

Abstract
Chromothripsis, the shattering and imperfect reassembly of one (or a few) chromosome(s)1, is an ubiquitous2 mutational process generating localized complex chromosomal rearrangements that drive genome evolution in cancer. Chromothripsis can be initiated by missegregation errors in mitosis3,4 or DNA metabolism5–7 that lead to entrapment of chromosomes within micronuclei and their subsequent fragmentation in the next interphase or upon mitotic entry6,8–10. Here, we use inducible degrons to demonstrate that chromothriptically produced pieces of a micronucleated chromosome are tethered together in mitosis by a protein complex consisting of Mediator of DNA damage checkpoint 1 (MDC1), DNA Topoisomerase II Binding Protein 1 (TOPBP1), and Cellular Inhibitor of PP2A (CIP2A), thereby enabling en masse segregation to the same daughter cell. Such tethering is shown to be crucial for the viability of cells undergoing chromosome missegregation and shattering after transient inactivation of the spindle assembly checkpoint. Transient, degron-induced reduction in CIP2A after chromosome micronucleation-dependent chromosome shattering is shown to drive acquisition of segmental deletions and inversions. Analyses of pancancer tumor genomes showed that expression of CIP2A and TOPBP1 was increased overall in cancers with genomic rearrangements, including copy number-neutral chromothripsis with minimal deletions, but comparatively reduced in cancers with canonical chromothripsis in which deletions were frequent. Thus, chromatin-bound tethers maintain proximity of fragments of a shattered chromosome enabling their re-encapsulation into, and religation within, a daughter cell nucleus to form heritable, chromothriptically rearranged chromosomes found in the majority of human cancers.
Elevated rates of chromosome missegregation, caused by errors in mitosis, are frequently observed in many cancers11. Chromosome missegregation can lead to numerical and structural chromosomal changes in cancer cells, resulting in increased phenotypic diversity, which provides a substrate for natural selection11. Defects in chromosome segregation can result in abnormal structures including micronuclei or chromatin bridges, which are prevalent across the spectrum of human neoplasia. Micronuclei and chromatin bridge formation trigger chromothripsis1,3–8,12, an event in which a chromosome is shattered into ten to hundreds of individual pieces, most of which are acentric. This shattering is followed by re-ligation in random order of individual pieces, with a few pieces missing, resulting in a massively rearranged chromosome(s). Such chromothriptic rearrangements can result in multiple oncogenic insults, including loss of tumor suppressors13, amplification of oncogenes1,13–15, and formation of oncogenic fusions15. Chromothripsis can also drive formation and evolution of circular extrachromosomal DNA (ecDNA)12, a major vehicle for oncogene amplification. How the pieces of a shattered chromosome are brought together and ligated to yield a chromothriptic chromosome is unknown16. Nevertheless, following micronucleation, newly generated structural variants are preferentially present in one of the two daughter cells3. Here, we identify a complex of MDC1, TOPBP1, and CIP2A whose recruitment to γH2AX-containing chromatin is responsible for tethering shattered chromosomal fragments to each other, thereby enabling their delivery en masse to a daughter cell nucleus and reassembly into a chromothriptically rearranged chromosome.
Mitotic clustering of broken chromosomes
To understand the behavior of a broken chromosome resulting from encapsulation into a micronucleus or a chromatin bridge, we generated fragmented chromosomes in RPE1 p53−/− cells using two approaches. Micronucleation was induced by Mps1 inhibitor (NMS-P715) treatment17, and chromatin bridge formation was induced by low dose inhibition of Topoisomerase II (with ICRF-193) was used to generate catenated sister chromatids to produce mitotic chromatin bridges7 (Fig. 1A). Since chromosomes within micronuclei undergo fragmentation before or upon entry into mitosis6–9, cells were arrested in the next mitosis using an inhibitor of the mitotic kinesin Eg5. To follow the fragmented chromosome pieces, we exploited that an initial DNA damage response to a double stranded break is phosphorylation of histone H2AX at S139A (γH2AX) throughout ~1 Mb of chromatin18–20 adjacent to the break. Generation of chromosome fragmentation by either method yielded γH2AX-containing chromosome fragments that remained tightly clustered in mitosis for large majorities (75.5%±0.5 or 65.5%±0.7, respectively) of fragmented chromosomes arising from Mps1 or Topoisomerase inhibition (Fig. 1B, C). Chromosome missegregation can cause p53 dependent cell cycle arrest in a subset of cells resulting in fewer cells entering a subsequent mitosis21,22. Correspondingly, we either used p53−/− RPE1 cells or p53 mutated DLD1 cells, or p53+/+ RPE1’s with p53 depleted using siRNA, as indicated. Presence or absence of p53 did not alter clustering frequency (Extended Data Fig. 1A).
Fig. 1. Chromosome fragments from abnormal nuclear structures are clustered during mitosis and segregated together to a daughter cell.
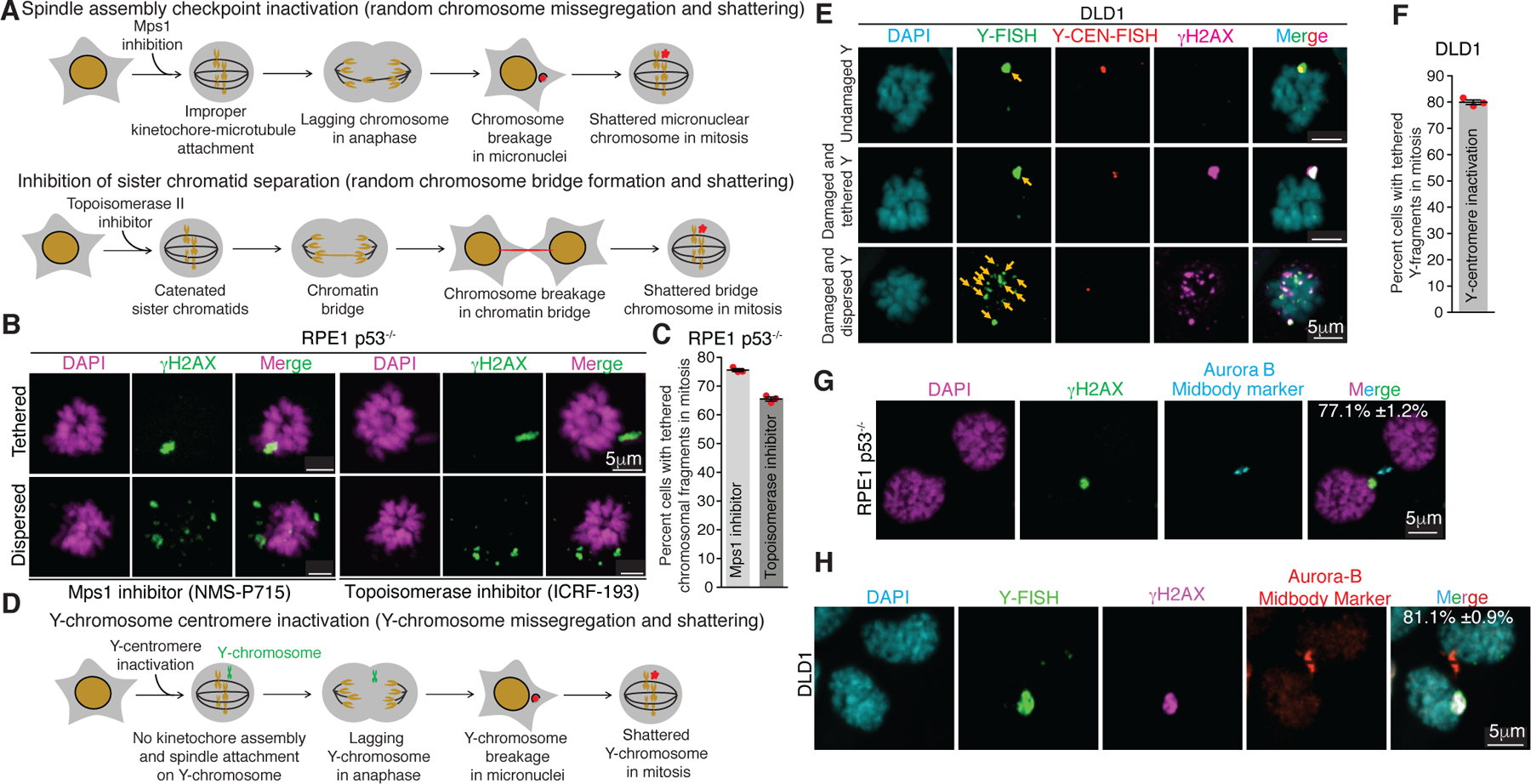
(A) Schematic of methods for generation of micronuclei and chromatin bridges in RPE1 p53−/− cells. (B) Representative images showing dispersed and tethered chromosomal fragments after micronuclei or chromatin bridge induction. (C) Quantitation of chromosomal fragment tethering from experiments in (A) and (B) (n=3 independent experiments, total 511 and 420 cells were analyzed for micronuclei and chromatin bridge induced condition, respectively). (D) Schematic for generation of Y-chromosome micronuclei in DLD1 cells. (E) Representative images showing different fates of micronuclear Y-chromosome in mitosis (yellow arrows point to the Y-chromosome fragments) (Y-FISH is Y-chromosome FISH and Y-CEN-FISH is Y-centromere FISH). (F) Quantitation of tethered Y-chromosome fragments during mitosis 3 days post Y-centromere inactivation (n=3 independent experiments, total 252 cells were analyzed). Representative images showing asymmetric inheritance of broken micronuclear chromosomes in (G) RPE1 p53−/− cells (n=3 independent experiments, total 201 were analyzed) and (H) DLD1 cells (n=3 independent experiments, total 143 were analyzed) (for (G) and (H), percentage of cells (mean +/− SEM) showing en masse inheritance of chromosomal fragments is indicated on the image). For graphs shown in (C) and (F), the mean +/− SEM are shown; individual data points are shown as solid red circles.
Next, we utilized a DLD1 cell model8 in which missegregation and micronucleation of the Y-chromosome could be triggered by selective inactivation of its centromere (Fig. 1D). As expected, Y-centromere inactivation resulted in damaged, γH2AX positive Y-chromosomes in the next mitosis, as determined by combined immunofluorescence for γH2AX and fluorescence in situ hybridization (FISH) for the Y-chromosome (Fig. 1E) or by Y-chromosome FISH on chromosome spreads (Extended Data Fig. 1B–D). The proportion of cells with a γH2AX positive Y-chromosome (Extended Data Fig. 2E) was similar to the proportion of fragmented Y-chromosomes (Extended Data Fig. 1D), confirming γH2AX to be a reliable marker for fragmented mitotic chromosomes. Once again, in a majority (79%±0.9) of cells, γH2AX positive fragments of the Y chromosome remain clustered even in extended mitoses (Fig. 1E, F). Clustering in mitosis of chromosomal fragments derived from micronuclei was not due to persistence in mitosis of a micronuclear envelope (Extended Data Fig. 2A, B). Asymmetric segregation of clustered fragments from an initially micronucleated chromosome to one daughter cell in the subsequent G1 phase was consistently observed, both in RPE1 p53−/− or DLD1 cells (Fig. 1G, H). Thus, shattered chromosome fragments produced from micronuclei or chromatin bridges remained clustered in the subsequent mitosis.
Clustering without PolD3, MRN, or PolQ
Micronuclei with an intact envelope have deficient nuclear import23 that delays replication9,10 and recruitment/retention of DNA repair proteins9,23, including 53BP1 and BRCA1 (Extended Data Fig. 3A, B). This DNA replication deficiency is compounded by micronuclear envelope rupture that exposes the micronuclear chromosome to cytoplasmic nucleases5,17. Under-replicated regions within a primary nucleus are known to undergo PolD3- (a subunit of polymerase Delta) mediated mitotic DNA repair synthesis (MIDAS)24. Chromosomes from micronuclei or an initial chromatin bridge have been reported to undergo a burst of DNA synthesis during mitosis7. We tested if replication intermediates formed as a result of PolD3-mediated mitotic DNA synthesis could facilitate the clustering of chromosomal pieces in mitosis. Although depletion of PolD3 or the key non-homologous end joining (NHEJ) ligase, Lig4, increased the percentage of cells with a broken mitotic Y-chromosome (Extended Data Fig. 2C–E) in the mitosis following Y-micronucleation, it did not alter the proportion of cells with clustered micronuclear chromosomal fragments (Extended Data Fig. 2F). PolD3’s involvement in damaged micronuclear chromosome repair was confirmed (Extended Data Fig. 2G–J) using “CEN-select”, a colony formation assay to monitor reassembly of a micronuclear chromosome by selecting for retention of a neomycin resistance gene inserted into an initially intact Y-chromosome4. Thus, PolD3 contributes to repair of fragmented micronuclear chromosomes, but does not play a role in mitotic clustering of the micronuclear chromosomal fragments.
The MRN complex (consisting of Mre11, Rad50, and Nbs1)25–28 and PolQ27 have been reported to be required for tethering an acentric piece of a broken chromosome to the centromere-containing fragment during mitosis and to be recruited to damaged micronuclear chromosomes in interphase4. We thus tested if the MRN complex or PolQ mediates tethering of micronuclear fragments during mitosis (Extended Data Fig. 3C). MRN complex or PolQ depletion did not affect the frequency of cells with tethered Y-pieces (Extended Data Fig. 3D–G). Thus, mitotic clustering of fragments from a micronucleated chromosome is not mediated by the MRN complex or PolQ.
MDC1, TOPBP1, CIP2A on broken chromosomes
TOPBP1 has also been proposed to maintain chromosomal integrity by tethering broken chromosomal pieces in mitosis after DNA damage resulting from ionizing radiation (IR)29,30 or replication stress caused by BRCA1/2 deficiency31 in the primary nucleus. TOPBP1 is recruited to some DNA damage lesions in mitosis through direct interaction with MDC130, which in turn directly binds γH2AX-containing chromatin assembled at double strand breaks (DSBs)18. CIP2A directly interacts with TOPBP1 and mediates its recruitment to DNA damage sites in mitosis and has been proposed to play a role in tethering broken chromosomal pieces29,31,32. Within micronuclei containing damaged chromosomes, detectable MDC1, TOPBP1, and CIP2A did not accumulate in the large majority (76.3±2.0%, 88.0±1.4%, and 82.4±0.3%, respectively) of interphase DLD1 or RPE1 p53−/− cells (Extended Data Fig. 4A–F). [For the minority of micronuclei in interphase with low level recruitment of the three proteins, micronuclear rupture was not a key determinant of recruitment (Extended Data Fig. 5A–E).]
Upon mitotic entry all three proteins were strongly recruited to γH2AX-containing micronuclei-derived chromatin (Fig. 2A–D), even in p53+/+ cells (Extended Data Fig. 5G–I). Live-cell imaging of GFPMDC1 or TOPBP1Clover showed their accumulation on the broken micronuclear chromosome concomitant with chromosome condensation at mitotic entry (Fig. 2E, F; Sup. Video 1 & 2). A similar accumulation of TOPBP1Clover on micronuclei with a ruptured micronuclear membrane (determined by the absence of 3xNLSmscarlet) was observed at the onset of mitosis (Extended Data Fig. 5F, Sup. Video 3). We conclude that robust recruitment of TOPBP1 to a damaged micronuclear chromosome is triggered by mitotic entry, when previously nuclear MDC1 and TOPBP1 are released into the mitotic cytoplasm. In contrast, damage-containing chromatin generated from chromatin bridge breakage recruited TOPBP1 (85.2±1.9%) and MDC1(91.3±0.3%), but not CIP2A, in interphase (Extended Data Fig. 5J–L). Consistent with this, the stubs of a broken bridge appeared to be encapsulated into a primary nucleus (Extended Data Fig. 5J–M). Live imaging revealed that once recruited, TOPBP1Clover remained on stubs for extended periods (≥11 hours of live imaging) (Extended Data Fig. 5M, Sup. Video 4).
Fig. 2. TOPBP1, MDC1, and CIP2A are recruited to a damaged micronuclear chromosome upon mitotic entry.
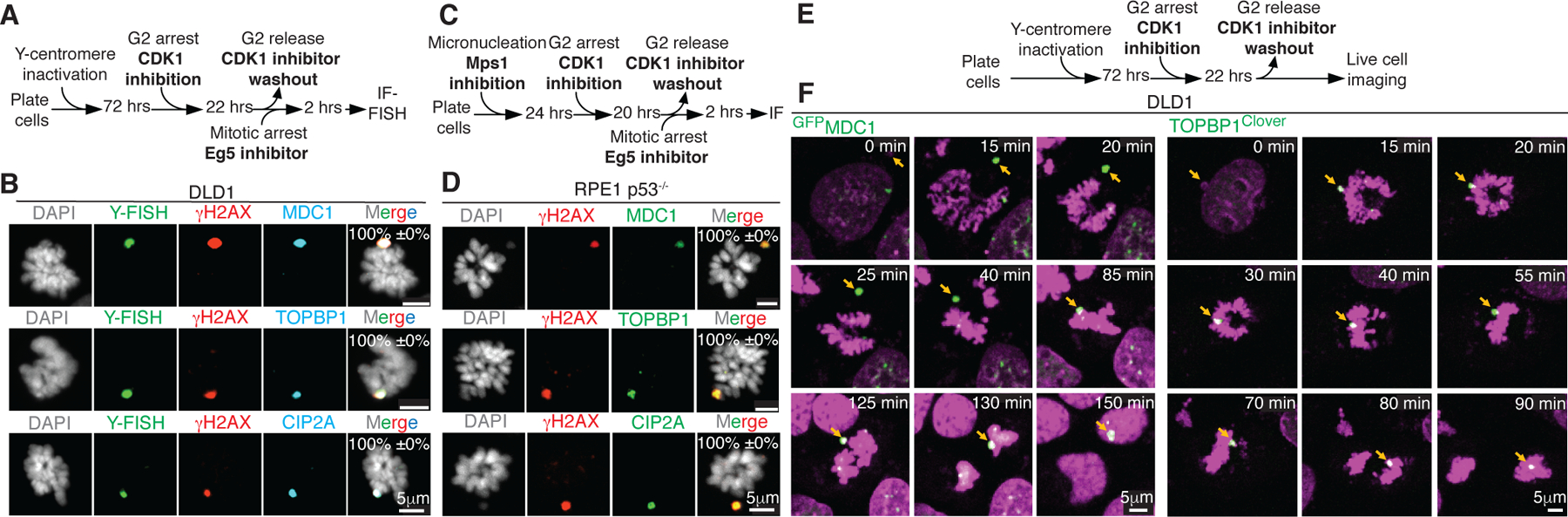
(A) Experimental outline for (B). (B) Representative images showing localization of MDC1, TOPBP1, and CIP2A on broken micronuclear Y-chromosome in mitosis (n=3 independent experiments, total 258, 252, and 263 DLD1 cells were analyzed for MDC1, TOPBP1, and CIP2A, respectively). (C) Experimental outline for (D). (D) Representative images showing localization of MDC1, TOPBP1, and CIP2A on broken micronuclear chromosome in mitosis (n=3 independent experiments, total 273, 265, and 271 RPE1 p53−/− cells were analyzed for MDC1, TOPBP1, and CIP2A, respectively). Mean percentage +/− SEM for localization of the indicated protein to fragmented (γH2AX positive) chromosome is indicated on the images in (B) and (D). (E) Schematics of experimental setup for (F). (F) Cropped frames of Sup. Videos 1 and 2 showing localization of GFPMDC1 and TOPBP1Clover to the micronuclear chromosome during mitosis.
Damaged micronuclear chromosomes were identified to undergo one of three fates while transitioning through mitosis. In the first, ~40% of the total damaged micronuclear chromosome fragments remained clustered throughout mitosis and were incorporated into only one of the daughter nuclei after cytokinesis (Fig. 2E, F; Sup. Video 1 & 2). A similar proportion of damaged and clustered micronuclear chromosome mass was encapsulated into a new micronucleus (Extended Data Fig. 6A; Sup. Video 5). In the final 18% of examples, damaged micronuclear chromosome fragments were clustered initially, but broke subsequently into smaller clusters from what appeared to be shear induced by mitotic spindle forces (Extended Data Fig. 6B; Sup. Video 6), a scenario likely responsible for the corresponding proportion of mitotic cells viewed in fixed imaging in which micronuclear chromosome fragments were dispersed.
MDC1, TOPBP1, CIP2A tether DNA fragments
We tested if MDC1, TOPBP1, and CIP2A mediate the tethering of fragmented micronuclear chromosomal pieces during mitosis by inducing Y-chromosome missegregation and then depleting MDC1, TOPBP1, or CIP2A using siRNA. Depletion of any of the three (MDC1, TOPBP1, or CIP2A) resulted in an increased proportion of cells with dispersed micronuclear chromosomal fragments in mitosis (Fig. 3A–C; Extended Data Fig. 7A). The increase in cells with dispersed Y fragments was not due to an increase in damaged Y-chromosomes as there was no statistically significant change in the frequency of cells with a damaged Y-chromosome in mitosis (Extended Data Fig. 7B). Similarly, homozygous deletion of MDC1 or CIP2A in RPE1 cells yielded an increase (compared to wildtype cells) in the percentage of cells with dispersed chromosomal fragments following micronucleation (Extended Data Fig. 7C, D). Thus, MDC1, TOPBP1, and CIP2A each may play a role in tethering chromosomal pieces during mitosis.
Fig. 3. MDC1, TOPBP1, and CIP2A mediate clustering of micronuclear chromosome fragments during mitosis.
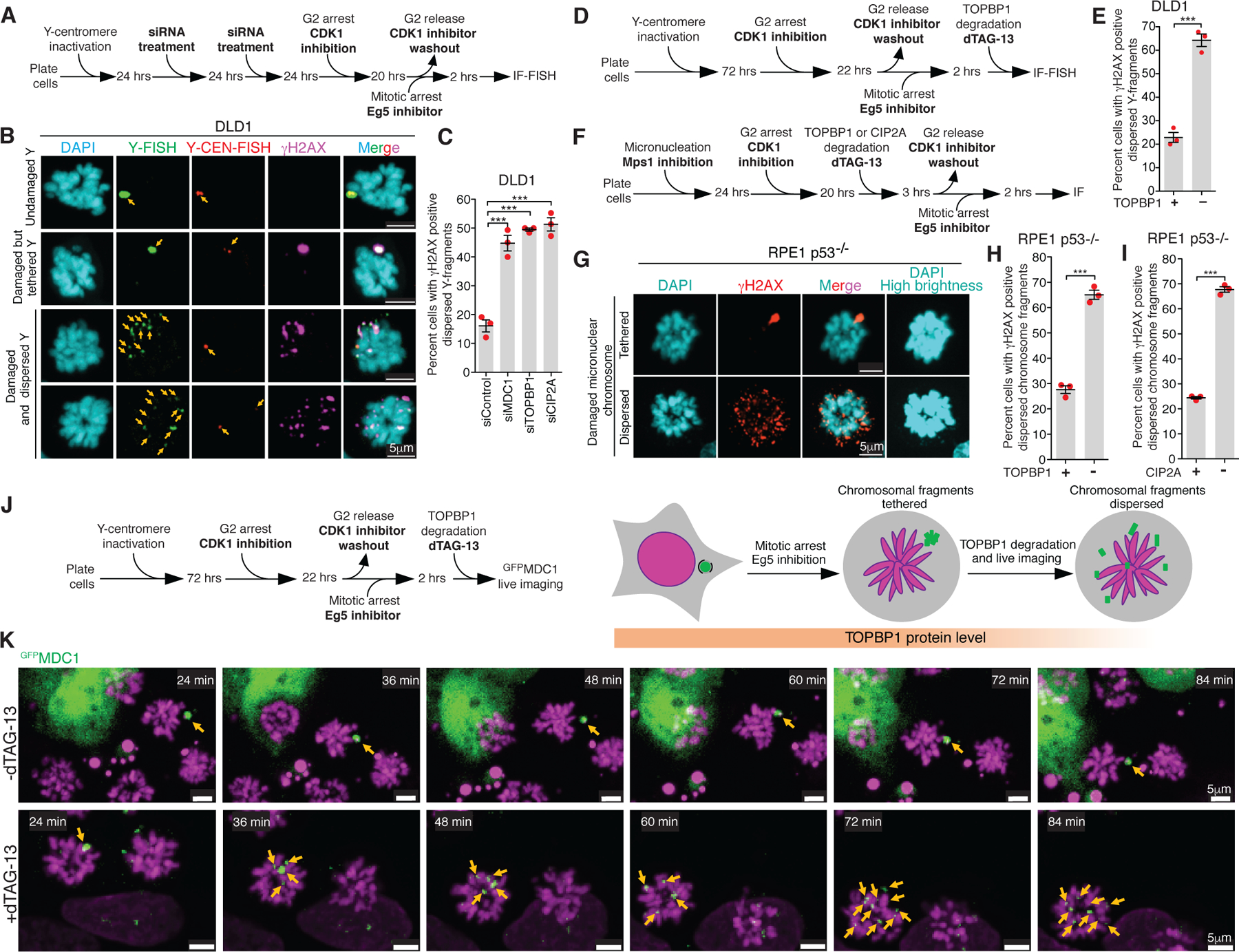
(A) Experimental outline for (B) and (C). (B) Representative images showing various fates of a micronucleated Y-chromosome in the subsequent mitosis. (C) Quantitation of cells with dispersed Y-chromosome fragments in mitosis upon siRNA mediated depletion of MDC1, TOPBP1, and CIP2A (n=3 independent experiments; a total of 770, 686, 662, and 667 cells were analyzed for control, siMDC1, siTOPBP1, and siCIP2A conditions, respectively). (One-way analysis of variance with Bonferroni’s multiple comparison test was applied, ***P<0.0001 and ns P>0.05.) (D) Schematic of experimental outline for (E). (E) Quantitation of cells with dispersed Y-chromosome fragments in mitosis upon degradation of TOPBP1dTag in mitosis (n=3 independent experiments; total 997 and 1131 cells were analyzed for control and TOPBP1dTag degradation condition, respectively). (F) Experimental outline for (G-I). (G) Representative images of RPE p53−/− cells showing tethered and dispersed damaged micronuclear fragments in mitosis after induction of micronucleation by Mps1 inhibition. Quantitation of cells with dispersed micronuclear fragments in mitosis upon degradation of (H) TOPBP1dTag (n=3 independent experiments; total 476 and 688 cells were analyzed for control and TOPBP1dTag degradation condition, respectively) or (I) dTagCIP2A (n=3 independent experiments; total 511 and 445 cells were analyzed for control and dTagCIP2A degradation conditions respectively). (J) Experimental outline and schematic for (K). (K) Frames of Sup. Video 7 and 8 showing behavior of GFPMDC1 labelled clustered damaged micronuclear chromosome in mitosis, when TOPBP1dTag degradation is induced (bottom) or not (top). Yellow arrows in (B) and (K) point to the fragments of micronuclear chromosomes. Two-tailed unpaired t-test was applied for data shown in (E) ***P=0.0003, (H) *** P<0.0001, and (I); *** P<0.0001. For graphs in (C), (E), (H) and (I), mean +/− SEM are shown, individual data points are shown as solid red circles.
TOPBP1 plays multiple distinct roles throughout the cell cycle33. To test if TOPBP1 acts as a protein tether for chromosomal fragments in mitosis or if the tethering of chromosomal pieces during mitosis is a consequence of its activity in the earlier cell cycle phase, we reduced TOPBP1 levels at specific cell cycle phases. To achieve this, we modified both endogenous TOPBP1 loci to encode a variant (TOPBP1dTag) containing a carboxyterminal degradation tag (dTag)34 (Extended Data Fig. 8A). TOPBP1dTag degradation was then induced by addition of the heterobifunctional small molecule dTAG-13 to mediate binding of the dTag to the E3 ubiquitin ligase cereblon (CRBN), producing rapid and efficient degradation of TOPBP1dTag with a half-life of 14 min (Extended Data Fig. 8C, D).
We tested if the S-phase role(s) of TOPBP1 in regulating DNA replication and ATR activation33,35 is sufficient for micronuclear fragment clustering in mitosis. We induced Y- micronucleation and induced TOPBP1 degradation in the next G2 followed by mitotic arrest (Extended Data Fig. 8F). Depletion of TOPBP1 in the G2 phase resulted in a three-fold increase in cells with dispersed Y fragments in mitosis (Extended Data Fig. 8G). Despite TOPBP1 depletion in G2, we did not observe any change in the percent of mitotic cells with a damaged Y chromosome, indicating that the DNA damage or repair frequency was not altered (Extended Data Fig. 8H). We conclude that beyond any G1 or S-phase roles of TOPBP1, tethering of chromosomal pieces in mitosis requires continued presence of TOPBP1 in G2.
TOPBP1 regulates mitotic DNA synthesis and the recruitment of various DNA repair proteins to some γH2AX positive chromatin sites during early mitosis36–38. We tested if chromosome fragment tethering is a consequence of repair events in early mitosis or if TOPBP1 acts as a protein tether for chromosomal fragments throughout mitosis. To do this, we induced Y-chromosome micronucleation and arrested cells in the subsequent mitosis by releasing G2 arrested cells for 2 hours. Induction of TOPBP1dTag loss during mitosis (Fig. 3D) produced a three-fold increase in the percentage of cells with dispersed of micronuclear fragments without any change in the proportion of mitotic cells with a damaged Y chromosome (Fig. 3E; Extended Data Fig. 8E, I).
We next followed the behavior during mitosis of clustered Y chromosomal fragments during the induced degradation of TOPBP1dTag (Fig. 3J). We used GFPMDC1 to follow micronuclear chromosomal fragments during mitosis, since its localization to damaged chromosome during mitosis is independent of TOPBP130. Live cell imaging revealed that in mitotically arrested cells all GFPMDC1 positive clusters of chromosomal fragments remained tethered for the full 2-hour span of filming (Fig. 3K, top panels; Sup. Video 7). However, when TOPBP1dTag degradation was induced during imaging, GFPMDC1-bound chromosome fragments dispersed within 30 minutes after the addition of dTAG-13 (Fig. 3K, bottom panels; Sup. Video 8). Thus, continuing presence of TOPBP1 is essential for maintaining micronuclear chromosome fragment clustering in mitosis.
Next, we modified both endogenous CIP2A or TOPBP1 loci of RPE p53−/− cells to encode dTagCIP2A or TOPBP1dTag. After induction of micronuclei or chromatin bridge formation (produced by Mps1 or Topoisomerase II inhibition, respectively), induced degradation in the subsequent G2 phase of dTagCIP2A or TOPBP1dTag led to loss of tethering of chromosome fragments marked by γH2AX (Fig. 3F–I; Extended Data Fig. 8B; Extended Data Fig. 8J–M). Micronuclear chromosomal fragments tethering in mitosis depends on functions of CIP2A beyond its more established role as a PP2A inhibitor39 (Extended Data Fig. 9A–C).
MDC1-TOPBP1-CIP2A mediates DNA tethering
Mitotic DNA lesions recruit TOPBP1 and CIP2A in MDC1 dependent29–31 and independent30,31 manners, implying the presence of MDC1-TOPBP1-CIP2A and TOPBP1-CIP2A subcomplexes. TOPBP1 interacts with MDC1 in mitosis through its BRCT1 and BRCT2 domains30 and with CIP2A through amino acids 756–89131. In cells in which both endogenous TOPBP1 alleles encoded degron tagged TOPBP1, we complemented induced TOPBP1dTag degradation with either wildtype TOPBP1 (TOPBP1WT) or TOPBP1 mutants that cannot interact with MDC1 (TOPBP1K155,250E) or CIP2A (TOPBP1Δ756−891) or both (TOPBP1K155,250E,Δ756−891) (Extended Data Fig. 10A). While complementation with TOPBP1WT restored fragment clustering, TOPBP1K155,250E, TOPBP1Δ756−891, or TOPBP1K155,250E, Δ756−891 were unable to rescue clustering after micronucleation (Extended Data Fig. 10B) or chromatin bridge formation (Extended Data Fig. 10C). In all cases, rescue required interactions among all three components, consistent with a MDC1-TOPBP1-CIP2A complex.
Moreover, the amount of TOPBP1 bound to a fragmented chromosome during mitosis was reduced to ~20% and ~2% in MDC1−/− and CIP2A−/− cells, respectively (Extended Data Fig. 10D, E), consistent with a major MDC1-TOPBP1-CIP2A complex and a smaller CIP2A-dependent, MDC1-independent pool of mitotic TOPBP1. To test if γH2AX is required for TOPBP1 recruitment and micronuclear fragment clustering, we utilized RPE1 H2AXS139A cells in which a serine to alanine (S-A) modification at position 139 of both H2AX alleles prevents formation of γH2AX. The absence of γH2AX produced a 50% reduction in the frequency of cells with TOPBP1 foci on damaged mitotic chromosomes (Extended Data Fig. 10F, G) and in those with TOPBP1 foci remaining, a ~70% reduction in TOPBP1 recruited onto previously micronucleated chromosomes (Extended Data Fig. 10H). Finally, the frequency of dispersed chromosome fragments in mitosis after micronucleation was increased by at least two-fold in the absence of γH2AX (Extended Data Fig. 10I, J). These data demonstrate a major γH2AX-dependent and a minor γH2AX-independent pathway in mitosis for TOPBP1/CIP2A recruitment to a fragmented chromosome.
DNA tethers enable en masse segregation
To test the consequence of disrupting chromosomal fragment clustering during mitosis (Fig. 4A), we induced micronucleation by Mps1 inhibition in RPE1 p53−/− cells and monitored the distribution of chromosome fragments using γH2AX in telophase or early G1 (Fig. 4B, C). Aurora-B staining at the midbody was used to identify paired daughter cell nuclei. Disruption of fragment clustering by degradation of TOPBP1dTag or dTagCIP2A resulted in random distributions of chromosomal fragments between the resulting daughter cells in a large (69.8±0.15% and 70.7±1.9% after TOPBP1dTag or dTagCIP2A degradation, respectively) majority of cases (Fig. 4C, D, Extended Data Fig. 11A) and with some fragments encapsulated within a main nucleus and others in the cytoplasm of daughter cells (Fig. 4C). The total number of distinct γH2AX positive fragments in both daughters increased by an order of magnitude (from a median of 1 for controls to 11 and 12) for TOPBP1dTag and dTagCIP2A degradation conditions, respectively (Extended Data Fig. 11B).
Fig. 4. CIP2A enables en masse transfer of fragments to the same daughter cell essential for viability of the resulting daughter cell.
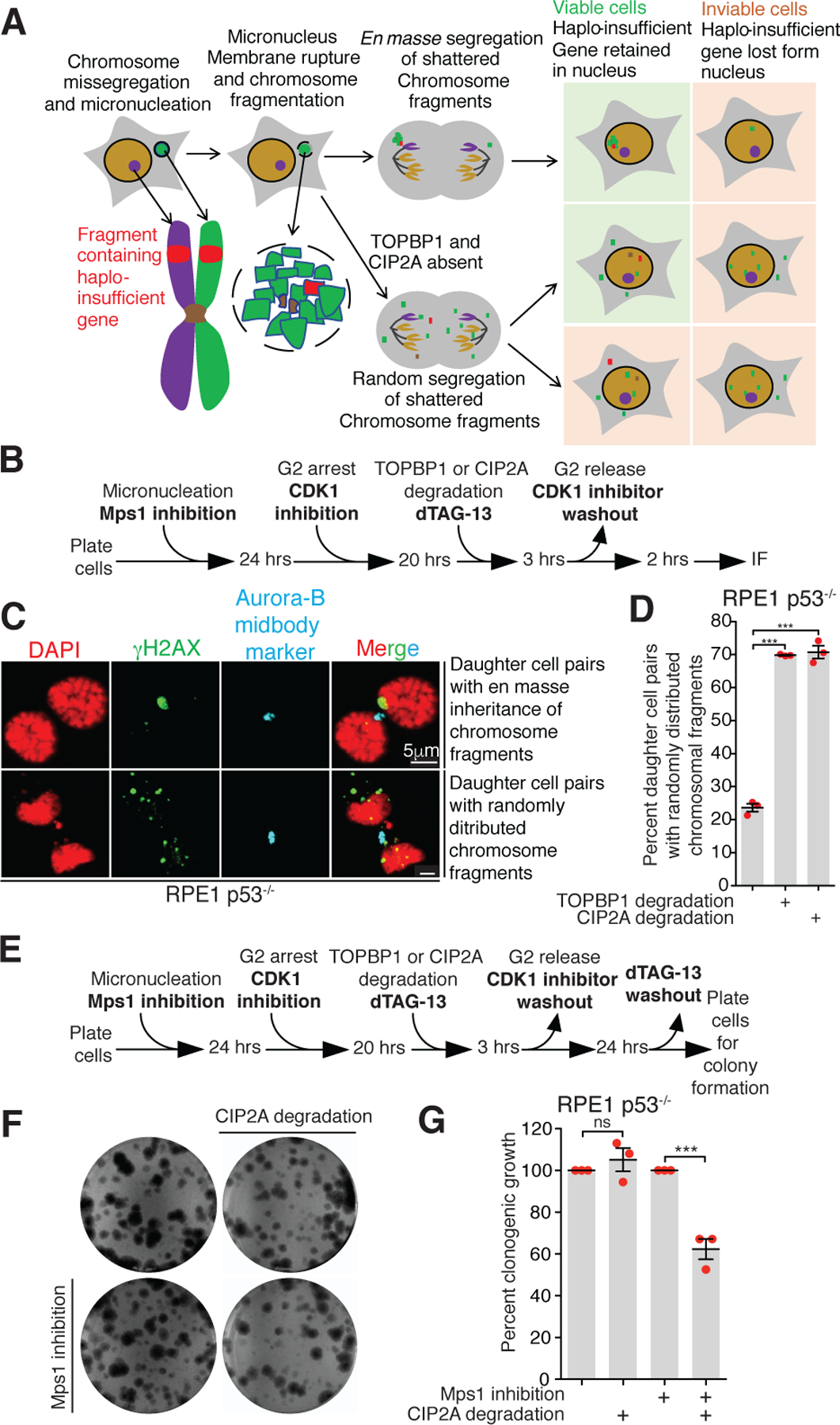
(A) Schematic showing sequence of events and fates of daughter cell post micronucleation in indicated conditions. (B) Experimental outline for (C) and (D). (C) Representative images showing various patterns of segregation of damaged micronuclear chromosome fragments in telophase or G1 phase. (D) Quantitation of cells with dispersed micronuclear fragments distributed between daughter cells following micronuclei formation and induced degradation in G2 of TOPBP1dTag or dTagCIP2A (n=3 independent experiments; total 306, 311, and 339 cells were analyzed for control, TOPBP1dTag degradation, and dTagCIP2A degradation conditions, respectively). (E) Experimental outline for (F) and (G). (F) Images of colony formed for experiment outlined in (E) for indicated conditions. (G) Quantitation of number of colonies formed for the experiment outlined in (E) for the indicated conditions (n=3 independent experiments). For (D) and (G), mean +/− SEM are shown; One-way analysis of variance with Bonferroni’s multiple comparison test was applied, *** P<0.0001 and ns P>0.05.
DNA tethering-mediated cell viability
Viability of cells forming frequent micronuclei will require continued expression in daughter cells of all essential genes (Fig. 4A), while dispersal of fragments and the loss of essential or haplo-insufficient genes/chromosomal regions (including centromeres or telomeres) would result in inviable daughter cells (Fig. 4A). We tested how viability of micronucleated cells was affected by the absence of mitotic fragment clustering by inducing micronuclei formation in one cell cycle in p53−/−, dTag/dTagCIP2A RPE1 cells and then inducing degradation of CIP2AdTag in the subsequent cell cycle (Fig. 4E). While CIP2A is not an essential gene, its transient depletion in the first mitosis after micronuclei formation reduced cell viability by 2-fold (Fig. 4F, G). Upon repeating the cell viability assay in TOPBP1dTag/dTag RPE1 p53−/− cells complemented with either TOPBP1WT or the CIP2A interaction mutant (TOPBP1Δ756−891), we observed a 2-fold reduction of cell viability in cells expressing TOPBP1Δ756−891 compared to TOPBP1WT (Extended Data Fig. 11C, D). We conclude that TOPBP1-CIP2A complexes, not other functions of CIP2A, enhance cell viability after micronucleation.
Genomic consequence of DNA de-tethering
The short-term consequences of the lack of fragment tethering were determined by inducing micronucleation in RPE1 p53−/− cells expressing CIP2A or dTagCIP2A, followed by transient CIP2A depletion (or not) throughout the next cell cycle and isolation of colonies grown from single cells. Whole genome sequencing was then conducted for 24 clones that had, and another 24 that had not, undergone transient CIP2A loss (Fig. 5A). Samples depleted in CIP2A demonstrated a significantly increased structural variant burden (non-depleted mean=8.4; CIP2A depleted mean=71.4), as well as a significantly increased copy number burden (non-depleted mean=57.8; CIP2A depleted mean=112.8) compared to CIP2A competent cells (Fig. 5B), supporting a role of CIP2A in protecting against structural alterations following chromosome mis-segregation.
Fig. 5. Transient loss of CIP2A following micronucleation leads to increased deletions and inversions.
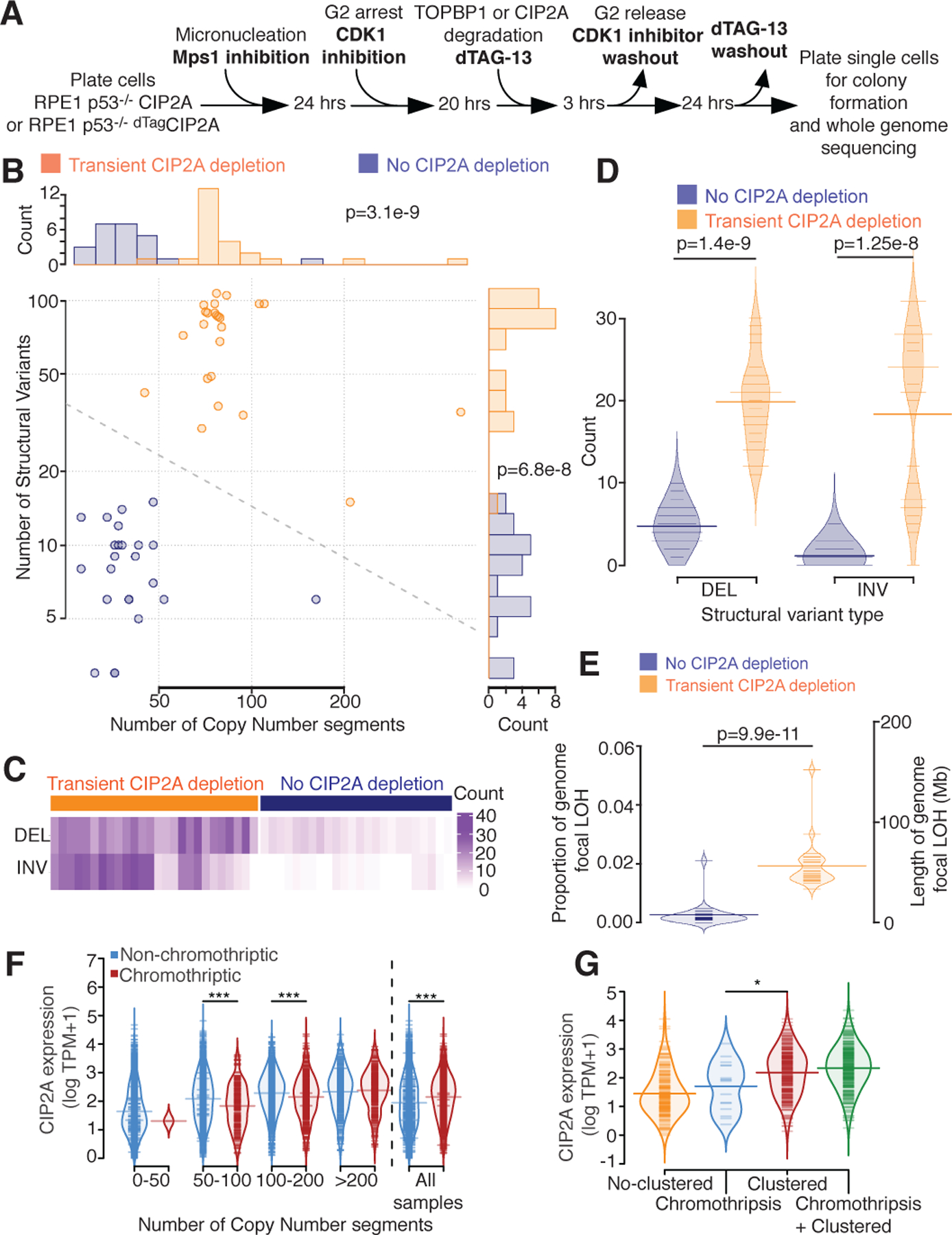
(A) Experimental outline for B-E. (B) Number of copy number segments (x-axis) and number of structural variants (y-axis) identified in 48 single cell clones with (orange, n=24) or without (blue, n=24) CIP2A depletion after micronucleation. Dotted line = decision boundary for a logistic regression predicting CIP2A condition from number of copy number segments and structural variants. Top panel: histogram of the number of copy number segments. Right panel: histogram of the number of structural variants. (C) Counts (purple) of deletions (DEL) and inversions (INV) split by CIP2A depleted (left, orange) or CIP2A unaltered (right, blue). (D) Number of deletion (DEL) or inversion (INV) identified (y-axis), split by CIP2A condition (color). (E) Proportion of the genome with focal loss of heterozygosity (LOH, <10 Megabase (Mb)) (left y-axis), or length of the genome LOH (right y-axis) in CIP2A unaltered (left, blue), or CIP2A depleted (right, orange) condition. Light colored distributions=density of proportions. For B, D, and E two-sided Mann-Whitney test was applied. (F) CIP2A gene expression (y-axis, log TPM+1) in cancer samples, stratified by copy number segmentation (x-axis) and chromothripsis status (CTLPscanner, color), or for all samples (right). ***=p<0.001, two-sided Mann-Whitney test. 50–100 p=9.7e-6. 100–200 p=7.4e-5. All samples p=1.4e-16. (G) CIP2A gene expression (y-axis) stratified by chromothripsis categories identified by ShatterSeek at a sample level (y-axis). No-clustered=no clustered event identified in the sample. Chromothripsis= only chromothriptic events identified. Clustered= only clustered events identified, may include ‘copy number neutral chromothripsis’. Chromothripsis + Clustered = both chromothriptic and clustered events found. *=p<0.05 (p=0.033), two-sided Mann-Whitney test. For F and G: log refers to natural log. For D, E, F and G: Light colored distributions=density of y-axis values. Thin colored lines=individual sample values. Thick colored lines=mean value.
A lack of tethering activity would posit an increase in the number of deletions and inversions following chromosomal mis-segregation. Indeed, we observed an increase in both the number of deletions (mean in non-depleted cells=4.8, mean in CIP2A depleted cells=19.8) and inversions (mean in non-depleted cells=1.1, mean in CIP2A depleted cells=18.3) in cells that had undergone transient CIP2A depletion after micronucleation compared to their non-depleted counterparts (Fig. 5C–D). Furthermore, copy number data corroborated this effect, with a significant increase in the proportion of the genome subject to focal loss of heterozygosity (LOH) upon CIP2A transient depletion (Fig. 5E).
Impact of tethering on the cancer genome
The impact of chromosomal fragment tethering on the cancer genome was first assessed by using 7,856 samples from The Cancer Genome Atlas (TCGA) project40 where cancer RNA expression data was associated with chromothripsis events identified using CTLPscanner41. An additional analysis was also performed using whole genome sequencing-derived chromothripsis events (determined with ShatterSeek2) in the 667 tumors with gene expression data from the Pan Cancer Analysis of Whole Genome (PCAWG) project42. Accumulation of RNAs encoding TOPBP1, CIP2A, and MDC1 was highly correlated with each other, with each more highly expressed in samples in which chromothripsis was identified by copy number or structural variants (Extended Data Fig. 12A–D). In low TOPBP1/CIP2A expressing chromothriptic samples in TCGA, there was a significant depletion of chromothripsis-amplification type copy number signatures43 and a trend towards enrichment in high expressing samples (Extended Data Fig. 12E, F).
We observed an overall increase in CIP2A expression in chromothriptic samples of TCGA (Fig. 5F; all samples); however, upon subsetting the data by copy number burden (defined as the count of copy number segments), we identified a stepwise increase in CIP2A expression as copy number burden increased. Within samples with similar levels of copy number burden, there was a concomitant decrease of CIP2A expression in canonical chromothriptic samples compared to non-chromothriptic samples (Fig. 5F). This effect was retained when expanding the analysis to account for both chromothripsis and copy number burden simultaneously (linear regression, Extended Data Fig. 12G). Similar effects were seen for TOPBP1 expression (Extended Data Fig. 12G), consistent with the role of CIP2A and TOPBP1 in promoting retention of chromothriptic fragments and copy number neutral chromothripsis. In contrast, MDC1 expression was found to have a positive association with chromothriptic samples, consistent with additional roles beyond tethering for MDC1 in DNA repair44 (Extended Data Fig. 12G).
In addition, we found that samples categorized as containing only ‘chromothriptic’ events by ShatterSeek using structural variant and copy number information2 displayed reduced levels of CIP2A in comparison to samples categorized as containing only ‘clustered’ events; clustered events do not show the oscillating copy number pattern indicative of canonical chromothripsis (Fig. 5G), thereby further supporting the role of CIP2A in tethering. Chromothripsis requires many steps beyond chromosome fragment tethering and thus unsurprisingly is associated with upregulation of many other DNA damage response genes, within which TOPBP1 and CIP2A are both significantly up-regulated in chromothriptic samples compared to non-chromothriptic samples (Extended Data Fig. 12H) and are amongst the most highly correlated genes with respect to gene expression, both in chromothriptic and non-chromothriptic samples (rchromo=0.71, rnonchromo=0.77; Extended Data Figure 17I).
Discussion
Here we identify that a complex of TOPBP1 and CIP2A recruited by MDC1 mediates the tethering of chromosomal fragments in mitosis generated as a result of either micronucleation or chromatin bridge formation. Such tethering is a crucial for the en masse inheritance of most or all micronuclear chromosome fragments by one of the daughter cells leading to production of rearranged chromothriptic chromosome with minimal loss of genomic content. Initial chromosome fragmentation within damaged or ruptured interphase micronuclei recruits megabase domains of γH2AX that rapidly recruit MDC1 released by disassembly of the main nuclear envelope. In turn, MDC1 assembled at or near each DSB directly recruits TOPBP1 and its binding partner CIP2A. The assemblies of TOPBP1 and CIP2A are essential for tethering of chromosomal fragments throughout mitosis and facilitate their incorporation into the primary nucleus of one daughter cell where they remain in close proximity, facilitating their ligation by NHEJ into a heritable chromothriptic chromosome (Extended Data Fig. 13). The nuclear foci resulting from inheritance of the clustered micronuclei fragments in the daughter nuclei may be similar to the recently described micronuclear body45. While the absence of rounded droplets in live cell imaging offers no support for chromosome fragment tethering through a mechanism involving TOPBP1 condensation46, mitotic chromosome fragment clustering may be mediated by the multiple phosphate-binding BRCT domains of TOPBP147 and/or by CIP2A oligomerization through its proposed coiled-coil domain.
While the mitotic clustering of the micronuclear-derived chromosome fragments may suggest minimal loss of DNA copy during chromothripsis, the chromothriptic signature in cancer genomes most often shows oscillation of copy number including loss of some DNA fragments48. Despite MDC1/TOPBP1/CIP2A tethering, losses may be mediated by multiple mechanisms, including (i) selective pressures, including the loss of tumor suppressor genes, (ii) spindle forces breaking initially clustered micronuclear chromosome fragments into smaller groups (as we observed in about 20% of cases in our live-cell imaging - Extended Data Fig. 6B), and (iii) altered gene copy numbers from defective DNA replication in micronuclei3,9 or exo-nuclease attack on the micronuclear DNA either in interphase5,6 or mitosis.
Transient, degron-induced reduction in CIP2A after chromosome micronucleation-dependent chromosome shattering drove acquisition of segmental deletions and inversions. Consistent with this, analyses of pan-cancer tumor genomes revealed that CIP2A and TOPBP1 expression was high in cancers with genomic rearrangements including chromothriptic cancers, but comparatively reduced in cancers with canonical chromothripsis in which deletions were frequent. We have shown that chromosome fragment clustering in mitosis affects viability of the resultant daughter cells (Fig. 4E–G, Extended Data Fig. 11C, D). Since CIP2A is non-essential to dividing cells and its expression is low in most normal human tissues but high in many cancers49, we propose that it is an attractive therapeutic target for chromosomally unstable tumors forming frequent micronuclei.
Methods
Cell culture
HEK293T (ATCC: CRL-11268), DLD-1, hTERT-RPE1 p53−/− (a kind gift from Prof. David Pellman), RPE1 MDC−/− (a kind gift from Prof. Manuel Stucki), RPE1 CIP2A−/− (a kind gift from Prof. Manuel Stucki), RPE1 H2AXS139A/S139A (a kind gift from Prof. Steve Jackson) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) or DMEM/F-12 (Gibco) (for hTERT-RPE1 p53−/−) media, supplemented with 10% tetracyclin-free fetal bovine serum (FBS), 2mM L-Glutamine, and 100ug/ml Normocin (Invivogen) at 37°C in presence of 5% CO2. Cell lines were periodically tested and confirmed free of mycoplasma.
Stable cell line generation and genome editing
For lentivirus production TOPBP1-clover cloned into LS135 or lenti-CMV/TO-GFP-MDC1 (779–2) (a gift from Eric Campeau, Addgene plasmid # 26285) were co-transfected with packaging plasmids pMD2.G and psPAX2 in HEK293T using TransIT-VirusGEN® Transfection Reagent (Mirus, MIR6705). Mutations (either S155,250E, or deletion of region encoding 756–891 amino acids, or both) were introduced in LS135-TOPBP1-Clover using site directed mutagenesis kit (Agilent) according to manufacturer’s protocol. The supernatant containing lentivirus was collected and filtered through a 0.45μm filter, 2–3 days after transfection. For generating stable cell lines expressing fluorescently tagged proteins, cells were infected with viral supernatant in presence of 5g/ml Polybrene (Santa Cruz). Single-cell clones expressing the protein of interest were isolated by fluorescence-activated cell sorting (FACS) (Sony SH800).
For tagging endogenous locus with dTAG, sgRNA sequences targeting the desired genomic region (shown in Extended Data Fig. 7A, B) were cloned in pSpCas9(BB)-2A-GFP (PX458) (a gift from Feng Zhang, Addgene plasmid # 48138). The repair template was created by cloning the left and right homology arm (~500bp) with 2xHA-dTAG (for N-term tagging) or dTAG-2xHA (for N-term tagging) into pUC19. Cas9 and sgRNA expressing plasmid (PX458) along with plasmid containing repair template were nucleofected in DLD1 or hTERT-RPE1 p53−/− using Cell Line Nucleofector™ Kit V (Lonza) and Nucleofector® 2b Device (Lonza) according to manufacturer’s protocol. Single cells expressing GFP were FACS sorted (Sony SH800) 2 days post nucleofection and grown to colonies. PCR and Immunoblotting were used to screen colonies derived from single cells to identify correctly edited colonies.
Following sgRNAs were used for editing and PCR primers were used for screening colonies:
sgTOPBP1: AGATGCGATTAGTGTACTCT
sgCIP2A: GGCAGTGGAGTCCATTGCAC
PCR primers for confirming editing at TOPBP1 locus:
For DLD1 cells:
TOPBP1fw-1: GGTGAGACTTTGTCCCACAGGGT
TOPBP1rv-1: AACCTTGTGCTCAGGCTCCTGTT
For RPE1 p53−/− cells:
TOPBP1fw-2: GAGACTTTGTCCCACAGGGTCCA
TOPBP1rv-2: AACCTTGTGCTCAGGCTCCTGTTA
PCR primers for confirming editing at CIP2A locus:
CIP2Afw: ATGCAGGCTCTGGCGGAGTG
CIP2Arv: ACGCTTACTAGGAAGGGGAAGTGC\
siRNA transfection
ON-TARGETplus SMARTpool siRNAs (Horizon Discovery) targeting either Lig4 (L-004254–00-0005), PolD3 (L-026692–01-0005), Mre11 (L-009271–00-0005), Rad50 (L-005232–00-0005), PolQ (L-015180–01-0005), MDC1 (L-003506–00-0005), TOPBP1 (L-012358–00-0005), CIP2A (L-014135–001-0005) or non-targeting pool (D-001810–10-05) were transfected using Lipofectamine RNAiMAX Transfection Reagent (ThermoFisher, 13778075) at the indicated timepoint according to manufacturer’s protocol.
Inhibitors and small molecules
Doxycycline was used at 1 μg/ml (Sigma), Indole-3-acetic acid (IAA) used at 500 μM (Sigma, I5148), CDK1 inhibitor RO-3306 used at 9 μM (Sigma-Aldrich, SML0569), NMS-P715 0.8 μM (Millipore Sigma, 475949), ICRF-193 used at 100 nM (Enzo life sciences, BML-GR332–0001) S-Trityl-L-cysteine (STLC) used at 5 μM (Enzo Life Sciences, ALX-105–011-M500), dTAG-13 used at 500 nM (Sigma-Aldrich, SML2601), Okadaic acid used at 500 nM (Fisher Scientific, 11–362-5U), Geneticin® Selective Antibiotic (G418 Sulfate) used at 300 μg/ml (Gibco, 10131035).
Immunoblot (IB)
Cells were lysed in SDS sample buffer and boiled at 95°C for 10 minutes. The whole cell lysate was resolved on SDS PAGE gel and transferred to Immobilon®-FL PVDF membrane (Millipore Sigma). The membrane was then blocked with Intercept® (TBS) Blocking Buffer (LI-COR) for 1 hour followed by overnight incubation at 4°C with primary antibody diluted in Intercept® (TBS) Blocking Buffer supplemented with 0.1% Tween-20. The membrane was washed 4 times (5min each) with TBST (TBS, 0.1% Tween-20) and then incubated for 1 hour at room temperature with fluorescent secondary antibody diluted in Intercept® (TBS) Blocking Buffer supplemented with 0.1% Tween-20. The membrane was washed 4 times (5 minutes each) with TBST (TBS, 0.1% Tween-20) and then imaged on Odyssey FC imager (LI-COR).
Immunofluorescence (IF)
Cells were plated on coverslips and treated as indicated. Cells were fixed with 4% PFA in PBS or 100% ice-cold methanol (for TOPBP1 staining) for 15 minutes at room temperature, followed by permeabilization with 0.5% Triton X-100 in PBS for 15 minutes at room temperature. Cells were then blocked with blocking buffer (0.2M glycine, 2.5% BSA, 0.1% Triton X-100, PBS) for 1 hour at room temperature. Coverslips were incubated with primary antibody diluted in blocking buffer for 1 hour at room temperature or overnight at 4°C. Coverslips were washed 4 times with PBST (PBS, 0.1% Tween-20), 10 minutes each at room temperature, followed by incubation with fluorescent secondary antibodies diluted in blocking buffer for 1 hour at room temperature. Coverslips were washed 4 times with PBST (PBS, 0.1% Tween-20), 10 minutes each at room temperature. Coverslips were then either processed for Fluorescent In-Situ Hybridization (FISH) or stained with DAPI and mounted on slides using Prolong Gold mounting media. For combined IF and FISH, cells were fixed with methanol/acetic acid (3:1) after IF procedure and then processed for FISH as described below.
Antibodies
Following antibodies were used in this study at the indicated concentration. Anti-phospho histone H2AX (Ser139) (γH2AX) (clone JBW301, EMD Millipore, 05–636, 1:1500 IF) (Cell Signaling Technologies, 2577, 1:1000 IF), anti-TOPBP1 (Bethyl Laboratories, A300–111A, 1:2000 IF, 1:2000 IB) (Abcam, ab2402, 1:1000 IF), anti-MDC1 (Abcam ab11171, 1:1000 IF, 1:1000 IB), anti-CIP2A (clone 2G10–3B5; Santa Cruz sc80659, 1:500 IF, 1:1000 IB), anti-GAPDH (clone 14C10, Cell Signaling Technologies, 1:5000 IB), anti-PolD3 (clone 3E2, Abnova, H00010714-M01, 1:1000 IB), anti-Lig4 (clone N2C2, GeneTex, GTX100100, 1:1000 IB), anti-Mre11 (clone 12D7, GeneTex, GTX70212, 1:1000), anti-Rad50 (Cell Signaling Technologies, 3427, 1:1000 IB), anti-HA-tag (Novus biologicals, NB600–363, 1:2000 IB), anti-Lamin A/C (clone E-1, Santa Cruz, sc-376248, 1:200 IF), anti-Lamin B1 (clone C-5, Santa Cruz, sc-365962, 1:200 IF), anti-53BP1 (Thermo Fisher Scientific, PA1–16565, 1:1000 IF), anti-BRCA1 (clone D-9, Santa Cruz, sc-6954, 1:1000 IF).
Chromosome spreads and Fluorescent In-Situ Hybridization (FISH)
For metaphase chromosome spreads, cells were arrested in mitosis for 4 hours using 100ng/ml colcemid (KaryoMAX, Thermo Fisher). The cells were then collected by trypsinization and incubated with hypotonic solution (25 mM KCl, 0.27% sodium citrate in distilled water) for 10–15 minutes at 37°C followed by fixation with cold methanol/acetic acid (3:1). Fixed cells were then pelleted and resuspended in appropriate concentration in fixative and dropped on the slides for chromosome spread preparation.
For DNA FISH, human Y-chromosome and human Y-centromere FISH probes (MetaSystems) were combined in a 1:1 ratio and applied to chromosome spreads and covered with coverslip or applied to cells on coverslips (for combined IF-FISH) followed by denaturation at 75°C for 5 minutes. The coverslips were sealed with Fixogum rubber cement (Marabu) and incubated overnight in a humidified chamber at 37°C for hybridization. Slides or coverslips were then washed first with 0.4% SSC buffer at 72°C for 2 minutes, and then by 2X SSC with 0.05% Tween-20 for 30 seconds at room temperature. Slides or coverslips were then washed with distilled water and counterstained with DAPI and mounted in Prolong Gold mounting media.
Fixed and Live cell Imaging
Fixed cell images were acquired on the DeltaVision Core system (Applied Precision) and maximum intensity projections were generated using the Fiji package of ImageJ. Fixed images were also acquired on the CQ1 benchtop spinning disk confocal high-content analysis system (Yokogawa). Mean fluorescence intensity for TOPBP1 and γH2AX signal was measured using Fiji package of ImageJ by manually drawing region of interest (ROI) around γH2AX signal on the damaged mitotic chromosome (for the experiment shown in extended data figure 14 C and E, ROI was drawn around TOPBP1 signal). The mean fluorescence intensity in the ROI was subtracted by the mean background signal intensity (background intensity was measured at an intercellular region free of damaged DNA). Mean TOPBP1 intensity was normalized to mean γH2AX intensity at a given ROI, and all the resulting normalized intensities were converted to percent intensity relative to the average normalized TOPBP1 intensity of the control group (for the experiment shown in extended data figure 14 C and E, mean TOPBP1 intensity in the ROI was converted into percent average TOPBP1 intensity relative to that of the control group).
For live-cell imaging, DLD1 or RPE1 p53−/− expressing the fluorescently tagged protein of interest were plated in 96 well glass-like polymer bottom plates (Cellvis) and treated as indicated. Live cell images were acquired for 4–24 hours CQ1 benchtop spinning disk confocal high-content analysis system (Yokogawa) (CQ1 software version 1.05.01.02). Cells were imaged at 40X or 60X dry objective and were maintained in humidified condition at 37°C in presence of 5% CO2 during imaging.
Clonogenic assay
Cells were treated as indicated in the experimental outline and the specified number of cells were plated. For the experiment shown in the Extended Data Fig. 2G–J, cells were grown in 300 μg/ml G418 containing media. After 10–15 days the cells were fixed with 100% methanol and stained with a solution of 0.5% crystal violet, 25% methanol. The colonies formed were manually quantified.
Statistical analysis
Three independent replicates were performed for each experiment unless otherwise stated. Prism v9.0 (GraphPad) was used for statistical analysis except for Fig. 4H-K and Extended Data Fig. 10 and 11. All graphs show mean and standard error with individual data points for each repeat. The statistical test used for analysis is indicated in figure legends.
Gene expression analysis:
Gene expression data (gene level counts and TPMs) for TCGA were downloaded from the GDC portal (https://portal.gdc.cancer.gov). Processed copy number profiles from SNP6 arrays for TCGA were obtained from: https://github.com/VanLoo-lab/ascat/tree/master/ReleasedData/TCGA_SNP6_hg19. Chromothripsis calls for PCAWG (ShatterSeek) were downloaded from: https://dcc.icgc.org/releases/PCAWG/evolution_and_heterogeneity/clustered_mut_processes. Chromothripsis was called from SNP6 copy number profiles using CTLPscanner41. Associations between log(TPM+1) CIP2A, TOPBP1, and MDC1 gene expression and chromothripsis were performed with two-sided Mann-Whitney tests between chromothriptic and non-chromothriptic samples. This was performed separately for chromothripsis defined by CTLPscanner (TCGA SNP6) and chromothripsis defined by ShatterSeek (PCAWG WGS). Expression of CIP2A and TOPBP1 was jointly categorized into lower, middle and upper by finding perpendicular lines to the line of best fit that pass through both the upper quartile and lower quartile for both TOPBP1 and CIP2A. In brief the line of best fit for log(TPM+1) gene expression of TOPBP1 and CIP2A is given as
where y is the gene expression for CIP2A, x is the gene expression for TOPBP1, a is the gradient and c is the intercept. A perpendicular line that passes through a point defined as [xi,yi] is then defined with coefficients:
Where [xi,yi] are given as the upper quartiles of TOPBP1 and CIP2A expression to determine a threshold for upper expression, and given as the lower quartiles of TOPBP1 and CIP2A expression to determine a threshold for lower expression.
Associations between categorized CIP2A/TOPBP1 expression and chromothriptic samples were performed using two-sided Fishers exact tests. Associations between categorized CIP2A/TOPBP1 expression and the number of segments in copy number profiles were tested with a Kruskal-Wallis rank-sum test, with subsequent pairwise two-sided Mann-Whitney tests. Associations between categorized CIP2A/TOPBP1 expression and the proportion of the genome as LOH were tested with a Kruskal-Wallis rank-sum test, with subsequent pairwise two-sided Mann-Whitney tests. Categorization and subsequent associations were performed for SNP6 data in all samples, for SNP6 data in only chromothriptic samples, and for WGS data in only chromothriptic samples. Copy number signature exposures for TCGA copy number profiles were downloaded from Steele et al. 202243. Associations between categorized CIP2A/TOPBP1 expression and chromothripsis-associated copy number signatures (CN4:9) were performed using Fisher’s exact tests. Correction for multiple testing was performed using the Benjamini-Hochberg method. A list of DNA repair genes was taken from Knijnenburg et al., 201850. Associations of repair gene expression (log(TPM+1) with chromothripsis (CTLPscanner) were performed with two-sided Mann-Whitney test. P-values were corrected for multiple testing using the Benjamini-Hochberg method. Correlations of gene expression (log(TPM+1)) between all pairwise combinations of DNA repair genes was evaluated with Pearson’s correlation coefficient, for chromothriptic and non-chromothriptic samples separately (CTLPscanner).
Associations of CIP2A gene expression (log(TPM+1)) with chromothriptic status (CTLPscanner) were performed for all samples, and within sets of samples with 0–50, 50–100, 100–200 and >200 copy number segments in their SNP6 copy number profiles. Linear regression models were then built separately for CIP2A, TOPBP1 and MDC1, with gene expression as the response variable (log(TPM+1)) and predictor variables of chromothriptic status (CTLPscanner) and copy number segmentaion groups (0–50, 50–100, 100–200, >200).
Samples were categorised by their ShatterSeek results. Samples with no putative chromothriptic/clustered regions identified were designated as No-clustered. Samples with only chromothriptic/clustered regions that were categorised as chromothriptic by ShatteSeek were designated Chromothriptic Samples with only chromothriptic/clustered regions that were categorized as clustered by ShatteSeek were designated Clustered Samples with both clustered and chromothriptic regions were designated as Chromothriptic + Clustered. Note that instances of ‘balanced chromothripsis’ will be categorized as clustered by ShatterSeek as they will lack the oscillating copy number that ShatterSeek requires to call chromothripsis. Association of CIP2A expression (log(TPM+1)) was tested between chromothriptic only and clustered only samples (two-sided Mann-Whitney test).
Cell line data processing:
Alignment of cell line FASTQ files was performed using the alignment steps of of the EnsembleVariantCallingPipeline without refinement (https://github.com/AlexandrovLab/EnsembleVariantCallingPipeline). In brief, alignment was performed with BWA-mem v0.7.17, BAM files were sorted using samtools v1.9, and duplicates were identified with Picard v2.18.15 MarkDuplicates. Reads were aligned to the human GRCh38.d1.vd1 reference sequence.
Copy number calling was performed using ASCAT v3.1.0 (https://github.com/VanLoo-lab/ascat), using SNP positions, GC content files, and replication timing files provided by the package for GRCh38. Parental clone 1 was used to determine heterozygous SNP positions to call copy number on for each of the experimental clones.
Structural variant calling was performed using delly v1.1.6 (https://github.com/dellytools/delly). Variant calling was performed using the somatic SV calling strategy, utilising parental clone 1 as the germline file for each experimental clone, and with parental clones 1–7 as a panel of controls. This involved somatic pre-filtering, genotype pre-filtering, and post-filtering as described in the delly manual. VCF files were filtered for PASS variants only, and were converted to bedpe files using bcftools v1.17–5-g36f1b0c.
Associations between experimental conditions (CIP2A depletion, no CIP2A depletion) for number of structural variants identified, number of deletion structural variants identified, number of inversion structural variants identified, number of copy number segments identified in copy number profiles and proportion of the genome identified as loss of heterozygosity were performed with two-sided Mann-Whitney tests.
Extended Data
Extended Data Fig. 1. Fragmentation of Y-chromosome after micronucleation.
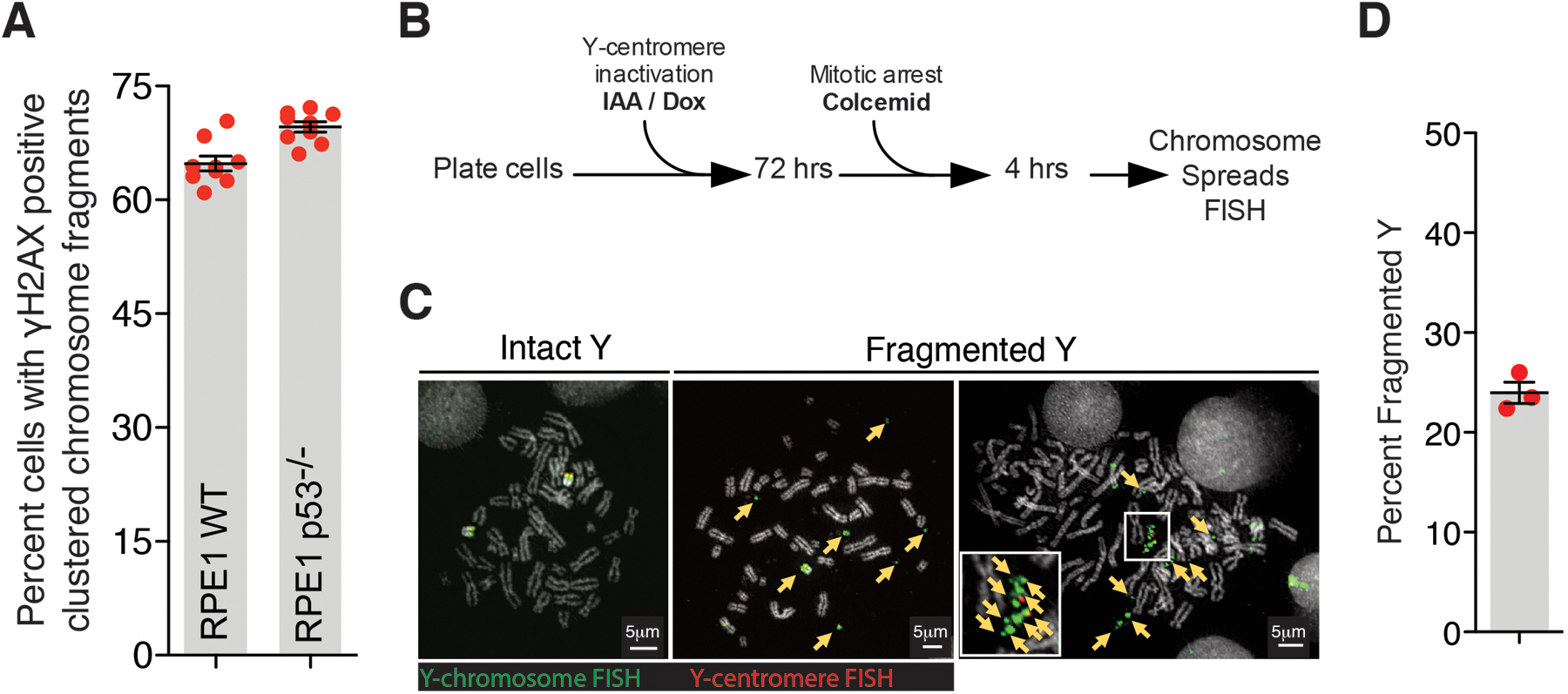
(A) Quantitation of chromosomal fragment tethering for RPE1 WT vs p53−/− cells (similar experimental setup as used in Fig.1C for Mps1i condition) (n=9 independent experiments, total 498 and 861 cells were analyzed for RPE1 WT and RPE1 p53−/− conditions, respectively). Two-tailed unpaired t-test was applied, P=0.0009. (B) Experimental schematic for (C) and (D). (C) Representative images of Y-chromosome in chromosome spreads from experiment outlined in (B). Yellow arrows point to Y-chromosome fragments. (D) Quantitation of fragmented Y-chromosome from experiments outlined in (B) (n=3 independent experiments, total 203 cells were analyzed). For (A) and (D), mean +/− SEM are shown.
Extended Data Fig. 2. Micronuclear envelopes do not persist in mitosis and PolD3 does not mediate clustering of broken chromosome fragments in mitosis.
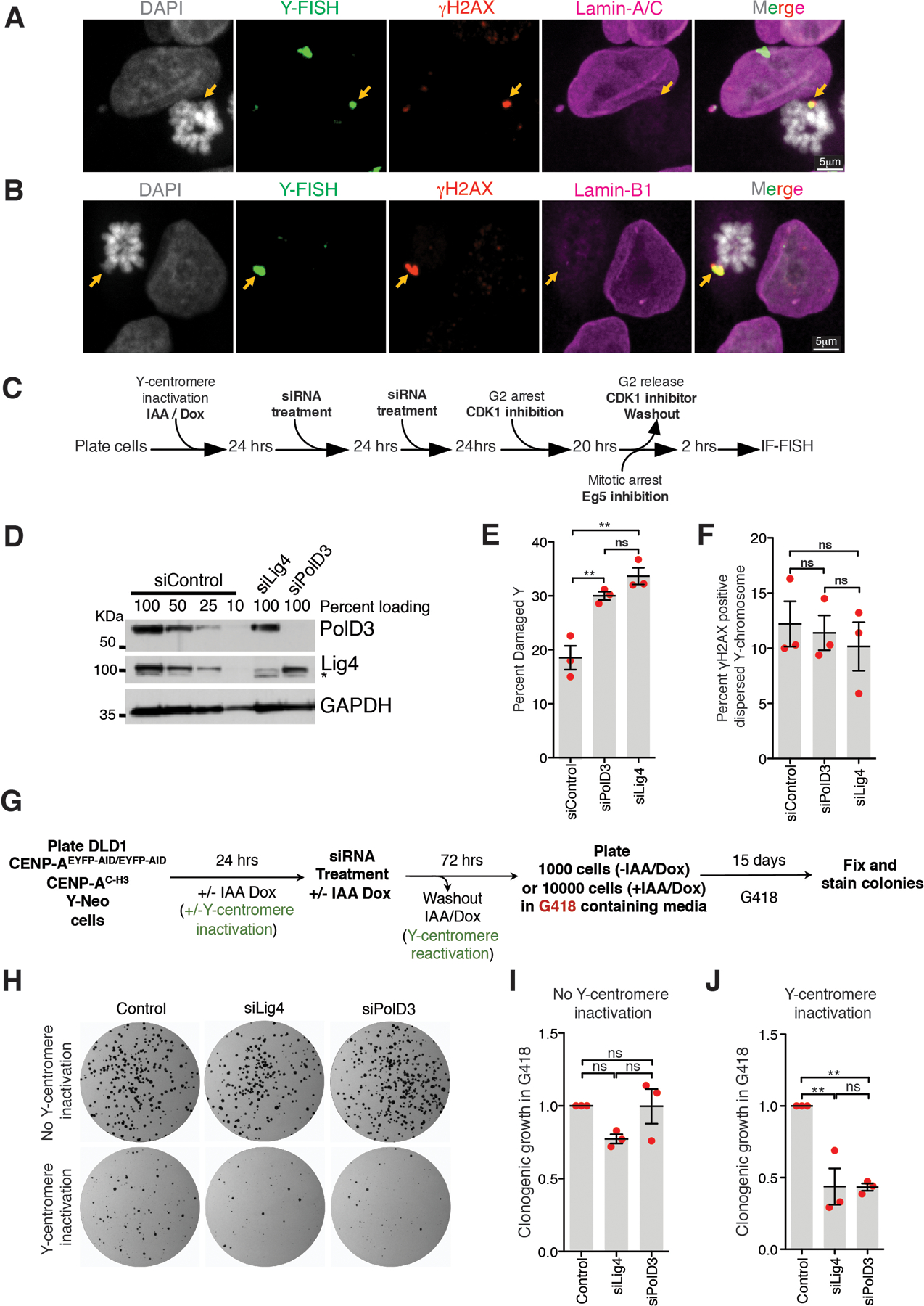
Representative image showing (A) Lamin A/C or (B) Lamin B1 staining on damaged and clustered Y-chromosome in mitosis (for A and B, 2 independent experiments were performed). (C) Schematic for experiment in (D), (E), and (F). (D) Immunoblots showing depletion of PolD3 and Lig4 from experiment described in (C) (uncropped blots in Supplementary Figure 1). (E) Quantitation of damaged Y-chromosome for experiment shown in (C). (F) Quantitation of fragment dispersal of a damaged Y-chromosome for experiment shown in (C) (for (E) and (F), n=3 independent experiments; total 651, 555, and 637 cells were analyzed for control, siPolD3, and siLig4 conditions, respectively). (One-way analysis of variance with Bonferroni’s multiple comparison test was applied, ** P<0.001 and ns P>0.05.) (G) Schematic of experimental setup for (H), (I), and (J). (H) Images of CEN-select colony formation assay from experiment outlines in (G). (I, J) Quantitation of clonogenic growth from the experiment outlined in (G) and shown in (H) (for (I) and (J), n=3 independent experiments were analyzed; one-way analysis of variance with Bonferroni’s multiple comparison test was applied, ** P<0.001 and ns P>0.05). For (E), (F), (I), and (G) the graphs mean +/− SEM are shown.
Extended Data Fig. 3. Neither the MRN complex nor PolQ mediates clustering of broken chromosome fragments in mitosis and micronuclei are defective in recruitment of DNA repair proteins.
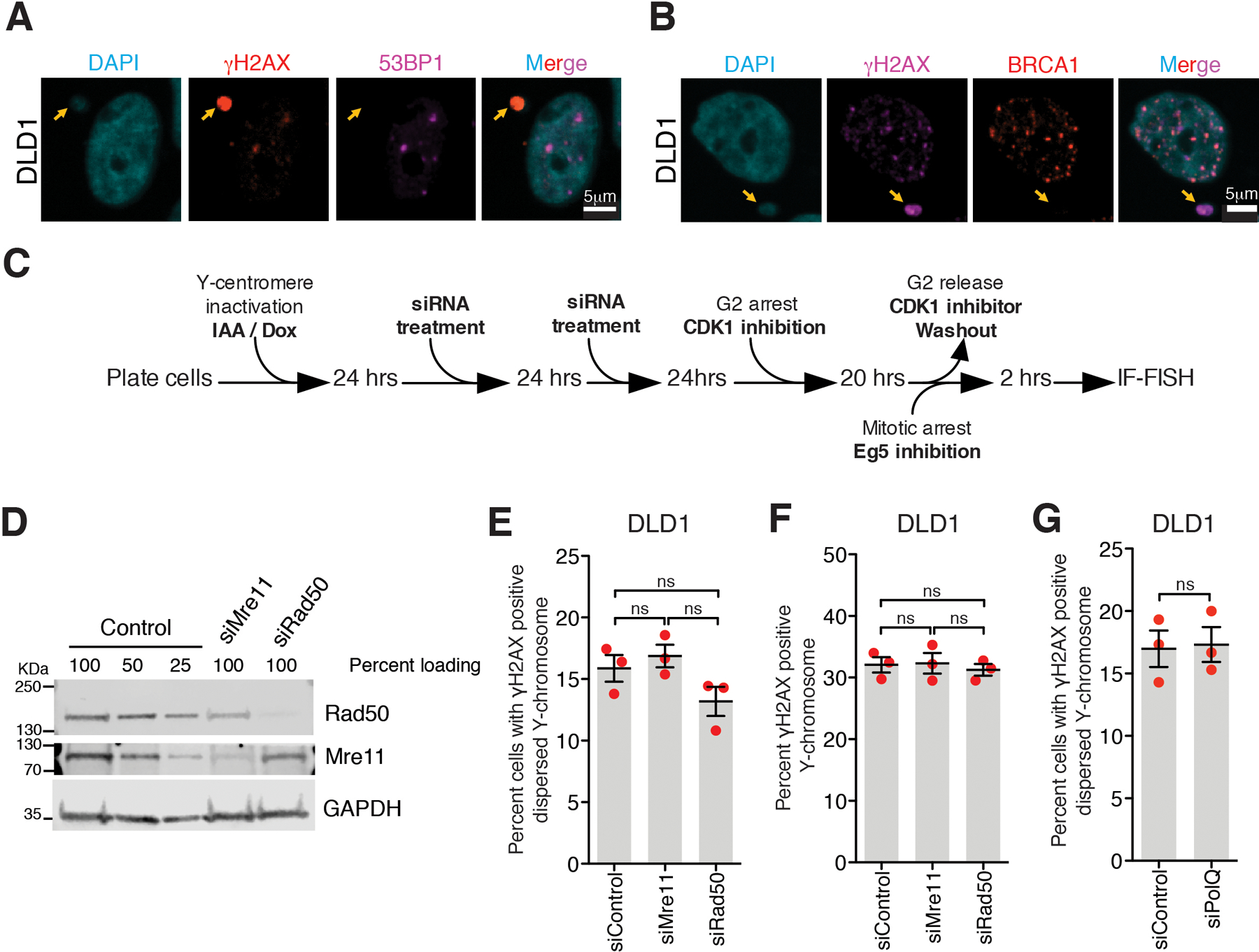
Representative images showing lack of recruitment to damaged micronuclei of (A) 53BP1 or (B) BRCA1 (for A and B, 2 independent experiments were performed). (C) Schematic of experimental setup for (D-G). (D) Immunoblot showing depletion of Rad50 and Mre11 for the experiment described in (C) (uncropped blots in Supplementary Figure 1). Quantitation of (E) dispersed Y-chromosome fragments or (F) damaged Y chromosomes from the experiments outlined in (C) after siRNA depletion of Mre11 or Rad50. (For (E) and (F), n=3 independent experiments; total 726, 742, and 727 cells were analyzed for control, siMre11, and siRad50 conditions, respectively. One-way analysis of variance with Bonferroni’s multiple comparison test was applied, ns P>0.05.) (G) Quantitation of damaged Y-chromosomes from the experiment outlined in (C) upon siRNA depletion of PolQ (n=3 independent experiments; total 240 and 217 cells were analyzed for control and PolQ depletion, respectively). (Two-tailed unpaired t-test was applied for data shown in (G); ns P>0.05.). For (E), (F), and (G) mean +/− SEM are shown.
Extended Data Fig. 4. Localization of MDC1, TOPBP1, and CIP2A to micronuclei.
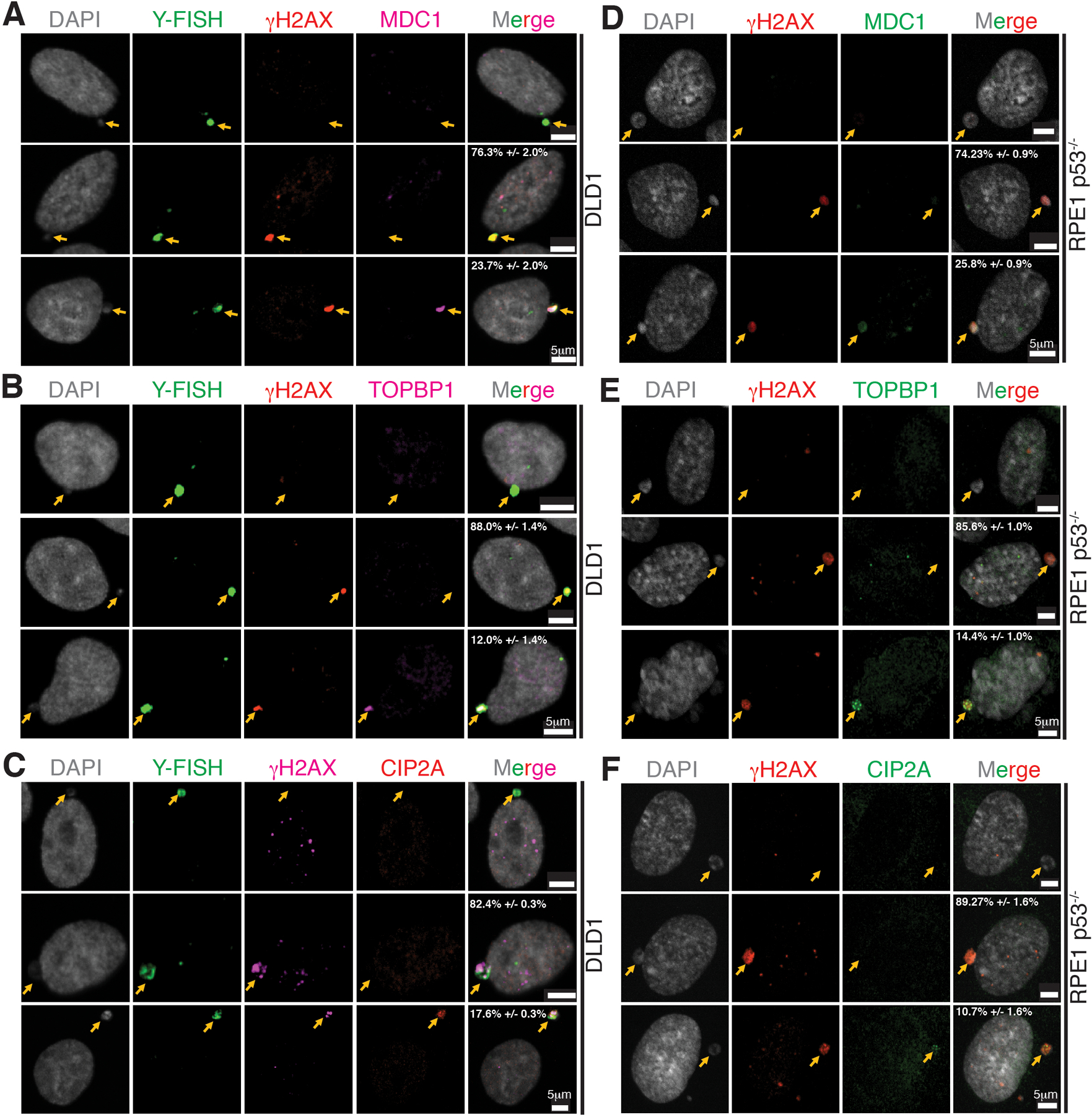
Representative images showing localization of (A, D) MDC1, (B, E) TOPBP1, and (C, F) CIP2A in micronuclei of DLD1 (n=3 independent experiments, total 482, 290, and 319 DLD1 cells were analyzed for MDC1, TOPBP1, and CIP2A, respectively) and RPE p53−/− (n=3 independent experiments, total 315, 360, and 299 RPE p53−/− cells were analyzed for MDC1, TOPBP1, and CIP2A, respectively) (percentage of cells (mean +/− SEM) with γH2AX positive micronuclei showing the phenotype in the images are indicated on the images). Yellow arrows point to the micronuclei.
Extended Data Fig. 5. Localization of MDC1, TOPBP1, and CIP2A to a broken chromatin bridge and micronuclear envelope integrity or p53 presence is not a major determinant of localization of MDC1, TOPBP1, and CIP2A to micronuclei.
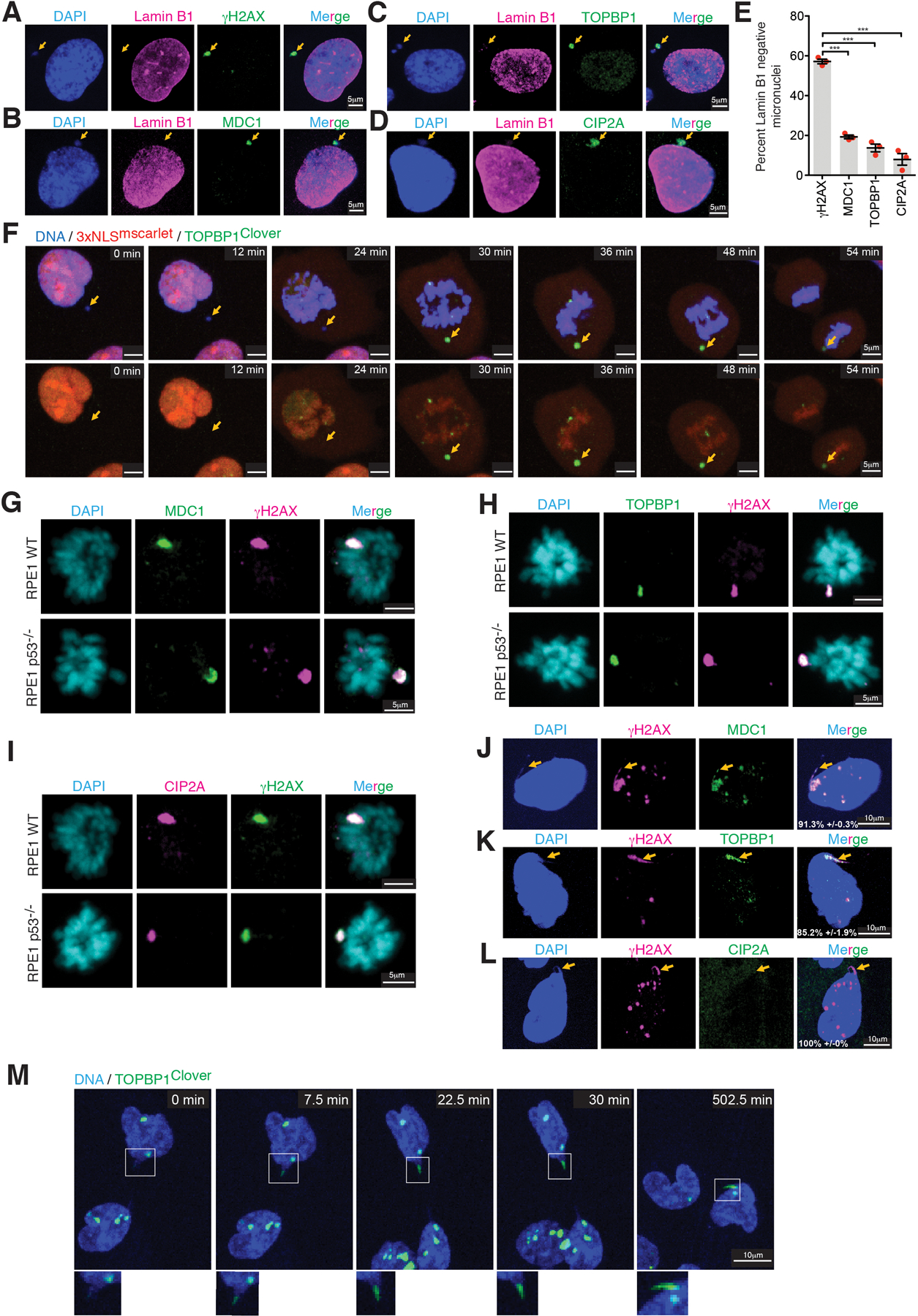
Representative images showing localization of Lamin B1 and (A)γH2AX, (D) MDC1, (C) TOPBP1, and (D) CIP2A in micronuclei of RPE1 p53-/−. (E) Quantitation of representative phenotypes shown in A-D (n=3 independent experiments, total 287, 307, 309, and 256 cells were analyzed for γH2AX, MDC1, TOPBP1, and CIP2A, respectively) (mean +/− SEM are shown). One-way analysis of variance with Bonferroni’s multiple comparison test was applied, *** P<0.0001. (F) Cropped frames of Sup. Videos 3 showing localization of TOPBP1Clover to the micronuclear chromosome which had nuclear envelope ruptured before entry into mitosis. From 2 independent experiments, a total of 60 mitotic chromosomes previously within micronuclei were identified. In all cases, TOPBP1Clover recruitment occurred at the onset of mitosis but did not have detectable 3XNLSmScarlet before mitosis entry. Representative images showing localization of (G) MDC1, (H) TOPBP1, and (I) CIP2A on broken micronuclear chromosome in mitosis in RPE1 WT and RPE1 p53−/− cells, same experimental setup as outline is Fig.2C was used (for G, H, and I, 2 independent experiments were performed). Representative images showing localization of (J) MDC1, (K) TOPBP1, and (L) CIP2A on the remnant of a broken chromatin bridge induced by topoisomerase inhibition in RPE1 p53−/− cells (n=3 independent experiments, a total of 161, 192, and 165 cells were measured for MDC1, TOPBP1, and CIP2A staining, respectively (percentage of cells (mean +/− SEM) with phenotype shown are indicated on the images). Yellow arrow points to the remnant of a broken bridge. (M) Cropped Frames of Sup. Videos 4 showing recruitment of TOPBP1Clover to the base of the chromatin bridge in interphase.
Extended Data Fig. 6. Fates of shattered micronuclear chromosomes during mitosis.
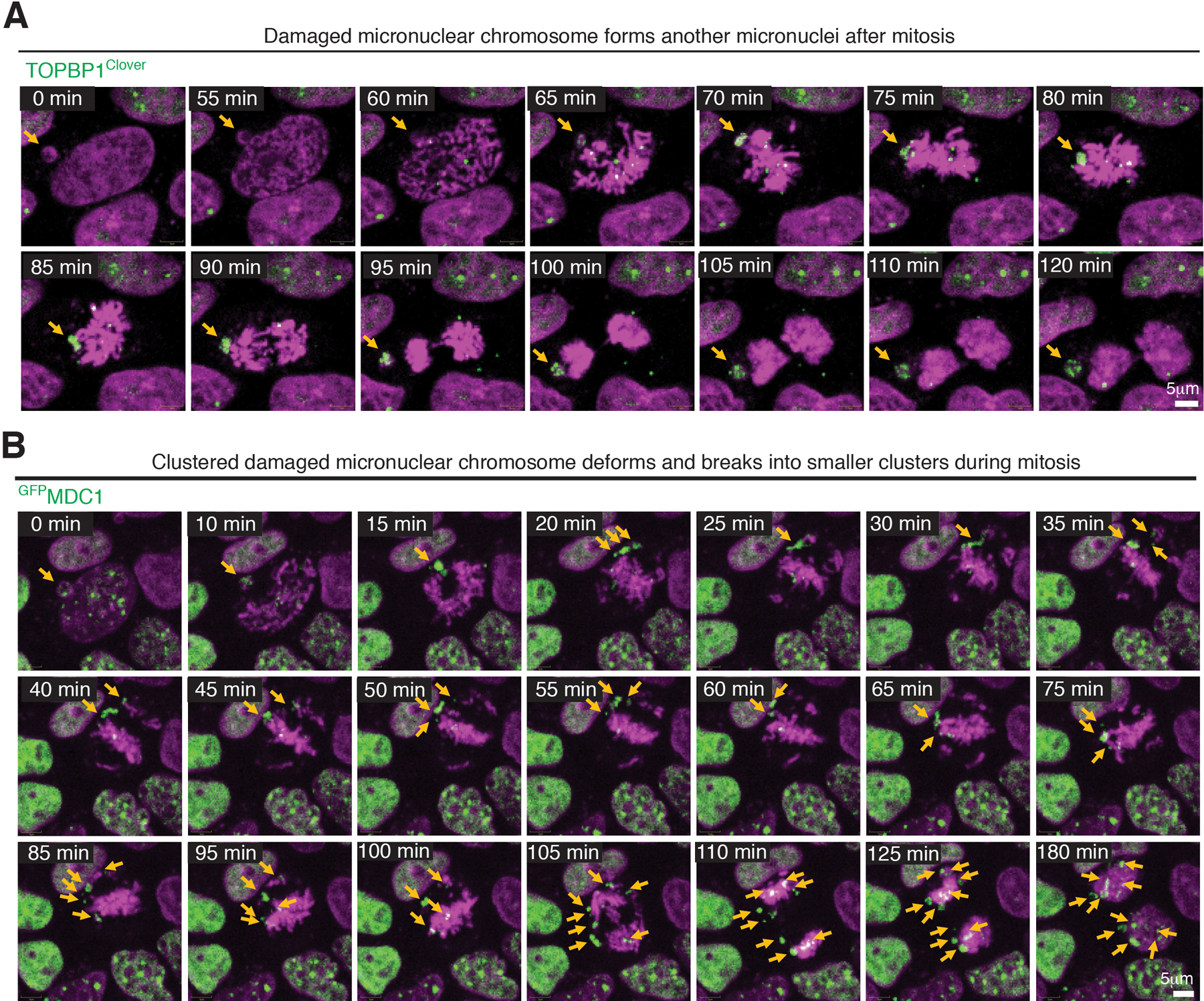
(A and B) Frames of Sup. Videos 5 and 6 showing different behaviors of micronuclear chromosomes throughout mitosis.
Extended Data Fig. 7. Role of MDC1, TOPBP1, and CIP2A in repair of a damaged micronuclear chromosome.
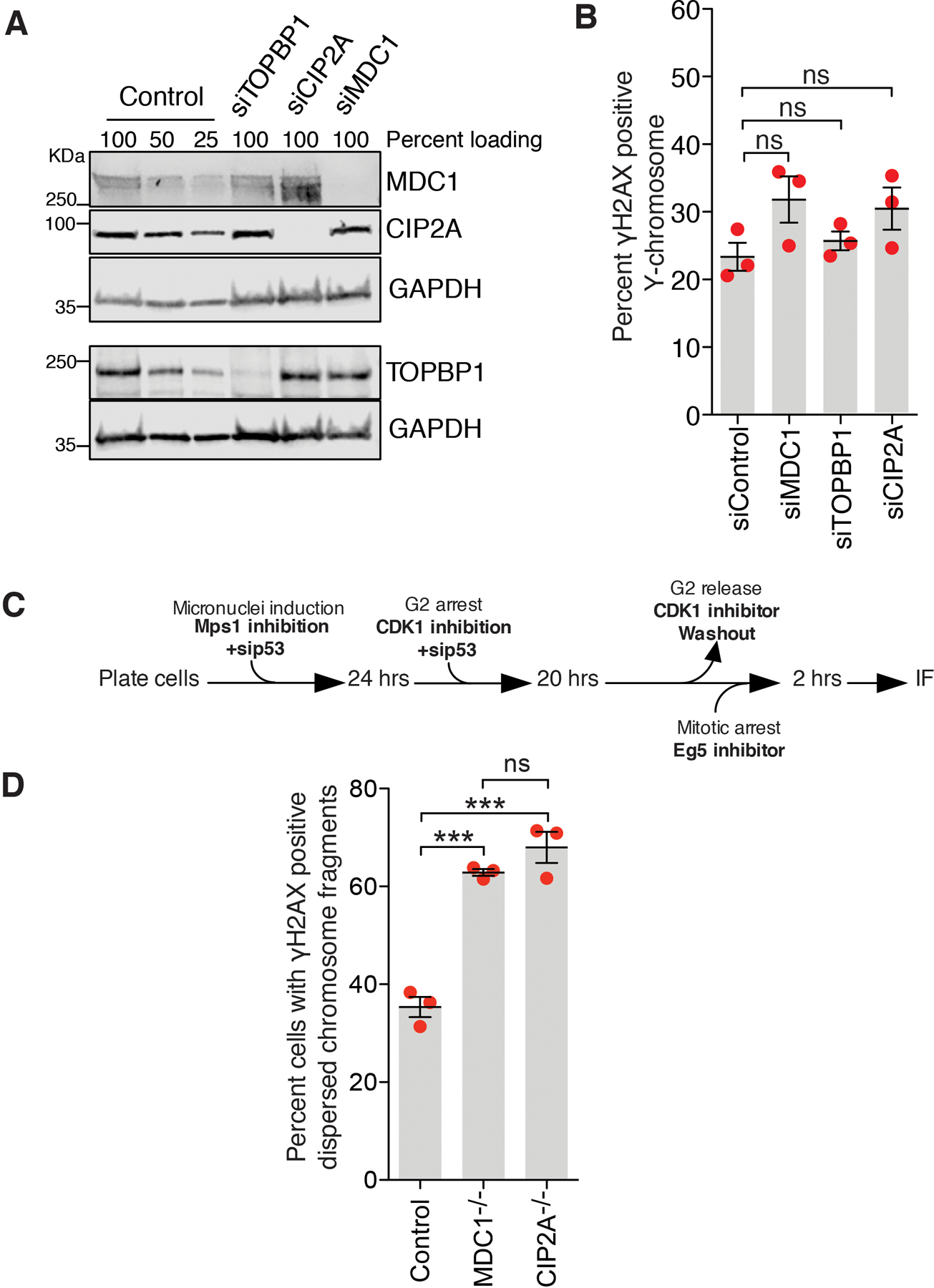
(A) Immunoblot showing depletion of TOPBP1, CIP2A, and MDC1 from the experiment shown in Fig. 3A–C (uncropped blots in Supplementary Figure 1). (B) Quantitation of damaged Y-chromosomes (tethered or dispersed) for the experiment outlined in Fig. 3A (n=3 independent experiments; number of cells analyzed is the same as in Fig. 3C). (C) Experimental outline for experiment shown in (D). (D) Quantitation of cells with dispersed micronuclear chromosome fragments in mitosis from the experiments outlined in (C). n=3 independent experiments; total 447, 345, and 385 cells were analyzed for control, MDC1−/−, and CIP2A−/− conditions, respectively. For both (B) and (D) One-way analysis of variance with Bonferroni’s multiple comparison test was applied, ns P>0.05, *** P<0.0001. For (B) and (D) mean +/− SEM are shown.
Extended Data Fig 8. Role of TOPBP1 and CIP2A in tethering fragments of damaged chromosome resulting from micronuclei or chromatin bridge during mitosis.
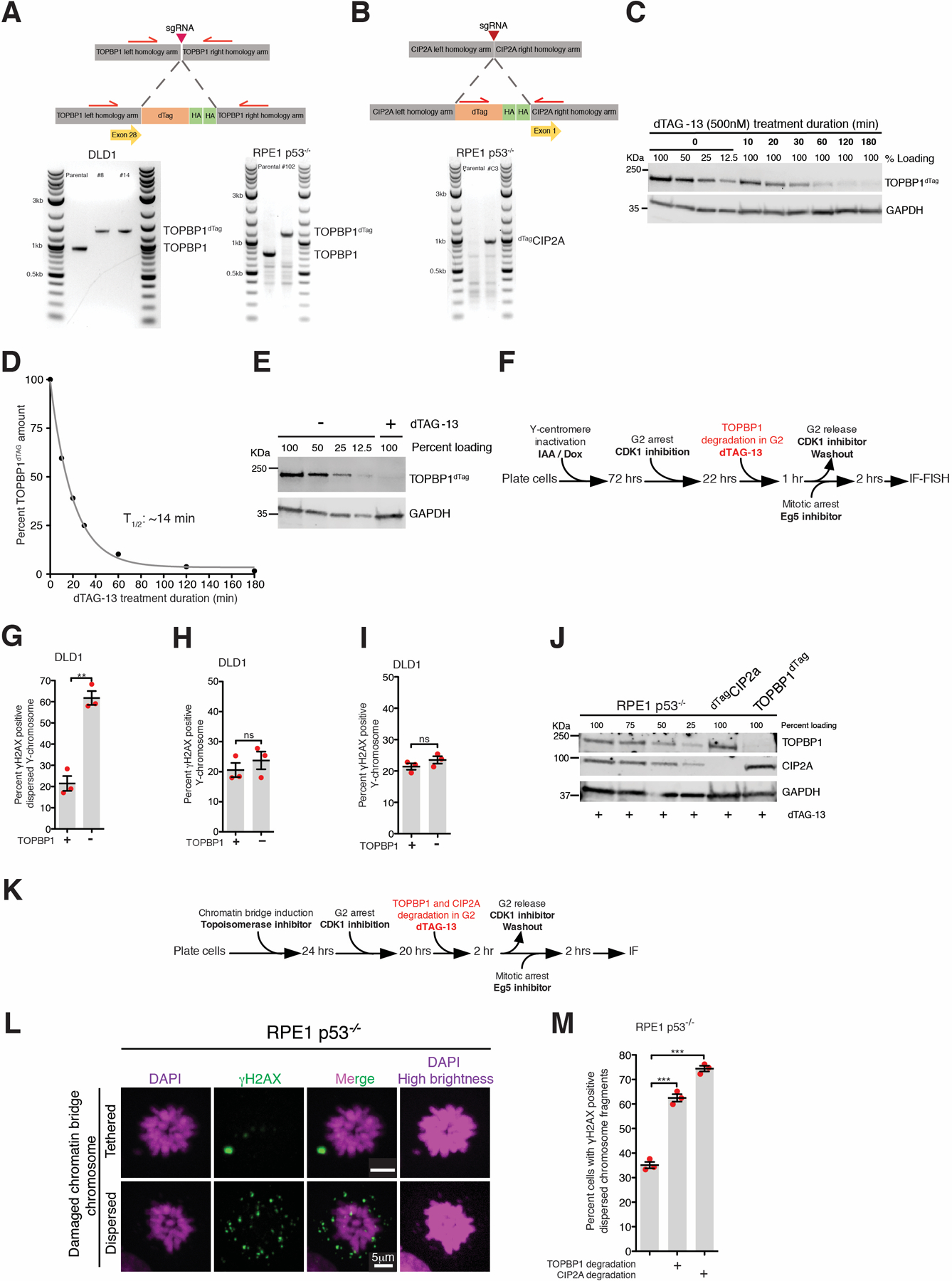
Schematic of (A) TOPBP1dTag and (B) dTagCIP2A tagging at genomic loci and an agarose gel image showing successful bi-allelic tagging. (C) Immunoblot showing dynamics of TOPBP1dTag degradation upon addition of dTAG-13 (for A, B, and C, the experiment was performed once). (D) Quantitation of TOPBP1dTag levels from (B). Data were fitted with single exponential decay curve, half-life 14 minutes. (E) Immunoblot showing depletion of TOPBP1dTag upon addition of dTAG-13 from the experiment outlined in Fig. 3D (two independent repeats were performed). (F) Experimental outline for (G) and (H). (G) Quantitation of cells with dispersed micronuclear Y-fragments in mitosis upon TOPBP1dTag degradation in G2. (H) Quantitation of cells with damaged Y-chromosome fragments (tethered or dispersed) in mitosis after degradation of TOPBP1dTag in G2 (for (G) and (H), n=3 independent experiments; total 693 and 672 cells were analyzed for control and TOPBP1dTag degradation conditions, respectively). (I) Quantitation of cells with damaged Y-fragments (tethered or dispersed) in mitosis upon degradation of TOPBP1dTag in mitosis for the experiment in Fig. 3D (the number of cells analyzed is the same as in Fig. 3E). For (G-I), mean +/− SEM are shown and two-tailed unpaired t-test was applied; *** P<0.0001 (for G P=0.0011) and ns P>0.05. (J) Immunoblot showing degradation of dTagCIP2A and TOPBP1dTag for the experiment outlined in Fig. 3F (immunoblot was performed for 2 of the 3 independent repeats). (for C, E, and J, uncropped blots in Supplementary Figure 1) (K) Experimental outline for (L) and (M). (L) Representative images of RPE p53−/− cells showing tethered and dispersed damaged micronuclear fragments in mitosis after induction of chromatin bridge by inhibition of Topoisomerase II. (M) Quantitation of cells with dispersed micronuclear fragments in mitosis following chromatin bridge induction upon degradation of TOPBP1dTag or dTagCIP2A (n=3 independent experiments; total 420, 518, and 396 cells were analyzed for control, TOPBP1dTag degradation, and dTagCIP2A degradation conditions, respectively). (Mean +/− SEM are shown and one-way analysis of variance with Bonferroni’s multiple comparison test was applied, *** P<0.0001.)
Extended Data Fig. 9. PP2A inhibition by CIP2A is not required for tethering damaged micronuclear chromosomal fragments during mitosis.
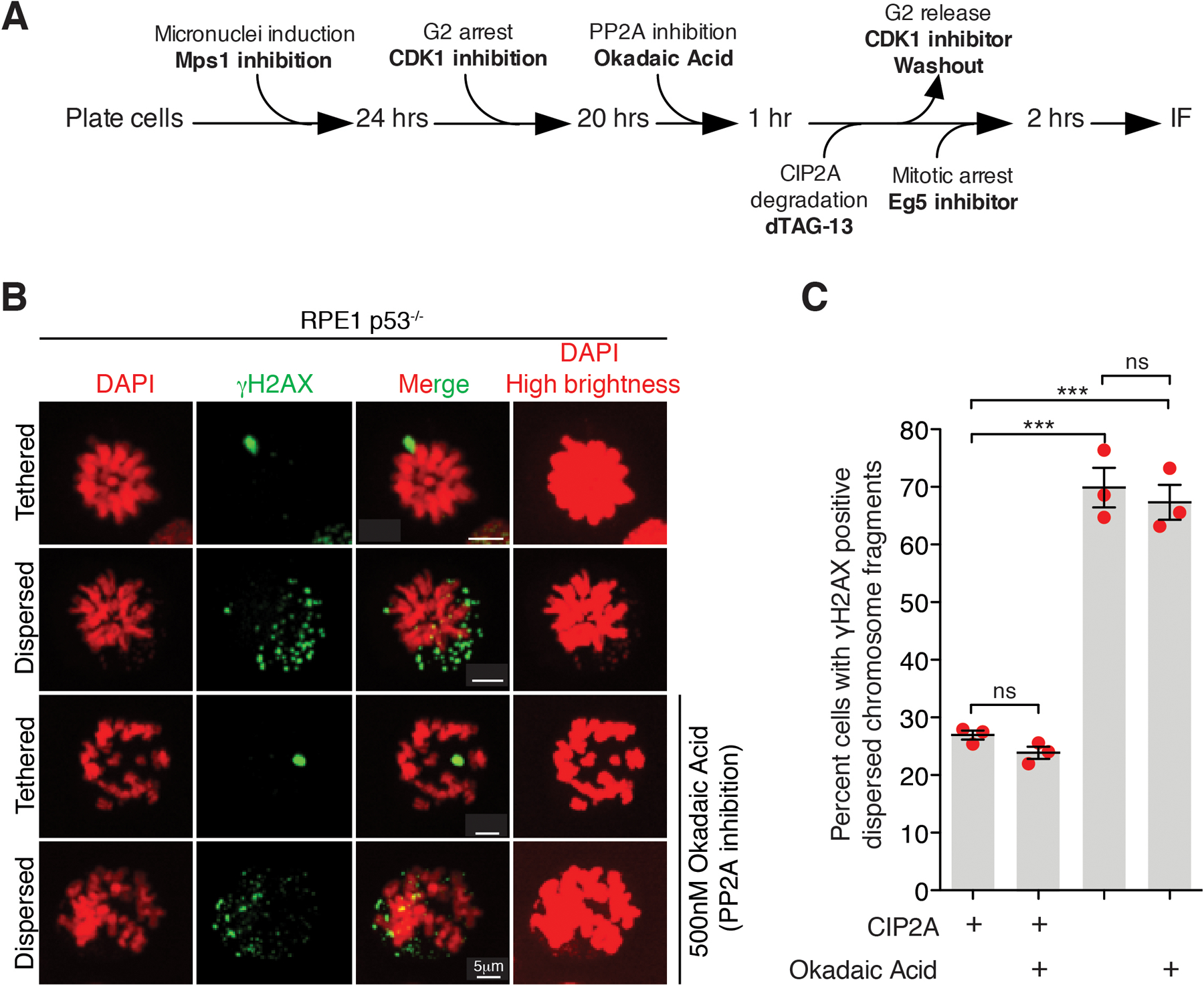
(A) Experimental outline for the experiment shown in (B) and (C). (B) Representative images of RPE p53−/− cells showing tethered and dispersed damaged micronuclear fragments in mitosis upon the indicated treatment(s). (C) Quantitation of cells with dispersed micronuclear fragments in mitosis following micronuclei induction upon degradation of dTagCIP2A (n=3 independent experiments; total 361, 281, 351, and 364 cells were analyzed for control, okadaic acid treated, dTagCIP2A degradation, and dTagCIP2A degradation with okadaic acid treated conditions, respectively). (Mean +/− SEM are shown and one-way analysis of variance with Bonferroni’s multiple comparison test was applied, *** P<0.0001 and ns P>0.05).
Extended Data Fig. 10. A complex of MDC1-TOPBP1-CIP2A recruited at γH2AX containing chromatin is the major mediator of tethering of chromosomal fragments generated from micronuclei or chromatin bridge during mitosis.
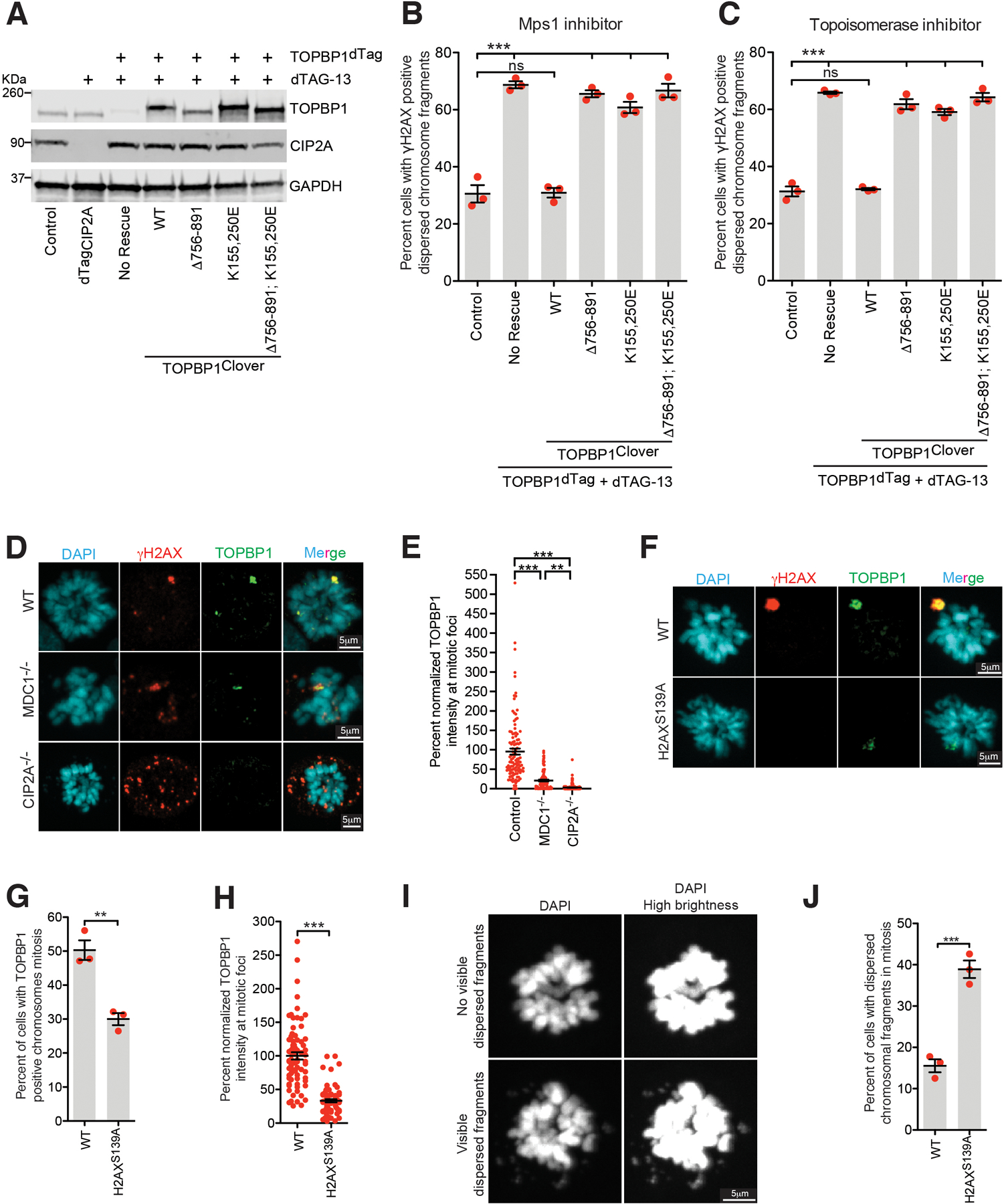
(A) Immunoblot showing degradation of dTagCIP2A and TOPBP1dTag and expression of various TOPBP1 mutants (uncropped blots in Supplementary Figure 1). Quantitation of cells with dispersed micronuclear fragments in mitosis following induction of (B) micronuclei or (C) chromatin bridge in indicated condition (n=3 independent experiments; a total 559, 608, 516, 530, 558, and 559 cells were analyzed for control, TOPBP1dTag degradation, TOPBP1WT, TOPBP1Δ756−891, TOPBP1K155,250E, and TOPBP1K155,250E; Δ756−891 conditions, respectively for (B); a total 302, 458, 190, 435, 456, and 301 cells were analyzed for control, TOPBP1dTag degradation, TOPBP1WT, TOPBP1Δ756−891, TOPBP1K155,250E, and TOPBP1K155,250E; Δ756−891 conditions, respectively for (C) ). (One-way analysis of variance with Bonferroni’s multiple comparison test was applied, ns P>0.05 *** P<0.0001.) Mean +/− SEM is shown on the graph. (D) Representative images showing localization of TOPBP1 on shattered chromosomal fragments in mitosis in RPE1 WT, MDC1−/−, and CIP2A−/− cells. (E) Graph showing TOPBP1 intensity normalized to γH2AX intensity in the indicated conditions (n= 27, 20, and 20 cells were analyzed for WT, MDC1−/−, and CIP2A−/− condition, respectively; a total of 113, 107, and 191 foci were measured for WT, MDC1−/−, and CIP2A−/− condition respectively). One-way analysis of variance with Bonferroni’s multiple comparison test was applied, ** P= 0.0035, *** P<0.0001. (F) Representative images showing localization of TOPBP1 on shattered chromosomal fragments in mitosis in RPE1 WT and H2AXS139A/S139A cells. (G) Graph showing percentage of cells showing TOPBP1 positive chromosome in mitosis following micronucleation (for (G) and (J) n=3 independent experiment, a total of 364 and 419 cells for WT and H2AXS139A/S139A condition, respectively were measured). Two-tailed unpaired t-test was applied for (G) P=0.0039, (H), and (J) P=0.0009, ** P>0.001 *** P<0.0001. (H) Graph showing TOPBP1 intensity in the indicated conditions (n= 20 cells were analyzed for WT and H2AXS139A/S139A condition, respectively; a total of 76, and 74 foci were measured for WT and H2AXS139A/S139A condition, respectively). (I) Representative images showing mitotic cell with presence or absence of dispersed chromosome fragments. (J) Graph showing percent mitotic cells with dispersed chromatin fragments in WT vs H2AXS139A/S139A cells. (We note that this is likely to be substantially underestimated since without γH2AX staining to mark chromosome fragmentation, the only available assay is observable DAPI-stained fragments found away from the bulk chromosome mass.) Mean +/− SEM is shown on the graph.
Extended Data Fig. 11. Asymmetric fragment segregation to daughter cells and fragment tethering requirement for cell viability after micronucleation.
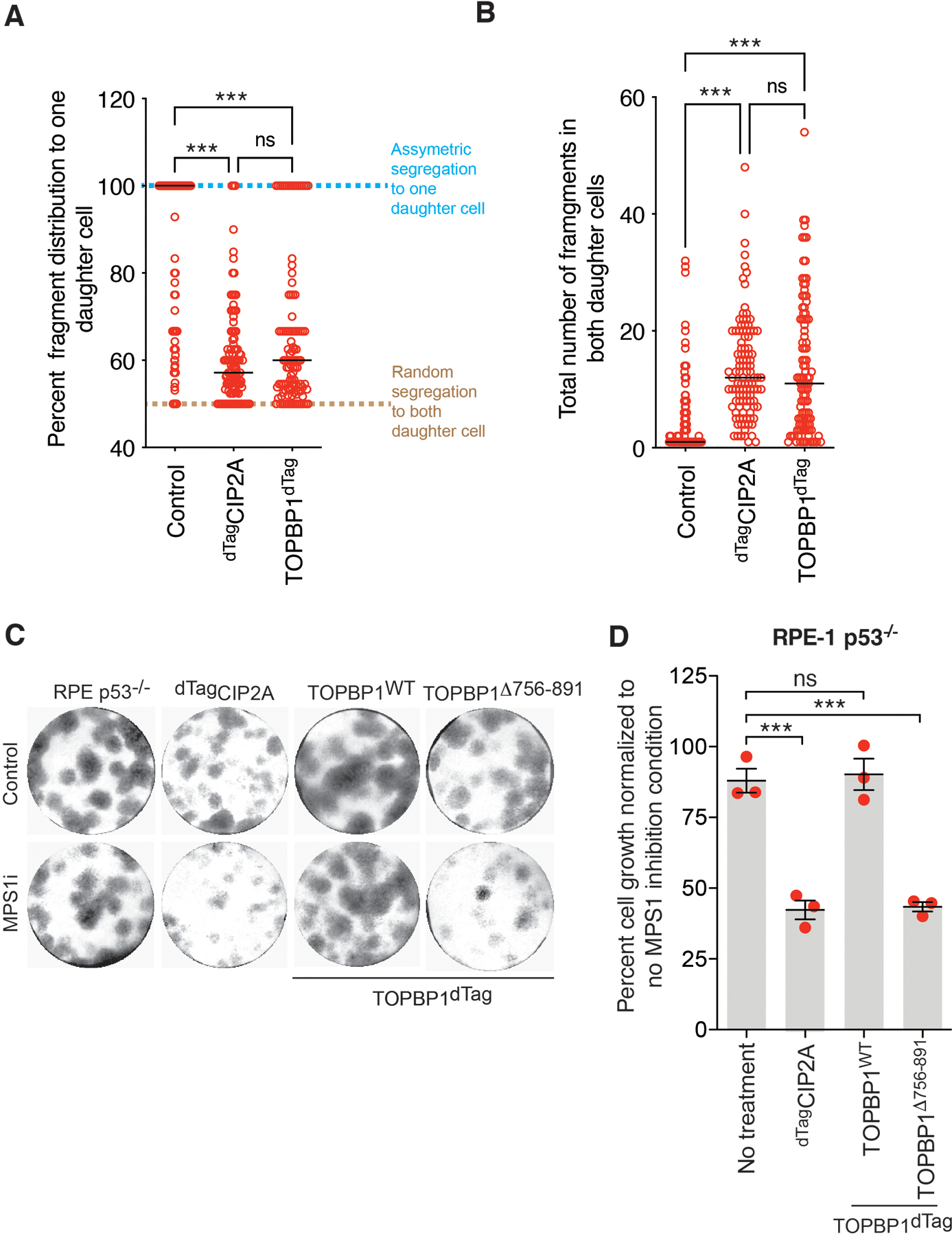
(A) Graph showing percent distribution of fragments between daughter cells in indicated condition from experiment shown in Fig. 4B–D. Percent distribution of fragments between daughter cells was calculated by dividing the number of fragments from the daughter that inherited the maximum number of fragments with the total number of fragments in both daughters and the resulting ratio was multiplied by 100. (B) Graph showing total number of fragments in both daughter cells for the indicated condition from experiment shown in Fig. 4B–D (for (A) and (B) n= 105, 107, and 107 daughter pairs for Control, dTagCIP2A, and TOPBP1dTag degradation condition, respectively were analyzed from one of the repeats of Fig. 4D). For A and B, Individual data points are shown in open red circles, median is shown as solid black line, and dashed black line shows 25th and 75th quartile. (C) Representative images of colony formed for indicated conditions. Same experimental setup as outlined in Fig.4E was used. (D) Quantitation of cell growth for the experiment in (C) (n=3 independent experiments), mean +/− SEM are shown. For (A), (B) and (D), One-way analysis of variance with Bonferroni’s multiple comparison test was applied, ns P>0.05, *** P<0.0001.
Extended Data Fig. 12. High expression of MDC1, TOPBP1, and CIP2A in tumors with a chromothriptically rearranged chromosome.
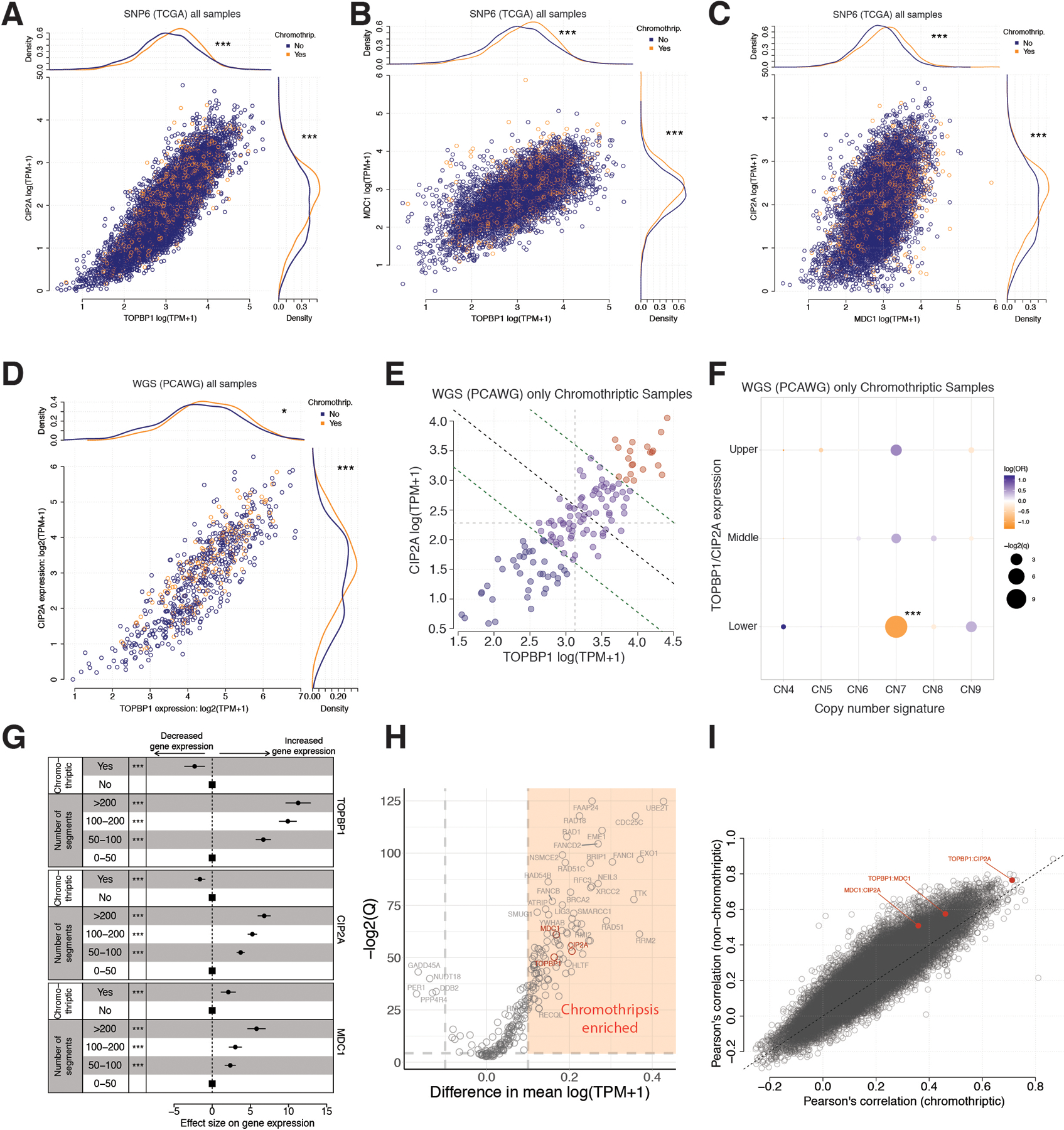
(A) Associations between gene expression (log(TPM+1)) of TOPBP1 (x-axis) and CIP2A (y-axis) as well as chromothriptic designation of samples (color) for 7,856 tumor samples from The Cancer Genome Atlas (TCGA). Main panel – scatter plot of TOPBP1 and CIP2A expression. Top panel – density of TOPBP1 expression in chromothriptic (orange) and non-chromothriptic samples (blue). Right panel – density of CIP2A expression in chromothriptic and non-chromothriptic samples. Difference in expression between chromothriptic and non-chromothriptic samples is tested with a two-sided Mann-Whitney test; ***p<0.001 (for TOPBP1 p=0, for CIP2A p=2.5e-14). Chromothripsis is defined by CTLPscanner from SNP-array derived copy number profiles. (B) Associations between gene expression (log(TPM+1)) of TOPBP1 (x-axis) and MDC1 (y-axis) as well as chromothriptic designation of samples (color) for 7,856 tumor samples from The Cancer Genome Atlas (TCGA). Main panel – scatter plot of TOPBP1 and MDC1 expression. Top panel – density of TOPBP1 expression in chromothriptic (orange) and non-chromothriptic samples (blue). Right panel – density of MDC1 expression in chromothriptic and non-chromothriptic samples. Difference in expression between chromothriptic and non-chromothriptic samples is tested with a two-sided Mann-Whitney test; ***p<0.001 (for TOPBP1 p=9.3e-57, for MDC1 p=1.1e-16). Chromothripsis is defined by CTLPscanner from SNP-array derived copy number profiles. (C) Associations between gene expression (log(TPM+1)) of MDC1 (x-axis) and CIP2A (y-axis) as well as chromothriptic designation of samples (color) for 7,856 tumor samples from The Cancer Genome Atlas (TCGA). Main panel – scatter plot of MDC1 and CIP2A expression. Top panel – density of MDC1 expression in chromothriptic (orange) and non-chromothriptic samples (blue). Right panel – density of CIP2A expression in chromothriptic and non-chromothriptic samples. Difference in expression between chromothriptic and non-chromothriptic samples is tested with a two-sided Mann-Whitney test; ***p<0.001 (for MDC1 p=2.0e-267, for CIP2A p=2.5e-14). Chromothripsis is defined by CTLPscanner from SNP-array derived copy number profiles. (D) Associations between gene expression (log(TPM+1)) of TOPBP1 (x-axis) and CIP2A (y-axis) as well as chromothriptic designation of samples (color) for 667 tumor samples from The Cancer Genome Atlas (TCGA) that overlap with Pan-cancer analysis of Whole Genomes (PCAWG) samples. Main panel – scatter plot of TOPBP1 and CIP2A expression. Top panel – density of TOPBP1 expression in chromothriptic (orange) and non-chromothriptic samples (blue). Right panel – density of CIP2A expression in chromothriptic and non-chromothriptic samples. Difference in expression between chromothriptic and non-chromothriptic samples is tested with a two-sided Mann-Whitney test; *p<0.05, ***p<0.001 (for TOPBP1 p=0.031, for CIP2A p=3.0e-4). Chromothripsis is defined by shatterSeek from WGS-derived copy number profiles. Log refers to natural log unless specified. (E) Gene expression of TOPBP1 and CIP2A (log(TPM+1)) in 135 chromothriptic tumor samples from The Cancer Genome Atlas (TCGA) that overlap with Pan-cancer Analysis of Whole Genomes (PCAWG) samples. Samples are categorized as joint upper (red), middle (purple) or lower (blue) expression of both genes by bisecting the line of best fit with a decision boundary at the upper quartile or lower quartiles of each gene (green dotted lines). Light grey dotted lines denote median values of gene expression, black dotted line indicates the bisecting line of the median. TPM=transcripts per million. Chromothriptic samples were determined by shatterSeek. (F) Associations between gene expression groups defined in (E) (y-axis), and chromothripsis associated copy number signatures (x-axis; CN4:9), using two-sided Fisher’s exact tests. Correction for multiple testing was performed using the Benjamini-Hochberg method. Color denotes odds ratio, size of points indicates the significance of the test. ***p<0.001. OR=odds ratio. CN7 is a signature associated with chromothripsis amplification. (G) Central coefficient estimates (circles) and 95% confidence intervals (horizontal lines) from linear regression models with the categorized number of segments and chromothriptic status (CTLPscanner) as predictor variables, and TOPBP1 (top), CIP2A (middle) and MDC1 (bottom) gene expression as response variables. Squares denote reference groups. X-axis=effect size of variables on gene expression. ***=p<0.001, two-sided Wald test, n=7,856. (H) Difference in gene expression (x-axis, log(TMP+1)) between chromothriptic and non-chromothriptic cancer samples from The Cancer Genome Atlas, as designated by CTLPscanner for 266 DNA repair genes (Knijnenburg et al., 2018). y-axis=significance (-log2(Q values), two-sided Mann-Whitney test). Genes labelled in red are genes of interest. Genes with >0.1 difference in mean expression and q<0.05 are considered enriched in chromothriptic samples (orange area). Q values are corrected for multiple testing using the Benjamini-Hochberg method. (I) Pearson’s correlation coefficient between all pairwise comparisons of gene pairs from 266 DNA repair genes (Knijnenburg et al., 2018) calculated in chromothriptic (x-axis) and non-chromothriptic (y-axis) samples separately, as defined by CTLPscanner. Red=gene pair involving two of TOPBP1, CIP2A or MDC1. Grey: all other gene pairs. Black dotted line: x=y. Log refers to natural log unless specified.
Extended Data Fig. 13. Model for chromothriptic chromosome formation.
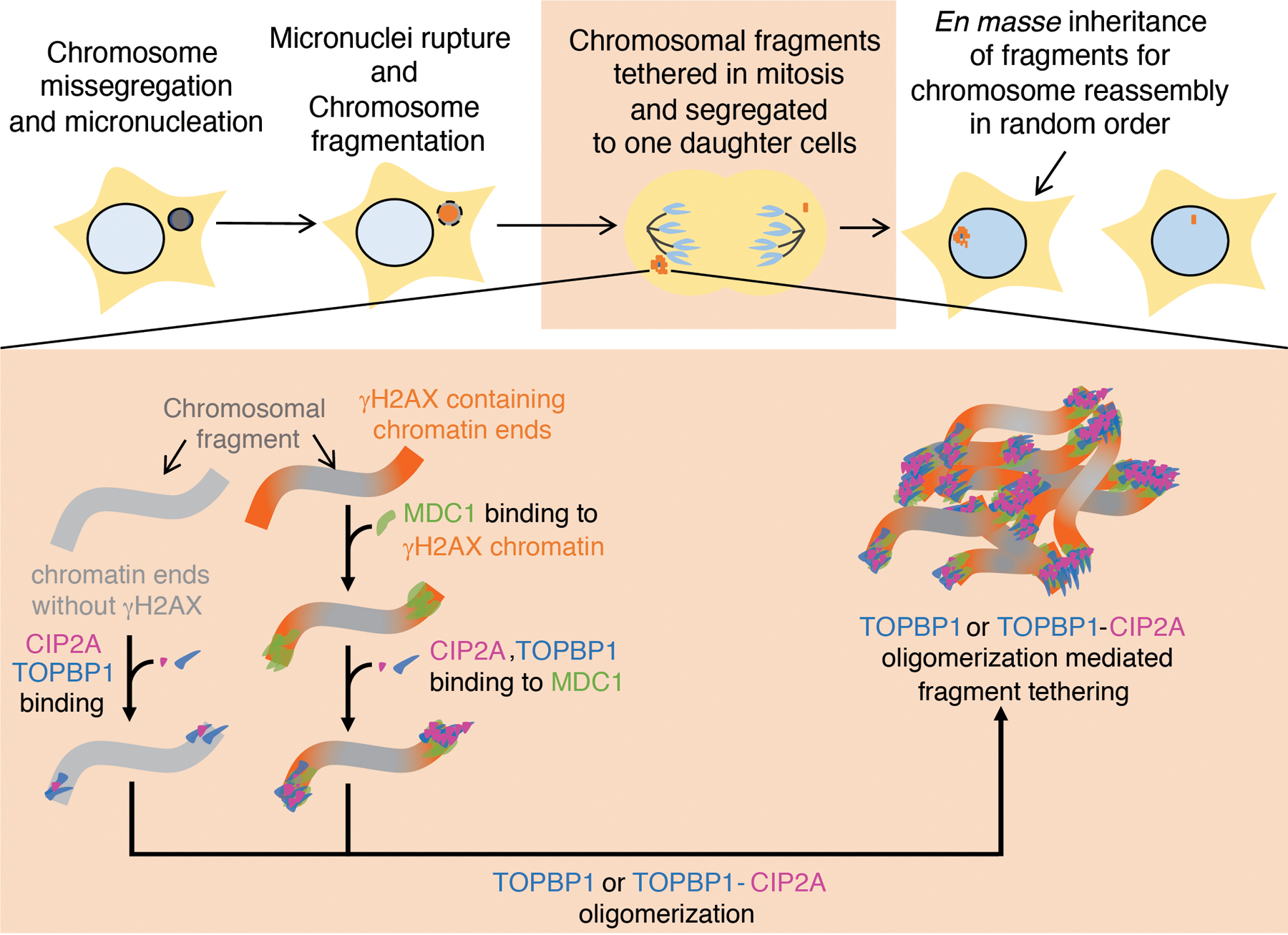
Chromosomes within micronuclei (that were formed as a result of errors in mitosis) are fragmented before or upon entry into mitosis. The chromosomal fragments (γH2AX positive or γH2AX negative) are tethered during mitosis by combined action of MDC1-TOPBP1-CIP2A or TOPBP1-CIP2A. Tethering of chromosomal fragments during mitosis ensures their transfer to the same daughter nucleus for subsequent ligation in random order, thereby resulting in reassembly of a heritable, highly rearranged chromosome.
Supplementary Material
Acknowledgements
This work was financially supported by grants from the US National Institutes of Health (R35 GM122476 to D.W.C. and R01ES030993–01A1, R01ES032547 and R01CA269919 to L.B.A.). P.T. is supported by a postdoctoral fellowship from the Hope Funds for Cancer Research (HFCR 21–04-02). L.B.A. is supported by a Packard Fellowship for Science and Engineering. The computational analyses reported in this manuscript have utilized the Triton Shared Computing Cluster at the San Diego Supercomputer Center of UC San Diego. We thank Prof. Manuel Stucki, Prof. David Pellman, and Prof. Steve Jackson for sharing reagents.
Footnotes
Competing Interests
LBA is a compensated consultant and has equity interest in io9, LLC. His spouse is an employee of Biotheranostics, Inc. LBA is an inventor of a US Patent 10,776,718 and he also declares U.S. provisional applications with serial numbers: 63/289,601; 63/269,033; 63/366,392 and 63/367,846. All other authors declare no competing interests.
Code availability
No custom algorithms were developed for the bioinformatic analysis. Data analysis code is available upon request.
Data availability
Whole genome sequencing data (FASTA files) from RPE1 cell lines generated for this study are available through SRA with accession number PRJNA961206. Gene expression data from TCGA used in this study were downloaded from the publicly available repository: https://portal.gdc.cancer.gov. Processed copy number profiles from SNP6 arrays for TCGA were downloaded from: https://github.com/VanLoo-lab/ascat/tree/master/ReleasedData/TCGA_SNP6_hg19. Chromothripsis was called from whole genome sequenced cancers using the shatterSeek algorithm by the PCAWG consortium, and downloaded from: https://dcc.icgc.org/releases/PCAWG/evolution_and_heterogeneity/clustered_mut_processes.
References:
- 1.Stephens PJ et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40, doi: 10.1016/j.cell.2010.11.055 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cortes-Ciriano I et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. N at Genet 52, 331–341, doi: 10.1038/s41588-019-0576-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang CZ et al. Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184, doi: 10.1038/nature14493 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ly P et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat Genet 51, 705–715, doi: 10.1038/s41588-019-0360-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maciejowski J et al. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat Genet 52, 884–890, doi: 10.1038/s41588-020-0667-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maciejowski J, Li Y, Bosco N, Campbell PJ & de Lange T Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163, 1641–1654, doi: 10.1016/j.cell.2015.11.054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umbreit NT et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 368, doi: 10.1126/science.aba0712 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ly P et al. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol 19, 68–75, doi: 10.1038/ncb3450 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crasta K et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58, doi: 10.1038/nature10802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato H & Sandberg AA Chromosome pulverization in human cells with micronuclei. J Natl Cancer Inst 40, 165–179 (1968). [PubMed] [Google Scholar]
- 11.Bakhoum SF & Cantley LC The Multifaceted Role of Chromosomal Instability in Cancer and Its Microenvironment. Cell 174, 1347–1360, doi: 10.1016/j.cell.2018.08.027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shoshani O et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature, doi: 10.1038/s41586-020-03064-z (2020). [DOI] [PMC free article] [PubMed]
- 13.Kloosterman WP et al. Chromothripsis is a common mechanism driving genomic rearrangements in primary and metastatic colorectal cancer. Genome Biol 12, R103, doi: 10.1186/gb-2011-12-10-r103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molenaar JJ et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589–593, doi: 10.1038/nature10910 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Teles Alves I et al. Gene fusions by chromothripsis of chromosome 5q in the VCaP prostate cancer cell line. Hum Genet 132, 709–713, doi: 10.1007/s00439-013-1308-1 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Ly P & Cleveland DW Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol 27, 917–930, doi: 10.1016/j.tcb.2017.08.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang S, Stokasimov E, Cui Y & Pellman D Breakage of cytoplasmic chromosomes by pathological DNA base excision repair. Nature, doi: 10.1038/s41586-022-04767-1 (2022). [DOI] [PMC free article] [PubMed]
- 18.Stucki M et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226, doi: 10.1016/j.cell.2005.09.038 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Clouaire T et al. Comprehensive Mapping of Histone Modifications at DNA Double-Strand Breaks Deciphers Repair Pathway Chromatin Signatures. Mol Cell 72, 250–262 e256, doi: 10.1016/j.molcel.2018.08.020 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iacovoni JS et al. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J 29, 1446–1457, doi: 10.1038/emboj.2010.38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soto M et al. p53 Prohibits Propagation of Chromosome Segregation Errors that Produce Structural Aneuploidies. Cell Rep 19, 2423–2431, doi: 10.1016/j.celrep.2017.05.055 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Santaguida S et al. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev Cell 41, 638–651 e635, doi: 10.1016/j.devcel.2017.05.022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hatch EM & Hetzer MW Linking Micronuclei to Chromosome Fragmentation. Cell 161, 1502–1504, doi: 10.1016/j.cell.2015.06.005 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Minocherhomji S et al. Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290, doi: 10.1038/nature16139 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Lobachev K, Vitriol E, Stemple J, Resnick MA & Bloom K Chromosome fragmentation after induction of a double-strand break is an active process prevented by the RMX repair complex. Curr Biol 14, 2107–2112, doi: 10.1016/j.cub.2004.11.051 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Kaye JA et al. DNA breaks promote genomic instability by impeding proper chromosome segregation. Curr Biol 14, 2096–2106, doi: 10.1016/j.cub.2004.10.051 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Clay DE, Bretscher HS, Jezuit EA, Bush KB & Fox DT Persistent DNA damage signaling and DNA polymerase theta promote broken chromosome segregation. J Cell Biol 220, doi: 10.1083/jcb.202106116 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Jager M et al. Human Rad50/Mre11 is a flexible complex that can tether DNA ends. Mol Cell 8, 1129–1135, doi: 10.1016/s1097-2765(01)00381-1 (2001). [DOI] [PubMed] [Google Scholar]
- 29.De Marco Zompit M et al. The CIP2A-TOPBP1 complex safeguards chromosomal stability during mitosis. Nat Commun 13, 4143, doi: 10.1038/s41467-022-31865-5 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leimbacher PA et al. MDC1 Interacts with TOPBP1 to Maintain Chromosomal Stability during Mitosis. Mol Cell 74, 571–583 e578, doi: 10.1016/j.molcel.2019.02.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adam S et al. The CIP2A-TOPBP1 axis safeguards chromosome stability and is a synthetic lethal target for BRCA-mutated cancer. Nat Cancer 2, 1357–1371, doi: 10.1038/s43018-021-00266-w (2021). [DOI] [PubMed] [Google Scholar]
- 32.Laine A et al. CIP2A Interacts with TopBP1 and Drives Basal-Like Breast Cancer Tumorigenesis. Cancer Res 81, 4319–4331, doi: 10.1158/0008-5472.CAN-20-3651 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wardlaw CP, Carr AM & Oliver AW TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair (Amst) 22, 165–174, doi: 10.1016/j.dnarep.2014.06.004 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Nabet B et al. The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol 14, 431–441, doi: 10.1038/s41589-018-0021-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JE, McAvoy SA, Smith DI & Chen J Human TopBP1 ensures genome integrity during normal S phase. Mol Cell Biol 25, 10907–10915, doi: 10.1128/MCB.25.24.10907-10915.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bagge J, Oestergaard VH & Lisby M Functions of TopBP1 in preserving genome integrity during mitosis. Semin Cell Dev Biol 113, 57–64, doi: 10.1016/j.semcdb.2020.08.009 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Gallina I, Christiansen SK, Pedersen RT, Lisby M & Oestergaard VH TopBP1-mediated DNA processing during mitosis. Cell Cycle 15, 176–183, doi: 10.1080/15384101.2015.1128595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pedersen RT, Kruse T, Nilsson J, Oestergaard VH & Lisby M TopBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J Cell Biol 210, 565–582, doi: 10.1083/jcb.201502107 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Junttila MR et al. CIP2A inhibits PP2A in human malignancies. Cell 130, 51–62, doi: 10.1016/j.cell.2007.04.044 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Hoadley KA et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–304 e296, doi: 10.1016/j.cell.2018.03.022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J et al. CTLPScanner: a web server for chromothripsis-like pattern detection. Nucleic Acids Res 44, W252–258, doi: 10.1093/nar/gkw434 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Consortium ITP-CA o. W. G. Pan-cancer analysis of whole genomes. Nature 578, 82–93, doi: 10.1038/s41586-020-1969-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steele CD et al. Signatures of copy number alterations in human cancer. Nature 606, 984–991, doi: 10.1038/s41586-022-04738-6 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Groelly FJ, Fawkes M, Dagg RA, Blackford AN & Tarsounas M Targeting DNA damage response pathways in cancer. Nat Rev Cancer 23, 78–94, doi: 10.1038/s41568-022-00535-5 (2023). [DOI] [PubMed] [Google Scholar]
- 45.Papathanasiou S et al. Transgenerational transcriptional heterogeneity from cytoplasmic chromatin. bioRxiv, 2022.2001.2012.475869, doi: 10.1101/2022.01.12.475869 (2022). [DOI]
- 46.Frattini C et al. TopBP1 assembles nuclear condensates to switch on ATR signaling. Mol Cell 81, 1231–1245 e1238, doi: 10.1016/j.molcel.2020.12.049 (2021). [DOI] [PubMed] [Google Scholar]
- 47.Kim A et al. Biochemical analysis of TOPBP1 oligomerization. DNA Repair (Amst) 96, 102973, doi: 10.1016/j.dnarep.2020.102973 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Korbel JO & Campbell PJ Criteria for inference of chromothripsis in cancer genomes. Cell 152, 1226–1236, doi: 10.1016/j.cell.2013.02.023 (2013). [DOI] [PubMed] [Google Scholar]
- 49.Khanna A & Pimanda JE Clinical significance of cancerous inhibitor of protein phosphatase 2A in human cancers. Int J Cancer 138, 525–532, doi: 10.1002/ijc.29431 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Knijnenburg TA et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep 23, 239–254 e236, doi: 10.1016/j.celrep.2018.03.076 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Whole genome sequencing data (FASTA files) from RPE1 cell lines generated for this study are available through SRA with accession number PRJNA961206. Gene expression data from TCGA used in this study were downloaded from the publicly available repository: https://portal.gdc.cancer.gov. Processed copy number profiles from SNP6 arrays for TCGA were downloaded from: https://github.com/VanLoo-lab/ascat/tree/master/ReleasedData/TCGA_SNP6_hg19. Chromothripsis was called from whole genome sequenced cancers using the shatterSeek algorithm by the PCAWG consortium, and downloaded from: https://dcc.icgc.org/releases/PCAWG/evolution_and_heterogeneity/clustered_mut_processes.