

Abstract
This article presents a comprehensive review on the synthesis and stability of ferrite nanoparticles such as nickel ferrite (NiFe2O4), zinc ferrite (ZnFe2O4), manganese ferrite (MnFe2O4), iron ferrite (Fe2O3), cobalt ferrite (CoFe2O4) and also mixed nanoparticles. Different synthetic methods for ferrite nanoparticles have been reviewed such as co-precipitation, thermal decomposition and hydrothermal, microwave-assisted and sonochemical methods. The effect on the stability of different capping agents like canola oil, glycerol, sodium dodecyl, sodium citrate, oleic acid, Triton-100 and sodium dodecyl benzene sulfonates has also been studied.
Keywords: Ferrite mixed nanoparticles, capping agent, hydrothermal method
Introduction
Nanotechnology includes the control and understanding of matter of 1–100 nm dimensions, where distinctive physical phenomena facilitate novel applications. The chemical and physical properties of nanomaterials are largely governed by their size, shape or morphology. A deviation in the size of nanomaterials can alter their physical properties without changing their composition. When the particle size falls below 100 nm, a large fraction of the component atoms is located on the surface of the nanocrystals, thus suggesting major changes in the magnetic structure and properties of the nanomaterials in contrast to their bulk counterparts. These nano- and micro-changes lead to new phenomena such as superparamagnetism, enhanced anisotropy and spin canting. Nanomaterials have been greatly utilized worldwide over the last 20 years due to their better thermophysical properties in engineering applications.1,2 This management of matter, with nanometre dimensions, has enabled the manufacture of new devices, structures and materials. Nanoparticles (NPs) offer an exceptional advance in certain sectors, as in medicine, consumer products, energy, materials and manufacturing.3–5 It has been shown that ferrites also act as a multifunctional detoxicant for soluble metal ions other than iron.1–3 Studies published in the 80s evidenced ferrites having in vivo binding potential for metals such as Cu2+, Cd2+, Al2+, Zn2+ and Be2+ after administration of their respective salts.4,5 The production of small magnetic NPs with sizes from 2 to 20 nm is important, because of their uses in multi-terabyte magnetic storage devices. 6 The magnetic properties of NPs originate because of the compact sizes of isolated NPs, and the influence of interparticle interactions is insignificant. The main issues in NP production are homogeneity of particle size, size control, crystal structure, shape control and arrangement for device applications. 7 Ferrites are superparamagnetic, hard and brittle ceramic materials. The general formula of ferrites is M1M2O4, where M2 is Fe and M1 is a transition metal which accounts for enhancing and elevating the magnetic properties of the ferrites. Depending upon coercivity, ferrites can be divided into soft ferrites and hard ferrites. Soft ferrites are distinguished as having a short period of magnetization and demagnetization, giving reduced hysteresis loss and low coercivity. The small loop area of soft ferrites opens its applications in switching circuits, magnetic drug delivery, treatment of bacterial infection and electromagnetic cores of various transformers. 8 In contrast, hard ferrites have a high magnetic strength value of 0.35 T − 160 kA m−1 for their cycle of magnetization and demagnetization – and have greater values of their coercivity and retentivity. These ferrites act as permanent magnets due to their high flux density and hysteresis loss, and are used for the preparation of data storage devices, microelectronics, radio formation and magnetic analysis.
Comparison of the syntheses of ferrite NPs
The co-precipitation method
The particles obtained with the co-precipitation method have a broad size distribution. 9 For the preparation of monodisperse NPs of magnetite, a number of methods are used as follows:
This is selected according to which a high temperature up to (265°C) is to be maintained: Fe(acac)3 (iron(III) trisacetylacetonate) is reacted with phenyl ether with alcohol, oleylamine and oleic acid. Using smaller NPs of magnetite as seeds, larger monodisperse magnetite NPs of up to 20 nm diameter can be synthesized and dispersed into a nonpolar solvent. The process does not require a size selection procedure and is readily scaled up for mass production. 10
The co-precipitation method is modified to synthesize iron oxide NPs: 0.074 g of ferric chloride and 0.190 g of ferrous chloride FeCl2 are used in a 2:1 ratio. These were dissolved in 20 mL of deionized water, stirred and heated at 60°C. To retard any unwanted oxidation reaction, the mixture was bubbled by Ar gas. Then, 10 mL of 2.5 M NaOH was added at 60°C for 20 min. Magnetic separation was used to remove NPs from the reaction mixture. To control particle size, the amount and concentration of NaOH solution were varied. 11
NiFe2O4 NPs are synthesized using the co-precipitation method. 20 mL of 0.1 M aqueous NiCl2·6H2O is added in 40 mL of aqueous 0.1 M FeCl3 in a round-bottomed flask. 4 g of urea and 50 mL of ethylene glycol are mixed and the whole refluxed for 6 h under nitrogen at 145°C. A brown-coloured precipitate appears. Centrifugation is used to collect the precipitate. To remove excess ethylene glycol, centrifugation is repeated several times using CH3OH and CH3OCH3. By changing the heating temperature (400°C, 500°C, 700°C, 800°C) in air for 4 h, NiFe2O4 NPs of various sizes can be prepared. 12
MnFe2O4 NPs were prepared by the co-precipitation method with canola oil as a surfactant as presented in Figure 1. 0.2 mol L−1 of MnCl2·6H2O and 0.4 mol L−1 of FeCl3·6H2O were reacted with steady magnetic stirring. An appropriate amount of canola oil was used as a capping agent to retard particle growth. To maintain pH (11–12), a 3 M aqueous NaOH was added dropwise.6,7 The reaction mixture was heated for 1 h at 80°C under constant stirring in order to convert the co-precipitate into spinel ferrite. The resulting mixture was cooled to room temperature. The precipitates were separated and washed with water and ethanol several times. The resulting powder was heated in air at 500°C for 6 h.
Figure 1.
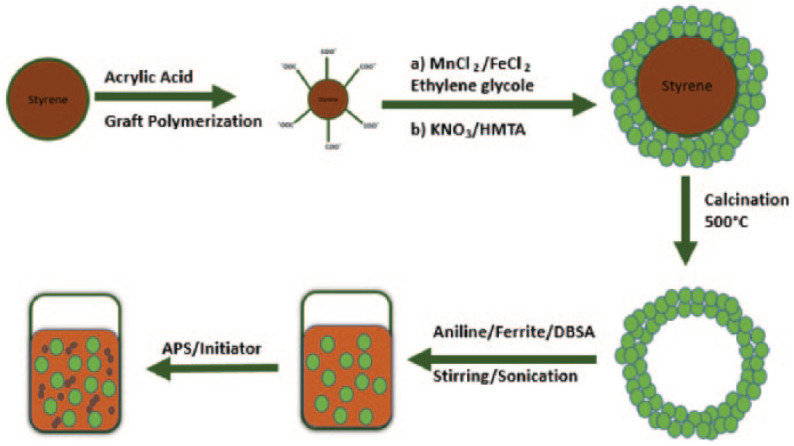
Schematic of the formation mechanism of the nanoporous polyaniline and hollow MnFe2O4 nanocomposite.
Nanocrystalline CoFe2O4 was synthesized by a simple co-precipitation method with canola oil being used as a surfactant. The stoichiometric quantities of analytical grade reagent (0.2 M CoCl2·6H2O and 0.4 M FeCl3·6H2O) were stirred magnetically. An appropriate quantity of surfactant (canola oil) solution was mixed in the above solution, in order to limit the particle growth and act as a capping agent. A solution of 3 M NaOH was added dropwise. The pH was adjusted to 11–12 during the reaction using aqueous NaOH. The details concerning the co-precipitation method have already been communicated.13,14 To convert the co-precipitate into spinel ferrite, the reaction mixture was heated and maintained for 1 h at 80°C with constant stirring. The reaction mixture was cooled down to room temperature. The precipitates were removed and washed with ethanol and water several times. The resulting ferrites were heated for 6 h at 500°C in air.
ZnFe2O4 NPs were prepared by the chemical co-precipitation method. Aqueous FeCl3·6H2O and ZnCl2 solutions were prepared by mixing suitable amounts of (Fe/Zn in 2:1) in 100 mL of distilled water. A 4 M solution of NaOH was used as a precipitating agent. Metal chlorides and NaOH solution were added dropwise (2 mL min−1) from two different burettes in the reaction vessel containing 100 mL of distilled water, and the solution was heated at 40°C with constant stirring. After 2 h, the precipitates that were obtained were centrifuged at 6000 r/min, washed repeatedly with acetone and distilled water and dried. 15
The combustion synthesis
The solution combustion synthesis of γ-Fe2O3: 0.140 mol of 96% Fe(NO3)3·9H2O and 0.179 mol of 99% D-(+)-C6H12O6 were mixed in 40 mL of distilled water in a round-bottomed flask. The flask was positioned in a heating mantle at 400°C. When the solution was heated, a smouldering combustion effect was observed between Fe(NO3)3·9H2O and glucose. A huge quantity of gases was evolved during this reaction. These gases were passed into a beaker of distilled water. 12 After 30 min, the gas development ceased and a black voluminous powder was formed. 50% of this black powder was further reacted with H2O2 with constant mechanical stirring, followed by evaporation to dryness. Each gram of the black powder was reacted with 50 mL of H2O2. Following evaporation towards dryness, a reddish brown powder was formed. 0.7 g of the reddish brown powder was sonicated for 2 h and the particles were covered with a bilayer of oleic acid. The oleic acid bilayer covering the NPs were diffused in 25 mL of saline solution, to achieve a stable colloidal suspension. 16
To synthesize CoFe2O4 NPs, a combustion method applied: aqueous solutions of Fe(NO3)3·9H2O and Co(NO3)2·6H2O were prepared and mixed. Then an appropriate amount of glycine was mixed in this solution. 17 The resultant solution was heated to a high temperature until combustion happened. Then sample was annealed at 200°C for 30 min.18,19 Combustion is an easy and quick mode of preparing NPs which provides high-value products. By altering the rate of fuel and the nitrates, the magnetic coercivity, size and magnetization can be varied.18,19
ZnFe2O4 NPs were prepared by the solution combustion technique with7.43 g of Zn(NO3)2·6H2O, 7.27 g of Fe(NO3)2·6H2O and 6.05 g of urea which act as the fuel. Urea has a high heat of combustion ∆Hc. In the first step, urea, Zn(NO3)2·6H2O and Fe(NO3)2·6H2O were taken in a 1:1:4 stoichiometric ratio and a uniform paste was prepared. A thick gel was obtained by heating this paste on a hot plate at 70°C–80°C. Then the gel was auto-combusted at 170°C–180°C on a hot plate to produce nanocrystalline ZnFe2O4 in 5 min. The nanocrystalline ZnFe2O4 powder was annealed at various temperatures, that is, 300°C, 500°C, 800°C and 1000°C for 4 h which provided a radiating powder of brown colour. 20
The sol–gel method
For the synthesis of nanostructured metal oxides, the sol–gel method is an appropriate wet route. 21 This process depends on hydroxylation and condensation of the molecular precursors in the solution state, initiating a ‘sol’ of nanometric particles. Solvent removal or chemical reaction is applied to the dried/gelled sol to obtain a three-dimensional (3D) metal oxide network. The gel properties are based on the arrangement formed in the sol phase of the sol–gel method. Water is generally used as a solvent but acid/base can also be used to hydrolyze the precursors. A colloidal gel is produced by acid catalysis, while base catalysis produced polymeric gel. Room temperature is used for these reactions but for the final crystalline state a heat treatment is applied.
The sol–gel method of preparing γ-Fe2O3 NPs is described as follows: 0.2 M ferric nitrate was dissolved in 100 mL ethylene glycol under constant stirring for 2 h at 401°C, and the sol was heated and maintained at 801°C to obtain a brown gel. The gel was ripened for 2 h and then dried at 1201°C for ca. 4 h. Subsequent to drying, the xerogel obtained was annealed at 200°C–400°C in vacuo. Finally, various-sized γ-Fe2O3 NPs were produced. 22
NiFe2O4 NPs. In the sol–gel method, aqueous 0.4 mol NiNO3 and 0.8 mol ferric nitrates were prepared and mixed in peracetic acid (PAA) solution. Phase separation occurred. A suitable amount of HNO3 was added dropwise to this solution till a clear green solution of pH 3.0 was obtained. The solution was evaporated at 50°C to obtain a transparent sol. Water was evaporated from the sol by heating at 50°C for 10 h as a result of which the sol changed into a thick brown gel. By varying the molar ratio of PAA and metal ions (0.5, 1.0, 1.5 and 2), different gels were prepared. To remove the organic content, the gels were calcined at 300°C/400°C for 2 h at a rate of 5°C min−1 and spinel NiFe2O4 NPs were obtained. 23
The sol–gel technique was used for the lower temperature preparation of glass, ceramics and others. In this technique, the precursor materials, Mn(CH3CO2·4H2O) and Fe(NO3)3·9H2O, were mixed in deionized water and ethylene glycol with constant stirring until a uniform mixture was obtained, which was then heated at 70°C for 12 h and dried at 80°C for 24 h. The resultant gel was finally ground, reheated at 900°C for 2 h and slowly cooled down to obtain MnFe2O4 NPs. Finally, sintering was conducted at 1200°C for 3 h at a rate of 2°C min−1 followed by slow cooling at room temperature at the same rate. 24
A solid-state technique was used to prepare ZnFe2O4 NPs. An appropriate quantity of 99.6% Fe2O3 and 99% ZnO powder were ground and calcined at 1200°C for 2 days. In the sol–gel technique, a clear solution of iron and zinc nitrates was prepared by dissolving in a small amount of deionized water. To prepare the gel, polyvinyl alcohol was used. An aqueous solution of polyvinyl alcohol was mixed with the solution of nitrates, and water was evaporated at 60°C–80°C. The gelation process was stopped and a dull red–coloured gel-type precursor was formed. ZnFe2O4 NPs were obtained on calcination of this precursor at 350°C–450°C. 25
Using the sol–gel method, stoichiometric amounts of Co(CH3CO2)2·4H2O (1/200 mol) and Fe(NO3)3·9H2O (1/100 mol) were dissolved in 100 cm3 of 2-methaoxyethanol and 2 cm3 of diethanolamine with the help of an ultrasonic cleaner. The reaction mixture was refluxed at 70°C for 12 h. The crystallization temperature of CoFe2O4 was determined by drying some portion of the solution for Mossbauer spectroscopy. CoFe2O4 films were spin-coated onto thermally oxidized Si wafers of ~700Å at 4000 r/min for 30 s. The substrate was washed with the standard cleaning silicon process before spin coating. 26 After spin coating, thin films were pyrolysed to 120°C and 270°C for 3 and 10 min, respectively. To gain the required thickness, the process of deposition was repeated. Spin-coated powders and samples were annealed at different temperatures from 350°C to 950°C for 3 h in air. 27
The sonochemical method
In the sonochemical method, structural hosts, polymers or organic capping agents, are applied to reduce NP growth. 21 Cavities are produced in the aqueous medium due to the ultrasonic irradiation, and the formation, growth and collapses of microbubbles take place. 28 The high temperatures and pressures generated by cavitations facilitate the occurrence of a number of remarkable chemical reactions. In some cases, thermally induced processes give crystalline NPs, while ultrasonic reactions produce amorphous products.
Using this method, the volatile organometallic compound, Fe(CO)5, was sonochemically decomposed with the addition of the stabilizer to prepare nanosized iron colloid. Ultrasonic irradiation of 0.2 mL of Fe(CO)5 in 20 mL of octanol with 1 g of polyvinylpyrrolidone (PVP) with (M ≈ 40,000) was performed. The reaction mixture was heated at 20°C in an O2-free Ar atmosphere, creating a black colloidal solution. Oleic acid can be used as a colloid stabilizer. 29 The tail of oleic acid (stabilizer) has a double bond, which is significant to its usefulness, as stearic acid cannot be used as a stabilizer. A hexadecane solution of 2 mol Fe(CO)5 and 0.3 mol oleic acid was sonicated at 30°C for 1 h, as a result of which the initial yellow-coloured solution changed to black. The resultant black solution of γ-Fe2O3 was evacuated to 50°C for 1 h to remove unreacted Fe(CO)5 and its isolation of an inert atmosphere system for further characterization. 30
The sonochemical method was used to prepare NiFe2O4 by reacting Ni(CO)4 and Fe(CO)5. Ni(CO)4 is a very poisonous liquid with an elevated vapour pressure. It is highly sensitive to moisture and so has to be handled carefully. Pentane (Fluka) and decalin (Sigma) were carefully dried with Na metal/4Å molecular sieve and stored in a glovebox. Ar gas was used to degas the precursor solution. The NiFe2O4 NPs were synthesized by ultrasonic irradiation of Ni(CO)4 and Fe(CO)5 in decalin at (273 K, 100–150 kPa) with a high-intensity ultrasonic probe. 3 h of irradiation provided a black powder which was then centrifuged and washed repeatedly with dry pentane in the glovebox. 31
In the sonochemical process, Fe(CO)5 was used without any purification. Co(CO)3(NO) was synthesized by the usual procedure. Pentane and decalin were dried by Na metal/4Å molecular sieve. The CoFe2O4 NPs were synthesized by ultrasonic irradiation of a solution of Co(NO)(CO)3 and Fe(CO)5 at 0°C, with 100–150 kPa pressure of O2 gas, by a high-strength ultrasonic probe (20 kHz, 100 W cm−2). A black powder was obtained after 3 h of irradiation; this was centrifuged and washed five times with dry pentane under vacuum. 32
Conductive polymer composites, materials encoded by inorganic NPs such as Al2O3, 33 Fe2O3, 33 RuO2 34 and TiO2, 35 are important due to their interactions with polymeric matrices to give novel properties. An MnFe2O4 nanocomposite was synthesized using 4 mL of aniline (0.044 M) and 10 mL of distilled H2O which were mixed and stirred for 15 min. In a separate container, 10 mL of deionized water was used to dilute 5 g of dodecylbenzenesulphonic acid (DBSA) to give a pulpy mixture. The pulpy mixture was then poured into aqueous aniline solution and the emulsion was agitated for 1 h to form a homogeneous phase. Next, 5 mL of an aqueous solution of 0.6 g hollow ferrite spheres was ultrasonicated for 10 min and poured into the above suspension. The mixture was sonicated for a further 15 min and agitated by stirring for 15 min; 10 mL of an aqueous solution of 5 g of ammonium persulphate (0.022 M) as an oxidizing agent was gradually added (for 15 min) with mechanical stirring to start the polymerization reaction. The green precipitate of polyaniline (PANi) and the MnFe2O4 nanocomposite was filtered, washed with distilled water and dried for 24 h in an oven at 601°C. 36 The formation of larger monodisperse magnetite NPs using smaller NPs of magnetite is illustrated in Figure 2.
Figure 2.
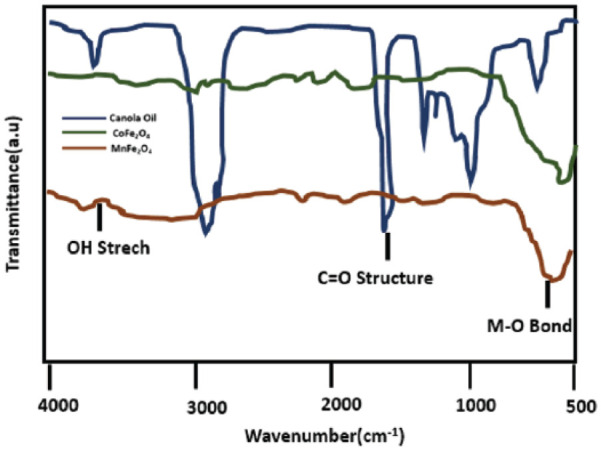
Illustration by the IR spectra of the formation of larger monodisperse magnetite nanoparticles using smaller nanoparticles of magnetite as seeds.
The microwave-assisted method
Microwave-assisted chemistry has gained much attention in recent years. The greatest benefit of microwave-assisted irradiation is its uniform heating of a substance in a glass or plastic reaction vessel, which is responsible for homogeneous nucleation and a smaller crystallization time as compared to conventional heating. This leads to the development of homogeneous colloidal materials.
In the microwave-assisted preparation of γ-Fe2O3 NPs, polyethylene glycol (PEG)-20000 (60 mg) and FeCl3·6H2O (76 mg) were mixed in 10 mL of distilled H2O. Then 0.4 mL of hydra and 1 mL of H2O2 were dissolved in this solution (note that heat is evolved during the reaction), and a red-coloured product was obtained. This suspension was heated at 100°C by microwave heating for 10 min and the resultant product was separated through centrifugation, washed twice with distilled H2O and absolute ethanol. The microwave oven used for the preparation of γ-Fe2O3 NPs (2.45 GHz, maximum power 300 W) was a fixed single-mode microwave system (Discover; CEM). The exclusive, spherical single-mode cavity makes sure that the sample was in very concentrated microwave field. The system featured a magnetic stirrer and a water-cooled condenser. The temperature was maintained by automatic adjustment of the microwave system. 37
An aqueous solution of vinyl pyrrolidone was prepared by mixing 4 g in 100 mL of deionized water at 363 K. 0.2 mM of Fe(NO3)2·6H2O and 0.1 mM of Zn(NO3)2·6H2O in the ratio of 2:1 were added to the polymer solution which was stirred vigorously for 2 h to give a colourless and transparent solution. A glass electrode was used to maintain the pH of solution at 1–2. The reaction mixture was annealed at 353 K in an oven for 48 h to evaporate H2O. The orange-coloured ZnFe2O4 was powdered in a mortar and calcined at 723, 773, 823 and 873 K for 3 h to decompose organic matter and crystallized. 38
The thermal decomposition method
The thermal decomposition method was used to synthesize NiFe2O4. 1 mM of Ni(acac)2 in 15 mL benzyl ether and 2 mM of Fe(acac)3 in 15 mL of oleylamine were dissolved under constant stirring to prepare a homogeneous solution. Water was evaporated at 110°C for 1 h under N2 (g), then the temperature was raised to 300°C for 1 h. The solution was then cooled to room temperature. The spinel ferrite was removed from the ethyl acetate–ethanol mixture, followed by centrifugation at 14,000 r/min for 30 min. The final product was washed with ethanol and centrifuged. The sample was dried at 150°C. In this procedure, oleylamine plays three roles, namely, as a reductant, as a solvent and as a surfactant. 39
The electrochemical method
NiFe2O4 was also prepared by an electrochemical method utilizing iron and nickel anodes of 99.5% purity. Ni(CH3)4Cl was used as a supporting electrolyte. The currents functional to the Ni and Fe anodes were 50 and 100 mA, respectively. The reaction proceeded for 30 min at 60°C. The resultant precipitate was magnetically separated, washed with distilled H2O and dried in vacuum at 60°C for 12 h.40,41
The solvothermal synthesis
A 15 mL solution of 0.54 g of FeCl3·6H2O and 0.14 g of ZnCl2 was prepared in glycerol and deionized H2O in a volume ratio of 1:1 and then 0.8 g of CH3COONa was agitated in the solution by stirring. The mixture was poured into a 40 mL Teflon-lined autoclave and heated at 200°C for 24 h in an oven. Then the autoclave was cooled to 25°C. The solid products were centrifuged and washed with ethanol and distilled water and dried in the oven at 60°C for 6 h. 42
The refluxing method
For the preparation of nanocrystalline Ni–Zn ferrite powders, the refluxing method was used. Ferric chloride, nickel sulphate and zinc sulphate were used as the starting agents in the aqueous solution. The salts were mixed in stoichiometric quantities (Ni:Zn:Fe = 1:1:4). An aqueous solution of NaOH was added to the starting solution under constant magnetic stirring until the pH became 9.5–12. The reaction mixture was constantly refluxed at about 107°C for a definite time. The precipitates formed were filtered after repeated washing with alcohol and water, and alcohol, respectively, and dried at 100°C for 6 h. 43 A schematic of the synthesis of metal ferrites is shown in Figure 3.
Figure 3.

General scheme for schematic synthesis of metal ferrite nanoparticles.
Effects of surfactant on metal ferrite NPs’ stability
Optical absorbance has been used to measure the stability of Fe2O3 NPs in the colloidal solution. Various surfactant concentrations were used to prepare colloidal solutions in quartz cuvettes. After the preparation of the suspension, the absorbance spectra of Fe2O3 NPs were obtained instantly (True = 0 h) and time (True = x), by keeping the colloidal suspension undisturbed. To measure the ultraviolet–visible (UV/VIS) absorbance spectra of the colloidal samples without saturation, dilutions of 1:100 and 1:200 were selected.
Initially, no specific UV–VIS peak appeared. However, with the passage of time, a decrease in absorbance was seen due to sedimentation of the Fe2O3 NPs. The sedimentation occurred at a higher rate in the initial period after sonication as shown by the UV/VIS spectrum. This effect appeared due to the absence of stabilizer in the colloidal solution, due to which aggregation of NPs occurred and their sizes increased in solution, which led to faster sedimentation. The use of a stabilizer avoids the nucleation of Fe2O3 NPs as well as impeding their agglomeration and growth. A specific absorbance peak of surfactants was observed when Fe2O3 NPs were layered with the tested surfactants; the absorbance spectra of magnetite NPs along with 8 mM of surfactant oleic acid were determined at True = 0 h and True = 24 h.
From a plot of absorbance at 224 nm (which is due to the surfactant peak) versus oleic acid concentration, a maximum absorbance was observed at concentrations greater than 50 mM. However, a minor decline in absorbance occurred after 24 h which might be due to some sort of agglomeration of Fe2O3 NPs which were not entirely broken by sonication, and settled down in the cuvette.
At concentrations beyond 64 mM, the maximum absorbance occurred because of the effective particle disaggregation of the particles and their stability over time. This study correlates with the results published by the other authors 44 inferring that beyond a specific concentration a double layer of oleic acid is formed around the Fe2O3 NPs to provide a highly hydrophilic surface on the NPs which stabilizes them in aqueous colloidal solutions. 45 At a lower concentration of sodium citrate (1.25 mM), a greater decline in peak intensity occurs; this occurs because the concentration of surfactant is insufficient to prevent the aggregation of NPs in colloidal suspension. 46 A small decline in peak intensity observed in the case of sodium citrate as compared to oleic acid,47,48 which proved sodium citrate to be a better surfactant for magnetite NPs.
The structure, morphology and size of the synthesized magnetite NPs were determined before and after surfactant coating. The structure was determined by X-ray powder diffraction (XRD) spectra of Fe2O3, while transmission electron microscopy (TEM) was used to determine the size and morphology.
Conclusion
Every synthesis method for ferrite NPs such as NiFe2O4, ZnFe2O4, MnFe2O4, Fe2O3, CoFe2O4 and mixed NPs, from the co-precipitation, thermal decomposition, hydrothermal, microwave-assisted and sonochemical methods, has its own pros and cons, but the co-precipitation method is considered the best because of the low temperature involved and the ease of process. The effects of different capping agents, namely, canola oil, recin oil, glycerol, sodium dodecyl, sodium citrate, oleic acid, Triton-100 and sodium dodecyl benzene sulphonates, on the properties of ferrites have been studied: these capping agents help in the size stability of NPs and reduce agglomeration.
Author biographies
Muhammad Imran Din received his PhD in Physical Chemistry from the Islamia University of Bahawalpur in 2013. He joined the Institute of Chemistry, University of the Punjab, Lahore, Pakistan, in November 2009. His field of interest is nanotechnology, theoretical chemistry, computational chemistry and material chemistry. Currently, he is working on energy from biomass material. His research work is published in different international journals and presented at various international conferences held worldwide. He has published over 50 research articles in leading international journals.
Faria Rafique holds an MSc as well as an MPhil degree – in Physical Chemistry – from the University of the Punjab. She has recently received a PhD in Physical Chemistry from Lahore University of Management Sciences. Her research focuses on the synthesis and characterization of biocompatible nanovectors as carriers of the anticancer drug Adriamycin and high-refractive-index photopolymerizable materials for optical applications.
Muhammad Sadaf Hussain holds an MSc degree – in Physical Chemistry – from the University of Agriculture, Faisalabad, as well as an MPhil in Physical Chemistry from the University of Punjab. His research focuses on enhanced photocatalytic performance and reusability of nano-ZnO-coated surfaces for degradation of methylene blue and synthesis and characterization of hybrid microgels for efficient catalytic degradation of Rhodamine B in aqueous medium.
Hafiz Arslan Mehmood is pursuing his MS in Biochemistry in the Institute of Chemistry, University of Punjab, Lahore, Pakistan. His research focuses on the fabrication of magnetite nanoparticles for catalytic pyrolysis applications.
Sadia Waseem is working as an Assistant Professor in the Institute of Chemistry, University of Punjab, Lahore, Pakistan. Her research interests include water quality, environmental issues, disinfection by products, haloacetic acids, trihalomethane and so on, modelling (predictive and time series), method development, monitoring of environmental pollutants, heavy metal studies, removal of heavy metals and treatment studies of biosorbents.
Footnotes
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship and/or publication of this article.
References
- 1. Volatron J, Kolosnjaj-Tabi J, Javed Y, et al. Sci Rep 2017; 7: 40075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Niederer W. Cell Mol Life Sci 1970; 26: 218–220. [Google Scholar]
- 3. Pead S, Durrant E, Webb B, et al. J Inorg Biochem 1995; 59: 15–27. [DOI] [PubMed] [Google Scholar]
- 4. Fleming J, Joshi J. Proc Natl Acad Sci 1987; 84: 7866–7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lindenschmidt R, Sendelbach L, Witschi H, et al. Toxicol Appl Pharmacol 1986; 82: 344–350. [DOI] [PubMed] [Google Scholar]
- 6. Pui A, Gherca D, Carja G. Dig J Nanomater Bios 2011; 6: 1783–1791. [Google Scholar]
- 7. Gherca D, Pui A, Cornei N, et al. J Magn Magn Mater 2012; 324: 3906–3911. [Google Scholar]
- 8. Sanpo N, Wen C, Berndt CC, et al. Microbial pathogens and strategies for combating them: science, technology and education. Spain: Formatex Research Center, 2013, pp. 239–250. [Google Scholar]
- 9. Park S-J, Kim S, Lee S, et al. J Am Chem Soc 2000; 122: 8581–8582. [Google Scholar]
- 10. Sun S, Zeng H. J Am Chem Soc 2002; 124: 8204–8205. [DOI] [PubMed] [Google Scholar]
- 11. Dozier D, Palchoudhury S, Bao Y. J Sci Health Univ 2010; 7: 16–18. [Google Scholar]
- 12. Ianoş R, Tăculescu A, Păcurariu C, et al. J Am Ceram Soc 2012; 95: 2236–2240. [Google Scholar]
- 13. Liu X, Yang J, Wang L, et al. Mater Sci Eng A 2000; 289: 241–245. [Google Scholar]
- 14. Dobryszycki J, Biallozor S. Corros Sci 2001; 43: 1309–1319. [Google Scholar]
- 15. Ebrahimi M, Shahraki RR, Ebrahimi SS, et al. J Supercond Novel Magn 2014; 27: 1587–1592. [Google Scholar]
- 16. Ianoş R, Tăculescu E-A, Păcurariu C, et al. Mater Chem Phys 2014; 148: 705–711. [Google Scholar]
- 17. Levine S, Bowen BD, Partridge SJ. Colloids Surf 1989; 38: 325–343. [Google Scholar]
- 18. Houshiar M, Zebhi F, Razi ZJ, et al. J Magn Magn Mater 2014; 371: 43–48. [Google Scholar]
- 19. Maaz K, Mumtaz A, Hasanain S, et al. J Magn Magn Mater 2007; 308: 289–295. [Google Scholar]
- 20. Sachin B, Sambaji B. Research J Recent Sci 2012: 2502. [Google Scholar]
- 21. Dai Z, Meiser F, Möhwald H. J Colloid Interface Sci 2005; 88: 298–300. [DOI] [PubMed] [Google Scholar]
- 22. Xu J, Yang H, Fu W, et al. J Magn Magn Mater 2007; 309: 307–311. [Google Scholar]
- 23. Chen D-H, He X-R. Mater Res Bull 2001; 36: 1369–1377. [Google Scholar]
- 24. Ahmed M, Okasha N, El-Dek S. Nanotechnol 2008; 19: 065603. [DOI] [PubMed] [Google Scholar]
- 25. Wang L, Zhou Q, Li F. Physica Status Solidi 2004; 241: 377–382. [Google Scholar]
- 26. Ghandhi SK. VLSI fabrication principles, New York: Wiley, 1983, pp. 401–411. [Google Scholar]
- 27. Lee J-G, Park JY, Oh Y-J, et al. J Appl Phys 1998; 84: 2801–2804. [Google Scholar]
- 28. Indira T, Lakshmi P. Int J Pharm Sci Nanotechnol 2010; 3: 1035–1042. [Google Scholar]
- 29. Fertman E. Magnetic fluids guidebook: Properties and applications. New York: Hemisphere, 1990, pp. 1–19. [Google Scholar]
- 30. Suslick KS, Fang M, Hyeon T. J Am Chem Soc 1996; 118: 11960–11961. [Google Scholar]
- 31. Shafi KV, Koltypin Y, Gedanken A, et al. J Phys Chem 1997; 101: 6409–6414. [Google Scholar]
- 32. Shafi KV, Gedanken A, Prozorov R, et al. Chem Mater 1998; 10: 3445–3450. [Google Scholar]
- 33. Teoh GL, Liew KY, Mahmood WA. Mater Lett 2007; 61: 4947–4949. [Google Scholar]
- 34. Rao CR, Vijayan M. Synth Met 2008; 158: 516–519. [Google Scholar]
- 35. Radhakrishnan S, Siju C, Mahanta D, et al. Electrochim Acta 2009; 54: 1249–1254. [Google Scholar]
- 36. Hosseini SH, Rahimi R, Kerdari H. Polym J 2011; 43: 745–750. [Google Scholar]
- 37. Wang W-W, Zhu Y-J, Ruan M-L. J Nanopart Res 2007; 9: 419–426. [Google Scholar]
- 38. Naseri MG, Saion EB, Hashim M, et al. Solid State Commun 2011; 151: 1031–1035. [Google Scholar]
- 39. Sarno M, Cirillo C, Ciambelli P. Chem Eng J 2014; 246: 27–38. [Google Scholar]
- 40. Galindo R, Mazario E, Gutiérrez S, et al. J Alloys Comp 2012; 536: S241–S244. [Google Scholar]
- 41. Cabrera L, Gutierrez S, Menendez N, et al. Electrochim Acta 2008; 53: 3436–3441. [Google Scholar]
- 42. Guo P, Cui L, Wang Y, et al. Langmuir 2013; 29: 8997–9003. [DOI] [PubMed] [Google Scholar]
- 43. Zhong Z, Li Q, Zhang Y, et al. Powder Technol 2005; 155: 193–195. [Google Scholar]
- 44. Yang K, Peng H, Wen Y, et al. Appl Surf Sci 2010; 256: 3093–3097. [Google Scholar]
- 45. Zhang H, Li L, Liu XL, et al. ACS Nano 2017; 11(4): 3614–3631. [DOI] [PubMed] [Google Scholar]
- 46. Imran M, Shaik AH, Ansari AR, et al. RSC Advances 2018; 8(25): 13970–13975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Biehl P, von der Lühe M, Dutz S, et al. Polymers 2018; 10(1): 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kumar JP, Prasad GK, Ramacharyulu PVRK, et al. J Alloys Comp 2017; 692: 833–840. [Google Scholar]