

Abstract

Mitochondrial dysfunction has been attributed to many disease indications, including metabolic, cardiovascular, neoplastic, and neurodegenerative diseases. Dynamin related protein 1 (DRP1) is crucial in regulating mitochondrial fission and maintaining mitochondrial homeostasis. MiD49 is a dynamic peripheral protein receptor on the surface of the mitochondrial membrane that recruits DRP1 protein to induce mitochondrial binary fission. By targeting the protein–protein interaction of DRP1/MiD49, we have discovered a novel and potent allosteric DRP1 inhibitor that inhibits mitochondria fragmentation in vitro. X-ray cocrystal structure revealed that it locked the closed DRP1 conformation by induced dimerization.
Keywords: DRP1, MiD49, Protein−protein interaction, Allosteric, inhibitor, Mitochondria
Mitochondria are highly dynamic and strongly connected organelles that continuously undergo fission and fusion, forming a tubulovesicular network within the cell. Mitochondrial dynamics are a balance of mitochondrial division and fusion that control the number, size, and distribution of mitochondria to meet the bioenergetic needs in cells. The two opposing processes of fission and fusion are critical events in numerous physiological processes including cell division, programmed cell death, calcium homeostasis, mitophagy, redox signaling, and mitochondrial DNA stability. Due to the essential role of mitochondrial dynamics in maintaining cell homeostasis, considerable research has focused on the role of mitochondrial dynamics in metabolic, cardiovascular, neoplastic, and neurodegenerative diseases.1−3 Excessive mitochondrial fragmentation has been shown to disrupt calcium homeostasis, decrease ATP production, and increase reactive oxygen species (ROS) production, leading to cell death.4
Mitochondrial fission is mediated through dynamin related protein 1 (DRP1), which is a member of the dynamin superfamily of GTPase. During the fission process, DRP1 is recruited to the outer mitochondrial membrane via its receptors mitochondrial elongation factor (MFF), mitochondrial dynamics protein of 49 and 51 kDa (MiD49 and MiD51). Once recruited, DRP1 self-assembles to form an oligomeric ring-shaped structure that wraps around the mitochondria and GTP hydrolysis and nucleotide exchange lead to conformational constriction by the DRP1 polymer.5−7 Inhibition of DRP1 using RNA interference or DRP1-dominant-negative mutations was shown to result in reduced mitochondrial fragmentation and improved mitochondrial function.8 Thus, these studies demonstrated that DRP1 is crucial in regulating mitochondrial fission and the therapeutic potential of inhibiting DRP1 to rebalance mitochondrial dynamics.
Multiple small molecule DRP1 inhibitors have been reported in the literature including mdivi-1,9 Drpitor1,10 DRP1i27,11 and DRP1 GTPase inhibitors.12 Mdivi-1 is widely reported to inhibit DRP1-dependent fission, elongate mitochondria, and mitigate brain injury. However, a recent report has indicated that mdivi-1 is not a specific DRP1 inhibitor.13 The ability of mdivi-1 to reversibly inhibit complex I and modify mitochondrial ROS production may contribute to the effects observed previously.9 Drpitor1 is a competitive GTPase inhibitor that reduces mitochondrial fragmentation.10 It reduces proliferation and induces apoptosis in cancer cells, as was demonstrated in a mouse xenograft model. Drpitor1 also inhibited mitochondrial ROS production, prevented mitochondrial fission, and improved right ventricular diastolic dysfunction during IR injury. Structurally, Drpitor1 is an analogue of ellipticine, a known DNA intercalator, and its applicability beyond oncology has yet to be fully explored. DRP1i27 is a human DRP1 isoform 3 binder with modest affinity binding to GTPase site of DRP1.11 The DRP1 inhibitors previously reported by Mitobridge were selective uncompetitive inhibitors of GTPase activity and served as unique tool compounds for exploring the roles of mitochondrial division in cells and assessing the potential therapeutic utility.12 However, these compounds did not exhibit a significant mitochondrial morphology change in cells, which limited its application. Herein, we report the discovery of a novel and potent allosteric DRP1 inhibitor that inhibits mitochondria fragmentation in vitro.
To date, multiple DRP1 protein structures have been resolved and deposited in the Protein Data Bank (PDB). The collection represents different aggregation states and conformations and sheds light on the assembly mechanism required for mitochondrial fission (Figure 1A–D).
Figure 1.

Different types of DRP1 structures reported in the literature.14,15 (A) Truncated DRP1, monomeric, closed conformation, apo, X-ray (PDB 4H1U). (B) Truncated DRP1, dimeric, open conformation, GMP-PNP bound, X-ray (PDB 3W6N). (C) Full-length DRP1, tetrameric, closed conformation, apo, X-ray (PDB 4BEJ). (D) Full-length DRP1, oligomeric, open conformation, GMP-PCP bound, complexed with MiD49, cryoEM (PDB 5WP9). (E) Hypothetical assembly mechanism where DRP1 undergoes conformational change upon GTP-binding, and subsequently MiD49 binds to the thus opened region. The key DRP1:MiD49 PPI triggers oligomerization and mitochondrial fission.
We contemplated that inhibiting the protein–protein interaction (PPI) between DRP1 and its adaptor proteins such as MiD49 would lead to suppression of mitochondrial fission; however, disrupting the protein–protein interaction with a small molecule poses challenges due to the large flat surface of the PPI interaction. Intriguingly, DRP1 undergoes a conformational change upon GTP binding that is required for MiD49 and MiD51 binding14,15 which suggested that a small molecule could potentially inhibit the protein–protein interaction by preferentially stabilizing the inactive closed conformation with allosteric binding (Figure 1E). Based on this hypothesis, we developed a high-throughput screen using AlphaLISA technology to evaluate the effect of compounds on the DRP1:MiD49 protein–protein interaction. AlphaLISA is a bead-based proximity technology and highly versatile to assay small molecule modulators of protein–protein interactions.16 Briefly, N-terminal His-tagged DRP1 (200 nM) and C-terminal GST-tagged Mid49 (20 nM) were mixed with GMP-PNP (100 μM) and allowed to incubate for 30 min at room temperature. Next, glutathione donor beads and nickel chelate acceptor beads were added to the protein mixture and incubated for 60 min at room temperature and protected from light. The AlphaLISA signal was detected on an Envision plate reader. The screening workflow is summarized in Figure 2. Due to the interest in targeting multiple disease indications including CNS disorders, we selected a library of 23000 compounds with predicted BBB penetration for the high-throughput screening. The 23K compounds were curated from ChemDiv’s collection17 of 1.6 M compounds by applying physicochemical-properties-based filters (CNS probabilistic MPO and BBB scores), followed by removing compounds with low predicted Caco-2/Pgp permeability using machine learning models while maximizing the structural diversity within the library. The high-throughput screening was performed in the following manner: (1) the primary screen of the 23K compounds with DRP1:MiD49 PPI AlphaLISA was performed at two concentrations (10 and 100 μM), (2) the compounds with >40% inhibition at 10 μM and >80% inhibition at 100 μM were further evaluated with a 11-point dose–response and counter-screened by TruHits for false positive removal, (3) the hits were further narrowed down by removing structural alerts and ranked by ligand efficiency and synthetic tractability, and (4) the hit expansion was conducted within the full compound collection at ChemDiv, procuring structural analogues of the hit compounds, and the analogues were further evaluated by AlphaLISA and TruHits. This series of exercises led to the discovery of compound 1 as a suitable medicinal chemistry starting point with an AlphaLISA IC50 of 3.6 μM.
Figure 2.

Screening workflow. A library of 23K compounds with predicted BBB penetration was selected by applying physicochemical-properties-based filters, followed by removing compounds with low predicted Caco-2/Pgp permeability using machine learning models. The 23K compounds were screened by AlphaLISA and counterscreened by TruHits. Further analysis and hit expansion campaign led to the discovery of the hit compound 1.
We then carried out ligand-based structural optimization and SAR analyses of hit compound 1 with a primary focus on improving the biochemical potency.
The SAR summary of the quinoline 4-position is shown in Table 1. Replacing alkoxy groups with a primary amine (2a) exhibited a similar potency as the corresponding alkoxy compound. Differently substituted secondary amines, however, showed potencies across a wide range (2b–k, IC50 from 0.12 to >100 μM). The azetidine substituted compound (2c) was potent (IC50 = 0.12 μM), but functionalization on the azetidine did not lead to further improvement in potency (2g, 2h). Other nitrogen-linked substitutions such as amide (2i) and heterocycles (2j,k) resulted in loss (IC50 > 10 μM) in potency.
Table 1. SAR Summary of the Quinoline 4-Position.


The data obtained from AlphaLISA in triplicates.
Next, additional compounds were synthesized to further improve the biochemical potency. Representative examples are summarized in Table 2. Investigation of the effect of substituent on the phenyl ring yielded a compound with an IC50 of 50 nM (3). However, this compound exhibited poor solubility (11 μM in PBS at pH 7.5) and cytotoxicity at 10 μM. Replacement of the isobutyramide with oxetane-3-carboxamide attenuated solubility (281 μM in PBS at pH 7.5) with retention of the potency (4a) (IC50 = 100 nM) by lowering the cLogP to 3.7. However, further modification to lower lipophilicity by substituents on the phenyl ring (4b) or azetidine (5) led to no further improvement.
Table 2. SAR Summary of the Additional Analogues.


The data obtained from AlphaLISA in triplicates.
Calculated values.
As all the previously reported DRP1 protein structures are either apo or bound to GTP analogues, and binding pocket besides the GTP-binding pocket has not been identified to understand the mechanism of PPI inhibition with our compounds and test our hypothesis that a small molecule allosteric binder can inhibit the PPI by stabilizing the closed conformation, cocrystal formation was attempted with compounds. Gratifyingly, DRP1–compound 3 cocrystal was obtained with a sitting-drop vapor diffusion method at 291 K in the presence of 50 mM potassium chloride, 10 mM magnesium chloride, and 15% (w/v) PEG6000, and the structure was solved in 2.4 Å resolution. The DRP1–compound 3 cocrystal structure highlights unprecedented structural features. Namely, the closed conformers of DRP1 form a C2 symmetric homodimer through the α-helixes of bundle signaling element (BSE), and thus the created allosteric pocket distinct from the GTP-binding pocket is filled with compound 3 (Figure 3A). Each unit comprising the homodimer overlaid well with the reported closed conformation of DRP1, with minor conformational changes in the BSE domain, as well as remote shift of an α helix in the GTP binding site (arrows 1, 3, and 2 in Figure 3B). Residue Gln314 from one monomer formed a hydrogen-bond network with Arg172 and Asp21 from the other monomer in the dimer interface (Figure 3C), which formed the induced binding pocket. Because compound 3 inhibits the conformational change prerequisite for MiD49 binding, this class of allosteric binders can potentially inhibit protein interactions with other mitochondrial outer membrane proteins such as MiD51, FIS1, and MFF. The key hydrogen bonds were observed at three sites: (1) the isobutyramide N–H with Gln314, (2) the isobutyramide oxygen lone pairs with Arg307 and a water molecule, and (3) quinoline nitrogen with another water molecule (Figure 3D). It is also noteworthy to mention the presence of the CH−π interaction between the azetidine methylene with Tyr315, which explains the marked difference in potencies between the azetidine compound (2c) and the dimethylamino compound (2b).
Figure 3.
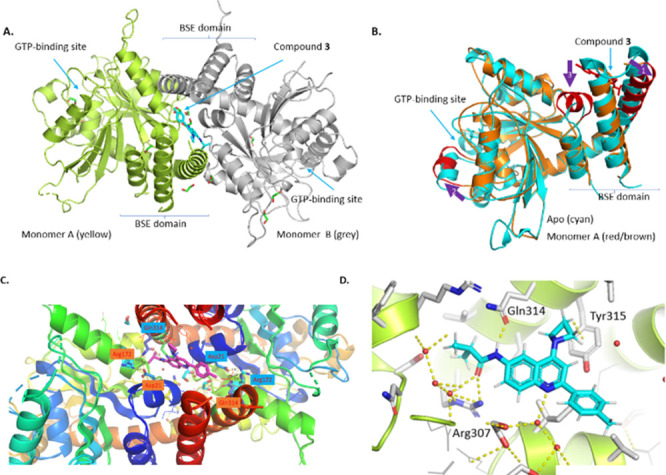
Analysis of DRP1–compound 3 cocrystal structure (PDB 8SKN). (A) The closed conformers of DRP1 form a C2 symmetric homodimer through the α-helixes of bundle signaling element (BSE), and thus created allosteric pocket distinct from the GTP-binding pocket is filled with compound 3. (B) Monomer A (red and brown) overlaid with the reported closed conformer (PDB 4H1U, cyan) with key conformational changes marked as arrows 1, 2, and 3. (C) Residue Gln314, Arg172, and Asp21 formed hydrogen-bond network in the dimer interface. (D) The key interactions identified in the binding pocket–hydrogen bonds with Gln314, Arg307, and water molecules, as well as the CH−π interaction with Tyr315.
Functional effects of the compounds on the mitochondrial morphology in cells were evaluated using high-content image analysis.18 First, supervised machine learning algorithms were trained with the images of mitochondria classified as: Normal, control siRNA; Fragmented, OPA1 siRNA; and Hypertubular, DRP1 siRNA as previously described.18 Next mitochondrial morphology controls were quantified from A549 cells with 5 μM carbonylcyanide-3-chlorophenylhydrazone (CCCP) and 50 μM cycloheximide (CHX), which induced fragmented and hypertubular mitochondria, respectively (Figure 4A). Next, we evaluated the effect of the compound on CCCP-induced mitochondrial fragmentation. The cells were treated with compounds for 1 h at 37 °C followed by cotreating with CCCP for additional 3 h at 37 °C. After the cells were fixed with paraformaldehyde and then stained with primary anti-TOM20 and secondary goat antimouse antibodies. The images were taken at 63× magnification using an Operetta CLS high-content analysis system. As compound 3 was limited due to solubility and cytotoxicity issues, compound 4a was chosen as an example. As shown in Figure 4B, compound 4a prevented mitochondrial fragmentation caused by CCCP in a dose-dependent manner. About half of fragmentation caused by CCCP was rescued by compound 4a at 0.37–- 3.3 μM, and the mitochondria showed morphology similar to the DMSO control at 10 μM.
Figure 4.

High-content imaging analysis of mitochondrial morphology affected by compound treatment. (A) CCCP treatment induced an increase in fragmented mitochondria, whereas CHX treatment increased hypertubular mitochondria as indicated by white arrows. (B) Pretreatment with compound 4a rescued CCCP-induced mitochondrial fragmentation in a dose-dependent manner. Mitochondrial morphology quantification shows dose-responsive increase in normal mitochondria and decrease in fragmented mitochondria.
To summarize, in this article, we present a novel strategy of hit finding targeting DRP1, and we have discovered a unique allosteric DRP1 inhibitor series disrupting DRP1/MiD49 protein–protein interaction. The unprecedented X-ray cocrystal structure revealed the dimerization of the closed DRP1 conformation which prevents the binding to MiD49. Representative compound 4a demonstrated inhibition of mitochondria fragmentation in cell. This provided a new strategy to inhibit DRP1 activity selectively.
Acknowledgments
This study was funded by Mitobridge, Inc., an Astellas company. We thank SAI for help on chemistry and biochemical and cellular assays. We thank Viva Biotech for help on protein production and X-ray cocrystallization.
Glossary
Abbreviations
- DRP1
dynamin-related protein 1
- MiD49
mitochondrial dynamics protein 49 kDa
- CNS
central nervous system
- MPO
multiparameter optimization score
- BBB
blood–brain barrier
- SAR
structure–activity relationship
- BSE
bundle-signaling element
- CCCP
carbonyl cyanide m-chlorophenyl hydrazone
- CHX
cycloheximide
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00223.
Experimental details for synthetic procedures and analytical data for key compounds and assay conditions, crystallographic data for compound 3 (PDF)
Author Present Address
† T.F.: Xontogeny, Boston, Massachusetts, 02116, United States
Author Present Address
‡ J.L.: Mitobridge, Astellas, Cambridge, Massachusetts 02138, United States.
Author Present Address
§ A.A.: Advanced Informatics & Analytics, Astellas, Tsukuba-shi, Ibaraki, 305–8585, Japan.
Author Present Address
∥ L.M.: Avilar Therapeutics, Waltham, Massachusetts, 02451, United States.
Author Present Address
⊥ B.L.: ArtBio, Inc. Cambridge, Massachusetts 02142, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): All authors were employees of Mitobridge, Inc., an Astellas company at the time of this study; some of the authors have held Mitobridge stock options in the past.
Supplementary Material
References
- Archer S. L. Mitochondrial dynamics--mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2013, 369, 2236–2251. 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- Youle R. J.; van der Bliek A. M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N.; Otera H.; Oka T.; Mihara K. Regulation and physiologic functions of GTPases in mitochondrial fusion and fission in mammals. Antioxid. Redox. Signal. 2013, 19, 389–399. 10.1089/ars.2012.4830. [DOI] [PubMed] [Google Scholar]
- Yen J. H.; Huang H. S.; Chuang C. J.; Huang S. T. Activation of dynamin-related protein 1 -dependent mitochondria fragmentation and suppression of osteosarcoma by cryptotanshinone. J. Exp. Clin. Cancer Res. 2019, 38, 42. 10.1186/s13046-018-1008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P. P.; Patterson A.; Stadler J.; Seeburg D. P.; Sheng M.; Blackstone C. Intra- and intermolecular domain interactions of the C-terminal GTPase effector domain of the multimeric dynamin-like GTPase Drp1. J. Biol. Chem. 2004, 279, 35967–35974. 10.1074/jbc.M404105200. [DOI] [PubMed] [Google Scholar]
- Lee Y. J.; Jeong S. Y.; Karbowski M.; Smith C. L.; Youle R. J. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. 10.1091/mbc.e04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs J. T.; Strack S. Functional characterization of phosphorylation sites in dynamin-related protein 1. Methods Enzymol 2009, 457, 231–253. 10.1016/S0076-6879(09)05013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca T. B.; Sánchez-Guerrero Á.; Milosevic I.; Raimundo N. Mitochondrial fission requires DRP1 but not dynamins. Nature 2019, 570 (7761), E34–E42. 10.1038/s41586-019-1296-y. [DOI] [PubMed] [Google Scholar]
- Cassidy-Stone A.; Chipuk J. E.; Ingerman E.; Song C.; Yoo C.; Kuwana T.; Kurth M. J.; Shaw J. T.; Hinshaw J. E.; Green D. R.; Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell. 2008, 14 (2), 193–204. 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D.; Dasgupta A.; Chen K. H.; Neuber-Hess M.; Patel J.; Hurst T. E.; Mewburn J. D.; Lima P. D. A.; Alizadeh E.; Martin A.; Wells M.; Snieckus V.; Archer S. L. Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: Therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury. FASEB J. 2020, 34 (1), 1447–1464. 10.1096/fj.201901467R. [DOI] [PubMed] [Google Scholar]
- Rosdah A. A.; Abbott B. M.; Langendorf C. G.; Deng Y.; Truong J. Q.; Waddell H. M. M.; Ling N. X. Y.; Smiles W. J.; Delbridge L. M. D.; Liu G. S.; Oakhill J. S.; Lim S. Y.; Holien J. K. A novel small molecule inhibitor of human Drp1. Sci. Rep. 2022, 12, 21531. 10.1038/s41598-022-25464-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallat A.; Uchiyama L. F.; Lewis S. C.; Fredenburg R. A.; Terada Y.; Ji N.; Nunnari J.; Tseng C. C. Discovery and characterization of selective small molecule inhibitors of the mammalian mitochondrial division dynamin, DRP1. Biochem. Biophys. Res. Commun. 2018, 499, 556–562. 10.1016/j.bbrc.2018.03.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordt E. A.; Clerc P.; Roelofs B. A.; Saladino A. J.; Tretter L.; Adam-Vizi V.; Cherok E.; Khalil A.; Yadava N.; Ge S. X.; Francis T. C.; Kennedy N. W.; Picton L. K.; Kumar T.; Uppuluri S.; Miller A. M.; Itoh K.; Karbowski M.; Sesaki H.; Hill R. B.; Polster B. M. The putative Drp1 inhibitor mdivi-1 is a reversible Complex I inhibitor that modulates reactive oxygen species. Dev. Cell. 2017, 40 (6), 583–594. 10.1016/j.devcel.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia R.; Wang R. Y. R.; Yusuf A.; Thomas P. V.; Agard D. A.; Shaw J. M.; Frost A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature 2018, 558, 401–405. 10.1038/s41586-018-0211-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Zhai Y.; Chen M.; Zhang K.; Chen Q.; Pang X.; Sun F. New interfaces on MiD51 for Drp1 recruitment and regulation. PLoS One 2019, 14 (1), e0211459. 10.1371/journal.pone.0211459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasgar A.; Jadhav A.; Simeonov A.; Coussens N. P. AlphaScreen-based assays: Ultra-high-throughput screening for small-molecule inhibitors of challenging enzymes and protein-protein interactions. Methods Mol. Biol. 2016, 1439, 77–98. 10.1007/978-1-4939-3673-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Integrated Drug Discovery CRO Services for Pharmaceutical R&D; ChemDiv: San Diego, CA, 2023, https://www.chemdiv.com.
- Cretin E.; Lopes P.; Vimont E.; Tatsuta T.; Langer T.; Gazi A.; Sachse M.; Yu-Wai-Man P.; Reynier P.; Wai T. High-throughput screening identifies suppressors of mitochondrial fragmentation in OPA1 fibroblasts. EMBO Mol. Med. 2021, 13, e13579. 10.15252/emmm.202013579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.