

Abstract
The tumor microenvironment (TME) directly determines patients' outcomes and therapeutic efficiencies. An in‐depth understanding of the TME is required to improve the prognosis of patients with cervical cancer (CC). This study conducted single‐cell RNA and TCR sequencing of six‐paired tumors and adjacent normal tissues to map the CC immune landscape. T and NK cells were highly enriched in the tumor area and transitioned from cytotoxic to exhaustion phenotypes. Our analyses suggest that cytotoxic large‐clone T cells are critical effectors in the antitumor response. This study also revealed tumor‐specific germinal center B cells associated with tertiary lymphoid structures. A high‐germinal center B cell proportion in patients with CC is predictive of improved clinical outcomes and is associated with elevated hormonal immune responses. We depicted an immune‐excluded stromal landscape and established a joint model of tumor and stromal cells to predict CC patients' prognosis. The study revealed tumor ecosystem subsets linked to antitumor response or prognosis in the TME and provides information for future combinational immunotherapy.
Keywords: cervical cancer, heterogeneity, single‐cell RNA sequencing, tumor microenvironment
Subject Categories: Cancer, Immunology
Single‐cell RNA and TCR sequencing of paired tumor and adjacent normal tissues highlights stromal immune features and prognostic cellular subsets in cervical squamous cell carcinoma.

Introduction
Despite tremendous progress achieved in cervical cancer (CC) prevention via widespread screening and prophylactic HPV vaccination, CC remains prevalent worldwide, with 604,000 new cases and 342,000 deaths in 2020 (Sung et al, 2021). Recently, an increasing number of studies have utilized immunotherapies, including immune checkpoint blockade (ICB), therapeutic vaccines and engineered T cells, to improve the survival of patients with CC. However, these regimens are usually limited to relatively modest efficacy or regression in a small group of patients (Youn et al, 2020; Colombo et al, 2021; Nagarsheth et al, 2021). Hence, the development of novel immunotherapeutic strategies or targeted therapies is urgently required.
Cervical cancer is characterized as an immune‐infiltrated but immunosuppressive cancer type, primarily due to the modulation of the TME by HPV (O'Donnell et al, 2019; Shamseddine et al, 2021). CC cells utilize multiple mechanisms to escape killing by cytotoxic T lymphocytes (CTL) and NK cells, including downregulation of major histocompatibility complex (MHC) genes, inhibition of the cGAS–STING pathway, and increased PD‐L1 expression (Ashrafi et al, 2005; Liu et al, 2016; Luo et al, 2020). Previous studies have indicated a distinct anti‐ or pro‐tumor immunity role of CD4+ T cells in CC. The CD4+CD161+ effector T cell subset correlates with prolonged survival, whereas regulatory T cells (Tregs) and T helper 17 cells (Th17) exhibit protumorigenic roles in precancerous lesions and CC (Heeren et al, 2015; Walch‐Ruckheim et al, 2015; Santegoets et al, 2019). Despite these findings, whether and how T cells recognize HPV‐specific antigens in CC is largely unknown. B‐cell signatures are associated with favorable clinical outcomes in CC (Kim et al, 2020). In addition, B‐cell‐mediated HPV‐specific antibody responses have been validated in HPV‐related head and neck squamous cell carcinoma (HNSCC), suggesting the potential role of B cells in HPV‐related cancers (Kim et al, 2020; Wieland et al, 2021). However, investigations of B cell subset functions in CC remain scarce. Finally, the studies above focused on specific cell subsets sorted by flow cytometry or were limited to classical immune markers. The cellular characterization, interactions and dynamic development of immune components in CC have not yet been investigated.
An in‐depth understanding of TME characterization can be instrumental in developing more efficacious therapeutic approaches. Single‐cell‐sequencing technology has created opportunities to deepen understanding of TME, specifically focusing on the phenotype and dynamics of immune and tumor stromal cells (Ren et al, 2021). To uncover the TME of CC, droplet‐based single‐cell RNA sequencing (scRNA‐seq) and single‐cell TCR‐sequencing (TCR‐seq) were conducted among six patients with cervical squamous cell carcinoma with matched primary tumor areas and adjacent normal tissues. By comparing the ecosystem between tumor and normal samples, a basal‐like epithelial subpopulation was identified that could be critical to the invasion and metastasis of CC cells. For the immune compartment, dynamic lineage tracking across different T and B cell phenotypes was observed, emphasizing the ongoing tumor‐specific immune‐exhausting or activating processes. Finally, the function of stromal subpopulations in the immune system was evaluated, and the co‐occurrence of inflammatory cancer‐associated fibroblasts (CAFs) and an immature endothelial subset that predicts poor outcomes were highlighted.
Results
Single‐cell transcriptomics and TCR profiles of CC
Eleven samples were collected from six patients who underwent hysterectomy (five pairs of tumor and adjacent normal tissues and one additional tumor sample, Table EV1). The gene expression profile and T‐cell receptor (TCR) repertoire of these samples were obtained at single‐cell resolution using the 10× Genomics Chromium platform (Fig 1A, Table EV2). To ensure that all the cells were of high quality and devoid of potential contaminants, we performed quality control, doublet removal (Table EV3), multiple cell‐type signature visualization, and batch correction using a streamed pipeline (Fig EV1A–D, Materials and Methods) (Wang et al, 2020). A total of 53,089 high‐quality cells were used for gene expression analysis, with an average of 5,486 reads and 1,678 genes detected per cell (Fig EV1B).
Figure 1. The single‐cell landscape for the cervical cancer.
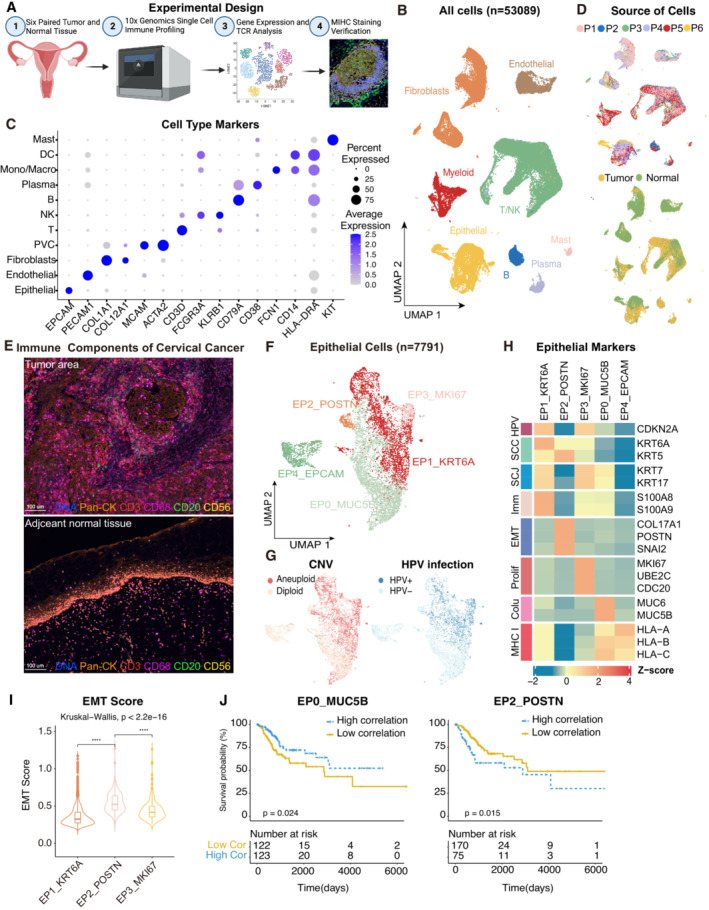
- Experimental design of primary cervix tumor collection, processing, sequencing and data analysis; Created with BioRender.com.
- UMAP plot of all the 53,089 single cells from 6 cervical cancer patients. Cells were annotated based on known lineage‐specific marker genes (denoted by colors).
- Dot plot showing the lineage‐specific marker genes of T and NK cells (T/NK), B cells, Plasma, Myeloid cells, Mast cells, Fibroblasts, Perivascular cells (PVC), Endothelial and Epithelial cells. The shade of color denotes the average gene expression level, the dot size denotes the percentage of gene expression in the corresponding lineage.
- UMAP plot of single cells profiled in the presenting work colored by patient (upper panel) and sample source (lower panel).
- Representative mIHC of immune components in CC tumors and adjacent normal tissues. T cells: CD3 (red), B cells: CD20 (green), macrophages: CD68 (pink), NK cells: CD56 (yellow), epithelial cells: Pan‐CK (orange). Scale bar = 100 μm.
- UMAP plot showing the subtypes of 7,791 epithelial cells. Cluster annotations are denoted and colored corresponding to the cell type in the figure.
- UMAP plot showing the distribution of CNV and HPV infection status (indicated by colors).
- Heatmap showing the expression of marker genes in each subtype of epithelial cells. SCC, squamous cell carcinoma; SCJ, squamocolumnar junction; Imm, immune‐related genes; Prolif, hyperproliferation; Colu, columnar epithelium; EMT, epithelial‐mesenchymal transition.
- Violin plot indicating the EMT Score of three malignant cell clusters; Differences between group were examined by Kruskal‐Wallis test. ****P < 0.0001 by pairwise Wilcoxon tests with the Benjamini‐Hochberg correction.
- Comparison of Overall survival (OS) rates for the high‐correlation and low‐correlation groups, stratified using the EP0_MUC5B (left panel) and EP2_POSTN (right panel) signatures in TCGA. P‐values are calculated using the log‐rank test (N = 255).
Figure EV1. ScRNA‐seq profiling of the ecosystem in CC samples.
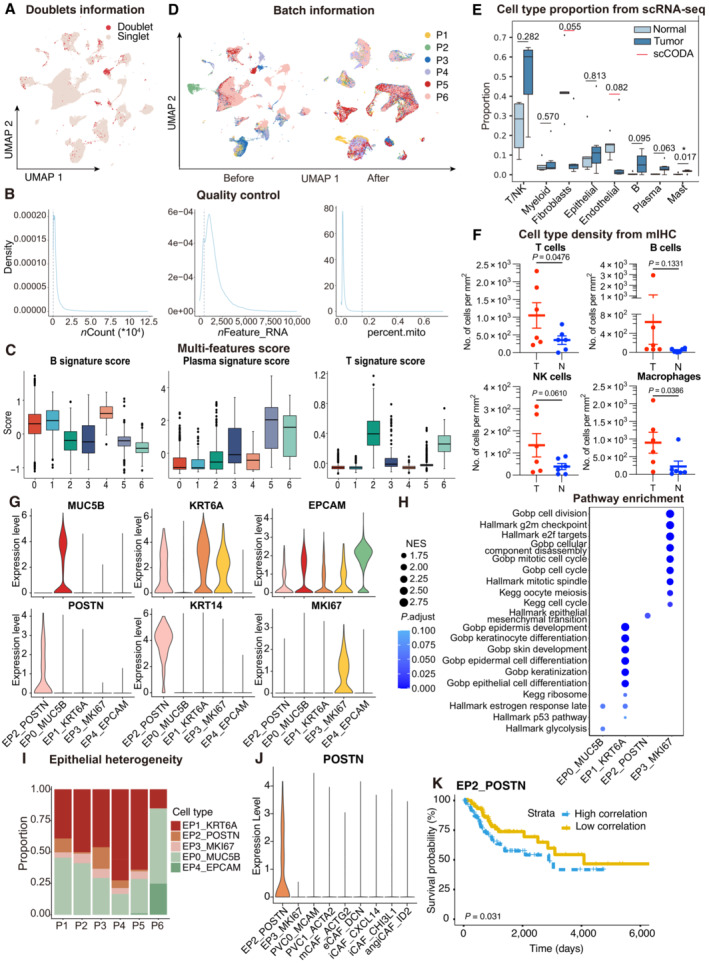
- UMAP plot showing the distribution of singlets (pink) and doublets (red) in CESC as evaluated using by R packages “DoubletFinder”.
- Density plots showing the distribution of UMIs (left panel), genes (middle panel), and mitochondrial percents (right panel) of all single cells. The light gray dotted line denotes the cut‐offs of quality control.
- Boxplots showing the T, Plasma and B cell three signature scores of B cell subtypes.
- UMAP plot showing the information of Batch from patients. Before (left panel) and after (right panel) batch effect removal.
- Boxplots showing the cell‐type proportions of six patients for matched tumor and normal samples. P < 0.05 was considered statistical significance; The scCODA model and the ALDEx2 model were used to examine the differences in cell‐type composition. Red bars indicate credible and significant results of scCODA. Stars indicate the significance calculated by ALDEx2 model (* P < 0.05).
- Scatter plots showing the quantification of T cells (CD3+), B cells (CD20+), macrophages (CD68+) and NK cells (CD56+) in tumor area (T) and adjacent normal tissue (N, n = 6). Represented Mean ± SEM. P‐value was measured by paired Student's t‐test.
- Violin plot showing the expression of marker genes in epithelial subclusters.
- Dot plot showing the selected signaling pathways (rows) with significant enrichment of GO, KEGG and Hallmark terms for four epithelial cell clusters.
- Stacked bar plot showing the epithelial cell clusters distribution among patients.
- Violin plot showing the expression of POSTN in EP2 and fibroblasts.
- OS rates for the high‐correlation and low‐correlation groups in TCGA, stratified using the EP2_POSTN signatures removed overlapped genes with fibroblasts. P‐values are calculated using the log‐rank test (N = 255).
Data information: Boxplots show the median and upper/lower quartiles (Related to Fig 1).
Seven major cell lineages were identified from the gene expression profiles and visualized using uniform manifold approximation and projection (UMAP, Fig 1B). Different cell lineages were annotated with typical cell markers as follows (Fig 1C): epithelial cells (EPCAM +), T cells (CD3D +), NK (natural killer) cells (KLRB1 +), B cells (CD79A +), and plasma cells (CD38 +), myeloid‐derived cells (FCN1 + for macrophages, CD14 + for dendritic cells, KIT + for mast cells), endothelial cells (PECAM1 +), and fibroblasts (COL1A1 + and COL12A1 + for fibroblasts, MCAM + and ACTA2 + for perivascular cells). Tumor samples exhibited a distinct cell lineage distribution from normal samples, with an increase in T/NK cells, B cells, plasma cells, and mast cells in the tumor samples, indicating the infiltration of immune cells in the CC microenvironment (Figs 1D and EV1E). The accumulation of immune cells in the CC microenvironment was further verified by multiplex immunohistochemistry (mIHC, Figs 1E and EV1F). In summary, the dataset demonstrated that the CC microenvironment harbors a complex ecosystem with increased immune cell infiltration and relatively fewer stromal cells.
POSTN + malignant cells manifest an invasive phenotype
The heterogeneity of epithelial cells was investigated by re‐clustering the cells into five different subtypes (Figs 1F and EV1G and H). To separate the malignant cells and normal epithelia, CopyKAT analysis was performed (Gao et al, 2021), which infers the malignant status of each cell based on copy number variation from scRNA‐seq data (Fig 1G). We also aligned the scRNA‐seq reads on the HPV genome (Serra & Chetty, 2018) and the HPV infection status of each cell was evaluated (Fig 1G). EP4_EPCAM and most cells from EP0_MUC5B were classified as normal epithelium with a diploid genome, whereas EP1_KRT6A, EP2_POSTN, and EP3_MKI67 were classified as malignant cells. As expected, the three malignant clusters harbored a high proportion of HPV‐infected cells, which is commonly observed in CC (Fig 1F and G), and although all five epithelial clusters were observed in the six patients, the proportion of malignant and normal clusters varied significantly, suggesting sizeable intertumoral heterogeneity between different patients (Fig EV1I).
Thereafter, focus was given to the characteristics of malignant cells based on their marker genes (Figs 1H and EV1G). EP3_MKI67 presented a hyperproliferative status, whereas EP1_KRT6A exhibited conventional CC cell features, such as high expression of KRT6A and CDKN2A and enrichment in the P53 pathway (Fig EV1H). In addition, EP1_KRT6A showed an immune‐related feature (S100A8 and S100A9) previously reported in Epstein–Barr virus‐associated nasopharyngeal carcinoma and downregulation of MHC class I genes, indicating that this subpopulation might play an essential role in immune cell dysfunction in the TME (Jin et al, 2020). Notably, EP2_POSTN displayed a hallmark signature of enrichment in epithelial‐mesenchymal transition (EMT) and unique expression of the metastasis‐related gene POSTN (Wei et al, 2021) among epithelial cells and fibroblasts (Figs 1I and EV1J). Survival analysis was performed using gene signatures from different epithelial subtypes (see Materials and Methods) on TCGA CESC patients to confirm their clinical relevance. As expected, patients with a high proportion of EP2_POSTN showed poor survival, whereas those with a high normal proportion of EP0_MUC5B cells had an improved prognosis (Fig 1J). The result remained consistent after removing the overlapping genes between EP2_POSTN and fibroblasts (Fig EV1K). According to the findings, EP2_POSTN may represent a subset of invasive malignant cells and is predictive of prognosis.
T and NK cells exhibit exhausted features in the TME
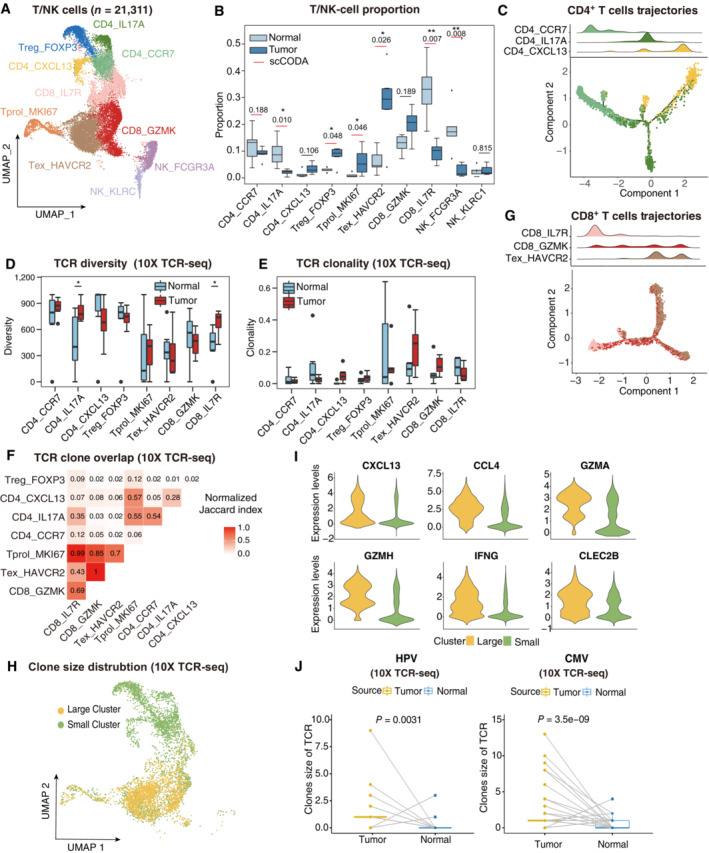
T and NK cells are vital effectors of tumor immunity. However, their dynamic status within the TME has not been completely investigated in CC. Therefore, 21,311 T and NK cells were isolated and 10 clusters derived (Fig 2A). In addition to proliferating T cells (Tprol_MKI67), CD8+ T cells were classified as memory T cells (CD8_IL7R), cytotoxic T cells (CD8_GZMK), and exhausted T cells (Tex_HAVCR2), whereas CD4+ T cells were separated into naïve CD4+ T cells (CD4_CCR7), Th17 cells (CD4_IL17A), follicular helper T cells (Tfh, CD4_CXCL13), and regulatory T cells (Tregs, Treg_FOXP3) according to the expression of canonical markers (Fig EV2A). The Tex_HAVCR2 subset expressed blended amounts of cytotoxic genes and inhibitory receptors (HAVCR2, LAG3, PDCD1, and TIGIT), whereas cytotoxic T cells expressed GZMK rather than GZMB. These observations roughly coincide with the general paradigm of other cancers (Ren et al, 2021). CD4+ T‐cell composition indicated a skewed response dominated by Th17 and Tfh subsets, in concordance with studies indicating a lack of cell‐mediated immunity (Th1‐type response) in cervical tumors (Shamseddine et al, 2021; Yuan et al, 2021). Moreover, the tumor area showed a high density of Tex_HAVCR2, Treg_FOXP3, and Tprol_MKI67 (Fig 2B), suggesting that the majority of tumor‐infiltrated T cells were exhausted and compromised by tumor antigens (Li et al, 2019). In contrast, adjacent normal tissues were enriched in pre‐stimulated phenotypes, such as CD4_CCR7 and CD8_IL7R. For NK cells, the NK_FCGR3A subset was excluded from the tumor area, corresponding to an activated cytotoxic phenotype (PRF1 and GNLY), whereas NK_KLRC1, a suppressive phenotype, was slightly enriched in the tumor area. In addition, the anti‐tumor activity of tumor‐infiltrating NK cells was not thoroughly dampened, as survival analysis revealed that both NK_FCGR3A and NK_KLRC1 signatures were associated with better outcomes of overall survival in the TCGA CC cohort (Fig EV2B), consistent with the observation that patients with CC high‐level intratumoral NK cells had an inferior risk of progression (Zhang et al, 2021). Overall, the data indicated that a large proportion of T/NK cells were exhausted, and the CD4 response was dominated by Th17 in the CC.
Figure 2. The dynamic subtypes of T/NK cells in TME and TCR repertoire profiling using 10× TCR‐seq data.
-
AUMAP plot showing the subtypes of 21,311 T/NK cells. Cluster annotations are denoted and colored corresponding to cell types in the figure.
-
BBoxplots showing the cell‐type proportions of T/NK cells for matched tumor and normal samples (n = 5). The scCODA model and the ALDEx2 model were used to examine the differences in T/NK cells' composition. Red bars indicate credible and significant results of scCODA. Stars indicate the significance calculated by ALDEx2 model (*P < 0.05, **P < 0.01).
-
C2D graph of T cells trajectories, from CD4+ T cell subsets. The cell density distribution, by the pseudo‐time, is shown at the top of the figure and colored corresponding to cell types, respectively.
-
D, EBoxplots showing each cell type TCR Diversity (D) and Clonality (E) level for matched tumor and normal samples (n = 5) by using 10× TCR data. Student's t‐test. *P < 0.05.
-
FTriangle heatmap showing the overlap of expanded TCR clonotypes across all possible combinations of T cell clusters. Data were aggregated for each of the indicated patient groups from 10× TCR data. Numbers indicate the normalized Jaccard index number of shared expanded TCR clonotypes for each cluster pair.
-
G2D graph of T cells trajectories, from CD8+ T cell subsets. The cell density distribution, by the pseudo‐time, is shown at the top of the figure and coloured corresponding to cell types, respectively.
-
HUMAP as in (A), but cells are colored corresponding to clone sizes, large (yellow) or small (green). Clones from 10× TCR data.
-
IViolin plots showing the differentially expressed genes between large and small clone (color‐coded as in H).
-
JBoxplots showing the clone size of HPV (left panel) and CMV (right panel) antigen‐specific TCR for matched tumor and normal samples by using 10× TCR data. Student's t‐test.
Data information: Boxplots show the median and upper/lower quartiles.
Figure EV2. The clinical function of NK cells and TCR repertoire profiling using TRUST predicted data.
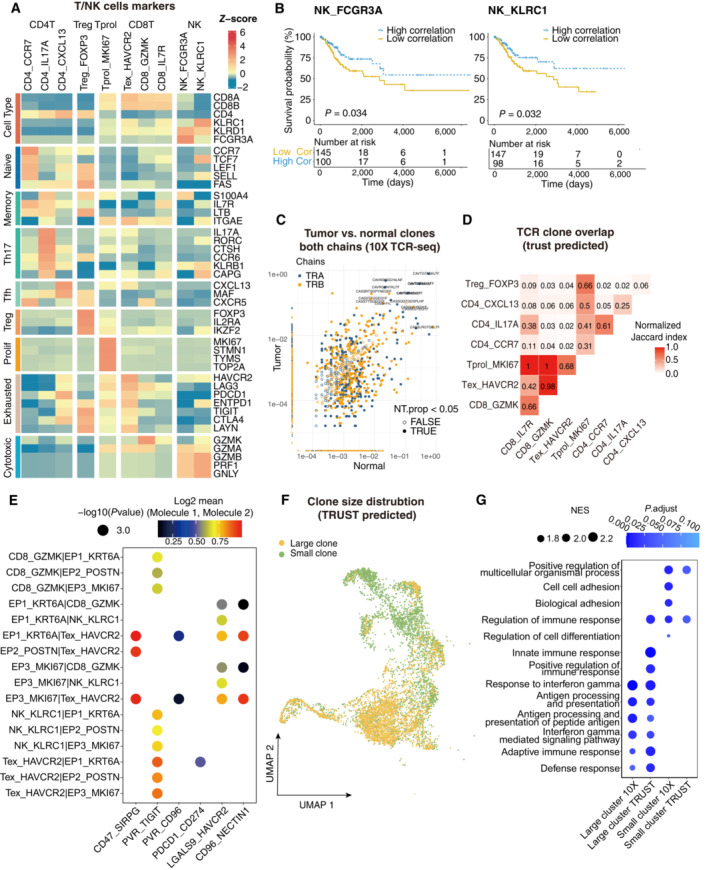
- Heatmap showing the expression of marker genes in each subtype of T/NK cells.
- Comparison of Overall survival (OS) rates for the high‐correlation and low‐correlation groups, stratified using the NK_FCGR3A (left panel) and NK_KLRC1 (right panel) signatures in TCGA. P‐values are calculated using the log‐rank test (N = 255).
- Representative example of tumor versus normal clones from 10× TCR data, showing both chains, with filled points representing clones suggesting a significant change in frequency.
- Triangle heatmap showing the overlap of expanded TCR clonotypes across all possible combinations of T cell clusters. Data were aggregated for each of the indicated patient groups from TRUST predicted TCR data. Numbers indicate the normalized Jaccard index number of shared expanded TCR clonotypes for each cluster pair.
- Bubble plots showing the interactions between epithelial cells and T/NK cell populations using CellPhoneDB.
- UMAP as in Fig 2A, but clones from TRUST predicted TCR data.
- Dot plot showing the selected signaling pathways (rows) with significant enrichment of GO terms for large clones and small clones identified by 10× or predicted by TRUST (Related to Fig 2).
Trajectory and TCR analyses indicated both transitioned and infiltrated sources of T cells
T‐cell responses to tumor immunity can be expanded from tissue‐resident memory cells or infiltrated from the lymph nodes or blood (Yost et al, 2019). Therefore, the origin and dynamic status of the T cell subsets were addressed. First, the pseudo‐time trajectory was reconstructed to understand the differentiation status of T cells using the Monocle2. Both CD4+ and CD8+ T cells are distributed in binary, branched structures. For CD4+ T cells, the root of the trajectory was CD4_CCR7, with CD4_IL17A and CD4_CXCL13 as the ending clusters (Fig 2C), indicating functional differentiation from naïve CD4 T cells to T helper cells. TCR similarity can be utilized as a natural marker to identify the dynamics of clonal T cells (Nikolich‐Zugich et al, 2004) TCR repertoire analysis was then conducted to understand the clonality and diversity of T cells in CC, to verify the TCR repertoire mapped from scRNA expression profiles using TRUST4 (Song et al, 2021). For single‐cell TCR‐seq data, 15,957 cells with sufficient read coverage were used for downstream analysis (Fig EV2C). CD4_CXCL13 showed reduced diversity and increased clonality in tumor samples compared with adjacent normal samples (Fig 2D and E), and CD4_IL17A was opposite, suggesting a potential clonal expansion of CD4_CXCL13 in response to tumor antigens. In addition, the similarity of complementarity‐determining region 3 (CDR3) sequences among differential T cell phenotypes (Glanville et al, 2017) were investigated, and the normalized Jaccard Index used to qualify T‐cell CDR3 similarity, where a greater value illustrated higher similarity between two clusters (see Materials and Methods). Notably, the intermediate to low Jaccard index across CD4+ T cells revealed that naïve T cells were not the sole origin of helper T cells (Figs 2F and EV2D). Treg_FOXP3 and CD4_CXCL13 showed extremely low TCR clone overlap across other subsets, indicating that these cells potentially infiltrated from an external environment, such as blood, instead of transitioning from naïve CD4+ T cells in CC. Of note, both trajectory analysis and Jaccard index revealed that naïve CD4+ T cells differentiated into Th17 cells rather than Tfh cells, consistent with research indicating that Th17 expansion was supported in the CC microenvironment (Walch‐Ruckheim et al, 2019) (Fig 2C and F). For CD8+ T cells, with CD8_IL7R memory T cells as the root, Tex_HAVCR2 assembles at the end of the branches and CD8_GZMK scatters around the trajectory, suggesting that the exhausted T cell phenotype was differentiated or switched from a cytotoxic phenotype (Fig 2G). Tex_HAVCR2 and CD8_GZMK showed higher clonal expansion levels in tumor samples than in adjacent normal samples (Fig 2E). Consistently, high‐level Jaccard indices among CD8_GZMK, Tex_HAVCR2, and Tprol_MKI67, the “hallmark” of ongoing expansion (Li et al, 2019), indicate that these cells were mainly from local expansion (Fig 2F). Moreover, the extremely high normalized Jaccard index between cytotoxic T cells and exhausted T cells illustrated that they represented a continuous status of T cell exhaustion instead of two distinct subsets (Fig 2F). To investigate the potential mechanism that induces the exhaustion of T/NK cells, cell–cell interaction analyses were performed using CellphoneDB (Fig EV2E), and poliovirus receptor (PVR)‐like protein signaling was the main co‐inhibitory interaction between T/NK cells (CD96 and ITGIT) and malignant cells (PVR and NECTIN1). Also observed were other interactions between malignant cells and T cells, including LGALS9:HAVCR2, CD47:SIRPG, and PDCD1:CD274. In summary, the findings suggest that CD8+ T cells exhibit a cytotoxic‐exhausted phenotype and a clear local expansion trajectory, and that the infiltrated Tregs and PVR‐like protein signaling may contribute to the immune‐resistance microenvironment of CC.
Cytotoxic and exhausted T cells form large T‐cell clones
Large clonal T cells shared identified TCR clonotypes as a result of clonal expansion; thus, these cells were considered as potentially tumor‐reactive T cells in the TME (Zheng et al, 2021). The phenotypes and abundance of large clone T cells also correlate with ICB therapy responses (Fairfax et al, 2020). Therefore, we investigated the phenotypes and gene expression of clonal T cells in CC with TCRs shared by more than five cells. Notably, large clones converged specifically in cytotoxic and exhausted T cell subsets, consistent with the T cell origin and trajectory (Figs 2H and EV2F, Dataset EV1). These large clone cells expressed a panel of cytotoxic genes (GZMA, GZMH, and IFNG) and chemokine genes (CXCL13 and CCL4), indicating an activated and lymphocyte recruitment phenotype (Fig 2I). In addition, large clones had a more pronounced enrichment of pathways related to interferon‐gamma than small clones, suggesting that large clones are more prone to cytotoxic immune responses (Fig EV2G). These results suggest that large clones of T cells may be critical effectors in the antitumor response, and that their specific antigens should be further investigated.
Although previous research has reported an unexpected magnitude of poised HPV‐specific T cells in CC after expansion, the natural status of HPV‐specific T cells has not been explored. TCRs' capacity to recognize HPV antigens was investigated by mapping the public clonotypes (Dataset EV2). Human cytomegalovirus (CMV), which can latently infect and elicit a robust T‐cell response, was used for comparison (van den Berg et al, 2019). The results showed enlarged clonal sizes of HPV and CMV in the tumor area compared with the adjacent normal area (Fig 2J, Dataset EV2), supporting the presence of HPV‐specific T‐cell responses. However, inconsistent with prior research (de Vos van Steenwijk et al, 2010), very few T cells were HPV‐specific, limiting further investigation of their phenotypes or expression profiles, perhaps due to heterogeneity among patients and unsorted cell capture strategies. Their dynamic states should also be further investigated during vaccination or immunotherapy to distinguish whether they were functional T cells or bystander clones, as previously reported (Simoni et al, 2018).
Germinal center responses are found in the tumor area
Currently, the anti‐tumor properties of B‐lymphocytes have been increasingly recognized (Liu et al, 2020; Lu et al, 2020), yet are less characterized in CC. To investigate the atlas and roles of B cells in CC, 3,107 B cells were separated (Fig 3A). Notably, B cells are predominantly derived from tumor samples instead of adjacent normal tissues, suggesting a consequent B cell response stimulated by the TME. Six clusters could be distinguished: activated B cells (ABC), memory B cells (MBC), germinal center B cells (GCB), plasma cells (PC), and transitional B cells (TC) (Fig EV3A). B cell subsets were also confirmed by a gene set enrichment analysis as previously reported (Fig 3B) (Cillo et al, 2020). The ABC cluster (B0_TNFRSF13B) was characterized by TNFRSF13B and CD83. MBC (B1_S1PR1) expressed BACH2 and KLF4, transcription factors for identifying germinal center‐derived memory B cells (Ripperger & Bhattacharya, 2021). GCB (B3_NEIL1) was identified by MME (encoding CD10), AICDA, and BCL6, while PC (PC_IGHA1 and PC_IGHG4) expressed MZB1 and XBP1 (Fig EV3A), suggesting an underlying germinal center (GC) response in the TME. Functional analyses suggested that the ABC gene signature is particularly concentrated in the antigen processing and presentation pathway, whereas the PCs' signature is enriched in complement activation and immunoglobulin production pathways, indicating the distinct anti‐tumor functions of different subsets of B cells (Fig 3C).
Figure 3. GC response identified in cervical cancer.
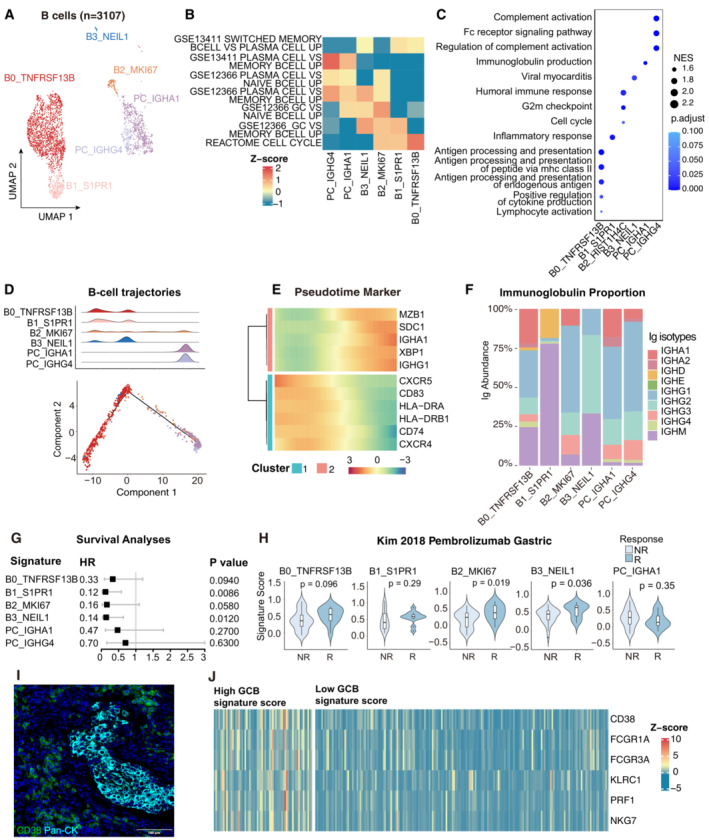
- UMAP plot showing the subtypes of 3,107 B cells. Cluster annotations are denoted and coloured corresponding to cell types in the figure.
- Heatmap showing the characterization of different B‐cell populations with B‐cell‐related pathways.
- Dot plot showing the selected signaling pathways (rows) with significant enrichment of GO terms for B‐cell and plasma clusters.
- 2D graph of B cells trajectories. The cell density distribution, by the pseudo‐time, is shown at the top of the figure and colored corresponding to cell types.
- Heatmap showing immune‐associated genes in the differentiation process.
- Stacked bar plot showing immunoglobulin (Ig) abundance of each B cell sub‐cluster.
- A forest plot showing the association of different B‐cell signatures with survival across the TCGA SCC cohort. Squares and lines indicate hazard ratios (HRs) and 95% confidence intervals (CIs), respectively. HRs were calculated using univariable Cox regression; P‐values were calculated using log‐rank test.
- Violin plots showing the association of different B‐cell signatures with the response (R, N = 15) and no response (NR, N = 42) to Pembrolizumab in the Kim cohort. P‐values calculated using the student's t‐test.
- Representative mIHC of plasma cells in CC tumor area. plasma cells: CD38+Pan‐CK−, scale bar = 100 μm.
- Expression of the genes that comprise the plasma cell and ADCC signatures in TCGA cohort.
Data information: Violin plots show the median and upper/lower quartiles.
Figure EV3. Clinical performance of B cell populations and BCR repertoire profiling using TRUST predicted data.
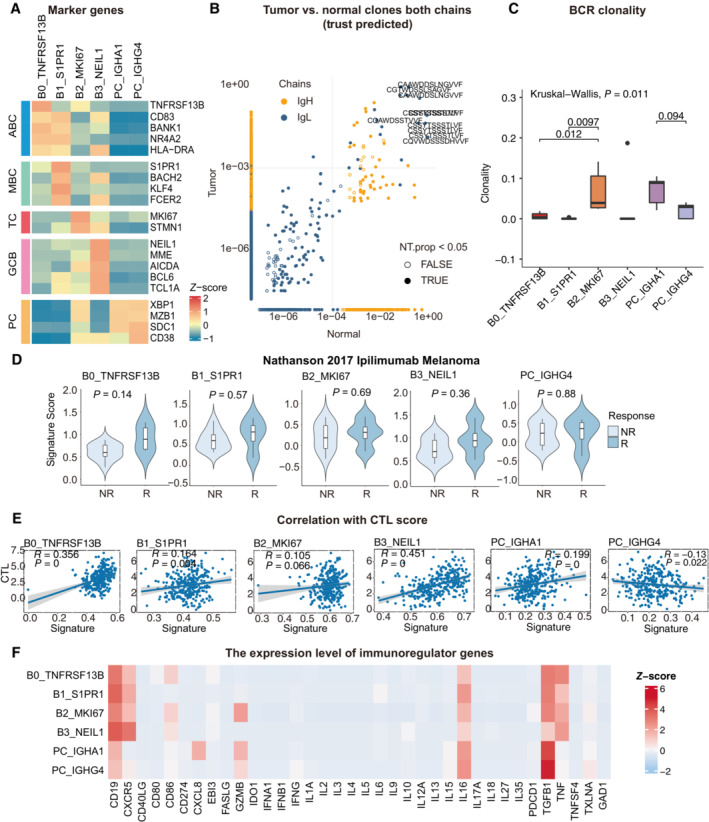
- Heatmap showing the expression of marker genes in each subtype of B cells.
- Representative example of tumor versus normal clones from TRUST predicted BCR data, showing both chains, with filled points representing clones suggesting a significant change in frequency.
- Boxplots showing each cell type BCR Clonality level for matched tumor and normal samples by using TRUST predicted BCR data. Student's t‐test, P < 0.05 was considered statistical significance.
- Violin plots showing the association of different B‐cell signatures with the response (R, N = 4) and no response (NR, N = 11) to Ipilimumab in the Nathanson 15 cohort. P‐values calculated using the student's t‐test.
- Scatter plot showing the correlation between the bulk sample with CTL score and the single‐cell B cell clusters.
- Heatmap showing the expression level of immunoregulator genes in B‐cell populations.
Data information: Boxplots show the median and upper/lower quartiles (Related to Fig 3).
The presence of GCB and GC‐derived MBC suggested activation of the GC response in CC. A pseudo‐time trajectory analysis of the B cells and PC was conducted to investigate the potential transitions. The trajectory path suggested a cell differentiation fate for B cells, starting at ABC (B0_TNFRSF13B), progressing towards GCB (B3_NEIL1) and GC‐derived MBC (B1_S1PR1), and terminating at PC (PC_IGHA1 and PC_IGHG4), with the TC cluster (B2_MKI67) spreading along the trajectory as a transitory status (Fig 3D). In addition, ABC marker genes (CD83 and TNFRSF13B) and GC‐related genes (CXCR4 and CXCR5) decreased continuously, whereas PC hallmark genes (MZB1, XBP1, SDC1, and IGHG1) increased, with an additional decrease in antigen‐presenting genes (Fig 3E). The trajectory of B‐cell subsets and gene expression illustrated a continuous B‐cell subset switch resembling the GC response. Furthermore, B cell receptor (BCR) data reconstructed via TRUST4 was used to investigate the clonal composition of B cells (Fig EV3B) (Song et al, 2021). The elevated clonality of the TC and PC subsets indicated that these subsets were derived from local expansion in the TME (Fig EV3C). The alterations of immunoglobulin (Ig) isotypes in GC response (Fig 3F), which arise from somatic hypermutation and class switch recombination, supported a tumor‐specific GC response in CC. In addition, IgG1 is the dominant subtype of IgG in GCB and PC, suggesting the potential effector activity of these subsets in antitumor immunity (Jacquelot et al, 2021). Overall, B cell subsets infiltrating the TME were characterized and germinal center reactions via BCR analysis and trajectory tracking were revealed, indicating ongoing tumor‐specific immunization with T‐dependent antigens in CC.
The infiltration of B cells is associated with increased patients' survival in CC
While B cells were highly heterogeneous in the CC TME, bulk RNA‐seq data from TCGA was used to evaluate whether B‐cell signatures were clinically significant. The GCB (HR = 0.14, P = 0.0120) and GC‐derived MBC (HR = 0.12, P = 0.0086) signatures were significantly associated with better outcomes (Fig 3G). It was then estimated whether responders who underwent immunotherapy had a higher density of B cells, as pembrolizumab (anti‐PD‐1 antibody) has been proven to benefit advanced CC patients (Colombo et al, 2021). All B cells exhibited a favorable trend, and the GCB and TC signatures were statistically significant in the gastric cancer cohort treated with pembrolizumab (Fig 3H). The trend was similar but did not reach statistical significance in another ipilimumab‐treated melanoma cohort (Fig EV3D). We also investigated the correlation between B‐cell subsets and cytotoxic T lymphocytes (CTL) (Jiang et al, 2018). Among the B cell and PC subsets, GCB (B3_NEIL1) showed the highest positive correlation with CTL (R = 0.451), indicating a potential interaction between GCB and T cells that could promote T‐cell infiltration in the TME (Fig EV3E). In addition, the expression levels of B cell‐related immunoregulatory genes were detected, as summarized in previous research (Wieland et al, 2021). Immunosuppressive genes related to B cells, including CD274, IL‐10, and IDO1, remained undetectable or at low levels apart from TGFB1 (Fig EV3F). Although TGFB1 is expressed in global B cell subsets, GCB maintains a relatively low level of TGFB1, suggesting multifaceted functions manifested in B cell subsets. A recent study by Meylan et al (2022) confirmed that tertiary lymphoid structures (TLS) could drive the in situ maturation of B cells, anti‐tumor antibody production, and antibody‐dependent cell‐mediated cytotoxicity (ADCC) in renal cell cancer. CD38+Pan‐CK− plasma cells were also consistently detected in GCB+ patients (Fig 3I), and patients with high CGB signatures showed higher expression of plasma markers (CD38), hallmarks of ADCC (FCGR1A and FCGR3A), and NK‐cell genes (Fig 3J). Collectively, the results revealed the heterogeneity of B‐cell subsets in anti‐tumor responses and emphasized the antineoplastic properties of GCB.
GCB subset is confined to tertiary lymphoid structures in TME
Considering the pivotal role of GCB, mIHC (n = 3) was performed to verify the localization of GCB in TME. Total B cells (CD20+) and PC (CD20−CD38+) were mostly stained adjacent to the PanCK+ tumor parenchyma (Figs 4A and B, and EV4A). GCB (CD20+BCL6+) was consistently present in well‐formed TLS rather than in poorly structured immune‐cell aggregates (Fig EV4B), and cell density statistics further verified the distribution of total B cells, PCs, and GCBs (Fig 4C). TLS constituted by a CD20+ B‐cell follicular and an outer CD4+ or CD8+ T cell was further observed by immunohistochemistry (IHC) in the same patient (Fig EV4A). These results indicate that the microenvironment supported by TLS might be necessary for the GC reaction in the TME. The presence of GC is a typical trait of mature TLS and is considered a favorable factor in prognosis and immunotherapy (Sautes‐Fridman et al, 2019; Cabrita et al, 2020). Thereafter, it was investigated whether TLS was associated with the outcomes of patients with CC. In the absence of transcriptomic signatures for TLS detection in CC, previously reported signatures were applied to TCGA patients (Sautes‐Fridman et al, 2019; Horeweg et al, 2022; Meylan et al, 2022). In addition, all prior signatures were associated with better outcomes apart from a single follicle dendritic cell marker L1CAM and chemokine signature (Fig EV4C), suggesting that TLS was beneficial for patient prognosis across cancers but had heterogeneity in neogenesis‐related chemokines. Notably, Tfh, a key regulator of GC reactions whose signature has optimal performance (HR = 0.092, P = 0.0028), highlights the crucial role of GC reaction‐related signatures in outcome prediction.
Figure 4. Localization and interactions of B cells and TLS in CC.
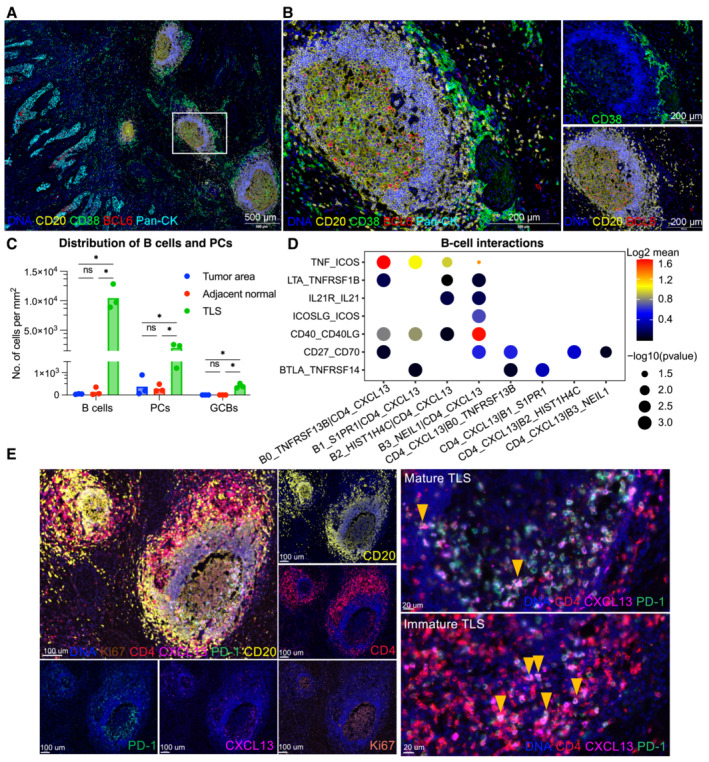
-
A, BB cell subsets localization in CC tissues are exhibited by mIHC via differential markers as follow: total B cell: CD20+, GCB: CD20+BCL6‐, PC: CD20‐CD38+; with scale =500 μm for (A), scale = 200 μm for (B).
-
CQuantification of B‐cell density in Pan‐CK+ tumor area, adjacent stroma and TLS. *P < 0.05, two‐way ANOVA following Tukey post‐hoc test.
-
DBubble plots showing the interactions between Tfh (CD4_CXCL13) and B‐cell populations using CellPhoneDB.
-
ERepresentative mIHC of CC tumor with CD4+PD‐1+CXCL13+ Tfh cells located in TLS (yellow arrows). scale bars = 100 μm for left panel, scale bars = 20 μm for right panel.
Figure EV4. Additional analysis of TLS.
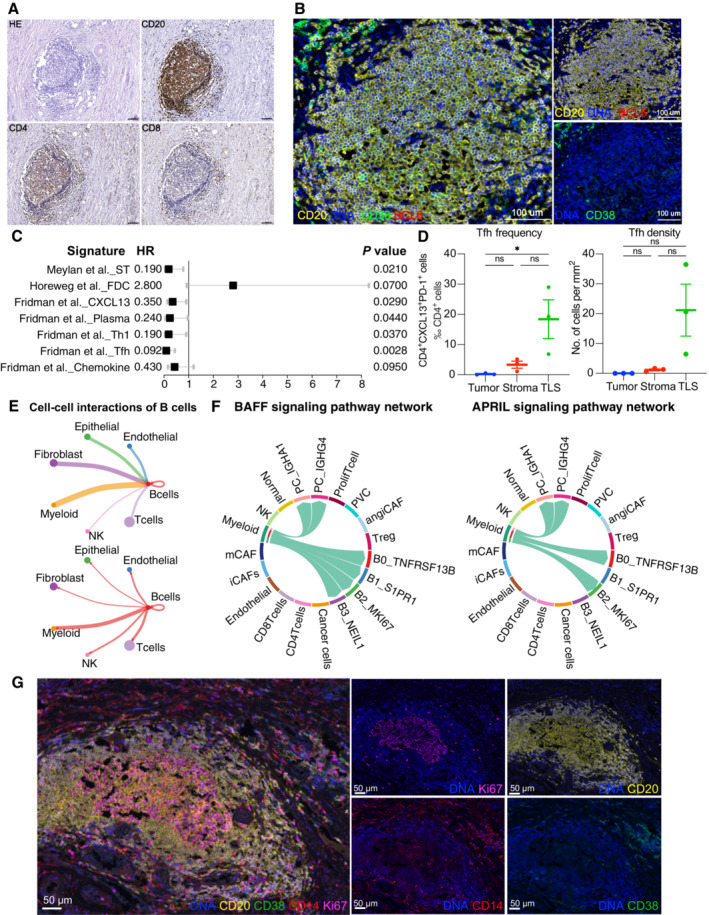
- Cellular compositions of TLS are exhibited by immunohistochemistry. Scale = 100 μm.
- B‐cell subsets are exhibited by mIHC via differential markers as follows: total B cell: CD20+, GCB: CD20+BCL6−, PC: CD20−CD38+. Scale = 100 μm.
- A forest plot showing the association of TLSs‐associated signatures with survival across the TCGA SCC cohort (n = 255). Squares and lines indicate hazard ratios (HRs) and 95% confidence intervals (CIs), respectively. HRs calculated using univariable Cox regression; P‐values calculated using log‐rank test; ST, spatial transcriptomics.
- Scatter plots showing the frequency and density of Tfh cells (CD4+PD‐1+CXCL13+) in TLSs, tumor and stroma (n = 3). Represented Mean ± SEM. P‐value was measured by the Holm–Sidak's multiple comparisons test, *P < 0.05.
- Circle plots showing the number of interaction weights between B cell groups and other cell groups. Edge colors are consistent with the sources as the sender, and edge weights are proportional to the interaction strength. A thicker edge line indicates a stronger signal.
- Chord diagram showing the communication networks of APRIL (left panel) and BAFF (right panel) signaling pathways. Arrows indicate the interactions (signaling pathways) from some cell groups to other cell groups.
- Representative mIHC of CD14+ cell localization in TLS. Scale bars are 50 μm (Related to Fig 4).
Since T‐B collaboration is an underlying mechanism for the anti‐tumor effects of B cells (Cui et al, 2021), the T cell component of TLS in CC was investigated by focusing on interactions between B cells and Tfh. Co‐stimulatory molecules (Cui et al, 2021) for B‐cell differentiation (ICOSLG‐ICOS) and proliferation (CD40‐CD40L) are prominent interactions between GCBs and Tfh cells (Fig 4D), as well as molecules that promote germinal center development (IL‐21R‐IL21) and T‐B cell activation (CD27‐CD70). Of note, the CD4_CXCL13 subset exhibited a PD‐1+CXCR5− Tfh‐CXCL13 cell phenotype identified in nasopharyngeal carcinoma (Fig EV2A) (Li et al, 2021). The spatial relevance of PD‐1+CD4+CXCL13+ Tfh and GCB were validated using mIHC (Fig 4E). Tfh cells colocalized with TLS restrictedly, particularly in immature TLS and germinal centers (Figs 4E and EV4B). Moreover, the quantification of cell density and frequency also indicated the aggregation of Tfh in TLS (Fig EV4D), suggesting its potential role in TLS maturation and T‐B collaboration. Similar to potential GCB‐Tfh interactions, possible cross‐talk among other compartments via CellChat (Fig EV4E) was evaluated, and myeloid cells showed significant cross‐talk with B cells compared to other cell lineages. More importantly, the B cell survival factors APRIL and BAFF signaling are predominant in B cell interactions with myeloid cells (Fig EV4F), resulting in B cell organization and survival in the tumor area. The localization of CD14+ myeloid cells in TLS was further vadiated with mIHC (Fig EV4G). In summary, the analyses suggest that TLS genesis and B‐cell responses could be orchestrated by the Tfh‐CXCL13 subset and myeloid cells.
Cellular heterogeneity of myeloid cells in CC
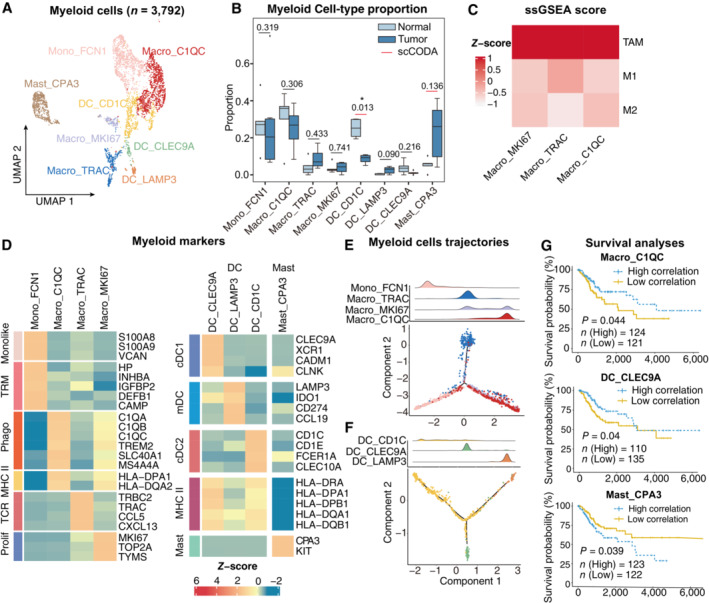
Despite the importance of tumor‐infiltrating myeloid cells in tumor progression, the subpopulations and properties of myeloid cells in CC have not been thoroughly investigated at the single‐cell level (Cheng et al, 2021). Three different types of myeloid cells were classified into cells from the mononuclear phagocyte system; dendritic cells (DCs), and mast cells (Fig 5A and B), according to canonical marker genes (Appendix Fig S1A) (Zilionis et al, 2019; Zhang et al, 2020a). Notably, all subtypes of macrophages resembled the signatures for tumor‐associated macrophages (TAMs) instead of M1 or M2 signatures (Fig 5C, Table EV4), indicating a more elaborate macrophage status in CC (Zhang et al, 2019; Hornburg et al, 2021). Specifically, Mono_FCN1 expressed the monocyte signature and a high level of tissue‐resident macrophage genes (Fig 5D), suggesting that the subset represented an intermediate state during the monocyte maturation of TAMs (Casanova‐Acebes et al, 2021). The presence of proliferating macrophages (Macro_MKI67) also indicated the potential renewal ability of macrophages in CC. Another tumor‐enriched subset, Macro_C1QC, expressed C1QC, TREM2, and MHC class II genes and was therefore annotated as a conventional phenotype of polarized phagocytosis and antigen presentation (Tirosh et al, 2016) Enrichment analyses also revealed that TAMs enriched complement activation pathways, and monocytes were enriched in humoral immune response (Appendix Fig S1B), suggesting that TAMs and monocytes may also be involved in antibody‐induced antitumor responses. Trajectory analysis confirmed the differentiation trend from monocytes to conventional macrophages (Fig 5E), suggesting that polarized macrophages might be derived from monocyte maturation and the proliferation of tissue‐resident macrophages. Interestingly, a unique subset of Macro_TRAC was independently distributed in the other branch of the trajectory. This subset showed features of TCR‐encoding genes (TRAC, CD247, and TRBC2) which could be identified as TCR+ macrophages and have been reported in the process of infection and pathological processes (Beham et al, 2011; Chavez‐Galan et al, 2015). Moreover, this subset expressed B cells, the TLS‐related chemokines CCL5, and CXCL13 (Fig 5D), indicating a potential role in B cell recruitment and TLS formation.
Figure 5. Phenotype diversity of myeloid cells in CC.
-
AUMAP plot showing the subtypes of 3,792 Myeloid cells. Cluster annotations are denoted and colored corresponding to cell types in the figure.
-
BBoxplots showing the cell‐type proportions of myeloid cells for matched tumor and normal samples (n = 5). The scCODA model and the ALDEx2 model were used to examine the differences in myeloid cells' composition. Red bars indicate credible and significant results of scCODA. Stars indicate the significance calculated by ALDEx2 model (*P < 0.05).
-
CHeatmap showing the characterization of different macrophage cell populations with M1, M2 and TAM scores.
-
DHeatmap showing the expression of marker genes in each subtype of myeloid cells; TRM, tissue‐resident macrophage; Phago, phagocytosis.
-
E, F2D graph of myeloid cells trajectories, from monocyte/macrophage cell (E) and Dendritic cell (F) subsets. The cell density distribution, by the pseudo‐time, is shown at the top of the figure and colored corresponding to cell types, respectively.
-
GComparison of Overall survival (OS) rates for the high‐correlation and low‐correlation groups, stratified using the Macro_C1QC (upper panel), and Mast_CPA3 (lower panel) signatures in TCGA (N = 255). P‐values are calculated using the log‐rank test.
Data information: Boxplots show the median and upper/lower quartiles.
For DC, conventional DCs (cDCs) were annotated using canonical markers such as CLEC9A + cDC1 and CD1C + cDC2. LAMP3 + mature DCs (mDCs) infiltrated mainly in tumor areas with the expression of immunoregulatory genes (IDO1 and CD274) and a lack of MHC class II‐encoding genes (Fig 5B and D), showing inhibitory roles in tumor‐infiltrating T cells. In addition, trajectory analysis delineated a conserved maturation module from conventional DCs to mature DCs (mDCs), suggesting that both cDC1 and cDC2 were the origins of mDCs (Fig 5F) (Ren et al, 2021). The majority of mast cells accumulated in the tumor area (Fig 5B), yet the role of mast cells in tumor progression remains unclear (Ribatti, 2016). According to their phenotypes, myeloid cells have distinct prognostic roles in CC patients. Macro_C1QC and DC_CLEC9A signatures were significantly correlated with a better outcome in TCGA patients, whereas highly infiltrating mast cells predicted a dismal prognosis (Fig 5G). In summary, the data illustrated the diversity of myeloid cells in CC and highlighted that these differential subsets have distinct anti‐ or pro‐tumor roles in tumor immunity.
Tumor‐specific iCAF and endothelial subset are associated with tumor progression and immunosuppression
Analysis of perivascular cells (PVCs) and CAFs (Fig 6A and B) was then conducted. PVCs with the robust PVC marker MCAM and angiogenic pericyte marker RGS5 (Fig 6C) were sorted (Sahai et al, 2020; Wu et al, 2020). Within CAFs, five subtypes with diverse characteristics were identified according to their marker genes in various cancers (Fig EV5A): myofibroblastic CAFs (mCAF_ACTG2), inflammatory CAFs (iCAF, iCAF_CHI3L1, and iCAF_CXCL14), extracellular matrix CAFs (eCAF_DCN) and angiogenesis‐associated CAFs (angiCAF_ID2) (Chen et al, 2020; Wu et al, 2020; Desbois & Wang, 2021). Expressing genes in cytokine‐mediated pathways, iCAF_CXCL14 and iCAF_CHI3L1 were identified as iCAFs (Fig 6C), which were further divided into cytokine‐related iCAF_CXCL14 and interleukin‐related iCAF_CHI3L1. Notably, compared with the adjacent normal tissues, the iCAF_CHI3L1 subset was predominately found in tumor samples, whereas the absence of cytokine‐related iCAFs in the tumor area suggests remodeling of iCAFs and restrained tumor control in the TME (Westrich et al, 2020). Importantly, the immunosuppressive mediator CHI3L1 and tumorigenic cytokines CXCL5 and IL11 were only expressed in iCAF_CHI3L1 (Fig EV5B). This unique subset was further verified by IHC staining and was similar to that of tumor cells (Fig EV5C). Surprisingly, eCAF_DCN, iCAF_CXCL14 and iCAF_CHI3L1 highly expressed extracellular matrix (ECM)‐associated genes (DCN, MMPs and collagens), suggesting their ability on ECM remodeling. CTL correlation analysis demonstrated that PVC0_MCAM, PVC1_ACTA2, eCAF_DCN and iCAF_CHI3L1 were inversely correlated with CTL (Fig 6D), partly as a result of ECM remodeling and immunomodulatory factors secreted by these subsets (Di Modugno et al, 2019). Correspondingly, the gene signatures of PVC0_MCAM and iCAF_CHI3L1 can also predict the response to ICB treatment in the melanoma or gastric cancer cohort (Fig EV5D). In summary, these results demonstrate heterogeneous CAFs subsets with distinct functions in CC and emphasize the potential tumorigenic and immunosuppressive roles of the iCAF_CHI3L1 subset.
Figure 6. Single‐cell resolution heterogeneity of stromal cells in cervical cancer.
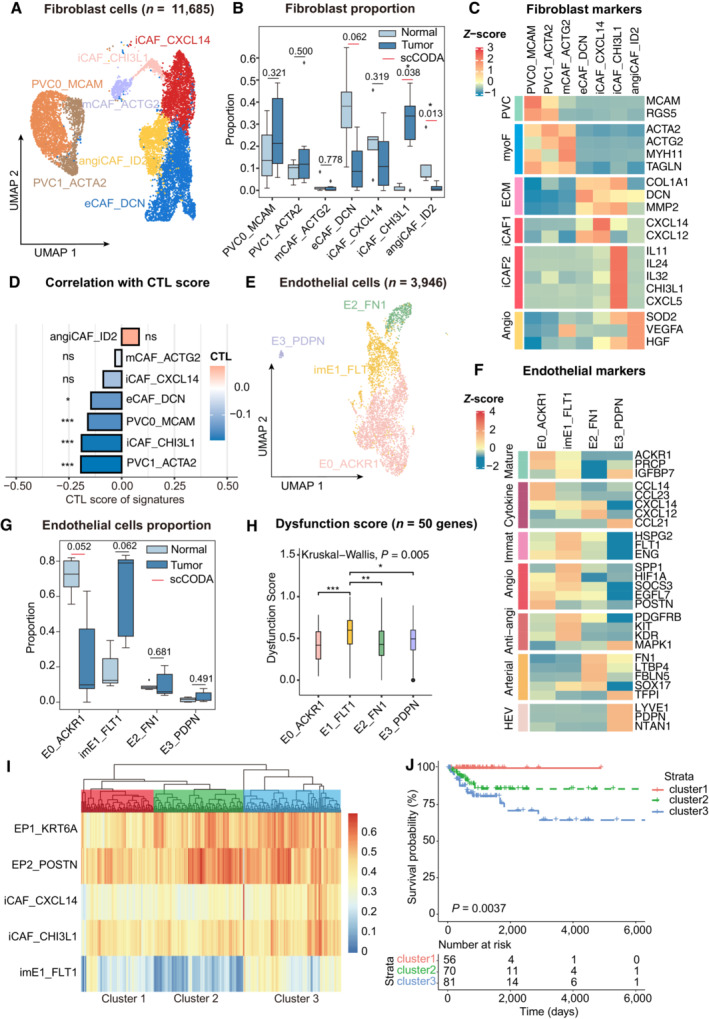
- UMAP plot showing the subtypes of 11,685 fibroblast cells. Cluster annotations are denoted and colored corresponding to cell types in the figure.
- Boxplots showing the cell‐type proportions of fibroblast cells for matched tumor and normal samples (n = 5).
- Heatmap showing the expression of marker genes in each subtype of fibroblasts; PVC, perivascular cells; myoF, myofibroblasts; ECM, extracellular matrix Angio, angiogenesis.
- Bar plot showing fibroblast signature associated with CTL exclusion. The bar width indicates the correlation between the bulk sample and the single‐cell fibroblast cluster. P‐values were computed using a two‐sided t‐test for correlation and were adjusted using the Benjamini–Hochberg procedure.
- UMAP plot showing the subtypes of 3,946 endothelial cells. Cluster annotations are denoted and coloured corresponding to cell types in the figure.
- Heatmap showing the expression of marker genes in each subtype of endothelial cells; Anti‐angi, anti‐angiogenic drug targets.
- Boxplots showing the cell‐type proportions of endothelial cells for matched tumor and normal samples.
- Boxplot showing the T‐cell dysfunction score of marker genes (n = 50) in endothelial cell populations; P‐values were caculated by Wilcox rank‐sum test with the Benjamini‐Hochberg correction.
- Unsupervised hierarchical clustering for patients from the TCGA dataset based on the correlation to EP1, EP2, iCAF_CXCL14, iCAF_CHI3L1 and imE1_FLT1.
- Comparison of Overall survival (OS) rates for three clusters identified in (I). P‐values are calculated using the log‐rank test (n = 255).
Data information: Boxplots show the median and upper/lower quartiles. The scCODA model and the ALDEx2 model were used to examine the differences in fibroblasts and endothelial cell‐type composition. Red bars indicate credible and significant results of scCODA. Stars indicate the significance calculated by ALDEx2 model. *P < 0.05, **P < 0.01, ***P < 0.001.
Figure EV5. Functional and clinical analysis of stromal cells.
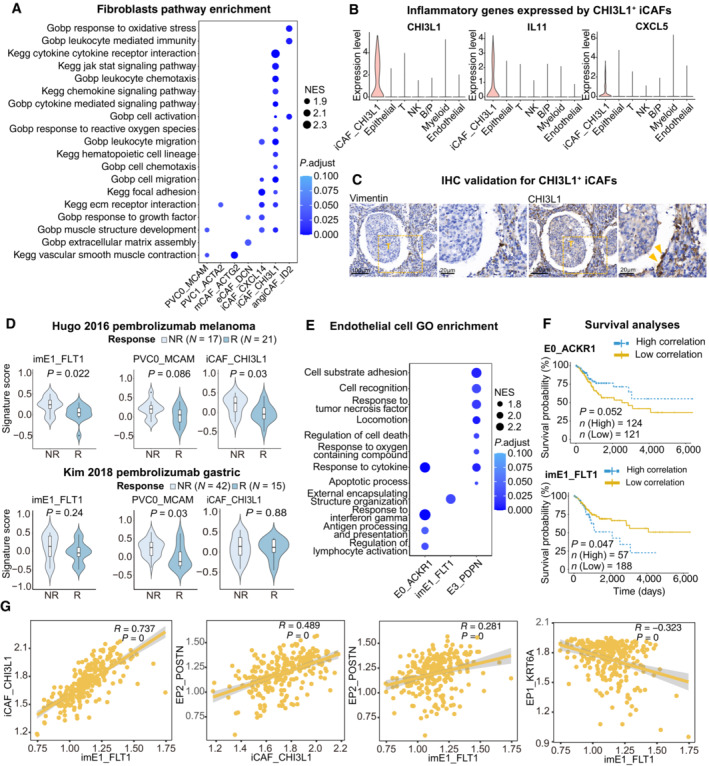
- Dot plot showing the selected signaling pathways (rows) with significant enrichment of GO, and KEGG terms for fibroblasts.
- Violin plot showing the expression of immune regulator genes in iCAF_CHI3L1 and other cell lineages.
- IHC staining of representative tumor section showing the expression of CHI3L1 in CAFs (yellow arrows) close to cancer cells; The scale bars for the left slide of each marker are 100 μm, and the scale bars for the right slide are 20 μm.
- Violin plots showing the association of different stromal cell signatures with the response (R) and no response (NR) to Pembrolizumab across on Hugo 26 cohort (upper panel) and Kim 45 cohort (lower panel). P‐values calculated using student's t‐test. Sample sizes are indicated in figure.
- Dot plot showing the selected signaling pathways (rows) with significant enrichment of GO, and endothelial cell clusters.
- Comparison of Overall survival (OS) rates for the high‐correlation and low‐correlation groups, stratified using the E0_ACKR1 (left panel) and imE1_FLT1 (right panel) signatures in TCGA (N = 255). P‐values are calculated using the log‐rank test.
- Scatter plot showing the correlation among imE1, EP1, EP2 and iCAF_CHI3L1 subsets.
Data information: Violin plots show the median and upper/lower quartiles (Related to Fig 6).
Angiogenesis is a validated target in the treatment of advanced CC. Previous research has indicated that angiogenesis occurs during the tumorigenesis of CC to ensure an adequate supply of nutrients during the development of cancer cells (Balasubramaniam et al, 2019). The dataset clustered four subpopulations of endothelial cells with apparent features (Fig 6E) (Goveia et al, 2020). imE1_FLT1, labeled by the VEGF receptor FLT1, expressed an immature gene signature (ENG, HSPG2 and KDR) and had an increasing trend in the tumor area, indicating a trait of neovascular endothelium (Fig 6F and G). Compared with other subpopulations, imE1_FLT1 enhanced the expression of angiogenesis‐related genes while dampening immunological surveillance via T cell dysfunction (Fig 6H). E0_ACKR1 was enriched in response to interferon‐gamma, antigen processing and presentation, and regulation of lymphocyte activation, which were absent in imE1_FLT1 enrichment (Fig EV5E). Of note, E3_PDPN were found to contain marker genes for high‐endothelial venules (HEV), including PDPN and NTAN1, which were adjacent to TLS and could recruit lymphocytes via CCL21 (Dieu‐Nosjean et al, 2016). In a trial by Lan et al (2020), a combination therapy of PD‐1 antibody and apatinib (anti‐VEGFR2) was applied in patients with CC, highlighting the power of vascular normalization in immunotherapy (Lan et al, 2020). We investigated if an abnormal tumor endothelial signature could predict patients' responses to immunotherapy. In the two pembrolizumab treatment cohorts, non‐responders showed accordant high‐imE1_FLT1 scores (Fig EV5D). This conclusion was also supported by survival analysis, which showed worse outcomes in patients highly associated with imE1_FLT1 compared with the E0_ACKR1 signature (Fig EV5F). Overall, imE1_FLT1 is a typical neovascular endothelial phenotype and thus could be considered a potential target for anti‐angiogenic regimens.
Co‐occurrence of stromal subsets predicts poor outcomes for patients with CC
To determine whether differential components could co‐occur in the same patient with CC and whether this co‐occurrence could predict prognosis, unsupervised hierarchical clustering was used to investigate the co‐occurrence of epithelial and stromal components. As shown in Fig 6I, a joint model incorporating classic and invasive tumor subsets, iCAFs and immature endothelial subsets provided a promising prognosis for patients with CC. Patients in cluster 3 showed a high correlation with EP2_POSTN, iCAF_CHI3L1 and imE1_FLT1, suggesting that they harbored invasion, immune suppression and angiogenesis characteristics. In cluster 2, only EP2 showed a significant correlation, whereas patients in cluster 1 showed no significant correlation within these subsets. Based on this, patients in cluster 1 had a better overall survival rate than those in clusters 2 and 3. There was an intermediate risk of death for those in cluster 2, while the poorest clinical outcomes were found in cluster 3 (Fig 6J). However, similar approaches failed to demonstrate significant results when immune components were used as predictors. This is partially due to the complicated status of immune cells in the TME which could not be categorized into specific patterns. Interestingly, signatures of imE1_FLT1, iCAF_CHI3L1 and EP2_POSTN were highly correlated in the subset of the TCGA cohort, whereas signatures of imE1_FLT1 and EP1_KRT6A were inversely correlated (Fig EV5G), suggesting the co‐occurrence of imE1_FLT1, EP2_POSTN and iCAF_CHI3L1 in patients with CC. According to these results, specific gene signatures derived from multiplex subsets of scRNA‐seq can be used as outcome predictors in patients with CC, further indicating that scRNA‐seq can provide profound insights into the co‐occurrence of cell types and offer clues for combinational therapy.
Discussion
In this study, single‐cell RNA and TCR repertoire sequencing of primary CC tissues and adjacent normal tissues were conducted to decode the heterogeneity of the TME. The intra‐ and inter‐tumoral heterogeneity of cancer cells was revealed, and POSTN+ cells were identified as a potential‐early invasive subtype. For the immune components, we elucidated the status transition of CD8+ T cells and Th17‐ and Tfh‐mediated CD4+ T‐cell responses in the TME. Large clonal cellular phenotypes and HPV antigen‐specific T‐cell responses were revealed that might underlie the cornerstone of immunotherapy targeting Tcells and/or HPV antigens. Notably, tumor‐specific B cell responses were identified, and the pivotal role of GCB in CC prognosis was revealed. The interactions between GCB cells and T cells in the TLS region provide potential strategies for inducing and maintaining TLS and B‐cell responses. Finally, we described the distinct phenotypes of CAFs and endothelial cells harbored in the tumor region and established a joint model that could segment patients with differential epithelial‐stromal co‐occurrence patterns and prognosis. Taken together, this study reveals the cellular features within the TME of CC and provides profound insights into potential therapeutic modalities or outcome prediction (Appendix Fig S2).
Analysis of T‐cell subpopulations revealed the dynamic status and differentiation of T cells in CC. The equivalent fraction of cytotoxic T cells and exhausted T cells, characterized by a mixture of dysfunctional and effector phenotypes, supports the generality of tumor‐infiltrating T cells between CC and other cancers (Ren et al, 2021). However, it was noticed that large clones aggregated in cytotoxic and exhausted T‐cell subsets but highly expressed a cytotoxic profile, which is consistent with previous reports in melanoma and pancreatic cancer that indicate high‐frequency clones are tumor‐reactive effector populations (Pasetto et al, 2016; Schalck et al, 2022). These results suggest the selected roles of anti‐tumor responses in tumor‐infiltrating T cells. HPV antigens are historical targets for antigen‐specific immunotherapies for CC, and their efficacy has been confirmed in a fraction of individuals (van der Burg & Melief, 2011; Stevanovic et al, 2017; Nagarsheth et al, 2021). However, the response of this key population to HPV‐based immunotherapies has not been thoroughly investigated. An enlarged HPV antigen‐specific T‐cell clone size was observed in the tumor area, whereas HPV‐specific clones were restricted to a limited number. We queried the TCRs in the VDJdb database to identify HPV‐specific T cells, but the epitopes collected in this database were limited to E7 protein, which might lead to a severe underestimation of HPV‐specific cells. PD‐1+TCF7+ HPV‐specific T‐cell subsets identified by scRNA‐seq could further proliferate and differentiate into effector T cells (Eberhardt et al, 2021). Hence, the phenotypes and dynamic status of HPV‐specific subsets in CC warrant further investigation. Moreover, sorted HPV‐specific T cells should be further investigated to distinguish the epitopes they identified and their subpopulations, so that strategies for immune checkpoint blockade and therapeutic vaccines can be developed precisely.
The anti‐tumor mechanism of B cells is a new frontier. This study's data highlight a GC response and showed that GCB predicts better clinical outcomes in patients with CC. As a transient structure within B‐cell follicles, GC are considered the hallmark of mature TLS, and the majority of tumor‐specific B‐cell responses are dependent on TLS (Fridman et al, 2022). In contrast, humoral immunity has been investigated in various cancers. Meylan et al (2022) demonstrated that TLS‐positive patients showed high‐density IgG‐stained cancer cells and cell apoptosis in renal cell carcinoma, and IGHG expression was only observed in the GC of TLS, suggesting that antibody production and the ADCC effect are anti‐tumor mechanisms of TLS. Similar results have been reported for HPV‐associated HNSCC (Cillo et al, 2020; Wieland et al, 2021). In contrast, T–B cell interactions may be another mechanism. Neoantigen‐derived GCB promotes effector functions in CD8 T cells in a Tfh‐IL‐21 manner, which has been demonstrated in a lung adenocarcinoma murine model, with TLS serving as a likely interaction location (Cui et al, 2021). IL‐21 is also significant for GC biology and plasma cell differentiation (Quast et al, 2022). In this study, it was noted that a subset of the CD4+CXCR5−PD‐1+CXCL13+ Tfh subset was restricted to TLS, particularly immature TLS and GC. This subset showed strong interactions with GCB, including IL21‐IL21R, ICOS‐ICOSL and CD70‐CD27. Thus, this subset should be considered a critical collaborator within TLS and warrants further investigation. In addition, ICOSL+ B cells within the TLS boost anti‐tumor immunity by increasing the effector to regulatory T‐cell ratio (Lu et al, 2020). However, whether GC is essential for TLS in anti‐tumor responses and how T–B cells collaborate to promote these processes in CC remain to be explored.
Recently, TLS and plasma cell signatures, rather than T cells, have been considered determinants of the ICB response. A phase 2 study in soft‐tissue sarcomas reported that TLS‐positive patients showed significantly higher progression‐free survival, and intratumoral plasma cells were strongly correlated with improved outcomes (Italiano et al, 2022). Likewise, large‐scale lung cancer cohorts verified that an increased plasma cell signature is associated with TLS and is significantly associated with extended OS with the PD‐L1 inhibitor atezolizumab (Patil et al, 2022). Given the vital role of TLS and B‐cell responses in ICB response and prediction, strategies that consider TLS as a predictive biomarker to select patients for ICB treatment and induce TLS neogenesis to sensitize patients to anti‐PD1/PD‐L1 regimens are attractive perspectives for improving the systematic treatment of CC. Of note, although multiple approaches such as adjuvant chemotherapy can be utilized to induce TLS, a study on cervical neoplastic lesions proved that a therapeutic HPV vaccine can develop TLS in intraepithelial lesions adjacent to the stroma (Maldonado et al, 2014), which appears to be a viable approach for HPV‐related cancers.
Cancer cells were distinguished from epithelial cells by integrating cell proportion, HPV mRNA expression, CDKN2A expression and genomic copy number profiles inferred by CopyKAT, which could serve as a pipeline for identifying HPV‐related cancer cells at single‐cell resolution. Although these classic features are lacking, it is interesting that a group of malignant cells show the hallmarks of EMT and basal epithelial cells (TP63 and KRT14). This is analogous to invasive leader cells reported in breast cancer and cutaneous SCC research, in which 3D organoid assays and spatial transcriptomics are exploited to substantiate the tumor leading‐edge localization of these basal‐EMT cells (Cheung et al, 2013; Ji et al, 2020). This study's results suggest the presence of these dangerous cancer leader cells in CC and depict the transcriptomic aspect. However, further experimental verification is needed to fully understand their biological behavior in CC invasion and metastasis. In addition, investigation of multiple component interactions in the TME is a unique advantage of scRNA‐seq. The joint model reported here showed that EP2_POSTN, imE1_FLT1 and iCAF_CHI3L1 could collectively predict inferior outcomes. However, their spatial relevance and whether they have mutually promoting mechanisms during tumorigenesis or progression warrant further investigation.
Several limitations remain in this research. We could not explore TME variations across HPV subtypes and histology due to the small sample size. Second, the use of biopsy forceps in tissue collection and a lack of cell sorting resulted in inadequate GCB cell numbers for further analysis. The data could serve as a benchmark for further immunotherapeutic investigation of CC at a single‐cell resolution.
Materials and Methods
Reagents and Tools table
| Reagent/resource | Reference or source | Identifier or catalog number |
|---|---|---|
| Experimental models | ||
| Details described in Table EV1 | ||
| Antibodies | ||
| PanCK(C11) | Cell Signaling Technology | Cat # 4545 |
| CD3(BP6027) | Biolynx | Cat # BX50022 |
| CD56(123C3) | Cell Signaling Technology | Cat # 3576 |
| CD68(BP6036) | Biolynx | Cat # BX50031 |
| CD20(E7B7T) | Cell Signaling Technology | Cat # 48750 |
| BCL6(EPR11410‐43) | Abcam | Cat # ab172610 |
| CD38(EPR4106) | Abcam | Cat # ab108403 |
| CD8(1G2B10) | Proteintech | Cat # 66868‐1‐Ig |
| CD4(2H4A2) | Proteintech | Cat # 67786‐1‐Ig |
| CD4(EPR6855) | Abcam | Cat # ab133616 |
| PD‐1(10377‐H08H) | Sinobiological | Cat # 10377‐MM23 |
| CXCL13(EPR23400‐92) | Abcam | Cat # ab246518 |
| CD14(4B4F12) | Abcam | Cat # ab182032 |
| Ki‐67(BP6045) | Biolynx | Cat # BX50040 |
| HRP‐labeled Goat Anti‐ RabbitIgG | Yuanxi Bio | Cat # A10012‐60 |
| Goat Anti‐Mouse IgG H&L (HRP) | Abcam | Cat # ab205719 |
| Chemicals, enzymes and other reagents | ||
| MACS Tissue Storage Solution | Miltenyi Biotec | Cat # 130‐100‐008 |
| RPMI medium | Gibco | Cat # C11875500BT |
| Fetal bovine serum | Gibco | Cat # A3161001C |
| Trypsin | ThermoFisher Scientific | Cat # 25200072 |
| Collagenase type IV | Worthington | Cat # LS004186 |
| Collagenase type I | Worthington | Cat # LS004194 |
| Dispase II | Roche | Cat # 4942078001 |
| Red blood cell lysis buffer | Gibco | Cat # A1049201 |
| DAB substrate kit | Abcam | Cat # ab64238 |
| Bovine serum albumin (BSA) | Sigma‐Aldrich | Cat # A9647 |
| Tris buffered saline with Tween 20 (TBST) | ThermoFisher Scientific | Cat # 37543 |
| 4′‐6′‐diamidino‐2‐phenylindole (DAPI) | Sigma‐Aldrich | Cat # D9542 |
| Software | ||
| Cell Ranger toolkit (version 5.0.0) | 10× Genomics | https://support.10xgenomics.com |
| MAESTRO v 1.5.1 | Wang et al (2020) | https://github.com/liulab‐dfci/MAESTRO |
| MAESTRO v 1.5.0 | Wang et al (2020) | https://github.com/liulab‐dfci/MAESTRO |
| DoubletFinder | McGinnis et al (2019) | https://github.com/ddiez/DoubletFinder |
| Python 3 | N/A | www.python.org |
| R 3.5.0 | R‐Project | https://cran.r‐project.org/mirrors.html |
| Seurat v2.3.4 | Satija et al (2015) | https://satijalab.org/seurat/ |
| ClusterProfiler v 3.18.1 | Yu et al (2012) | https://guangchuangyu.github.io/software/clusterProfiler/ |
| scCODA | Buttner et al (2021) | https://github.com/theislab/scCODA |
| TRUST4 v1.0.5.1 | Song et al (2021) | https://github.com/milaboratory/mixcr‐rna‐seq‐paper |
| VDJmatch v1.2.2 | Shugay et al (2018) | https://vdjdb.cdr3.net |
| ggplot2 | Hadley Wickham et al (2016) | https://cran.r‐project.org/web/packages/ggplot2/index.html |
| ggpubr v0.4 | R‐Project | https://cran.r‐project.org/web/packages/ggpubr/index.html |
| CopyKat | Gao et al (2021) | https://github.com/navinlabcode/copykat |
| GSVA v1.38.2 | Hanzelmann et al (2013) | https://www.bioconductor.org/packages/release/bioc/html/GSVA.html |
| CellPhoneDB v0.22 | Efremova et al (2020) | www.cellphonedb.org |
| CellChat v1.1.0 | Jin et al (2021) | http://www.cellchat.org/ |
| Monocle2 v4.0.5 | Qiu et al (2017) | https://cole‐trapnell‐lab.github.io/monocle‐release/ |
| GraphPad Prism 7.0 | GraphPad | https://www.graphpad.com/ |
| Halo10 software | Indica Labs | https://indicalab.com/halo/ |
| Other | ||
| Illumina NexSeq 500 | Illumina | |
| Chromium Next GEM Single Cell 5′ Kit v1.1 | 10× Genomics | PN‐1000165 |
| PanoVIEW VS200 slide scanner | Panovue | |
| TSA 5‐color kit | Yuanxi Bio | Cat # H‐D110051 |
| PDOne 5‐plex TSA‐RM kit | Panovue | Cat # 10144100100 |
Methods and Protocols
Patients and sample preparation
All the samples were collected from the Shanghai East Hospital, Shanghai, China. Patients with CC were selected based on the following criteria: (i) diagnosed with CC by pathological colposcopy biopsy; (ii) CT‐ or MRI‐detected resectable tumor mass with a maximum diameter of more than 2 cm; (iii) no previous medical history of other cancers and (iv) no other anti‐tumor treatment before surgery. All participants provided written informed consent, and this study was supervised and assessed by the Ethics Committee Board of Tongji University‐affiliated East Hospital. The experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
After surgery, the tumor and adjacent normal tissues (more than 2 cm away from the tumor edge with visual observation) were collected immediately. Biopsy forceps were used to grab the tissues and acquire accurate tissue types in the terminated cervical area. After washing with phosphate‐buffered saline (PBS) three times, the tissues were transported into containers containing ice‐cold tissue storage solution (Miltenyi Biotec) and sent to the laboratory within 4 h. In the laboratory, the tissues were cut into 1 mm pieces, digested with 0.25% trypsin (ThermoFisher Scientific) at 37°C for 30 min, and terminated with RPMI medium (Gibco) containing 10% fetal bovine serum (Gibco). Tissues were then transported into a dissociation cocktail containing collagenase type IV (100 U/ml, Worthington), collagenase type I (100 U/ml, Worthington) and dispase (0.6 U/ml, Roche). After filtering through a 70‐um nylon mesh, the cells were centrifuged and suspended in a red blood cell lysis buffer (Gibco). Finally, the collected cells were suspended in 1 ml of PBS for counting and other processes.
Single‐cell RNA‐seq library and TCR‐seq library preparation and sequencing
Single‐cell RNA‐seq libraries were prepared using the Chromium Next GEM Single Cell 5′ Kit v1.1, from 10× Genomics, to generate single‐cell gel beads in the emulsion (GEMs), following the manufacturer's protocol. Briefly, single cells were suspended in PBS containing 0.04% bovine serum albumin (BSA, Sigma‐Aldrich). Approximately, 10,000 cells were captured in droplets to generate nanoliter‐scale gel beads during emulsion (GEMs). Reverse transcription was performed in a thermal cycler (ThermoFisher Scientific) at 53°C for 45 min, 85°C for 5 min, and held at 4°C. Complementary DNA was generated and amplified, emulsions were broken, and cDNA was isolated and purified using Dynabeads Cleanup Mix. The amplified cDNA was then used for both 5′ gene expression library construction and V(D)J library construction. Single‐cell RNA and TCR V(D)J libraries were sequenced using an Illumina Nova‐seq 6000 with 150 bp paired‐end (PE150) reads.
Single‐cell RNA‐Seq data processing
Raw gene expression matrices were generated for each sample using the Cell Ranger toolkit (version 5.0.0) provided by 10× Genomics. This pipeline was coupled with the human reference genome (GRCh38). Doublets were removed before the data were integrated (see the following method for more detail). Multiple plain text count tables were converted and merged into an HDF5 format using the python‐based package MAESTRO (version 1.5.1) (Wang et al, 2020) and subsequent clustering analysis for a dataset using the RNARunSeurat function of the R‐based package MAESTRO (version 1.5.0), including normalization, feature selection, dimension reduction, clustering and UMAP visualization. Thereafter, low‐quality cells were processed to filter out if they met the following criteria: (i) < 500 unique molecular identifiers (UMIs), (ii) < 1,000 genes or (iii) > 15% UMIs derived from the mitochondrial genome. The samples were processed independently, and so high‐dimensional variables, common in single‐cell sequencing data, may have introduced potential batch effects. To alleviate this, canonical correlation analysis (CCA) was used, and the RNABatchCorrect function was implemented in MAESTRO to remove the batch. Clustering was performed again with 3,000 features used in the CCA analysis, 1:30 dimensions used for the UMAP analysis, a clustering resolution parameter of 1, and default parameters used otherwise.
Doublet removal
The R package DoubletFinder (version 4.0.5) (McGinnis et al, 2019) was applied to each sequencing library to remove potential doublets with an expected doublet rate of 6%. In addition, clusters with multiple well‐defined marker genes from different cell types were considered doublets and removed in the downstream analysis. For each cluster of a major cell type, the scores of the functional modules for the cell cluster were calculated using the AddModuleScore function in Seurat (Table EV5). The average expression levels of the corresponding clusters were subtracted from the aggregated expression of the control feature sets. All the analyzed genes were binned based on their averaged expression, and the control features were randomly selected from each bin. Functional modules include T cells, B cells, plasma cells, myeloid cells, mast cells, epithelial cells, fibroblasts and endothelial cell signature scores. The genes involved are listed in the Table EV5. The clusters (such as C2 and C6) of a major cell type (e.g., B cells, Fig EV1D) with multiple cell‐type features were removed.
Cell‐type proportion analysis
In each patient, cell‐type proportions were calculated by dividing the number of cells in a cluster by the total number of cells in the sample. The compositional data analysis tool scCODA (Buttner et al, 2021) and ALDEx2 (Fernandes et al, 2014) was used to compare the differential relative abundance between tumor and adjacent normal tissue derived from scRNA‐seq data. For scCODA, the reference of compositional changes was alternatively selected, and the false discovery rate (FDR) level was 0.2 for all cell lineages. For AlDEx2, the P‐value derived from for a comparison of two conditions was determined by the Welch's t test (Wi.ep).
Differential expression analyses and cell cluster annotation
The clusters were identified as different major cell lineages based on the average gene expression of well‐known markers, including T/NK cells (CD3D), B cells (CD79A), myeloid cells (CD68 and CPA3), epithelial cells (KRT family genes KRT5 and KRT17), fibroblast cells (COL1A1) and endothelial cells (PECAM1). Sub‐cluster annotation of cell lineages refers to the specific genes listed in Dataset EV3.
Repeating the process (normalization, dimensionality reduction and clustering), sub‐clusters were further identified and annotated as different specific cell subtypes based on the average expression of respective gene sets in each major cell type. All the differential expression analyses were conducted using the FindAllMarkers function in Seurat (version 4.0.5) (Hao et al, 2021). A significantly differentially expressed gene (DEG) was determined if it had a Bonferroni‐adjusted P‐value lower than 0.05 and an average natural logarithm (ln) fold‐change of expression of at least 0.15 and 0.25 for T cells and other cells, respectively. The DEGs of each subcluster were listed in Dataset EV1.
Pathway enrichment analysis
To gain functional and mechanistic insights into a cell cluster, Gene Ontology (GO), KEGG and Hallmark Pathway enrichment analyses were performed using the Molecular Signatures Database (MSigDB v7.1) (Liberzon et al, 2015) to identify biological pathways that were enriched in a certain gene list from a specific cell subtype. The DEGs from each cluster were then passed on to the ClusterProfiler (version 3.18.1) package for functional enrichment.
TCR sequence assembly
To assemble the TCR sequences, the Cell Ranger toolkit (version 5.0.0) provided by 10× Genomics was used to align TCR‐seq reads to the human reference genome (GRCh38) (refdata‐cellranger‐vdj‐GRCh38‐alts‐ensembl‐2.0.0).
The TRUST4 algorithm was applied (version 1.0.5.1) (Song et al, 2021) to infer TCR and BCR clonotypes from the scRNA‐seq data of CESC patients. TRUST4 performs de novo assembly on V, J and C genes, including the hypervariable complementarity‐determining region 3 (CDR3), and reports the consensus of BCR/TCR sequences. It then realigns the contigs to the IMGT reference gene sequences (Song et al, 2021) to report the corresponding information.
The clones were identified based on the CDR3 nucleotide sequence. TCR/BCR clones whose barcode did not match the barcode of T cell/B cell scRNA‐seq data were removed, and complementarity‐determining region 3 (CDR3) gene regions were then used to quantify TCR clonotypes. We identified a median of 870 α (IQR, 350–1,885) and 969 β (IQR, 531–2,208) unique chains per sample from the 10× TCR‐seq dataset, 230 α (IQR, 122–808) and 535 β (IQR, 360–1,853) unique chains per sample from the TRUST4‐predicted TCR data, and 95 IGH (IQR, 17–215.5) unique chains per sample from the TRUST4‐predicted BCR data.
Single‐cell TCR‐seq clonotype analysis
Information about clonotypes was extracted with default parameters and processed in R, and clonal indices, including clonality and diversity, were calculated using the function trust‐stats.py of the TRUST package.
To quantify the TCR/BCR similarity between clusters, the Jaccard Index was calculated among the CDR3aa sequences of each of the two T‐/B‐cell subclusters and the result normalized to the 0–1 range.
| (1) |
In Equation (1), A is the CDR3aa number of subcluster A of T‐/B‐cells and B is the CDR3aa number of subcluster B of T‐/B‐cells.
| (2) |
In Equation (2), Shannon entropy is the normalized entropy over the number of unique clones and N is the number of unique clones.
| (3) |
In Equation (3), CDR3aa is the number of unique CDR3 calls for a chain and is the total number of reads for that chain.
Predicting T‐cell receptor antigen specificity
To check the specificity of the T‐cell receptor (TCR), the VDJdb database was queried (Shugay et al, 2018) using the standalone VDJmatch software version 1.2.2. CDR3 sequence search parameters were set that allowed two substitutions, one insertion, two deletions, or indels for HPV antigen search and default parameters for CMV antigen search. The TCR TRB chain was used to align TCR sequences in the database.
Modeling and comparison of small and large TCR clonotypes
Clones were labeled as large or small according to the number of unique CD3 amino acid sequences larger than five for TCR or one for BCR, with a significant correlation between the number of large clones identified in single‐cell data.
An integrated analysis of all the merged data was performed. Following the identification of cellular subgroups based on TCR clone size, conserved marker‐defining subgroups were identified using the FindMarkers function with a default two‐sided Wilcoxon rank‐sum test. Plots were generated using ggpubr (v.0.4) and customized ggplot2.
HPV reference genome construction and HPV virus read verification
An HPV reference genome, including HPV 16, 18, 31, 45 and 66 genome sequences was built to check HPV infection status. The reference genome files, including FASTA and GTF files, were aligned using Cell Ranger. FASTA and GTF files for the HPV reference genome were built to refer to the cell range process. Briefly, the complete HPV 16, 18, 31, 45, 66 genome sequences (HPV16REF, HPV18REF, HPV31REF, HPV45REF and HPV66REF) were downloaded from the Papillomavirus genome database (PaVE) (Van Doorslaer et al, 2017), and then multiple HPV.fa files and HPV.gtf files were used as input to run cellranger mkref. Finally, the cellranger count was run to aggregate raw data from the samples and to align reads to the HPV reference genome.
Copy number variation analysis
To distinguish cancer cells from non‐malignant cell types, CopyKat (Gao et al, 2021) was applied using the integrative Bayesian approaches to identify genome‐wide aneuploidy at 5 MB resolution in single cells to separate tumor cells from normal cells. Raw single‐cell gene expression data were extracted from the Seurat object. Epithelial cells from normal samples were used as the control reference. The following CopyKat parameters were used for analysis: two genes per chromosome for cell filtering, 0.04 fractions of genes for smoothing, 0.1 fractions of genes and 25 window sizes for segmentation, with default parameters used otherwise.
Gene set enrichment analysis
For a specific cell subtype, scRNA signatures were applied to bulk RNA‐seq samples using single‐sample gene set enrichment analysis (ssGSEA) (Subramanian et al, 2005; Barbie et al, 2009). ssGSEA scores were calculated using the GSVA package (version 1.38.2) (Hanzelmann et al, 2013) in Bioconductor. P‐values were calculated by a one‐sided hypergeometric test and adjusted for multiple comparisons.
Survival analyses and ICB response prediction
In the TCGA CESC cohort, patients receiving bulk RNA sequences with various treatments and endpoints were analyzed for clinical and survival outcomes (cancer‐specific survival and recurrence). Survival analyses for each cell type subset were performed based on gene signature ratios (Details in Appendix Fig S3). After grouping, the Cox proportional hazards model implemented in the R package survival (version 3.2‐11) was used to correct disease state for survival analyses and Kaplan–Meier survival curves were plotted using the R function ggsurvplot. Hazard ratios were calculated using Cox regression, and P‐values were calculated using the log‐rank test. Dataset EV4 summarizes the gene signatures used for the survival analyses.
The ICB prediction was conducted as previously reported (Jia et al, 2018). Briefly, the sub‐cluster single sample gene set enrichment analysis (ssGSEA) signature scores calculated for each tumor sample in the immunotherapy cohort are used as biomarkers to predict the response to ICB. For comparisons of sub‐cluster ssGSEA signature scores between responders (CR/PR) and non‐responders (PD) in bulk RNA sequencing, a two‐sided Wilcoxon test was used. Dataset EV4 summarizes the gene signatures used for the ICB prediction.
Cellular communication analysis
Potential cell–cell communication between any two different cell types in CC, was investigated via ligand‐receptor analyses using CellPhoneDB software (version 0.22) (Efremova et al, 2020) and R packages CellChat (Version 1.1.0) (Jin et al, 2021) with default parameters. The gene expression matrices of all cells were selected as inputs for cell–cell interaction analysis. The most relevant cell type‐specific ligand‐receptor interactions and specific signaling pathways (BAFF and APRIL) were identified in the corresponding sub‐clusters.
Developmental trajectory inference
To identify the transcriptional changes in immune cell types, trajectory analyses were performed for CD4+ T, CD8+ T, B, macrophages and DCs using Monocle2 (version 4.0.5) (Qiu et al, 2017). The data for the indicated clusters were calculated using Seurat as the input for Monocle2 analysis. The Monocle algorithm was applied to the top 50–1,000 signature genes, calculated using the differential GeneTest function. The trajectory of the immune cells was constructed using the default parameters of Monocle after dimensionality reduction and cell ordering.
T‐cell dysfunction and exclusion analysis
Cytotoxic T lymphocyte (CTLs) dysfunction and exclusion analysis were performed using a strategy similar to that of TIDE (Jiang et al, 2018) to investigate the immunomodulatory roles of different stromal subsets. To estimate CTL levels, TIDE uses pivot genes such as CD8A, CD8B, GZMA, GZMB and PRF1 and calculates the average expression of these genes. Patients with CESC from the TCGA cohort were grouped by CTL level, with the mean value as a cutoff point. For CTL dysfunction analysis, TIDE evaluates the associations between stromal subset signature genes and survival outcome with the CTL level corrected in Cox–PH regression. Genes with higher CTL dysfunction scores indicate an antagonistic interaction with CTL levels, where the survival benefit of patients with high CTL is lost, thus indicating that it is related to CTL dysfunction. Then, T‐cell dysfunction scores of marker genes (top 50) among subsets were compared. The statistical significance was determined with the Kruskal–Wallis test followed by a t‐test with the Benjamini–Hochberg correction.
For CTL exclusion analysis, Pearson correlations between the pivot (CTL level) and other signature genes were examined, with survival as the response. In this study, gene signatures were defined by the average gene expression value, where the cells in the cluster of interest were compared to all cells.
Immunohistochemistry and multiplex immunohistochemistry
IHC staining was performed as previously reported (Sun et al, 2020; Zhang et al, 2020b). Briefly, formalin fixation and paraffin embedding (FFPE) specimens from patients were stained with IHC to identify B cells (CD20, E7B7T, 1:200, and CST), CD4+ T cells (CD4, 2H4A2, 1:450, and Proteintech), and CD8+ T cells (CD8, 1G2B10, 1:200 and Proteintech). The tissue sections were then stained with hematoxylin and diaminobenzidine (DAB, abcam). The mIHC staining was performed according to the manufacturer's instructions. Briefly, 4‐μm‐thick FFPE sections were stained with primary antibodies (see Reagents and Tools table) sequentially and paired with tyramide signal amplification (TSA) staining kits. Then, cell nuclei were stained with 4′‐6′‐diamidino‐2‐phenylindole (DAPI, Sigma‐Aldrich). For example, in a panel aimed at validating B‐cell subsets, slides were incubated for half an hour with anti‐CD20 antibodies (E7B7T, CST), followed by 10 min with a horseradish peroxidase‐conjugated anti‐rabbit antibody (A10012‐60, Yuanxi Bio). In order to label fluorescence, a 10‐min incubation was developed using TSA 570. After washing in TBST buffer (ThermoFisher Scientific), slides were transferred to a preheated citrate solution (90°C) before being microwaved for 15 min. Slides were cooled in the same solution to room temperature. All the slides were washed in Tris buffer between each step. The same process was repeated for the remaining antibodies, including BCL6, Pan‐CK and CD38. The slides were then treated with two drops of DAPI, washed with distilled water and air dried. Slides were scanned using a PanoVIEW VS200 slide scanner (Panovue) with an Olympus 20× lens. Cell density (positive cells mm−2), frequency, and distance analysis were performed using Halo10 software (Indica Labs). Multiplex IHC and IHC‐staining assays were performed with at least three biological replicates.
Joint analysis of the multiple predictors
To examine the prognostic value of gene signatures derived from multiple subsets, specific gene signatures derived from multiple subsets of scRNA‐seq were used as predictors, and patients with TCGA were divided into multiple groups using unsupervised hierarchical clustering. Finally, the Cox proportional hazards model was used to perform survival analysis to predict the patient's prognosis, as described above.
Statistics analysis
All the statistical analyses were performed in R (version 4.0.5) or GraphPad Prism (version 7.0), including the two‐sided paired Student's t‐test, the two‐sided Wilcoxon test, the two‐sided Pearson correlation test, the two‐way ANOVA with Tukey's post hoc test, the one‐way ANOVA with Holm–Sidak's multiple comparisons test, and the two‐sided Kruskal–Wallis test. Statistical significance was determined at P < 0.05. There were no statistical methods used to predetermine the sample size. During experiments and the assessment of results, investigators were not blinded to allocation.
Author contributions
Fang Li: Conceptualization; resources; data curation; supervision; funding acquisition; project administration. Jinli Gao: Investigation; methodology. Xuejun Wen: Supervision. Quan Gu: Investigation; writing – review and editing. Jiuhong Kang: Writing – review and editing. Chenfei Wang: Conceptualization; data curation; software; formal analysis; supervision; funding acquisition; project administration. Qingfeng Zhang: Supervision; writing – review and editing. Jin Wang: Writing – review and editing. Yetian Ruan: Investigation. Ya Han: Software; investigation; methodology. Guangxu Cao: Resources; validation; investigation; visualization; methodology; writing – original draft; writing – review and editing. Jiali Yue: Software; investigation; visualization; methodology; writing – original draft. Min Liu: Investigation. Xinxin Xu: Investigation. Yong Zhi: Investigation; visualization. Jianqiao Lu: Investigation.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
PDF+
Acknowledgements
This study was supported by the National Key R&D Program of China (No. 2021YFC2701200, 2022YFA1106000), the National Natural Science Foundation of China (No. 82273091, 32170660, 31801059, 81872290, 82203512), the Shanghai Science and Technology Development Foundation (No. 21S31905100), the East Hospital Initial Foundation (No. DFRC201917), the Shanghai Municipal Health Commission Foundation (202150001), the Shanghai Rising Star Program (No. 21QA1408200), and the Natural Science Foundation of Shanghai (No. 21ZR1467600). Quan Gu is funded by the Medical Research Council (MC_UU_1201412). We thank Prof. Zhaohui Huang (Jiangnan University, China) and Prof. Xiaoming Zhang (Institute Pasteur of Shanghai, China) for their critical reading and comments.
The EMBO Journal (2023) 42: e110757
Contributor Information
Chenfei Wang, Email: 08chenfeiwang@tongji.edu.cn.
Fang Li, Email: fang_li@tongji.edu.cn.
Data availability
The codes used in this study are available upon request from the corresponding authors. The datasets and computer code produced in this study are available in the following databases: Single‐cell sequencing data is available on the Genome Sequence Archive (GSA) repository (HRA001742). For ICB response prediction analysis, RNA‐seq data from the following accessions were used: GSE78220 (Melanoma) and PRJEB25780 (gastric cancer). For survival analysis, bulk RNA‐seq data from the following study was used: the TCGA CESC database.
References
- Ashrafi GH, Haghshenas MR, Marchetti B, O'Brien PM, Campo MS (2005) E5 protein of human papillomavirus type 16 selectively downregulates surface HLA class I. Int J Cancer 113: 276–283 [DOI] [PubMed] [Google Scholar]
- Balasubramaniam SD, Balakrishnan V, Oon CE, Kaur G (2019) Key molecular events in cervical cancer development. Medicina (Kaunas) 55: 384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C et al (2009) Systematic RNA interference reveals that oncogenic KRAS‐driven cancers require TBK1. Nature 462: 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beham AW, Puellmann K, Laird R, Fuchs T, Streich R, Breysach C, Raddatz D, Oniga S, Peccerella T, Findeisen P et al (2011) A TNF‐regulated recombinatorial macrophage immune receptor implicated in granuloma formation in tuberculosis. PLoS Pathog 7: e1002375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner M, Ostner J, Muller CL, Theis FJ, Schubert B (2021) scCODA is a Bayesian model for compositional single‐cell data analysis. Nat Commun 12: 6876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon‐Christersson J et al (2020) Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577: 561–565 [DOI] [PubMed] [Google Scholar]
- Casanova‐Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, Brown M, Chang C, Troncoso L, Chen ST et al (2021) Tissue‐resident macrophages provide a pro‐tumorigenic niche to early NSCLC cells. Nature 595: 578–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez‐Galan L, Olleros ML, Vesin D, Garcia I (2015) Much more than M1 and M2 macrophages, there are also CD169+ and TCR+ macrophages. Front Immunol 6: 263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhou L, Liu L, Hou Y, Xiong M, Yang Y, Hu J, Chen K (2020) Single‐cell RNA sequencing highlights the role of inflammatory cancer‐associated fibroblasts in bladder urothelial carcinoma. Nat Commun 11: 5077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, Qin S, Zhang L, Ouyang H, Du P et al (2021) A pan‐cancer single‐cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184: 792–809 [DOI] [PubMed] [Google Scholar]
- Cheung KJ, Gabrielson E, Werb Z, Ewald AJ (2013) Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 155: 1639–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cillo AR, Kurten CHL, Tabib T, Qi Z, Onkar S, Wang T, Liu A, Duvvuri U, Kim S, Soose RJ et al (2020) Immune landscape of viral‐ and carcinogen‐driven head and neck cancer. Immunity 52: 183–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo N, Dubot C, Lorusso D, Caceres MV, Hasegawa K, Shapira‐Frommer R, Tewari KS, Salman P, Hoyos Usta E, Yanez E et al (2021) Pembrolizumab for persistent, recurrent, or metastatic cervical cancer. N Engl J Med 385: 1856–1867 [DOI] [PubMed] [Google Scholar]
- Cui C, Wang J, Fagerberg E, Chen PM, Connolly KA, Damo M, Cheung JF, Mao T, Askari AS, Chen S et al (2021) Neoantigen‐driven B cell and CD4 T follicular helper cell collaboration promotes anti‐tumor CD8 T cell responses. Cell 184: e6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vos van Steenwijk PJ, Heusinkveld M, Ramwadhdoebe TH, Lowik MJ, van der Hulst JM, Goedemans R, Piersma SJ, Kenter GG, van der Burg SH (2010) An unexpectedly large polyclonal repertoire of HPV‐specific T cells is poised for action in patients with cervical cancer. Cancer Res 70: 2707–2717 [DOI] [PubMed] [Google Scholar]
- Desbois M, Wang Y (2021) Cancer‐associated fibroblasts: key players in shaping the tumor immune microenvironment. Immunol Rev 302: 241–258 [DOI] [PubMed] [Google Scholar]
- Di Modugno F, Colosi C, Trono P, Antonacci G, Ruocco G, Nistico P (2019) 3D models in the new era of immune oncology: focus on T cells, CAF and ECM. J Exp Clin Cancer Res 38: 117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieu‐Nosjean MC, Giraldo NA, Kaplon H, Germain C, Fridman WH, Sautes‐Fridman C (2016) Tertiary lymphoid structures, drivers of the anti‐tumor responses in human cancers. Immunol Rev 271: 260–275 [DOI] [PubMed] [Google Scholar]
- Eberhardt CS, Kissick HT, Patel MR, Cardenas MA, Prokhnevska N, Obeng RC, Nasti TH, Griffith CC, Im SJ, Wang X et al (2021) Functional HPV‐specific PD‐1+ stem‐like CD8 T cells in head and neck cancer. Nature 597: 279–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremova M, Vento‐Tormo M, Teichmann SA, Vento‐Tormo R (2020) CellPhoneDB: inferring cell‐cell communication from combined expression of multi‐subunit ligand‐receptor complexes. Nat Protoc 15: 1484–1506 [DOI] [PubMed] [Google Scholar]
- Fairfax BP, Taylor CA, Watson RA, Nassiri I, Danielli S, Fang H, Mahe EA, Cooper R, Woodcock V, Traill Z et al (2020) Peripheral CD8+ T cell characteristics associated with durable responses to immune checkpoint blockade in patients with metastatic melanoma. Nat Med 26: 193–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes AD, Reid JN, Macklaim JM, McMurrough TA, Edgell DR, Gloor GB (2014) Unifying the analysis of high‐throughput sequencing datasets: characterizing RNA‐seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2: 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman WH, Meylan M, Petitprez F, Sun CM, Italiano A, Sautes‐Fridman C (2022) B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat Rev Clin Oncol 19: 441–457 [DOI] [PubMed] [Google Scholar]
- Gao R, Bai S, Henderson YC, Lin Y, Schalck A, Yan Y, Kumar T, Hu M, Sei E, Davis A et al (2021) Delineating copy number and clonal substructure in human tumors from single‐cell transcriptomes. Nat Biotechnol 39: 599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, Ji X, Han A, Krams SM, Pettus C et al (2017) Identifying specificity groups in the T cell receptor repertoire. Nature 547: 94–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goveia J, Rohlenova K, Taverna F, Treps L, Conradi LC, Pircher A, Geldhof V, de Rooij L, Kalucka J, Sokol L et al (2020) An integrated gene expression landscape profiling approach to identify lung tumor endothelial cell heterogeneity and angiogenic candidates. Cancer Cell 37: e13 [DOI] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinformatics 14: 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen‐Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M et al (2021) Integrated analysis of multimodal single‐cell data. Cell 184: 3573–3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeren AM, Koster BD, Samuels S, Ferns DM, Chondronasiou D, Kenter GG, Jordanova ES, de Gruijl TD (2015) High and interrelated rates of PD‐L1+CD14+ antigen‐presenting cells and regulatory T cells mark the microenvironment of metastatic lymph nodes from patients with cervical cancer. Cancer Immunol Res 3: 48–58 [DOI] [PubMed] [Google Scholar]
- Horeweg N, Workel HH, Loiero D, Church DN, Vermij L, Leon‐Castillo A, Krog RT, de Boer SM, Nout RA, Powell ME et al (2022) Tertiary lymphoid structures critical for prognosis in endometrial cancer patients. Nat Commun 13: 1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornburg M, Desbois M, Lu S, Guan Y, Lo AA, Kaufman S, Elrod A, Lotstein A, DesRochers TM, Munoz‐Rodriguez JL et al (2021) Single‐cell dissection of cellular components and interactions shaping the tumor immune phenotypes in ovarian cancer. Cancer Cell 39: 928–944 [DOI] [PubMed] [Google Scholar]
- Italiano A, Bessede A, Pulido M, Bompas E, Piperno‐Neumann S, Chevreau C, Penel N, Bertucci F, Toulmonde M, Bellera C et al (2022) Pembrolizumab in soft‐tissue sarcomas with tertiary lymphoid structures: a phase 2 PEMBROSARC trial cohort. Nat Med 28: 1199–1206 [DOI] [PubMed] [Google Scholar]
- Jacquelot N, Tellier J, Nutt SL, Belz GT (2021) Tertiary lymphoid structures and B lymphocytes in cancer prognosis and response to immunotherapies. Onco Targets Ther 10: 1900508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, Guo MG, George BM, Mollbrink A, Bergenstrahle J et al (2020) Multimodal analysis of composition and spatial architecture in human squamous cell carcinoma. Cell 182: e422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Q, Wu W, Wang Y, Alexander PB, Sun C, Gong Z, Cheng JN, Sun H, Guan Y, Xia X et al (2018) Local mutational diversity drives intratumoral immune heterogeneity in non‐small cell lung cancer. Nat Commun 9: 5361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B et al (2018) Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 24: 1550–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Li R, Chen MY, Yu C, Tang LQ, Liu YM, Li JP, Liu YN, Luo YL, Zhao Y et al (2020) Single‐cell transcriptomic analysis defines the interplay between tumor cells, viral infection, and the microenvironment in nasopharyngeal carcinoma. Cell Res 30: 950–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Guerrero‐Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, Nie Q (2021) Inference and analysis of cell‐cell communication using CellChat. Nat Commun 12: 1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SS, Shen S, Miyauchi S, Sanders PD, Franiak‐Pietryga I, Mell L, Gutkind JS, Cohen EEW, Califano JA, Sharabi AB (2020) B cells improve overall survival in HPV‐associated squamous cell carcinomas and are activated by radiation and PD‐1 blockade. Clin Cancer Res 26: 3345–3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan C, Shen J, Wang Y, Li J, Liu Z, He M, Cao X, Ling J, Huang J, Zheng M et al (2020) Camrelizumab plus Apatinib in patients with advanced cervical cancer (CLAP): a multicenter, open‐label, single‐arm, phase II trial. J Clin Oncol 38: 4095–4106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard‐Solodkin D, van Akkooi ACJ, van den Braber M, Rozeman EA, Haanen J, Blank CU et al (2019) Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 176: 775–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JP, Wu CY, Chen MY, Liu SX, Yan SM, Kang YF, Sun C, Grandis JR, Zeng MS, Zhong Q (2021) PD‐1+CXCR5−CD4+ Th‐CXCL13 cell subset drives B cells into tertiary lymphoid structures of nasopharyngeal carcinoma. J Immunother Cancer 9: e002101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P (2015) The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst 1: 417–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Lu J, Tian H, Du W, Zhao L, Feng J, Yuan D, Li Z (2016) Increased expression of PD‐L1 by the human papillomavirus 16 E7 oncoprotein inhibits anticancer immunity. Mol Med Rep 15: 1063–1070 [DOI] [PubMed] [Google Scholar]
- Liu Y, Wang L, Lo KW, Lui VWY (2020) Omics‐wide quantitative B‐cell infiltration analyses identify GPR18 for human cancer prognosis with superiority over CD20. Commun Biol 3: 234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhao Q, Liao JY, Song E, Xia Q, Pan J, Li Y, Li J, Zhou B, Ye Y et al (2020) Complement signals determine opposite effects of B cells in chemotherapy‐induced immunity. Cell 180: 1081–1097 [DOI] [PubMed] [Google Scholar]
- Luo X, Donnelly CR, Gong W, Heath BR, Hao Y, Donnelly LA, Moghbeli T, Tan YS, Lin X, Bellile E et al (2020) HPV16 drives cancer immune escape via NLRX1‐mediated degradation of STING. J Clin Invest 130: 1635–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado L, Teague JE, Morrow MP, Jotova I, Wu TC, Wang C, Desmarais C, Boyer JD, Tycko B, Robins HS et al (2014) Intramuscular therapeutic vaccination targeting HPV16 induces T cell responses that localize in mucosal lesions. Sci Transl Med 6: 221ra213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, Gartner ZJ (2019) DoubletFinder: doublet detection in single‐cell RNA sequencing data using artificial nearest neighbors. Cell Syst 8: 329–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan M, Petitprez F, Becht E, Bougouin A, Pupier G, Calvez A, Giglioli I, Verkarre V, Lacroix G, Verneau J et al (2022) Tertiary lymphoid structures generate and propagate anti‐tumor antibody‐producing plasma cells in renal cell cancer. Immunity 55: 527–541 [DOI] [PubMed] [Google Scholar]
- Nagarsheth NB, Norberg SM, Sinkoe AL, Adhikary S, Meyer TJ, Lack JB, Warner AC, Schweitzer C, Doran SL, Korrapati S et al (2021) TCR‐engineered T cells targeting E7 for patients with metastatic HPV‐associated epithelial cancers. Nat Med 27: 419–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolich‐Zugich J, Slifka MK, Messaoudi I (2004) The many important facets of T‐cell repertoire diversity. Nat Rev Immunol 4: 123–132 [DOI] [PubMed] [Google Scholar]
- O'Donnell JS, Teng MWL, Smyth MJ (2019) Cancer immunoediting and resistance to T cell‐based immunotherapy. Nat Rev Clin Oncol 16: 151–167 [DOI] [PubMed] [Google Scholar]
- Pasetto A, Gros A, Robbins PF, Deniger DC, Prickett TD, Matus‐Nicodemos R, Douek DC, Howie B, Robins H, Parkhurst MR et al (2016) Tumor‐ and neoantigen‐reactive T‐cell receptors can be identified based on their frequency in fresh tumor. Cancer Immunol Res 4: 734–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil NS, Nabet BY, Muller S, Koeppen H, Zou W, Giltnane J, Au‐Yeung A, Srivats S, Cheng JH, Takahashi C et al (2022) Intratumoral plasma cells predict outcomes to PD‐L1 blockade in non‐small cell lung cancer. Cancer Cell 40: e284 [DOI] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma YA, Trapnell C (2017) Single‐cell mRNA quantification and differential analysis with Census. Nat Methods 14: 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast I, Dvorscek AR, Pattaroni C, Steiner TM, McKenzie CI, Pitt C, O'Donnell K, Ding Z, Hill DL, Brink R et al (2022) Interleukin‐21, acting beyond the immunological synapse, independently controls T follicular helper and germinal center B cells. Immunity 55: 1414–1430 [DOI] [PubMed] [Google Scholar]
- Ren X, Zhang L, Zhang Y, Li Z, Siemers N, Zhang Z (2021) Insights gained from single‐cell analysis of immune cells in the tumor microenvironment. Annu Rev Immunol 39: 583–609 [DOI] [PubMed] [Google Scholar]
- Ribatti D (2016) Mast cells as therapeutic target in cancer. Eur J Pharmacol 778: 152–157 [DOI] [PubMed] [Google Scholar]
- Ripperger TJ, Bhattacharya D (2021) Transcriptional and metabolic control of memory B cells and plasma cells. Annu Rev Immunol 39: 345–368 [DOI] [PubMed] [Google Scholar]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, Fearon D, Greten FR, Hingorani SR, Hunter T et al (2020) A framework for advancing our understanding of cancer‐associated fibroblasts. Nat Rev Cancer 20: 174–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santegoets SJ, van Ham VJ, Ehsan I, Charoentong P, Duurland CL, van Unen V, Hollt T, van der Velden LA, van Egmond SL, Kortekaas KE et al (2019) The anatomical location shapes the immune infiltrate in tumors of same etiology and affects survival. Clin Cancer Res 25: 240–252 [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, Regev A (2015) Spatial reconstruction of single‐cell gene expression data. Nat Biotechnol 33: 495–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sautes‐Fridman C, Petitprez F, Calderaro J, Fridman WH (2019) Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer 19: 307–325 [DOI] [PubMed] [Google Scholar]
- Schalck A, Sakellariou‐Thompson D, Forget MA, Sei E, Hughes TG, Reuben A, Bai S, Hu M, Kumar T, Hurd MW et al (2022) Single cell sequencing reveals trajectory of tumor‐infiltrating lymphocyte states in pancreatic cancer. Cancer Discov 12: 2330–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serra S, Chetty R (2018) p16. J Clin Pathol 71: 853–858 [DOI] [PubMed] [Google Scholar]
- Shamseddine AA, Burman B, Lee NY, Zamarin D, Riaz N (2021) Tumor immunity and immunotherapy for HPV‐related cancers. Cancer Discov 11: 1896–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugay M, Bagaev DV, Zvyagin IV, Vroomans RM, Crawford JC, Dolton G, Komech EA, Sycheva AL, Koneva AE, Egorov ES et al (2018) VDJdb: a curated database of T‐cell receptor sequences with known antigen specificity. Nucleic Acids Res 46: D419–D427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H et al (2018) Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature 557: 575–579 [DOI] [PubMed] [Google Scholar]
- Song L, Cohen D, Ouyang Z, Cao Y, Hu X, Liu XS (2021) TRUST4: immune repertoire reconstruction from bulk and single‐cell RNA‐seq data. Nat Methods 18: 627–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, Robins HS, Robbins PF, Klebanoff CA, Rosenberg SA et al (2017) Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science 356: 200–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Wu L, Zhong Y, Zhou K, Hou Y, Wang Z, Zhang Z, Xie J, Wang C, Chen D et al (2020) Single‐cell landscape of the ecosystem in early‐relapse hepatocellular carcinoma. Cell 184: 404–421 [DOI] [PubMed] [Google Scholar]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71: 209–249 [DOI] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G et al (2016) Dissecting the multicellular ecosystem of metastatic melanoma by single‐cell RNA‐seq. Science 352: 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg SPH, Pardieck IN, Lanfermeijer J, Sauce D, Klenerman P, van Baarle D, Arens R (2019) The hallmarks of CMV‐specific CD8 T‐cell differentiation. Med Microbiol Immunol 208: 365–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg SH, Melief CJ (2011) Therapeutic vaccination against human papilloma virus induced malignancies. Curr Opin Immunol 23: 252–257 [DOI] [PubMed] [Google Scholar]
- Van Doorslaer K, Li Z, Xirasagar S, Maes P, Kaminsky D, Liou D, Sun Q, Kaur R, Huyen Y, McBride AA (2017) The papillomavirus episteme: a major update to the papillomavirus sequence database. Nucleic Acids Res 45: D499–D506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walch‐Ruckheim B, Mavrova R, Henning M, Vicinus B, Kim YJ, Bohle RM, Juhasz‐Boss I, Solomayer EF, Smola S (2015) Stromal fibroblasts induce CCL20 through IL6/C/EBPbeta to support the recruitment of Th17 cells during cervical cancer progression. Cancer Res 75: 5248–5259 [DOI] [PubMed] [Google Scholar]
- Walch‐Ruckheim B, Stroder R, Theobald L, Pahne‐Zeppenfeld J, Hegde S, Kim YJ, Bohle RM, Juhasz‐Boss I, Solomayer EF, Smola S (2019) Cervical cancer‐instructed stromal fibroblasts enhance IL23 expression in dendritic cells to support expansion of Th17 cells. Cancer Res 79: 1573–1586 [DOI] [PubMed] [Google Scholar]
- Wang C, Sun D, Huang X, Wan C, Li Z, Han Y, Qin Q, Fan J, Qiu X, Xie Y et al (2020) Integrative analyses of single‐cell transcriptome and regulome using MAESTRO. Genome Biol 21: 198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei WF, Chen XJ, Liang LJ, Yu L, Wu XG, Zhou CF, Wang ZC, Fan LS, Hu Z, Liang L et al (2021) Periostin+ cancer‐associated fibroblasts promote lymph node metastasis by impairing the lymphatic endothelial barriers in cervical squamous cell carcinoma. Mol Oncol 15: 210–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westrich JA, Vermeer DW, Colbert PL, Spanos WC, Pyeon D (2020) The multifarious roles of the chemokine CXCL14 in cancer progression and immune responses. Mol Carcinog 59: 794–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016) Ggplot2: elegant graphics for data analysis. New York, NY: Springer‐Verlag; [Google Scholar]
- Wieland A, Patel MR, Cardenas MA, Eberhardt CS, Hudson WH, Obeng RC, Griffith CC, Wang X, Chen ZG, Kissick HT et al (2021) Defining HPV‐specific B cell responses in patients with head and neck cancer. Nature 597: 274–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS, Murphy KJ, Pereira B, Al‐Eryani G, Bartonicek N et al (2020) Stromal cell diversity associated with immune evasion in human triple‐negative breast cancer. EMBO J 39: e104063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, McNamara KL, Granja JM, Sarin KY, Brown RA et al (2019) Clonal replacement of tumor‐specific T cells following PD‐1 blockade. Nat Med 25: 1251–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn JW, Hur SY, Woo JW, Kim YM, Lim MC, Park SY, Seo SS, No JH, Kim BG, Lee JK et al (2020) Pembrolizumab plus GX‐188E therapeutic DNA vaccine in patients with HPV‐16‐positive or HPV‐18‐positive advanced cervical cancer: interim results of a single‐arm, phase 2 trial. Lancet Oncol 21: 1653–1660 [DOI] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16: 284–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Cai X, Shen F, Ma F (2021) HPV post‐infection microenvironment and cervical cancer. Cancer Lett 497: 243–254 [DOI] [PubMed] [Google Scholar]
- Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, Modak M, Carotta S, Haslinger C, Kind D et al (2019) Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell 179: 829–845 [DOI] [PubMed] [Google Scholar]
- Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O'Brien SA, He Y, Wang L, Zhang Q, Kim A et al (2020a) Single‐cell analyses inform mechanisms of myeloid‐targeted therapies in colon cancer. Cell 181: 442–459 [DOI] [PubMed] [Google Scholar]
- Zhang WH, Wang WQ, Han X, Gao HL, Xu SS, Li S, Li TJ, Xu HX, Li H, Ye LY et al (2020b) Infiltrating pattern and prognostic value of tertiary lymphoid structures in resected non‐functional pancreatic neuroendocrine tumors. J Immunother Cancer 8: e001188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yu M, Jing Y, Cheng J, Zhang C, Cheng L, Lu H, Cai MC, Wu J, Wang W et al (2021) Baseline immunity and impact of chemotherapy on immune microenvironment in cervical cancer. Br J Cancer 124: 414–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, Ren X, Wang L, Wu X, Zhang J et al (2021) Pan‐cancer single‐cell landscape of tumor‐infiltrating T cells. Science 374: abe6474 [DOI] [PubMed] [Google Scholar]
- Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, Krishnan I, Maroni G, Meyerovitz CV, Kerwin CM et al (2019) Single‐cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 50: 1317–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]