6 Hz, 1H), 2.62 (dd, J = 12.1, 2.9 Hz, 1H), 2.53 (dd, J = 12.0, 4.4 Hz, 1H), 1.74–1.62 (m, 4H), 1.48 (s, 3H), 1.47 (s, 3H), 0.86 (s, 9H), −0.02 (s, 3H), −0.05 (s, 3H). 13C NMR (101 MHz, methanol-d4): δ 169.3, 169.0, 161.3, 160.3, 149.6, 141.2, 139.8, 138.8, 136.63, 136.58, 131.0, 130.9, 130.5, 130.4, 129.9, 129.5, 129.4, 128.9, 128.8, 128.6, 128.4, 127.2, 122.7, 119.4, 116.5, 114.3, 113.5, 112.5, 73.9, 72.1, 71.0, 70.8, 58.2, 56.7, 55.6, 45.8, 40.9, 38.7, 29.9, 29.2, 28.2, 27.3, 26.4, 18.9, −4.5, −4.6. HRMS-ESI (m/z) calcd for C52H68N3O6Si [M + H]+ 858.4877. Found 858.4862. [α]D20 = −40.7 (c 1.1, CHCl3).
N1-(4-(Benzyloxy)butyl)-N3-((2S,3R)-1-(3-(benzyloxy)phenyl)-3-((tert-butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)-N1-methylisophthalamide (28b)
The hydrolysis of methyl benzoate was performed using compound 27b (45 mg, 0.127 mmol, 1 equiv) and 1 M NaOH solution (254 μL, 0.254 mmol, 2 equiv), following general procedure D. Full conversion was achieved after stirring at 50 °C for 4 h. Crude acid was used in the next step without further purification. The title compound was obtained as a colorless oil (68 mg, 61% yield over 2 steps) from amine 18 (105 mg, 0.191 mmol, 1.5 equiv), HBTU (73 mg, 0.19 mmol, 1.5 equiv), DIPEA (66 μL, 0.38 mmol, 3 equiv), and crude acid from the previous step, following general procedure G. A pure material was obtained by column chromatography on silica gel using gradient elution from 10% EtOAc in hexanes to 100% EtOAc. 1H NMR (400 MHz, chloroform-d): δ 9.60 (d, J = 6.9 Hz, 1H), 7.96–7.86 (m, 2H), 7.57–7.48 (m, 1H), 7.48–7.43 (m, 1H), 7.43–7.38 (m, 2H), 7.38–7.27 (m, 8H), 7.26–7.23 (m, 1H), 7.21–7.11 (m, 1H), 7.01–6.91 (m, 2H), 6.84–6.74 (m, 4H), 5.02 (s, 2H), 4.56–4.31 (m, 3H), 3.75 (s, 3H), 3.66–3.60 (m, 1H), 3.59–3.47 (m, 2H), 3.35 (t, J = 6.1 Hz, 1H), 3.31–3.15 (m, 2H), 3.05 (s, 1.5H), 2.92 (s, 1.5H), 2.68 (d, J = 11.8 Hz, 1H), 2.54 (dd, J = 11.8, 4.6 Hz, 1H), 2.39 (dd, J = 13.7, 10.3 Hz, 1H), 1.80–1.57 (m, 3H), 1.52 (s, 3H), 1.50 (s, 3H), 1.46–1.38 (m, 1H), 0.79 (s, 9H), −0.13 (s, 3H), −0.17 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 171.3, 170.6, 166.4, 166.3, 159.9, 159.0, 148.2, 139.9, 138.7, 138.5, 137.34, 137.25, 137.17, 135.5, 129.7, 129.5, 128.8, 128.7, 128.5, 128.0, 127.8, 127.7, 127.6, 125.7, 125.5, 121.8, 118.3, 115.5, 113.2, 112.6, 111.4, 73.1, 70.0, 69.6, 68.6, 58.0, 55.7, 55.3, 51.3, 47.4, 44.1, 38.6, 37.6, 32.8, 29.8, 29.4, 27.3, 26.8, 25.9, 25.4, 23.9, −4.8, −5.1. HRMS-ESI (m/z) calcd for C53H70N3O6Si [M + H]+ 872.5034. Found 872.5039. [α]D20 = −36.2 (c 1.2, CHCl3).
N1-(4-(Benzyloxy)butyl)-N3-((2S,3R)-1-(3-(benzyloxy)phenyl)-3-((tert-butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)-N1-(3,3,3-trifluoropropyl)isophthalamide (28c)
The hydrolysis of methyl benzoate was performed using compound 27c (190 mg, 0.434 mmol, 1 equiv) and 1 M NaOH solution (2.61 mL, 2.61 mmol, 6 equiv), following general procedure D. Full conversion was achieved after stirring at room temperature for 4 h. Crude acid was used in the next step without further purification. The title compound was obtained as a colorless oil (240 mg, 58% yield over 2 steps) from amine 18 (239 mg, 0.435 mmol, 1 equiv), HBTU (165 mg, 0.435 mmol, 1 equiv), DIPEA (150 μL, 0.869 mmol, 2 equiv), and crude acid from the previous step, following general procedure G. A pure material was obtained by column chromatography on silica gel using gradient elution from 10% EtOAc in hexanes to 100% EtOAc. 1H NMR (400 MHz, chloroform-d): δ 9.63 (d, J = 6.8 Hz, 1H), 7.94–7.87 (m, 2H), 7.51–7.43 (m, 2H), 7.43–7.39 (m, 2H), 7.39–7.35 (m, 2H), 7.35–7.30 (m, 3H), 7.30–7.24 (m, 4H), 7.20–7.14 (m, 1H), 7.00–6.91 (m, 2H), 6.84–6.74 (m, 4H), 5.02 (s, 2H), 4.55–4.33 (m, 3H), 3.75 (s, 3H), 3.70–3.42 (m, 4H), 3.40–3.24 (m, 3H), 3.21 (dd, J = 13.7, 5.2 Hz, 1H), 2.68 (d, J = 12.1 Hz, 1H), 2.62–2.16 (m, 4H), 1.66–1.58 (m, 2H), 1.52 (s, 3H), 1.50 (s, 3H), 1.47–1.36 (m, 2H), 0.79 (s, 9H), −0.13 (s, 3H), −0.16 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 171.3, 166.2, 159.9, 159.0, 148.2, 139.8, 138.4, 137.2, 136.8, 135.8, 129.7, 129.5, 129.2, 128.9, 128.7, 128.5, 128.0, 127.7, 127.6, 126.7 (q, J = 255.1 Hz), 121.8, 118.2, 115.5, 113.1, 112.6, 111.4, 73.1, 70.0, 69.5, 68.6, 58.0, 55.7, 55.2, 50.2, 44.1, 39.5, 38.5, 31.9 (q, J = 27.3 Hz), 29.8, 29.4, 26.8, 26.0, 25.8, 18.1, −4.9, −5.1. HRMS-ESI (m/z) calcd for C55H71N3O6F3Si [M + H]+ 954.5064. Found 954.5062. [α]D20 = −29.8 (c 1.1, CHCl3).
N1-(4-(Benzyloxy)butyl)-N3-((2S,3R)-1-(3-(benzyloxy)phenyl)-3-((tert-butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)-N1-(2-(dimethylamino)ethyl)isophthalamide (28d)
The hydrolysis of methyl benzoate was performed using compound 27d (250 mg, 0.606 mmol, 1 equiv) and 1 M NaOH solution (1.21 mL, 1.21 mmol, 2 equiv), following general procedure D. Full conversion was achieved after stirring at 60 °C for 2 h. Crude acid was used in the next step without further purification. The title compound was obtained as a colorless oil (305 mg, 55% yield over 2 steps) from amine 18 (331 mg, 0.603 mmol, 1 equiv), HBTU (228 mg, 0.602 mmol, 1 equiv), DIPEA (208 μL, 1.20 mmol, 2 equiv), and crude acid from previous step, by following general procedure G. A pure material was obtained by column chromatography on silica gel using gradient elution from 10% EtOAc in hexanes to 100% EtOAc. 1H NMR (400 MHz, methanol-d4) for 2 rotamers: δ 7.87–7.81 (m, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.60–7.45 (m, 2H), 7.39–7.33 (m, 2H), 7.33–7.26 (m, 5H), 7.26–7.18 (m, 4H), 7.18–7.11 (m, 1H), 7.02–6.94 (m, 2H), 6.84–6.74 (m, 4H), 5.06–4.93 (m, 2H), 4.58–4.46 (m, 1H), 4.42 (td, J = 7.7, 4.3 Hz, 1H), 4.33 (s, 1H), 3.76–3.66 (m, 4H), 3.62 (t, J = 7.4 Hz, 1H), 3.53 (q, J = 7.8, 6.8 Hz, 2H), 3.35–3.32 (m, 1H), 3.29–3.22 (m, 2H), 2.86 (dd, J = 13.8, 7.6 Hz, 1H), 2.81 (s, 3H), 2.66 (dd, J = 13.7, 7.7 Hz, 1H), 2.62–2.55 (m, 2H), 2.55–2.48 (m, 1H), 2.29 (s, 3H), 1.96 (s, 3H), 1.80–1.72 (m, 1H), 1.72–1.64 (m, 1H), 1.64–1.54 (m, 1H), 1.52–1.42 (m, 6H), 1.41–1.32 (m, 1H), 0.84 (s, 9H), −0.05 (s, 3H), −0.07 (s, 3H). 13C NMR (101 MHz, methanol-d4) for 2 rotamers: δ 173.1, 168.5, 161.3, 160.2, 155.6, 149.5, 141.2, 139.6, 138.74, 138.71, 138.35, 138.28, 136.3, 130.63, 130.57, 130.47, 130.2, 129.5, 129.4, 129.2, 128.9, 128.8, 128.7, 128.4, 126.2, 122.7, 119.4, 116.6, 114.4, 113.6, 112.5, 74.0, 73.9, 72.0, 71.0, 70.8, 70.6, 58.3, 58.2, 57.3, 56.7, 55.7, 50.2, 46.7, 45.8, 45.6, 44.1, 39.0, 38.7, 30.4, 30.3, 29.3, 29.2, 28.1, 27.6, 26.7, 26.5, 26.4, 25.6, 18.9, −4.5, −4.6. HRMS-ESI (m/z) calcd for C56H77N4O6Si [M + H]+ 929.5612. Found 929.5617. [α]D20 = −44.9 (c 1.0, CHCl3).
N1-((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-1-(3-hydroxyphenyl)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)-N3-(4-hydroxybutyl)isophthalamide (29a)
The title compound was obtained as a colorless oil (52 mg, 73% yield) from compound 28a (90 mg, 0.10 mmol, 1 equiv) and 10% palladium on carbon (22 mg, 0.021 mmol, 0.2 equiv), following general procedure H. 1H NMR (400 MHz, methanol-d4): δ 8.22 (td, J = 1.8, 0.5 Hz, 1H), 7.95 (ddd, J = 7.8, 1.8, 1.1 Hz, 1H), 7.87 (ddd, J = 7.7, 1.9, 1.1 Hz, 1H), 7.54 (td, J = 7.8, 0.6 Hz, 1H), 7.21 (t, J = 8.0 Hz, 1H), 7.09–7.03 (m, 1H), 6.99 (ddd, J = 7.7, 1.8, 0.9 Hz, 1H), 6.97 (t, J = 2.1 Hz, 1H), 6.77 (ddd, J = 8.2, 2.5, 0.8 Hz, 1H), 6.69–6.64 (m, 2H), 6.61 (ddd, J = 8.1, 2.4, 1.1 Hz, 1H), 4.42 (td, J = 7.6, 4.4 Hz, 1H), 3.78 (td, J = 4.3, 2.9 Hz, 1H), 3.71 (s, 3H), 3.60 (t, J = 6.3 Hz, 2H), 3.41 (t, J = 6.9 Hz, 2H), 2.85 (dd, J = 13.6, 7.5 Hz, 1H), 2.71–2.61 (m, 2H), 2.58 (dd, J = 12.0, 4.4 Hz, 1H), 1.75–1.65 (m, 2H), 1.65–1.56 (m, 2H), 1.50 (s, 3H), 1.49 (s, 3H), 0.86 (s, 9H), −0.01 (s, 3H), −0.04 (s, 3H). 13C NMR (101 MHz, methanol-d4): δ 169.3, 169.0, 161.3, 158.6, 149.5, 141.1, 136.6, 131.0, 130.8, 130.5, 130.4, 129.8, 127.2, 121.3, 119.3, 116.9, 114.4, 113.4, 112.6, 62.6, 58.2, 56.8, 55.6, 45.8, 40.9, 38.7, 31.0, 29.9, 29.1, 27.0, 26.4, 18.9, −4.5, −4.7. HRMS-ESI (m/z) calcd for C39H58N3O6Si [M + H]+ 692.4095. Found 692.4100. [α]D20 = −52.8 (c 1.0, CHCl3).
N1-((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-1-(3-hydroxyphenyl)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)-N3-(4-hydroxybutyl)-N3-methylisophthalamide (29b)
The title compound was obtained as a colorless oil (75 mg, 64% yield) from compound 28b (147 mg, 0.169 mmol, 1 equiv) and 10% palladium on carbon (36 mg, 0.034 mmol, 0.2 equiv), following general procedure H. 1H NMR (400 MHz, methanol-d4): δ 7.91–7.83 (m, 1H), 7.79 (br s, 1H), 7.64–7.55 (m, 2H), 7.26 (t, J = 8.0 Hz, 1H), 7.09 (t, J = 8.1 Hz, 1H), 7.06–7.00 (m, 2H), 6.83 (dd, J = 8.1, 2.4 Hz, 1H), 6.71–6.66 (m, 2H), 6.64 (ddd, J = 8.1, 2.3, 1.2 Hz, 1H), 4.45 (td, J = 7.6, 4.6 Hz, 1H), 3.82 (td, J = 4.4, 2.9 Hz, 1H), 3.76 (s, 3H), 3.66 (t, J = 6.4 Hz, 1H), 3.61 (t, J = 7.3 Hz, 1H), 3.46 (t, J = 6.3 Hz, 1H), 3.33–3.30 (m, 1H), 3.11 (s, 1.5H), 2.99 (s, 1.5H), 2.87 (dd, J = 13.7, 7.7 Hz, 1H), 2.75–2.66 (m, 2H), 2.62 (dd, J = 12.0, 4.4 Hz, 1H), 1.84–1.73 (m, 1H), 1.73–1.59 (m, 2H), 1.54 (s, 3H), 1.53 (s, 3H), 1.37 (dt, J = 9.0, 6.5 Hz, 1H), 0.90 (s, 9H), 0.03 (s, 3H), 0.00 (s, 3H). 13C NMR (101 MHz, methanol-d4): δ 173.1, 172.58, 168.8, 161.3, 158.6, 149.3, 141.1, 138.2, 138.1, 136.4, 130.8, 130.5, 130.4, 130.2, 130.1, 129.4, 129.3, 126.32, 126.28, 121.3, 119.3, 116.9, 114.4, 113.5, 112.6, 72.1, 62.5, 62.1, 58.1, 56.9, 55.6, 52.4, 45.8, 38.6, 38.0, 33.2, 30.8, 30.3, 30.0, 29.0, 26.5, 26.4, 25.7, 24.4, 18.9, −4.5, −4.7. HRMS-ESI (m/z) calcd for C38H56N3O6Si [M + H]+ 678.3938. Found 678.3958. [α]D20 = −37.0 (c 2.0, CHCl3).
N1-((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-1-(3-hydroxyphenyl)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)-N3-(4-hydroxybutyl)-N3-(3,3,3-trifluoropropyl)isophthalamide (29c)
The title compound was obtained as a colorless oil (123 mg, 69% yield) from compound 28c (220 mg, 0.231 mmol, 1 equiv) and 10% palladium on carbon (49 mg, 0.046 mmol, 0.2 equiv), following general procedure H. 1H NMR (400 MHz, methanol-d4): δ 7.90–7.83 (m, 1H), 7.82–7.78 (m, 1H), 7.63–7.50 (m, 2H), 7.23 (t, J = 7.9 Hz, 1H), 7.01–6.94 (m, 2H), 6.89 (t, J = 7.6 Hz, 1H), 6.79 (ddd, J = 8.2, 2.5, 0.9 Hz, 1H), 6.48–6.42 (m, 2H), 6.31 (dt, J = 7.4, 1.4 Hz, 1H), 4.42 (ddd, J = 8.9, 6.7, 3.9 Hz, 1H), 3.82 (td, J = 4.4, 2.6 Hz, 1H), 3.79–3.74 (m, 1H), 3.72 (s, 3H), 3.67–3.36 (m, 3H), 3.31–3.26 (m, 1H), 2.88 (dd, J = 13.6, 6.8 Hz, 1H), 2.72 (dd, J = 12.0, 2.6 Hz, 1H), 2.68–2.40 (m, 4H), 1.83–1.55 (m, 3H), 1.50 (s, 3H), 1.49 (s, 3H), 1.36–1.30 (m, 1H), 0.84 (s, 9H), −0.04 (s, 3H), −0.08 (s, 3H). 13C NMR (101 MHz, methanol-d4): δ 173.3, 168.44, 168.40, 161.3, 149.5, 139.7, 137.9, 136.8, 130.6, 130.4, 130.2, 130.1, 129.3, 127.9 (q, J = 272.5 Hz), 126.2, 120.9, 119.3, 118.2, 115.5, 113.3, 112.7, 71.4, 62.0, 58.6, 56.7, 55.7, 51.1, 45.6, 40.3, 39.1, 32.3 (q, J = 28.0 Hz), 30.9, 30.4, 30.22, 29.3, 26.6, 26.4, 26.2, 24.9, 18.9, −4.6. HRMS-ESI (m/z) calcd for C41H59N3O6Si [M + H]+ 774.4125. Found 774.4122. [α]D20 = −24.4 (c 1.0, CHCl3).
N1-((2S,3R)-3-((tert-butyldimethylsilyl)oxy)-1-(3-hydroxyphenyl)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)-N3-(2-(dimethylamino)ethyl)-N3-(4-hydroxybutyl)isophthalamide (29d)
The title compound was obtained as a colorless oil (292 mg, 68% yield) from compound 28d (534 mg, 0.575 mmol, 1 equiv) and 10% palladium on carbon (245 mg, 0.115 mmol, 0.2 equiv), following general procedure H. 1H NMR (400 MHz, methanol-d4): δ 7.89–7.85 (m, 1H), 7.84–7.77 (m, 1H), 7.62–7.53 (m, 2H), 7.23 (t, J = 8.0 Hz, 1H), 7.02–6.94 (m, 2H), 6.89 (t, J = 7.7 Hz, 1H), 6.79 (ddd, J = 8.2, 2.6, 0.8 Hz, 1H), 6.48–6.41 (m, 2H), 6.31 (dt, J = 7.5, 1.3 Hz, 1H), 4.42 (ddd, J = 10.2, 6.6, 3.8 Hz, 1H), 3.84–3.80 (m, 1H), 3.73 (s, 3H), 3.70–3.59 (m, 2H), 3.59–3.49 (m, 1H), 3.45–3.39 (m, 1H), 3.39–3.34 (m, 1H), 3.29–3.26 (m, 1H), 2.94–2.84 (m, 1H), 2.77–2.68 (m, 1H), 2.66–2.59 (m, 1H), 2.59–2.52 (m, 1H), 2.51–2.39 (m, 2H), 2.39–1.99 (m, 6H), 1.82–1.69 (m, 1H), 1.69–1.56 (m, 2H), 1.50 (s, 6H), 1.36–1.31 (m, 1H), 0.83 (s, 9H), −0.05 (s, 3H), −0.08 (s, 3H). 13C NMR (101 MHz, methanol-d4): δ 173.2, 168.4, 168.30, 161.3, 149.5, 139.7, 138.3, 136.6, 130.5, 130.4, 130.2, 129.2, 129.1, 126.22, 126.17, 120.9, 119.3, 118.2, 115.4, 113.3, 112.6, 71.3, 62.5, 62.1, 58.6, 58.3, 57.3, 56.7, 55.7, 50.9, 45.8, 45.6, 44.1, 39.2, 31.0, 30.8, 30.5, 30.3, 30.2, 29.4, 26.4, 26.3, 25.2, 18.9, −4.58, −4.60. HRMS-ESI (m/z) calcd for C42H65N4O6Si [M + H]+ 794.4673. Found 794.4667. [α]D20 = −44.7 (c 0.9, CHCl3).
(S)-4-((R)-1-Hydroxy-2-((2-(3-methoxyphenyl)propan-2-yl)amino)ethyl)-7-oxa-3,12-diaza-1,6(1,3)-dibenzenacyclotridecaphane-2,13-dione (7b)
The intramolecular Mitsunobu reaction (cyclization) was performed using compound 29a (61 mg, 0.090 mmol, 1 equiv), DTBAD (41 mg, 0.18 mmol, 2 equiv), and PPh3 (47 mg, 0.18 mmol, 2 equiv), following general procedure I. The desilylation was performed using NH4F (73 mg, 2.7 mmol, 30 equiv), following procedure F. Full conversion was achieved after stirring at 40 °C overnight. The crude product was purified by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM. Additional purification by preparative reverse phase HPLC using gradient elution from 10 to 95% MeCN in 0.1% aqueous HCOOH solution afforded product 7b as an amorphous solid (13 mg, 26% yield). 1H NMR (400 MHz, methanol-d4): δ 7.84 (dt, J = 7.7, 1.5 Hz, 1H), 7.64 (dt, J = 7.7, 1.4 Hz, 1H), 7.49 (t, J = 7.7 Hz, 1H), 7.36–7.34 (m, 1H), 7.21 (t, J = 7.9 Hz, 1H), 7.15 (t, J = 7.9 Hz, 1H), 7.05–6.99 (m, 3H), 6.86 (dt, J = 7.9, 1.2 Hz, 1H), 6.80–6.71 (m, 2H), 4.22 (dddd, J = 18.9, 11.6, 6.7, 4.3 Hz, 2H), 4.15–4.06 (m, 1H), 3.76 (s, 3H), 3.64–3.56 (m, 2H), 3.46 (ddd, J = 13.7, 6.9, 3.8 Hz, 1H), 3.24 (dd, J = 13.7, 4.1 Hz, 1H), 2.72–2.59 (m, 1H), 2.54–2.41 (m, 2H), 2.03–1.92 (m, 2H), 1.92–1.84 (m, 2H), 1.49 (s, 3H), 1.48 (s, 3H). 13C NMR (101 MHz, methanol-d4): δ 170.6, 169.8, 161.2, 159.6, 149.8, 141.8, 136.9, 136.2, 131.7, 131.6, 131.0, 130.3, 130.2, 125.5, 122.2, 119.3, 118.0, 113.7, 113.3, 112.4, 74.6, 68.3, 56.9, 55.7, 55.6, 47.3, 41.3, 37.7, 30.1, 29.1, 28.8, 26.5. HRMS-ESI (m/z) calcd for C32H40N3O5 [M + H]+ 546.2968. Found 546.2956. [α]D20 = −30.3 (c 1.0, CHCl3).
(S)-4-((R)-1-Hydroxy-2-((2-(3-methoxyphenyl)propan-2-yl)amino)ethyl)-12-methyl-7-oxa-3,12-diaza-1,6(1,3)-dibenzenacyclotridecaphane-2,13-dione (7c)
The intramolecular Mitsunobu reaction (cyclization) was performed using compound 29b (33 mg, 0.048 mmol, 1 equiv), DTBAD (22 mg, 0.10 mmol, 2 equiv), and PPh3 (25 mg, 0.10 mmol, 2 equiv), following general procedure I. The desilylation was performed using NH4F (53 mg, 1.43 mmol, 30 equiv), following procedure F. Full conversion was achieved after stirring at 40 °C overnight. Purification by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM afforded product 7c as an amorphous solid (15 mg, 56% yield). 1H NMR (400 MHz, methanol-d4) for the mixture of rotamers: δ 7.73–7.24 (m, 4H), 7.24–6.98 (m, 4H), 6.97–6.64 (m, 4H), 4.68–4.03 (m, 2H), 3.94–3.59 (m, 5H), 3.31–2.86 (m, 5H), 2.72–2.19 (m, 4H), 2.03–1.83 (m, 1H), 1.82–1.71 (m, 1H), 1.70–1.39 (m, 8H). 13C NMR (101 MHz, methanol-d4) for the mixture of rotamers: δ 173.12, 173.05, 171.2, 169.0, 161.41, 161.35, 159.6, 159.1, 147.7, 141.8, 141.6, 138.1, 138.0, 136.6, 135.3, 131.0, 130.9, 130.69, 130.65, 130.57, 130.51, 130.3, 130.1, 129.8, 125.5, 124.9, 122.6, 121.6, 119.29, 119.24, 118.1, 116.0, 115.6, 113.9, 113.6, 113.4, 113.34, 113.25, 113.16, 73.8, 73.4, 67.8, 67.1, 58.9, 58.4, 56.2, 56.1, 55.72, 55.67, 51.9, 47.9, 47.4, 38.2, 38.1, 37.3, 37.2, 33.0, 28.9, 28.4, 28.31, 28.25, 26.4, 26.0, 22.4. HRMS-ESI (m/z) calcd for C33H42N3O5 [M + H]+ 560.3124. Found 560.3129. [α]D20 = 24.5 (c 0.7, CHCl3).
(S)-4-((R)-1-Hydroxy-2-((2-(3-methoxyphenyl)propan-2-yl)amino)ethyl)-12-(3,3,3-trifluoropropyl)-7-oxa-3,12-diaza-1,6(1,3)-dibenzenacyclotridecaphane-2,13-dione (7d)
The intramolecular Mitsunobu reaction (cyclization) was performed using compound 29c (100 mg, 0.129 mmol, 1 equiv), DTBAD (89 mg, 0.39 mmol, 3 equiv), and PPh3 (102 mg, 0.388 mmol, 3 equiv), following general procedure I. The desilylation was performed using NH4F (104 mg, 3.87 mmol, 30 equiv), following procedure F. Full conversion was achieved after stirring at 40 °C overnight. Purification by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM afforded product 7d as an amorphous solid (31 mg, 47% yield). 1H NMR (400 MHz, methanol-d4) for the mixture of rotamers: δ 7.67 (dt, J = 7.7, 1.6 Hz, 1H), 7.61–7.55 (m, 1H), 7.51 (t, J = 7.6 Hz, 1H), 7.48–7.42 (m, 1H), 7.16 (t, J = 8.0 Hz, 1H), 7.14–7.08 (m, 1H), 7.06–6.94 (m, 2H), 6.85–6.79 (m, 1H), 6.74–6.69 (m, 1H), 6.68–6.61 (m, 2H), 4.16 (ddd, J = 11.3, 7.8, 3.4 Hz, 1H), 3.84–3.74 (m, 4H), 3.72 (s, 3H), 3.71–3.66 (m, 1H), 3.29–3.10 (m, 3H), 2.72–2.57 (m, 3H), 2.54 (dd, J = 11.9, 3.3 Hz, 1H), 2.42 (dd, J = 11.9, 7.5 Hz, 1H), 1.84–1.68 (m, 1H), 1.68–1.53 (m, 2H), 1.53–1.39 (m, 7H). 13C NMR (101 MHz, methanol-d4) for a major rotamer: δ 173.3, 168.9, 161.2, 159.1, 149.9, 141.9, 137.8, 135.9, 130.7, 130.43, 130.37, 130.29, 129.7, 127.9 (q, J = 276.1 Hz), 124.9, 122.8, 119.4, 117.7, 113.7, 113.2, 112.5, 74.2, 67.6, 56.9, 56.4, 55.7, 50.4, 47.4, 40.1 (q, J = 4.4 Hz), 37.5, 32.3 (q, J = 28.0 Hz), 30.1, 28.9, 26.5, 26.4. HRMS-ESI (m/z) calcd for C35H43N3O5F3 [M + H]+ 642.3155. Found 642.3151. [α]D20 = −19.7 (c 1.0, CHCl3).
(S)-12-(2-(Dimethylamino)ethyl)-4-((R)-1-hydroxy-2-((2-(3-methoxyphenyl)propan-2-yl)amino)ethyl)-7-oxa-3,12-diaza-1,6(1,3)-dibenzenacyclotridecaphane-2,13-dione (7f)
The intramolecular Mitsunobu reaction (cyclization) was performed using compound 29d (114 mg, 0.152 mmol, 1 equiv), DTBAD (88 mg, 0.38 mmol, 2.5 equiv), and PPh3 (100 mg, 0.381 mmol, 2 equiv), following general procedure I. The desilylation was performed using NH4F (149 mg, 4.56 mmol, 30 equiv), following procedure F. Full conversion was achieved after stirring at 40 °C overnight. The crude product was purified by column chromatography on silica gel using gradient elution from DCM to 50% MeOH in DCM. Additional purification by column chromatography on amino-functionalized silica gel using DCM afforded product 7f as an amorphous solid (33 mg, 40% yield). 1H NMR (400 MHz, methanol-d4) for the mixture of rotamers: δ 7.75–7.64 (m, 1H), 7.64–7.54 (m, 1H), 7.54–7.46 (m, 2H), 7.27–7.08 (m, 2H), 7.08–6.94 (m, 2H), 6.91–6.80 (m, 1H), 6.80–6.72 (m, 1H), 6.72–6.60 (m, 2H), 4.54–4.07 (m, 2H), 3.94–3.50 (m, 7H), 3.29–2.93 (m, 3H), 2.72–2.19 (m, 10H), 2.08–1.85 (m, 2H), 1.81–1.70 (m, 1H), 1.69–1.38 (m, 8H). 13C NMR (101 MHz, methanol-d4) for a major rotamer: δ 171.8, 167.4, 159.8, 157.7, 148.0, 140.4, 136.7, 134.1, 129.6, 129.0, 128.9, 128.3, 123.4, 121.3, 117.9, 116.7, 114.4, 112.2, 111.8, 111.2, 72.6, 66.4, 55.8, 55.7, 54.9, 54.2, 49.0, 45.9, 44.3, 44.0, 42.4, 36.0, 28.5, 27.3, 25.3, 25.1. HRMS-ESI (m/z) calcd for C36H49N4O5 [M + H]+ 617.3703. Found 617.3723. [α]D20 = 18.7 (c 1.0, CHCl3).
tert-Butyl ((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-1-(3-hydroxyphenyl)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)carbamate (30)
To a solution of compound 16 (440 mg, 0.678 mmol, 1 equiv) in MeOH (12 mL), 10% palladium on carbon (72 mg, 0.068 mmol, 0.1 equiv) was added. The reaction mixture was stirred under a H2 atmosphere (1 atm) for 3 h. Carbon was filtered using a syringe filter (Teflon; 0.45 μm, washed with MeOH). The filtrate was evaporated under reduced pressure. Purification by column chromatography on silica gel using gradient elution from 10 to 50% EtOAc in hexanes afforded product 30 as a colorless oil (304 mg, 80% yield). 1H NMR (400 MHz, chloroform-d): δ 7.26 (t, J = 8.0 Hz, 1H), 7.09 (t, J = 7.7 Hz, 1H), 7.06–6.96 (m, 2H), 6.78 (ddd, J = 8.2, 2.5, 0.9 Hz, 1H), 6.75–6.65 (m, 2H), 6.62 (t, J = 1.9 Hz, 1H), 6.44–6.02 (m, 1H), 4.02–3.90 (m, 1H), 3.82 (s, 3H), 3.73–3.57 (m, 1H), 2.83–2.65 (m, 1H), 2.56–2.39 (m, 3H), 1.46 (s, 3H), 1.43 (s, 3H), 1.42–1.22 (m, 9H), 0.87 (s, 9H), −0.03 (s, 3H), −0.07 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 159.6, 156.3, 156.2, 149.3, 140.4, 129.6, 129.3, 121.2, 118.6, 116.1, 113.4, 112.7, 111.4, 78.9, 71.4, 56.6, 55.6, 55.4, 44.7, 37.4, 29.7, 29.4, 28.6, 26.0, 18.2, −4.7, −4.8. HRMS-ESI (m/z) calcd for C31H51N2O5Si [M + H]+ 559.3567. Found 559.3563. [α]D20 = −22.3 (c 1.3, CHCl3).
3-((2S,3R)-2-((tert-Butoxycarbonyl)amino)-3-((tert-Butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butyl)phenyl trifluoromethanesulfonate (31)
To a solution of compound 30 (1.12 g, 2.00 mmol, 1 equiv) in DCM (25 mL), triethylamine (1.12 mL, 8.02 mmol, 4 equiv) was added, followed by N-phenyl-bis(trifluoromethanesulfonimide) (1.43 g, 4.01 mmol, 2 equiv). The reaction mixture was stirred at room temperature for 1 h. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in EtOAc (40 mL), washed sequentially with 1 M aqueous HCl (20 mL), 1 M aqueous NaOH (20 mL), and brine (20 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification by column chromatography on silica gel using gradient elution from 1 to 5% MeOH in DCM afforded product 31 as a colorless oil (1.16 g, 84% yield). 1H NMR (400 MHz, chloroform-d): δ 7.33 (t, J = 8.0 Hz, 1H), 7.26 (d, J = 7.8 Hz, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.13–7.07 (m, 1H), 7.06–7.04 (m, 1H), 7.03–6.99 (m, 2H), 6.77 (ddd, J = 8.1, 2.5, 0.9 Hz, 1H), 4.03–3.89 (m, 1H), 3.80 (s, 3H), 3.72–3.58 (m, 1H), 2.77 (dd, J = 14.0, 8.5 Hz, 1H), 2.64 (dd, J = 14.1, 6.5 Hz, 1H), 2.59–2.44 (m, 2H), 1.45 (s, 3H), 1.44 (s, 3H), 1.41–1.25 (m, 9H), 0.87 (s, 9H), −0.02 (s, 3H), −0.05 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 159.8, 155.8, 149.6, 149.2, 142.3, 130.1, 129.4, 129.3, 122.2, 119.0, 118.5, 112.7, 111.3, 78.9, 71.8, 56.2, 55.6, 55.3, 45.0, 37.0, 29.8, 29.2, 28.5, 25.9, 18.2. HRMS-ESI (m/z) calcd for C32H50N2O7SiSF3 [M + H]+ 691.3060. Found 691.3070. [α]D20 = −28.8 (c 1.2, CHCl3).
tert-Butyl ((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)-1-(3-vinylphenyl)butan-2-yl)carbamate (32)
To a solution of compound 31 (600 mg, 0.869 mmol, 1 equiv) in DMF (20 mL, sparged with argon), Pd(PPh3)4 (251 mg, 0.217 mmol, 0.25 equiv) was added, followed by 0.5 M LiCl solution in THF (5.21 mL, 2.61 mmol, 3 equiv). Tri-n-butyl(vinyl)tin (508 μL, 1.74 mmol, 2 equiv) was added, and the reaction mixture was stirred at 60 °C for 20 h. The solvent was evaporated under reduced pressure, and the residue was dissolved in diethyl ether (30 mL) and water (30 mL). The aqueous phase was separated and washed with diethyl ether (30 mL). The combined organic phase was washed with brine (20 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification by column chromatography on silica gel/powdered K2CO3 mixture (9:1 w/w) using 10% EtOAc in hexanes afforded product 32 as a colorless oil (390 mg, 79% yield). 1H NMR (400 MHz, chloroform-d): δ 7.25–7.12 (m, 4H), 7.07–6.96 (m, 3H), 6.77 (ddd, J = 8.2, 2.4, 1.0 Hz, 1H), 6.67 (dd, J = 17.6, 10.9 Hz, 1H), 6.48–6.20 (m, 1H), 5.72 (dd, J = 17.6, 1.0 Hz, 1H), 5.21 (dd, J = 10.9, 1.0 Hz, 1H), 4.05–3.93 (m, 1H), 3.80 (s, 3H), 3.72–3.59 (m, 1H), 2.83 (dd, J = 14.0, 7.7 Hz, 1H), 2.57 (dd, J = 13.7, 7.7 Hz, 1H), 2.50 (d, J = 4.2 Hz, 2H), 1.45 (s, 3H), 1.43 (s, 3H), 1.41–1.26 (m, 9H), 0.87 (s, 9H), −0.03 (s, 3H), −0.08 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 159.8, 155.9, 149.4, 139.1, 137.7, 137.0, 129.3, 128.7, 128.6, 127.2, 124.1, 118.5, 113.8, 112.5, 111.4, 78.6, 71.4, 56.5, 55.6, 55.3, 44.8, 37.4, 29.8, 29.3, 28.6, 26.0, 18.2, −4.7, −4.8. HRMS-ESI (m/z) calcd for C33H53N2O4Si [M + H]+ 569.3775. Found 569.3773. [α]D20 = −16.9 (c 1.2, CHCl3).
tert-Butyl ((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-1-(3-(2-hydroxyethyl)phenyl)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butan-2-yl)carbamate (33)
A solution of compound 32 (160 mg, 0.281 mmol, 1 equiv) in THF (10 mL) was cooled to 0 °C. Borane-dimethyl sulfide complex (160 μL, 1.69 mmol, 6 equiv) was added dropwise. The ice bath was removed, and the reaction mixture was left stirring at room temperature for 2 h. Then, 1 M aqueous NaOH solution (2.8 mL, 2.8 mmol, 10 equiv) and water (2.5 mL) were added, followed by 35% aqueous hydrogen peroxide solution (745 μL, 8.44 mmol, 30 equiv). The reaction mixture was stirred at room temperature for 10 min and then at 50 °C for 1 h. The reaction mixture was extracted with DCM (3 × 20 mL). The combined organic phase was washed with brine (30 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification by column chromatography on silica gel using gradient elution from DCM to 5% MeOH in DCM afforded product 33 as a colorless oil (130 mg, 79% yield). 1H NMR (400 MHz, chloroform-d): δ 7.26 (t, J = 7.9 Hz, 1H), 7.22–7.13 (m, 1H), 7.13–7.05 (m, 1H), 7.05–6.95 (m, 4H), 6.77 (ddd, J = 8.2, 2.5, 0.9 Hz, 1H), 6.12 (d, J = 8.5 Hz, 1H), 4.07–3.93 (m, 1H), 3.92–3.73 (m, 5H), 3.72–3.57 (m, 1H), 2.89–2.73 (m, 2H), 2.73–2.56 (m, 2H), 2.51 (d, J = 4.3 Hz, 2H), 1.46 (s, 3H), 1.44 (s, 3H), 1.33 (s, 9H), 0.87 (s, 9H), −0.01 (s, 3H), −0.05 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 159.7, 155.9, 149.4, 139.0, 138.8, 130.1, 129.3, 128.6, 127.7, 127.0, 118.5, 112.5, 111.3, 71.9, 63.8, 56.5, 55.6, 55.3, 45.0, 39.4, 37.5, 29.7, 29.3, 28.5, 26.0, 18.2, −4.6, −4.7. HRMS-ESI (m/z) calcd for C33H55N2O5Si [M + H]+ 587.3880. Found 587.3882. [α]D20 = −34.7 (c 1.0, CHCl3).
tert-Butyl ((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)-1-(3-(2-(2-oxo-2-(propylamino)ethoxy)ethyl)phenyl)butan-2-yl)carbamate (35)
A solution of compound 33 (1.4 g, 2.4 mmol, 1 equiv) in THF (50 mL) was cooled to −78 °C. Then, 1 M KHMDS solution in THF (3.58 mL, 3.58 mmol, 1.5 equiv) was added dropwise. The reaction mixture was stirred for 5 min, and then a solution of bromide 34 (644 mg, 3.58 mmol, 1.5 equiv) in THF (10 mL) was added. The reaction mixture was stirred for 1 h at −78 °C. The reaction mixture was quenched with water (10 mL) and extracted with EtOAc (2 × 30 mL). The combined organic phase was washed with brine (30 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification by column chromatography on silica gel using gradient elution from 10% EtOAc to 50% EtOAc in hexanes afforded product 35 as a colorless oil (1.20 g, 73% yield). 1H NMR (400 MHz, chloroform-d): δ 7.24 (t, J = 7.9 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.07–6.94 (m, 5H), 6.76 (ddd, J = 8.2, 2.5, 1.0 Hz, 1H), 6.45–6.24 (m, 2H), 4.00–3.93 (m, 1H), 3.98 (s, 1H), 3.91 (s, 2H), 3.80 (s, 3H), 3.72–3.59 (m, 3H), 3.19–3.07 (m, 2H), 2.85 (t, J = 6.7 Hz, 2H), 2.82–2.75 (m, 1H), 2.59–2.52 (m, 1H), 2.52–2.45 (m, 2H), 1.51–1.23 (m, 17H), 0.86 (t, J = 7.3 Hz, 3H), 0.86 (s, 9H), −0.05 (s, 3H), −0.10 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 169.7, 159.8, 156.0, 149.3, 139.3, 138.6, 129.7, 129.3, 128.6, 127.5, 126.8, 118.5, 112.5, 111.3, 78.6, 72.7, 71.3, 70.3, 56.7, 55.5, 55.3, 44.7, 40.6, 37.5, 36.3, 29.7, 29.4, 28.6, 26.0, 22.9, 18.2, 11.5, −4.7, −4.8. HRMS-ESI (m/z) calcd for C38H64N3O6Si [M + H]+ 686.4564. Found 686.4579. [α]D20 = −20.6 (c 1.0, CHCl3).
Methyl 3-((2-(3-((2S,3R)-2-((tert-Butoxycarbonyl)amino)-3-((tert-butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)butyl)phenethoxy)ethyl)(propyl)carbamoyl)benzoate (36)
To a solution of compound 35 (1.20 g, 1.75 mmol, 1 equiv) in THF (100 mL), borane-dimethyl sulfide complex was added. The reaction mixture was stirred at 75 °C for 4 h. The reaction mixture was cooled to 0 °C and was quenched by dropwise addition of MeOH (50 mL). Volatiles were evaporated under reduced pressure. The residue was dissolved in MeOH (30 mL) and evaporated again. The residue was dissolved in MeOH (50 mL), and 10% aqueous Rochelle′s salt solution (80 mL) was added. The mixture was stirred at 70 °C for 120 h. The reaction mixture was concentrated under reduced pressure, and the aqueous residue was extracted with EtOAc (3 × 40 mL). The combined organic phase was washed with brine (30 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM afforded crude amine (620 mg), which was used in the next step without further purification. Amide coupling was performed using crude amine from the previous step (620 mg, 0.92 mmol, 1 equiv), acid 19 (199 mg, 1.11 mmol, 1.2 equiv), DIPEA (319 μL, 1.84 mmol, 2 equiv), and HBTU (420 mg, 1.11 mmol, 1.2 equiv), following procedure G. Purification by column chromatography on silica gel using gradient elution from 10% EtOAc to 50% EtOAc in hexanes afforded product 36 as a colorless oil (480 mg, 33% yield over 2 steps). 1H NMR (400 MHz, methanol-d4) for 2 rotamers: δ 8.13–7.93 (m, 2H), 7.62–7.42 (m, 2H), 7.23 (t, J = 8.2 Hz, 1H), 7.15 (t, J = 7.5 Hz, 1H), 7.11–6.93 (m, 5H), 6.83–6.71 (m, 1H), 3.94–3.83 (m, 4H), 3.77 (s, 3H), 3.75–3.63 (m, 4H), 3.57 (t, J = 7.0 Hz, 1H), 3.52–3.44 (m, 2H), 3.44–3.37 (m, 1H), 3.18–3.11 (m, 1H), 2.91–2.78 (m, 2H), 2.75–2.64 (m, 1H), 2.60–2.33 (m, 3H), 1.73–1.59 (m, 1H), 1.54–1.40 (m, 7H), 1.39–1.20 (m, 9H), 0.96 (t, J = 7.4 Hz, 1.6H), 0.90 (s, 9H), 0.68 (t, J = 7.4 Hz, 1.4H), 0.03 (s, 3H), −0.02 (s, 3H). 13C NMR (101 MHz, methanol-d4) for 2 rotamers: δ 173.0, 167.6, 161.3, 157.8, 150.0, 140.5, 140.3, 140.2, 138.5, 138.4, 134.8, 132.7, 132.1, 131.6, 131.4, 131.3, 130.9, 130.8, 130.3, 130.1, 129.9, 129.31, 129.25, 128.5, 128.1, 127.8, 119.4, 113.4, 112.5, 79.8, 74.1, 73.2, 69.5, 68.9, 66.4, 59.6, 57.3, 56.8, 56.6, 55.7, 53.2, 52.9, 49.5, 46.3, 46.2, 38.0, 37.3, 37.2, 29.9, 29.1, 28.9, 28.5, 26.5, 22.7, 21.6, 19.0, 11.7, 11.3, −4.2, −4.5. HRMS-ESI (m/z) calcd for C47H72N3O8Si [M + H]+ 834.5089. Found 834.5102. [α]D20 = −11.6 (c 1.1, CHCl3).
(S)-4-((R)-1-Hydroxy-2-((2-(3-methoxyphenyl)propan-2-yl)amino)ethyl)-12-propyl-9-oxa-3,12-diaza-1,6(1,3)-dibenzenacyclotridecaphane-2,13-dione (7i)
To a solution of compound 36 (440 mg, 0.528 mmol, 1 equiv) in methanol (15 mL), 1 M aqueous NaOH solution (1.05 mL, 1.05 mmol, 2 equiv) was added, and the reaction mixture was stirred at 55 °C overnight. The solvent was evaporated under reduced pressure, and the residue was dissolved in 4 M HCl solution in dioxane (2.64 mL, 10.5 mmol, 20 mmol) and 6 M aqueous HCl solution (8.80 mL, 52.7 mmol, 100 equiv). The heterogeneous reaction mixture was stirred at 60 °C overnight. The resulting homogenous mixture was evaporated under reduced pressure. The residue was dissolved in DCM (40 mL). DIPEA (365 μL, 2.11 mmol, 4 equiv) was added, followed by HBTU (300 mg, 0.80 mmol, 1.5 equiv). The reaction mixture was washed with water (30 mL) and brine (30 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Purification by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM afforded product 7i as a colorless oil (156 mg, 49% yield). 1H NMR (400 MHz, methanol-d4) for a mixture of rotamers (0.55:0.35:0.1): δ 7.67–7.35 (m, 3.6H), 7.28–6.99 (m, 7H), 6.82–6.71 (m, 1H), 6.61–6.43 (m, 0.4H)4.55–3.84 (m, 2H), 3.83–3.56 (m, 8H), 3.44–3.35 (m, 2H), 3.29–2.99 (m, 2H), 2.96–2.64 (m, 3H), 2.58–2.41 (m, 2H), 1.73 (dddt, J = 27.1, 13.3, 8.5, 6.9 Hz, 1H), 1.50 (dd, J = 8.0, 3.9 Hz, 6H), 1.43–1.20 (m, 1H), 1.03 (t, J = 7.4 Hz, 1.65H), 0.74 (t, J = 7.4 Hz, 0.25H), 0.62 (t, J = 7.4 Hz, 1.1H). 13C NMR (101 MHz, methanol-d4) for two major rotamers: δ 173.8, 173.0, 171.1, 170.3, 161.2, 150.0, 149.9, 141.3, 140.4, 140.2, 140.0, 138.3, 137.7, 136.83, 136.80, 131.6, 131.5, 130.4, 130.29, 130.25, 130.20, 130.0, 129.7, 129.53, 129.49, 129.0, 128.8, 127.8, 127.0, 125.88, 125.86, 125.79, 119.3, 113.3, 113.2, 112.5, 112.4, 75.0, 74.6, 73.3, 72.7, 69.8, 67.7, 56.86, 56.84, 56.1, 55.7, 54.1, 53.5, 47.3, 46.7, 46.2, 37.6, 37.5, 37.4, 37.1, 30.2, 30.1, 29.0, 28.8, 22.5, 21.5, 11.7, 11.1. HRMS-ESI (m/z) calcd for C35H46N3O5 [M + H]+ 588.3437. Found 588.3450. [α]D20 = 34.7 (c 0.9, CHCl3).
(2R,3S)-2-((tert-Butyldimethylsilyl)oxy)-N1-(2-(3-methoxyphenyl)propan-2-yl)-4-(3-vinylphenyl)butane-1,3-diamine (37)
To a solution of compound 32 (400 mg, 0.703 mmol, 1 equiv) in DCM (1 mL), trifluoroacetic acid (1.08 mL, 14.1 mmol, 20 equiv) was added. The reaction mixture was stirred for 1 h. Volatiles were evaporated under reduced pressure. Diethyl ether (25 mL) and 1 M aqueous NaOH (20 mL) were added. The aqueous phase was extracted with diethyl ether (25 mL). The combined organic phase was washed with brine (20 mL), dried over anhydrous Na2SO4, and evaporated under reduced pressure. Crude product 37 (314 mg, 95% yield) was used in the next step without further purification. 1H NMR (400 MHz, methanol-d4): δ 7.32–7.21 (m, 4H), 7.05 (dt, J = 7.0, 1.8 Hz, 1H), 7.03–6.97 (m, 2H), 6.78 (ddd, J = 8.2, 2.5, 1.0 Hz, 1H), 6.71 (dd, J = 17.6, 10.9 Hz, 1H), 5.76 (dd, J = 17.6, 1.1 Hz, 1H), 5.22 (dd, J = 10.9, 1.1 Hz, 1H), 3.77 (s, 3H), 3.65 (td, J = 5.7, 4.1 Hz, 1H), 3.17 (ddd, J = 8.3, 6.3, 4.2 Hz, 1H), 2.72 (dd, J = 13.5, 6.3 Hz, 1H), 2.53–2.45 (m, 3H), 1.45 (s, 3H), 1.42 (s, 3H), 0.92 (s, 9H), 0.05 (s, 3H), 0.03 (s, 3H). 13C NMR (101 MHz, methanol-d4): δ 161.3, 150.2, 140.6, 139.3, 138.1, 130.3, 130.0, 129.7, 128.0, 125.3, 119.3, 114.2, 113.4, 112.4, 75.9, 57.6, 56.8, 55.7, 45.0, 40.0, 30.5, 28.7, 26.5, 19.0, −4.1, −4.4. HRMS-ESI (m/z) calcd for C28H45N2O2Si [M + H]+ 469.3251. Found 469.3256. [α]D20 = 4.3 (c 1.0, CHCl3).
N1-(But-3-en-1-yl)-N3-((2S,3R)-3-((tert-butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)-1-(3-vinylphenyl)butan-2-yl)-N1-propylisophthalamide (39a)
The title compound was obtained as a colorless oil (280 mg, 60% yield) from amine 37 (305 mg, 0.651 mmol, 1 equiv), HBTU (247 mg, 0.651, 1 equiv), DIPEA (225 μL, 1.30 mmol, 2 equiv), and acid 38a (170 mg, 0.651 mmol, 1 equiv), following general procedure G. A pure material was obtained by column chromatography on silica gel using 2% MeOH in DCM. 1H NMR (400 MHz, chloroform-d) for 2 rotamers: δ 9.56 (d, J = 6.7 Hz, 1H), 7.92–7.85 (m, 2H), 7.51–7.42 (m, 2H), 7.29–7.24 (m, 1H), 7.23–7.18 (m, 3H), 7.07 (dt, J = 7.0, 1.8 Hz, 1H), 7.00–6.93 (m, 2H), 6.78 (ddd, J = 8.2, 2.5, 0.9 Hz, 1H), 6.66 (dd, J = 17.6, 10.9 Hz, 1H), 5.93–5.78 (m, 0.5H), 5.71 (dd, J = 17.7, 1.0 Hz, 1H), 5.61–5.46 (m, 0.5H), 5.21 (dd, J = 10.8, 0.9 Hz, 1H), 5.18–4.91 (m, 2H), 4.51–4.41 (m, 1H), 3.75 (s, 3H), 3.65 (ddd, J = 4.7, 3.1, 1.3 Hz, 1H), 3.61–3.50 (m, 1H), 3.50–3.41 (m, 1H), 3.33–3.10 (m, 3H), 2.70 (dd, J = 11.8, 1.5 Hz, 1H), 2.57 (ddd, J = 11.8, 4.8, 1.2 Hz, 1H), 2.48–2.36 (m, 2H), 2.29–2.17 (m, 1H), 1.75–1.61 (m, 1H), 1.53 (s, 3H), 1.51 (s, 3H), 1.02–0.92 (m, 1H), 0.82–0.68 (m, 11H), −0.14 (s, 3H), −0.18 (s, 3H). 13C NMR (101 MHz, chloroform-d) for 2 rotamers: δ 171.1, 166.4, 159.9, 148.2, 138.6, 137.9, 137.8, 135.6, 134.2, 129.5, 129.4, 128.8, 128.7, 128.6, 127.6, 127.1, 125.3, 124.4, 118.3, 117.7, 116.9, 114.1, 112.6, 111.4, 68.7, 57.9, 55.7, 55.3, 51.1, 48.7, 46.7, 44.3, 44.2, 38.3, 33.3, 32.1, 29.8, 29.5, 25.9, 22.1, 20.8, 18.1, 11.6, 11.2, −4.9, −5.1. HRMS-ESI (m/z) calcd for C43H62N3O4Si [M + H]+ 712.4510. Found 712.4526. [α]D20 = −46.8 (c 1.4, CHCl3).
N1-((2S,3R)-3-((tert-Butyldimethylsilyl)oxy)-4-((2-(3-methoxyphenyl)propan-2-yl)amino)-1-(3-vinylphenyl)butan-2-yl)-N3-(pent-4-en-1-yl)-N3-propylisophthalamide (39b)
The title compound was obtained as a colorless oil (550 mg, 83% yield) from amine 37 (426 mg, 0.908 mmol, 1 equiv), HBTU (344 mg, 0.908 mmol, 1 equiv), DIPEA (314 μL, 1.82 mmol, 2 equiv), and acid 38b (250 mg, 0.908 mmol, 1 equiv), following general procedure G. A pure material was obtained by column chromatography on silica gel using gradient elution from 10 to 50% EtOAc in hexanes. 1H NMR (400 MHz, chloroform-d) for 2 rotamers: δ 9.57 (d, J = 6.9 Hz, 1H), 7.93–7.83 (m, 2H), 7.51–7.42 (m, 2H), 7.30–7.18 (m, 4H), 7.06 (dt, J = 6.8, 1.8 Hz, 1H), 7.00–6.92 (m, 2H), 6.81–6.75 (m, 1H), 6.66 (dd, J = 17.6, 10.9 Hz, 1H), 5.95–5.76 (m, 1H), 5.84 (d, J = 7.2 Hz, 0.5H), 5.71 (dd, J = 17.6, 1.0 Hz, 1H), 5.64–5.54 (m, 0.5H), 5.21 (dd, J = 10.9, 1.0 Hz, 1H), 5.03–4.95 (m, 1H), 4.95–4.78 (m, 1H), 4.46 (tt, J = 8.8, 5.4 Hz, 1H), 3.75 (s, 3H), 3.68–3.62 (m, 1H), 3.56–3.35 (m, 2H), 3.26–3.09 (m, 3H), 2.70 (d, J = 11.7 Hz, 1H), 2.62–2.54 (m, 1H), 2.43 (dd, J = 13.8, 10.2 Hz, 1H), 2.16–2.08 (m, 1H), 1.91–1.82 (m, 1H), 1.80–1.70 (m, 1H), 1.70–1.62 (m, 1H), 1.62–1.58 (m, 1H), 1.58–1.55 (m, 1H), 1.53 (s, 3H), 1.51 (s, 3H), 1.00–0.94 (m, 1H), 0.82–0.69 (m, 11H), −0.14 (s, 3H), −0.18 (s, 3H). 13C NMR (101 MHz, chloroform-d) for 2 rotamers: δ 171.0, 166.4, 159.9, 148.2, 138.6, 137.96, 137.92, 137.8, 137.1, 136.8, 135.5, 129.5, 129.3, 128.8, 128.7, 127.6, 127.1, 125.3, 124.4, 118.3, 115.6, 115.2, 114.0, 112.6, 111.4, 68.7, 57.9, 55.7, 55.2, 51.0, 48.7, 46.7, 44.6, 44.1, 38.3, 31.4, 30.7, 29.8, 29.4, 27.9, 26.8, 25.8, 22.1, 20.9, 18.1, 11.6, 11.2, −4.9, −5.1. HRMS-ESI (m/z) calcd for C44H64N3O4Si [M + H]+ 726.4666. Found 726.4677. [α]D20 = −35.3 (c 2.5, CHCl3).
(S)-4-((R)-1-Hydroxy-2-((2-(3-methoxyphenyl)propan-2-yl)amino)ethyl)-11-propyl-3,11-diaza-1,6(1,3)-dibenzenacyclododecaphane-2,12-dione (7j)
Ring-closing metathesis and hydrogenation of the double bond were performed using compound 39a (250 mg, 0.351 mmol, 1 equiv), Zhan Catalyst-1B (26 mg, 0.035 mmol, 0.1 equiv), and 10% palladium on carbon (37 mg, 0.035 mmol, 0.1 equiv), following general procedure J. Crude macrocycle 40a (210 mg, 87% yield) was used in the next step without purification.
The desilylation was performed using crude macrocycle 40a from the previous step (210 mg, 0.306 mmol, 1 equiv), NH4F (340 mg, 9.18 mmol, 30 equiv), following general procedure F. Full conversion was achieved after stirring at 40 °C overnight. Purification by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM afforded product 7j as an amorphous solid (114 mg, 65% yield). 1H NMR (400 MHz, methanol-d4) δ 7.70 (dt, J = 7.8, 1.5 Hz, 1H), 7.50 (t, J = 7.7 Hz, 1H), 7.46–7.39 (m, 2H), 7.23–7.09 (m, 3H), 7.04–6.97 (m, 2H), 6.96–6.89 (m, 2H), 6.75 (ddd, J = 8.2, 2.5, 0.9 Hz, 1H), 4.15 (ddd, J = 10.8, 8.5, 3.5 Hz, 1H), 3.78–3.71 (m, 4H), 3.58–3.45 (m, 1H), 3.42–3.32 (m, 1H), 3.24 (dd, J = 13.6, 3.5 Hz, 1H), 3.05–2.92 (m, 2H), 2.68 (dd, J = 13.6, 11.0 Hz, 1H), 2.60–2.42 (m, 4H), 1.71 (h, J = 6.8, 6.2 Hz, 2H), 1.67–1.57 (m, 1H), 1.51 (s, 6H), 1.48–1.38 (m, 1H), 1.38–1.19 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (101 MHz, methanol-d4): δ 173.2, 168.9, 161.3, 148.3, 142.8, 139.9, 138.2, 135.3, 131.0, 130.5, 130.3, 130.2, 129.6, 129.5, 128.0, 127.7, 124.9, 119.2, 113.2, 112.9, 73.3, 57.9, 56.6, 55.6, 50.9, 48.6, 47.4, 37.5, 35.9, 29.7, 29.30, 29.0, 28.4, 21.8, 11.7. HRMS-ESI (m/z) calcd for C36H48N3O4 [M + H]+ 572.3488. Found 572.3496. [α]D20 = −17.2 (c 0.5, CHCl3).
(S)-4-((R)-1-Hydroxy-2-((2-(3-methoxyphenyl)propan-2-yl)amino)ethyl)-12-propyl-3,12-diaza-1,6(1,3)-dibenzenacyclotridecaphane-2,13-dione (7k)
Ring-closing metathesis and hydrogenation of the double bond were performed using compound 39b (430 mg, 0.592 mmol, 1 equiv), Zhan Catalyst-1B (43 mg, 0.059 mmol, 0.1 equiv), and 10% palladium on carbon (63 mg, 0.059 mmol, 0.1 equiv), following general procedure J. Crude macrocycle 40b (250 mg, 60% yield) was used in the next step without purification.
The desilylation was performed using crude macrocycle 40b from the previous step (250 mg, 0.357 mmol, 1 equiv), NH4F (397 mg, 10.7 mmol, 30 equiv), following general procedure F. Full conversion was achieved after stirring at 50 °C for 16 h. Purification by column chromatography on silica gel using gradient elution from DCM to 10% MeOH in DCM afforded product 7k as an amorphous solid (140 mg, 67% yield). 1H NMR (400 MHz, methanol-d4) for a mixture of rotamers (0.80:0.13:0.07): δ 7.71–7.51 (m, 2H), 7.48 (t, J = 7.7 Hz, 1H), 7.39 (dt, J = 7.6, 1.4 Hz, 1H), 7.25–7.09 (m, 2H), 7.09–6.96 (m, 4H), 6.92 (d, J = 7.7 Hz, 1H), 6.82–6.56 (m, 1H), 4.45–4.17 (m, 1H), 3.80–3.71 (m, 3H), 3.71–3.58 (m, 2H), 3.34–3.28 (m, 1H), 3.24 (dd, J = 14.3, 3.7 Hz, 1H), 3.19–3.08 (m, 1H), 3.04 (dd, J = 14.5, 7.8 Hz, 1H), 2.73–2.61 (m, 2H), 2.54–2.36 (m, 3H), 1.85–1.61 (m, 3H), 1.53–1.35 (m, 9H), 1.15–1.05 (m, 1H), 0.99 (t, J = 7.4 Hz, 2.4H), 0.96–0.84 (m, 1H), 0.71 (t, J = 7.3 Hz, 0.2H), 0.63 (t, J = 7.4 Hz, 0.4H). 13C NMR (101 MHz, methanol-d4) for a major rotamer: δ 173.3, 169.0, 161.2, 149.9, 142.9, 140.1, 138.3, 135.8, 131.1, 130.5, 130.3, 130.2, 129.6, 129.5, 127. 4, 126.9, 125.3, 119.3, 113.2, 112.5, 74.6, 56.8, 55.9, 55.6, 47.2, 46.8, 37.3, 36.5, 31.8, 30.1, 29.1, 28.9, 26.1, 21.5, 11.7. HRMS-ESI (m/z) calcd for C36H48N3O4 [M + H]+ 586.3645 Found 586.3651. [α]D20 = −9.5 (c 0.8, CHCl3)
Acknowledgments
This work was supported by the Latvian Investment and Development Agency (Agreement No: KC-PI-2020); Vadims Kovada acknowledges the LIOS student grant (grant No IG-2020-08), and Raitis Bobrovs acknowledges the European Regional Development Fund (Project No. 1.1.1.2/VIAA/2/18/379) for financial support. This work was also supported by funding to M.J.B. from the Wellcome Trust (220318/A/20/Z) and the Francis Crick Institute (https://www.crick.ac.uk/), which receives its core funding from Cancer Research UK (CC2129), the UK Medical Research Council (CC2129), and the Wellcome Trust (CC2129). For the purpose of Open Access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. The authors thank Iveta Kanepe-Lapsa for conducting PMIV and BACE1 biochemical assays.
Glossary
Abbreviations
- CLint
intrinsic clearance
- DoR
Day of Recrudescence
- GSK
GlaxoSmithKline
- ND
not determined
- PPB
plasma protein binding
- RBC
red blood cell
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00812.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO, World Malaria Report 2022, World Health Organ: Geneva, 2016. https://www.who.int/publications/i/item/9789240064898. [Google Scholar]
- Sachs J.; Malaney P. The economic and social burden of malaria. Nature 2002, 415, 680–685. 10.1038/415680a. [DOI] [PubMed] [Google Scholar]
- Wells T. N. C.; Hooft van Huijsduijnen R.; Van Voorhis W. C. Malaria medicines: a glass half full?. Nat. Rev. Drug Discovery 2015, 14, 424–442. 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- Hyde J. E. Drug-resistant malaria–an insight. FEBS J. 2007, 274, 4688–4698. 10.1111/j.1742-4658.2007.05999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells T. N. C.; Alonso P. L.; Gutteridge W. E. New medicines to improve control and contribute to the eradication of malaria. Nat. Rev. Drug Discovery 2009, 8, 879–891. 10.1038/nrd2972. [DOI] [PubMed] [Google Scholar]
- Dondorp A. M.; Nosten F.; Yi P.; Das D.; Phyo A. P.; Tarning J.; Lwin K. M.; Ariey F.; Hanpithakpong W.; Lee S. J.; et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. 10.1056/NEJMoa0808859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows J. N.; Duparc S.; Gutteridge W. E.; Hooft van Huijsduijnen R.; Kaszubska W.; Macintyre F.; Mazzuri S.; Möhrle J. J.; Wells T. N. C. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017, 16, 26 10.1186/s12936-016-1675-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicines for malaria venture, MMV′s pipeline of antimalarial drugs. Acessed 02.02.2023. https://www.mmv.org/research-development/mmvs-pipeline-antimalarial-drugs.
- Haque T. S.; Skillman A. G.; Lee C. E.; Habashita H.; Gluzman I. Y.; Ewing T. J.; Goldberg D. E.; Kuntz I. D.; Ellman J. A. Potent, low-molecular-weight non-peptide inhibitors of malarial aspartyl protease plasmepsin II. J. Med. Chem. 1999, 42, 1428–1440. 10.1021/jm980641t. [DOI] [PubMed] [Google Scholar]
- Ersmark K.; Feierberg I.; Bjelic S.; Hamelink E.; Hackett F.; Blackman M. J.; Hultén J.; Samuelsson B.; Åqvist J.; Hallberg A. Potent inhibitors of the Plasmodium falciparum enzymes plasmepsin I and II devoid of cathepsin D inhibitory activity. J. Med. Chem. 2004, 47, 110–122. 10.1021/jm030933g. [DOI] [PubMed] [Google Scholar]
- Boss C.; Corminboeuf O.; Grisostomi C.; Meyer S.; Jones A. F.; Prade L.; Binkert C.; Fischli W.; Weller T.; Bur D. Achiral, cheap, and potent inhibitors of plasmepsins I, II, and IV. ChemMedChem 2006, 1, 1341–1345. 10.1002/cmdc.200600223. [DOI] [PubMed] [Google Scholar]
- Meyers M. J.; Goldberg D. E. Recent advances in plasmepsin medicinal chemistry and implications for future antimalarial drug discovery efforts. Curr. Top. Med. Chem. 2012, 12, 445–455. 10.2174/156802612799362959. [DOI] [PubMed] [Google Scholar]
- Liu J.; Istvan E. S.; Gluzman I. Y.; Gross J.; Goldberg D. E. Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 8840–8845. 10.1073/pnas.0601876103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo I.; Babbitt S.; Muralidharan V.; Butler T.; Oksman A.; Goldberg D. E. Plasmepsin V licenses Plasmodium proteins for export into the host erythrocyte. Nature 2010, 463, 632–636. 10.1038/nature08726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasamu A. S.; Glushakova S.; Russo I.; Vaupel B.; Oksman A.; Kim A. S.; Fremont D. H.; Tolia N.; Beck J. R.; Meyers M. J.; et al. Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science 2017, 358, 518–522. 10.1126/science.aan1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pino P.; Caldelari R.; Mukherjee B.; Vahokoski J.; Klages N.; Maco B.; Collins C. R.; Blackman M. J.; Kursula I.; Heussler V.; et al. A multistage antimalarial targets the plasmepsins IX and X essential for invasion and egress. Science 2017, 358, 522–528. 10.1126/science.aaf8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers M. J.; Tortorella M. D.; Xu J.; Qin L.; He Z.; Lang X.; Zeng W.; Xu W.; Qin L.; Prinsen M. J.; et al. Evaluation of aminohydantoins as a novel class of antimalarial agents. ACS Med. Chem. Lett. 2014, 5, 89–93. 10.1021/ml400412x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe M. A.; Cardenas A.; Valentin J.-P.; Zhu Z.; Abendroth J.; Castro J. L.; Class R.; Delaunois A.; Fleurance R.; Gerets H.; et al. Discovery and characterization of potent, efficacious, orally available antimalarial plasmepsin X inhibitors and preclinical safety assessment of UCB7362. J. Med. Chem. 2022, 65, 14121–14143. 10.1021/acs.jmedchem.2c01336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favuzza P.; de Lera Ruiz M.; Thompson J. K.; Triglia T.; Ngo A.; Steel R. W. J.; Vavrek M.; Christensen J.; Healer J.; Boyce C.; Guo Z.; Hu M.; Khan T.; Murgolo N.; Zhao L.; Penington J. S.; Reaksudsan K.; Jarman K.; Dietrich M. H.; Richardson L.; Guo K.-Y.; Lopaticki S.; Tham W.-H.; Rottmann M.; Papenfuss T.; Robbins J. A.; Boddey J. A.; Sleebs B. E.; Sabroux H. J.; McCauley J. A.; Olsen D. B.; Cowman A. F. Dual plasmepsin-targeting antimalarial agents disrupt multiple stages of the malaria parasite life cycle. Cell Host Microbe 2020, 27, 642–658. 10.1016/j.chom.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F. J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J. L.; Vanderwall D. E.; Green D. V.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. 10.1038/nature09107. [DOI] [PubMed] [Google Scholar]
- Jaudzems K.; Tars K.; Maurops G.; Ivdra N.; Otikovs M.; Leitans J.; Kanepe-Lapsa I.; Domraceva I.; Mutule I.; Trapencieris P.; Blackman M. J. A.; Jirgensons A. Plasmepsin inhibitory activity and structure-guided optimization of a potent hydroxyethylamine-based antimalarial hit. ACS Med. Chem. Lett. 2014, 5, 373–377. 10.1021/ml4004952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zogota R.; Kinena L.; Withers-Martinez C.; Blackman M. J.; Bobrovs R.; Pantelejevs T.; Kanepe-Lapsa I.; Ozola V.; Jaudzems K.; Suna E.; Jirgensons A. Peptidomimetic plasmepsin inhibitors with potent anti-malarial activity and selectivity against cathepsin D. Eur. J. Med. Chem. 2019, 163, 344–352. 10.1016/j.ejmech.2018.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidumniece E.; Withers-Martinez C.; Hackett F.; Blackman M. J.; Jirgensons A. Subtilisin-like serine protease 1 (SUB1) as an emerging antimalarial drug target: current achievements in inhibitor discovery. J. Med. Chem. 2022, 65, 12535–12545. 10.1021/acs.jmedchem.2c01093. [DOI] [PubMed] [Google Scholar]
- Driggers E. M.; Hale S. P.; Lee J.; Terrett N. K. The exploration of macrocycles for drug discovery — an underexploited structural class. Nat. Rev. Drug Discovery 2008, 7, 608–624. 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- Marsault E.; Peterson M. L. Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J. Med. Chem. 2011, 54, 1961–2004. 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- Mallinson J.; Collins I. Macrocycles in new drug discovery. Future Med. Chem. 2012, 4, 1409–1438. 10.4155/fmc.12.93. [DOI] [PubMed] [Google Scholar]
- Cummings M. D.; Sekharan S. Structure-based macrocycle design in small-molecule drug discovery and simple metrics to identify opportunities for macrocyclization of small-molecule ligands. J. Med. Chem. 2019, 62, 6843–6853. 10.1021/acs.jmedchem.8b01985. [DOI] [PubMed] [Google Scholar]
- Buesking A. W.; Baguley T. D.; Ellman J. A. Asymmetric synthesis of amines by the Knochel-type MgCl2-enhanced addition of benzyl zinc reagents to N-tert-butanesulfinyl aldimines. Org. Lett. 2011, 13, 964–967. 10.1021/ol103002t. [DOI] [PubMed] [Google Scholar]
- Liu G.; Cogan D. A.; Ellman J. A. Catalytic asymmetric synthesis of tert-butanesulfinamide. Application to the asymmetric synthesis of amines. J. Am. Chem. Soc. 1997, 119, 9913–9914. 10.1021/ja972012z. [DOI] [Google Scholar]
- Wolleb H.; Ogawa S.; Schneider M.; Shemet A.; Muri J.; Kopf M.; Carreira E. M. Synthesis and structure–activity relationship studies of anti-inflammatory epoxyisoprostane analogues. Org. Lett. 2018, 20, 3014–3016. 10.1021/acs.orglett.8b01042. [DOI] [PubMed] [Google Scholar]
- Jiménez-Díaz M. B.; Mulet T.; Viera S.; Gómez V.; Garuti H.; Ibáñez J.; Alvarez-Doval A.; Shultz L. D.; Martínez A.; Gargallo-Viola D.; Angulo-Barturen I. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD- scid IL2R mice engrafted with human erythrocytes. Antimicrob. Agents Chemother. 2009, 53, 4533–4536. 10.1128/AAC.00519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajid M.; Withers-Martinez C.; Blackman M. J. Maturation and specificity of Plasmodium falciparum subtilisin-like protease-1, a malaria merozoite subtilisin-like serine protease. J. Biol. Chem. 2000, 275, 631–641. 10.1074/jbc.275.1.631. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Nasamu A. S.; Rubiano K. C.; Goldberg D. E. Activation of the Plasmodium egress effector subtilisin-like protease 1 is mediated by Plasmepsin X destruction of the prodomain. mBio 2023, 14, e0067323 10.1128/mbio.00673-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarta-Gatsi C.; Andenmatten N.; Jimenez-Diaz M. B.; Gobeau N.; Cherkaoui-Rabti M. H.; Fuchs A.; Diaz P.; Berja S.; Sanchez R.; Gomez H.; Ruiz E.; Sainz P.; Salazar E.; Gil-Merino R.; Mendoza L. M.; Eguizabal C.; Leroy D.; Moehrle J. J.; Tornesi B.; Angulo-Barturen I. Predicting Optimal Antimalarial Drug Combinations from a standardized Plasmodium falciparum humanized mouse model. Antimicrob. Agents Chemother. 2023, 67, e0157422 10.1128/aac.01574-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers-Martinez C.; Saldanha J. W.; Ely B.; Hackett F.; O’Connor T.; Blackman M. J. Expression of recombinant Plasmodium falciparum subtilisin-like protease-1 in insect cells: characterization, comparision with the parasite protease, and homology mdoling. J. Biol. Chem. 2002, 277, 29698–29709. 10.1074/jbc.M203088200. [DOI] [PubMed] [Google Scholar]
- Hall R.; McBride J.; Morgan G.; Tait A.; Werner Zolg J.; Walliker D.; Scaife J. Antigens of the erythrocytic stages of the human malaria parasite Plasmodium falciparum detected by monoclonal antibodies. Mol. Biochem. Parasitol. 1983, 7, 247–265. 10.1016/0166-6851(83)90025-7. [DOI] [PubMed] [Google Scholar]
- Donald R. G.; Zhong T.; Wiersma H.; Nare B.; Yao D.; Lee A.; Allocco J.; Liberator P. A. Anticoccidial kinase inhibitors: identification of protein kinase targets secondary to cGMP-dependent protein kinase. Mol. Biochem. Parasitol. 2006, 149, 86–98. 10.1016/j.molbiopara.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Collins C. R.; Hackett F.; Strath M.; Penzo M.; Withers-Martinez C.; Baker D. A.; Blackman M. J. Malaria parasite cGMP-dependent protein kinase regulates blood stage merozoite secretory organelle discharge and egress. PLoS Pathog. 2013, 9, e1003344 10.1371/journal.ppat.1003344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris P. K.; Yeoh S.; Dluzewski A. R.; O’Donnell R. A.; Withers-Martinez C.; Hackett F.; Bannister L. H.; Mitchell G. H.; Blackman M. J. Molecular identification of a malaria merozoite surface sheddase. PLoS Pathog. 2005, 1, e29 10.1371/journal.ppat.0010029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abagyan R.; Totrov M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. 10.1006/jmbi.1994.1052. [DOI] [PubMed] [Google Scholar]
- Hill J. R. In vitro drug metabolism using liver microsomes. Curr. Protoc. Pharmacol. 2004, 7, Unit7.8 10.1002/0471141755.ph0708s23. [DOI] [PubMed] [Google Scholar]
- Jiménez-Díaz M. B.; Mulet T.; Gómez V.; Viera S.; Alvarez A.; Garuti H.; Vázquez Y.; Fernández A.; Ibáñez J.; Jiménez M.; Gargallo-Viola D.; Angulo-Barturen I. Quantitative measurement of Plasmodium-infected erythrocytes in murine models of malaria by flow cytometry using bidimensional assessment of SYTO-16 fluorescence. Cytometry, Part A 2009, 75A, 225–235. 10.1002/cyto.a.20647. [DOI] [PubMed] [Google Scholar]
- Fontenelle C. Q.; Conroy M.; Light M.; Poisson T.; Pannecoucke X.; Linclau B. Stereoselectivity of the Honda–Reformatsky reaction in reactions with ethyl bromodifluoroacetate with α-oxygenated sulfinylimines. J. Org. Chem. 2014, 79, 4186–4195. 10.1021/jo500396p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.