

Abstract

Glutamate plays a key role in cognition and mood, and it has been shown that inhibiting ionotropic glutamate receptors disrupts cognition, while enhancing ionotropic receptor activity is pro-cognitive. One approach to elevating glutamatergic tone has been to antagonize presynaptic metabotropic glutamate receptor 2 (mGluR2). A desire for selectivity over the largely homologous mGluR3 motivated a strategy to achieve selectivity through the identification of mGluR2 negative allosteric modulators (NAMs). Extensive screening and optimization efforts led to the identification of a novel series of 4-arylquinoline-2-carboxamides. This series was optimized for mGluR2 NAM potency, clean off-target activity, and desirable physical properties, which resulted in the identification of improved C4 and C7 substituents. The initial lead compound from this series was Ames-positive in a single strain with metabolic activation, indicating that a reactive metabolite was likely responsible for the genetic toxicity. Metabolic profiling and Ames assessment across multiple analogs identified key structure–activity relationships associated with Ames positivity. Further optimization led to the Ames-negative mGluR2 negative allosteric modulator MK-8768.
Keywords: mGluR2, glutamate, Ames, cognition
The role of glutamate as a major excitatory neurotransmitter in the CNS1 is well-known, and the effects of glutamate are mediated primarily through ionotrophic and metabotropic glutamate receptors (mGluRs).2−4 Inhibiting the ionotropic glutamate receptors (NMDA and AMPA receptors) can disrupt cognition,5 and activation of these receptors as a mechanism to enhance cognition6−8 is associated with a number of challenges including desensitization and adverse events. The challenge of avoiding these unwanted effects while still elevating glutamatergic tone is potentially ameliorated through antagonism of presynaptic glutamate receptors.9 Group II mGluRs (mGluR2, mGluR3) are presynaptic10 and are autoinhibitory GPCRs such that inhibition of mGluR2 has been shown to increase synaptic glutamate, increase postsynaptic activity and plasticity, and potentially improve learning and memory.11,12 Promisingly, antagonists13 of mGluR2/3 show efficacy in preclinical cognition assays,14,15 but mGluR3-driven hyperlocomotion and increased wake16 have motivated the desire for mGluR2 selective ligands. Unfortunately, obtaining selective orthosteric antagonists has been elusive, presumably due to the high level of homology between mGluR2 and mGluR3. Therefore, selective inhibition of mGluR2 may be more achievable with allosteric ligands. The potential impact of developing a safe mGluR2 negative allosteric modulator (NAM) is significant, as a drug with this mechanism of action could have positive effects on a range of CNS disorders including Alzheimer’s disease cognition17 and depression18−20—both of which have significant unmet medical need.
In addition to a number of publications from other institutions on their efforts21−24 toward identifying selective mGluR2 inhibitors, our company recently published25 an approach to identification of selective mGluR2 NAMs, which led to the discovery of a 4-arylquinoline-2-carboxamide series. A high throughput screen identified compound 1 (Figure 1); initial SAR studies demonstrated that C2-carboxamide was required for potency, the C4 vector preferred lipophilic groups, and C7 tolerated polar groups. Compound 2 was identified following these early studies, and it demonstrated the properties commensurate with an in vivo tool compound.
Figure 1.

Screening hit 1 and tool compound 2.
Compound 2 has good mGluR2 inhibition potency (FLIPR IC50 = 9 nM), full selectivity against mGluR3 (FLIPR IC50 ≥ 10 000 nM), and excellent rat oral pharmacokinetics (100%F). Compound 2 also was used to demonstrate that a selective mGluR2 NAM could show desirable activity in a mouse delayed nonmatch to position (DNMTP)26 cognition assay. While 2 proved to be a good tool compound, it did have several properties that required optimization. The major areas we looked to improve were brain penetration, solubility, and off-target selectivity. 2 is a substrate for P-gp active transport27 (rat P-gp BA:AB = 7.0), and thus the goal to advance an improved analog with optimum brain penetration was to eliminate this liability. The off-target challenges with 2 were primarily ion channels (hERG-binding28 IC50 = 27 μM) and pregnane X receptor (PXR) agonism29 (EC50 = 0.9 μM, 158% max agonism), which can lead to CYP induction and significant drug–drug interactions.
The initial strategy to identify an improved mGluR2 NAM was to further expand the C4 and C7 SAR to balance the overall properties with a focus on four identified key issues (P-gp, solubility, PXR, hERG). The synthetic tractability of the carboxamide series was a key feature that we were able to exploit to broadly explore both C4 and C7 substitutions with chemical libraries which enabled many analogs to be prepared quickly and a more rapid advancement of the series (Scheme 1).30 In order to explore C4 diversity, approach A was used for many analogs—in this sequence, the C7 benzyl halide of intermediate A1 was initially functionalized (SN2, Suzuki, etc.), and the resulting intermediate A2 was then subsequently reacted at C4 with Suzuki or similar cross-couplings. C7-SAR could be expanded using approach B, which simply flips the order of operations—starting from B1/B2, the initial Suzuki reaction to give B3/B4 is followed by halogenation and functionalization of the C7-methyl group (R = Me) or direct functionalization if R = Cl. Both approaches pivoted on a late-stage conversion of 2-cyanoquinolines A3 and B5/B6 to the desired primary carboxamides.
Scheme 1. General Approach to C4 and C7 Libraries.

RC7 amine or heterocycle, DIEA or K2CO3, DMF or CH3CN.
Pd(dppf)Cl2, RC7-B(OH)2, K3PO4, dioxane/water, 100 °C.
RC4-B(OH)2, Pd(PPh3)4, Na2CO3, dioxane/water, 100 °C.
B5—NBS, AIBN, CCl4, then RC7 amine or heterocycle, DIEA or K2CO3, DMF or CH3CN.
B6–potassium vinyltrifluoroborate, Pd(OAc)2, RuPhos, Cs2CO3, dioxane, 60 °C.
9-BBN, THF, 60 °C, then Pd2 (dba)3, cataCXium, K2CO3, water.
Examination of the C7 vector with multiple libraries and targeted singletons further validated the previously published25 early SAR with a wide variety of polar functionality being tolerated including amides, carbamates, heterocycles, and amines (Table 1). Reducing the number of carbonyl groups and removing the amide of 2 did lead to improved solubility and reduced P-gp efflux but led to a large reduction in mGluR2 FLIPR potency and no improvement in other properties (3a). Acyclic amides were broadly tolerated with a ∼10-fold loss in potency and either no change in solubility (3b) or improved solubility with a large increase in P-gp efflux (3c). Complete removal of carbonyl functionality to give either homobenzylic (3d, 3e) or benzylic (3f, 3g) C- or N-linked aromatic heterocycles maintained much of the potency of 2 and generally provided analogs with no P-gp-transport liability, but analogs were poorly soluble and had low micromolar inhibition in the hERG binding assay. In an attempt to improve solubility, amino heterocycles were examined (3h, 3i) and were generally similar to the related homobenzylic heterocycles but did not lead to significant improvements in solubility or in other parameters, with the exception of 3i, which had greatly reduced PXR activity. Reduction of aromatic content is a well-established strategy31 to improve solubility and other properties, and while the simple primary benzyl amine was not potent (data not shown), a variety of alkyl and, in particular, fluoroalkyl substituted amines (3j–n) maintained reasonable potency. Fluoroalkyl amines were not included to address any specific liability but in order to increase library diversity. The methyl-substituted morpholine 3k improved solubility and PXR activity significantly, and the corresponding trifluoromethyl-piperazine 3m improved potency and was a P-gp nonsubstrate.
Table 1. C7 SAR.

| # | mGluR2 IC50 (nM) | pH 7 soly (μM) | PXR (EC50 μM,% max) | Rat P-gp (BA:AB)/Papp (10–6cm/s)b | hERGc IC50 (μM) |
|---|---|---|---|---|---|
| 2 | 4 | 4 | 0.9, 158% | 7.0/33 | 27 |
| 3a | 123 | 127 | 9.7, 95% | 3.3/36 | 1432 |
| 3b | 37 | <2 | 3.5, 110% | 5.0/38 | 40 |
| 3c | 40 | 143 | 0.3, 122% | 45/32 | 20.6 |
| 3d | 7 | <1 | >30, 47% | 1.0/31 | >60 |
| 3e | 15 | <1 | 9.2, 97% | 0.6/25 | 2.9 |
| 3f | 13 | 4 | >30, 21% | 0.7/31 | 3.5 |
| 3g | 27 | <2 | 16.7, 53% | 0.9/27 | 1.9 |
| 3h | 15 | 3 | >30, 49% | 1.5/30 | 3.3 |
| 3i | 19 | 7 | >30, 1% | 13/31 | 6.2 |
| 3j | 22 | 6 | >30, 50% | 0.2/24 | 10.2 |
| 3ka | 43 | 103 | >30, 26% | NT | 5.5 |
| 3la | 24 | 2 | 16.8, 73% | 0.6/24 | 5.5 |
| 3ma | 12 | 105 | 7.1, 99% | 0.3/27 | 11.2 |
| 3na | 41 | <2 | 4.9, 184% | 1.4/28 | 11.2 |
Mixture of enantiomers.
P-gp transport ratio BA/AB determined using LLC-MDR1 cells.
hERG-[35S]-MK-499 binding assay28
Overall, the extensive exploration of C7 with a C4-fluorophenyl led to diverse effects on properties, but it was challenging to balance multiple properties in parallel.
Examining the impact on properties in the same way for the C4 vector again was enabled with a library-focused approach (Table 2). As was communicated in the initial report,25 lipophilic groups were preferred at this position, and while substitution of the fluorophenyl ring was tolerated, the impact on other properties was generally minimal. With the primary goal of improving solubility, the sp3 content of the molecules was increased with the introduction of saturated cycloalkyl and heterocycles (4a–e). 4-Methylcyclohexene derivative 4a displayed excellent mGluR2 NAM potency and was a non-P-gp substrate, but the solubility was not improved. Attempts to introduce heteroatoms into targeted positions either negatively impacted P-gp (4b, 4d) or significantly reduced potency (4c, 4e). This strategy did improve solubility, but the improvement was only modest for the analogs that maintained some potency (4b, 4d).
Table 2. C4 SAR.

| # | mGluR2 IC50 (nM) | pH 7 soly (μM) | PXR (EC50 μM,% max) | Rat P-gp (BA:AB)/Papp (10–6cm/s) | hERGc IC50 (μM) |
|---|---|---|---|---|---|
| 2 | 4 | 4 | 0.9, 158% | 7.0/33 | 27 |
| 4aa | 8 | <1 | 0.6, 115% | 1.4/32 | >60 |
| 4ba | 68 | 125 | NT | 19/38 | NT |
| 4c | 4030 | 203 | NT | NT | NT |
| 4d | 18 | <2 | 1.0, 152% | 11/35 | 17 |
| 4e | 3170 | 157 | 2.8, 87% | NT | >60 |
| 4f | 41 | 64 | 6.0, 137% | 18/30 | >60 |
| 4g | >5000 | 46 | NT | NT | NT |
| 4h | 74 | 99 | >30, 54% | 17/30 | NT |
| 4i | 67 | 162 | 20, 70% | 22/24 | 27 |
| 4j | 107 | 150 | 30, 30% | NT | NT |
Mixture of enantiomers.
P-gp transport ratio BA/AB determined using LLC-MDR1 cells.
hERG-[35S]-MK-499 binding assay
A wide range of heteroaromatic groups was then explored. Methylthiazole analogue 4f maintained reasonable potency (41 nM) and pH 7 solubility (41 μM) but did not positively impact PXR or P-gp.
3-Methylpyrazole 4g lost all potency, but isomeric 4h was much more active (74 nM) and also showed some improvement in pH 7 solubility (99 μM) and PXR (>30 μM, 54%). N-methyl analogue 4i also improved PXR and solubility while maintaining similar potency (67 nM). Further extending the N-alkyl group led to a loss in potency, but 4j did maintain the positive impact on the other key optimization parameters.
Given the benefits of multiple parameters and the demonstrated (Table 1) ability to reduce P-gp efflux with C7 substituents, N-methylpyrazole was selected to explore further, with the primary goals being to increase potency and decrease P-gp efflux while maintaining or improving the positive impacts on solubility, PXR, and hERG binding. As before, libraries were used to expand on the C7 SAR with the C4 N-methylpyrazole-containing core (Table 3). With the C4, fluorophenyl, homobenzylic, and benzylic heterocycles were highly potent. However, when combined with the C4 N-methylpyrazole (5a,b), no significant increase in potency compared to 4i was observed, and moderate (53–56 nM) potency was maintained. Promisingly, the key off-targets (PXR and hERG) were significantly improved, and the P-gp transport was also reduced compared to 4i. Potency could be improved with additional substitution (5c), but activity on PXR and hERG was significantly increased, and the P-gp-transport also increased. Interestingly, while the C4-fluorophenyl containing C7-trifluoromethylpiperazine (3m) was one of the more potent early analogs, the corresponding C4-N-methylpyrazole (5d) lost significant potency. In contrast, the related morpholines maintained (5e) or improved (5f) upon the potency present with the C4-fluorophenyl, and both analogs also had promising profiles with low PXR activity and excellent solubility. 5f was not a substrate for P-gp. Additional 3-substituted morpholine analogs were then explored, but the SAR was quite sensitive, with small alkyl groups (5g,h) eroding the PXR selectivity and other small structural modifications (5i) leading to a loss in potency. While both enantiomers of the racemic trifluoromethyl-containing analogue (5f) maintained good potency (mGluR2 NAM IC50: S-isomer = 31 nM/R-isomer = 11 nM), the more potent R-isomer ((R)-5f) was selected for further profiling. (R)-5f maintained the excellent selectivity (mGluR3 NAM IC50 = 7800 nM) of 2 and showed significant improvement in solubility (pH7 = 142 μM), PXR (EC50 ≥ 30 μM, 9% max), and P-gp ((BA:AB; rat/human) = 1.7/0.8; Papp = 29). (R)-5f also possessed an excellent ion channel profile (hERG/CaV1.2/NaV1.5 (IC50) ≥ 60/22/>30 μM) as well as a high level of general off-target selectivity (no hits in Panlabs33 panel of >120 targets tested at 30 μM). (R)-5f was also orally bioavailable in rodents (rat = 100%F @ 20 mpk) and demonstrated activity in the mouse DMNTP assay (data not shown). While (R)-5f appeared to have a very attractive profile, it was discovered in an exploratory 5-strain Ames assessment34 that in the presence of metabolic activation (rat S9 fraction) in a single strain (TA97a), (R)-5f was Ames-positive (>2-fold increase in revertants at ≥300 μg/plate), thus indicating a risk of mutagenicity. This result prevented (R)-5f from advancing further, but since the Ames-positive result was only in the presence of metabolic activation, we explored the sites of metabolism with hopes to identify a way to mitigate the problematic pathway.
Table 3. C4 N-Methylpyrazole SAR.

| # | mGluR2 IC50 (nM) | pH 7 soly (μM) | PXR (EC50 μM,% max) | rat P-gp (BA:AB)/Papp (10–6cm/s) | hERGc IC50 (μM) |
|---|---|---|---|---|---|
| 5a | 56 | 141 | >30, 8% | 9.6/30 | >60 |
| 5b | 53 | 105 | >30, 2% | 6.6/36 | >60 |
| 5ca | 11 | 107 | 19, 75% | 18/29 | 10 |
| 5da | 122 | 175 | >30, 5% | 4.2/29 | >20 |
| 5ea | 166 | 175 | >30, 1% | NT | >60 |
| 5fa | 12 | 132 | >30, 24% | 1.3/30 | 19 |
| (R)-5f | 11 | 142 | >30, 9% | 1.7/29 | >60 |
| 5ga | 32 | 175 | 1.7, 100% | 3.4/29 | 51 |
| 5ha | 23 | 142 | 29, 51% | 1.3/34 | 23 |
| 5ia | 67 | 193 | NT | NT | NT |
Mixture of enantiomers.
P-gp transport ratio BA/AB determined using LLC-MDR1 cells.
hERG-[35S]-MK-499 binding assay
In human, dog, and rat hepatocytes as well as in rat and dog in vivo, the metabolism of (R)-5f features several C7-trifluoromethyl morpholine oxidation products as well as demethylation of the C4-N-methylpyrazole (Supporting Information S2). We then assessed the single strain Ames profile of analogs with changes in the C4 and C7 region using a screening paradigm in TA97a with rat S9 only. The results from this effort (Supporting Information S3) revealed that changes in the C4 vector did not lead to changes in Ames-positivity in TA97a with rat S9, but minor or major changes in the C7 substituent result in single-strain Ames-negative analogs, indicating the likely cause of the Ames positivity in (R)-5f is an unidentified/unconfirmed reactive metabolite or intermediate on the C7-trifluoromethylmorpholine. While all di- or trisubstituted C7-trifluoromethylmorpholines (S2–7 to S2–13) were Ames negative in TA97a with rat S9, the (2R,6R)-2-methyl-6-(trifluoromethyl) morpholine isomer possessed the best combination of potency, P-gp, solubility, and hERG inhibition and advanced to become MK-8768.
A more complete profile of MK-8768 is shown in Table 4. MK-8768 is a 9.6 nM inhibitor of mGluR2 with complete selectivity against mGluR1,3,4,5,6,8 (IC50 > 10 000 nM). MK-8768 also demonstrated an excellent overall selectivity profile with weak to no activity on the hERG, IKs, and Nav1.5 ion channels and no confirmed activities < 10 μM in a Panlabs screen of >120 off-targets. MK-8768 is a nonsubstrate for rat, human, and monkey P-gp; has high passive permeability; and demonstrated good brain penetration in rats (Kpu,u > 1; CSF:[plasma]u = 1). A lack of significant activity at PXR or inhibition (reversible and time-dependent) of cytochrome P450s indicates that MK-8768 has a low risk for being a perpetrator of drug–drug interactions. In human hepatocytes, the major routes of metabolism were hydroxylation of the morpholine ring region and dehydrogenation of the morpholine region. N-Demethylation of the pyrazole was also observed in human hepatocytes but was a more significant pathway in rats and monkeys. Interestingly, similar to (R)-5f, metabolism on the C7-morpholine was the major metabolic pathway (Supporting Information S4), but MK-8768 was Ames negative in a full five-strain Ames assay. In rats, dogs, and monkeys, MK-8768 exhibited moderate clearance, and the effective half-life ranged from 1.7 h in monkeys to 3.3 h in rats. The oral bioavailability was 32% and 34% in rats and dogs, respectively. The in vivo efficacy of MK-8768 was then assessed in a rhesus monkey object retrieval detour (ORD) task,35,36 which is an assessment of executive function and attention. In the ORD assay, performance on difficult, but not easy, trials is disrupted by scopolamine, ketamine, and PFC lesions. MK-8768 was coadministered (Figure 2) with a dose of scopolamine titrated to produce the targeted level of impairment (∼90% correct difficult trials reduced to ∼60% correct difficult trials), and a significant reversal of the scopolamine deficit was achieved by intramuscular doses from 0.03 to 1 mg/kg (see Supporting Information S1 for details). The efficacious doses produced unbound plasma concentrations from 3–65 nM, which represents 0.3–6.5-fold the in vitro mGluR2 IC50. Higher doses of MK-8768 (3 mg/kg) were not effective in improving difficult trial performance, indicative of an inverted U-shaped dose–effect curve that oftentimes accompanies pro-cognitive mechanisms in preclinical studies.
Table 4. Profile of MK-8768.

| mGluR2 FLIPR IC50 | 9.6 nM |
| mGluR1,3,4,5,6,8 FLIPR IC50 | >10000 nM |
| P-GPa (BA:AB; rat/human/monkey); Papp | 2.1/1.5/1.4; 32 |
| PXR EC50 | >30 μM, 12% @ 30 μM |
| hERG/IKs/NaV1.5IC50b | 22/>30/>30 μM |
| five strain Ames | negative |
| CYP IC50 (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4) | >26 μM, no time dependent inhibition |
| tosylate salt xtal soly (SGF/FaSSIF/FeSSIF) | 4.1/0.44/2.1 mg/mL |
| plasma fraction unbound (human/rat/dog/monkey) | 2.5%/4%/4.5%/4.3% |
| rat PK (CL; Vd; T1/2,eff; bioavailability)c | 36 mL/min/kg; 5.7 L/kg; 1.8 h; 32% F |
| dog PK (CL; Vd; T1/2,eff; bioavailability)d | 24 mL/min/kg; 7.3 L/kg; 3.3 h; 34% F |
| monkey PK (CL; Vd; T1/2,eff)e | 22 mL/min/kg; 3.2 L/kg; 1.7 h |
P-gp transport ratio BA/AB determined using LLC-MDR1 cells.
Determined using PatchXpress automated patch-clamp system.
Rat: 1 mg/kg IV, 2 mg/kg PO.
Dog: 0.25 mg/kg IV, 0.5 mg/kg PO.
Monkey: 0.5 mg/kg IV.
Figure 2.
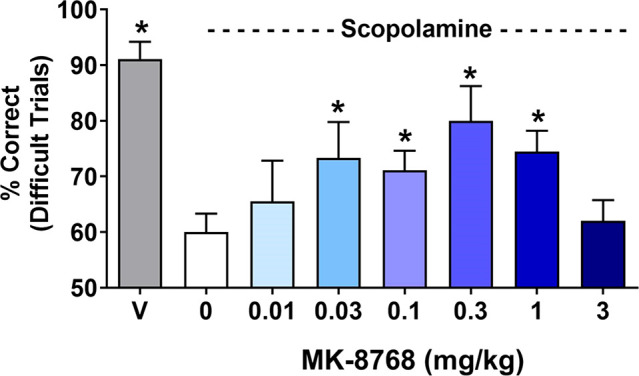
MK-8768 efficacy in the rhesus ORD task. MK-8768 improved object retrieval in rhesus monkeys following scopolamine impairment. *Indicates significantly different than animals given vehicle prior to scopolamine (white bar).
MK-8768 can be prepared as a single enantiomer starting from (R)-3,3,3-trifluoropropylene oxide (TFPO) and (S)-2-chloropropanoic acid in the longest linear sequence of eight steps in 31% overall yield (Scheme 2).
Scheme 2. Synthesis of MK-8768.

In summary, initial screening for mGluR2 inhibition identified a series of mGluR2-selective quinoline carboxamides that were optimized for potency, brain penetration, solubility, and off-target profile. This led to the identification of (R)-5f, which has an excellent all-around profile but was Ames-positive in a single strain with metabolic activation. SAR studies established the problematic functionality as the monosubstituted C7-trifluoromethylmorpholine, and Ames-negative disubstituted analogue MK-8768 was subsequently identified as the optimum analog. MK-8768 is a potent, selective, highly soluble mGluR2 NAM with good oral pharmacokinetics and demonstrated efficacy in a rhesus monkey model of executive function and attention. Further investigations of MK-8768 in additional preclinical or clinical settings will be published at a later date.
Acknowledgments
We would like to thank all mGluR2 NAM project members who contributed to this work.
Glossary
Abbreviations
- CNS
central nervous system
- mGluR2
metabotropic glutamate receptor 2
- NAM
negative allosteric modulator
- P-gp
P-glcoprotein
- HPLC
high-performance liquid chromatography
- DIEA
diisopropylethyl amine
- RuPhos
2-dicyclohexylphosphino-2′,6′-diisopropoxybiphenyl
- cataCXium
Di(1-adamantyl)-n-butylphosphine
- NMDA
N-methyl-d-aspartate
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- FLIPR
fluorescence imaging plate reader
- DNMTP
delayed nonmatch to position
- hERG
human ether-a-go-go related gene
- PXR
pregnane X receptor
- SAR
structure–activity relationship
- CaV1.2
calcium channel, voltage-dependent, L type, alpha 1C subunit
- NaV1.5
sodium channel protein type 5 subunit alpha
- CYP
cytochrome P450
- PK
pharmacokinetics
- CL
clearance
- Vd
volume of distribution
- ORD
object retrieval detour
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00210.
Experimental details and HRMS data of the synthesis of MK-8768; HRMS data for examples 1–5i and S3–1 to S3–13; in vitro metabolism profile for (R)-5f and MK-8768; Ames TA97a (with rat S9) SAR (PDF)
Author Contributions
The design, synthesis, and characterization of compounds was conducted by M.T.R., P.J.M., B.H., Z.M., Y.S., P.d.L., J.L.F., Y.H., J.M.-C.W., Z.-Q.Y., J.J.P., D.M.H., J.J.M., H.Z., C.J.B., and A.C. The compounds were analyzed and further characterized by M.L.C., I.H., L.M., J.O., V.N.U., J.B.S., D.C.D., V.P., D.M.E., J.D.V., and J.M.U. The DMPK characterization of compounds was conducted by KLF, BL, YL. The work was managed by R.E.D., J.T.K., J.M.U., and R.S.M. M.T.R. wrote the manuscript with help in review from all authors.
These studies were funded by Merck Sharp and Dohme LLC, a subsidiary of Merck and Co., Inc., Rahway, NJ, USA.
The authors declare the following competing financial interest(s): All authors were employees of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA at the time of their contribution to this work.
Supplementary Material
References
- Fonnum F. Glutamate: a neurotransmitter in mammalian brain. J. Neurochem. 1984, 42 (1), 1–11. 10.1111/j.1471-4159.1984.tb09689.x. [DOI] [PubMed] [Google Scholar]
- Kew J. N. C.; Kemp J. A. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology (Berlin, Ger.) 2005, 179 (1), 4–29. 10.1007/s00213-005-2200-z. [DOI] [PubMed] [Google Scholar]
- Reiner A.; Levitz J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98 (6), 1080–1098. 10.1016/j.neuron.2018.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender C. M.; Conn P. J. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keifer J.; Zheng Z. AMPA receptor trafficking and learning. Eur. J. Neurosci 2010, 32 (2), 269–277. 10.1111/j.1460-9568.2010.07339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch G. Memory enhancement: the search for mechanism-based drugs. Nat. Neurosci. 2002, 5 (Suppl), 1035–1038. 10.1038/nn935. [DOI] [PubMed] [Google Scholar]
- Tang Y.-P.; Shimizu E.; Dube G. R.; Rampon C.; Kerchner G.; Zhuo M.; Liu G.; Tsien J. Z. Genetic enhancement of learning and memory in mice. Nature (London) 1999, 401 (6748), 63–69. 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Morrow J. A.; Maclean J. K. F.; Jamieson C. Recent advances in positive allosteric modulators of the AMPA receptor. Curr. Opin. Drug Discovery Dev. 2006, 9 (5), 571–579. [PubMed] [Google Scholar]
- Conn P. J.; Christopoulos A.; Lindsley C. W. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discovery 2009, 8 (1), 41–54. 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto R.; Kinoshita A.; Wada E.; Nomura S.; Ohishi H.; Takada M.; Flor P. J.; Neki A.; Abe T.; Nakanishi S.; Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J. Neurosci. 1997, 17 (19), 7503–7522. 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z.-X.; Baker D. A.; Shen H.; Carson D. S.; Kalivas P. W. Group II metabotropic glutamate receptors modulate extracellular glutamate in the nucleus accumbens. J. Pharmacol. Exp. Ther. 2002, 300 (1), 162–171. 10.1124/jpet.300.1.162. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Steele J. W.; Lee S. W.; Clemenson G. D.; Carter T. A.; Treuner K.; Gadient R.; Wedel P.; Glabe C.; Barlow C.; Ehrlich M. E.; Gage F. H.; Gandy S. Proneurogenic Group II mGluR antagonist improves learning and reduces anxiety in Alzheimer Aβ oligomer mouse. Mol. Psychiatry 2014, 19 (11), 1235–1242. 10.1038/mp.2014.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qunies A. a. M.; Emmitte K. A. Negative allosteric modulators of group II metabotropic glutamate receptors: A patent review (2015 - present). Expert Opin. Ther. Pat. 2021, 31 (8), 687–708. 10.1080/13543776.2021.1903431. [DOI] [PubMed] [Google Scholar]
- Shimazaki T.; Kaku A.; Chaki S. Blockade of the metabotropic glutamate 2/3 receptors enhances social memory via the AMPA receptor in rats. Eur. J. Pharmacol. 2007, 575 (1–3), 94–97. 10.1016/j.ejphar.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Woltering T. J.; Wichmann J.; Goetschi E.; Knoflach F.; Ballard T. M.; Huwyler J.; Gatti S. Synthesis and characterization of 1,3-dihydro-benzo[b][1, 4]diazepin-2-one derivatives: Part 4. In vivo active potent and selective non-competitive metabotropic glutamate receptor 2/3 antagonists. Bioorg. Med. Chem. Lett. 2010, 20 (23), 6969–6974. 10.1016/j.bmcl.2010.09.125. [DOI] [PubMed] [Google Scholar]
- Wood C. M.; Wafford K. A.; McCarthy A. P.; Hewes N.; Shanks E.; Lodge D.; Robinson E. S. J. Investigating the role of mGluR2 versus mGluR3 in antipsychotic-like effects, sleep-wake architecture and network oscillatory activity using novel Han Wistar rats lacking mGluR2 expression. Neuropharmacology 2018, 140, 246–259. 10.1016/j.neuropharm.2018.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis P. T. Glutamatergic Approaches to the Treatment of Cognitive and Behavioural Symptoms of Alzheimer’s Disease. Neurodegener. Dis. 2008, 5 (3–4), 241–243. 10.1159/000113713. [DOI] [PubMed] [Google Scholar]
- Campo B.; Kalinichev M.; Lambeng N.; El Yacoubi M.; Royer-Urios I.; Schneider M.; Legrand C.; Parron D.; Girard F.; Bessif A.; Poli S.; Vaugeois J.-M.; Le Poul E.; Celanire S. Characterization of an mGluR2/3 Negative Allosteric Modulator in Rodent Models of Depression. J. Neurogenet. 2011, 25 (4), 152–166. 10.3109/01677063.2011.627485. [DOI] [PubMed] [Google Scholar]
- Krystal J. H.; Mathew S. J.; D’Souza D. C.; Garakani A.; Gunduz-Bruce H.; Charney D. S. Potential psychiatric applications of metabotropic glutamate receptor agonists and antagonists. CNS Drugs 2010, 24 (8), 669–693. 10.2165/11533230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Sanacora G.; Treccani G.; Popoli M. Towards a glutamate hypothesis of depression. Neuropharmacology 2012, 62 (1), 63–77. 10.1016/j.neuropharm.2011.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts A. S.; Rodriguez A. L.; Smith K. A.; Engers J. L.; Morrison R. D.; Byers F. W.; Blobaum A. L.; Locuson C. W.; Chang S.; Venable D. F.; Niswender C. M.; Daniels J. S.; Conn P. J.; Lindsley C. W.; Emmitte K. A. Design of 4-Oxo-1-aryl-1,4-dihydroquinoline-3-carboxamides as Selective Negative Allosteric Modulators of Metabotropic Glutamate Receptor Subtype 2. J. Med. Chem. 2015, 58 (22), 9027–9040. 10.1021/acs.jmedchem.5b01371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Xiao Z.; Kumata K.; Yamasaki T.; Josephson L.; Zhang M.-R.; Wang L.; Liang S. H. Positron Emission Tomography (PET) Imaging of Metabotropic Glutamate Receptor Subtype 2 (mGlu2) Based on a Negative Allosteric Modulator Radioligand. Neuromethods 2021, 164, 23–37. 10.1007/978-1-0716-1107-4_2. [DOI] [Google Scholar]
- Childress E. S.; Wieting J. M.; Felts A. S.; Breiner M. M.; Long M. F.; Luscombe V. B.; Rodriguez A. L.; Cho H. P.; Blobaum A. L.; Niswender C. M.; Emmitte K. A.; Conn P. J.; Lindsley C. W. Discovery of Novel Central Nervous System Penetrant Metabotropic Glutamate Receptor Subtype 2 (mGlu2) Negative Allosteric Modulators (NAMs) Based on Functionalized Pyrazolo[1, 5-a]pyrimidine-5-carboxamide and Thieno[3, 2-b]pyridine-5-carboxamide Cores. J. Med. Chem. 2019, 62 (1), 378–384. 10.1021/acs.jmedchem.8b01266. [DOI] [PubMed] [Google Scholar]
- Bollinger K. A.; Felts A. S.; Brassard C. J.; Engers J. L.; Rodriguez A. L.; Weiner R. L.; Cho H. P.; Chang S.; Bubser M.; Jones C. K.; Blobaum A. L.; Niswender C. M.; Conn P. J.; Emmitte K. A.; Lindsley C. W. Design and Synthesis of mGlu2 NAMs with Improved Potency and CNS Penetration Based on a Truncated Picolinamide Core. ACS Med. Chem. Lett. 2017, 8 (9), 919–924. 10.1021/acsmedchemlett.7b00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y.; Diamond T. L.; Hershey J. C.; Huang S.; Magliaro B. C.; O’Brien J. A.; Schlegel K.-A. S.; Puri V.; Uebele V. N.; Uslaner J. M.; Wang C.; Converso A. Discovery of 4-arylquinoline-2-carboxamides, highly potent and selective class of mGluR2 negative allosteric modulators: From HTS to activity in animal models. Bioorg. Med. Chem. Lett. 2020, 30 (9), 127066. 10.1016/j.bmcl.2020.127066. [DOI] [PubMed] [Google Scholar]
- Dunnett S. B.; Evenden J. L.; Iversen S. D. Delay-dependent short-term memory deficits in aged rats. Psychopharmacology (Berlin) 1988, 96 (2), 174. 10.1007/BF00177557. [DOI] [PubMed] [Google Scholar]
- Doan K. M. M.; Humphreys J. E.; Webster L. O.; Wring S. A.; Shampine L. J.; Serabjit-Singh C. J.; Adkison K. K.; Polli J. W. Passive permeability and P-glycoprotein-mediated efflux differentiate central nervous system (CNS) and non-CNS marketed drugs. J. Pharmacol. Exp. Ther. 2002, 303 (3), 1029–1037. 10.1124/jpet.102.039255. [DOI] [PubMed] [Google Scholar]
- Priest B. T.; Bell I. M.; Garcia M. L. Role of hERG potassium channel assays in drug development. Channels (Austin) 2008, 2 (2), 87–93. 10.4161/chan.2.2.6004. [DOI] [PubMed] [Google Scholar]
- Sinz M. W. Evaluation of pregnane X receptor (PXR)-mediated CYP3A4 drug-drug interactions in drug development. Drug Metab. Rev. 2013, 45 (1), 3–14. 10.3109/03602532.2012.743560. [DOI] [PubMed] [Google Scholar]
- Bungard C. J.; Converso A.; De Leon P.; Hanney B.; Hartingh T. J.; Manikowski J. J.; Manley P. J.; Meissner R.; Meng Z.; Perkins J. J.; Rudd M. T.; Shu Y.. Preparation of quinoline carboxamide and quinoline carbonitrile derivatives as mGluR2-negative allosteric modulators. WO2013066736, 2013.
- Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52 (21), 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Zhang Y.; Chen Z.; Shao T.; Van R.; Kumata K.; Deng X.; Fu H.; Yamasaki T.; Rong J.; Hu K.; Hatori A.; Xie L.; Yu Q.; Ye W.; Xu H.; Sheffler D. J.; Cosford N. D. P.; Shao Y.; Tang P.; Wang L.; Zhang M.-R.; Liang S. H. Synthesis and preliminary studies of 11C-labeled tetrahydro-1,7-naphthyridine-2-carboxamides for PET imaging of metabotropic glutamate receptor 2. Theranostics 2020, 10 (24), 11178–11196. 10.7150/thno.42587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eurofins Discovery - Your Novel Molecule Can Change the World.https://www.eurofinsdiscovery.com/.
- Escobar P. A.; Kemper R. A.; Tarca J.; Nicolette J.; Kenyon M.; Glowienke S.; Sawant S. G.; Christensen J.; Johnson T. E.; McKnight C.; Ward G.; Galloway S. M.; Custer L.; Gocke E.; O’Donovan M. R.; Braun K.; Snyder R. D.; Mahadevan B. Bacterial mutagenicity screening in the pharmaceutical industry. Mutat. Res., Rev. Mutat. Res. 2013, 752 (2), 99–118. 10.1016/j.mrrev.2012.12.002. [DOI] [PubMed] [Google Scholar]
- Diamond A.; Zola-Morgan S.; Squire L. R. Successful performance by monkeys with lesions of the hippocampal formation on AB and object retrieval, two tasks that mark developmental changes in human infants. Behav Neurosci 1989, 103 (3), 526–537. 10.1037/0735-7044.103.3.526. [DOI] [PubMed] [Google Scholar]
- Taylor J. R.; Elsworth J. D.; Roth R. H.; Sladek J. R. Jr.; Redmond D. E. Jr., Cognitive and motor deficits in the acquisition of an object retrieval/detour task in MPTP-treated monkeys. Brain 1990, 113 (3), 617–637. 10.1093/brain/113.3.617. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.