

SUMMARY
To disseminate through the body, Zika virus (ZIKV) is thought to exploit the mobility of myeloid cells, in particular monocytes and dendritic cells. However, the timing and mechanisms underlying shuttling of the virus by immune cells remains unclear. To understand the early steps in ZIKV transit from the skin, at different time points, we spatially mapped ZIKV infection in lymph nodes (LNs), an intermediary site en route to the blood. Contrary to prevailing hypotheses, migratory immune cells are not required for the virus to reach the LNs or blood. Instead, ZIKV rapidly infects a subset of sessile CD169+ macrophages in the LNs, which release the virus to infect downstream LNs. Infection of CD169+ macrophages alone is sufficient to initiate viremia. Overall, our experiments indicate that macrophages that reside in the LNs contribute to initial ZIKV spread. These studies enhance our understanding of ZIKV dissemination and identify another anatomical site for potential antiviral intervention.
In brief
Reynoso et al. show that macrophages in the draining lymph nodes are rapidly infected following footpad inoculation of mice with Zika virus. These nodal macrophages serve as a source of the virus for early ZIKV dissemination.
Graphical Abstract

INTRODUCTION
Flaviviruses are single-stranded, positive-sense RNA viruses that cause a spectrum of severe neurotropic and viscerotropic human diseases and have high epidemic potential.1 Zika virus (ZIKV) is a mosquito-borne flavivirus that explosively emerged in the Americas in 2015.2 Although most ZIKV infections during this epidemic were mild and self-limiting, some individuals experienced prolonged viremia with viral RNA detectable in the serum for weeks to months.3 This epidemic revealed new clinical features of infection, including congenital neurodevelopmental disease and microcephaly.4 Concerns over ZIKV-induced disease spurred the rapid development of animal models to evaluate ZIKV pathogenesis, vaccine candidates, and therapeutics (reviewed in Shan et al.5). Neutralizing antibodies (NAbs) have been identified as a correlate of protection after viral challenge.6–9 No drugs or vaccines are currently approved for treatment of ZIKV infection.
ZIKV induces pathogenesis after dissemination from the skin into the blood, after which the virus gains access to many different tissues, including the brain and, in pregnant women, the decidua, placenta, and fetus.10–12 Viral replication in the tissue as well as induction of antiviral immune responses, including type I interferons (IFN-I), can cause tissue damage and fetal demise.13,14 The importance of dissemination in ZIKV-induced disease has led to investigation of the cellular targets for ZIKV infection and the routes of dissemination into peripheral tissues. The TAM family of receptor tyrosine kinases (including Tyro3, Axl, and Mertk [TAM]) were among the first proteins identified that could promote ZIKV infection in vitro.15–17 Because these proteins are highly expressed on endothelial cells,18 ZIKV infection of vascular endothelial cells via TAM receptors provides one explanation for the ability of ZIKV to access many different tissues from the blood. However, mice deficient in AXL and MERTK expression have been shown to exhibit similar levels of ZIKV infection and viral distribution as wild-type mice.19 A second possible route for ZIKV movement into the tissues is through mobile immune cells. In human blood, circulating monocytes have been identified as the primary cell type infected by ZIKV, and human monocytes also can be infected in vitro.20–22 Using a ZIKV engineered to only infect myeloid cells, one study showed that monocytes represent the major myeloid population to disseminate ZIKV in mice.23 Indeed, monocytes are an attractive conduit for viral movement because these cells are abundant in the blood and migrate into even immune-privileged tissues.24 Thus, monocytes could serve as “Trojan horses” that distribute ZIKV throughout the body.22,25 Many other myeloid cells in the skin, including dermal dendritic cells (DCs) and Langerhans cells, also are permissive for ZIKV23,26–30 and have been proposed to facilitate its dissemination.
Viral dissemination is often viewed as the sum of its products (e.g., cumulative peripheral organ infection) because this is ultimately what results in virus-induced disease. However, dissemination is a complex, multi-step process, and disruption of early dissemination events could prevent or limit downstream viral entry into distant tissues. The cells responsible for early viral movement, particularly within the lymph node (LN), remain unclear. Additionally, it is unknown whether the LN serves to amplify virus draining from the infection site or simply acts as a passageway for virus produced in the skin.
In this study, we examined the early events after ZIKV infection to determine the mechanisms allowing the initial movement of ZIKV from skin to LN to blood. Using mouse models of ZIKV infection, we demonstrate that mobile immune cells do not initially act as Trojan horses of viral distribution while executing their immune functions. Instead, we show that LNs are seeded by ZIKV within minutes of inoculation via lymphatic vessel transport rather than by cellular migration. Moreover, immobile macrophages in the LNs are readily infected by ZIKV before viremia is detectable. Furthermore, viremia can be established without monocyte infection or DC migration from the skin. Thus, LN macrophages are a key link in the chain of ZIKV dissemination that could be targeted by antiviral therapeutics.
RESULTS
ZIKV is captured by the local draining LNs before systemic dissemination
To understand how ZIKV disseminates from the skin, we used an established mouse model of ZIKV infection, footpad inoculation of interferon alpha receptor 1 (Ifnar1)−/− mice.12 Although ZIKV efficiently antagonizes human IFN-I responses, it cannot bind to or inhibit murine STAT2, necessitating circumvention of IFN-I signaling to support murine infection.31 We first examined the early kinetics of viral delivery to the LN draining the hindfoot, the popliteal LN (PLN), reasoning that we should not detect high levels of virus within an hour if skin cells were needed to replicate the virus (Figure 1). We inoculated mice in the hind footpad with 104 focus-forming units (FFUs) of ZIKV H/PF/2013 and harvested PLNs from 5 min to 1 h post infection (p.i.) for infectious viral titers, as determined by a focus formation assay (FFA) (Figure 1A). Within the first hour, we detected infectious ZIKV in PLN homogenates but not in the serum, indicating that the virus reaches the PLN after initial inoculation, likely via the lymphatics. The initial entry of infectious virus in the PLN peaked between 10 and 30 min after inoculation before dropping to levels just above the limit of detection at 1 h.
Figure 1. ZIKV replicates in the PLN before systemic dissemination.

(A) Viral titers (in focus-forming units [FFUs] per milliliter) in the PLN (left, blue dots) or serum (right, gray dots) harvested at the indicated time (in minutes) during the first hour following footpad (FP) injection of Ifnar1−/− mice with 104 FFUs of ZIKV H/PF/2013. Dots represent individual mice (either pooled PLNs or separate sera) and the average of technical replicates in the focus formation assay (FFA). A dashed line shows the limit of detection (LOD) of the FFA. Values below the LOD are reported as half the LOD (125 FFU/mL). PLNs and sera were harvested from the same mice. Data are shown from 1 of 2 complete time-course experiments.
(B) Viral titers in the PLN (left, blue dots) or serum (right, gray dots) at the indicated time point during the first 32 h p.i. of Ifnar1−/− mice. Data are shown from 1 of 2 complete time-course experiments.
(C) Illustration of the route of lymphatic draining from the FP. Lymph first flows to the popliteal LN (PLN), followed by the iliac LN (ILN). Downstream of the nodes, lymph enters the thoracic duct, followed by the subclavian vein (where it enters the blood).
(D) Viral titers in PLNs (blue bars) and ILNs (red bars) of Ifnar1−/− mice at the indicated time p.i. (in hours). PLNs and ILNs were harvested from the same mice. Data are shown from two pooled experiments with 4 mice/group.
All experiments were repeated 2–3 times with 3–4 mice per group. Dots represent individual mice (either pooled LNs or separate sera) and the average of technical replicates in the FFA. Error bars, SEM. Dashed line, LOD for the assay. Statistics, one-way ANOVA. Exact p values are shown in relation to uninfected controls.
We next quantified infectious virus in the PLN or serum over a longer period, every 4 h for the first 32 h p.i. (Figure 1B). At 8 h, we did not detect infectious virus in the PLN. However, viral titers rebounded to greater levels than the initial input virus by 16 h p.i. Viremia was detected at 12 h p.i. and was elevated by 16 h p.i. We obtained similar results in wild-type C57BL/6 mice receiving an anti-IFNAR1 Ab (MAR1–5A3, described in Lazear et al.11) (Figure S1). In C57BL/6 mice with intact IFN-I signaling, similar levels of infectious virus were measured at the PLN 30 min post-inoculation, but titers never increased, indicating that viral replication drives the higher viral titers seen in the PLN at later time points in Ifnar1−/− mice (Figure S1).
The LNs act as a system of sequential filters that remove viruses from the lymph flow.32 From the PLN, lymph fluid passes to the iliac LN (ILN) and then to the renal LN (RLN) before emptying into the bloodstream via the thoracic duct (depicted in Figure 1C).33 Thus, the ILN should have the opportunity to sequester virus not captured by the PLN. Infectious virus was not detected in the ILN before 16 h p.i., indicating that the PLN captures virtually all lymph-borne ZIKV at this initial inoculation dose (Figure 1D). Together, these data demonstrate that infectious ZIKV inoculated into the skin via injection passes to the draining LN, where it is captured.
ZIKV replicates in distinct LN macrophage niches
LNs contain at least five distinct populations of macrophages, which form a network to carry out specific immune functions.34 Macrophages present in nodal sinuses represent an immobile population of phagocytic cells that are situated with their somas in the sinusoidal space and projections through the lymphatic endothelial cells that form the sinus floor and provide cytokines necessary for macrophage survival.35,36 Sinus-resident macrophages can be subdivided further based on their location within the LN and expression of cell-surface molecules.37 Sinus-resident macrophages nearest the afferent (incoming) lymph vessel have been termed subcapsular sinus macrophages (SSMs), whereas those nearest the efferent (exiting) lymph vessels are called medullary sinus macrophages (MSMs). These populations access lymph-borne particulates sequentially as lymph enters the node through the subcapsular sinus and flows around to the medullary sinus.38
Because of their location, we first investigated whether lymphoid sinus-resident macrophages capture ZIKV. We imaged frozen cross-sections of the PLN harvested from 8–24 h p.i. from C57BL/6 mice, C57BL/6 mice treated with an anti-IFNAR1 Ab, or Ifnar1−/− mice (Figure 2). LNs were oriented so that they were sectioned from the top of the LN (near the afferent lymphatics) through the medulla and hilum, as described previously,39 thus affording a complete view of the major anatomical subdivisions of the LN. We distinguished CD169+ sinusoidal macrophage populations by their location and morphology, as described previously.37,39,40 At 8 h p.i., we detected ZIKV E protein that colocalized with small patches of SSMs, regardless of the type I IFN signaling capacity of the recipient mice and, thus, likely reflecting binding of the input virus (Figure 2A). We did not detect ZIKV E protein staining in negative control LNs infected with vaccinia virus (VACV), which induces nodal inflammation and increases background Ab staining (Figure S2). We identified ZIKV-infected cells by staining for the non-structural protein NS2b, which is not incorporated into virions (Figures 2B–2D). By 8 h p.i., NS2b staining was detected at low levels in the LN and restricted to SSMs. By 16 h, we detected prominent NS2b staining in MSMs located in medullary sinuses (Figures 2C and 2D). Most of the NS2b-infected cells in Ifnar1−/− mice expressed CD11b and SIGNR1, but not CD11c, at 16 h p.i., consistent with previous analyses demonstrating a paucity of infected DCs in ZIKV-infected LNs at early time points39 (Figures S2D–S2F). Staining of the ILN revealed a similar but delayed pattern of infection, with SSMs being productively infected at 16 h p.i., followed by MSMs at 24 h p.i. (Figure S2).
Figure 2. ZIKV infects macrophages in LN sinuses.

(A) Confocal images of frozen PLN sections from nodes harvested 8 h p.i. from C57BL/6 mice (top panels), C57BL/6 mice given the anti-IFNAR1 Ab MAR1–5A3 (center panels), and Ifnar1−/− mice (bottom panels). Blue, B cells; green, CD169+ macrophages; red, ZIKV E protein; white, CD31. The far right panels show a higher-magnification view of the SCS. Dashed ovals show specific areas with ZIKV E protein staining. An asterisk indicates patches of SSMs with E protein staining.
(B) Confocal images of frozen PLN sections from nodes harvested 8 h p.i. from C57BL/6 mice (top panels), C57BL/6 mice given the anti-IFNAR1 Ab MAR1–5A3 (center panels), and Ifnar1−/− mice (bottom panels). Blue, B220; green, CD169; red, ZIKV NS2b protein; white, CD31. The far right panel shows a higher-magnification view of the SCS. Dashed ovals show specific areas with ZIKV NS2b protein staining.
(C) As in (B), but PLNs were harvested 16 h p.i.
(D) Confocal images of frozen PLN sections from nodes harvested at the indicated time (shown in hours p.i. in the top right corners) from Ifnar1−/− mice. The top panels show macrophages in the SCS (SCS macrophages [SSMs]), and the bottom panels show macrophages in medullary sinuses (MS macrophages [MSMs]). Green, CD169; red, ZIKV NS2b protein.
(E) Confocal images of frozen PLN sections from nodes harvested 16 h p.i. from hStat2-KI mice infected with mouse-adapted ZIKV. Green, CD169; red, ERTR-7 (LN stroma); white, ZIKV NS2b protein; blue, B220. Higher-magnification images on the right show NS2b+ SSMs (top panel) and MSMs (bottom panel). Images are representative of 6–10 PLNs/time point/condition harvested from 3–5 mice. Scale bars are in micrometers.
In these studies, both murine models of ZIKV infection utilized mice that were deficient in type I IFN signaling either genetically or through Ab treatment. To understand whether LN macrophages were also infected in a more immunocompetent animal model, we inoculated human STAT2 knockin (hStat2-KI) mice with a mouse-adapted strain of ZIKV (MA Dakar), as described previously.10 Confocal imaging of the PLN 16 h p.i. revealed ZIKV NS2b+ SSMs and MSMs (Figure 2E). Together, these data reveal that, in mice, footpad-inoculated ZIKV transits through and infects LN macrophages.
ZIKV infection disrupts LN macrophages
After inflammatory stimulation by either live or dead virus, SSMs undergo attrition because of inflammasome activation and pyroptosis or through necroptosis.41–43 We exploited this feature as a surrogate measure of LN-macrophage sensing of lymph-borne ZIKV without a requirement for direct infection. We performed flow cytometry on single-cell suspensions of PLNs or ILNs harvested 72 h p.i. from mice with or without IFN-I signaling (Figures 3A and 3B). We gated on live CD45+CD11cloCD11b+CD169+ cells and further separated macrophage populations using F4/80 (present on MSMs only37). SSMs were absent in the PLN and ILN at this time point; this did not require productive ZIKV infection because SSMs were also ablated in wild-type (WT) mice. In contrast to SSMs, MSMs are not thought to undergo p.i. attrition.41 However, by 72 h p.i., MSMs were largely absent in Ifnar1−/− mice, although some MSMs were still detectable in C57BL/6 mice with intact IFN-I signaling (Figure 3). The prominent MSM ablation in Ifnar1−/− mice indicates that the MSM compartment has access to virus or viral antigen produced after viral replication in these animals.
Figure 3. ZIKV disrupts LN macrophage networks.

(A) Flow plots generated from single-cell suspensions of PLNs harvested from uninfected C57BL/6 mice (left panel), ZIKV-infected C57BL/6 mice (center left), C57BL/6 + anti-IFNAR1 Ab MAR1–5A3 mice (center right), and Ifnar1−/− mice (right) 72 h p.i. Cells were first gated on CD45+ B220— CD3— CD11b+ CD11clow cells. Gating indicates SSMs (left gates, CD169+ F4/80—) and MSMs (right gates, CD169+ F4/80+).
(B) Frequency of CD169+ SSMs (far left and center right panels) or MSMs (center left and far right panels) in PLNs (blue bars) and ILNs (red bars) as a percentage of total LN macrophages from the experiment shown in flow plots in (A). Statistics, one-way ANOVA. Dots show pooled LNs from individual mice. Error bars, SEM. The experiment was repeated 3 times with 3–4 mice/group.
(C) Confocal images of frozen PLN sections harvested from C57BL/6 mice (top panels), C57BL/6 mice given the anti-IFNAR1 Ab MAR1–5A3 (center panels), or Ifnar1−/− mice (bottom panels) at the indicated time p.i. Blue, B cells; green, CD169+ macrophages; red, stromal cells (ERTR-7+). The far right panel shows a higher-magnification view of the medullary sinus.
Images in (C) are representative of 6–10 LNs/time point/condition harvested from 3–5 mice. Scale bars are in micrometers.
LN macrophages are difficult to remove by enzymatic tissue dissociation and may be overestimated by flow cytometry approaches because of lymphocyte binding of CD169+ cell debris.44 As a second measure, we also examined LNs over the same time frame using confocal microscopy (Figure 3C). Imaging at 72 h p.i. recapitulated the SSM and MSM ablation in Ifnar1−/− mice observed using flow cytometry. We also noted little overt change in LN architecture, as judged by staining of LN stromal and vascular cells. The LN macrophage network remained largely intact at 8 h and partially intact at 24 h (Figure 3C). Together, these data show that footpad inoculation with ZIKV results in infection and disruption of the PLN macrophage network.
CD169+ macrophage infection alone allows systemic viral dissemination
To more definitively understand the role of LN macrophages in ZIKV dissemination, we crossed Siglec1-cre mice (with Cre recombinase expression in CD169+ cells45) with Ifnar1-floxed mice46 to conditionally delete IFN-I signaling in CD169+ macrophages (Siglec1-cre Ifnar1fl/fl mice, hereafter referred to as CD169 conditional knockout (cKO); Figure 4). Viral titers in the PLN or ILN at 24 h p.i. infection were unexpectedly high even though viral replication was restricted to only CD169+ macrophages (titers were statistically similar to Ifnar1−/− mice) (Figure 4A). Moreover, the levels of infectious ZIKV in the serum were similar in Ifnar1−/− and CD169 cKO mice, indicating that infection of CD169+ macrophages alone could lead to high levels of disseminated virus by 24 h p.i. Confocal analyses using ZIKV NS2b protein staining confirmed productive infection of LN macrophages in the PLN and ILN of CD169 cKO mice (Figures 4B and 4C). These data indicate that CD169+ macrophages, including those in the LNs, serve as targets for early ZIKV replication.
Figure 4. CD169+ macrophage infection supports systemic dissemination.
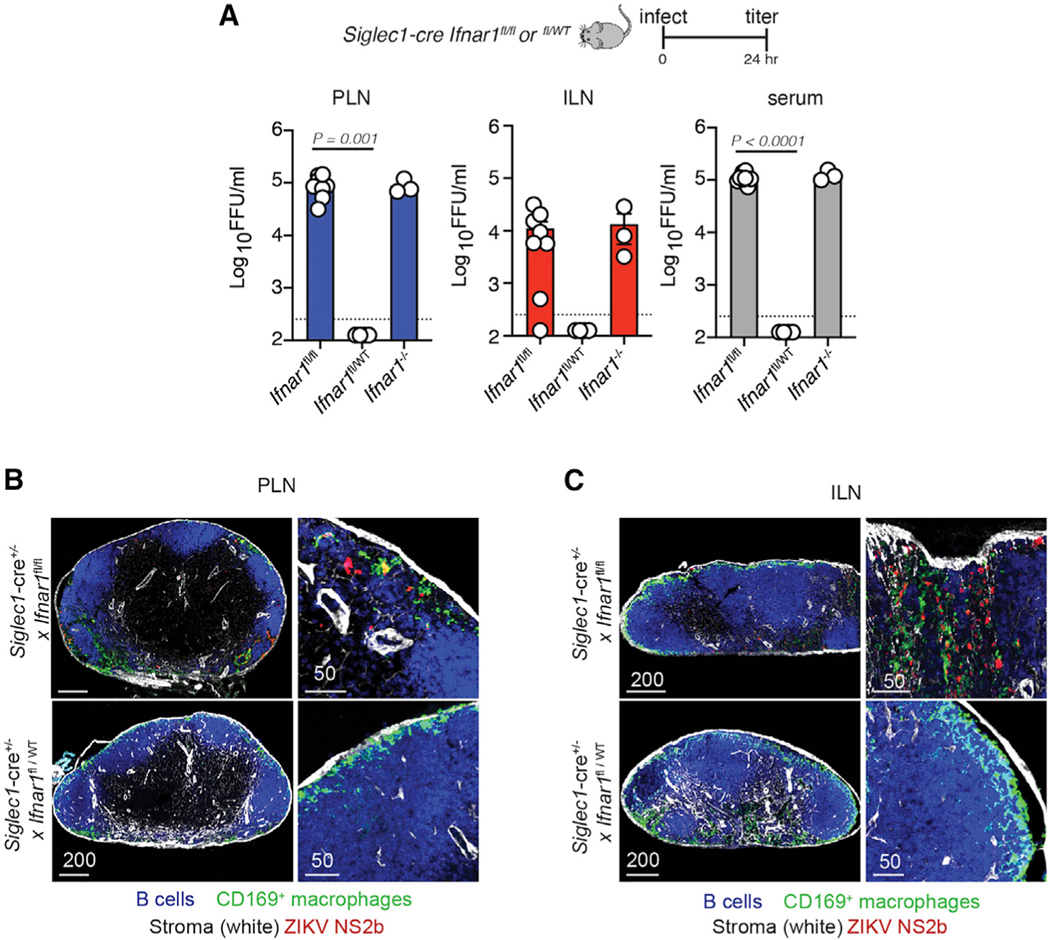
(A) Viral titers (FFUs per milliliter) in the PLN (left, blue bars), ILN (center, red bars), and serum (right, gray bars) harvested 24 h p.i. from Siglec1-cre Ifnarfl/fl (homozygous Ifnar1 knockout in CD169+ cells [cKO]) mice, Siglec1-cre Ifnarfl/WT (heterozygous knockout in CD169+ cells), and Ifnar1−/− mice with 104 FFUs of ZIKV. The experiment was repeated 3 times with 3–4 mice per group. Results shown are pooled from two independent experiments. Dots represent individual mice (either pooled LNs or separate serum) and the average of technical replicates. Dashed line, LOD for the assay. Values below the LOD are reported as half the LOD (125 FFU/mL).
(B) Confocal images of frozen PLN sections harvested 24 h p.i. from Siglec1-cre Ifnarfl/fl (homozygous cKO) mice (top panels) and Siglec1-cre Ifnarfl/WT (heterozygous cKO mice, bottom panels) stained for B cells (using the B220 Ab, blue), CD169+ macrophages (green), stromal cells (using ERTR7, white), and ZIKV NS2b protein (red). The right panels show higher-magnification images. Scale bars are in micrometers.
(C) As in (B) but showing the ILN instead of PLN.
Dermal dendritic cell migration is not required for viral transit to the blood after footpad inoculation
Although dermal DCs are thought to carry infectious virus to the LNs to initiate nodal infection and adaptive immune responses, our kinetic examination of nodal macrophage infection suggested that macrophages were infected before DC trafficking from the skin, which takes at least 6 h to occur.47 Therefore, we next quantitated virus at the inoculation site (foot), LNs, and serum of CD169 cKO mice at 12 h p.i. (Figure 5A). Although we detected viral dissemination in the serum by 12 h in Ifnar1−/− and CD169 cKO mice, we did not yet detect infectious virus in foot homogenates (Figure 5A). Viral titers were statistically similar between Ifnar1−/− and CD169 cKO mice at this early time point.
Figure 5. Migrating dendritic cells are dispensable for ZIKV dissemination after FP inoculation.

(A) Viral titers (FFUs per milliliter) in the PLN (left, blue bars), serum (center, gray bars), and FP (right, yellow bars) harvested 12 h p.i. from Siglec1-cre Ifnarfl/fl (homozygous Ifnar1 knockout in CD169+ cells [cKO]) mice, Siglec1-cre Ifnarfl/WT (heterozygous cKO) mice, and Ifnar1−/− mice inoculated with 104 FFUs of ZIKV. The experiment was repeated 2 times with 3 mice per group. Results shown are pooled from two independent experiments. Dots represent individual mice (either pooled LNs or separate serum or feet) and the average of technical replicates. Dashed line, LOD for the assay. Values below the LOD are reported as half the LOD (125 FFU/mL).
(B) Viral titers (FFUs per milliliter) in the PLN (left, blue bars), ILN (center, red bars), and serum (right, gray bars) harvested 12 h p.i. from C57BL/6 or Ccr7−/− mice treated with the anti-IFNAR1 Ab MAR1–5A3 and inoculated with 104 FFUs of ZIKV. The experiment was repeated 2 times with 3 mice per group. Results shown are pooled from two independent experiments. Dots represent individual mice (either pooled LNs or separate serum) and the average of technical replicates. Dashed line, LOD for the assay. Values below the LOD are reported as half the LOD (125 FFU/mL).
(C) Frequency of CD169+ SSMs (left) and MSMs (right) in the either PLNs (blue bars) or ILNs (red bars) 72 h p.i. of C57BL/6 or Ccr7−/− mice as a percentage of total LN macrophages Statistics, one-way ANOVA. Dots show pooled LNs from individual mice. Error bars, SEM. The experiment was repeated 2 times with 3 mice/group.
(D) Confocal images of frozen LN sections from nodes harvested 16 h p.i. from C57BL/6 or Ccr7−/− mice treated with an anti-IFNAR1 Ab. Blue, B cells; green, CD169+ macrophages; red, ZIKV NS2b; white, Lyve1. Right panels show a higher magnification of infected SSMs. Scale bars are in micrometers.
Skin-resident dermal DCs and Langerhans cells use the chemokine receptor CCR7 to migrate via the lymphatics to the skin-draining LN.48 To dissect the contribution of DC/Langerhans cell migration from the skin to LN infection, we treated Ccr7−/− mice (lacking DC lymphatic migration; Figure S3) with an anti-IFNAR1 Ab and quantitated infectious virus at 24 h p.i. (Figure 5B). Viral titers in the PLN were not statistically different regardless of DC migration, and mice still developed similar levels of viremia. Notably, CCR7 deficiency significantly decreased viral titers in the ILN, suggesting that cellular movement either from or within the PLN impacts viral transport to downstream LNs. Flow cytometry at 72 h revealed attrition of SSMs and MSMs in the PLN and ILN (Figure 5C), indicating that DC migration is not required for disruption of nodal macrophage networks. We confirmed productive infection of LN macrophages in anti-IFNAR1 Ab-treated Ccr7−/− mice through confocal microscopy and staining for NS2b (Figure 5D). Thus, our data suggest that macrophages can produce the virus needed for early systemic dissemination in the absence of a significant contribution of infectious virus from migratory DCs.
Infected monocytes do not account for high levels of virus in the blood
Monocytes are mobile myeloid cells rapidly recruited to sites of infection, where they typically die or mature into sedentary tissue macrophages.49 Flavivirus infection of mice and humans results in monocyte mobilization from the bone marrow into the blood and then the skin, where they are thought to serve as targets of infection.20,21,28 Furthermore, monocytes have been proposed to become “Trojan horses” for ZIKV and act as a major cell population disseminating the virus in mice.23 Based on these studies, we assessed the role of Ly6chigh monocytes during early ZIKV dissemination (Figures 6A and 6B). ZIKV infection mobilized monocytes into the blood within 24 h of infection. (Figure 6B, left). Although monocytes were detected in the infected PLN, treatment with an anti-IFNAR1 Ab greatly reduced monocyte nodal numbers (Figure 6B, right). To deplete monocytes, we treated mice with the depleting monoclonal Ab (mAb) GR1 (against Ly6C and Ly6G) as described previously50 (Figure S4). GR1 treatment eliminated most Ly6Chigh monocytes and neutrophils in the blood and PLN before and after infection.
Figure 6. Monocytes are not needed for early ZIKV dissemination.

(A) Flow cytometry plots generated from single-cell suspensions of PLNs harvested from naive C57BL/6 mice (left) and infected C57BL/6 mice at 24 h p.i. (all other panels). Where indicated, mice were given the MAR1–5A3 (anti-IFNAR1) or GR1 Ab before infection. An additional dose of MAR1–5A3 was given at the time of infection. Cells were first gated on CD45+ cells. Gating indicates Ly6C+ monocytes (top boxes) and Ly6G+ neutrophils (center boxes).
(B) Frequency of Ly6C+ monocytes in the blood (left) and PLN (right) of the indicated mice 24 h p.i. Statistics, one-way ANOVA. Dots show pooled LNs from individual mice. Error bars, SEM. The experiment was repeated 2 times with 3–5 mice/group.
(C) Confocal images of frozen PLN sections from nodes harvested 24 h p.i. from LysM-eGFP mice treated with MAR1–5A3. B220, blue; LysM-eGFP+ cells, green; blood vessel (CD31), white; ZIKV NS2b protein, red. The right panels omit B cells for clarity. Scale bars are in micrometers.
(D) Viral titers (FFUs per milliliter) in the PLN (left, blue bars) and serum (right, gray bars) harvested 24 h p.i. from Ifnar1−/− mice inoculated with 104 FFUs of ZIKV. Mice were given the GR1 Ab prior to infection. The experiment was repeated 3 times with 3–4 mice per group. Dots represent individual mice (either pooled LNs or separate serum) and the average of technical replicates. Dashed line, LOD for the assay. Statistics, unpaired t test.
Using confocal and multiphoton microscopy, we examined PLN sections (either live or frozen fixed) for the presence of monocytes in LysM-eGFP reporter mice, which possess green myelomonocytes of varying GFP intensity (neutrophils [bright], monocytes [bright intermediate], and macrophages [dim]).51 Whereas eGFP-bright neutrophils accumulated in and near the SCS around ZIKV-infected and dying macrophages, we did not detect NS2b-expressing eGFP+ cells at the time points examined (8–24 h p.i.) (Figures 6C and S4). Furthermore, depletion of monocytes did not impact infectious viral titers in the PLN or serum at 24 h p.i. (Figure 6D).
Collectively, these data indicate that Ly6Chigh monocytes are not trafficking infectious virus from the skin into the LNs or blood early during infection. Furthermore, infection of blood monocytes is not required for the high levels of blood-borne ZIKV in these mouse models.
CD169+ macrophage infection alone does not induce systemic disease
We next examined the contribution of CD169+ macrophage infection to the sustained viremia detected in Ifnar1−/− mice through 5 days p.i. (Figure 7A). Notably, viral titers in the serum were low in CD169 cKO mice on day 3 p.i., and little infectious ZIKV was present in the serum on day 5 p.i. (Figure 7B). Similarly, viral titers in the PLN were low in CD169 cKO mice on day 3 p.i., but not in Ifnar1−/− mice, suggesting that there are additional targets for ZIKV infection in the LNs leading to sustained viral production. Consistent with reduced nodal and serum viral levels, CD169 cKO mice exhibited no weight loss or mortality after inoculation with the same dose of virus that resulted in marked virus-induced morbidity in Ifnar1−/− mice (Figure 7C). Collectively, our data indicate that nodal macrophages contribute to viral dissemination from the LNs; however, infection of other cells is needed for virus-induced morbidity.
Figure 7. CD169+ macrophage infection does not result in morbidity.

(A) Viral titers (FFUs per milliliter) in the PLN (left, blue dots) and serum (right, gray bars) harvested at the indicated day p.i. from Ifnar1−/− mice. The experiment was repeated 2 times with 3–4 mice per group. Results shown are pooled from two experiments. Dots represent individual mice (either pooled LNs or separate serum) and the average of technical replicates. Dashed line, LOD for the assay. Values below the LOD are reported as half the LOD (125 FFU/mL).
(B) As in (A) but in Siglec1-cre Ifnarfl/fl (cKO) mice.
(C) Weight loss (as a percentage of starting weight) in Ifnar1−/− and Siglec1-cre Ifnarfl/fl (cKO) mice. Dots show average weight per day p.i. (indicated on the x axis). The experiment was repeated twice with 4–5 mice/group. Scale bars, SEM. Statistics, one-way ANOVA.
DISCUSSION
Many medically important viral pathogens, including ZIKV and monkeypox virus, follow a similar initial distribution route through the host during infection. After viral entry via a breach in an epithelial barrier, virus travels to and replicates in the draining LN. After the LN, virus can be detected in the blood and eventually in distal tissues, where the consequences of viral infection are often observable as host pathology. Each step in this process involves movement of the virus into a new part of the body against physical and immunological barriers. For example, upon tissue entry, phagocytes of the innate immune system can capture the virus to eliminate spread and produce cytokines that limit viral replication in the tissue.52 When the virus reaches the blood, endothelial cells form a primary barrier preventing diffusion of the virus into the tissue.53 While mechanisms enabling viral circumvention of some of these important controls of systemic homeostasis are well established, our knowledge of events unfolding in some areas, such as the LNs, is still limited.
One strategy allowing a virus to bypass normal barriers against spread is to transit with immune cells that migrate throughout the body. The number of migratory immune cells, particularly blood monocytes, increases by orders of magnitude during infection. Numerous studies in mice and humans have identified ZIKV-infected monocytes in the blood or in peripheral organs.20–23,25 The results of these studies, along with others during infections with related flaviviruses, have led to the idea that myeloid cells are critical for viral dissemination from the skin. However, monocytes could function to spread the virus at multiple time points during infection. Our studies refine the events leading to ZIKV in the tissues to include LN macrophages as cells that are responsible for the initial movement from the LNs. Monocyte involvement likely occurs as ZIKV spreads from the blood to distal tissues. Depletion of monocytes did not impact early viral burden in the serum, suggesting that monocytes are not the source of early virus in the blood. Monocyte infection could occur as a result of viremia and might be enhanced by the increased monocyte mobilization seen during infection.
In addition to monocytes, migratory dermal DCs are implicated in the spread of ZIKV and other flaviviruses from the skin.23,27,30,54–60 Given the right stimulation, DCs clearly possess the capacity to migrate from virally infected tissues to the LNs.48 To our knowledge, however, ZIKV-infected migratory DCs have not been isolated from or identified in the LNs. In a previous study, we microscopically examined ZIKV-infected LNs 24 h after infection and did not identify infected migratory DCs.39 Our data here indicate that ZIKV is present in the blood before large numbers of DCs migrate into the LNs. Future experiments will be required to understand the role of infected DCs at later time points in the pathogenesis sequence. Recent studies with the poxvirus vaccinia virus have demonstrated that the immune system can halt viral spread after cutaneous viral replication by shutting off lymphatic transport of the virus or by preventing DC trafficking to the LNs.61,62 If these skin immune-defense mechanisms also occur during ZIKV infection, then viral replication in the skin might not directly translate to infectious virus in the blood. Nonetheless, local viral replication in the skin is poised to influence viremia through indirect mechanisms, such as pro-inflammatory cytokine production.
The lymphatics can deliver a first bolus of virus to the LNs for capture by strategically positioned macrophages and DCs.39,63,64 This early virus delivery initiates the adaptive immune response by providing antigen for B cell activation and allowing priming of CD8+ T cells by LN-resident DCs.39,64,65 However, given the small number of SSMs in the LNs (with some estimates of only 200 SSMs per PLN43), we evaluated whether LN macrophages could produce sufficient virus for downstream infection. Our data in CD169 cKO mice suggest that this small group of LN cells can produce enough virus to result in detectable viremia by 12 h p.i. Future studies will be needed to determine the sources for continued virus output in CD169 cKO mice (for example, other LNs in the chain or CD169+ macrophages in other tissues, such as the spleen). Although high levels of blood-borne virus could be detected in Ifnar1−/− animals 5 days p.i., viremia waned in CD169 cKO mice. This result suggests that there is a finite population of CD169+ macrophages that can produce ZIKV.
SSMs undergo attrition during LN infection, vaccination, and inflammation.41 The mechanisms leading to SSM death have been debated (e.g., pyroptosis versus necroptosis)41–43 because SSMs that are not directly infected also die after stimulation. This observation is perplexing because LN macrophage death impairs LN filtration as well as development of humoral immune responses to secondary pathogens. Based on our data, we propose that LN macrophage death represents a programmed response to limit virus production by eliminating uninfected cells that could serve as vessels to amplify and disseminate the virus.
Collectively, our data reveal that lymphatics and LN sinusoidal macrophages are key participants in dissemination of ZIKV to and from the LNs. Early viral spread to the blood occurred in the absence of cellular transport by DCs or monocytes. Together, our data illuminate a time point during ZIKV dissemination that could be targeted to prevent downstream infection before viral movement into peripheral tissues.
Limitations of the study
Our studies used murine models of ZIKV infection where the virus was delivered by an injection. This route of inoculation likely differs from human infection after a mosquito bite in terms of cell types infected and the kinetics of infection. The volume of inoculation also differs from that delivered by a mosquito and may particularly affect the requirement for different cell types to carry the virus to the LNs from the skin. Future studies will be needed to determine whether migratory DCs are required for early ZIKV dissemination after a mosquito bite but not footpad inoculation. Additionally, mosquito saliva has immunomodulatory effects that are not accounted for in our models.56,66,67
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the lead contact: Heather Hickman (hhickman@mail.nih.gov).
Materials availability
All materials developed in this study will be available from the lead contact upon reasonable request.
Data and code availability
This paper does not report original code.
Unprocessed data underlying the display items in the manuscript, related to the figures, are available from the lead contact upon request.
Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Ifnar1−/− (Line 314), Ccr7−/− (Line 8453), and LysM-eGFP (Line 342) were obtained from the National Institute of Allergy and Infectious Diseases intramural research repository at Taconic Farms. Wild-type C57BL/6N mice were obtained from Taconic Farms. Ifnar1fl/fl mice (B6(Cg)-Ifnar1tm1.1Ees/J; #28256) were obtained from Jackson Laboratories. CD169-cre mice have been previously reported.68 Embryos were obtained from Riken, rederived and bred in-house to generate CD169-cre Ifnar1fl/fl conditional knockout (cKO) mice. hStat2-KI mice in a C57BL/6 background have been reported previously and were bred at Washington University School of Medicine.10 Male and female mice from 6–12 weeks of age were used for experiments. Mice were housed under specific pathogen-free conditions and provided standard rodent chow and sterile water as necessary. All animal studies were approved by and performed in accordance with the Animal Care and Use Committee of the National Institute of Allergy and Infectious Diseases or Washington University School of Medicine.
Viruses
ZIKV strains H/PF2013 and MA-Dakar10 were used as indicated.
METHOD DETAILS
Viral infections and titers
Mice were anesthetized using isoflurane and infected in both hind footpads with 1×104 FFU ZIKV H/PF/2013 unless otherwise indicated (for some experiments, only one footpad was infected). In Figure 2E, mice were inoculated with 1×104 FFU MA-Dakar.10 To determine viral titers, LNs or hind feet were collected at various times p.i. and placed in 250 mL of RPMI +2% FBS + HEPES/LN or 500 μL per foot in metal bead lysing matrix tubes (MP Biomedicals). Samples were homogenized using a Fastprep-24 (MP Biomedicals). Alternatively, blood was collected at the indicated time p.i. into serum gel Z/1.1 tubes (Sarstedt) and serum separated by centrifugation. To measure ZIKV-induced morbidity, mice were weighed before infection and daily thereafter, and the percent starting weight was calculated for each mouse.
Infectious virus titers were determined using a focus-forming assay (FFA).69 Vero cells were seeded at a concentration of 2 × 104 per well in 96-well plates and incubated at 37°C until 90–100% confluency was reached. Confluent monolayers were inoculated in duplicate with LN homogenates or serum and incubated at 37°C for 4 h. Cells then were overlaid with 1% (wt/vol) methylcellulose (Sigma) in Opti-MEM supplemented (Gibco) with 1% Pen-strep (Gibco) and incubated at 37°C. After 48 h, cells were fixed with 4% paraformaldehyde for 30 min at room temperature. Cells were incubated for 2 h with ZV-67 mAb70 in PBS supplemented with 0.1% saponin and 0.1% BSA. Plates were washed and stained with HRP-conjugated anti-mouse IgG (Jackson laboratories). Virus-infected foci were visualized using TrueBlue peroxidase substrate (KPL) for 20 min and counted by an ImmunoSpot 5.0.37 micro-analyzer (CTL).
Antibody treatment
MAR1–5A3 Ab71 was administered intraperitoneally at 1 mg in 1 mL of sterile PBS 12 h before and the day of footpad infection. For inflammatory monocyte depletion, GR1 Ab was administered intraperitoneally at 0.25 mg in 1 mL of PBS 12–24 h before infection.
Flow cytometric analyses after enzymatic tissue dissociation
LNs were collected at various times p.i. and single-cell suspensions prepared by digestion with Liberase (Roche) + DNAse (Worthington) for ~1 h at 37°C. Cells were disrupted by 3 rounds of vigorous pipetting, suspensions were filtered through 60 mm nylon-filter capped FACS tubes. LN cells were stained for 20 min on ice with CD45 (clone 30-F11), CD3 (clone 17A2), B220 (clone RA3–6B2), CD11c (clone N418), CD11b (clone M1/70, BioLegend), CD169 (clone 3D6.112), F4/80 (clone BM8), Gr-1(clone RB6–8C5) and/or (HK1.4) and fixable viability dye Ghost Dye UV450 or Zombie Aqua. Samples were washed twice post-staining in Hanks Balanced Salt Solution +0.1% BSA and fixed for 20 min with 3.2% paraformaldehyde. Samples were washed twice in PBS and acquired on a Fortessa flow cytometer (BD Biosciences). Flow data were analyzed using FlowJo Software (TreeStar).
Confocal microscopy of frozen LN sections
LNs were removed at the indicated time p.i., fixed in periodate-lysine-paraformaldehyde (PLP) for 48 h, and moved to 30% sucrose/PBS solution for 24 h. Tissues were embedded in optimal-cutting-temperature (OCT) medium (Electron Microscopy Sciences), oriented to cut through both the B cell follicles and the medullary sinuses, and frozen in dry-ice-cooled isopentane. 16-mm sections were cut on a Leica cryostat (Leica Microsystems). Sections were blocked with 5% goat, donkey, bovine, rat, or rabbit serum and then stained with one or more of the following Abs: ZIKV NS2b protein Ab (polyclonal, Genetex), ZIKV E protein Ab (polyclonal, Genetex), ERTR7 (rat monoclonal, Abcam), B220 (clone RA3–6B2, Thermo Fisher Scientific), CD169 (clone 3D6.112, BioLegend), Lyve-1 (clone ALY7, Thermo Fisher Scientific), CD11c (clone N418, Thermo Fisher Scientific), SIGN-R1 (clone eBio22D1, Thermo Fisher Scientific), CD31 (clone MEC13.3, BioLegend). Sections were incubated with secondary antibodies as needed and as controls, and images were acquired on a Leice SP8 microscope using identical PMT (photomultiplier tube) and laser settings. Images were processed and analyzed using Imaris software (Oxford Instruments).
QUANTIFICATION AND STATISTICAL ANALYSES
Significances were calculated using Prism V 8.3.0 (Graphpad Software) using unpaired two-tailed Mann-Whitney t-tests (when only two groups were present) or using a one-way ANOVA as indicated in the Figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-ZIKV E protein (polyclonal) | GeneTex | Cat# GTX 133314; RRID: AB_2747413 |
| Anti-ZIKV NS2b protein (polyclonal) | GeneTex | GTX133308; RRID:AB_2715494 |
| CD169 (clone 3D6.112) | BioLegend | Cat# 142419; RRID: AB_2566436 |
| B220 (clone RA3–6B2) | Thermo Fisher Scientific | Cat# 48–0452-82; RRID: AB_1548761 |
| CD31 (clone MEC13.3) | BioLegend | Cat# 102516; RRID: AB_2161029 |
| Lyve1 (clone ALY7) | Thermo Fisher Scientific | Cat# 50–0443-82; RRID_0597449 |
| ER-TR7 (rat monoclonal) | Abcam | Cat# ab51824; RRID: AB_881651 |
| SIGNR1 (clone eBio22D1) | Thermo Fisher Scientific | Cat# 14–2093-82; RRID: AB_795885 |
| CD11c (clone N418) | Thermo Fisher Scientific | Cat# 14–0114-85; RRID: AB_467116 |
| Ly6c APC/Cyanine7 (clone HK1.4) | BioLegend | Cat# 128026; RRID:AB_10640120 |
| Gr-1 eFluor 450 (clone RB6–8C5) | Thermo Fisher Scientific | Cat# 48–5931-82; RRID:AB_1548788 |
| CD45 700 (clone 30-F11) | BioLegend | Cat# 103128; RRID: AB_493715 |
| F4/80 PE (clone BM8) | Thermo Fisher Scientific | Cat# 12–4801-82; RRID: AB_465923 |
| CD11b 594 (clone M1/70) | BioLegend | Cat# 101254; RRID: AB_2563231 |
| CD11c PerCP-Cyanine 5.5 (clone N418) | Thermo Fisher Scientific | Cat# 45–0114-82: RRID: AB_925727 |
| CD3 PE/Cyanine 7 (clone 17A2) | BioLegend | Cat#100220; RRID:AB_1732057 |
| B220 PE/Cyanine 7 (clone RA3–6B2) | Thermo Fisher Scientific | Cat# 25–0452-82; RRID:AB_469627 |
| MAR1–5A3 | BioXcell | Cat# BE0241; RRID:AB_2687723 |
| GR1 | BioXcell | Cat# BE0075; RRID:AB_10312146 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Zika virus H/PF/2013 | Pierson laboratory | N/A |
| Zika virus MA-Dakar | Diamond laboratory | N/A |
|
| ||
| Chemicals peptides and recombinant proteins | ||
|
| ||
| Zombie Aqua™ Fixable Viability Kit | BioLegend | Cat# 423102 |
| Spectral DAPI | Perkin Elmer | FP1490 |
| Ghost dye UV450 | Tonbo Biosciences | Cat# 13–0868-T500 |
| BSA | Sigma-Aldrich | Cat# A3059–500G |
| FBS | Hyclone | Cat# SH30070.03 |
| Triton X- | Sigma-Aldrich | Cat# t9284 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15714-S |
| Sodium (meta)periodate | Sigma-Aldrich | Cat# S1878–500G |
| Lysine | Sigma-Aldrich | Cat# L5501–100G |
| 2-Methylbutane (Isopentane) | Sigma-Aldrich | Cat# M32631–500ML |
| Optimal-cutting-temperature (OCT) compound | Electron Microscopy Sciences | Cat# 62550–01 |
| Methyl cellulose | Sigma-Aldrich | Cat# M0512–250G |
| Gibco™ Opti-MEM™ Reduced Serum Medium, GlutaMAX™ Supplement |
Thermo Fisher Scientific | Cat# 51–985-034–500ML |
| Penicillin-Streptomycin | Thermo Fisher Scientific | Cat# 15070063 |
| Peroxidase AffiniPure Donkey Anti-Mouse IgG(H + L) |
Jackson Laboratories | Cat# 715–035-150 |
| TrueBlue™ Horseradish Peroxidase (HRP) substrate,KPL | VWR | Cat# 95059–168 |
|
| ||
| Critical commercial reagents | ||
|
| ||
| Lysing Matric S (1/8″) metal beads | MP Biomedicals | Cat# 116925100 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Vero | ATCC | Cat# ATCC CRL-1587 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mice | ||
| Mouse: C57BL/6N | Taconic | Cat #C57BL/6N |
| Mouse: Ifnar1−/− | NIAID Taconic Research Repository | Cat #314 |
| Mouse: Siglec1-cre | Riken | RBRC06239 |
| Mouse: B6(Cg)-Ifnar1tm1.1Ees/J (Ifnar1-fl) | Jackson Laboratories | Cat# 028256; RRID: IMSR_JAX:028,256 |
| Mouse: Siglec1-cre Ifnar1fl/fl | Bred in Hickman laboratory | N/A |
| Mouse: Ccr7−/− | NIAID Taconic Research Repository | Cat# 8853 |
| Mouse: hSTAT2-KI | Jackson Laboratory | Strain #:031,630 RRID:IMSR_JAX:031,630 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo 10 | TreeStar | RRID: SCR_008520 |
| Prism 7 and 8 | Graphpad | RRID: SCR_002798 |
| Imaris 9.0.0 | Bitplane | RRID: SCR_007370 |
Highlights.
ZIKV infects and replicates in distinct LN macrophage populations
LN macrophage infection precedes the development of viremia
Virus reaches the blood in the absence of DC migration or monocyte infection
CD169+ macrophage infection alone does not induce morbidity
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the NIAID and NIH (R01AI073755 to M.S.D.). The graphical abstract was drawn by Ethan Tyler (NIH Medical Arts Branch).
DECLARATION OF INTERESTS
M.S.D. is a consultant for Inbios, Vir Biotechnology, Senda Biosciences, Moderna, and Immunome. The Diamond laboratory has received unrelated funding support in sponsored research agreements from Vir Biotechnology, Emergent BioSolutions, Generate Biomedicines, and Moderna.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112126.
REFERENCES
- 1.Pierson TC, and Diamond MS (2020). The continued threat of emerging flaviviruses. Nat. Microbiol 5, 796–812. 10.1038/s41564-020-0714-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diamond MS, Ledgerwood JE, and Pierson TC (2019). Zika virus vaccine development: progress in the face of new challenges. Annu. Rev. Med 70, 121–135. 10.1146/annurev-med-040717-051127. [DOI] [PubMed] [Google Scholar]
- 3.Paz-Bailey G, Rosenberg ES, Doyle K, Munoz-Jordan J, Santiago GA, Klein L, Perez-Padilla J, Medina FA, Waterman SH, Gubern CG, et al. (2018). Persistence of Zika virus in body fluids - final report. N. Engl. J. Med 379, 1234–1243. 10.1056/NEJMoa1613108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mlakar J, Korva M, Tul N, Popović M, Poljšak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodušek V, et al. (2016). Zika virus associated with microcephaly. N. Engl. J. Med 374, 951–958. 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- 5.Shan C, Xie X, and Shi PY (2018). Zika virus vaccine: progress and challenges. Cell Host Microbe 24, 12–17. 10.1016/j.chom.2018.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dowd KA, Ko SY, Morabito KM, Yang ES, Pelc RS, DeMaso CR, Castilho LR, Abbink P, Boyd M, Nityanandam R, et al. (2016). Rapid development of a DNA vaccine for Zika virus. Science 354, 237–240. 10.1126/science.aai9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Rompay KKA, Keesler RI, Ardeshir A, Watanabe J, Usachenko J, Singapuri A, Cruzen C, Bliss-Moreau E, Murphy AM, Yee JL, et al. (2019). DNA vaccination before conception protects Zika virus-exposed pregnant macaques against prolonged viremia and improves fetal outcomes. Sci. Transl. Med 11, eaay2736. 10.1126/scitranslmed.aay2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abbink P, Larocca RA, Visitsunthorn K, Boyd M, De La Barrera RA, Gromowski GD, Kirilova M, Peterson R, Li Z, Nanayakkara O, et al. (2017). Durability and correlates of vaccine protection against Zika virus in rhesus monkeys. Sci. Transl. Med 9, eaao4163. 10.1126/scitranslmed.aao4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, et al. (2017). Modified mRNA vaccines protect against Zika virus infection. Cell 168, 1114–1125.e10. 10.1016/j.cell.2017.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gorman MJ, Caine EA, Zaitsev K, Begley MC, Weger-Lucarelli J, Uccellini MB, Tripathi S, Morrison J, Yount BL, Dinnon KH 3rd., et al. (2018). An immunocompetent mouse model of Zika virus infection. Cell Host Microbe 23, 672–685.e6. 10.1016/j.chom.2018.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, and Diamond MS (2016). A mouse model of Zika virus pathogenesis. Cell Host Microbe 19, 720–730. 10.1016/j.chom.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrison TE, and Diamond MS (2017). Animal models of Zika virus infection, pathogenesis, and immunity. J. Virol 91, e00009–17. 10.1128/JVI.00009-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yockey LJ, Jurado KA, Arora N, Millet A, Rakib T, Milano KM, Hastings AK, Fikrig E, Kong Y, Horvath TL, et al. (2018). Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol 3, eaao1680. 10.1126/sciimmunol.aao1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miner JJ, Cao B, Govero J, Smith AM, Fernandez E, Cabrera OH, Garber C, Noll M, Klein RS, Noguchi KK, et al. (2016). Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 165, 1081–1091. 10.1016/j.cell.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, DeLalio LJ, Isakson BE, and Wang TT (2016). AXL-mediated productive infection of human endothelial cells by Zika virus. Circ. Res 119, 1183–1189. 10.1161/circresaha.116.309866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richard AS, Shim BS, Kwon YC, Zhang R, Otsuka Y, Schmitt K, Berri F, Diamond MS, and Choe H.(2017). AXL-dependent infection of human fetal endothelial cells distinguishes Zika virus from other pathogenic flaviviruses. Proc. Natl. Acad. Sci. USA 114, 2024–2029. 10.1073/pnas.1620558114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mladinich MC, Schwedes J, and Mackow ER (2017). Zika virus persistently infects and is basolaterally released from primary human brain microvascular endothelial cells. mBio 8, e00952–17. 10.1128/mBio.00952-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lemke G.(2013). Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol 5, a009076. 10.1101/cshperspect.a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hastings AK, Yockey LJ, Jagger BW, Hwang J, Uraki R, Gaitsch HF, Parnell LA, Cao B, Mysorekar IU, Rothlin CV, et al. (2017). TAM receptors are not required for Zika virus infection in mice. Cell Rep. 19, 558–568. 10.1016/j.celrep.2017.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Michlmayr D, Andrade P, Gonzalez K, Balmaseda A, and Harris E.(2017). CD14(+)CD16(+) monocytes are the main target of Zika virus infection in peripheral blood mononuclear cells in a paediatric study in Nicaragua. Nat. Microbiol 2, 1462–1470. 10.1038/s41564-017-0035-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foo SS, Chen W, Chan Y, Bowman JW, Chang LC, Choi Y, Yoo JS, Ge J, Cheng G, Bonnin A, et al. (2017). Asian Zika virus strains target CD14(+) blood monocytes and induce M2-skewed immunosuppression during pregnancy. Nat. Microbiol 2, 1558–1570. 10.1038/s41564-017-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ayala-Nunez NV, Follain G, Delalande F, Hirschler A, Partiot E, Hale GL, Bollweg BC, Roels J, Chazal M, Bakoa F, et al. (2019). Zika virus enhances monocyte adhesion and transmigration favoring viral dissemination to neural cells. Nat. Commun 10, 4430. 10.1038/s41467-019-12408-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonald EM, Anderson J, Wilusz J, Ebel GD, and Brault AC (2020). Zika virus replication in myeloid cells during acute infection is vital to viral dissemination and pathogenesis in a mouse model. J. Virol 94, e00838–20. 10.1128/JVI.00838-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Teh YC, Ding JL, Ng LG, and Chong SZ (2019). Capturing the fantastic voyage of monocytes through time and space. Front. Immunol 10, 834. 10.3389/fimmu.2019.00834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jurado KA, and Iwasaki A.(2017). Zika virus targets blood monocytes. Nat. Microbiol 2, 1460–1461. 10.1038/s41564-017-0049-7. [DOI] [PubMed] [Google Scholar]
- 26.Cerny D, Haniffa M, Shin A, Bigliardi P, Tan BK, Lee B, Poidinger M, Tan EY, Ginhoux F, and Fink K.(2014). Selective susceptibility of human skin antigen presenting cells to productive dengue virus infection. PLoS Pathog. 10, e1004548. 10.1371/journal.ppat.1004548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duangkhae P, Erdos G, Ryman KD, Watkins SC, Falo LD Jr., Marques ETA Jr., and Barratt-Boyes SM (2018). Interplay between keratinocytes and myeloid cells drives dengue virus spread in human skin. J. Invest. Dermatol 138, 618–626. 10.1016/j.jid.2017.10.018. [DOI] [PubMed] [Google Scholar]
- 28.Schmid MA, and Harris E.(2014). Monocyte recruitment to the dermis and differentiation to dendritic cells increases the targets for dengue virus replication. PLoS Pathog. 10, e1004541. 10.1371/journal.ppat.1004541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu SJ, Grouard-Vogel G, Sun W, Mascola JR, Brachtel E, Putvatana R, Louder MK, Filgueira L, Marovich MA, Wong HK, et al. (2000). Human skin Langerhans cells are targets of dengue virus infection. Nat. Med 6, 816–820. 10.1038/77553. [DOI] [PubMed] [Google Scholar]
- 30.Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, et al. (2015). Biology of Zika virus infection in human skin cells. J. Virol 89, 8880–8896. 10.1128/JVI.00354-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sánchez-Seco MP, Evans MJ, Best SM, and García-Sastre A.(2016). Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19, 882–890. 10.1016/j.chom.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozawa M, Nakajima S, Kobayashi D, Tomii K, Li NJ, Watarai T, Suzuki R, Watanabe S, Kanda Y, Takeuchi A, and Katakai T.(2022). Micro- and macro-anatomical frameworks of lymph nodes indispensable for the lymphatic system filtering function. Front. Cell Dev. Biol 10, 902601. 10.3389/fcell.2022.902601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harrell MI, Iritani BM, and Ruddell A.(2008). Lymph node mapping in the mouse. J. Immunol. Methods 332, 170–174. 10.1016/j.jim.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bellomo A, Gentek R, Bajénoff M, and Baratin M.(2018). Lymph node macrophages: scavengers, immune sentinels and trophic effectors. Cell. Immunol 330, 168–174. 10.1016/j.cellimm.2018.01.010. [DOI] [PubMed] [Google Scholar]
- 35.Gray EE, and Cyster JG (2012). Lymph node macrophages. J. Innate Immun 4, 424–436. 10.1159/000337007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mondor I, Baratin M, Lagueyrie M, Saro L, Henri S, Gentek R, Suerinck D, Kastenmuller W, Jiang JX, and Bajénoff M.(2019). Lymphatic endothelial cells are essential components of the subcapsular sinus macrophage niche. Immunity 50, 1453–1466.e4. 10.1016/j.immuni.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phan TG, Green JA, Gray EE, Xu Y, and Cyster JG (2009). Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat. Immunol 10, 786–793. 10.1038/ni.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jafarnejad M, Woodruff MC, Zawieja DC, Carroll MC, and Moore JE Jr. (2015). Modeling lymph flow and fluid exchange with blood vessels in lymph nodes. Lymphat. Res. Biol 13, 234–247. 10.1089/lrb.2015.0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reynoso GV, Weisberg AS, Shannon JP, McManus DT, Shores L, Americo JL, Stan RV, Yewdell JW, and Hickman HD (2019). Lymph node conduits transport virions for rapid T cell activation. Nat. Immunol 20, 602–612. 10.1038/s41590-019-0342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sung JH, Zhang H, Moseman EA, Alvarez D, Iannacone M, Henrickson SE, de la Torre JC, Groom JR, Luster AD, and von Andrian UH (2012). Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell 150, 1249–1263. 10.1016/j.cell.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaya M, Castello A, Montaner B, Rogers N, Reis e Sousa C, Bruckbauer A, and Batista FD (2015). Host response. Inflammation-induced disruption of SCS macrophages impairs B cell responses to secondary infection. Science 347, 667–672. 10.1126/science.aaa1300. [DOI] [PubMed] [Google Scholar]
- 42.Sagoo P, Garcia Z, Breart B, Lemaître F, Michonneau D, Albert ML, Levy Y, and Bousso P.(2016). In vivo imaging of inflammasome activation reveals a subcapsular macrophage burst response that mobilizes innate and adaptive immunity. Nat. Med 22, 64–71. 10.1038/nm.4016. [DOI] [PubMed] [Google Scholar]
- 43.Chatziandreou N, Farsakoglu Y, Palomino-Segura M, D’Antuono R, Pizzagalli DU, Sallusto F, Lukacs-Kornek V, Uguccioni M, Corti D, Turley SJ, et al. (2017). Macrophage death following influenza vaccination initiates the inflammatory response that promotes dendritic cell function in the draining lymph node. Cell Rep. 18, 2427–2440. 10.1016/j.celrep.2017.02.026. [DOI] [PubMed] [Google Scholar]
- 44.Gray EE, Friend S, Suzuki K, Phan TG, and Cyster JG (2012). Subcapsular sinus macrophage fragmentation and CD169+ bleb acquisition by closely associated IL-17-committed innate-like lymphocytes. PLoS One 7, e38258. 10.1371/journal.pone.0038258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karasawa K, Asano K, Moriyama S, Ushiki M, Monya M, Iida M, Kuboki E, Yagita H, Uchida K, Nitta K, and Tanaka M.(2015). Vascular-resident CD169-positive monocytes and macrophages control neutrophil accumulation in the kidney with ischemia-reperfusion injury. J. Am. Soc. Nephrol 26, 896–906. 10.1681/ASN.2014020195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prigge JR, Hoyt TR, Dobrinen E, Capecchi MR, Schmidt EE, and Meissner N.(2015). Type I IFNs act upon hematopoietic progenitors to protect and maintain hematopoiesis during pneumocystis lung infection in mice. J. Immunol 195, 5347–5357. 10.4049/jimmunol.1501553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomura M, Hata A, Matsuoka S, Shand FHW, Nakanishi Y, Ikebuchi R, Ueha S, Tsutsui H, Inaba K, Matsushima K, et al. (2014). Tracking and quantification of dendritic cell migration and antigen trafficking between the skin and lymph nodes. Sci. Rep 4, 6030. 10.1038/srep06030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Platt AM, and Randolph GJ (2013). Dendritic cell migration through the lymphatic vasculature to lymph nodes. Adv. Immunol 120, 51–68. 10.1016/B978-0-12-417028-5.00002-8. [DOI] [PubMed] [Google Scholar]
- 49.Italiani P, and Boraschi D.(2014). From monocytes to M1/M2 macrophages: phenotypical vs. Functional differentiation. Front. Immunol 5, 514. 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hickman HD, Reynoso GV, Ngudiankama BF, Rubin EJ, Magadán JG, Cush SS, Gibbs J, Molon B, Bronte V, Bennink JR, and Yewdell JW (2013). Anatomically restricted synergistic antiviral activities of innate and adaptive immune cells in the skin. Cell Host Microbe 13, 155–168. 10.1016/j.chom.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Faust N, Varas F, Kelly LM, Heck S, and Graf T.(2000). Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood 96, 719–726. [PubMed] [Google Scholar]
- 52.Shi C, and Pamer EG (2011). Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol 11, 762–774. 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mehta D, and Malik AB (2006). Signaling mechanisms regulating endothelial permeability. Physiol. Rev 86, 279–367. 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 54.Lei V, Petty AJ, Atwater AR, Wolfe SA, and MacLeod AS (2020). Skin viral infections: host antiviral innate immunity and viral immune evasion. Front. Immunol 11, 593901. 10.3389/fimmu.2020.593901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.King CA, Wegman AD, and Endy TP (2020). Mobilization and activation of the innate immune response to dengue virus. Front. Cell. Infect. Microbiol 10, 574417. 10.3389/fcimb.2020.574417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schneider CA, Calvo E, and Peterson KE (2021). Arboviruses: how saliva impacts the journey from vector to host. Int. J. Mol. Sci 22, 9173. 10.3390/ijms22179173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Castillo JA, Naranjo JS, Rojas M, Castañ o D, and Velilla PA (2019). Role of monocytes in the pathogenesis of dengue. Arch. Immunol. Ther. Exp 67, 27–40. 10.1007/s00005-018-0525-7. [DOI] [PubMed] [Google Scholar]
- 58.Troupin A, Shirley D, Londono-Renteria B, Watson AM, McHale C, Hall A, Hartstone-Rose A, Klimstra WB, Gomez G, and Colpitts TM (2016). A role for human skin mast cells in dengue virus infection and systemic spread. J. Immunol 197, 4382–4391. 10.4049/jimmunol.1600846. [DOI] [PubMed] [Google Scholar]
- 59.Begum F, Das S, Mukherjee D, Mal S, and Ray U.(2019). Insight into the tropism of dengue virus in humans. Viruses 11, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rathore APS, and St John AL (2018). Immune responses to dengue virus in the skin. Open Biol. 8, 180087. 10.1098/rsob.180087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aggio JB, Krmeska V, Ferguson BJ, Wowk PF, and Rothfuchs AG (2021). Vaccinia virus infection inhibits skin dendritic cell migration to the draining lymph node. J. Immunol 206, 776–784. 10.4049/jimmunol.2000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Churchill MJ, du Bois H, Heim TA, Mudianto T, Steele MM, Nolz JC, and Lund AW (2022). Infection-induced lymphatic zippering restricts fluid transport and viral dissemination from skin. J. Exp. Med 219, e20211830. 10.1084/jem.20211830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hickman HD, Takeda K, Skon CN, Murray FR, Hensley SE, Loomis J, Barber GN, Bennink JR, and Yewdell JW (2008). Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat. Immunol 9, 155–165. 10.1038/ni1557. [DOI] [PubMed] [Google Scholar]
- 64.Junt T, Moseman EA, Iannacone M, Massberg S, Lang PA, Boes M, Fink K, Henrickson SE, Shayakhmetov DM, Di Paolo NC, et al. (2007). Subcapsular sinus macrophages in lymph nodes clear lymphborne viruses and present them to antiviral B cells. Nature 450, 110–114. 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- 65.Hickman HD, Li L, Reynoso GV, Rubin EJ, Skon CN, Mays JW, Gibbs J, Schwartz O, Bennink JR, and Yewdell JW (2011). Chemokines control naive CD8+ T cell selection of optimal lymph node antigen presenting cells. J. Exp. Med 208, 2511–2524. 10.1084/jem.20102545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pingen M, Bryden SR, Pondeville E, Schnettler E, Kohl A, Merits A, Fazakerley JK, Graham GJ, and McKimmie CS (2016). Host inflammatory response to mosquito bites enhances the severity of arbovirus infection. Immunity 44, 1455–1469. 10.1016/j.immuni.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pingen M, Schmid MA, Harris E, and McKimmie CS (2017). Mosquito biting modulates skin response to virus infection. Trends Parasitol. 33, 645–657. 10.1016/j.pt.2017.04.003. [DOI] [PubMed] [Google Scholar]
- 68.Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, Kanagawa O, Fujii S.i., and Tanaka M.(2011). CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity 34, 85–95. 10.1016/j.immuni.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 69.Brien JD, Lazear HM, and Diamond MS (2013). Propagation, quantification, detection, and storage of West Nile virus. Curr. Protoc. Microbiol 31, 15d.13.11–15d.13.18. 10.1002/9780471729259.mc15d03s31. [DOI] [PubMed] [Google Scholar]
- 70.Zhao H, Fernandez E, Dowd KA, Speer SD, Platt DJ, Gorman MJ, Govero J, Nelson CA, Pierson TC, Diamond MS, and Fremont DH (2016). Structural basis of Zika virus-specific antibody protection. Cell 166, 1016–1027. 10.1016/j.cell.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sheehan KCF, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, Dungo-Arthur C, Carrero JA, White JM, Hertzog PJ, and Schreiber RD (2006). Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J. Interferon Cytokine Res 26, 804–819. 10.1089/jir.2006.26.804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper does not report original code.
Unprocessed data underlying the display items in the manuscript, related to the figures, are available from the lead contact upon request.
Any additional information required to analyze the data reported in this paper is available from the lead contact upon request.