

Abstract
Naked mole‐rats (NMRs) have exceptional longevity and are resistant to age‐related physiological decline and diseases. Given the role of cellular senescence in aging, we postulated that NMRs possess unidentified species‐specific mechanisms to prevent senescent cell accumulation. Here, we show that upon induction of cellular senescence, NMR fibroblasts underwent delayed and progressive cell death that required activation of the INK4a‐retinoblastoma protein (RB) pathway (termed “INK4a‐RB cell death”), a phenomenon not observed in mouse fibroblasts. Naked mole‐rat fibroblasts uniquely accumulated serotonin and were inherently vulnerable to hydrogen peroxide (H2O2). After activation of the INK4a‐RB pathway, NMR fibroblasts increased monoamine oxidase levels, leading to serotonin oxidization and H2O2 production, which resulted in increased intracellular oxidative damage and cell death activation. In the NMR lung, induction of cellular senescence caused delayed, progressive cell death mediated by monoamine oxidase activation, thereby preventing senescent cell accumulation, consistent with in vitro results. The present findings indicate that INK4a‐RB cell death likely functions as a natural senolytic mechanism in NMRs, providing an evolutionary rationale for senescent cell removal as a strategy to resist aging.
Keywords: cell death, cellular senescence, INK4a, monoamine oxidase, naked mole‐rat
Subject Categories: Autophagy & Cell Death, Metabolism, Respiratory System
Long‐lived naked mole‐rats have a unique response to cellular senescence induction that may underlie their ability to prevent their senescent cell accumulation, a phenomenon associated with longevity.

Introduction
The naked mole‐rat (Heterocephalus glaber, NMR; Fig 1A) is an African mammal that forms a eusocial colony in subterranean environments (Jarvis, 1981). The NMR has extraordinary longevity, with a maximum life span of more than 37 years, although its body mass is similar to that of the laboratory mouse (Buffenstein, 2008; Lee et al, 2020; Oka et al, 2023). Moreover, the NMR shows a unique delayed aging phenotype (Buffenstein, 2008). The mortality rate of the NMR does not increase during its lifetime, and physiological functions, including fecundity, are maintained until near the end of its life (Ruby et al, 2018; Brieño‐Enríquez et al, 2023). In addition, the NMR is unusually resistant to several age‐related diseases such as metabolic disorders, neurodegenerative diseases, and cancer (Ungvari et al, 2008; Finch, 2009; Edrey et al, 2011).
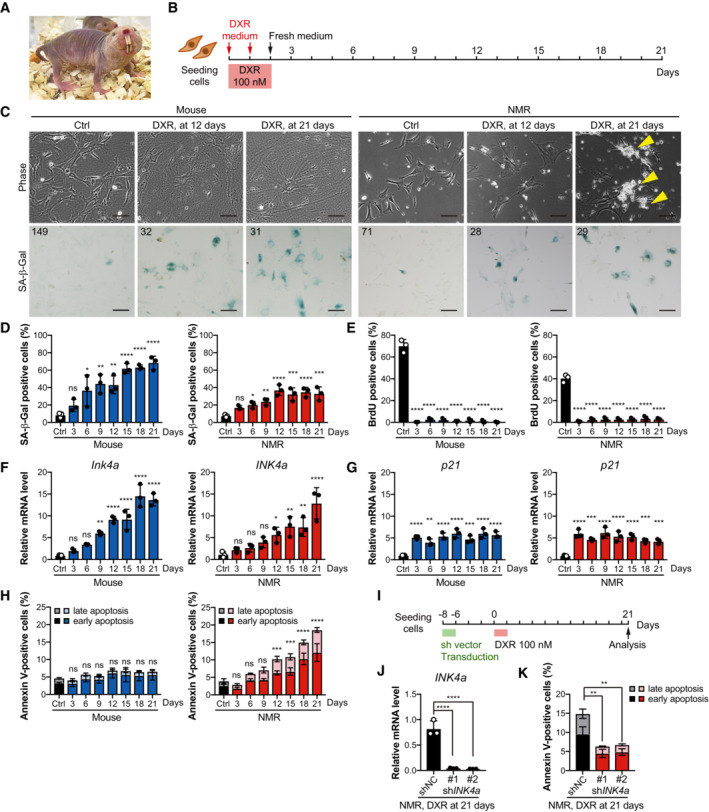
Figure 1. Induction of cellular senescence by DNA damage leads to progressive cell death, a delayed response after upregulation of INK4a in NMR fibroblasts.
-
AAdult naked mole‐rats (NMRs).
-
BScheme for doxorubicin (DXR) treatment and time‐course analysis.
-
CCell morphology and SA‐β‐Gal activity of mouse or NMR fibroblasts 12 or 21 days after DXR treatment. Arrowheads indicate dying cells. The number in the upper left corner indicates Hoechst‐positive nuclei. Scale bar, 100 μm.
-
D–HTime‐course analysis of mouse or NMR fibroblasts after DXR treatment: quantification of SA‐β‐Gal‐positive cells (%) (D); quantification of BrdU‐positive cells (%) (E); qRT‐PCR analysis of the expression of INK4a (F) or p21 (G) normalized to ACTB mRNA levels; quantification of Annexin V‐positive cells (%) (Annexin V+/PI− as early apoptotic and Annexin V+/PI+ double‐positive as late apoptotic) (H).
-
IScheme for DXR treatment after INK4a knockdown in NMR fibroblasts.
-
J, KqRT‐PCR analysis of the expression of INK4a (J) and quantification of Annexin V‐positive cells (%) (K) in shINK4a‐transduced NMR fibroblasts at 21 days after DXR treatment.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, not significant. One‐way ANOVA followed by Dunnett's multiple comparison test for (D–H, J, and K). Data are expressed as the mean ± SD from n = 3 biological replicates.
Recently, several mechanisms that may contribute to the NMR's longevity, delayed aging, and cancer‐resistance phenotypes have been proposed. These mechanisms include a high DNA repair capacity (Evdokimov et al, 2018; Tian et al, 2019; Yamamura et al, 2021), low L1 retrotransposition activity (Yamaguchi et al, 2021), high translational fidelity (Azpurua et al, 2013), high protein stability (Perez et al, 2009), high signaling activity of nuclear factor erythroid 2‐related factor 2 (NRF2) (Lewis et al, 2015), production of high molecular mass hyaluronan (Tian et al, 2013), a dampened tissue inflammatory response and inability to induce necroptosis (Oka et al, 2022), and resistance to oncogenic genes transduction and in vitro reprogramming (Tian et al, 2013; Miyawaki et al, 2016; Tan et al, 2017).
Cellular senescence is an irreversible cell proliferation arrest that is induced in response to stresses such as telomere shortening, genotoxic stress, and oncogene activation (Gorgoulis et al, 2019). In mammals, stressed cells activate p16INK4a (INK4a), which is a cyclin‐dependent kinase inhibitor (CKI). INK4a inhibits the cyclin‐dependent kinases CDK4/6, and the inactivation of CDK4/6 results in activation of the tumor suppressor retinoblastoma (RB) protein family. Stressed cells also activate p53, which in turn upregulates another CKI, p21CIP1 (p21). p21 inhibits CDK2, resulting in activation of RB (Herranz & Gil, 2018). Persistent activation of RB is essential for irreversible cell cycle arrest in senescent cells (Gorgoulis et al, 2019).
Senescent cells acquire resistance to apoptosis and secrete various proteins, a response known as the senescence‐associated secretory phenotype (SASP), which includes secretion of pro‐inflammatory cytokines and chemokines (Coppé et al, 2008, 2010; Gorgoulis et al, 2019; Birch & Gil, 2020). During aging, senescent cells accumulate in tissues and promote inflammation, resulting in age‐related physiological decline and disorders including cancer (Baker et al, 2011, 2016; Calcinotto et al, 2019; Gasek et al, 2021).
Although NMR fibroblasts do not exhibit replicative senescence (Seluanov et al, 2008), they become senescent after DNA damage or oncogene activation (Zhao et al, 2018). Our group and Chee et al. showed that suppression of alternative reading frame or catenin beta 1 induces cellular senescence in NMR fibroblasts (Miyawaki et al, 2016; Chee et al, 2021). These observations indicate that NMR cells can undergo cellular senescence in several situations. However, the fate of senescent NMR cells remains poorly characterized. In contrast to these in vitro observations, Lee et al. recently showed that INK4a expression in NMR brain tissues only increases slightly during aging (Lee et al, 2020). This observation suggests that NMRs possess unidentified mechanism(s) to prevent the accumulation of senescent cells.
In mice, targeted removal of senescent cells (senolysis) inhibits age‐related deterioration and diseases (Baker et al, 2011, 2016), prompting clinical trials in humans (Calcinotto et al, 2019; Gasek et al, 2021); however, whether the findings in mice are generalizable remains an open question (Gasek et al, 2021). In this study, we investigated the fate of NMR cells after induction of cellular senescence. The results showed that upon cellular senescence induction, NMR cells underwent delayed, progressive cell death through activation of the INK4a‐RB pathway (termed “INK4a‐RB cell death”) and a unique regulation of serotonin metabolism by monoamine oxidases (MAOs). This INK4a‐RB cell death in NMRs can be regarded as “natural senolysis” and provides an evolutionary rationale for removing senescent cells as a therapeutic strategy to prevent aging.
Results
Induction of cellular senescence by DNA damage leads to progressive cell death, a delayed response after upregulation of INK4a in NMR fibroblasts
Doxorubicin (DXR), a DNA‐damaging agent, is often used to induce cellular senescence at low doses (100–250 nM) in human and mouse fibroblasts (Petrova et al, 2016; Baar et al, 2017; Demaria et al, 2017; Omori et al, 2020). To determine the fate of NMR cells after induction of cellular senescence, mouse (6‐week‐old, young adult) and NMR (1‐year‐old, young adult) primary skin fibroblasts were treated twice with 100 nM of DXR for 24 h, and a time‐course analysis was performed (Fig 1B). By Day 12 after DXR treatment, both mouse and NMR cells became flattened and enlarged (Fig 1C) and showed activation of markers related to cellular senescence, such as increased senescence‐associated β‐galactosidase (SA‐β‐Gal) activity (Fig 1C and D), decreased BrdU incorporation (Fig 1E), and increased expression of CKIs, that is, INK4a and p21 (Fig 1F and G). Progressive cell death, including Annexin V‐positive apoptosis, was observed only in NMR fibroblasts (Fig 1H). After cell cycle arrest, as indicated by the cessation of BrdU incorporation observed on Day 3 (Fig 1E), NMR cell death slowly increased, became significant at approximately Day 12, and continued to increase (Fig 1H). These suggest that delayed, progressive cell death occurs in NMR fibroblasts during the cellular senescence process after cell cycle arrest.
To investigate the effect of INK4a upregulation on progressive cell death, senescence was induced in NMR fibroblasts by exposure to DXR either before or after INK4a knockdown (Fig 1I, Appendix Fig S1A). In each experiment, cell proliferation increased, and the number of dead cells decreased markedly on Day 21 or Day 24 after DXR treatment (Fig 1J and K, Appendix Fig S1B–E). These results suggest that similar to mouse fibroblasts, senescence induction by DXR in NMR fibroblasts leads to INK4a upregulation, cell cycle arrest, and senescence. However, in contrast to mouse cells, NMR cells activate delayed and progressive cell death after INK4a activation and cell cycle arrest.
INK4a transduction leads to progressive cell death in NMR cells
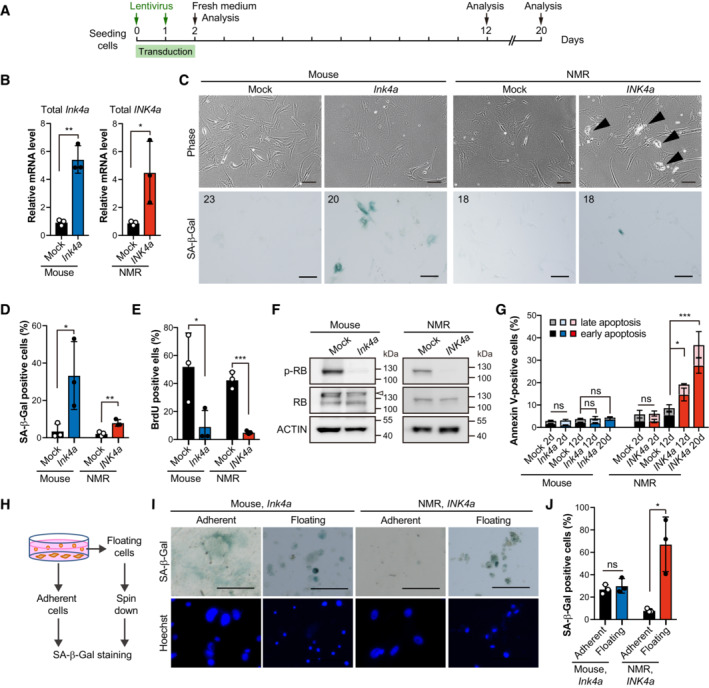
In general, upregulation of INK4a in mouse and human fibroblasts leads to cell cycle arrest and plays an important role in maintaining the senescent state, although it does not typically result in cell death (Al‐Mohanna et al, 2004; Mirzayans et al, 2012; Childs et al, 2014). To investigate whether upregulation of INK4a results in cell death in NMR skin fibroblasts, INK4a was upregulated in NMR fibroblasts by lentiviral transduction (Fig 2A and B). After Ink4a or INK4a transduction, both mouse and NMR fibroblasts showed increased SA‐β‐Gal activity (Fig 2C and D), decreased BrdU incorporation (Fig 2E), and activation (dephosphorylated) of the RB protein (Fig 2F). However, only NMR fibroblasts showed delayed, progressive cell death, including Annexin V‐positive apoptosis, on Day 12 after INK4a transduction, and cell death further increased on Day 20 (Fig 2G). When NMR INK4a was introduced into mouse fibroblasts, or mouse Ink4a into NMR fibroblasts, only NMR cells activated cell death, indicating that the sequence difference in Ink4a/INK4a (Miyawaki et al, 2015) did not affect cell death (Appendix Fig S2A and B). SA‐β‐Gal‐positive cells were significantly enriched in the floating dead cell population of INK4a‐transduced NMR fibroblasts, but not in the live adherent cell population, indicating that cell death occurs predominantly in SA‐β‐Gal‐positive cells (Fig 2H–J). In addition, we confirmed that dead senescent mouse fibroblasts killed by intense UV‐C irradiation maintained SA‐β‐Gal activity, whereas nonsenescent proliferating mouse and NMR fibroblasts killed by UV‐C did not show an increase in SA‐β‐Gal activity (Appendix Fig S2C). NMR fibroblasts derived from lung tissue also showed activation of cell death after transduction with INK4a (Appendix Fig S2D–F). These results demonstrate that INK4a upregulation in NMR fibroblasts leads to delayed and progressive activation of cell death.
Figure 2. INK4a transduction leads to progressive cell death in NMR cells.
-
AScheme for INK4a transduction.
-
BqRT‐PCR analysis of the expression of INK4a at 12 days after INK4a or mock transduction, normalized to ACTB mRNA levels.
-
CCell morphology and SA‐β‐Gal activity of mouse or NMR fibroblasts 12 days after INK4a transduction. Arrowheads indicate dying cells. The number in the upper left corner indicates Hoechst‐positive nuclei. Scale bar, 100 μm.
-
D, EQuantification of SA‐β‐Gal‐positive cells (%) (D) and BrdU‐positive cells (%) (E) at 12 days after INK4a transduction.
-
FWestern blot analysis of RB in mouse or NMR fibroblasts at 12 days after INK4a transduction (p, phospho‐specific antibody). ACTIN was used as a loading control. The arrowhead indicates a nonspecific band.
-
GQuantification of Annexin V/PI‐positive cells (%) (Annexin V+/PI− as early apoptotic and Annexin V+/PI+ double‐positive as late apoptotic) at 2, 12, or 20 days after INK4a transduction.
-
HScheme for SA‐β‐Gal staining of adherent and floating cells.
-
ISA‐β‐Gal activity in the adherent living cell population and floating dead cell population in mouse or NMR fibroblast cultures at 12 days after INK4a transduction. Scale bar, 100 μm.
-
JQuantification of SA‐β‐Gal‐positive cells in the adherent living cell population and floating dead cell population after the same treatment as in (I).
Data information: *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant. Unpaired t‐test versus control for (B, D, E, day 2 in G, and J). One‐way ANOVA followed by Dunnett's multiple comparison test for (day 12–20 in G). Data are expressed as the mean ± SD from n = 3 biological replicates.
Source data are available online for this figure.
Upregulation of INK4a results in progressive cell death via RB, independent of p53 in NMR fibroblasts
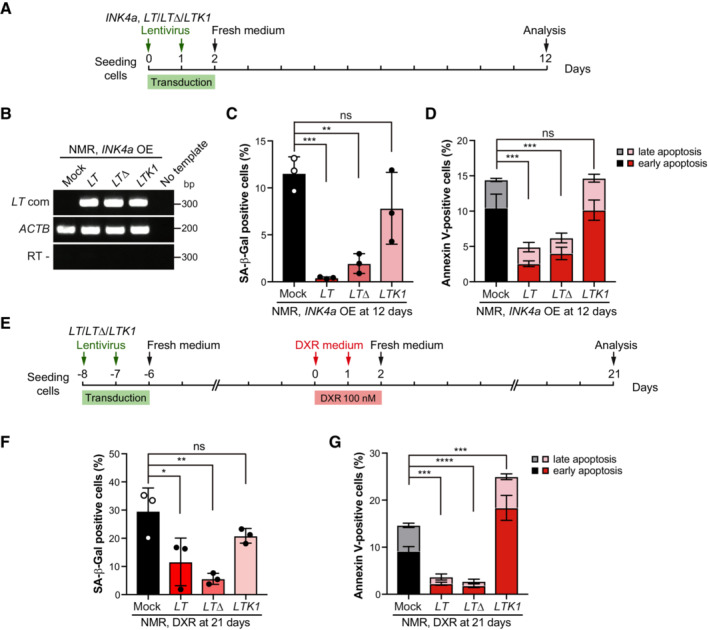
In mammalian cells, upregulation of INK4a results in activation of RB but not p53 (Ohtani et al, 2004). High levels of p53 activation lead to apoptotic gene transcription and ensuing apoptosis (Childs et al, 2014). To determine whether activation of the RB or p53 is required for the activation of progressive cell death in INK4a‐transduced NMR cells, we used the viral oncoprotein SV40 Large T antigen (LT) and its derivatives to suppress RB and p53 activities. Wild‐type LT inactivates both p53 and RB (p53‐/RB‐), the LTΔ434–444 (LTΔ) mutant inactivates only RB (p53+/RB−), and the LTK1 mutant inactivates only p53 (p53−/RB+) (Hahn et al, 2002). Each of these expression constructs, together with INK4a, was transduced into NMR fibroblasts (Fig 3A and B). Wild‐type LT (p53−/RB−) and LTΔ (p53+/RB−) strongly suppressed both SA‐β‐Gal activity and cell death in INK4a‐transduced cells (Fig 3C and D). By contrast, inactivation of p53 alone by LTK1 (p53−/RB+) had no effect on cell death in INK4a‐transduced cells (Fig 3D). This suggests that the induction of progressive cell death in INK4a‐transduced NMR fibroblasts requires RB activation and is independent of p53 activity.
Figure 3. Upregulation of INK4a results in progressive cell death via RB, independent of p53 in NMR fibroblasts.
-
AScheme for transduction of INK4a and different forms of SV40 Large T antigen (LT, LTΔ, and LTK1).
-
BRT‐PCR analysis of SV40 Large T expression in NMR‐fibroblasts transduced with different forms of SV40 Large T antigen (LT, LTΔ, and LTK1) and INK4a.
-
C, DQuantification of SA‐β‐Gal‐positive cells (%) (C) and Annexin V‐positive cells (%) (Annexin V+/PI− as early apoptotic and Annexin V+/PI+ double‐positive as late apoptotic) (D) in NMR‐fibroblasts transduced with different forms of SV40 Large T antigen (LT, LTΔ, and LTK1) and INK4a. OE; overexpression.
-
EScheme for doxorubicin (DXR) treatment following transduction of different forms of SV40 Large T antigen (LT, LTΔ, and LTK1).
-
FQuantification of SA‐β‐Gal‐positive cells (%) at 21 days after DXR treatment in NMR‐fibroblasts transduced with different forms of SV40 Large T antigen (LT, LTΔ, and LTK1).
-
GQuantification of Annexin V‐positive cells after the same treatment as in (F). Annexin V+/PI− cells were counted as early apoptotic cells and Annexin V+/PI− cells as late apoptotic cells.
Data information: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns, not significant. One‐way ANOVA followed by Dunnett's multiple comparison test. Data are expressed as the mean ± SD from n = 3 biological replicates.
Source data are available online for this figure.
Similar to the response in INK4a‐transduced cells, mild DXR treatment to induce cellular senescence in NMR fibroblasts resulted in increased SA‐β‐Gal activity and progressive cell death (Fig 1D and H). DXR treatment‐induced SA‐β‐Gal activity and progressive cell death were significantly suppressed by transduction of wild‐type LT (p53‐/RB‐) and LTΔ (p53+/RB−), but not by LTK1 (p53−/RB+) (Fig 3E–G). This result supports that RB, but not p53, is required for the progressive cell death induced by senescence stimuli.
To determine whether the progressive cell death in NMR cells after senescence induction by DXR is independent of p53, mRNA sequencing (mRNA‐seq) was performed in DXR‐treated NMR fibroblasts on Day 21. Gene set enrichment analysis (GSEA) (Mootha et al, 2003; Subramanian et al, 2005) showed that genes related to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway “p53 signaling pathway” were not enriched for differentially expressed genes in DXR‐treated NMR fibroblasts (Appendix Fig S3A). The expression of p53 target genes related to apoptosis (Fischer, 2017) did not show consistent changes in DXR‐treated NMR fibroblasts (Appendix Fig S3B). Consistent with the mRNA‐seq data, p53 protein levels were not altered in DXR‐treated NMR fibroblasts (Appendix Fig S3C). Taken together, these results indicate that INK4a and RB contribute to the delayed, progressive cell death in NMR cells after senescence induction by DXR, and that this process is independent of p53 activity (Figs 1K and 3G, Appendix Figs S1D and S3).
Seluanov et al. previously reported a unique phenotype for NMR fibroblasts: unlike mouse fibroblasts, NMR fibroblasts stop proliferating at an earlier, semiconfluent stage (early contact inhibition) via upregulation of INK4a (Seluanov et al, 2009). In this study, INK4a upregulation by DXR treatment or INK4a transduction caused progressive cell death in NMR fibroblasts (Fig 1H and 2G). To investigate how INK4a‐upregulated NMR cells behave after contact inhibition (CI), NMR and mouse fibroblasts were seeded at high density and placed under long‐term observation (Fig EV1A and B). On Days 14 and 28 after the start of culture, both mouse and NMR fibroblasts showed increased SA‐β‐Gal activity (Fig EV1C and D), decreased BrdU incorporation (Fig EV1E), and increased INK4a expression (Fig EV1F). We observed a significant increase in cell death in NMR fibroblasts, but not in mouse fibroblasts, on Days 14 and 28 (Fig EV1B and G). Knockdown of INK4a significantly reduced cell death induced by continuous culture after CI in NMR fibroblasts (Fig EV1H and I), as well as inhibiting cell death in DXR‐treated cells (Fig 1K and Appendix Fig S1D).
Figure EV1. Continuous cultivation after contact inhibition results in progressive cell death in NMR fibroblasts through INK4a upregulation.

-
AScheme for continuous cultivation after induction of contact inhibition (CI).
-
B, CCell morphology (B) and SA‐β‐Gal staining (C) in mouse or NMR fibroblasts at 14 or 28 days after induction of CI. Scale bar, 100 μm. The number in the upper left corner indicates Hoechst‐positive nuclei.
-
D, EQuantification of SA‐β‐Gal‐positive cells (%) (D), and BrdU‐positive cells (%) (E) at 14 or 28 days after induction of CI.
-
FqRT‐PCR analysis of the expression of INK4a in mouse or NMR fibroblasts at 14 or 28 days after induction of CI, normalized to ACTB mRNA levels.
-
GQuantification of Annexin V‐positive (early apoptotic; Annexin V+/PI− and late apoptotic; Annexin V+/PI+ double‐positive) cells (%) at 14 or 28 days after induction of CI.
-
H, IqRT‐PCR analysis of the expression of INK4a normalized to ACTB mRNA levels (H), and quantification of Annexin V‐positive (early apoptotic; Annexin V+/PI− and late apoptotic; Annexin V+/PI+ double‐positive) cells (%) (I) in shINK4a‐transduced NMR‐fibroblasts at 21 days after induction of CI.
-
JMouse fibroblasts were passaged 28 days after induction of CI and subjected to cell proliferation analysis.
-
KNMR fibroblasts were passaged 28 days after induction of CI and subjected to cell proliferation analysis.
Data information: *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, not significant. One‐way ANOVA followed by Dunnett's multiple comparison test for (D–I). Data are expressed as the mean ± SD from n = 3 biological replicates except for (G) (n = 5).
Considering that CI typically induces a quiescent state in mouse and human cells (Imai et al, 2014), we investigated whether long‐term culture of NMR cells at high density induces cellular senescence or quiescence. To this end, NMR cells were passaged at 28 days after long‐term culture to induce CI and observed to determine whether the cells could recover proliferation. Two out of three NMR primary fibroblast cultures resumed proliferation and one did not, in contrast to mouse primary fibroblast cultures, all of which resumed proliferation (Fig EV1J and K). This suggests that NMR fibroblasts cultured long term with upregulated INK4a after induced CI presumably contain both senescent and quiescent cells. Taken together, these results demonstrate that, in NMR fibroblasts, stimuli that activate the INK4a‐RB pathway and induce senescence, and in some cases quiescence, result in delayed and progressive cell death. The characteristic activation of cell death observed in NMR cells was termed “INK4a‐RB cell death.”
INK4a‐upregulated NMR cells activate serotonin metabolism that produces hydrogen peroxide
The mechanism of INK4a‐RB cell death in NMR cells was investigated by performing mRNA‐seq of NMR fibroblasts in which delayed, progressive cell death was induced by either DXR treatment, INK4a transduction, or CI. We then performed enrichment analysis using Metascape on genes that were commonly upregulated (> 1.5‐fold) in DXR‐treated, INK4a‐transduced, or CI‐induced NMR fibroblasts (Appendix Fig S4 and Table EV1). Pathway analysis revealed that genes associated with several Gene Ontology (GO) terms, such as “Regulation of collagen metabolic process,” “Lysosome,” and “Response to oxidative stress,” were enriched. However, genes associated with cell death‐related GO terms were not enriched (Appendix Fig S4B). Next, we focused on the mRNA‐seq data of INK4a‐transduced NMR fibroblasts (Fig 4A). Metascape was used to perform enrichment analysis of genes upregulated > 1.5‐fold in INK4a‐transduced NMR fibroblasts compared with control NMR fibroblasts (Fig 4B and Table EV2). In INK4a‐transduced NMR cells, the enrichment of genes related to the GO terms “SASP,” “Aging,” and “Positive regulation of cell death” may reflect cellular senescence and INK4a‐RB cell death. However, we did not identify genes directly related to cell death, including apoptosis, corresponding to the term “positive regulation of cell death.” Genes related to the GO term “Hydrogen peroxide metabolic process” were also enriched in INK4a‐transduced NMR fibroblasts. NMR fibroblasts are highly susceptible and prone to cell death upon hydrogen peroxide (H2O2) treatment (Salmon et al, 2008), which is supported by our data (Fig 4C). However, the biological significance of the susceptibility of NMR fibroblasts to H2O2 remains to be elucidated.
Figure 4. INK4a‐transduced NMR cells activate serotonin metabolism that produce hydrogen peroxide.

- Venn diagram showing the differentially expressed genes (DEGs, > 1.5‐fold in INK4a‐transduced fibroblasts) identified from comparisons of mock‐transduced and INK4a‐transduced NMR fibroblasts 12 days after transduction. Data were obtained from n = 3 biological replicates.
- Top 20 enriched gene ontology (GO) terms and KEGG pathways obtained using Metascape analysis of the 165 common genes in (A).
- Quantification of PI‐positive cells in NMR and mouse fibroblasts 18 h after 6 h of treatment with the indicated doses of hydrogen peroxide (H2O2). Data are expressed as the mean ± SD from n = 3 biological replicates.
- Levels of serotonin and 5‐hydroxyindoleacetic acid (5‐HIAA) measured by liquid chromatography–tandem mass spectrometry (LC–MS/MS) in mouse or NMR fibroblasts at 12 days after INK4a transduction. Data are expressed as the mean ± SD from two technical replicates for each fibroblasts culture (n = 3 biological replicates).
Data information: *P < 0.05; **P < 0.01; ****P < 0.0001; ns, not significant. Two‐way ANOVA followed by Sidak's multiple comparisons test for (C). Unpaired t‐test versus control for (D).
To compare the metabolite profiles of NMR and mouse cells and to examine changes in these cells after INK4a‐transduction, we conducted metabolomic mass spectrometry analysis (Appendix Fig S5). In mock‐treated control cells, a high level of serotonin was observed specifically in NMR fibroblasts but not in mouse fibroblasts (Fig 4D). Furthermore, serotonin was largely absent in NMR fibroblasts at 12 days after INK4a‐transduction (Fig 4D). Serotonin is converted to 5‐hydroxyindoleacetic acid (5‐HIAA) by MAO, accompanied by the production of a large amount of H2O2, which can lead to intracellular oxidative damage and cell death (Trouche et al, 2010; Edmondson, 2014). In INK4a‐transduced cells, a decrease in serotonin was associated with the accumulation of 5‐HIAA (Fig 4D).
Consistent with the data from the mRNA‐seq and metabolome analyses, the level of intracellular reactive oxygen species (ROS) measured by 2′,7′‐dihydrodichlorofluorescin diacetate (DCFH‐DA) (Suematsu et al, 1992) was significantly increased in INK4a‐transduced NMR fibroblasts on Day 12 (Fig 5A). Accordingly, the lipid peroxidation level was also significantly increased in INK4a‐transduced NMR fibroblasts (Fig 5B). Treatment of INK4a‐transduced NMR fibroblasts with anti‐oxidants such as N‐acetyl L‐cysteine (NAC) (Ezeriņa et al, 2018), Trolox (Forrest et al, 1994), and Tempol (Wilcox, 2010) significantly reduced cell death (Fig 5C), indicating that increased ROS contributes to INK4a‐RB cell death.
Figure 5. Activation of monoamine oxidases plays a crucial role in the induction of INK4a‐RB cell death in NMR cells.

- Quantification of reactive oxygen species (ROS) using 2′,7′‐dihydrodichlorofluorescin diacetate (DCFH‐DA) in mock‐ or INK4a‐transduced NMR fibroblasts at 12 days after transduction. Data are expressed as the mean ± SD from n = 3 biological replicates.
- Left, representative images of lipid peroxidation (green) staining in NMR fibroblasts at 12 days after INK4a‐transduction. Scale bar, 50 μm. Right, quantification of signal intensity of lipid peroxidation staining. Data are expressed as the mean ± SD from n = 3 biological replicates.
- Quantification of PI‐positive cells in NMR fibroblasts treated for 24 h with N‐acetyl L‐cysteine (NAC), Trolox, or Tempol at 20 days after INK4a transduction (%). Data are expressed as the mean ± SD from n = 6 biological replicates except for Trolox and Tempol (n = 4).
- Western blot of monoamine oxidase (MAO)‐A and MAO‐B in mouse or NMR fibroblasts at 20 days after INK4a transduction. ACTIN was used as a loading control. Numbers below the gel images indicate quantification of MAO‐A or ‐B/ACTIN intensity (n = 3 average).
- Quantification of ROS using DCFH‐DA in INK4a‐transduced NMR fibroblasts at 20 days after transduction. Cells were treated with each inhibitor for 24 h prior to analysis; Clorgyline (MAO‐A inhibitor, 10 μM), Rasagiline (MAO‐B inhibitor, 10 μM), Phenelzine (inhibitor of both MAO‐A and MAO‐B, 50 μM). Data are expressed as the mean ± SD from n = 4 biological replicates.
- Quantification of PI‐positive cells in NMR fibroblasts treated for 24 h with Clorgyline, Rasagiline, or Phenelzine at 20 days after INK4a transduction (%). Data are expressed as the mean ± SD from n = 3 biological replicates except for phenelzine (n = 5).
- Western blot of MAO‐A and MAO‐B in mouse or NMR fibroblasts at 21 days after doxorubicin (DXR) treatment. ACTIN was used as a loading control. Numbers below the gel images indicate quantification of MAO‐A or ‐B/ACTIN intensity (n = 3 average).
- Quantification of PI‐positive cells in NMR fibroblasts treated for 24 h with the indicated dose of Phenelzine (Phe) at 21 days after DXR treatment (%). Data are expressed as the mean ± SD from n = 3 biological replicates.
Data information: *P < 0.05; **P < 0.01; ****P < 0.0001; ns, not significant; Unpaired t‐test versus control for (A, B, NAC of C, and F). One‐way ANOVA followed by Dunnett's multiple comparison test for (Trolox and Tempol of C, E, and H).
Source data are available online for this figure.
Consistent with the metabolome analysis, transcriptomic analysis of INK4a‐transduced NMR cells (Fig 4B) showed that MAOB was one of the enriched genes related to the GO term “hydrogen peroxide metabolic process” (Table EV2). Accordingly, INK4a‐transduced NMR fibroblasts showed increased protein levels of MAO‐A and MAO‐B, whereas no such increase was observed in mouse fibroblasts after Ink4a transduction (Fig 5D). These results suggest that upregulation of INK4a in NMR cells, which are inherently susceptible to H2O2, leads to activation of serotonin metabolism via activation of MAO proteins, resulting in the production of large amounts of H2O2, and the resulting oxidative damage may contribute to INK4a‐RB cell death.
Activation of MAOs plays a crucial role in the induction of INK4a‐RB cell death in NMR cells
To determine whether activation of MAO‐A or ‐B is involved in INK4a‐RB cell death, INK4a‐transduced NMR fibroblasts were treated with the inhibitors clorgyline for MAO‐A, rasagiline for MAO‐B, and phenelzine for both MAO‐A and ‐B. Treatment with each of these inhibitors decreased DCF fluorescence intensity (Fig 5E), indicating a decrease in intracellular ROS, and significantly inhibited cell death in INK4a‐transduced NMR fibroblasts (Fig 5F). We next examined the contribution of MAOs to the activation of cell death in NMR cells subjected to cellular senescence induction by DXR‐treatment. MAO protein levels were increased in DXR‐treated NMR fibroblasts but not in mouse fibroblasts (Fig 5G). In DXR‐treated NMR fibroblasts, MAO inhibitors significantly suppressed cell death (Fig 5H). These results suggest that activation of monoamine oxidation by elevated MAOs plays a crucial role in the induction of INK4a‐RB cell death in NMR cells.
To investigate whether further upregulation of INK4a expression could enhance cell death in senescent NMR cells, DXR‐treated NMR fibroblasts were transduced with INK4a, and the levels of cell death, ROS, and MAO proteins were assessed, which showed no significant increase (Appendix Fig S6A–E). Furthermore, no significant difference in cell death rates was observed between expression vectors with different levels of INK4a expression (Appendix Fig S6F–I). These results support our conclusion that exceeding a certain threshold level of INK4a, subsequent cell cycle arrest, and the resulting cellular changes including senescence, are important for the activation of INK4a‐RB cell death.
Induction of cellular senescence leads to delayed, progressive cell death via MAO activation in NMR lungs
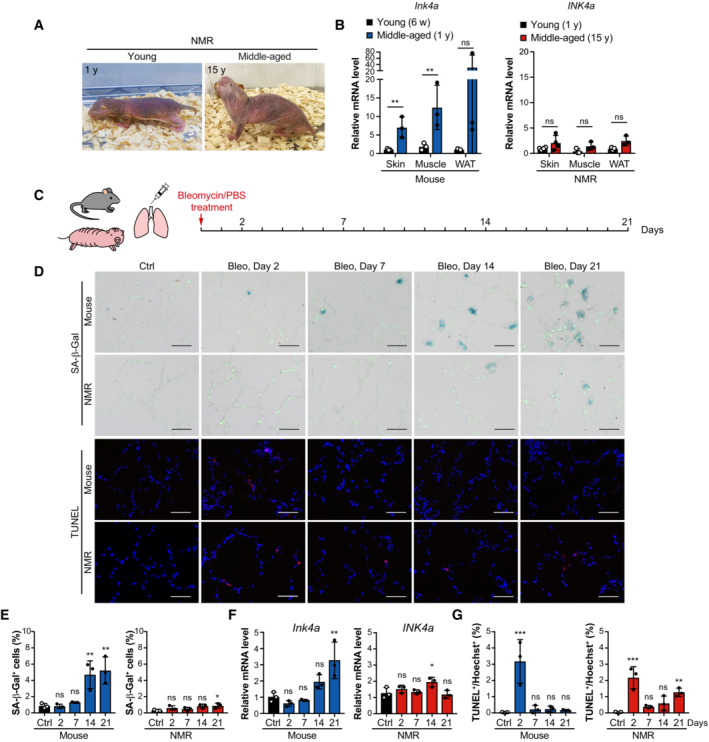
Lee et al. showed that the NMR brain only shows a slight increase in INK4a expression with age (Lee et al, 2020). We obtained similar results for the NMR skin, abdominal muscle, and inguinal white adipose tissue; the increase in INK4a expression in the tissues from 15‐year‐old (middle‐aged) compared with 1‐year‐old (young adult) NMRs was less pronounced than that in the tissues of 1‐year‐old (middle‐aged) compared with 6‐week‐old (young adult) mice (Fig 6A and B). This observation suggests that INK4a‐upregulated senescent cells are less likely to accumulate in NMR tissues than in those of mice in vivo.
Figure 6. Induction of cellular senescence leads to less accumulation of senescent cells and causes delayed, progressive cell death in NMR lungs.
-
ALeft, young NMR (one‐year‐old). Right, middle‐aged NMR (15‐year‐old).
-
BqRT‐PCR analysis of INK4a expression in the skin, muscle, and white adipose tissue (WAT) of 6‐week‐old mice (young; 6 w), 1‐year‐old mice (middle‐aged; 1 y), 1‐year‐old NMRs (young; 1 y), and 15‐year‐old NMRs (middle‐aged; 15 y).
-
CScheme for bleomycin (Bleo) treatment.
-
DSA‐β‐Gal activity (SA‐β‐Gal, blue; nuclei, green) and TUNEL staining (TUNEL, red; nuclei, blue) in lungs of mice or NMRs at 2, 7, 14, and 21 days after Bleo administration.
-
E–GTime‐course analysis of mice or NMR lungs after Bleo administration: quantification of SA‐β‐Gal‐positive cells (%) (E); qRT‐PCR analysis of the expression of INK4a normalized to ACTB mRNA levels (F); quantification of TUNEL‐positive cells (%) (G).
Data information: Scale bar, 50 μm. *P < 0.05, **P < 0.01, ***P < 0.001; ns, not significant. Unpaired t‐test versus young for (B). One‐way ANOVA followed by Dunnett's multiple comparison test for (E–G). Data are expressed as the mean ± SD from n = 3 biological replicates except for NMR skin in (B) (n = 4).
To investigate the potential occurrence of INK4a‐RB cell death in NMR tissue in vivo, cellular senescence was induced in mouse and NMR lungs using bleomycin, a DNA‐damaging agent (Aoshiba et al, 2003). Young adult mice (8‐week‐old) and young adult NMRs (1‐year‐old) received bleomycin by intratracheal administration, and a time‐course tissue analysis was performed (Fig 6C). In mouse lungs, the number of SA‐β‐Gal positive cells and Ink4a expression increased significantly on Day 21 (Fig 6D–F), whereas in NMR lungs, the increase in the number of SA‐β‐Gal positive cells and INK4a expression was lower than that in mice even on Day 21 (Fig 6D–F). These results suggest that NMR lungs are less prone to accumulate senescent cells in response to senescence‐inducing stimuli. TUNEL staining showed that both mouse and NMR lung tissues exhibited acute cell death on Day 2 after bleomycin treatment, and only NMR lungs exhibited delayed and progressive cell death that became significant on Day 21 (Fig 6D and G).
To investigate the role of MAOs in the progressive cell death observed in bleomycin‐treated NMR lungs, NMRs were treated with phenelzine for 5 consecutive days starting on Day 16 after bleomycin administration (Fig 7A). Phenelzine administration increased blood serotonin levels in mice and NMRs (Appendix Fig S7). After phenelzine administration, in bleomycin‐treated NMR lungs, the number of SA‐β‐Gal‐positive cells and the expression of INK4a significantly increased, and the number of TUNEL‐positive cells significantly decreased, in contrast to those in mice (Fig 7B–E). It was confirmed that phenelzine administration alone did not cause these changes in NMR lungs (Appendix Fig S8). These results indicate that, consistent with the in vitro findings, the induction of cellular senescence in NMR tissue results in delayed, progressive cell death that occurs through the activation of MAOs and contributes to the suppression of senescent cell accumulation.
Figure 7. Induction of cellular senescence leads to delayed, progressive cell death via MAO activation and the resulting suppression of senescent cell accumulation in the NMR lungs.

-
AScheme for bleomycin (Bleo) treatment and additional phenelzine (Phe) treatment. Mouse or NMR lungs were treated with Phe for 5 days starting at 16 days after Bleo administration.
-
BSA‐β‐Gal activity (SA‐β‐Gal, blue; nuclei, green) and TUNEL staining (TUNEL, red; nuclei, blue) in lungs of mice or NMRs at 21 days after Bleo administration, with or without Phe are shown.
-
C–EQuantification of SA‐β‐Gal‐positive cells (%) (C), qRT‐PCR analysis of INK4a expression normalized to ACTB mRNA levels (D), quantification of TUNEL‐positive cells (%) (E) in mouse or NMR lungs at 21 days after Bleo administration, with or without Phe.
Data information: Scale bar, 50 μm. *P < 0.05; ns, not significant. Unpaired t‐test versus Phe‐ for (C–E). Data are expressed as the mean ± SD from n = 3 biological replicates.
Discussion
In this study, we found that INK4a‐RB cell death is uniquely activated after induction of cellular senescence in NMRs in vitro, and this conclusion was supported by in vivo data. We also showed that this type of cell death is regulated by the activation of MAO proteins and serotonin metabolism in an NMR‐specific manner (Fig 8). INK4a‐RB cell death in NMRs may serve as a “natural senolysis” process that inhibits the accumulation of senescent cells, which in turn may contribute to delayed aging and resistance to age‐related diseases such as cancer and Alzheimer's disease, both of which are poorly observed in NMRs (Edrey et al, 2011, 2013). On the contrary, senescent cells play a crucial role in the maintenance of tissue homeostasis, developmental processes, and tissue repair (Muñoz‐Espín et al, 2013; Storer et al, 2013; Demaria et al, 2014; Reyes et al, 2022). Therefore, the safety of senolytic drugs that kill senescent cells remains controversial. This study strongly suggests that the NMR has a “natural senolytic” phenotype, which supports, from an evolutionary perspective, the elimination of senescent cells and the development of safer senolytic drugs. Future studies on INK4a‐RB cell death in NMRs may help to identify which senescent cells are suitable for elimination. INK4a‐RB cell death was observed not only in DXR‐treated senescent cell culture, but also in cells continuously cultured after CI, where both senescent cells and quiescent cells may be present. The INK4a‐RB pathway is activated under various conditions of cell stress (Gorgoulis et al, 2019). It is possible that the NMR induces INK4a‐RB cell death in dysfunctional and precancerous cells in which INK4a is persistently upregulated as a result of various stresses. Such elimination of aberrant cells may contribute to long‐term tissue homeostasis and resistance to carcinogenesis in the NMR, a possibility that requires future analysis.
Figure 8. The molecular mechanism of cell death upon cellular senescence induction in NMR.

Graphical illustration showing the main findings of this study. NMR fibroblasts uniquely accumulate serotonin and are inherently vulnerable to hydrogen peroxide (H2O2). Upon induction of cellular senescence, the activated INK4a‐RB pathway causes NMR fibroblasts to increase monoamine oxidase (MAO) levels, leading to serotonin oxidization and H2O2 production, resulting in increased oxidative damage and activation of cell death.
In this study, INK4a upregulation by various stimuli such as DXR treatment, INK4a transduction, and long‐term culture after CI resulted in delayed, progressive cell death in NMR fibroblasts but not in mouse fibroblasts. These findings, combined with the results of INK4a knockdown and transduction experiments, suggest that INK4a upregulation, subsequent cell cycle arrest, and the resulting cellular changes including senescence are important for the activation of INK4a‐RB cell death (Figs 1 and 2). In the process of INK4a‐RB cell death, activation of serotonin metabolism increased oxidative damage, leading to the activation of cell death, including apoptosis. However, analysis of mRNA‐seq data from INK4a‐upregulated (DXR‐treated, INK4a‐transduced, or CI‐induced) NMR cells did not identify any genes directly responsible for the induction of apoptosis. Furthermore, the present results indicate that INK4a‐RB cell death is independent of p53 activity (Fig 3 and Appendix Fig S3). Further analysis is needed to identify death‐inducing factors that may contribute to INK4a‐RB cell death in NMR cells, such as p53‐independent apoptosis‐related proteins.
NMR fibroblasts uniquely accumulate serotonin in their normal, nonsenescent state. Following INK4a‐RB activation, which leads to MAO activation, serotonin is oxidized to produce H2O2, which plays a significant role in increasing oxidative damage and inducing INK4a‐RB cell death (Figs 4 and 5). The accumulation of serotonin in NMR cells may act as a mechanism to prepare for the induction of INK4a‐RB cell death in response to stress, and the activation of serotonin metabolism via MAO activation may serve as a “senolytic metabolic switch” to kill NMR cells as they become senescent. However, further investigation is needed to understand the molecular mechanisms underlying MAO activation after INK4a‐RB activation in NMR fibroblasts. In addition to its role in INK4a‐RB cell death, serotonin accumulation in NMR fibroblasts may have other functions. For example, extracellularly secreted serotonin binds to G protein‐coupled receptors on the cell membrane of fibroblasts, thereby activating cell proliferation and the production of extracellular matrix proteins such as collagen (Mann & Oakley, 2013; Sadiq et al, 2018). It remains to be investigated whether the serotonin that accumulates in NMR fibroblasts is secreted extracellularly and has other roles in these cells. In addition, detailed analyses of the in vivo dynamics of serotonin in NMR tissues, including the lung, is also needed.
NMR fibroblasts exhibit complex behaviors and are susceptible or resistant to oxidative stress depending on the type of intervention (e.g., they are more resistant to paraquat, an oxidative stressor, whereas they are more susceptible to H2O2 than mouse cells) (Salmon et al, 2008; Munro et al, 2019; Takasugi et al, 2020). The result shown in Fig 4C is consistent with that of a previous study reporting that NMR fibroblasts are highly susceptible to H2O2 (Salmon et al, 2008). The markedly low activity of glutathione peroxidase reported in NMRs may be related to the vulnerability of NMR cells to H2O2 (Andziak et al, 2005, 2006; Kasaikina et al, 2011). NMR cells have low mitochondrial activity, a low oxygen consumption rate, and high mitochondrial ROS scavenging activity (Munro et al, 2019; Lau et al, 2020). Therefore, their vulnerability to intracellular H2O2 may not be a concern because ROS leakage from mitochondria is low in the normal state. Until now, the biological significance of the vulnerability of NMR cells to H2O2 remained unclear. The present results suggest that this vulnerability likely contributes to INK4a‐RB cell death in conjunction with the unique regulation of serotonin metabolic switch, which produces H2O2 when NMR cells become senescent.
Zhao et al. reported that NMR fibroblasts require a higher dose of gamma‐ray irradiation to induce cellular senescence compared with mouse fibroblasts (Zhao et al, 2018). Although they did not perform a time‐course analysis of cell death after induction of cellular senescence, these authors showed that NMR fibroblasts have a lower rate of irradiation‐induced acute cell death than mouse fibroblasts at 3 days after the same dose of irradiation. We have previously shown that NMR neural stem/progenitor cells have a lower rate of irradiation‐induced acute cell death compared with mouse neural stem/progenitor cells (Yamamura et al, 2021). This phenotype may be due to the efficient double‐strand break repair system in NMRs as previously reported (Tian et al, 2019). Taken together, these findings suggest that NMR cells may have a dual protective system that inhibits both the emergence and the accumulation of senescent cells through efficient DNA double‐strand break repair at an earlier stage and INK4a‐RB cell death at a later stage.
INK4a‐RB cell death was observed in skin and lung NMR fibroblasts under different conditions (i.e., DXR treatment, transduction of INK4a, and continuous culture after CI). This phenomenon was likewise observed in cultures using different lots of fetal bovine serum (FBS). In the future studies, it will be important to clarify whether INK4a‐RB cell death is induced under other conditions and in different cell types, including neural stem cells (Yamamura et al, 2021), macrophages (Wada et al, 2019), and adipocytes (Oiwa et al, 2020; Cheng et al, 2021), as well as in other organs besides the lungs in vivo. In this study, we developed a method to induce cellular senescence in the NMR lung in vivo. The development of genetic modification techniques for NMR individuals and the generation of INK4a reporter NMRs may allow detailed analysis of the dynamics of INK4a‐upregulated cells, including senescent cells, in NMR tissues.
Because senescent cells secrete SASP factors and promote tissue inflammation, INK4a‐RB cell death may contribute to the suppression of tissue inflammation in NMRs. Recent research from our group has shown that NMRs have lost the ability to induce necroptosis, a type of programmed necrosis that strongly induces tissue inflammation by releasing cellular components, and exhibit a dampened inflammatory response to carcinogenic stimuli, which likely contributes to their resistance to carcinogenesis (Oka et al, 2022). Taken together with the present findings, these data suggest that NMRs may have evolved to attenuate tissue inflammatory responses through various systems, including species‐specific regulation of cell death and cellular senescence. In another long‐lived and cancer‐resistant rodent, the blind mole‐rat, the expression of SASP genes is suppressed upon induction of cellular senescence, which may also contribute to the attenuation of inflammatory responses in their tissues (Odeh et al, 2020). On the contrary, carcinogen‐treated blind mole‐rat tissues undergo inflammatory, severe necrotic cell death that is markedly different from the carcinogen‐treated NMRs tissues (Gorbunova et al, 2012; Zhao et al, 2021). To the best of our knowledge, there are few reports with a detailed characterization of senescent cells in other long‐lived or carcinogenesis‐resistant animals, such as bats, elephants, and bowhead whales, and future studies are needed.
The present study has elucidated a unique mechanism of INK4a‐RB cell death upon induction of cellular senescence in NMRs. Further analysis of species‐specific responses to the induction of cellular senescence in long‐lived and cancer‐resistant species, including NMRs, may lead to the development of novel anti‐aging and anti‐cancer strategies in humans.
Materials and Methods
Animals
The Ethics Committees of Kumamoto University (approval no. A30‐043, A2020‐042 and A2022‐079), Hokkaido University (approval no. 14‐0065), and Keio University (approval no. 12024) approved all procedures, which were in accordance with the Guide for the Care and Use of Laboratory Animals (United States National Institutes of Health, Bethesda, MD). NMRs were maintained in Kumamoto University. All NMRs used in this study were raised in rooms that were maintained at 30°C ± 0.5°C and 55% ± 5% humidity with 12‐h light and 12‐h dark cycles (Oka et al, 2022). C57BL/6N mice were purchased from CLEA Japan, Inc. Mice were kept in rooms that were maintained at 24.5°C ± 1.5°C and 50% ± 10% humidity with 12‐h light and 12‐h dark cycles. The information on NMRs used in this study is listed in Appendix Table S1. Cells and tissues were obtained from at least three animals.
Cell culture and drug treatment
Primary NMR or mouse fibroblasts were isolated from back skin or the lungs of 1–2‐year‐old young adult male and female NMRs or 6‐week‐old young adult male C57BL/6N mice. Primary fibroblasts prepared from tissues of at least three different animals were used as biological replicates. Tissues were washed with ice‐cold phosphate‐buffered saline (PBS; Nacalai‐Tesque) containing 1% penicillin/streptomycin (FUJIFILM Wako) and 2.5 μg/ml amphotericin B (FUJIFILM Wako). Tissues were minced, and then suspended in the culture medium (contents described below), plated on gelatin‐coated 10‐cm cell culture dishes (IWAKI), and cultured at 32°C in a humidified atmosphere containing 5% CO2 and 5% O2. Cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Sigma) supplemented with 15% fetal bovine serum (FBS) (for NMR fibroblasts) or 10% FBS (for mouse fibroblasts) (BioWest or Gibco, at least three lots), 1% penicillin/streptomycin, 2 mM L‐glutamine (Nacalai‐Tesque or FUJIFILM Wako), and 0.1 mM nonessential amino acids (NEAA, Nacalai‐Tesque or FUJIFILM Wako). Fibroblasts were used within five passages. The medium was replaced every 2 days. Fibroblasts were routinely tested for mycoplasma and tested negative. For induction of CI, fibroblasts were seeded at 6 × 105 cells/10‐cm dish and the medium was replaced every 2 days. The cells were cultured for 28 days. For drug treatments, a 1 M stock solution of N‐acetyl L‐cysteine (NAC, FUJIFILM Wako) was prepared in 20 mM HEPES buffer (Nacalai‐Tesque), titrated with NaOH to pH 7.4, and filter‐sterilized and used at 0.5 mM. Trolox (Sigma) was dissolved in ethanol at 0.5 M as a stock solution and used at 1 mM. Tempol (R&D Systems) was dissolved in PBS at 100 mM as a stock solution and used at 2 mM. Clorgyline (Cayman Chemical) and rasagiline (Abcam) were dissolved in dimethyl sulfoxide (DMSO; Sigma) at 10 mM as a stock solution and used at 10 μM. Phenelzine (MedChemExpress) was dissolved in DMSO at 50 mM as a stock solution and used at 50 μM for INK4a‐transduced NMR cells and at 10–100 μM for DXR‐treated NMR cells. NAC, Trolox, Tempol, clorgyline, rasagiline, and phenelzine were added during the 24‐h period before cell death analysis. All experiments were performed in triplicate or greater.
Doxorubicin treatment
Mouse and NMR fibroblasts were exposed twice to doxorubicin (DXR; FUJIFILM Wako) at 100 nM for 24 h. DXR‐containing medium was added to subconfluent fibroblasts. After 24 h, the medium was replaced with freshly prepared DXR‐containing medium for an additional 24 h. The cells were then washed and cultured in fresh medium for 21 days with medium changes every 2 days.
SA‐β‐Gal activity analysis
For measuring cellular senescence, SA‐β‐Gal staining was performed using a Senescence Detection Kit (BioVision). Cells or fresh‐frozen lung sections were stained according to the manufacturer's instructions for 48 h at 37°C. The cells or lung sections were washed with PBS and stained with Hoechst 33258 (Sigma‐Aldrich, 1 mg/ml; 1:1000 in PBS) for 10 min at room temperature in the dark. The cells were washed with PBS and analyzed by microscopy (Keyence). Entire cell populations in three to four random microscope fields (at least 150 cells) were analyzed for perinuclear blue staining indicative of SA‐β‐gal activity and Hoechst‐positive nuclei. At least four random microscopic fields of each animal were obtained for analysis of lung sections; three animals were used per experiment. To stain floating cells, the culture supernatant was spun down, and the cell pellet was resuspended in Smear Gell kit (GenoStaff) and spread on the slide surface. The slides were stained and analyzed in the same way as adherent cells.
5‐bromo‐2'‐deoxyuridine (BrdU) incorporation assay
To analyze cell proliferation, BrdU labelling was performed for 2 days for mouse fibroblasts and 4 days for NMR fibroblasts as previously described (Seluanov et al, 2009). Cells were then fixed with 4% paraformaldehyde (PFA; FUJIFILM Wako) in PBS and subjected to immunostaining. BrdU was detected using primary sheep antibody against BrdU (Fitzgerald; 20‐BS17; 1:200) and Alexa Fluor 555 anti‐sheep IgG (A11015; Life Technologies; 1:500) secondary antibody. Cell nuclei were stained with 1 μg/ml Hoechst 33258. Cells were observed under a BZ‐X 710 fluorescence microscope (Keyence) and counted using a BZ‐X image analyzer (Keyence). Entire cell populations in four random microscope fields (at least 150 cells) of each primary fibroblast culture were analyzed for BrdU‐positive and Hoechst‐positive nuclei. Three mouse or NMR primary fibroblast cultures were used.
RNA isolation, RT‐PCR, and quantitative RT‐PCR (qRT‐PCR)
Total RNA was extracted using the RNeasy Plus Mini Kit (Qiagen, for fibroblasts) or TRIzol (Thermo Fisher Scientific, for tissue samples) according to the manufacturer's protocol. To remove genomic DNA, the gDNA Eliminator Spin Column (Qiagen) or the TURBO DNA‐free™ Kit (Invitrogen) was used according to the manufacturer's protocol. Reverse transcription (RT) reactions were performed with the ReverTra Ace qPCR RT Master Mix (TOYOBO) using 400 ng of total RNA input. The resulting cDNA was used for RT‐PCR and quantitative RT‐PCR (qRT‐PCR). For RT‐PCR, 30 cycles of amplification were performed under the following conditions using ExTaqHS (TaKaRa): denaturing at 95°C for 5 s, annealing at 62°C for 10 s, and extension at 72°C for 20 s. The DNA fragments were electrophoresed in 2% agarose gels. Uncropped images of all the gels are shown in the Source data file. qRT‐PCR reactions were set up in triplicate using SYBR Premix Ex Taq™ II (Tli RNaseH Plus) (TaKaRa), Fast SYBR Green Master Mix (Invitrogen), PowerUp SYBR Green Master Mix (Thermo Fisher Scientific), or THUNDERBIRD SYBR qPCR Mix (TOYOBO), and run on a ViiA 7, StepOne plus Real‐Time PCR System (Applied Biosystems) or CFX384 Touch Real‐Time PCR Detection System (Bio‐Rad). Primer sequences are listed in Appendix Table S2.
Flow cytometry analysis for cell death detection
Cell death was examined using a FITC Annexin V Apoptosis Detection kit (BD Biosciences or BioLegend). Adherent cells were harvested and stained according to the manufacturer's protocols. Flow cytometry was performed with a FACSCalibur or FACSVerse (BD Biosciences) flow cytometer. These data were analyzed using the FlowJo 10 software (BD Biosciences). The experiments were performed in triplicate.
Lentivirus preparation and transduction
The lentiviral vectors pCSII‐EF‐NMR‐INK4a‐TK‐hyg, pCSII‐EF‐NMR‐INK4a, pCSII‐EF‐mouse‐Ink4a‐TK‐hyg, pCSII‐EF‐mouse‐Ink4a, and pCSII‐EF‐LT/LTΔ434–444/LTK1 were used for ectopic expression, and H1 promoter‐driven vectors previously described were used for shRNA expression (Miyoshi et al, 1998). The pCSII‐EF‐TK‐Hyg vector was used as the mock vector. The backbone vectors for ectopic expression (pCSII‐EF‐RfA) and shRNA expression were purchased from RIKEN BioResource Research Center. The backbone vector for ectopic expression (pCSII‐EF‐RfA‐TK‐Hyg) was kindly provided by Dr. Hayato Naka‐Kaneda (Shiga University of Medical Science). The knockdown vectors expressing shRNA against NMR‐INK4a (Miyawaki et al, 2015) were generated. The sequences of the siRNA oligos were as follows: shINK4a‐1, 5′‐GGUCCAGGAGGUACGCGAGCU‐3′ and shINK4a‐2, 5′‐GCCCAAUGCCCGGAACCGUUU‐3′. Plasmid and packaging vectors (pCMV‐VSV‐G‐RSV‐Rev and pCAG‐HIVgp) were used to transfect HEK293T cells with Polyethylenimine MAX transfection reagent (Polysciences) according to the manufacturer's instructions. At 9 h after transfection, the medium was replaced, and the conditioned medium containing viral particles was collected twice every 24 h.
For lentiviral transduction, cells were seeded at 3 × 105 cells/10‐cm dish 1 day before transduction. The conditioned medium containing lentivirus was filtered through a 0.45‐μm syringe filter (Sartorius), diluted twofold in growth medium, and used for viral transduction. At 24 h after the first transduction, the medium was replaced with a second conditioned medium containing lentivirus. After viral transduction, the medium was replaced with growth medium, which was changed every 2 days.
For DXR treatment and INK4a knockdown experiments, NMR fibroblasts were treated with DXR either (i) before (Appendix Fig S1A) or (ii) after (Fig 1I) INK4a knockdown. For (i), cells were seeded at 3 × 105 cells/10‐cm dish and treated with DXR (100 nM) for 2 days starting the next day. Twelve days after the DXR treatment, cells were lentivirally transduced with INK4a knockdown vector. Twenty‐four days after the DXR treatment, cell death was examined. For (ii), cells were seeded at 3 × 105 cells/10‐cm dish 1 day before transduction. Then INK4a knockdown vector was lentivirally transduced (on −8 days in Fig 1I). Eight days after the transduction, cells were treated with DXR (100 nM) for 2 days. Twenty‐one days after the DXR treatment, cell death was examined.
Western blotting
The cells were washed with PBS, lysed in cell‐lysis buffer (125 mM Tris–HCl, pH 6.8, 4% sodium dodecyl sulphate [SDS], and 10% sucrose), and boiled for 5 min. Protein concentrations were determined using the BCA Protein Assay Kit (TaKaRa). The samples were subjected to SDS–PAGE, and transferred to a PVDF membrane using a Trans‐Blot Turbo Transfer System (Bio‐Rad). Membranes were incubated with primary antibodies against RB (CST; 9,309; 1:1,000 for NMR, CST; 9,313; 1:1,000 for mouse), pRB (CST; 8,516; 1:1,000), MAO‐A (Abcam; ab126751; 1:1,000), MAO‐B (Novus Biologicals; NBP1‐87493; 1:1,000), p53 (Santa Cruz; sc‐126; 1:1,000), and β‐actin (CST; 4,970; 1:2,000). The membranes were incubated with HRP‐conjugated anti‐rabbit (CST; 7,074; 1:1,000) or anti‐mouse (CST; 7,076; 1:1,000) IgG secondary antibodies and visualized using the ECL Western Blotting Detection System or ECL Prime Western Blotting Detection Reagent (Amersham). The LAS‐4000 mini imaging system (FUJIFILM) was used for signal detection and Multi Gauge V3.0 software (FUJIFILM) was used for data analysis. The experiments were performed in triplicate. Uncropped images of all the blots are shown in the Source data file.
Hoechst‐propidium iodide (PI) staining assay for cell death detection
Cells were seeded in 24‐well plates or 60‐mm dishes and stained with Hoechst 33342 (DOJINDO; 1 mg/ml; 1:1000 in growth medium) for 10 min at 32°C. Then, the cells were stained with PI (10 mg/ml; FUJIFILM Wako; 1:1000 in growth medium) for 5 min at 32°C. Images were captured with a BZ‐X 710 fluorescence microscope (Keyence), and positive cells were counted using a BZ‐X image analyzer (Keyence). Entire cell populations in 8–12 random microscope fields (at least 350 cells) of each primary fibroblast culture were analyzed for PI‐positive and Hoechst‐positive nuclei. Three mouse or NMR primary fibroblast cultures were used.
H2O2 sensitivity test
Cells in a 24‐well plate were incubated with the indicated doses of H2O2 (Nacalai‐Tesque) for 6 h in DMEM. Cells were then washed and incubated with growth medium for 18 h. To evaluate cell death, a Hoechst‐PI staining assay was performed. Entire cell populations in six random microscope fields (at least 150 cells) of each primary fibroblast culture were analyzed for PI‐positive and Hoechst‐positive nuclei. Three mouse or NMR primary fibroblast cultures were used.
Measurement of intracellular ROS
For detection of cellular ROS, the DCFDA/H2DCFDA Cellular ROS Assay Kit (Abcam; ab113851) was used according to the manufacturer's protocol. Briefly, fibroblasts were plated on a black 96‐well plate with a clear bottom overnight. Cells were treated with 25 mM DCF‐DA solution for 45 min at 32°C in the dark. After washing once with PBS, the plate was analyzed using a fluorescence plate reader (GloMax; Promega) at Ex/Em = 485/535 nm. The experiments were performed in triplicate.
Detection of lipid peroxidation
For detection of lipid peroxidation, the Click‐iT lipid peroxidation imaging kit (Invitrogen) was used according to the manufacturer's protocol. Briefly, cells were plated on coverslips in a 24‐well plate and treated with Click‐it linoleamide alkyne for 24 h at 32°C. After washing with PBS, cells were fixed in 4% PFA for 15 min at room temperature, washed with PBS, permeabilized with 0.05% TritonX‐100 in PBS for 10 min, and blocked with 1% BSA in PBS for 30 min. Cells were washed, and the click reaction was performed with 5 μM Alexa Fluor 488 azide for 30 min. After washing, cells were stained with Hoechst 33258. Images were captured using the BZ‐X 710 fluorescence microscope (Keyence) and analyzed using a BZ‐X image analyzer (Keyence). Entire cell populations in 12 random microscope fields (at least 150 cells) of each primary fibroblast culture were analyzed for green fluorescence and Hoechst‐positive nuclei. Three mouse or NMR primary fibroblast cultures were used.
RNA‐sequencing
NMR fibroblasts were homogenized in TRIzol reagent (Invitrogen). RNA extraction and library preparation were performed at the Novogene Bioinformatics Institute. NMR samples were sequenced using the Novaseq 6000 (150 bp paired‐end). NMR reference genome assemblies (GRCm38 and HetGla_female_1.0) and the corresponding annotation files were obtained from Ensembl release 92 (Yates et al, 2019). Raw reads were trimmed using Trim Galore (ver. 0.5.0, https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), and the transcript abundance was calculated using Kallisto (ver. 0.48.0) (Bray et al, 2016) as transcripts per million (TPM) and estimated counts. Differentially expressed genes in DXR‐treated, Ink4a‐transduced, or CI‐induced cells were identified by comparing upregulated genes (> 0.5 in TPM and > 1.5‐fold change from control cells) among three primary fibroblast cultures. Count data were processed using the integrated Differential Expression and Pathway analysis (iDEP) (iDEP.96) (Ge et al, 2018). GSEA of the KEGG pathway “p53 signaling pathway” was performed using processed data for DXR‐treated cells and control cells.
Metabolome analysis
Metabolite extraction from cultured cells was performed as described previously (Semba et al, 2016). Briefly, frozen cells were lysed and scraped with ice‐cold methanol (500 μl) together with internal standard (IS) compounds (see below), followed by the addition of an equal volume of ultrapure water and 0.4 times the volume of chloroform (LC/MS grade, FUJIFILM Wako). The suspension was then centrifuged at 15,000 g for 15 min at 4°C. After centrifugation, the aqueous phase was ultrafiltered using an ultrafiltration tube (Ultrafree MC‐PLHCC, Human Metabolome Technologies). The filtrate was concentrated with a vacuum concentrator (SpeedVac, Thermo). The concentrated filtrate was dissolved in 50 μl of ultrapure water and used for liquid chromatography–tandem mass spectrometry (LC–MS/MS) and ion chromatography‐mass spectrometry (IC‐MS) analyses. The internal standard (IS) compounds used were 2‐morpholinoethanesulfonic acid (MES) and L‐methionine sulfone as ISs for anionic and cationic metabolites, respectively. Because these compounds are not present in the tissues, they serve as ideal standards. Loss of endogenous metabolites during sample preparation was corrected by calculating the recovery rate (%) for each sample measurement.
For metabolome analysis, anionic metabolites were measured using an orbitrap‐type MS (Q‐Exactive focus, Thermo Fisher Scientific) connected to a high‐performance ion‐chromatography system (ICS‐5000+, Thermo Fisher Scientific) that enables highly selective and sensitive metabolite quantification owing to the IC‐separation and Fourier Transfer MS principle. The IC was equipped with an anion electrolytic suppressor (Thermo Scientific Dionex AERS 500) to convert the potassium hydroxide gradient into pure water before the sample enters the mass spectrometer. The separation was performed using a Thermo Scientific Dionex IonPac AS11‐HC, 4‐μm particle size column. The IC flow rate was 0.25 ml/min supplemented postcolumn with 0.18 ml/min makeup flow of MeOH. The potassium hydroxide gradient conditions for IC separation were as follows: from 1 mM to 100 mM (0–40 min), 100 mM (40–50 min), and 1 mM (50.1–60 min), at a column temperature of 30°C. The Q Exactive focus mass spectrometer was operated under ESI‐negative mode for all detections. Full mass scan (m/z 70–900) was used at a resolution of 70,000. The automatic gain control (AGC) target was set at 3 × 106 ions, and the maximum ion injection time (IT) was 100 ms. The source ionization parameters were optimized with the spray voltage at 3 kV and other parameters were as follows: transfer temperature at 320°C, S‐Lens level at 50, heater temperature at 300°C, Sheath gas at 36, and Aux gas at 10.
Cationic metabolites (amino acids) were quantified using LC–MS/MS. Briefly, a triple‐quadrupole mass spectrometer equipped with an ESI ion source (LCMS‐8060, Shimadzu Corporation) was used in the positive and negative‐ESI and multiple reaction monitoring (MRM) modes. The samples were resolved on the Discovery HS F5‐3 column (2.1 mm I.D. x 150 mm L, 3 μm particle, Sigma‐Aldrich), using a step gradient with mobile phase A (0.1% formate) and mobile phase B (0.1% acetonitrile) at ratios of 100:0 (0–5 min), 75:25 (5–11 min), 65:35 (11–15 min), 5:95 (15–20 min), and 100:0 (20–25 min), at a flow rate of 0.25 ml/min and a column temperature of 40°C. MRM conditions for each amino acid were described previously (Oka et al, 2017).
Tissue biopsy
To obtain skin samples without sacrifice, mice (C57BL/6N; young [6‐week‐old] and middle‐aged [1‐year‐old], female) and NMRs (young [1‐year‐old] and middle‐aged [15‐year‐old, the currently available oldest animals in our laboratory], male and female) were anesthetized with isoflurane (FUJIFILM Wako). A 5–7‐mm incision was made at the base of the hind leg, and a 2 mm × 5 mm piece of skin, inguinal white adipose tissue, and abdominal muscle were collected (three animals per experimental group). Biopsy samples of skin, abdominal muscle, and inguinal white adipose tissue were subjected to RNA isolation.
In vivo drug treatments
Mice (C57BL/6N, 8‐week‐old, male) and NMRs (1–2‐year‐old, male) were anesthetized with 0.3 μg/g medetomidine hydrochloride (Dorbene Vet; Kyoritsu Seiyaku Co.), 4 μg/g midazolam (Dormicum; Asteras Pharma Inc.), and 5 μg/g butorphanol (Vetorphale; Meiji Seika Pharma Co.). In a supine position, a 1‐cm incision was made on the neck skin, and the sternohyoid muscles were separated to expose the trachea. Using a 29 G needle, 100 μl of PBS containing bleomycin hydrochloride (2.5 mg/kg body weight; Nippon Kayaku) was injected intratracheally with air. On Days 2, 7, 14, and 21 after the bleomycin injection, the animals were sacrificed humanely using isoflurane anesthesia. One bronchus was tied with a string, a lung was inflated with Super Cryoembedding Medium (SECTION‐LAB) for histological analysis, and the opposite side was collected as frozen tissue for RNA extraction. Phenelzine sulfate salt (Cayman Chemical) was freshly diluted each day in saline. A 30 mg/kg/day dose of phenelzine was intraperitoneally administered for 5 consecutive days, as it took 5 consecutive days for serotonin levels to increase following phenelzine administration in mice and NMR. Whole blood was collected from the heart, and after adding EDTA‐2 K (1–1.5 mg/ml blood, DOJINDO), the blood was inverted, mixed, and stored at −80°C. Blood serotonin levels were measured by Japan Institute for the Control of Aging, NIKKEN SEIL.
TUNEL assay
TUNEL staining (for quantifying cell death) was performed using the TUNEL Assay Kit BrdU‐Red (Abcam; ab66110) according to the manufacturer's instructions. Fresh‐frozen lung sections (10 μm) were fixed with 4% PFA for 15 min at room temperature, washed with PBS, and then treated with 10 μg/ml Proteinase K (Kanto Chemical) solution for 5 min at room temperature. Sections were washed with PBS, fixed with 4% PFA for 5 min at room temperature, and washed with PBS. Sections were washed twice for 5 min each in Wash buffer (Abcam; ab66110). Sections were labeled with DNA labelling solution in a dark humidified 37°C incubator for 1 h. Sections were washed with PBS and then stained with anti‐BrdU‐Red antibody for 30 min at room temperature. Nuclei were counterstained with Hoechst 33258. At least four random microscopic fields of each animal were obtained for analysis of lung sections; three animals were used per experiment. Images were captured using a BZ‐X 710 or a BZ‐X 800 fluorescence microscope and analyzed using a BZ‐X image analyzer (KEYENCE).
Statistical analysis
Prism 7 software (GraphPad) was used for statistical analysis. Data were analyzed using two‐way ANOVA followed by Sidak's multiple comparisons test, or one‐way ANOVA followed by Sidak's multiple comparisons test, Tukey's multiple comparison test or Dunnett's multiple comparison test. Unpaired t‐tests were used to compare the two groups. Each data point was presented as the mean ± standard deviation (SD) derived from at least three animals or biological replicates. P‐values < 0.05 were considered statistically significant. The sample sizes and the number of replicates (at least three individuals or three independent experiments) are described in the figure legends.
Author contributions
Yoshimi Kawamura: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; validation; investigation; visualization; methodology; writing – original draft; project administration; writing – review and editing. Kaori Oka: Data curation; software; formal analysis; validation; investigation; visualization; writing – review and editing. Takashi Semba: Investigation; methodology; writing – review and editing. Mayuko Takamori: Data curation; validation; investigation. Yuki Sugiura: Resources; data curation; visualization; methodology; writing – original draft. Riyo Yamasaki: Investigation. Yusuke Suzuki: Investigation. Takeshi Chujo: Writing – original draft; writing – review and editing. Mari Nagase: Investigation. Yuki Oiwa: Investigation. Shusuke Fujioka: Investigation. Sayuri Homma: Investigation. Yuki Yamamura: Investigation. Shingo Miyawaki: Investigation. Minoru Narita: Investigation. Takaichi Fukuda: Investigation; methodology. Yusuke Sakai: Validation; methodology. Takatsugu Ishimoto: Resources; investigation. Kazuhito Tomizawa: Investigation. Makoto Suematsu: Writing – original draft; writing – review and editing. Takuya Yamamoto: Resources; data curation; software. Hidemasa Bono: Resources; data curation; software; formal analysis; writing – original draft. Hideyuki Okano: Resources; supervision; writing – original draft; writing – review and editing. Kyoko Miura: Conceptualization; resources; supervision; funding acquisition; investigation; methodology; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
PDF+
Source Data for Appendix
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Acknowledgments
We thank Drs. J. Kohyama, K. Seino, H. Wada, M. Nakanishi, Y. Johmura, A. Shimizu, K. Murano, and H. Siomi for their scientific discussion and administrative support, M. Kobe, Y. Tanabe, M. Nagata, N. Onoue, Y. Fujimura, N. Arai, and C. Fukaya for their help with animal maintenance, Y. Tanoue, Y. Takahashi, and all members of the K.M. laboratories for technical assistance and scientific discussion, T. Moroishi for cell death inhibitors, H. Miyoshi for lentiviral vectors, and H. Naka‐Kaneda for pCSII‐EF‐RfA‐TK‐hyg vector. We thank to Jane Doe of the Liaison Laboratory Research Promotion Center for technical support. This work was supported in part by JST FOREST Program (JPMJFR216C), AMED under Grant Numbers JP21gm5010001, JP21bm0704040, and JST PRESTO Program (JPMJPR12M2) to KM, JST JSPS KAKENHI Grant Number to KM (JP21H02392, JP21H05143, JP22K18355, JP19K22370, JP18H02365, JP16H06279 (PAGS)) and YK (JP25430116, JP19K06469), Tenure‐Track Grant of Kumamoto University to YK. JST COI‐NEXT JPMJPF2010 to K.M. and H.B. KM was also supported by the Takeda Science Foundation, Mitsubishi Foundation, Kanzawa Medical Research Foundation, Japan Foundation For Aging And Health, KOSÉ Cosmetology Research Foundation, Research Grant of the Princess Takamatsu Cancer Research Fund, The Nakatomi Foundation, and Naito Foundation, Foundation for Promotion of Cancer Research, Kato Memorial Bioscience Foundation, MSD Life Science Foundation, Inamori Foundation, SGH Foundation, Terumo Life Science Foundation, The Ichiro Kanehara Foundation, and Frontier Salon Foundation. This work was also supported, in part, by ROIS‐DS‐JOINT (012RP2018) to KM. M.S. is supported by AMED‐Moonshot for Microbiome (Lead: Professor. Kenya Honda).
The EMBO Journal (2023) 42: e111133
Contributor Information
Yoshimi Kawamura, Email: kawamuray@kumamoto-u.ac.jp.
Kyoko Miura, Email: miurak@kumamoto-u.ac.jp.
Data availability
RNA‐seq data are deposited in the DDBJ under accession number DRA016018.
References
- Al‐Mohanna MA, Manogaran PS, Al‐Mukhalafi Z, Al‐Hussein A, Aboussekhra KA (2004) The tumor suppressor p16INK4a gene is a regulator of apoptosis induced by ultraviolet light and cisplatin. Oncogene 23: 201–212 [DOI] [PubMed] [Google Scholar]
- Andziak B, O'Connor TP, Buffenstein R (2005) Antioxidants do not explain the disparate longevity between mice and the longest‐living rodent, the naked mole‐rat. Mech Ageing Dev 126: 1206–1212 [DOI] [PubMed] [Google Scholar]
- Andziak B, O'Connor TP, Qi W, DeWaal EM, Pierce A, Chaudhuri AR, Van Remmen H, Buffenstein R (2006) High oxidative damage levels in the longest‐living rodent, the naked mole‐rat. Aging Cell 5: 463–471 [DOI] [PubMed] [Google Scholar]
- Aoshiba K, Tsuji T, Nagai A (2003) Bleomycin induces cellular senescence in alveolar epithelial cells. Eur Respir J 22: 436–443 [DOI] [PubMed] [Google Scholar]
- Azpurua J, Ke Z, Chen IX, Zhang Q, Ermolenko DN, Zhang ZD, Gorbunova V, Seluanov A (2013) Naked mole‐rat has increased translational fidelity compared with the mouse, as well as a unique 28S ribosomal RNA cleavage. Proc Natl Acad Sci U S A 110: 17350–17355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA et al (2017) Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169: 132–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM (2011) Clearance of p16Ink4a‐positive senescent cells delays ageing‐associated disorders. Nature 479: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A et al (2016) Naturally occurring p16Ink4a‐positive cells shorten healthy lifespan. Nature 530: 184–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birch J, Gil J (2020) Senescence and the SASP: many therapeutic avenues. Genes Dev 34: 1565–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L (2016) Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol 34: 525–527 [DOI] [PubMed] [Google Scholar]
- Brieño‐Enríquez MA, Faykoo‐Martinez M, Goben M, Grenier JK, McGrath A, Prado AM, Sinopoli J, Wagner K, Walsh PT, Lopa SH et al (2023) Postnatal oogenesis leads to an exceptionally large ovarian reserve in naked mole‐rats. Nat Commun 14: 670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffenstein R (2008) Negligible senescence in the longest living rodent, the naked mole‐rat: insights from a successfully aging species. J Comp Physiol B 178: 439–445 [DOI] [PubMed] [Google Scholar]
- Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A (2019) Cellular senescence: aging, cancer, and injury. Physiol Rev 99: 1047–1078 [DOI] [PubMed] [Google Scholar]
- Chee W‐Y, Kurahashi Y, Kim J, Miura K, Okuzaki D, Ishitani T, Kajiwara K, Nada S, Okano H, Okada M (2021) β‐Catenin‐promoted cholesterol metabolism protects against cellular senescence in naked mole‐rat cells. Commun Biol 4: 357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Sebaa R, Malholtra N, Lacoste B, El Hankouri Z, Kirby A, Bennett NC, van Jaarsveld B, Hart DW, Tattersall GJ et al (2021) Naked mole‐rat brown fat thermogenesis is diminished during hypoxia through a rapid decrease in UCP1. Nat Commun 12: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM, Van DJM (2014) Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep 15: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé J‐P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez P‐Y, Campisi J (2008) Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé J‐P, Desprez P‐Y, Krtolica A, Campisi J (2010) The senescence‐associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5: 99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, VanSteeg H, Dollé MET et al (2014) An essential role for senescent cells in optimal wound healing through secretion of PDGF‐AA. Dev Cell 31: 722–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, O'Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM et al (2017) Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov 7: 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmondson D (2014) Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: biological implications. Curr Pharm Des 20: 155–160 [DOI] [PubMed] [Google Scholar]
- Edrey YH, Hanes M, Pinto M, Mele J, Buffenstein R (2011) Successful aging and sustained good health in the naked mole rat: a long‐lived mammalian model for biogerontology and biomedical research. ILAR J 52: 41–53 [DOI] [PubMed] [Google Scholar]
- Edrey YH, Medina DX, Gaczynska M, Osmulski PA, Oddo S, Caccamo A, Buffenstein R (2013) Amyloid beta and the longest‐lived rodent: the naked mole‐rat as a model for natural protection from alzheimer's disease. Neurobiol Aging 34: 2352–2360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evdokimov A, Kutuzov M, Petruseva I, Lukjanchikova N, Kashina E, Kolova E, Zemerova T, Romanenko S, Perelman P, Prokopov D et al (2018) Naked mole rat cells display more efficient excision repair than mouse cells. Aging (Albany NY) 10: 1454–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezeriņa D, Takano Y, Hanaoka K, Urano Y, Dick TP (2018) N‐acetyl cysteine functions as a fast‐acting antioxidant by triggering intracellular H2S and sulfane sulfur production. Cell Chem Biol 25: 447–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch CE (2009) Update on slow aging and negligible senescence – a mini‐review. Gerontology 55: 307–313 [DOI] [PubMed] [Google Scholar]
- Fischer M (2017) Census and evaluation of p53 target genes. Oncogene 36: 3943–3956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest VJ, Kang Y, McClain DE, Robinson DH, Ramakrishnan N (1994) Oxidative stress‐induced apoptosis prevented by Trolox. Free Radic Biol Med 16: 675–684 [DOI] [PubMed] [Google Scholar]
- Gasek NS, Kuchel GA, Kirkland JL, Xu M (2021) Strategies for targeting senescent cells in human disease. Nat Aging 1: 870–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge SX, Son EW, Yao R (2018) iDEP: an integrated web application for differential expression and pathway analysis of RNA‐seq data. BMC Bioinformatics 19: 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbunova V, Hine C, Tian X, Ablaeva J, Gudkov AV, Nevo E, Seluanov A (2012) Cancer resistance in the blind mole rat is mediated by concerted necrotic cell death mechanism. Proc Natl Acad Sci U S A 109: 19392–19396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G et al (2019) Cellular senescence: defining a path forward. Cell 179: 813–827 [DOI] [PubMed] [Google Scholar]
- Hahn WC, Dessain SK, Brooks MW, King JE, Elenbaas B, Sabatini DM, DeCaprio JA, Weinberg RA (2002) Enumeration of the simian virus 40 early region elements necessary for human cell transformation. Mol Cell Biol 22: 2111–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz N, Gil J (2018) Mechanisms and functions of cellular senescence. J Clin Invest 128: 1238–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Takahashi A, Hanyu A, Hori S, Sato S, Naka K, Hirao A, Ohtani N, Hara E (2014) Crosstalk between the Rb pathway and AKT signaling forms a quiescence‐senescence switch. Cell Rep 7: 194–207 [DOI] [PubMed] [Google Scholar]
- Jarvis JUM (1981) Eusociality in a mammal: cooperative breeding in naked mole‐rat colonies. Science 212: 571–573 [DOI] [PubMed] [Google Scholar]
- Kasaikina MV, Lobanov AV, Malinouski MY, Lee BC, Seravalli J, Fomenko DE, Turanov AA, Finney L, Vogt S, Park TJ et al (2011) Reduced utilization of selenium by naked mole rats due to a specific defect in GPx1 expression. J Biol Chem 286: 17005–17014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau GY, Milsom WK, Richards JG, Pamenter ME (2020) Heart mitochondria from naked mole‐rats (Heterocephalus glaber) are more coupled, but similarly susceptible to anoxia‐reoxygenation stress than in laboratory mice (Mus musculus). Comp Biochem Physiol B Biochem Mol Biol 240: 110375 [DOI] [PubMed] [Google Scholar]
- Lee BP, Smith M, Buffenstein R, Harries LW (2020) Negligible senescence in naked mole rats may be a consequence of well‐maintained splicing regulation. GeroScience 42: 633–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis KN, Wason E, Edrey YH, Kristan DM, Nevo E, Buffenstein R (2015) Regulation of Nrf2 signaling and longevity in naturally long‐lived rodents. Proc Natl Acad Sci U S A 112: 3722–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DA, Oakley F (2013) Serotonin paracrine signaling in tissue fibrosis. Biochim Biophys Acta 1832: 905–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzayans R, Andrais B, Hansen G, Murray D (2012) Role of p16INK4A in replicative senescence and DNA damage‐induced premature senescence in p53‐deficient human cells. Biochem Res Int 2012: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyawaki S, Kawamura Y, Hachiya T, Shimizu A, Miura K (2015) Molecular cloning and characterization of the INK4a and ARF genes in naked mole‐rat. Inflamm Regen 35: 42–50 [Google Scholar]
- Miyawaki S, Kawamura Y, Oiwa Y, Shimizu A, Hachiya T, Bono H, Koya I, Okada Y, Kimura T, Tsuchiya Y et al (2016) Tumour resistance in induced pluripotent stem cells derived from naked mole‐rats. Nat Commun 7: 11471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Blömer U, Takahashi M, Gage FH, Verma IM (1998) Development of a self‐inactivating lentivirus vector. J Virol 72: 8150–8157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K‐F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E et al (2003) PGC‐1α‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34: 267–273 [DOI] [PubMed] [Google Scholar]
- Muñoz‐Espín D, Cañamero M, Maraver A, Gómez‐López G, Contreras J, Murillo‐Cuesta S, Rodríguez‐Baeza A, Varela‐Nieto I, Ruberte J, Collado M et al (2013) Programmed cell senescence during mammalian embryonic development. Cell 155: 1104–1118 [DOI] [PubMed] [Google Scholar]
- Munro D, Baldy C, Pamenter ME, Treberg JR (2019) The exceptional longevity of the naked mole‐rat may be explained by mitochondrial antioxidant defenses. Aging Cell 18: e12916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odeh A, Dronina M, Domankevich V, Shams I, Manov I (2020) Downregulation of the inflammatory network in senescent fibroblasts and aging tissues of the long‐lived and cancer‐resistant subterranean wild rodent, Spalax. Aging Cell 19: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani N, Yamakoshi K, Takahashi A, Hara E (2004) The p16INK4a‐RB pathway: molecular link between cellular senescence and tumor suppression. J Med Invest 51: 146–153 [DOI] [PubMed] [Google Scholar]
- Oiwa Y, Oka K, Yasui H, Higashikawa K, Bono H, Kawamura Y, Miyawaki S, Watarai A, Kikusui T, Shimizu A et al (2020) Characterization of brown adipose tissue thermogenesis in the naked mole‐rat (Heterocephalus glaber), a heterothermic mammal. Sci Rep 10: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka M, Hashimoto K, Yamaguchi Y, Saitoh S, Sugiura Y, Motoi Y, Honda K, Kikko Y, Ohata S, Suematsu M et al (2017) Arl8b is required for lysosomal degradation of maternal proteins in the visceral yolk sac endoderm of mouse embryos. J Cell Sci 130: 3568–3577 [DOI] [PubMed] [Google Scholar]
- Oka K, Fujioka S, Kawamura Y, Komohara Y, Chujo T, Sekiguchi K, Yamamura Y, Oiwa Y, Omamiuda‐Ishikawa N, Komaki S et al (2022) Resistance to chemical carcinogenesis induction via a dampened inflammatory response in naked mole‐rats. Commun Biol 5: 287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka K, Yamakawa M, Kawamura Y, Kutsukake N, Miura K (2023) The naked mole‐rat as a model for healthy aging. Annu Rev Anim Biosci 11: 207–226 [DOI] [PubMed] [Google Scholar]
- Omori S, Wang T‐W, Johmura Y, Kanai T, Nakano Y, Kido T, Susaki EA, Nakajima T, Shichino S, Ueha S et al (2020) Generation of a p16 reporter mouse and its use to characterize and target p16high cells in vivo . Cell Metab 32: 814–828.e6 [DOI] [PubMed] [Google Scholar]
- Perez VI, Buffenstein R, Masamsetti V, Leonard S, Salmon AB, Mele J, Andziak B, Yang T, Edrey Y, Friguet B et al (2009) Protein stability and resistance to oxidative stress are determinants of longevity in the longest‐living rodent, the naked mole‐rat. Proc Natl Acad Sci U S A 106: 3059–3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrova NV, Velichko AK, Razin SV, Kantidze OL (2016) Small molecule compounds that induce cellular senescence. Aging Cell 15: 999–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes NS, Krasilnikov M, Allen NC, Lee JY, Hyams B, Zhou M, Ravishankar S, Cassandras M, Wang C, Khan I et al (2022) Sentinel p16 INK4a+ cells in the basement membrane form a reparative niche in the lung. Science 378: 192–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Smith M, Buffenstein R (2018) Naked mole‐rat mortality rates defy gompertzian laws by not increasing with age. Elife 7: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadiq A, Shah A, Jeschke MG, Belo C, Hayat MQ, Murad S, Amini‐Nik S (2018) The role of serotonin during skin healing in post‐thermal injury. Int J Mol Sci 19: 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon AB, Akha AAS, Buffenstein R, Miller RA (2008) Fibroblasts from naked mole‐rats are resistant to multiple forms of cell injury, but sensitive to peroxide, ultraviolet light, and endoplasmic reticulum stress. J Gerontol A Biol Sci Med Sci 63: 232–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seluanov A, Hine C, Bozzella M, Hall A, Sasahara THC, Ribeiro AACM, Catania KC, Presgraves DC, Gorbunova V (2008) Distinct tumor suppressor mechanisms evolve in rodent species that differ in size and lifespan. Aging Cell 7: 813–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seluanov A, Hine C, Azpurua J, Feigenson M, Bozzella M, Mao Z, Catania KC, Gorbunova V (2009) Hypersensitivity to contact inhibition provides a clue to cancer resistance of naked mole‐rat. Proc Natl Acad Sci U S A 106: 19352–19357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, Miyazawa H, Yamaguchi Y, Miura M, Jenkins DMR et al (2016) HIF‐1α‐PDK1 axis‐induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun 7: 11635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storer M, Mas A, Robert‐Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J et al (2013) Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155: 1119–1130 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suematsu M, Suzuki H, Ishii H, Kato S, Yanagisawa T, Asako H, Suzuki M, Tsuchiya M (1992) Early midzonal oxidative stress preceding cell death in hypoperfused rat liver. Gastroenterology 103: 994–1001 [DOI] [PubMed] [Google Scholar]
- Takasugi M, Firsanov D, Tombline G, Ning H, Ablaeva J, Seluanov A, Gorbunova V (2020) Naked mole‐rat very‐high‐molecular‐mass hyaluronan exhibits superior cytoprotective properties. Nat Commun 11: 2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Ke Z, Tombline G, Macoretta N, Hayes K, Tian X, Lv R, Ablaeva J, Gilbert M, Bhanu NV et al (2017) Naked mole rat cells have a stable epigenome that resists iPSC reprogramming. Stem Cell Reports 9: 1721–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Azpurua J, Hine C, Vaidya A, Myakishev‐Rempel M, Ablaeva J, Mao Z, Nevo E, Gorbunova V, Seluanov A (2013) High‐molecular‐mass hyaluronan mediates the cancer resistance of the naked mole rat. Nature 499: 346–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Firsanov D, Zhang Z, Cheng Y, Luo L, Tombline G, Tan R, Simon M, Henderson S, Steffan J et al (2019) SIRT6 is responsible for more efficient DNA double‐strand break repair in long‐lived species. Cell 177: 622–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trouche E, Mias C, Seguelas MH, Ordener C, Cussac D, Parini A (2010) Characterization of monoamine oxidases in mesenchymal stem cells: role in hydrogen peroxide generation and serotonin‐dependent apoptosis. Stem Cells Dev 19: 1571–1578 [DOI] [PubMed] [Google Scholar]
- Ungvari Z, Buffenstein R, Austad SN, Podlutsky A, Kaley G, Csiszar A (2008) Oxidative stress in vascular senescence: lessons from successfully aging species. Front Biosci 13: 5056–5070 [DOI] [PubMed] [Google Scholar]
- Wada H, Shibata Y, Abe Y, Otsuka R, Eguchi N, Kawamura Y, Oka K, Baghdadi M, Atsumi T, Miura K et al (2019) Flow cytometric identification and cell‐line establishment of macrophages in naked mole‐rats. Sci Rep 9: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CS (2010) Effects of tempol and redox‐cycling nitroxides in models of oxidative stress. Pharmacol Ther 126: 119–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi S, Nohara S, Nishikawa Y, Suzuki Y, Kawamura Y, Miura K, Tomonaga K, Ueda K, Honda T (2021) Characterization of an active LINE‐1 in the naked mole‐rat genome. Sci Rep 11: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura Y, Kawamura Y, Oiwa Y, Oka K, Onishi N, Saya H, Miura K (2021) Isolation and characterization of neural stem/progenitor cells in the subventricular zone of the naked mole‐rat brain. Inflamm Regen 41: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez‐Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R et al (2019) Ensembl 2020. Nucleic Acids Res 48: D682–D688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Tyshkovskiy A, Muñoz‐Espín D, Tian X, Serrano M, de Magalhaes JP, Nevo E, Gladyshev VN, Seluanov A, Gorbunova V (2018) Naked mole rats can undergo developmental, oncogene‐induced and DNA damage‐induced cellular senescence. Proc Natl Acad Sci U S A 115: 1801–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Oreskovic E, Zhang Q, Lu Q, Gilman A, Lin YS, He J, Zheng Z, Lu JY, Lee J et al (2021) Transposon‐triggered innate immune response confers cancer resistance to the blind mole rat. Nat Immunol 22: 1219–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]