

Abstract
Ventricular tachycardia (VT) and ventricular fibrillation (VF) are the most frequent causes of death in the first 24 hours after myocardial infarction. Previous studies showed that depleting TRPV1 receptors with resiniferatoxin (RTX) led to a reduced risk of VT and VF post-myocardial infarction. Therefore, the question of resiniferatoxin as a cardioprotector against myocardial infarction (MI)-induced VT and VF was raised. The RNA sequence data from 3 groups of pigs, each having 4 animals (4 controls, 4 myocardial infarction - MI, and 4 RTX + MI) was analyzed through the lens of differentially expressed genes. The differential expression comparison was conducted in two ways: MI versus Control and RTX+MI versus MI. The results showed the downregulation of deleterious genes involved in inflammation and future plaque instability in the RTX group compared with the MI group. In the case of some of the genes, these findings were reinforced by obtaining the same trends in the MI versus Control group. All in all, we propose further investigation of RTX as a prophylactic method against cardiovascular complications of MI.
Keywords: Myocardial infarction, TRPV1, RNA sequence analysis, resiniferatoxin, LIPG, pentraxin 3, ADAM thrombospondinin family.
INTRODUCTION
Cardiac transient receptor potential channel 1 (TRPV1) afferent signaling plays a key role in arrhythmogenesis post-myocardial infarction. TRPV1 is a receptor located on the afferent fibers and it partly mediates the cardiac sympathetic afferent reflex (CSAR)1. TRPV1 is activated by heat, capsaicin, metabolites of ischemia (e.g., bradykinin), and mechanical transduction2. During and after a myocardial infarction the CSAR is activated, contributing to the persistence of reflex adrenergic activation. This enhances ventricular tachycardia (VT) or ventricular fibrillation (VF) occurrence.
Interestingly, other data on capsaicin-induced activation of TRPV1 in cardiomyocytes enhanced their apoptosis, presumably via increased intracellular levels of calcium following TRPV1 stimulation3. Another way to induce apoptosis was represented by the higher than normal mitochondrial superoxide levels3. In contrast, adding a TRPV1 antagonist such as capsazepine led to improved cell viability following hypoxia and ischemia of the heart3.
It is proposed that altering the cardiac sympathetic afferent reflex post-myocardial infarction (MI) will improve the prognosis of patients. After MI, there is augmented CSAR4. A study conducted in 2019 revealed that the denervation of the afferent component reduced the number of ventricular tachycardia (VT)/ventricular fibrillation (VF) experienced4. This finding was related to the suppression of stellate ganglion activity, and presumably, cardiac adrenergic tone. In turn, the electrophysiologic stability of the myocardium was enhanced, protecting the heart from developing VT/VF.
Resiniferatoxin (RTX) depletes TRPV1 in the epicardium through direct application, exerting beneficial effects post-MI. A previous experiment found that CaMK II and RYR2 channel expression was reduced in subjects with HF treated with RTX5. Both are associated with impaired ventricular function. Furthermore, subjects with MI present with higher than normal tyrosine hydroxylase, connexin 43, TGF β1, and growth-associated protein 436. Such markers are associated with cardiac remodeling and electrical instability of the heart. In the case of subjects treated with RTX after the MI episode, these four variables were lower than expected. Consequently, improvements in the LV function and the ventricular electrophysiologic properties were recorded6.
How cardiac afferent depletion by RTX impacts myocardial gene expression to impart its beneficial effects remains unknown.
MATERIALS AND METHODS
The study was performed with the required approval from the host institution, using three groups of pigs, each group containing 4 subjects. The first group is a control group. The second one had a MI event induced. Lastly, the third group got both a MI and an RTX treatment. The cardiac tissue was obtained after cardiac dissection. The 12 samples were randomly arranged (control, infarcted, and treated-infarct animals). The specific pathways of interest researched were the GPCR signaling pathways for both M2 signaling pathway as well as the beta 1 pathway, the ion channel expression and regulation, the myocardial stress response pathways, and the inflammation pathways. The purification method used was a total RNA isolation using Qiagen RNAeasy Column. The concentration was measured via Nanodrop.
The data was obtained through Illumina sequencing (HiSeq 3000), which uses sequencing by synthesis mechanism. The kit used for the RNASeq libraries was a KAPA Stranded RNA-Seq Kit. A flow cell had the adapter (sequence primer) bound to the original sequence. After the polymerase elongated the complementary chain (i.e.: the reverse strand), the original strand was washed away. Now, the sequence primer complementary to the other end of the reverse strand caused the bending over and bridge amplification, leading to a forward strand alongside the reverse strand. This process was repeated until the PCR colony was obtained. Lastly, a sequence primer was added to the cluster. The cluster was stimulated by a light source and a fluorescent color was emitted each time the 4 competing nucleotides were added to the complementary strand. The depth of coverage was two-lane for all, with approximately 50 million reads. The read length was 1x50. The data quality check was done with Illumina SAV.
The data processing consisted of two major steps. Firstly, the data mapping was carried out using Bowtie2 version 2.1.0. Secondly, the gene expression level was estimated with RSEM v1.2.15. The gene expression was normalized using a trimmed mean of M-values. The results identified differentially expressed genes using EdgeR. The genes were filtered for a a fold change of > 1.5 and a p-value > 1 * 10e-5. RStudio was employed for the differentially expressed genes (Figure 1), for data visualization of outliers (Figure 2), and for the generation of heatmaps (Figure 3). The pathway enrichment analysis was conducted with the support of the gProfiler database. To better grasp the outliers and the genes differentially expressed in the RTX+MI vs. MI comparison, RStudio was used for data visualization. The ggplot library was part of the code (Figure 1 and Figure 2).
Figure 1. RTX+MI vs. MI - differentially expressed genes.

Figure 2. Outliers in the differential gene expression analysis of RTX+MI vs. MI.
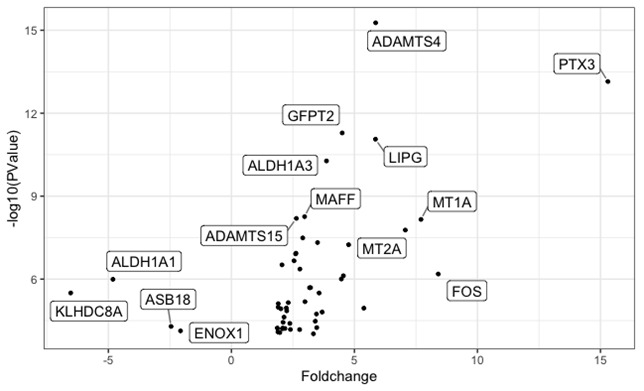
Figure 3. Gene Counts for MI+RTX vs. MI and MI vs. Control.
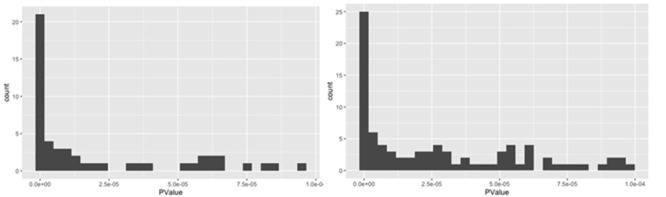
(A) MI + RTX vs. MI Counts of genes with P-values below 10e-5 (B) MI vs. Control Counts of genes with P values below 10e-5.
There were 85 differentially expressed genes (Figure 1) in the comparison between the MI and the control group, with a P value greater than 10e-5. In contrast, in the comparison conducted between the MI group and the MI group treated with resiniferatoxin only 51 genes had a P value greater than 10e-5 which were differentially expressed (see Supplementary Information). Some genes or families of genes were found differentially expressed in both comparisons, requiring further investigation. Table 1 below summarizes them. Moreover, tumoral genes which have an effect on cardiovascular disease were found to change in the RTX+MI group in comparison with the MI group (Table 2). The outliers in the DGE of RTX+MI vs. MI were plotted (Figure 2), alongside the pathways they are involved.
Table 1. Family of genes and genes differentially expressed in-between groups and common to both comparisons.
| Gene | RTX+MI vs. MI | MI vs. Control | ||
|---|---|---|---|---|
| Fold change | P value | Fold change | P value | |
| LIPG | 5.85 | 8.72*10e-12 | -2.58 | 1.56e-06 |
| ANGPTL4 vs. ANGPT2 | 2.54 | 2.16e-07 | -1.87 | 9.49e-06 |
| ADAMTS (-1&-4 vs. -2) | 2.78 & 5.86 | 4.34e-07 & 5.37e-16 | 2.63 | 2.81e-07 |
| MMP (-25 vs. -15) | 3.45 | 1.82e-05 | 2.23 | 1.96e-05 |
Table 2. Tumoral genes differentially expressed in RTX+MI vs. MI comparison.
| Tumoral genes | Foldchange | P-Value |
|---|---|---|
| CSRNP1 | 2.62 | 1.158e-07 |
| MYC | 2.61 | 1.219e-07 |
| FOS | 8.40 | 6.554e-07 |
RESULTS
There were 85 differentially expressed genes (Figure 1) in the comparison between the MI and the control group, with a P value greater than 10e-5. In contrast, in the comparison conducted between the MI group and the MI group treated with resiniferatoxin only 51 genes had a P value greater than 10e-5 which were differentially expressed (see Supplementary Information). Some genes or families of genes were found differentially expressed in both comparisons, requiring further investigation. Table 1 below summarizes them. Moreover, tumoral genes which have an effect on cardiovascular disease were found to change in the RTX+MI group in comparison with the MI group (Table 2). The outliers in the DGE of RTX+MI vs. MI were plotted (Figure 2), alongside the pathways they are involved.
DISCUSSION
Our study opens new questions. Resiniferatoxin seemed to modulate expression of both deleterious and beneficial genes involved in the complications following myocardial infarction. Thus, we discussed which genes of interest need further investigation to prove the beneficial effects of resiniferatoxin post-MI.
Family of genes and genes commonly found in both differential expression analyses
Between the genes differentially expressed in both comparisons (MI+RTX vs. MI and MI vs. control), we found lipase G, endothelial type (LIPG) is an enzyme that plays a key role in both atherosclerosis and oncogenesis. Its deleterious effects on atherosclerosis have been proven previously7. Moreover, patients with atherosclerosis have high levels of LIPG expression7. Its role in inflammation is described by LIPG's relationship with cytokines. More precisely, IL6 is a potent activator of atherosclerosis, while being an up regulator of LIPG8. On the other hand, previous studies used LIPG inhibitors to prevent and treat cardiovascular diseases9. The therapeutic result was an increased expression of cardioprotective HDL-C9.
Our research results reveal that RTX modulates LIPG expression, which in turn may potentially explain the previously observed beneficial roles of RTX, through a cardioprotective role. In our MI group, an almost 6-fold increase in LIPG expression was measured in comparison with the RTX+MI group. On the other hand, the control group had 2.58-fold lower levels in their LIPG expression, demonstrating the role of MI in upregulating LIPG expression.
ADAMTS-1, alongside ADAMTS-4,-5,-8, and -15, is one of the proteoglycanases responsible for cleaving proteoglycans10. ADAMTS-1 is responsible for cleaving versican, a proteoglycan of early and late atherosclerotic disease. Its cleavage allows vascular smooth muscle cells to invade the intima and proliferate, leading to neovascularization. Moreover, its decomposition compromises the fibrous cap integrity11-13. Overexpression of ADAMTS-1 was found with the neointimal formation in a murine model of carotid artery stenosis14. In a previous GWAS, the heterozygous and homozygous genes encoding for a proline amino acid were shown to increase the risk of an acute coronary syndrome15. It was shown that pravastatin therapy reduced considerably the risk in these patients.
RTX modulates ADAMTS-1 in patients that suffered from myocardial infarction. Our findings revealed that RTX-treated subjects post-MI had a 2.78-fold decrease in ADAMTS1 in comparison with the MI subjects. This suggests a potentially protective role of RTX similar to pravastatin for future ACS, although further experiments need to be performed for validation.
ADAMTS-2 plays a key role in cardiac remodeling, functioning as a procollagen N-propeptidase16. It was found that patients with HCM have ADAMTS-2 upregulated to promote myocardial repair and scarring17. However, overexpression of ADAMTS-2 leads to inhibition of the PI3K/AKT pathway involved in cardiac remodeling. In our cohort, the MI patients had increased gene expression levels of ADAMTS-2 in comparison with the controls, indicating remodeling impairments.
Our findings suggest an almost 6-fold increase in ADAMTS-4 in patients with MI without the RTX treatment. ADAMTS-4 is a proteinase responsible for the degradation of versican, an extracellular matrix protein. Recent reports indicate a higher ADAMTS-4 expression in patients with unstable atherosclerotic plaques as well as ACS18-22. A previous baboon aortoiliac graft study identified the role ADAMTS-4 plays in vascular smooth muscle cell death23. Therefore, the decreased ADAMTS-4 expression of the RTX-treated pigs correlates with a reduced vascular smooth muscle cell death. In a mice model, ADAMTS-4 knock-out mice showed less lipid and macrophage accumulation with a more stable plaque, comprising a thicker fibrous cap with more collagen deposition24. Since RTX has a similar effect to ADAMTS-4 reduction, RTX could be translated into the clinic as a plaque stabilizer for preventing postMI complications.
Another pair differentially expressed in the two comparative groups was the angiopoietin family. In the RTX+MI vs. MI comparison, the ANGPTL4 gene was found to be 2.54 fold more expressed in the MI group. Inactivating mutations in ANGPTL4 led to lower levels of triglycerides and higher levels of HD18. Moreover, monoclonal antibody inhibition of ANGPTL4 in monkey and rat models significantly reduced triglycerides. All in all, inactivating variants of ANGPTL4 are associated with reduced CAD in humans18.
ANGPT2 is the other member of the angiopoietin family that showed a differential expression in the MI group in comparison with the control group. In the literature, ANGPT2 transcription is usually elevated in endothelial cells at the infarct border zone19. Its expression enhances hypoxia and neutrophil infiltration as well as vascular permeability. Our findings suggest an upregulation of the ANGPT2 gene in the MI group. The control group had a -1.84 fold change in its expression.
All in all, resiniferatoxin seems to modulate genes involved in CAD and the atherosclerotic plaque. Thus, it could play a role in modulating reinfarction following stent treatment.
Possible resiniferatoxin effects on cardiovascular gene expression
Although it remains to be proved whether RTX had a significant clinical effect, the differential gene expression analysis between the MI and MI+RTX group highlighted significant differences in gene expression for genes linked to cardiovascular disease.
A pair of highly expressed transcripts in the MI group in comparison with the RTX+MI group was MMP 25 and TIMP1. Research shows that pharmacologic inhibition of MMPs contributes to LV dilation attenuation. This process was proposed as a potential therapy for HF after MI20. TIMP1 plays two key roles in the relationship with MMPs. Firstly, TIMPs inhibit MMPs21. Secondly, TIMP1 inhibits angiogenesis required for revascularization of the infarcted area21. Our research shows that patients with MI had a 3.45-fold increase in the expression of MMP 25 and a 4.46-fold higher level of TIMP1. These findings may potentially suggest that neovascularization is more impaired in the MI group, although further experiments are required to have a definitive conclusion.
A potent and beneficial effect resiniferatoxin exerts on PTX3 gene expression. Pentraxin 3 is responsible for angiogenesis and tissue remodeling. The PLATO trial25 showed that admission serum levels of PTX3 are correlated with cardiovascular death and spontaneous MI. PTX3 has a higher sensitivity than CRP and a strong association with NT-proBNP and troponin T levels. Thus, as resiniferatoxin subjects showed a 15-fold decrease, RTX could be used as a lowering agent during the admission process for a STEMI.
The HUNT2 study26 highlighted the role PTX3 had as a biomarker for a first MI event in a study population observed for 10 years. There are already trials that use other agents, such as rosuvastatin, to decrease PTX327. The literature on PTX3 finds that estimating the cardiovascular mortality risk with PTX3 and NT-proBNP increases the accuracy of the estimation28. Thus, we propose a future study to verify the translation of the resiniferatoxin benefits into a protocol consisting of PTX3 determination and cardiovascular risk evaluation, followed by RTX treatment for patients at high risk of a first MI event.
Under hypoxic strain, cardiomyocytes undergo activation of the Wnt/β-catenin pathway. The ortholog in humans of CSRNP1 is Axud129. Axud1 is a proapoptotic protein. In the literature, Axud1-knockdown mice had less activation of the Wnt/β-catenin pathway and reduced apoptosis30. Thus, by reducing the levels of CSRNP1, RTX may have some inhibitory roles on apoptosis following MI that needs to be further investigated.
A previous differential gene expression study demonstrated higher levels of FOS expression in patients who developed HF post-MI compared to patients who had a MI without HF31. Since in the RTX+MI group the expression was lower, these subjects can be protected against future HF. FOS was found to be upregulated in hypertrophied infarcted myocardium32. As such RTX decreasing FOS might provide a protective role against excessive hypertrophy following MI, an idea which needs further validation.
A previous study highlighted how miR-142-5p and miR-212-5p regulate the c-MYC pathway during myocardial infarction33. The overexpression of miRNA led to the suppression of proliferation and collagen formation. Thus, the mice which had a hypoxic-ischemic injury improved their heart function. Since RTX suppresses MYC as miR-142-5p and miR-212-5p do, it can be hypothesized that RTX improves cardiac function through suppression of c-Myc, a hypothesis that needs to be further studied.
Overexpression of THBS1 (thrombospondinin-1) was shown in literature to have deleterious cardiac effects and its expression was inhibited by RTX. A differential gene expression study34 identified THBS-1 as a candidate biomarker for cardiac hypertrophy. Moreover, THBS-1 gene was overexpressed in endothelin-stimulated cardiomyocytes, leading to hypertrophy. Another instance when THBS-1 was overexpressed was in the case of autophagy-mediated cardiac atrophy35. To corroborate these aspects with our data, RTX might protect against excessive hypertrophy post-MI as well as against cardiomyocyte atrophy following infarction. The MI group had a 3.56 fold increase in the THBS1 expression in comparison with the RTX+MI group. Further investigation is warranted to verify this hypothesis.
RTX has an effect on the extracellular space, by reducing the expression of SEMA3F (2.9 fold). The previous research36 showed that SEMA3F has a key role in upregulating PECAM1 and leukocyte extravasation. Furthermore, the serum levels of cardiac arrest patients were found to be elevated after cardiac arrest. These levels were associated with decreased survival, myocardial dysfunction and prolonged vasopressor activity. By modulating SEMA3F, RTX could potentially reduce the risk of HF, prolong survival, prevent fatal arrhythmias that cause cardiac arrest and most certainly reduce the inflammation after MI.
RTX impacts the NAD+ pathway as well. In vitro studies37 identified that highly expressing ALDH1A3 cardiomyocytes are prone to excessive proliferation into the atrium. These cells were collected from patients with ischemic disease. By decreasing 3.86 fold, RTX could potentially protect against the electrical instability of the atria post-MI, a hypothesis which remains to be further investigated.
CONCLUSION
All in all, resiniferatoxin modulates the expression of important genes that are potentially related with beneficial outcomes following myocardial infarction. By downregulating the expression of genes involved in inflammation, plaque instability, and inappropriate angiogenesis, resiniferatoxin is a potential prophylactic method for future ACS in myocardial infarction patients. Additional detailed investigations have to be performed to confirm the beneficial role of the indicated gene expression changes post-MI.
Acknowledgments
We thank the Cardiac Arrhythmia Center (Neurocardiology Laboratory) from UCLA for providing the data used in this analysis.AAM was funded by George E. Palade fellowship during his research internship at the UCLA, CA, USA.
Footnotes
Conflict of interests: The author declares no conflicts of interest
Abbreviations: Ventricular tachycardia (VT); ventricular fibrillation (VF); transient receptor potential channel 1 (TRPV1); myocardial infarction (MI); resiniferatoxin (RTX); heart failure (HF); left ventricle (LV), cardiac sympathetic afferent reflex (CSAR), hypertrophic cardiomyopathy (HCM), acute coronary syndrome (ACS), matrix metalloproteinase MMP.
DISCOVERIES is a peer-reviewed, open access, online, multidisciplinary and integrative journal, publishing high impact and innovative manuscripts from all areas related to MEDICINE, BIOLOGY and CHEMISTRY
References
- 1.Mechanosensitive TRP channels in cardiovascular pathophysiology. Inoue Ryuji, Jian Zhong, Kawarabayashi Yasuhiro. Pharmacology & Therapeutics. 2009;123(3):371-385. doi: 10.1016/j.pharmthera.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Resiniferatoxin reduces cardiac sympathetic nerve activation to exert a cardioprotective effect during myocardial infarction. Su Ludefu, Liu Yu, Tang Yanhong, Zhou Mingmin, Xiong Liang, Huang Congxin. International journal of clinical and experimental pathology. 2021;14(4):408–416. [PMC free article] [PubMed] [Google Scholar]
- 3.Capsaicin-Sensitive Sensory Nerves and the TRPV1 Ion Channel in Cardiac Physiology and Pathologies. Szabados Tamara, Gömöri Kamilla, Pálvölgyi Laura, Görbe Anikó, Baczkó István, Helyes Zsuzsanna, Jancsó Gábor, Ferdinandy Péter, Bencsik Péter. International journal of molecular sciences. 2020;21(12) doi: 10.3390/ijms21124472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Resiniferatoxin reduces ventricular arrhythmias in heart failure via selectively blunting cardiac sympathetic afferent projection into spinal cord in rats. Wu Yong, Hu Zhengtao, Wang Deguo, Lv Kun, Hu Nengwei. European journal of pharmacology. 2020;867:172836. doi: 10.1016/j.ejphar.2019.172836. [DOI] [PubMed] [Google Scholar]
- 5.Inhibition of DRG-TRPV1 upregulation in myocardial ischemia contributes to exogenous cardioprotection. Xu Shijin, Xu Yan, Cheng Xueying, Huang Cheng, Pan Yonglu, Jin Shiyun, Xiong Wei, Zhang Li, He Shufang, Zhang Ye. Journal of molecular and cellular cardiology. 2020;138:175–184. doi: 10.1016/j.yjmcc.2019.12.003. [DOI] [PubMed] [Google Scholar]
- 6.Cardiac TRPV1 afferent signaling promotes arrhythmogenic ventricular remodeling after myocardial infarction. Yoshie Koji, Rajendran Pradeep S, Massoud Louis, Mistry Janki, Swid M Amer, Wu Xiaohui, Sallam Tamer, Zhang Rui, Goldhaber Joshua I, Salavatian Siamak, Ajijola Olujimi A. JCI insight. 2020;5(3) doi: 10.1172/jci.insight.124477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Endothelial lipase plasma levels are increased in both sexes in stable coronary artery disease and only in women with acute coronary syndrome but not associated with the severity of coronary artery disease. Trbušić M, Potočnjak I, Tiran B, Bodrožić-Džakić T, Milošević M, Degoricija V, Frank S. Croat Med J. 2016 Oct 31;57(5):482-492. 2016;57(5):482–492. [PubMed] [Google Scholar]
- 8.Endothelial lipase is upregulated by interleukin-6 partly via the p38 MAPK and p65 NF-κB signaling pathways. Yue Xin, Wu Minghui, Jiang Hong, Hao Jing, Zhao Qinghao, Zhu Qing, Saren Gaowa, Zhang Yun, Zhang Xiaoli. Molecular Medicine Reports. 2016;14(3):1979-1985. doi: 10.3892/mmr.2016.5457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Discovery and synthesis of tetrahydropyrimidinedione-4-carboxamides as endothelial lipase inhibitors. Hu Carol H., Wang Tammy C., Qiao Jennifer X., Haque Lauren, Chen Alice Y.A., Taylor David S., Ying Xiaohong, Onorato Joelle M., Galella Michael, Shen Hong, Huang Christine S., Toussaint Nathalie, Li Yi-Xin, Abell Lynn, Adam Leonard P., Gordon David, Wexler Ruth R., Finlay Heather J. Bioorganic & Medicinal Chemistry Letters. 2018;28(23-24):3721-3725. doi: 10.1016/j.bmcl.2018.10.022. [DOI] [PubMed] [Google Scholar]
- 10.ADAMTS proteases in cardiovascular physiology and disease. Santamaria Salvatore, de Groot Rens. Open Biology. 2020;10(12):200333. doi: 10.1098/rsob.200333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. Sandy J D, Westling J, Kenagy R D, Iruela-Arispe M L, Verscharen C, Rodriguez-Mazaneque J C, Zimmermann D R, Lemire J M, Fischer J W, Wight T N, Clowes A W. The Journal of biological chemistry. 2001;276(16):13372–8. doi: 10.1074/jbc.M009737200. [DOI] [PubMed] [Google Scholar]
- 12.Proteoglycans in Atherosclerosis and Restenosis. Wight Thomas N., Merrilees Mervyn J. Circulation Research. 2004;94(9):1158-1167. doi: 10.1161/01.RES.0000126921.29919.51. [DOI] [PubMed] [Google Scholar]
- 13.Versican: a versatile extracellular matrix proteoglycan in cell biology. Wight Thomas N. Current Opinion in Cell Biology. 2002;14(5):617-623. doi: 10.1016/s0955-0674(02)00375-7. [DOI] [PubMed] [Google Scholar]
- 14.Role of ADAMTS-1 in atherosclerosis: remodeling of carotid artery, immunohistochemistry, and proteolysis of versican. Jönsson-Rylander Ann-Cathrine, Nilsson Tina, Fritsche-Danielson Regina, Hammarström Anette, Behrendt Margareta, Andersson Jan-Olof, Lindgren Kerstin, Andersson Ann-Katrin, Wallbrandt Pia, Rosengren Birgitta, Brodin Peter, Thelin Anders, Westin Annika, Hurt-Camejo Eva, Lee-Søgaard Chung-Hyun. Arteriosclerosis, thrombosis, and vascular biology. 2005;25(1):180–5. doi: 10.1161/01.ATV.0000150045.27127.37. [DOI] [PubMed] [Google Scholar]
- 15.Association between ADAMTS1 matrix metalloproteinase gene variation, coronary heart disease, and benefit of statin therapy. Sabatine Marc S, Ploughman Lynn, Simonsen Katy L, Iakoubova Olga A, Kirchgessner Todd G, Ranade Koustubh, Tsuchihashi Zenta, Zerba Kim E, Long Diane U, Tong Carmen H, Packard Christopher J, Pfeffer Marc A, Devlin James J, Shepherd James, Campos Hannia, Sacks Frank M, Braunwald Eugene. Arteriosclerosis, thrombosis, and vascular biology. 2008;28(3):562–7. doi: 10.1161/ATVBAHA.107.156653. [DOI] [PubMed] [Google Scholar]
- 16.Human Ehlers-Danlos syndrome type VII C and bovine dermatosparaxis are caused by mutations in the procollagen I N-proteinase gene. Colige A, Sieron A L, Li S W, Schwarze U, Petty E, Wertelecki W, Wilcox W, Krakow D, Cohn D H, Reardon W, Byers P H, Lapière C M, Prockop D J, Nusgens B V. American journal of human genetics. 1999;65(2):308–17. doi: 10.1086/302504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Critical Role of ADAMTS2 (A Disintegrin and Metalloproteinase With Thrombospondin Motifs 2) in Cardiac Hypertrophy Induced by Pressure Overload. Wang Xiaodi, Chen Wen, Zhang Jie, Khan Aiman, Li Liangpeng, Huang Fuhua, Qiu Zhibing, Wang Liming, Chen Xin. Hypertension. 2017;69(6):1060-1069. doi: 10.1161/HYPERTENSIONAHA.116.08581. [DOI] [PubMed] [Google Scholar]
- 18.Inactivating Variants in <i>ANGPTL4</i> and Risk of Coronary Artery Disease. Dewey Frederick E., Gusarova Viktoria, O’Dushlaine Colm, Gottesman Omri, Trejos Jesus, Hunt Charleen, Van Hout Cristopher V., Habegger Lukas, Buckler David, Lai Ka-Man V., Leader Joseph B., Murray Michael F., Ritchie Marylyn D., Kirchner H. Lester, Ledbetter David H., Penn John, Lopez Alexander, Borecki Ingrid B., Overton John D., Reid Jeffrey G., Carey David J., Murphy Andrew J., Yancopoulos George D., Baras Aris, Gromada Jesper, Shuldiner Alan R. New England Journal of Medicine. 2016;374(12):1123-1133. doi: 10.1056/NEJMoa1510926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Angiopoietin-2 exacerbates cardiac hypoxia and inflammation after myocardial infarction. Lee Seung-Jun, Lee Choong-kun, Kang Seok, Park Intae, Kim Yoo Hyung, Kim Seo Ki, Hong Seon Pyo, Bae Hosung, He Yulong, Kubota Yoshiaki, Koh Gou Young. Journal of Clinical Investigation. 2018;128(11):5018-5033. doi: 10.1172/JCI99659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matrix Metalloproteinase Inhibition After Myocardial Infarction. Creemers Esther E.J.M., Cleutjens Jack P.M., Smits Jos F.M., Daemen Mat J.A.P. Circulation Research. 2001;89(3):201-210. doi: 10.1161/hh1501.094396. [DOI] [PubMed] [Google Scholar]
- 21.Matrix metalloproteinases in cardiovascular disease. Liu Peter, Sun Mei, Sader Sawsan. Canadian Journal of Cardiology. 2006;22:25B-30B. doi: 10.1016/s0828-282x(06)70983-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Relationship between ADAMTS4 and carotid atherosclerotic plaque vulnerability in humans. Dong Honglin, Du Tian, Premaratne Shyamal, Zhao Cynthia X., Tian Qinqin, Li Yongjun, Yan Sheng, Zhang Wayne W. Journal of Vascular Surgery. 2018;67(4):1120-1126. doi: 10.1016/j.jvs.2017.08.075. [DOI] [PubMed] [Google Scholar]
- 23.Cell Death–associated ADAMTS4 and Versican Degradation in Vascular Tissue. Kenagy Richard D., Min Seung-Kee, Clowes Alexander W., Sandy John D. Journal of Histochemistry & Cytochemistry. 2009;57(9):889-897. doi: 10.1369/jhc.2009.953901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loss of ADAMTS4 reduces high fat diet-induced atherosclerosis and enhances plaque stability in ApoE−/− mice. Kumar Saran, Chen Mo, Li Yan, Wong Fiona H. S., Thiam Chung Wee, Hossain Md Zakir, Poh Kian Keong, Hirohata Satoshi, Ogawa Hiroko, Angeli Véronique, Ge Ruowen. Scientific Reports. 2016;6(1) doi: 10.1038/srep31130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pentraxin-3 vs C-reactive protein and other prognostic biomarkers in acute coronary syndrome: A substudy of the Platelet Inhibition and Patients Outcomes (PLATO) trial. Kontny Frederic, Andersen Thomas, Ueland Thor, Åkerblom Axel, Lakic Tatevik G, Michelsen Annika E, Aukrust Pål, Bertilsson Maria, Becker Richard C, Himmelmann Anders, James Stefan K, Siegbahn Agneta, Storey Robert F, Wallentin Lars. European Heart Journal: Acute Cardiovascular Care. 2019;9(4):313-322. doi: 10.1177/2048872619846334. [DOI] [PubMed] [Google Scholar]
- 26.Pentraxin 3, ficolin-2 and lectin pathway associated serine protease MASP-3 as early predictors of myocardial infarction - the HUNT2 study. Vengen Inga Thorsen, Enger Tone Bull, Videm Vibeke, Garred Peter. Scientific reports. 2017;7:43045. doi: 10.1038/srep43045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Effects of rosuvastatin on pentraxin 3 level and platelet aggregation rate in elderly patients with acute myocardial infarction undergoing elective interventional therapy: a double-blind controlled study. Li Y-H, Wang L-H, Li Q, Yu D-W, Gai Y-S, Cai S-X, Tian F, Zhou H-Y. European review for medical and pharmacological sciences. 2017;21(16):3730–3735. [PubMed] [Google Scholar]
- 28.Combining Novel Biomarkers for Risk Stratification of Two-Year Cardiovascular Mortality in Patients with ST-Elevation Myocardial Infarction. Zagidullin Naufal, Motloch Lukas J, Gareeva Diana, Hamitova Aysilu, Lakman Irina, Krioni Ilja, Popov Denis, Zulkarneev Rustem, Paar Vera, Kopp Kristen, Jirak Peter, Ishmetov Vladimir, Hoppe Uta C, Tulbaev Eduard, Pavlov Valentin. Journal of clinical medicine. 2020;9(2) doi: 10.3390/jcm9020550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Characterization of a family of novel cysteine- serine-rich nuclear proteins (CSRNP). Gingras Sébastien, Pelletier Stéphane, Boyd Kelli, Ihle James N. PloS one. 2007;2(8):e808. doi: 10.1371/journal.pone.0000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Axin1 up-regulated 1 accelerates stress-induced cardiomyocytes apoptosis through activating Wnt/β-catenin signaling. Ye Xing, Lin Junyi, Lin Zebin, Xue Aimin, Li Liliang, Zhao Ziqin, Liu Li, Shen Yiwen, Cong Bin. Experimental cell research. 2017;359(2):441–448. doi: 10.1016/j.yexcr.2017.08.027. [DOI] [PubMed] [Google Scholar]
- 31.Identification of potentially critical genes in the development of heart failure after ST-segment elevation myocardial infarction (STEMI). Qian Cheng, Chang Danqi, Li Hang, Wang Yanggan. Journal of cellular biochemistry. 2019;120(5):7771–7777. doi: 10.1002/jcb.28051. [DOI] [PubMed] [Google Scholar]
- 32.Alterations in cardiac gene expression during ventricular remodeling following experimental myocardial infarction. Gidh-Jain M, Huang B, Jain P, Gick G, El-Sherif N. Journal of molecular and cellular cardiology. 1998;30(3):627–37. doi: 10.1006/jmcc.1997.0628. [DOI] [PubMed] [Google Scholar]
- 33.miR-142-5p and miR-212-5p cooperatively inhibit the proliferation and collagen formation of cardiac fibroblasts by regulating c-Myc/TP53INP1. Wang Zhiqian, Fu Mingming, Li Yongjun. Canadian journal of physiology and pharmacology. 2020;98(5):314–323. doi: 10.1139/cjpp-2019-0495. [DOI] [PubMed] [Google Scholar]
- 34.Data Mining Identifies CCN2 and THBS1 as Biomarker Candidates for Cardiac Hypertrophy. Johansson Markus, Tangruksa Benyapa, Heydarkhan-Hagvall Sepideh, Jeppsson Anders, Sartipy Peter, Synnergren Jane. Life (Basel, Switzerland) 2022;12(5) doi: 10.3390/life12050726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated autophagy. Vanhoutte Davy, Schips Tobias G, Vo Alexander, Grimes Kelly M, Baldwin Tanya A, Brody Matthew J, Accornero Federica, Sargent Michelle A, Molkentin Jeffery D. Nature communications. 2021;12(1):3928. doi: 10.1038/s41467-021-24215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Semaphorin 3F Promotes Transendothelial Migration of Leukocytes in the Inflammatory Response After Survived Cardiac Arrest. Reichert Stephanie, Scheid Stefanie, Roth Tina, Herkel Marius, Petrova Diana, Linden Alexandra, Weberbauer Miki, Esser Jennifer, Diehl Philipp, Grundmann Sebastian, Busch Hans-Jörg, Fink Katrin, Bode Christoph, Moser Martin, Helbing Thomas. Inflammation. 2019;42(4):1252–1264. doi: 10.1007/s10753-019-00985-4. [DOI] [PubMed] [Google Scholar]
- 37.ALDH1A3 Is the Key Isoform That Contributes to Aldehyde Dehydrogenase Activity and Affects in Vitro Proliferation in Cardiac Atrial Appendage Progenitor Cells. Puttini Stefania, Plaisance Isabelle, Barile Lucio, Cervio Elisabetta, Milano Giuseppina, Marcato Paola, Pedrazzini Thierry, Vassalli Giuseppe. Frontiers in cardiovascular medicine. 2018;5:90. doi: 10.3389/fcvm.2018.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]