

Abstract

Toll-like receptor (TLR) 7 and TLR8 are endosomal sensors of the innate immune system that are activated by GU-rich single stranded RNA (ssRNA). Multiple genetic and functional lines of evidence link chronic activation of TLR7/8 to the pathogenesis of systemic autoimmune diseases (sAID) such as Sjögren’s syndrome (SjS) and systemic lupus erythematosus (SLE). This makes targeting TLR7/8-induced inflammation with small-molecule inhibitors an attractive approach for the treatment of patients suffering from systemic autoimmune diseases. Here, we describe how structure-based optimization of compound 2 resulted in the discovery of 34 (MHV370, (S)-N-(4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)morpholine-3-carboxamide). Its in vivo activity allows for further profiling toward clinical trials in patients with autoimmune disorders, and a Phase 2 proof of concept study of MHV370 has been initiated, testing its safety and efficacy in patients with Sjögren’s syndrome and mixed connective tissue disease.
Keywords: Toll-like receptor 7, toll-like receptor 8, lupus, innate immunity, SAR
Toll-like receptors (TLRs) are evolutionarily conserved type I transmembrane proteins of the innate immune system that recognize conserved molecular patterns (pathogen associated molecular patterns, PAMPs), which are present in proteins, lipids, and nucleic acids from pathogens. Activation of TLR pathways leads to host defense mechanisms such as secretion of inflammatory cytokines, induction of interferon pathways and activation of B cells and neutrophils.1 The endosomal TLRs, TLR3, TLR7/8 and TLR9 recognize double stranded RNA (dsDNA), GU-rich single stranded RNA (ssRNA) and nonmethylated single stranded DNA (ssDNA), respectively. Aberrant activation of these nucleic acid sensing endosomal TLRs has been invoked as a potential driver of autoimmune diseases such as systemic lupus erythematosus (SLE) and Sjögren’s syndrome (SjS).2,3
Genetic and functional evidence suggests chronic hyperactivation of TLR7/8 by endogenous ssRNA as a driver of several systemic autoimmune diseases. For example, TLR7 gain-of-function mutations were found in children suffering from severe autoimmunity, and expression of one such mutant in mice causes lethal lupus-like disease,4 as does overexpression of TLR75 or human TLR8.6
Increased activation of TLR7 and TLR8 drives multiple cellular mechanisms of systemic autoimmune diseases. For example, RNA-containing immune complexes (ICs) between autoantibodies of patients and ribonucleoproteins (RNPs) drive secretion of type I interferon (IFNs) from plasmacytoid dendritic cells (pDCs) in a TLR7-dependent manner,7 and IFNs8 along with an elevated IFN stimulated gene signature9 are characteristic features of SLE patients. Neutrophils get activated via TLR810 autoreactive B cells internalize RNA-containing autoantigens for TLR7 activation11 and complexed RNA-containing autoantigens are engulfed by monocytes/DCs for activation of TLR8.12 Together, enhanced activation of autoreactive B cells, pDCs and monocytes/DCs via TLR7/8 increases the secretion of antineuclear antibodies (ANAs) and sustains a self-perpetuating feedback loop of inflammation and tissue damage (Figure 1).
Figure 1.
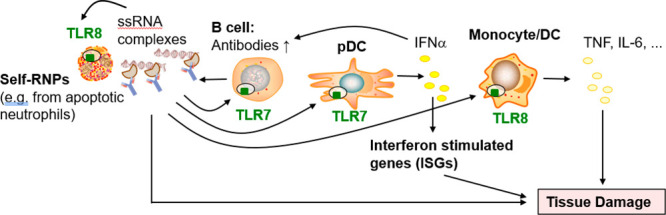
Role of TLR7 and TLR8 in the pathology of SLE and related chronic inflammatory diseases. ssRNA complexes are immune complexes of RNPs and autoantibodies or complexes of ssRNA with amphiphilic peptides.
Consequently, TLR7/8 antagonists could serve to break this vicious loop in patients and developing small-molecule inhibitors is an attractive approach for the treatment of such autoimmune indications13−15
We recently described a series of potent TLR7/8 antagonists discovered in a phenotypic screen.13 This effort was focused around modulating the amine moiety to address the liabilities of the piperazine group in the primary hit and on potency optimization through identification of the most adequate “head group” (4-substituted 1,6-dimethyl-1H-pyrazolo[3,4-b]pyridine) moiety to optimize potency; compound 1, Figure 2. Here we expand on our SAR optimization of a subseries of 1 that allowed us to identify the clinical candidate compound 34 ((S)-N-(4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)morpholine-3-carboxamide, MHV370),
Figure 2.

Discovery of compound 2 by SAR database scouting TLR7 cellular assays in human PBMCs and mouse splenocytes; details in the Supporting Information. aIC50 ± SD, numbers in parentheses denote the number of independent experiments.
Although 1 (Figure 2) was a potent TLR7 antagonist, it was not suitable as a drug candidate due to its physicochemical properties (low solubility) and hERG inhibition. These properties were evident across the entire series and as a consequence impossible to improve by staying within the current compound configuration (i.e., aromatic or heteroaromatic ring appended to the bicyclic 1,6-dimethyl-1H-pyrazolo[3,4-b]pyridine moiety). For this reason, we made selected changes to the scaffold focusing on decreasing sp2 character. This resulted in compound 2, which had adequate potency on TLR7 in human PBMCs (IC50 of 0.018 ± 0.023 μM, n = 210) and mouse splenocytes (IC50 of 0.074 ± 0.11 μM, n = 98) and displayed an improved solubility and hERG inhibition profile (IC50: 29 μM), as shown in Figure 2.
The synthesis of 2 is depicted in Scheme 1. Alkylation of 3 with 4 using Cs2CO3 in DMSO at 120 °C for 18 h afforded an easily separable mixture of 5 and 6. Hydrogenation (H cube) of 5 afforded 7 quantitatively. Buchwald coupling of 7 and 8 in the presence of Pd2dba3, RuPhos, and Cs2CO3 in THF at 80 °C afforded the Boc derivative 9. Boc deprotection (4 N HCl/dioxane) followed by recrystallization in IPA and trituration in a slurry of Ambersep9000OH in MeOH at rt afforded 2 in 90% yield.
Scheme 1. Synthesis of Compound 2.

Reagents and conditions. a. Cs2CO3, DMSO, 120°C, 18 h. b. H-cube. c. Pd2dba3, RuPhos, Cs2CO3, THF, 80°C, 18 h. d. 4 N HCl/dioxane, MeOH, RT, 1h. e. Crystallization in IPA. f. Ambersep900OH, MeOH, rt, 1 h.
When profiled for its ADME properties, 2 showed poor permeability (log PAMPA = −5.1) associated with a strong efflux in the Caco2 assay (ratio A-B/B-A = 15.2). When 2 was tested for its PK properties in mice at doses of 5 mg/kg IV and 20 mg/kg PO as a solution (formulation PEG300/D5W, 3:1), it displayed a moderate clearance (24 mL/min/kg), high Vss (4.1 L/kg), short MRT (2.9 h), and low oral bioavailability of 4%, consistent with the observed permeability and efflux data. The data collected on 2 are displayed in Table 1.
Table 1. ADME/Physicochemical and Mouse PK Profiles of Compound 2.
| ADME
profile of 2 |
mouse
PK profile of 2 |
||||
|---|---|---|---|---|---|
| CYP 3A4 (μM) | >50a | IV dosing (3 mg/kg) | PO dosing (20 mg/kg) | ||
| h, m, r Met CL (μL/min/mg) | 19, 15, 36 | AUC (hanM) | 5005 | AUC (h·nM) | 1146 |
| Caco2 (A-B/B-A); ratio | 2.1/15.7; 7.5 | CL (mL/min/kg) | 24 | Cmax (nM) | 321 |
| LogPAMPA (cm/s) | –5.1 | Cmax (nM) | 321 | Clast; Tlast (nM; h) | 35; 7 |
| CYP 3A4 TDI (Ki/Kinact) | 0.002 | Vss (L/kg) | 4.1 | MRT (h) | 2.9 |
| PXR (CYP induction) | 7.9%b | T1/2 (h) | 2.1 | %F | 4 |
Inhibition of CYP 2D9 and CYP 2D6 with IC50 of >25 μM.
Rifampicin was used as standard (set at 100%). IV and PO formulation: 2.9 mg/mL in PEG300/D5W, 3:1; solution.
We hypothesized that the free amine appended to the octabicyclic system was responsible for the lack of permeability and therefore evaluated several amine-capping substitutions (mostly mono- and disubstituted alkyl and heteroalkyl derivatives). Scheme 2 depicts a sample of the first round of SAR. The monomethyl amine derivative 11 was synthesized by condensation of 10 on 8 using Pd2dba3, Cs2CO3, RuPhos in dioxane at reflux, followed by boc deprotection (4 N HCl in dioxane at RT). Compounds 12–14 were synthesized by reductive amination of 2 using formaldehyde, cyclobutanone, and oxetan-3-one, respectively. Sulfonylation (DIPEA, MeSO2Cl, DCM, 0 °C to RT) or acylation (DIPEA, AcCl, DCM, RT) of 2 afforded the corresponding sulfonamide 15 and acetamide 16. A simple alkylation reaction (K2CO3, EtOH, 2-methoxyethyl bromide, 1,4-dibromobutane, 1-bromo-2-(2-bromoethoxy)ethane, 1,3-dibromopropane, and (S)-1-bromo-2-(((R)-1-bromopropan-2-yl)oxy)propane) was used to synthesize compounds 17–21, respectively. Compound 22 was synthesized by condensation of 2,2-difluoroethyl trifluoromethanesulfonate in the presence of DIPEA in refluxing THF overnight.
Scheme 2. Synthetic Route of Alkyl-Substituted Amines.

Reagents and conditions: a. Pd2dba3, Cs2CO3, RuPhos, dioxane, 10, reflux 18 h. b. HCl-dioxane, RT. c. Paraformaldehyde, DIEPA, NaBH3CN, THF/MeOH, RT. d. Cyclobutanone, NaBH3CN, AcOH, MeOH, RT to 75°C. e. Oxetan-3-one, NaBH3(OAc)3, DCE, AcOH, RT. f, AcCl or MsCl, DIPEA, DCM, 0°C to RT. g. 1-Bromo-2-methoxyethane, K2CO3, IPA, reflux. h/k. Corresponding dialkyl bromide, K2CO3, EtOH, 120°C, MW irradiation, 30 min. l. (S)-1-(4-Methylbenzenesulfonate)-2-(((R)-1-(4-methylbenzenesulfonate)-2-yl)oxy)propane, DIEPA, DMA, 100°C, 18h. m. 2,2-Difluoroethyl trifluoromethanesulfonate, DIPEA, THF, reflux, ON.
Table 2 summarizes the findings associated with our first round of SAR using various amine-capping groups.
Table 2. Profiling Data of Compounds 2, 11–22a.
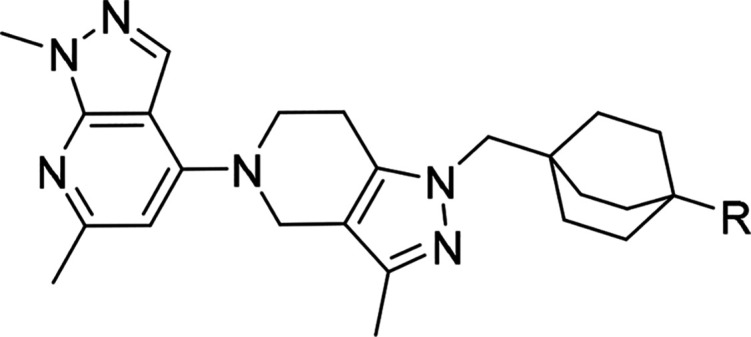

Potency in cellular assays for TLR7, TLR8, and TLR9 (PBMCs) as described in the Supporting Information. IC50 in μM ± geomeans; numbers in brackets denote number of independent experiments. HT solubility and CYP3A4 inhibition (μM), TDI: Kobs (min–1), hERG: 3H-dofetilite displacement assay (μM). Metabolic clearance: CLint (μL/min/mg). ND = not determined.
As predicted, most substitutions had a positive effect on permeability as measured using the PAMPA assay (Caco-2 followed a similar trend), and this first round of SAR provided interesting results. Alkyl, heteroalkyl and fluoroalkyl substituents (compounds 11–14 and 17–22) were well tolerated to maintain potent TLR7/8 inhibition and improved their permeability. However, most compounds exhibited high human metabolic clearance in vitro (14–16, 18–22) and this trend was consistent across most compounds synthesized. Time-dependent inhibition of CYP3A4 enzyme (as measured by high throughput time dependent enzymatic activity assay) also appeared to be high for some compounds (12, 13, 15, 16, 18–22). Surprisingly, the sulfonamide 15 and the carboxamide 16 displayed an excellent TLR7 inhibition profile (0.008 ± 0.007 and 0.004 ± 0.002 μM, respectively) but were inactive toward inhibition of TLR8 responses (>10 μM and 5.2 ± 4.5 μM, respectively). We hypothesized that TLR7/8 dual inhibition required a higher basicity than TLR7 inhibition and that therefore introducing an amine at a proximal distance to the amide would be a good strategy to maintain dual potency. To achieve this, we introduced simple capping groups like substituted glycine or alanine derivatives (resulting in 23–29) or alkylamines possessing an alpha carboxamide (resulting in 30) (Scheme 3). In our derivatization efforts, we used only a set of small amino acids to keep the molecular weight/PSA and cLogP under control. In parallel, we also tried to move the amide away from the octabicyclic amine moiety (only one example is shown, compound 30) and found that these derivatives retained activity. Piperazinone (compounds 31–32) and morpholinamide substituents in the octabicyclo system (compounds 33–35) (Scheme 4) also retained activity.
Scheme 3. Synthetic Route for Compounds 23–30.

Reagents and conditions: a. Corresponding amino acid (boc-protected where required), HATU, DIEPA, DCM, RT, then HCl-dioxane. b. (30) 2-bromo-N,N-dimethylacetamide, Cs2CO3, DMF, RT.
Scheme 4. Synthetic Route for Compounds 31–35.

Reagents and conditions: a. tert-Butyl methyl(2-oxoethyl)carbamate, NaBH(OAc)3, CPME, RT. b. 2-Bromoacetate, K2CO3, EtOH, MW irradiation, 150°C, 1 h then 1 N NaOH, RT. c. HCl-dioxane, RT. d. HATU, DIEPA, DMF, 0 °C. e 2-Bromoacetylbromide, DIEPA, DCM, 0°C to RT followed by HCl-dioxane, RT. f. K2CO3, EtOH, MW irradiation, 120 °C, 40 min followed 1 N aqueous HCl and lyophilization. g. Corresponding morpholino carboxylic acid, HATU, DIEPA, DCM, RT, then HCl-dioxane.
The cellular potency on the TLR pathways and the properties for compounds 23–35 are shown in Table 3. In general, all compounds were potent TLR7 antagonists (IC50 < 0.07 μM). However, only a few derivatives showed sufficient TLR8 inhibition (27, 28, 31, and 36). TLR9 pathway inhibition was generally weak across the series, as previously described.9 Compounds 23–35 all showed good solubility (>175 μM). While most compounds had acceptable CYP inhibition profiles (CYP2D6, CYP3A1 comparable to CYP3A4, data not shown), one liability of this series was unpredictable human clearance in liver microsomes (25–30 and 31–33). Compound 30 had an interesting overall profile but showed time dependent CYP inhibition. Several derivatives were selected for PK profiling in parallel to evaluate in vitro-in vivo correlations. The mouse and rat PK profiles of compounds 23, 28, and 34 are illustrated in Table 4.
Table 3. Profiling Data of Compoundsa.
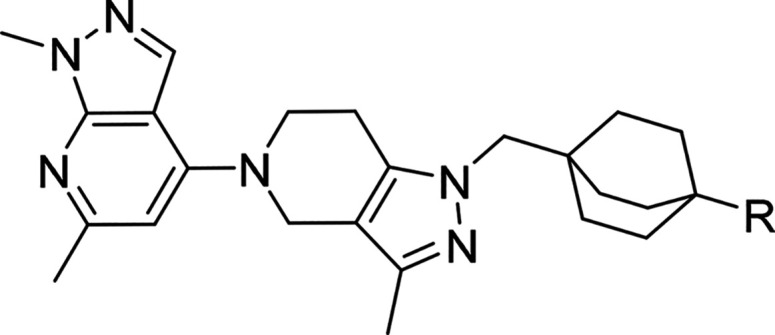

Potency (IC50 in μM) within cellular assays for TLR7, TLR8, and TLR9 (PBMCs) as described in the Supporting Information. HT solubility and CYP3A4 inhibition (μM), TDI: Kobs (min–1), hERG: 3H-dofetilite displacement assay (μM). Metabolic clearance: CLint (μL/min/mg). ND = not determined.
Table 4. Mouse and Rat PK Profiles of Compounds 23, 28, and 34a.
|
23 |
28 |
34 |
||||
|---|---|---|---|---|---|---|
| mouse | rat | mouse | rat | mouse | rat | |
| CL (mL·min–1·kg–1) | 7 | 19 | 37 | 19 | 11 | 18 |
| Vss (L kg–1) | 1 | 3 | 3.5 | 2 | 0.7 | 1.3 |
| IV AUC (nM·h) | 9276 | 1782 | 1790 | 1784 | 5672 | 1776 |
| MRT (h) | 2.4 | 2.8 | 1.6 | 1.6 | 1 | 1.2 |
| PO Cmax (nM) | 515 | 48 | 237 | 31 | 1858 | 282 |
| PO AUC (nM·h) | 4629 | 618 | 2336 | 483 | 9160 | 2817 |
| %F | 10 | 12 | 26 | 9 | 32 | 52 |
Formulation for IV and PO: 1 mg/mL in PEG300/D5W, 3:1; solution.
Of all these derivatives, the PK profile of compound 34 was most attractive. Compound 34 possessed adequate PK properties in mouse and rat, as it showed moderate clearance (11 and 18 mL.min–1.kg–1 respectively) and low Vss (0.7 and 1.3 L kg–1 respectively). The relatively short mean residence time (MRT) of 1 and 1.2 h, respectively, as well as moderate bioavailability (%F: 32 and 52 respectively) were considered sufficient for translation into acceptable parameters in human. It is notable that while the PAMPA score of 34 was similar to 2, the Caco2 assay (ratio A-B/B-A = 1.92) was vastly superior, and this was reflected in better exposure.
Compound 34 (MHV370) was profiled for inhibition of TLR7 and TLR8 responses and selectivity against TLR4 and TLR9 pathway activation in human PBMCs and whole blood. Compound 34 (MHV370) showed a slightly higher potency on TLR7 compared to TLR8 in both assay matrices (Table 5). In contrast, compound 34 (MHV370) showed very weak inhibition of TLR9 and was inactive against TLR4 (Table 5). Furthermore, compound 34 (MHV370) inhibited murine TLR7, yet it did not inhibit mouse TLR4 or TLR9, as shown on mouse splenocytes. In the preparation of in vivo experiments in mice, we also demonstrated potent and selective TLR7 inhibition in murine whole blood (Table 5). It was not possible to study TLR8 responses in murine assays, as rodent TLR8 does not recognize ssRNA ligands.16 In addition, compound 34 (MHV370) showed high selectivity against other TLRs (Hawtin et al., under revision) and did not show any inhibition of catalytic activity on 46 kinases in a biochemical assay panel (assay details in the Supporting Information).
Table 5. Cellular Profile of Compound 34 (MHV370) on the Indicated TLRsa.
| TLR7 (IFNα) | TLR8 (IL-6) | TLR9 (IFNα) | TLR4 (IL-6) | |
|---|---|---|---|---|
| hPBMC | 0.004 ± 0.002 (39) | 0.068 ± 0.032 (16) | 1.0 ± 0.77 (20) | >10 (10) |
| huWB | 0.007 ± 0.004 (16) | 0.074 ± 0.047 (4) | 1.2 ± 0.52 (12) | >10 (4) |
| msSpleno | 0.009 ± 0.003 (6) | NA | >1 (6) | >10 (4) |
| mWB | 0.051 ± 0.027 (7) | NA | >10 (4) | >10 (4) |
Assay conditions are described in the Supporting Information. Geometric means of IC50 (μM) ± SD, number of individual experiments, or donors are indicated in parentheses. NA = Not applicable since mouse TLR8 does not recognize ssRNA ligands.
An X-ray cocrystal structure of compound 34 (MHV370) with the endosomal domain of human TLR8 confirmed their direct interaction (Figure 3). Similar to published structures (Tanji, Science2013) two TLR8 protomers build a homodimeric complex with an axial symmetry, forming at their interface two symmetrical binding pockets. A key interaction is the hydrogen bond between the pyrazolo-pyridine of compound 34 (MHV370) and the NH of Gly351. In addition, the central part of compound 34 (MHV370) is embedded in the hydrophobic pocket formed by the aromatic and aliphatic side chains of the two TLR8 moieties.
Figure 3.
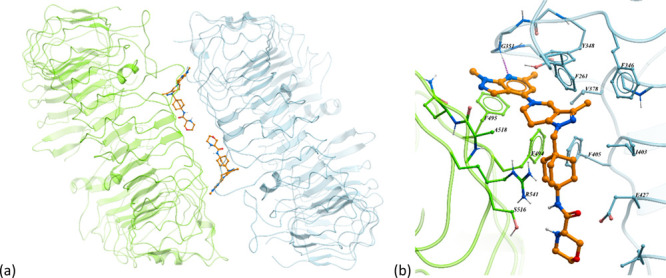
Cocrystal structure of compound 34 (MHV370) with TLR8 (PDB 8PFI). (a) Full view of the homodimeric structure and the two symmetrical binding sites formed between thae two TLR8 chains (one TLR8 monomer in light green and the second monomer in light blue), carbon atoms of compound 34 (MHV370) in orange. (b) Close-up view of one binding site showing amino acids in close contact with compound 34 (MHV370). Notable interactions include a hydrogen bond interaction (represented magenta dotted line) between the pyrazolo-pyridine moiety and Gly351 and many hydrophobic interactions between the central part of compound 34 (MHV370) and residues Phe261, Phe346, Val378, Ile403, Phe405, and Phe495.
Based on the above data as well as superior PK and CYP inhibition profiles relative to 35, we evaluated compound 34 (MHV370) in vivo in an acute PK/PD model. Oral dosing of MHV370 showed a dose-dependent suppression of IFNα, IL-6 and TNF in mice following challenge with the TLR7 ligand ssRNA/DOTAP. Near-complete inhibition of IFNα, IL-6, and TNF in plasma was observed with 10 mg/kg of compound 34 (MHV370) at 2 h post challenge (Figure 4).
Figure 4.
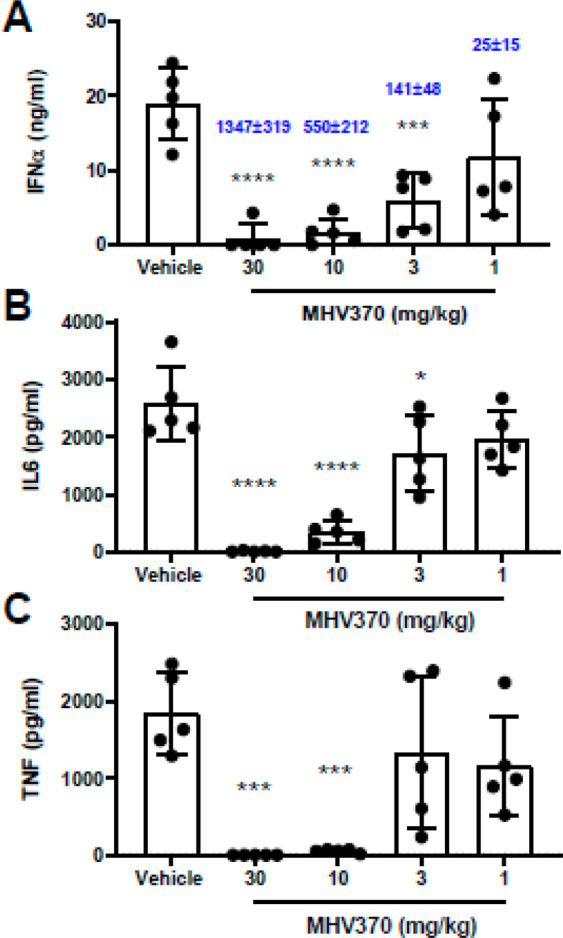
In vivo efficacy of compound 34 (MHV370). Compound 34 (MHV370) suppresses serum levels of multiple proinflammatory cytokines in mice after an acute TLR7 challenge. Bars represent means and SD, dots represent individual mice. Blue numbers represent blood exposures in nM (±SD), ***p > 0.001, ****p > 0.0001, ANOVA with Dunnett’s post test.
These results encouraged us to further characterize 34 (MHV370) in more complex, chronic in vivo mouse models.17 The safety and efficacy profiles in these studies were excellent, and thus a clinical trial has been initiated for MHV370 in patients suffering from Sjögren’s syndrome (SjS) and mixed connective tissue disease (MCTD, NCT04988087). Two other TLR7/8 antagonists are in clinical development, afimetoran/BMS-986256 by BMS and enpatoran/M5049 by Merck, and the in vitro and in vivo profile of enpatoran has been published.18 Both enpatoran and MHV370 show a higher potency on TLR7 than on TLR8, and MHV370 shows a higher potency on TLR8 than that reported for enpatoran. Both compounds were only weakly active or inactive on TLR9 and both can be safely dosed in mouse models of chronic inflammation.17,18
In conclusion, a SAR campaign on compound 2 led to substitution of the amine present on 2 in order to address its deficiencies (weak potency and poor permeability, leading to poor oral exposure). Systematic SAR on that amide led to the identification of compound 34 (MHV370; (S)-N-(4-((5-(1,6-dimethyl-1H-pyrazolo[3,4-b]pyridin-4-yl)-3-methyl-4,5,6,7-tetrahydro-1H-pyrazolo[4,3-c]pyridin-1-yl)methyl)bicyclo[2.2.2]octan-1-yl)morpholine-3-carboxamide) which possesses a favorable balance of potency/PK and physicochemical properties. In the meantime, a Phase 2 clinical proof of concept study has been initiated for compound 34 (MHV370) for systemic autoimmune diseases with high unmet medical need [NCT04988087].
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00136.
Author Present Address
§ Kumquat Biosciences, 10770 Wateridge Circle, Ste. 120, San Diego, CA 92121, USA
Author Present Address
∥ Odyssey Therapeutics, 4242 Campus Point Court, San Diego, CA 92121, USA
Author Contributions
PM, PA, JD, ToJu, and SH designed experiments and wrote the manuscript. CA, TB, GR, GZ, DC, DH, TaJa, JM, DM, WP, PS, GZ, and YZ conducted experiments. CB supported manuscript writing.
The authors declare no competing financial interest.
Supplementary Material
References
- Kawai T.; Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature immunology 2010, 11 (5), 373–84. 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Marshak-Rothstein A.; Rifkin I. R. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annual review of immunology 2007, 25, 419–41. 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- Junt T.; Barchet W. Translating nucleic acid-sensing pathways into therapies. Nature reviews. Immunology 2015, 15 (9), 529–44. 10.1038/nri3875. [DOI] [PubMed] [Google Scholar]
- Brown G. J.; Canete P. F.; Wang H.; Medhavy A.; Bones J.; Roco J. A.; He Y.; Qin Y.; Cappello J.; Ellyard J. I.; Bassett K.; Shen Q.; Burgio G.; Zhang Y.; Turnbull C.; Meng X.; Wu P.; Cho E.; Miosge L. A.; Andrews T. D.; Field M. A.; Tvorogov D.; Lopez A. F.; Babon J. J.; Lopez C. A.; Gonzalez-Murillo A.; Garulo D. C.; Pascual V.; Levy T.; Mallack E. J.; Calame D. G.; Lotze T.; Lupski J. R.; Ding H.; Ullah T. R.; Walters G. D.; Koina M. E.; Cook M. C.; Shen N.; de Lucas Collantes C.; Corry B.; Gantier M. P.; Athanasopoulos V.; Vinuesa C. G. TLR7 gain-of-function genetic variation causes human lupus. Nature 2022, 605 (7909), 349–356. 10.1038/s41586-022-04642-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane J. A.; Pisitkun P.; Barrett R. S.; Feigenbaum L.; Town T.; Ward J. M.; Flavell R. A.; Bolland S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 2007, 27 (5), 801–10. 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiducci C.; Gong M.; Cepika A. M.; Xu Z.; Tripodo C.; Bennett L.; Crain C.; Quartier P.; Cush J. J.; Pascual V.; Coffman R. L.; Barrat F. J. RNA recognition by human TLR8 can lead to autoimmune inflammation. Journal of experimental medicine 2013, 210 (13), 2903–19. 10.1084/jem.20131044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat F. J.; Meeker T.; Gregorio J.; Chan J. H.; Uematsu S.; Akira S.; Chang B.; Duramad O.; Coffman R. L. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. Journal of experimental medicine 2005, 202 (8), 1131–9. 10.1084/jem.20050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero M. P.; Decalf J.; Bondet V.; Hunt D.; Rice G. I.; Werneke S.; McGlasson S. L.; Alyanakian M. A.; Bader-Meunier B.; Barnerias C.; Bellon N.; Belot A.; Bodemer C.; Briggs T. A.; Desguerre I.; Fremond M. L.; Hully M.; van den Maagdenberg A.; Melki I.; Meyts I.; Musset L.; Pelzer N.; Quartier P.; Terwindt G. M.; Wardlaw J.; Wiseman S.; Rieux-Laucat F.; Rose Y.; Neven B.; Hertel C.; Hayday A.; Albert M. L.; Rozenberg F.; Crow Y. J.; Duffy D. Detection of interferon alpha protein reveals differential levels and cellular sources in disease. Journal of experimental medicine 2017, 214, 1547. 10.1084/jem.20161451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau R.; Hong S.; Cantarel B.; Baldwin N.; Baisch J.; Edens M.; Cepika A. M.; Acs P.; Turner J.; Anguiano E.; Vinod P.; Kahn S.; Obermoser G.; Blankenship D.; Wakeland E.; Nassi L.; Gotte A.; Punaro M.; Liu Y. J.; Banchereau J.; Rossello-Urgell J.; Wright T.; Pascual V. Personalized Immunomonitoring Uncovers Molecular Networks that Stratify Lupus Patients. Cell 2016, 165 (3), 551. 10.1016/j.cell.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herster F.; Bittner Z.; Archer N. K.; Dickhofer S.; Eisel D.; Eigenbrod T.; Knorpp T.; Schneiderhan-Marra N.; Loffler M. W.; Kalbacher H.; Vierbuchen T.; Heine H.; Miller L. S.; Hartl D.; Freund L.; Schakel K.; Heister M.; Ghoreschi K.; Weber A. N. R. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat. Commun. 2020, 11 (1), 105. 10.1038/s41467-019-13756-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos A. M.; Busconi L.; Marshak-Rothstein A. Regulation of autoreactive B cell responses to endogenous TLR ligands. Autoimmunity 2010, 43 (1), 76–83. 10.3109/08916930903374618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly D.; Chamilos G.; Lande R.; Gregorio J.; Meller S.; Facchinetti V.; Homey B.; Barrat F. J.; Zal T.; Gilliet M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. Journal of experimental medicine 2009, 206 (9), 1983–94. 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper P. B.; Deane J.; Betschart C.; Buffet D.; Collignon Zipfel G.; Gordon P.; Hampton J.; Hawtin S.; Ibanez M.; Jiang T.; Junt T.; Knoepfel T.; Liu B.; Maginnis J.; McKeever U.; Michellys P. Y.; Mutnick D.; Nayak B.; Niwa S.; Richmond W.; Rush J. S.; Syka P.; Zhang Y.; Zhu X. Discovery of potent, orally bioavailable in vivo efficacious antagonists of the TLR7/8 pathway. Bioorg. Med. Chem. Lett. 2020, 30 (17), 127366. 10.1016/j.bmcl.2020.127366. [DOI] [PubMed] [Google Scholar]
- Knoepfel T.; Nimsgern P.; Jacquier S.; Bourrel M.; Vangrevelinghe E.; Glatthar R.; Behnke D.; Alper P. B.; Michellys P. Y.; Deane J.; Junt T.; Zipfel G.; Limonta S.; Hawtin S.; Andre C.; Boulay T.; Loetscher P.; Faller M.; Blank J.; Feifel R.; Betschart C. Target-Based Identification and Optimization of 5-Indazol-5-yl Pyridones as Toll-like Receptor 7 and 8 Antagonists Using a Biochemical TLR8 Antagonist Competition Assay. Journal of medicinal chemistry 2020, 63 (15), 8276–8295. 10.1021/acs.jmedchem.0c00130. [DOI] [PubMed] [Google Scholar]
- Vlach J.; Bender A. T.; Przetak M.; Pereira A.; Deshpande A.; Johnson T. L.; Reissig S.; Tzvetkov E.; Musil D.; Morse N. T.; Haselmayer P.; Zimmerli S. C.; Okitsu S. L.; Walsky R. L.; Sherer B. Discovery of M5049: A Novel Selective Toll-Like Receptor 7/8 Inhibitor for Treatment of Autoimmunity. J. Pharmacol Exp Ther 2021, 376 (3), 397–409. 10.1124/jpet.120.000275. [DOI] [PubMed] [Google Scholar]
- Forsbach A.; Nemorin J. G.; Montino C.; Muller C.; Samulowitz U.; Vicari A. P.; Jurk M.; Mutwiri G. K.; Krieg A. M.; Lipford G. B.; Vollmer J. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J.Immunol. 2008, 180 (6), 3729–3738. 10.4049/jimmunol.180.6.3729. [DOI] [PubMed] [Google Scholar]
- Hawtin S.; André C.; Collignon-Zipfel G.; Appenzeller S.; Bannert B.; Baumgartner L.; Beck D.; Betschart C.; Boulay T.; Brunner H. I.; Ceci M.; Deane J.; Feifel R.; Ferrero E.; Kyburz D.; Lafossas F.; Loetscher P.; Merz-Stoeckle C.; Michellys P.; Nuesslein-Hildesheim B.; Raulf F.; Rush J. S.; Ruzzante G.; Stein T.; Zaharevitz S.; Wieczorek G.; Siegel R.; Gergely P.; Shisha T.; Junt T. Preclinical characterization of the Toll-like receptor 7/8 antagonist MHV370 for lupus therapy. Cell Rep. Med. 2023, 4, 101036. 10.1016/j.xcrm.2023.101036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlach J.; Bender A. T.; Przetak M.; Pereira A.; Deshpande A.; Johnson T. L.; Reissig S.; Tzvetkov E.; Musil D.; Morse N. T.; Haselmayer P.; Zimmerli S. C.; Okitsu S. L.; Walsky R. L.; Sherer B. Discovery of M5049: A Novel Selective Toll-Like Receptor 7/8 Inhibitor for Treatment of Autoimmunity. J. Pharmacol. Exp. Ther. 2021, 376 (3), 397–409. 10.1124/jpet.120.000275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.