

Abstract
Amyloid oligomers and fibrils are protein aggregates that cause an onset and progression of many neurodegenerative diseases, diabetes type 2 and systemic amyloidosis. Although a growing body of evidence shows that oligomers and fibrils trigger mitochondrial dysfunction simultaneously enhancing production of reactive oxygen species, exact mechanisms by which these protein aggregates exert their toxicities remain unclear. In this study, we used advanced microscopic and spectroscopic methods to examine topography and structure of insulin aggregates grown in the lipid-free environment, as well as in the presence of major classes of phospho- and sphingolipids. We also employed a set of molecular markers to determine the extent to which insulin aggregates induce a damage of cell endoplasmic reticulum (ER), an important cell organelle used for calcium storage, protein synthesis and folding. Our results show that insulin aggregates activate the expression of Activating Transcription Factor 6 (ATF6), a transmembrane protein that is involved in unfolded protein response (UPR) of the stressed ER. At the same time, two other ER transmembrane proteins, Inositol Requiring 1 (IRE1α) and eLF2a, the product of PKR-like ER kinase (PERK), exhibited very low expression levels. Furthermore, amyloid aggregates trigger an expression of the 78-kDa glucose-regulated protein GRP78, which is also involved in the UPR. We also observed UPR-induced expression of a proapoptotic transcription factor CHOP, which, in turn, regulates expression of caspase 3 kinase and BCL2 protein family members, including the ER localized Bax. These findings show that insulin oligomers and fibrils induce UPR-associated ER stress and ultimately fatal changes in cell homeostasis.
Keywords: Endoplasmic reticulum, Amyloids, Unfolded protein response, Lipids, AFM-IR
1. Introduction
Abrupt aggregation misfolded proteins into oligomers and fibrils is the molecular signature of a large group of pathologies that include type 2 diabetes [1], Alzheimer [2], Parkinson [3]and Huntington diseases [4–7]. A growing body of evidence suggests that such aggression is taken place in endosomes that accumulate misfolded proteins in high concentrations [8–10]. These cell incisions have acidic pH that favors formation of highly toxic oligomers that decrease spontaneous calcium oscillations in neurons [10]. Amyloid oligomers and fibrils can be endocytosed by cells and appear in the cytosol by the direct permeabilization of cell membranes [11,12]. This results in mitochondrial disfunction and enhanced levels of reaction oxygen species, which ultimately leads to the cell apoptosis [13–16].
Endoplasmic reticulum (ER) plays vitally important role in protein synthesis and folding; it also serves as the calcium storage of the cell [17]. Endosomal localization of both endocytosed and endosome-formed amyloids, as well as previously reported ER-endosomes contact sites, suggest that amyloid toxicity can induce the ER stress and disfunction, which, in turn, can lead to the cell death [18,19]. The ER stress activates a set of signaling pathways, known as the unfolded protein response (UPR), to mitigate the ER stress and restore homeostasis, Fig. 1 [20]. The UPR is induced by three ER transmembrane proteins: Inositol Requiring 1 (IRE1), PKR-like ER kinase (PERK), and Activating Transcription Factor 6 (ATF6) [21–23]. These proteins are regulated by 78-kDa glucose-regulated chaperon GRP78, also known as BiP [18,19]. Under ER stress, GRP78 dissociates from IRE1, PERK and ATF6, which results in their activation [24,25].
Fig. 1.
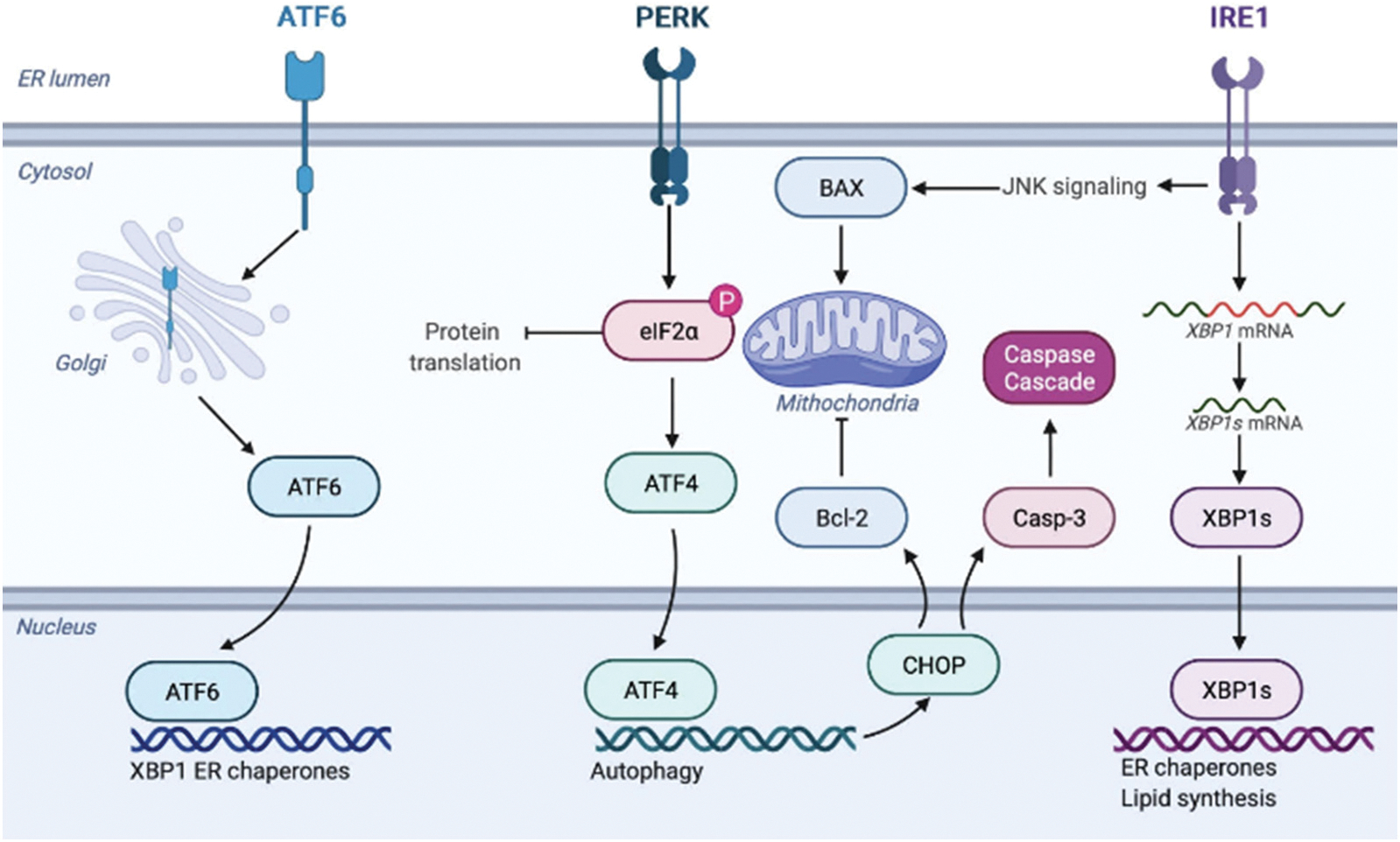
Schematic illustration of UPR stress response by ER.
Alpha isoform of IRE1 (IRE1α) is a type I ER transmembrane kinase that senses unfolded or misfolded proteins by its N-terminal luminal domain [26]. Upon activation, IRE1α dimerizes and auto-phosphorylates splicing X-box binding protein 1 (XBP-1) mRNA [27–29]. Spliced XBP1 mRNA encodes a basic leucine zipper (b-ZIP) transcription factor that upregulates UPR target genes, as well as genes that encode folding proteins such as protein disulfide isomerase and chaperones [27–29]. XBP1 also activates the expression of a proapoptotic activating transcription factor ATF4, which in turn activates C/EBP Homologous Protein (CHOP) that regulates the expression of caspase 3 kinase and BCL2 protein family members [30,31]. It should be noted that IRE1α also activates c-Jun N-terminal protein kinase (JNK), which in turn, induces cell apoptosis by regulating the BCL2 family of proteins, including the ER- and mitochondria-localized Bax [31,32].
ER stress also induces oligomerization and auto-phosphorylation of PERK, a type I ER transmembrane kinase, which engages subunit of eukaryotic initiation factor 2 (eIF2a) by phosphorylation of its Ser51 residue [26–29]. Phosphorylated eIF2a prevents the formation of ribosomal complexes that are involved in initiation of mRNA translation [26]. This suppresses protein expression, and consequently, ER workload. Furthermore, ER stress activates expression of type II ER transmembrane transcription factor, ATF6 that migrates to the Golgi where it is cleaved by proteases to generate bZIP factor [18,19]. BZIP factor then translocates to the nucleus, where it activates the UPR genes involved in the protein folding, processing, and degradation [18,19].
Quantitative polymerase chain reaction (qPCR) can be used to monitor the level of expression of the described above ER stress factors [33]. For instance, using this approach, Fonseca and co-workers were able to quantify the expression levels of spliced and unspliced XBP-1 in different cell lines [34]. In this study, we used qPCR approach to examine the levels of ER stress caused by insulin fibrils grown in the lipid-free environment, as well as insulin aggregates grown in the presence of phospho- and sphingolipids. In our previous study, we demonstrated that phospholipids cardiolipin (CL) and phosphatidylcholine (PC) drastically altered secondary structure and toxicity of insulin aggregates [35]. Specifically, in the presence of PC, insulin yielded only small spherical aggregates that had predominantly unordered protein secondary structure and significantly lower cell toxicity compared to insulin fibrils grown in the lipid-free environment. Insulin aggregation in the presence of CL enables integration of the lipid into the structure of corresponding oligomers and fibrils which lowers their toxicity. These findings show that lipids can uniquely alter the secondary structure and consequently toxicity of amyloid aggregates.
In this work, we examine the extent to which insulin fibrils grown in the lipid-free environment, as well as in the presence of PC, CL, phosphatidylserine (PS), ceramide (CER) and sphingomyelin (SM) exert the ER stress in rat dopaminergic neural cells. These lipids occupy a significant part of lipid membranes in most of eukaryotic cells b [36–39]. Specifically, PC is the most observed lipid that takes up to 47 % of the membranes, whereas the ~10 % of the organelle and plasma membranes are occupied by PS. Although the contributions of CER and SM are smaller, these lipids take up to 6 % and 4 % of eukaryotic plasma membranes, respectively [36–39]. CL is unique to the inner mitochondrial membranes where it constitutes about 20 % of all present lipids. It should be noted that all chosen lipids for this work possessed C16-C18 saturated fatty acids (FA). Although lipids with unsaturated FA also play an important role in membrane biophysics, elucidation of their impact on insulin aggregation is the subject for a separate study. We also examine the levels of ER stressed caused by insulin aggregates that were formed in the mixture of PC and CL (1:1 and 4:1 M rations). These ratios correspond to the relative amount of PC and CL in inner mitochondrial membrane. In this study, we also determine molecular mechanisms that are involved in the amyloid-induced stress.
2. Experimental section
2.1. Materials
1,2-Ditetradecanoyl-sn-glycero-3-phospho-l-serine (DMPS or PS), 1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC or PC), 1’,3′-bis [1,2-distearoyl-sn-glycero-3-phospho]-glycerol (18:0 cardiolipin (CL)), sphingomyelin (SM) and ceramide (CER) were purchased from Avanti (Alabaster, AL, USA).
2.2. Liposome and fibril preparation
DMPS, DMPC and CL large unilamellar vesicles (LUVs) were prepared accordingly to the method reported by Galvagnion et al. [40]. Briefly, 0.6 mg of the lipid were dissolved in 2.6 ml of phosphate buffered saline (PBS) pH 7.4. Lipid solutions were heated in water bath to ~50 °C for 30 min and then placed into liquid nitrogen for 3–5 min. This procedure was repeated 10 times. After this, lipid solutions were passed 15 times through a 100 nm membrane that was placed into the extruder (Avanti, Alabaster, AL, USA). LUV sizes were determined by dynamic light scattering. Due to the poor assembly properties, no LUVs for SM and CER were prepared; lipids used as received.
In the lipid-free environment, 400 μM of insulin was dissolved in PBS; solution pH was adjusted to pH 3.0 using concentrated HCl. For Ins: CL, Ins:CER, Ins:PS and Ins:SM, as well as for Ins:PC:CL (1:0.5:0.5) and Ins:PC:CL (1:0.8:0.2), 400 μM of insulin was mixed with an equivalent concentration of the corresponding lipid; solution pH was adjusted to pH 3.0 using concentrated HCl. Next, the solutions were placed in the plate reader (Tecan, Männedorf, Switzerland) and incubated at 37 °C under 510 rpm for 24 h.
2.3. Atomic force microscopy (AFM) imaging
AFM imaging was performed using silicon AFM probes with related parameters force constant 2.7 N/m and resonance frequency 50–80 kHz were purchased from Appnano (Mountain View, CA, USA) on AIST-NT-HORIBA system (Edison, NJ). For each measurement, an aliquot of the sample was diluted with 1× PBS, pH 3.0, deposited onto pre-cleaned silicon wafer and dried under a flow of dry nitrogen. At least two independently prepared protein samples were measured by AFM to ensure full reproducibility of the reported results. We analyzed at least 4 large areas (5 × 5 μm) on each sample to ensure that reported AFM results are representative for the sample. This large-field imaging is also performed to ensure that we do not overlook any long fibrils or fibril bundles that can be present in the samples of protein aggregates. For each sample, at heights and lengths of at least 100 different aggregates were measured using AIST-NT software (Edison, NJ, USA).
2.4. Atomic force microscopy – infrared (AFM-IR) spectroscopy
AFM-IR imaging was conducted using a Nano-IR3 system (Bruker, Santa Barbara, CA, USA). The IR source was a QCL laser. Contact-mode AFM tips (ContGB-G AFM probe, NanoAndMore, Watsonville, CA, USA) were used to obtain all spectra. From 18 to 30 individual aggregates were analyzed for each sample. Treatment and analysis of collected spectra was performed in Matlab (The Mathworks, Inc. Natick, Massachusetts, USA).
2.5. QPCR
N27 Rat dopaminergic neural cells were grown in RPMI 1640 Medium (Thermo Fisher Scientific, Waltham, MA, USA) with 10 % fetal bovine serum (FBS) (Invitrogen, Waltham, MA, USA) in 96 well-plate (5000 cells per well) at 37 °C under 5 % CO2. After 24 h, the cells were found to fully adhere to the wells reaching ~70 % confluency. Next, insulin aggregates were added and incubated for 4 h, 14 h and 24 h.
After incubation, the cells were collected for the isolation of ribonucleic acid (RNA) by extraction using Trizol (Invitrogen). To determine the presence and amount of RNA, we used agarose gel electrophoresis and the agarose gel documentation system, as well as NanoDrop. Next, we synthesized coding deoxyribonucleic acid (DNA) using reverse transcriptase, a set of SuperScript III (Invitrogen). A real-time polymerase chain reaction (PCR) was performed with primers corresponding to the selected genes. SYBR Master Mix (Thermo Fisher Scientific) was used for PCR of the mixture. PCR was performed with primers from Table 1 in the CFX96 Real-Time System (Bio-Rad) device.
Table 1.
Primer sequences used for qPCR analysis of changes in the expression of ER-stress associated genes.
| Gene | Primers 3′-5′ |
|---|---|
|
| |
| ATF6_F | CAAGACCGAAGATGTCCATTGTG |
| ATF6_R | ATCCTGGTGTCCATGACCTGA |
| BAXa_F | TGACATGTTTTCTGACGGC |
| BAXa_R | CAGCCCATCTTCTTCCAGA |
| GRP78_F | CGGCAGCTGCTATTGCTTA |
| GRP78_R | CCATGACACGCTGGTCAAA |
| XBP1_F | CTGGTTGCTGAAGAGGAGG |
| XBP1_R | CATGGGGAGATGTTCTGGAG |
| ATF4_F | CTAAGGCGGGCTCCTCCGA |
| ATF4_R | CCCAACAGGGCATCCAAGTCG |
| CHOP_F | ACTCTCCAGATTCCAGTCAG |
| CHOP_R | GCCGTTCATTCTCTTCAGC |
| IRE1a_F | AAACTACGCCTCCCCTGTG |
| IRE1a_R | CTAGATAGCGCAGGGTCTC |
| eIf2a_F | GCATATAGTGGAAAGGTGAGGT |
| eIf2a_R | GAGGTCCGACAGCTCTAAC |
| GAPDH_F | GCACAGTCAAGGCTGAGAATG |
| GAPDH_R | TGGTGGTGAAGACGCCAGTA |
| β-Actin_F | TGAAGTCTGACGTGGACATC |
| β-Actin_R | ACTCGTCATACTCCTGCTTG |
Each sample was performed in a triplet. Then we plotted the average CT values from each sample compared to the absolute amount of the control gene, the so-called housekeeping genes, in our case it was GAPDH and β-Actin, to create a standard curve. This comparison of experimental CT data with the control gene gives the value of the number of genes of interest to us that are present in the cells. For data analysis, the values of samples were selected that exhibited a growth of up to 35 cycles, control genes - up to 25 cycles. In each triplet, the variation between repetitions was up to 0.5 cycles. When analyzing the data, we used the −2ΔCT method.
3. Results
3.1. Microscopic analysis of insulin aggregates
Using AFM, we performed morphologic characterization of insulin aggregates grown in the presence of lipids, as well as in the lipid-free environment, Fig. 2. Insulin fibrils grown in the lipid-free environment (Ins) stretch microns in length and have 10–12 nm in height, Supporting Figs. 1 and 2. Insulin aggression in the presence of CL yields short (30–60 nm) spherical aggregates and ~200 nm long fibrils that had 6–8 nm in height, Supporting Figs. 1 and 2. Similar aggregates (5–11 nm in height) were found in Ins:CER, Ins:PS and Ins:SM. At the same time, we found that insulin aggression in the presence of PC yields only small (4–6 nm in height) oligomers. Similar structures were observed in Ins: PC:CL (1:0.8:0.2). Nevertheless, in Ins:PC:CL (1:0.8:0.2), we also observed short fibril-like structures. We also found that with an increase in the concentration of CL relative to PC (Ins:PC:CL (1:0.5:0.5)), short (40–70 nm) spherical aggregates and 90–160 nm long fibrils that have ~12 nm in height were grown, Supporting Figs. 1 and 2. These fibrils exhibited similar topologies with those formed by insulin in the presence of CL itself. Summarizing, we found that lipids drastically alter the morphology of insulin aggregates if present at the stage of protein aggregation. Lipids either shorten the length of the grown fibrils (CL, SP, CER and SM) or fully inhibit (PC) fibril formation. PC also exerts the inhibitory activity in the mixtures with CL. We found that with an increase in the relative ration of PC relative to CL (Ins:PC:CL (1:0.8:0.2) vs Ins:PC:CL (1:0.5:0.5)), stronger inhibition of fibril formation is observed.
Fig. 2.

Lipids uniquely alter morphologies of insulin aggregates. AFM images of Ins (A), Ins:PS (B), Ins:PC (C), Ins:CL (D), Ins:PC:CL (1:0.5:0.5) (E), Ins:PC:CL (1:0.8:0.2) (F), Ins:SM (G) and Ins:CER (H) aggregates. Scale bars are 200 nm.
3.2. Structural analysis of protein aggregates
One may expect that lipids not only alter the morphology of insulin aggregates but also change their secondary structure. To answer this question, we used AFM-IR spectroscopy. In AFM-IR, a metalized scanning probe can be placed on the surface of individual aggregates. Illumination of the sample with pulsed tunable IR light induces thermal expansions in the sample are recorded by the scanning probe [41–43]. In addition to elucidation of the secondary structure of individual oligomers, this innovative analytical approach can be also used to reveal presence of lipids in their structure [44–47].
AFM-IR spectra collected from individual insulin fibrils that were formed in the lipid-free environment exhibited amide I (1630–1690 cm−1) and amide II (1500–1570 cm−1) which originated from the peptide bond vibrations [14]. The position of amide I band can be used for a quantitative analysis of the secondary structure of protein aggregates. If the amide I is centered around 1655 cm−1 and ~1695 cm−1, high α-helical and anti-parallel β-sheet secondary structure can be expected, respectively [14,48,49], Supporting Fig. 3. If the secondary structure of the aggregates is dominated by parallel β-sheet, amide I band in the corresponding AFM-IR spectra will be centered ~1620 cm−1, whereas the shift of this vibration to 1660 cm−1 is indicative of the unordered protein [14]. In the AFM-IR spectra collected form insulin fibrils that were grown in the lipid-free environment, amide I band was centered around 1625 cm−1, which indicated the predominance of the parallel β-sheet in their structure, Supporting Figs. 3 and 4. AFM-IR analysis of Ins:PC and PC:CL (1:0.8:0.2) oligomers revealed that their secondary structure is dominated by unordered protein. We also found that AFM-IR spectra collected from individual Ins:PC and PC:CL (1:0.8:0.2) oligomers exhibited vibrational bands centered ~800 and 1000–1200 cm−1, Fig. 3. These vibrational bands correspond to C–H and vibration respectively [50]. These findings show that Ins:PC and PC:CL (1:0.8:0.2) oligomers possess lipids in their structure. Similar conclusions about the lipid content could be made for all analyzed insulin aggregates that were grown in the presence of lipids (Ins:PS, Ins: CL, Ins:CER, Ins:SM and Ins:PC:CL (1:0.5:0.5)) [51]. However, these samples exhibited higher degree of variability of the aggregates from perspective of their secondary structure. Specifically, we found that Ins: PS oligomers exhibited high amount of unordered protein and α-helix in their secondary structure, whereas Ins:PS fibrils were primarily composed of parallel β-sheet, Supporting Fig. 3. Similar structural variability between oligomers and fibrils has been observed for Ins:CL, Ins:SM, Ins:CER and Ins:PC:CL (1:0.5:0.5). Specifically, we found that Ins:CL, Ins:SM, Ins:CER and Ins:PC:CL (1:0.5:0.5) oligomers had predominantly unordered protein and α-helix in their secondary structure, whereas fibrils observed in these samples were dominated by parallel β-sheet, Supporting Fig. 3.
Fig. 3.
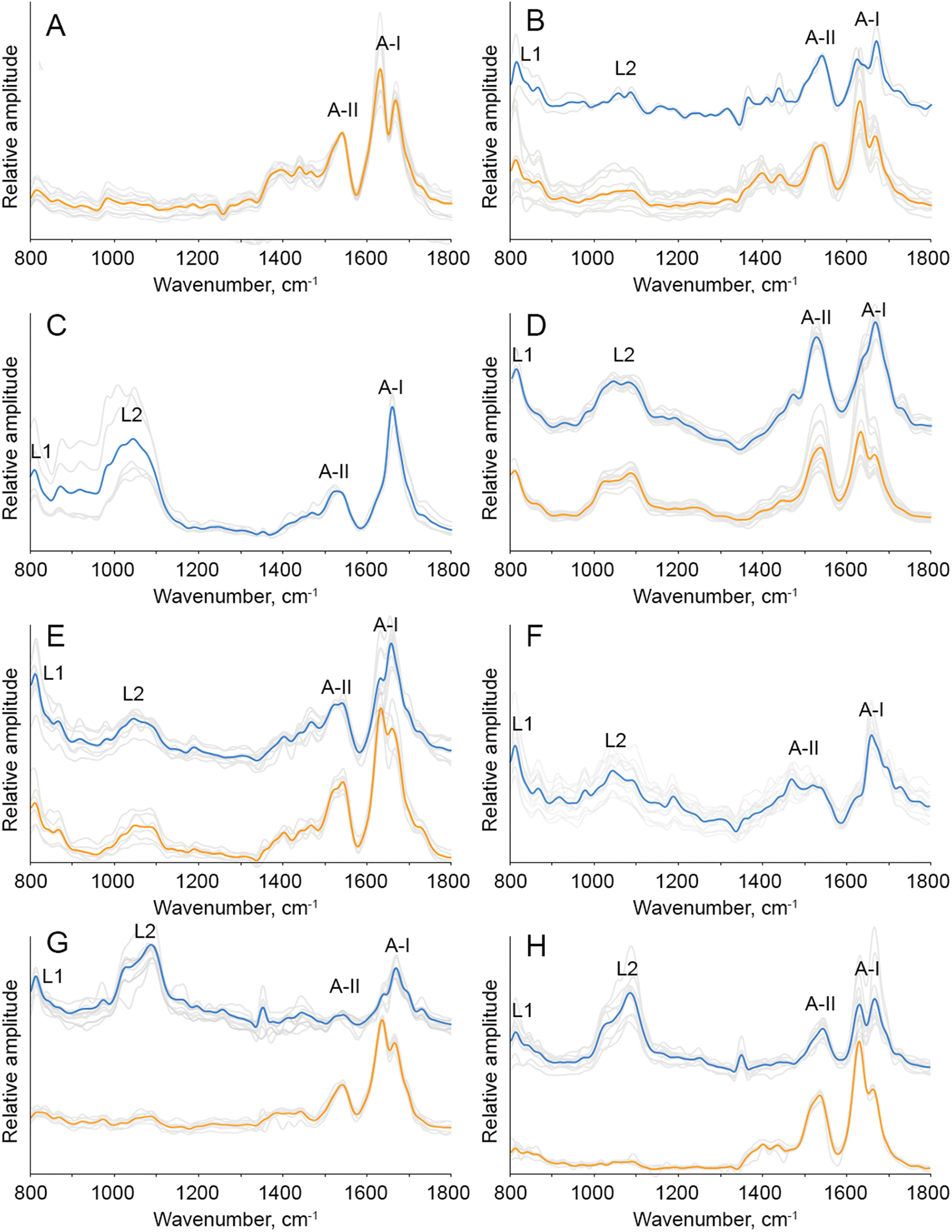
Nanoscale analysis of lipid content of insulin aggregates. AFM-IR spectra of insulin aggregates grown in the absence of lipids (Ins) and in the presence of PS, PC, CL, as Ins:PC:CL (1:0.5:0.5), as Ins:PC:CL (1:0.8:0.2), SM and CER. Spectra collected from individual aggregates are in gray; the corresponding average spectra of the oligomers are in blue and fibrils are in red.
3.3. Elucidation of the ER stress caused by insulin aggregates
Analysis of the expression levels of UPR-related genes showed strong increase in the intensity of GRP78 already 4 h (Fig. 4A) after cell exposition to insulin aggregates that were grown in the presence of lipids and insulin fibrils grown in the lipid-free environment (Ins). We found that Ins:CL, Ins:PC:CL (1:0.8:0.2) and Ins:PC:CL (1:0.5:0.5) exerted significantly higher GRP78 expression levels comparing to Ins, Ins:PC, Ins:PS, Ins:CER and Ins:SM. At the same time, Ins exerted the highest level of ATF6 expression that was not observed for any other type of insulin aggregates. Similar to GRP78, Ins:CL, Ins:PC:CL (1:0.8:0.2) and Ins:PC:CL (1:0.5:0.5) demonstrated stronger activation of ATF6 than Ins: PC, Ins:PS, Ins:CER and Ins:SM. We also found that none of the protein samples induced any change in the expression eIF2a, whereas only Ins: CER induced an increase in the expression level of IRE1α. These findings show that UPD-related activation of GRP78, which is triggered by protein aggregates, primarily induces activation of ATF6, with very low activation of IRE1 and PERK signaling pathways. These conclusions are further supported by a very low if any XBP-1 expression.
Fig. 4.
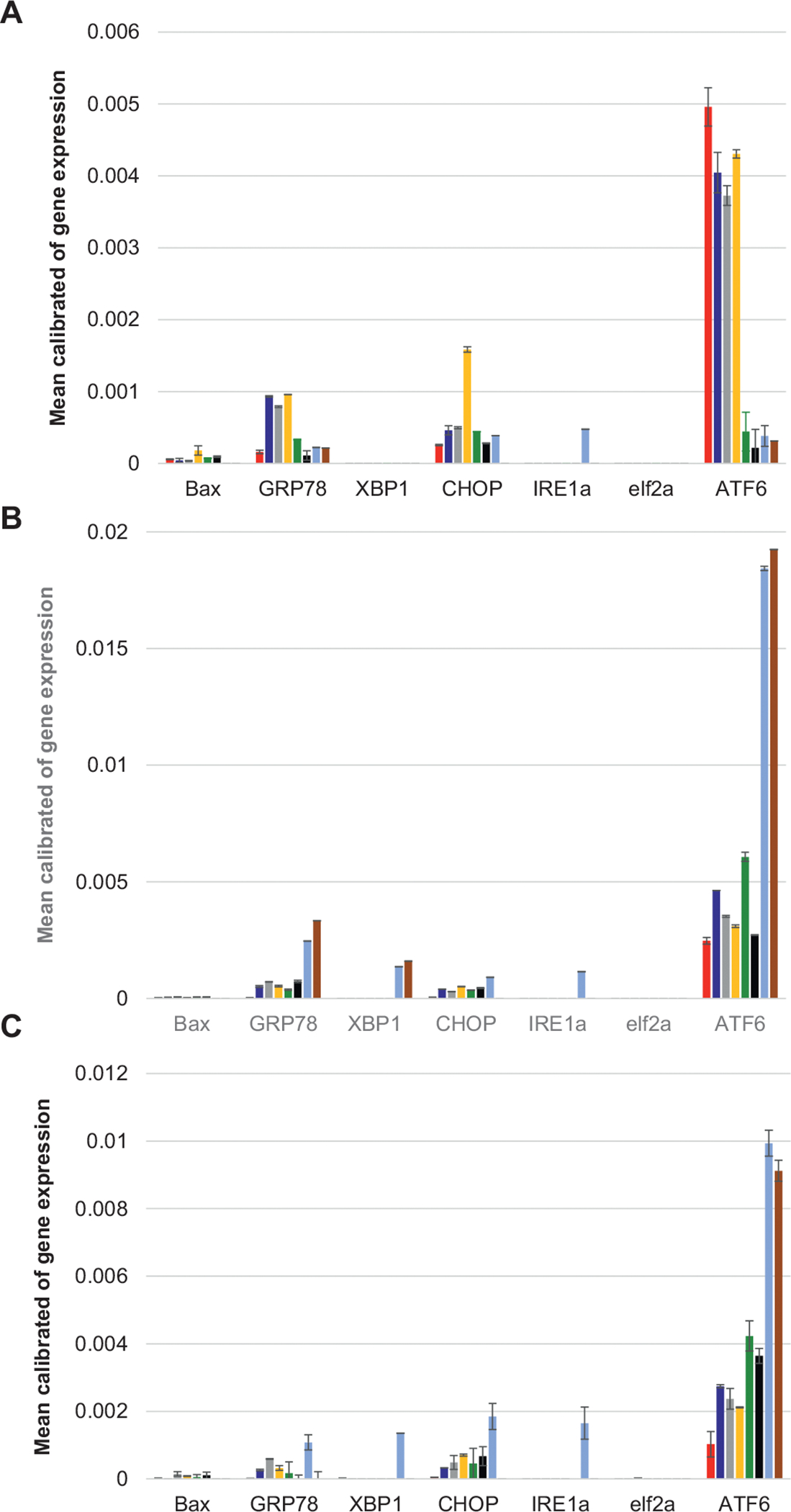
Insulin aggregates induce ER stress in rat dopaminergic neural cells. qPCR results of changes in the expression of Bax, GRP78, XBP1, CHOP, IRE1α, eIF2a and ATF6 at the early (4 h) (A), middle (14 h) (B) and late (24 h) (C) stages of cell exposition to the amyloid aggregates. Ins (red), Ins:CL (blue), Ins: PC:CL (1:0.5:0.5) (gray), Ins:PC:CL (1:0.8:0.2) (yellow), Ins:PC (green), Ins: PS (black), Ins:SM (light blue) and Ins:CER (brown).
At the same time, we observed an increase in the expression of CHOP. This suggests that protein aggregates trigger an alternative to XBP-1 mechanisms of CHOP activation. As expected, we found that all analyzed protein aggregates engaged CHOP-mediated expression of Bax. Furthermore, direct relationship between the levels of expression of CHOP and BAX were observed. Specifically, we found that Ins:PC:CL (1:0.5:0.5) induced the highest comparing to other protein aggregates expression of both CHOP and Bax. These findings show that different insulin aggregates although induce different levels of expression of UPR-related genes, molecular mechanisms activated by all aggregates are very similar.
We also found that levels of expression of the discussed above of UPR-related genes drastically changed from early (4 h) to middle (14 h) and late (24 h) stages of the neuronal cell stress, Fig. 2B–C. Specifically, we observed a drastic decrease in the expression of CHOP and ATF6 caused by Ins relative to the expression levels of these genes caused by insulin aggregates grown in the presence of lipids. We also observed no change in the expression of eIF2a for all analyzed protein aggregates. At the same time, we observed a significant increase in the expression of XBP1 caused by Ins:CER and Ins:SM. These protein aggregates also induced a drastic increase in the expression of GRP78 and ATF6, which was not observed at early stages of the UPR stress caused by these protein aggregates (Fig. 2A). It should be noted that at 14 h, Ins:SM exerted the highest from all other protein samples levels of GRP78, XBP1, CHOP, IRE1α and ATF6. These findings show that presence of SM in the structure of the protein aggregates drastically increases levels of the UPR-related stress response on such stressors.
At 24 h, Ins:SM induced high level of expression of only ATF6, whereas Ins:CER demonstrated the highest expression levels of GRP78, XBP1, CHOP, IRE1α and ATF6. We also found that Ins:CL, Ins:PC:CL (1:0.8:0.2) and Ins:PC:CL (1:0.5:0.5), Ins:PS and Ins:PC exerted lower than Ins:CER and Ins:SM levels of ATF6 expression, however, they were significantly higher than the levels of ATF6 expression caused by Ins. These findings show that presence of lipids in the structure of the protein aggregates results in higher levels of the ER stress compared to the levels of the stress caused by insulin aggregates grown in the lipid-free environment.
4. Discussion
A growing body of evidence shows that lipids change aggregation properties of amyloidogenic proteins and peptides. For finance, Zhang et al. showed that low levels of anionic lipids promoted islet amyloid precursor protein (IAPP) aggregation and enhanced membrane permeabilization potential of these aggregates [52]. At the same time, zwitterionic lipid did not alter the rate of IAPP aggregation, whereas cholesterol at or below physiological levels significantly decelerated IAPP amyloid formation, as well as lowered the propensity of IAPP aggregates to cause membrane leakage. Lipids can also uniquely alter the structure of amyloid β1–40 (Aβ1–40) and α-synuclein aggregates [53]. Furthermore, aggregation of insulin in the presence of CL yields structurally different aggregates from the oligomers that were formed in the presence of PC. Both Ins:CL and Ins:PC aggregates exerted significantly lower cell toxicity compared to the insulin fibrils that were grown in the lipid-free environment. These findings raised a question whether Ins:PC and Ins:CL, as well as other insulin aggregates that were formed in the presence of lipids activate distinctly different mechanisms of cell damage comparing to the pathways activated by the lipid-free insulin fibrils. Furthermore, one may wonder whether lipids determine the degree of the cell damage caused by amyloid aggregates.
There are several mechanisms by which protein aggregates can exert the cell damages [10,11]. Srinivasan and co-workers showed that amyloid fibrils damage eukaryotic plasma membranes, whereas experimental findings reported by Schützmann and co-workers suggest that amyloid toxicity is associated with irreversible damages of late endosomes [10,11]. Furthermore, numerous studies show that amyloid aggregates cause mitochondrial dysfunction and ROS stress [15,54]. At the same time, the role of ER in such amyloid-related stresses remained unclear.
Our current findings show that insulin aggregates induce the ER stress, which triggers (i) autophagy, (ii) suppression of protein expression, and (iii) enhancement of chaperon activity. These conclusions can be made by the activation of the UPR of the ER of cells exposed to amyloid aggregates. Specifically, we observed an enhancement in the expression of GRP78, protein that regulates the downstream regulation of ATF6, PERK and IRE1α. We also observed that exposition of cells to amyloid aggregates is primarily associated with an enhancement of the expression of ATF6, whereas two other ER transmembrane proteins, IRE1α and eLF2a exhibited very low expression levels. We also observed UPR-induced expression of a proapoptotic transcription factor CHOP, which, in turn, regulates expression of caspase 3 kinase and BCL2 protein family members, including the ER localized Bax. These findings suggest that amyloid-induced ER stress strongly affects the functionality of Golgi via AFT6, as well as cell mitochondria (BCL2) and, ultimately, transcription in the cell nuclei (ATF6, CHOP, and XBP1).
We also found that levels of the expression of the discussed above UPR-related proteins directly depend on the secondary structure of the aggregates and on the chemical nature of the lipid present in such aggregates. It should be noted that lipids themselves induce no ER stress. Previously reported NMR results showed that α-Syn develops strong electrostatic interactions with polar headgroups of lipids simultaneously building up strong hydrophobic interactions with aliphatic chains of the lipids. Similar findings were recently reported by Rizevsky and co-workers for insulin. Specifically, the researchers examined interactions between PC, CL and insulin using AFM-IR. It has been found that vibrational bands of lipids in the spectra collected from Ins:PC and Ins: CL aggregates shifted compared to the vibrations of lipids in LUVs themselves. These shifts pointed on the interactions between lipids and the protein. These results are in a good agreement with the recently proposed lipid-chaperone hypothesis [55–57]. According to this hypothesis, membrane lipids interact with amyloidogenic proteins forming stable lipid-protein complexes These species can be easily transported across the lipid bilayer into the cytosol where they exert mitochondrial dysfunction, ROS and ER stresses. Furthermore, membranes containing lipid with high critical micelle concentration (cmc) repress fibril formation and promote pore formation, whereas lipid having low cmc suppress pore and favor fibril formation [58,59]. Thus, the chemical structure of the lipid strongly correlates with the toxicity that the corresponding lipid:protein aggregates can exert.
5. Conclusions
Our experimental findings show that insulin fibrils grown in the lipid-free environment, as well as insulin aggregates induce a strong ER stress, which, in turn, activates the expression of UPR-related genes. Using qPCR, we were able to determine changes in the expression rates of proteins that are linked to the UPR stress. We found that all analyzed aggregates enhance the expression of ATP6, as well as GRP78, CHOP and Bax, with very little changes in the expression of IRE1α and eLF2a, the product of PERK. This triggers an autophagy, suppression of protein expression, and enhancement of chaperon activity that aim to mitigate the ER stress and restore normal cell physiology.
Supplementary Material
Acknowledgment
We are grateful to the National Institute of General Medical Sciences for the provided financial support (R35GM142869).
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
M.M. conceptualization, methodology, investigation, visualization, validation, writing; S.R. conceptualization, methodology, investigation, visualization, data curation, validation, writing; D.K. conceptualization, resources, project administration, supervision, funding acquisition.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bbadis.2022.166485.
Data availability
Data will be made available on request.
References
- [1].Milardi D, Gazit E, Radford SE, Xu Y, Gallardo RU, Caflisch A, Westermark GT, Westermark P, Rosa C, Ramamoorthy A, Proteostasis of islet amyloid polypeptide: a molecular perspective of risk factors and protective strategies for type II diabetes, Chem. Rev. 121 (2021) 1845–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Roda AR, Serra-Mir G, Montoliu-Gaya L, Tiessler L, Villegas S, Amyloid-beta peptide and tau protein crosstalk in Alzheimer’s disease, Neural Regen. Res. 17 (2022) 1666–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, Lang AE, Parkinson disease, Nat. Rev. Dis. Primers 3 (2017) 17013. [DOI] [PubMed] [Google Scholar]
- [4].McColgan P, Tabrizi SJ, Huntington’s disease: a clinical review, Eur. J. Neurol. 25 (2018) 24–34. [DOI] [PubMed] [Google Scholar]
- [5].Chiti F, Dobson CM, Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade, Annu. Rev. Biochem. 86 (2017) 27–68. [DOI] [PubMed] [Google Scholar]
- [6].Knowles TP, Vendruscolo M, Dobson CM, The amyloid state and its association with protein misfolding diseases, Nat. Rev. 15 (2014) 384–396. [DOI] [PubMed] [Google Scholar]
- [7].Iadanza MG, Jackson MP, Hewitt EW, Ranson NA, Radford SE, A new era for understanding amyloid structures and disease, Nat. Rev. Mol. Cell. Biol. 19 (2018) 755–773. [DOI] [PubMed] [Google Scholar]
- [8].Marshall KE, Vadukul DM, Staras K, Serpell LC, Misfolded amyloid-beta-42 impairs the endosomal-lysosomal pathway, Cell. Mol. Life Sci. 77 (2020) 5031–5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zheng L, Cedazo-Minguez A, Hallbeck M, Jerhammar F, Marcusson J, Terman A, Intracellular distribution of amyloid beta peptide and its relationship to the lysosomal system, Transl. Neurodegener. 1 (2012) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schutzmann MP, Hasecke F, Bachmann S, Zielinski M, Hansch S, Schroder GF, Zempel H, Hoyer W, Endo-lysosomal Abeta concentration and pH trigger formation of Abeta oligomers that potently induce Tau missorting, Nat. Commun. 12 (2021) 4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Srinivasan S, Patke S, Wang Y, Ye Z, Litt J, Srivastava SK, Lopez MM, Kurouski D, Lednev IK, Kane RS, Colon W, Pathogenic serum amyloid a 1.1 shows a long oligomer-rich fibrillation lag phase contrary to the highly amyloidogenic non-pathogenic SAA2.2, J. Biol. Chem. 288 (2013) 2744–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yin H, Zheng LQ, Fang W, Lai YH, Porenta N, Goubert G, Zhang H, Su HS, Ren B, Richardson JO, Li JF, Zenobi R, Nanometre-scale spectroscopic visualization of catalytic sites during a hydrogenation reaction on a Pd/Au bimetallic catalyst, Nat. Catal. 3 (2020) 834–842. [Google Scholar]
- [13].Paravastu AK, Qahwash I, Leapman RD, Meredith SC, Tycko R, Seeded growth of beta-amyloid fibrils from Alzheimer’s brain-derived fibrils produces a distinct fibril structure, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 7443–7448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kurouski D, Van Duyne RP, Lednev IK, Exploring the structure and formation mechanism of amyloid fibrils by Raman spectroscopy: a review, Analyst 140 (2015) 4967–4980. [DOI] [PubMed] [Google Scholar]
- [15].Cataldi R, Chia S, Pisani K, Ruggeri FS, Xu CK, Sneideris T, Perni M, Sarwat S, Joshi P, Kumita JR, Linse S, Habchi J, Knowles TPJ, Mannini B, Dobson CM, Vendruscolo M, A dopamine metabolite stabilizes neurotoxic amyloid-beta oligomers, Commun. Biol. 4 (2021) 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Novitskaya V, Bocharova OV, Bronstein I, Baskakov IV, Amyloid fibrils of mammalian prion protein are highly toxic to cultured cells and primary neurons, J. Biol. Chem. 281 (2006) 13828–13836. [DOI] [PubMed] [Google Scholar]
- [17].Placido AI, Pereira CM, Duarte AI, Candeias E, Correia SC, Santos RX, Carvalho C, Cardoso S, Oliveira CR, Moreira PI, The role of endoplasmic reticulum in amyloid precursor protein processing and trafficking: implications for Alzheimer’s disease, Biochim. Biophys. Acta 2014 (1842) 1444–1453. [DOI] [PubMed] [Google Scholar]
- [18].Adams CJ, Kopp MC, Larburu N, Nowak PR, Ali MMU, Structure and molecular mechanism of ER stress signaling by the unfolded protein response signal activator IRE1, Front. Mol. Biosci. 6 (2019) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kopp MC, Larburu N, Durairaj V, Adams CJ, Ali MMU, UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor, Nat. Struct. Mol. Biol. 26 (2019) 1053–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Oslowski CM, Urano F, The binary switch that controls the life and death decisions of ER stressed beta cells, Curr. Opin. Cell Biol. 23 (2011) 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Urano F, Bertolotti A, Ron D, IRE1 and efferent signaling from the endoplasmic reticulum, J. Cell Sci. 113 (Pt 21) (2000) 3697–3702. [DOI] [PubMed] [Google Scholar]
- [22].Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D, IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA, Nature 415 (2002) 92–96. [DOI] [PubMed] [Google Scholar]
- [23].Shen X, Ellis RE, Lee K, Liu CY, Yang K, Solomon A, Yoshida H, Morimoto R, Kurnit DM, Mori K, Kaufman RJ, Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development, Cell 107 (2001) 893–903. [DOI] [PubMed] [Google Scholar]
- [24].Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D, Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response, Nat. Cell Biol. 2 (2000) 326–332. [DOI] [PubMed] [Google Scholar]
- [25].Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D, Perk is essential for translational regulation and cell survival during the unfolded protein response, Mol. Cell 5 (2000) 897–904. [DOI] [PubMed] [Google Scholar]
- [26].Harding HP, Zhang Y, Ron D, Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase, Nature 397 (1999) 271–274. [DOI] [PubMed] [Google Scholar]
- [27].Lee AH, Iwakoshi NN, Glimcher LH, XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response, Mol. Cell. Biol. 23 (2003) 7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Iwakoshi NN, Lee AH, Glimcher LH, The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response, Immunol. Rev. 194 (2003) 29–38. [DOI] [PubMed] [Google Scholar]
- [29].Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH, Proteasome inhibitors disrupt the unfolded protein response in myeloma cells, Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 9946–9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sok J, Wang XZ, Batchvarova N, Kuroda M, Harding H, Ron D, CHOP-dependent stress-inducible expression of a novel form of carbonic anhydrase VI, Mol. Cell. Biol. 19 (1999) 495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D, Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1, Science 287 (2000) 664–666. [DOI] [PubMed] [Google Scholar]
- [32].Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB, Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis, J. Cell Biol. 162 (2003) 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Oslowski CM, Urano F, Measuring ER stress and the unfolded protein response using mammalian tissue culture system, Method. Enzymol. 490 (2011) 71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fonseca SG, Ishigaki S, Oslowski CM, Lu S, Lipson KL, Ghosh R, Hayashi E, Ishihara H, Oka Y, Permutt MA, Urano F, Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells, J. Clin. Invest. 120 (2010) 744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rizevsky S, Matveyenka M, Kurouski D, Nanoscale structural analysis of a lipid-driven aggregation of insulin, J. Phys. Chem. Lett. (2022), 10.1021/acs.jpclett.1c04012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].van Meer G, Voelker DR, Feigenson GW, Membrane lipids: where they are and how they behave, Nat. Rev. Mol. Cell Biol. 9 (2008) 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CR, Shimizu T, Spener F, van Meer G, Wakelam MJ, Dennis EA, Update of the LIPID MAPS comprehensive classification system for lipids, J. Lipid Res. 50 (Suppl) (2009) S9–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fitzner D, Bader JM, Penkert H, Bergner CG, Su M, Weil MT, Surma MA, Mann M, Klose C, Simons M, Cell-type- and brain-region-resolved mouse brain lipidome, Cell Rep. 32 (2020), 108132. [DOI] [PubMed] [Google Scholar]
- [39].Michaelson DM, Barkai G, Barenholz Y, Asymmetry of lipid organization in cholinergic synaptic vesicle membranes, Biochem. J. 211 (1983) 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Galvagnion C, Buell AK, Meisl G, Michaels TCT, Vendruscolo M, Knowles TPJ, Dobson CM, Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation, Nat. Chem. Biol. 11 (2015) 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dazzi A, Glotin F, Carminati R, Theory of infrared nanospectroscopy by photothermal induced resonance, J. Appl. Phys. 107 (2010), 124519. [Google Scholar]
- [42].Dazzi A, Prater CB, AFM-IR: technology and applications in nanoscale infrared spectroscopy and chemical imaging, Chem. Rev. 117 (2017) 5146–5173. [DOI] [PubMed] [Google Scholar]
- [43].Kurouski D, Dazzi A, Zenobi R, Centrone A, Infrared and Raman chemical imaging and spectroscopy at the nanoscale, Chem. Soc. Rev. 49 (2020) 3315–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Katzenmeyer AM, Aksyuk V, Centrone A, Nanoscale infrared spectroscopy: improving the spectral range of the photothermal induced resonance technique, Anal. Chem. 85 (2013) 1972–1979. [DOI] [PubMed] [Google Scholar]
- [45].Katzenmeyer AM, Holland G, Kjoller K, Centrone A, Absorption spectroscopy and imaging from the visible through mid-infrared with 20 nm resolution, Anal. Chem. 87 (2015) 3154–3159. [DOI] [PubMed] [Google Scholar]
- [46].Ruggeri FS, Mannini B, Schmid R, Vendruscolo M, Knowles TPJ, Single molecule secondary structure determination of proteins through infrared absorption nanospectroscopy, Nat. Commun. 11 (2020) 2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lu F, Jin MZ, Belkin MA, Tip-enhanced infrared nanospectroscopy via molecular expansion force detection, Nat. Photon. 8 (2014) 307–312. [Google Scholar]
- [48].Kurouski D, Deckert-Gaudig T, Deckert V, Lednev IK, Structure J Am. Chem. Soc. 134 (2012) 13323–13329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kurouski D, Dukor RK, Lu X, Nafie LA, Lednev IK, Spontaneous inter-conversion of insulin fibril chirality, Chem. Commun. (Camb.) 48 (2012) 2837–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dou T, Li Z, Zhang J, Evilevitch A, Kurouski D, Nanoscale structural characterization of individual viral particles using atomic force microscopy infrared spectroscopy (AFM-IR) and tip-enhanced Raman spectroscopy (TERS), Anal. Chem. 92 (2020) 11297–11304. [DOI] [PubMed] [Google Scholar]
- [51].Rizevsky S, Matveyenka M, Kurouski D, Nanoscale structural analysis of a lipid-driven aggregation of insulin, J. Phys. Chem. Lett. 13 (2022) 2467–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang X, St Clair JR, London E, Raleigh DP, Islet amyloid polypeptide membrane interactions: effects of membrane composition, Biochemistry 56 (2017) 376–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Avdulov NA, Chochina SV, Igbavboa U, Warden CS, Vassiliev AV, Wood WG, Lipid binding to amyloid beta-peptide aggregates: preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids, J. Neurochem. 69 (1997) 1746–1752. [DOI] [PubMed] [Google Scholar]
- [54].Chen SW, Drakulic S, Deas E, Ouberai M, Aprile FA, Arranz R, Ness S, Roodveldt C, Guilliams T, De-Genst EJ, Klenerman D, Wood NW, Knowles TP, Alfonso C, Rivas G, Abramov AY, Valpuesta JM, Dobson CM, Cremades N, Structural characterization of toxic oligomers that are kinetically trapped during alpha-synuclein fibril formation, Proc. Natl. Acad. Sci. U. S. A. 112 (2015) E1994–E2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tempra C, Scollo F, Pannuzzo M, Lolicato F, La Rosa C, A unifying framework for amyloid-mediated membrane damage: the lipid-chaperone hypothesis, Biochim. Biophys. Acta Proteins Proteom. 1870 (2022), 140767. [DOI] [PubMed] [Google Scholar]
- [56].La Rosa C, Scalisi S, Lolicato F, Pannuzzo M, Raudino A, Lipid-assisted protein transport: a diffusion-reaction model supported by kinetic experiments and molecular dynamics simulations, J. Chem. Phys. 144 (2016), 184901. [DOI] [PubMed] [Google Scholar]
- [57].Korshavn KJ, Satriano C, Lin Y, Zhang R, Dulchavsky M, Bhunia A, Ivanova MI, Lee YH, La Rosa C, Lim MH, Ramamoorthy A, Reduced lipid bilayer thickness regulates the aggregation and cytotoxicity of amyloid-beta, J. Biol. Chem. 292 (2017) 4638–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Scollo F, Tempra C, Lolicato F, Sciacca MFM, Raudino A, Milardi D, La Rosa C, Phospholipids critical micellar concentrations trigger different mechanisms of intrinsically disordered proteins interaction with model membranes, J. Phys. Chem. Lett. 9 (2018) 5125–5129. [DOI] [PubMed] [Google Scholar]
- [59].Sciacca MF, Lolicato F, Tempra C, Scollo F, Sahoo BR, Watson MD, Garcia-Vinuales S, Milardi D, Raudino A, Lee JC, Ramamoorthy A, La Rosa C, Lipid-chaperone hypothesis: a common molecular mechanism of membrane disruption by intrinsically disordered proteins, ACS Chem. Neurosci. 11 (2020) 4336–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.