

Abstract
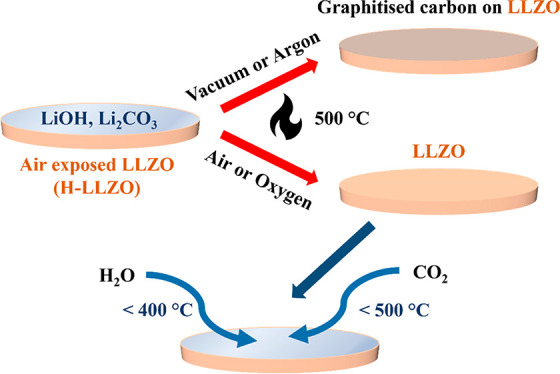
Garnet solid-electrolyte-based Li-metal batteries can be used in energy storage devices with high energy densities and thermal stability. However, the tendency of garnets to form lithium hydroxide and carbonate on the surface in an ambient atmosphere poses significant processing challenges. In this work, the decomposition of surface layers under various gas environments is studied by using two surface-sensitive techniques, near-ambient-pressure X-ray photoelectron spectroscopy and grazing incidence X-ray diffraction. It is found that heating to 500 °C under an oxygen atmosphere (of 1 mbar and above) leads to a clean garnet surface, whereas low oxygen partial pressures (i.e., in argon or vacuum) lead to additional graphitic carbon deposits. The clean surface of garnets reacts directly with moisture and carbon dioxide below 400 and 500 °C, respectively. This suggests that additional CO2 concentration controls are needed for the handling of garnets. By heating under O2 along with avoiding H2O and CO2, symmetric cells with less than 10 Ωcm2 interface resistance are prepared without the use of any interlayers; plating currents of >1 mA cm–2 without dendrite initiation are demonstrated.
Solid-electrolyte (SE)-based Li-metal batteries can enable high-energy storage devices due to their potential compatibility with a Li metal anode and high-voltage cathodes.1−3 They offer greater thermal stability than the current state-of-the-art liquid-electrolyte-based Li-ion batteries.4−6 Among various SEs explored so far, doped LLZO (Li7La3Zr2O12) garnets have high room-temperature (RT) ionic conductivities of 0.1–1 mS cm–1 and comparatively wide electrochemical stability, which make them promising candidates for commercial applications.7−10
It is well known that LLZO reacts with trace moisture and carbon dioxide in the atmosphere.11−15 This results in the exchange of Li+ in the lattice with H+ to form protonated LLZO (HxLi7–xLa3Zr2O12) and lithium hydroxide and carbonate surface layers. Protonation leads to lattice contraction and a change of symmetry from Ia3̅d to I4̅3d,16−20 and the resulting heterogeneous surface layers have very low Li-ion conductivity and thus increase the interfacial resistance when LLZO is, for example, paired with a Li metal anode.11,12,21,22 This leads to non-uniform current distribution at the Li–LLZO–Li metal interface, which decreases the critical current density (ICCD) at which Li metal dendrites nucleate and short-circuit the cell.13,14,23
Different protocols for regeneration of LLZO have been reported, wherein the samples have been treated under a variety of gases and temperatures.21,24−27 Despite this, the minimum temperature needed to regenerate the surface has not been definitively established, and the effect of different gases has not yet been systematically studied. It has also been reported that excessive heating of LLZO results in pyrochlore formation. This irreversible decomposition has been studied using bulk X-ray diffraction (XRD), and a range of onset temperatures have been reported.25,28,29 Since the decomposition starts at the surface of LLZO, careful investigation with surface-sensitive techniques is needed to determine the onset temperature for this reaction accurately. Finally, even though the composition of the surface layers has been characterized, the onset temperature and the reaction mechanisms leading to formation of surface layers are poorly understood; for example, contradicting reports exist on the direct reactivity of LLZO with CO2.11,12,30,31 The extreme sensitivity of the LLZO surface demands not only surface sensitivity but also in situ techniques to understand the regeneration and reactivity of LLZO.
In this study, air-exposed LLZO pellets were heated under different gas environments (vacuum, argon, static air, and flowing air) to study the regeneration process, and in situ grazing incidence X-ray diffraction (GIXRD) patterns were collected to capture the structural changes at the surface. To map the chemical composition of the surface, in situ near-ambient-pressure X-ray photoelectron spectroscopy (NAP-XPS) spectra were then collected while the samples were heated under vacuum, argon, dry air, and oxygen, with heating under oxygen resulting in complete regeneration and a clean LLZO surface. The samples were then freshly regenerated by heating under oxygen, and in situ NAP-XPS spectra were collected while the samples were cooled under H2O vapors, CO2, and a mixture of H2O vapors + CO2 to understand the formation of surface layers on LLZO. Finally, by heating air-exposed LLZO samples under oxygen and avoiding CO2 and H2O during cooling, low Li–LLZO interfacial resistances (<10 Ωcm2) were achieved, and dendrite-free plating was obtained at currents above 1 mA cm–2.
GIXRD Measurements under Different Gas Environments
Al-LLZO (with composition Al0.36Li5.92La3Zr2O12) powder was synthesized using a solid-state method (see methods in the Supporting Information (SI)), hot-pressed, and cut into pellets (∼99% relative density). The phase purity of the pellets was confirmed via synchrotron XRD (Figure S1). GIXRD was then performed in different gas environments. Due to the limitation of the setup, heating could not be performed under pure oxygen, but vacuum (0.01 mbar), argon, static air, and flowing air (1 atm) environments were tested. All samples were first exposed to air for 20 min at RT and then placed into the GIXRD setup in the beamline. A grazing incidence angle of 0.1° was chosen, which corresponds to a probing depth of about ∼3 nm (for LLZO). The samples were heated in controlled gas environments to 800 °C in steps of 100 °C, and GIXRD patterns were collected at each temperature and then on cooling to RT (see experimental details in the SI).
Lithium carbonate and lithium hydroxide were observed in all air-exposed samples at RT (Figure 1). On heating under vacuum, the Li2CO3 and LiOH on the LLZO pellets were observed to decompose above 500 °C (reflections marked by ▼ and ●, respectively, Figure 1, top) and almost completely disappeared by 700 and 800 °C, respectively, in line with previous observations of pure Li2CO3 decomposition under vacuum.32 At 800 °C, formation of Li2ZrO3 and LaAlO3 was observed (reflections marked by ⧫ and *, respectively, Figure 1, top), although no pyrochlore La2Zr2O7 was observed.
Figure 1.

GIXRD (λ = 0.8856 Å) patterns of air-exposed samples heated under different gas environments from RT to 800 °C in 100 °C increments and then cooled to RT. The second and third columns of images are the enlarged versions of the regions corresponding to Li2CO3 (110) and La2Zr2O7 (222) reflections, respectively. The black dotted-line box and ■ represent LLZO reflections, ⧫ represents Li2ZrO3, * represents LaAlO3, ● represents LiOH, and ▼ represents Li2CO3. The peaks represented by ★ in the vacuum case at 800 °C could not be indexed to any known compound, and these peaks disappear upon cooling to RT. The shift in the LLZO (211) peaks toward lower 2θ as the sample is heated is due to thermal lattice expansion; sharp discontinuities in the reflections of all the phases are seen between the patterns collected at 800 °C and RT due to rapid lattice contractions.
Upon heating under argon, Li2CO3 and LiOH were observed to decompose at the lower temperatures of 400 and 500 °C, respectively. Heating to 800 °C did not result in any observable LLZO decomposition, but the pyrochlore, La2Zr2O7, was detected after the samples were cooled to RT, suggesting LLZO decomposes at elevated temperatures under argon. Under flowing air, Li2CO3 and LiOH were observed to decompose above 400 and 600 °C, respectively, and La2Zr2O7 pyrochlore was again observed after the samples were cooled to RT as in the argon case, suggesting a similar decomposition mechanism is occurring under flowing air. Interestingly, when samples were heated under static air (under conditions similar to those in a box furnace), Li2CO3 and LiOH were observed to decompose only above 600 °C, as in the vacuum case (Figure S2). Extensive decomposition of LLZO and pyrochlore formation was also observed upon heating above 500 °C in static air, and the LLZO signal completely disappeared above 600 °C, suggesting that trace moisture in gas environments not removed by gas flow or by pulling a vacuum induces more rapid decomposition of LLZO. We note that Li2O evaporation is significantly enhanced by the presence of water (forming the more volatile product, LiOH),33 accounting for many of the degradation products seen by GIXRD on heating in static air.
NAP-XPS Measurements under Different Gas Environments
NAP-XPS was performed to complement the GIXRD observations and map the evolution of the chemical composition of the surface of LLZO pellets during heating. The pellets were polished and stored in a glovebox and were then exposed to air for 20 min before being pumped into the NAP-XPS instrument on the beamline. The incident energy was tuned in a way that the kinetic energy of the ejected photoelectrons probed was ∼200 eV; this corresponds to a probing depth of ∼3 nm for all XPS measurements. The samples were heated in controlled gas (1 mbar, vacuum: 2 × 10–8 mbar) environments to 500 °C in steps of 100 °C and then cooled to RT, and XPS spectra were collected during these processes (see experimental details in the SI). The evolution of C 1s, O 1s, and La 3d spectra is presented in Figure 2, while the Li 1s and Zr 3d XPS spectra are shown in Figure S4. The C 1s XPS spectra showed two signals at RT for all samples (Figure 2). Since lithium carbonate is expected to form on the surface of the air-exposed samples, the peak at higher eV was aligned to the Li2CO3 peak at 289.9 eV.26 The XPS spectra of the other elements were shifted by the same amount. Significant asymmetric broadening and shifting of peaks and variations in intensities were observed for all samples due to charging, especially at low temperatures, complicating analysis; thus the 500 °C data are also compared in Figure 2, as they are the most straightforward to analyze.
Figure 2.

C 1s, O 1s, and La 3d XPS spectra of the air-exposed samples heated under different gas environments from RT to 500 °C in 100 °C increments and then cooled to RT. The gray dotted line and the gray dotted-line box represent Li2CO3, the golden dotted line represents LLZO, and the black dotted-line box represents surface-adsorbed hydrocarbons and graphitized carbon. The last row compares the C 1s, O 1s, and La 3d XPS spectra at 500 °C under different gas environments.
An additional peak at ∼285 eV was observed in the C 1s spectra in all environments when the samples were heated (Figure 2). This peak disappeared as the samples were heated to 500 °C under dry air and oxygen. By contrast, this peak remained for samples heated under vacuum and argon and stayed even after cooling to RT. This suggests that this peak originates from surface-adsorbed (sp3-containing) hydrocarbons, likely present in the vacuum chamber or in the glovebox, which oxidize in dry air and oxygen but graphitize under low oxygen partial pressures (vacuum and argon), resulting in a shift of this peak to lower eV (284.1 eV; see comparison at 500 °C, Figure 2). Similar observations have been made in previous in situ XPS studies performed under vacuum.25,26 Our results show that heating under argon can also result in graphitic carbon on the surface of LLZO.
As the samples were heated from RT to 100 °C, the broad O 1s peak shifted toward higher eV. The shift toward higher eV can be attributed to surface -HCO3– species34 which are enhanced due to surface desorption of H2O. Above 100 °C, sharpening of the peak and a shift toward higher eV were observed. The sharpening can be attributed to better charge compensation during XPS measurements at elevated temperatures and the completion of surface-adsorbed water release process. The peak now seen can be assigned to a CO32– species. An additional small peak emerged at 500 °C around ∼529 eV in the O 1s XPS spectra in all gas environments (Figure 2). Sharp peaks were similarly observed in the La 3d and Zr 3d spectra at 500 °C that could not be seen at RT (Figures 2 and S5). Thus, the peak at 529 eV in the O 1s spectra is assigned to the LLZO lattice, confirming the regeneration of LLZO at 500 °C (although the presence of a C 1s Li2CO3 peak at 500 °C shows incomplete decomposition of the surface layers). The LLZO O 1s peak remains even after cooling to RT (Figure S6) under vacuum, argon, and oxygen but not under dry air.
The GIXRD and XPS results are summarized in Tables S1–S3 and Figure S3. The GIXRD observations suggest that heating until 500 °C can decompose the surface layers on LLZO if the samples are treated under either argon or flowing air (and by extension oxygen). Although the XPS spectra showed incomplete decomposition of the surface layers at 500 °C, this discrepancy might be due to the differences in the way the temperatures of the samples are measured in the two setups. Additional graphitic formation was observed when samples were heated under vacuum and argon. Though recent studies have shown that carbon interlayers reduce Li–LLZO interface resistance and improve performance,35 the graphitization on the surface due to heating under argon and vacuum need not be uniform, and any heterogeneity could lead to current focusing when LLZO is used in a SE against Li metal. Finally, avoiding temperatures above 500 °C will prevent pyrochlore formation irrespective of the environments under which samples are heated to regenerate LLZO.
NAP-XPS Measurements to Study the Onset Temperature for Formation of Surface Layers
To prevent any reformation of surface layers during cooling, it is important to determine the onset temperature for the reaction of LLZO with moisture and CO2. LLZO samples were first treated under oxygen at 500 °C for 1 h to allow for surface regeneration, the LLZO lattice peak being observed in O 1s spectra at ∼529 eV, confirming the removal of most of the surface contaminants. Next, H2O vapor was introduced into the reaction chamber by manually opening a valve which was connected to a quartz tube containing water (see experimental details in the SI). XPS spectra were then collected while the samples were cooled from 500 °C to RT (Figure 3). The LLZO O 1s lattice peak remained on introduction of H2O vapor, and the C 1s spectrum showed a single peak which is attributed to residual Li2CO3 as discussed above.
Figure 3.

C 1s and O 1s XPS spectra of LLZO during cooling from 500 °C to RT under different gas environments. The gray dotted line represents Li2CO3, the golden dotted line represents LLZO, the black dotted-line box represents additional hydrocarbon-related species on the surface, and the dark blue dotted line represents LiOH.
As the sample was cooled to 400 °C, an additional peak appeared at around ∼286 eV in the C 1s spectra along with the continued reduction in the intensity of the Li2CO3 peak. In the O 1s spectra, the Li2CO3 peak broadened whereas the LLZO lattice peak remained. The additional peak is attributed to oxidized surface-adsorbed hydrocarbons (the shift corresponds to ether-containing hydrocarbons) as observed in the literature when H2O is introduced into the XPS chamber.36 At 300 °C, the LLZO peak in the O 1s spectra shifted to higher eV, suggesting protonation of LLZO. The Li 1s peak broadened, which is attributed to LiOH formation. The intensity of the additional (∼286 eV) peak in the C 1s spectra increased and was now comparable to that of the Li2CO3 peak. As the sample was cooled below 200 °C, the LLZO lattice peak completely disappeared, suggesting extensive formation of LiOH on the surface of LLZO, in line with a recent study:37
| 1 |
The additional peak in the C 1s spectra remained even after cooling to RT.
To probe the reaction of carbon dioxide with LLZO, the samples were heated under O2 as earlier and CO2 was introduced into the reaction chamber (see experimental details in the SI). XPS spectra were collected as the samples were cooled to RT. At 500 °C, the LLZO lattice peak was observed in the O 1s region at ∼529 eV, the peak remaining on introduction of CO2. The C 1s spectrum showed a single peak corresponding to Li2CO3.
Upon cooling to 400 °C, the LLZO peak intensity dropped significantly, suggesting a direct reaction of LLZO with CO2. This can be expressed as
| 2 |
A reaction of this form requires the extraction of oxygen anions, formally via the extraction of Li2O. Thus, this reaction is likely localized just at the surface, as a significant amount of oxygen vacancies need to be generated in the lattice for this reaction to proceed into the bulk of the sample. The reactions with CO2 are acid–base reactions, with the basicity of the parent oxide phase Li2O (and the Li+ mobility) driving the reaction to form Li2CO3. However, the parent oxide, La2O3 is more basic, forming La2(CO3)3, which decomposes at high temperatures via the formation of La2O2CO3; La2O2CO3 does not decompose to form La2O3 until 950 °C under 1 atm of CO2.38 Thus, reactions of the following form can also occur, written here for the parent LLZO phase to illustrate one possible decomposition pathway:
| 3 |
While this reaction likely only occurs at the surface, as it involves migration of La3+ and Zr4+, it does not require the formation of oxygen vacancies in LLZO. It also represents a plausible mechanism in the formation of the pyrochlore phase at higher temperatures (as seen by GIXRD; Table S1), where La3+/Zr4+ migration can occur. Furthermore, when heated in static air (or any closed vessel), any CO2 released from the decomposition of Li2CO3, which occurs at a lower temperature, can then react to form lanthanum (oxy) carbonate (La2O2CO3).
The presence of any trace water will also result in reactions not requiring the generation of multiple oxygen vacancies:
| 4 |
or
| 5 |
As the sample was cooled below 300 °C, the LLZO lattice peak completely disappeared, and a new additional peak was observed in the C 1s spectra around ∼284.8 eV, which can be attributed to surface-adsorbed hydrocarbons. As the sample cooled to 100 °C, this additional peak completely disappeared in the C 1s spectra and a significant broadening of the Li2CO3 peak was observed. This was accompanied by broadening of the Li2CO3 O 1s peak, which suggests either extensive reaction of LLZO with CO2 or severe charging of the sample. These results also explain the broadening of the O 1s spectra observed at RT after regeneration of LLZO by heating under dry air (Figure S6): the trace amounts of CO2 in dry air will react with LLZO below 400 °C and passivate the surface.
In an ambient atmosphere, both H2O (∼30 mbar) and CO2 (∼0.42 mbar) are present in trace amounts. To check the reactivity of LLZO in this case, LLZO was first heat-treated under O2 as in the earlier cases and then cooled to RT under a 1:1 mixture of H2O and CO2 (see experimental details in the SI). At 400 °C, a reduction and a shift in the LLZO peak toward higher eV were observed, indicating protonation. An additional peak in the C 1s spectra was observed as in the H2O and CO2 case. Upon further cooling, the Li 1s, C 1s, and O 1s peak evolution was essentially a combination of that seen in the pure H2O and pure CO2 cases, suggesting reactions described by eqs 3, 4, and 5 are occurring. The summarized results are shown in Table S4.
These results suggest that CO2 levels in the environment where LLZO regeneration is performed need to be controlled along with H2O levels. This poses a unique challenge for handling LLZO, since CO2 levels are not usually monitored and controlled.
Our results should be compared with recent reports in which LLZO/thin-film cathode model systems (with either LiNi0.6Mn0.2Co0.2O2 or LiCoO2 as the cathode) were sintered in the presence of CO2, resulting in enhanced decomposition of both the cathode and LLZO;39−41 these results highlight the strong driving force for carbonate formation, even when the LLZO surface is protected via the formation of a LLZO–cathode interface. In contrast, heating to 700 °C under either pure oxygen or an inert atmosphere (N2) was shown to result in a low LLZO–cathode interface resistance. A higher interfacial resistance was seen on heating in humidified oxygen at 500 °C, the resistance dropping to a value comparable to the pure O2 results, however, on heating to 700 °C. In the current study, by contrast, any moisture was found to result in extensive decomposition of LLZO above 500 °C; the lack of degradation in the presence of moisture in the previous cathode−LLZO systems is ascribed to the protection of the LLZO surface by the cathode film, which helps to reduce LiOH evaporation even in moist environments. In our studies of the bare pellets, the inert atmosphere (argon) led to graphitic deposits on the surface of LLZO, and only oxygen was found to be the ideal gas to regenerate LLZO.
Electrochemical Studies
Since the NAP-XPS and GIXRD measurements suggested that heating under oxygen (partial pressures of 1 mbar and above) results in the decomposition of surface layers and can lead to complete and clean regeneration of LLZO, the LLZO pellets were treated under oxygen and then transferred to a glovebox without any exposure to air via a custom setup that was purged with argon (Figure S10). Li–LLZO–Li symmetric cells were assembled, impedance measurements were conducted (Figure 4), and the data were fit by using an equivalent circuit model (Figure S7). The Li–LLZO interfacial resistance was found to be <10 Ωcm2, in comparison to >500 Ωcm2 interfacial resistance shown by samples that had just been polished inside the glovebox (Figure S9). To estimate the ICCD before Li dendrites are formed, unidirectional currents were applied to the symmetric cell (see experimental details in the SI). The ICCD was found to be >1 mA cm–2 (Figure S8), in comparison to the reported ICCD of <0.5 mA cm–2 for cells with interface resistance >10 Ωcm2,42,43 suggesting that with an optimized protocol to reduce interface resistance, high plating current densities can be achieved in LLZO garnets.
Figure 4.

Impedance spectra of (left) a LLZO pellet under blocking conditions, with fits to an equivalent circuit model to determine the bulk and grain boundary contributions, and (right) a Li–LLZO–Li symmetric cell showing the contributions from bulk, grain boundary, and Li–LLZO interface.
In conclusion, this work demonstrates that the surface layers (LiOH and Li2CO3) formed upon exposure of garnets to ambient air can be decomposed by heating to 500 °C, irrespective of the gas environment. Additionally, graphitic carbons have been found to form during the heating of LLZO under vacuum and argon. Though argon is the most common environment for the regeneration of LLZO, heterogeneous graphitization can lead to current focusing and affect the performance of LLZO when used as a SE. Heating above 600–700 °C was found to result in decomposition of LLZO into pyrochlores under argon and flowing air but not under vacuum. A clean LLZO surface was found to react with moisture at temperatures below 400 °C, and direct evidence for reaction of LLZO with CO2 was shown below 500 °C. An optimized protocol for regeneration of LLZO is presented, and it is shown that interfacial resistances below 10 Ω·cm2 can be achieved for LLZO pellets without the use of any interlayers between Li metal and LLZO. These results mean that the handling of LLZO will require more specialized CO2 level controls in addition to the dry rooms used for assembly of liquid-electrolyte-based Li-ion batteries.
Acknowledgments
S.V. acknowledges funding from the Cambridge Commonwealth European and International Trust, Faraday Institution (SOLBAT, FIRG007), and Royal Society (RP/R1/180147). F.N.S. also acknowledges funding from The Faraday Institution CATMAT project (FIRG016). S.N. thanks the Royal Society (United Kingdom) and Science and Engineering Research Board (Government of India) for the award of Newton-Bhabha International Fellowship (NIF/R1/180075). C.P.G. thanks the EU via an Advanced EU ERC grant (EC H2020 835073). Professor Norman Fleck and Professor Vikram Deshpande are thanked for access to their laboratories for sample preparation and for helpful discussions. We thank Simon Marshall and Graham Smith for assistance in operating the hot-press and Anthony Dennis and Harry Druiff for assistance in cutting hot-pressed samples. We thank the Diamond Light Source, UK, for access to beamline B07 (SI29728) for NAP-XPS measurements and DELTA, Germany, for providing synchrotron radiation at beamline BL9 for grazing incidence X-ray diffraction measurements. We also acknowledge the I11 beamline for synchrotron XRD at the Diamond Light Source, UK, under BAG proposal CY28349.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsenergylett.3c01042.
Experimental methods, XRD of samples, additional XPS and GIXRD data, tables summarizing observations from XPS and GIXRD data, equivalent circuit used for fitting impedance and electrochemistry data, and setup used for thermal treatment of samples (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Janek J.; Zeier W. G. A Solid Future for Battery Development. Nat. Energy 2016, 1 (9), 16141. 10.1038/nenergy.2016.141. [DOI] [Google Scholar]
- Gao Z.; Sun H.; Fu L.; Ye F.; Zhang Y.; Luo W.; Huang Y. Promises, Challenges, and Recent Progress of Inorganic Solid-State Electrolytes for All-Solid-State Lithium Batteries. Adv. Mater. 2018, 30 (17), 1705702. 10.1002/adma.201705702. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Shao Y.; Lotsch B.; Hu Y.-S.; Li H.; Janek J.; Nazar L. F.; Nan C.-W.; Maier J.; Armand M.; Chen L. New Horizons for Inorganic Solid State Ion Conductors. Energy Environ. Sci. 2018, 11 (8), 1945–1976. 10.1039/C8EE01053F. [DOI] [Google Scholar]
- Inoue T.; Mukai K. Are All-Solid-State Lithium-Ion Batteries Really Safe?–Verification by Differential Scanning Calorimetry with an All-Inclusive Microcell. ACS Appl. Mater. Interfaces 2017, 9 (2), 1507–1515. 10.1021/acsami.6b13224. [DOI] [PubMed] [Google Scholar]
- Famprikis T.; Canepa P.; Dawson J. A.; Islam M. S.; Masquelier C. Fundamentals of Inorganic Solid-State Electrolytes for Batteries. Nat. Mater. 2019, 18 (12), 1278–1291. 10.1038/s41563-019-0431-3. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Wang S.; Li H.; Chen L.; Wu F. Progress in Thermal Stability of All-Solid-State-Li-Ion-Batteries. InfoMat 2021, 3 (8), 827–853. 10.1002/inf2.12224. [DOI] [Google Scholar]
- Zhu Y.; He X.; Mo Y. Origin of Outstanding Stability in the Lithium Solid Electrolyte Materials: Insights from Thermodynamic Analyses Based on First-Principles Calculations. ACS Appl. Mater. Interfaces 2015, 7 (42), 23685–23693. 10.1021/acsami.5b07517. [DOI] [PubMed] [Google Scholar]
- Thompson T.; Yu S.; Williams L.; Schmidt R. D.; Garcia-Mendez R.; Wolfenstine J.; Allen J. L.; Kioupakis E.; Siegel D. J.; Sakamoto J. Electrochemical Window of the Li-Ion Solid Electrolyte Li7La3Zr2O12. ACS Energy Lett. 2017, 2, 462–468. 10.1021/acsenergylett.6b00593. [DOI] [Google Scholar]
- Han F.; Zhu Y.; He X.; Mo Y.; Wang C. Electrochemical Stability of Li10GeP2S12 and Li7La3Zr2O12 Solid Electrolytes. Adv. Energy Mater. 2016, 6 (8), 1501590. 10.1002/aenm.201501590. [DOI] [Google Scholar]
- Abouali S.; Yim C. H.; Merati A.; Abu-Lebdeh Y.; Thangadurai V. Garnet-Based Solid-State Li Batteries: From Materials Design to Battery Architecture. ACS Energy Lett. 2021, 6, 1920–1941. 10.1021/acsenergylett.1c00401. [DOI] [Google Scholar]
- Cheng L.; Crumlin E. J.; Chen W.; Qiao R.; Hou H.; Franz Lux S.; Zorba V.; Russo R.; Kostecki R.; Liu Z.; Persson K.; Yang W.; Cabana J.; Richardson T.; Chen G.; Doeff M. The Origin of High Electrolyte-Electrode Interfacial Resistances in Lithium Cells Containing Garnet Type Solid Electrolytes. Phys. Chem. Chem. Phys. 2014, 16 (34), 18294–18300. 10.1039/C4CP02921F. [DOI] [PubMed] [Google Scholar]
- Sharafi A.; Yu S.; Naguib M.; Lee M.; Ma C.; Meyer H. M.; Nanda J.; Chi M.; Siegel D. J.; Sakamoto J. Impact of Air Exposure and Surface Chemistry on Li-Li7La3Zr2O12 Interfacial Resistance. J. Mater. Chem. A Mater. 2017, 5 (26), 13475–13487. 10.1039/C7TA03162A. [DOI] [Google Scholar]
- Hofstetter K.; Samson A. J.; Narayanan S.; Thangadurai V. Present Understanding of the Stability of Li-Stuffed Garnets with Moisture, Carbon Dioxide, and Metallic Lithium. J. Power Sources 2018, 390, 297–312. 10.1016/j.jpowsour.2018.04.016. [DOI] [Google Scholar]
- Wang C.; Fu K.; Kammampata S. P.; McOwen D. W.; Samson A. J.; Zhang L.; Hitz G. T.; Nolan A. M.; Wachsman E. D.; Mo Y.; Thangadurai V.; Hu L. Garnet-Type Solid-State Electrolytes: Materials, Interfaces, and Batteries. Chem. Rev. 2020, 120 (10), 4257–4300. 10.1021/acs.chemrev.9b00427. [DOI] [PubMed] [Google Scholar]
- Huo H.; Luo J.; Thangadurai V.; Guo X.; Nan C. W.; Sun X. Li2CO3: A Critical Issue for Developing Solid Garnet Batteries. ACS Energy Lett. 2020, 5 (1), 252–262. 10.1021/acsenergylett.9b02401. [DOI] [Google Scholar]
- Galven C.; Suard E.; Mounier D.; Crosnier-Lopez M. P.; le Berre F. Structural Characterization of a New Acentric Protonated Garnet: Li6-xHxCaLa2Nb2O12. J. Mater. Res. 2013, 28 (16), 2147–2153. 10.1557/jmr.2013.209. [DOI] [Google Scholar]
- Orera A.; Larraz G.; Rodríguez-Velamazán J. A.; Campo J.; Sanjuán M. L. Influence of Li+ and H+ Distribution on the Crystal Structure of Li7-xHxLa3Zr2O12 (0 ≤ x ≤ 5) Garnets. Inorg. Chem. 2016, 55 (3), 1324–1332. 10.1021/acs.inorgchem.5b02708. [DOI] [PubMed] [Google Scholar]
- Redhammer G. J.; Badami P.; Meven M.; Ganschow S.; Berendts S.; Tippelt G.; Rettenwander D. Wet-Environment-Induced Structural Alterations in Single- And Polycrystalline LLZTO Solid Electrolytes Studied by Diffraction Techniques. ACS Appl. Mater. Interfaces 2021, 13 (1), 350–359. 10.1021/acsami.0c16016. [DOI] [PubMed] [Google Scholar]
- Redhammer G. J.; Tippelt G.; Portenkirchner A.; Rettenwander D. Aging Behavior of Al- and Ga- Stabilized Li7La3Zr2O12 Garnet-Type, Solid-State Electrolyte Based on Powder and Single Crystal X-Ray Diffraction. Crystals (Basel) 2021, 11 (7), 721. 10.3390/cryst11070721. [DOI] [Google Scholar]
- Redhammer G. J.; Tippelt G.; Rettenwander D. Deep Hydration of an Li7–3xLa3Zr2MIIIxO12 Solid-State Electrolyte Material: A Case Study on Al- and Ga-Stabilized LLZO. Acta Crystallogr. C Struct Chem. 2022, 78 (1), 1–6. 10.1107/S2053229621012250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharafi A.; Kazyak E.; Davis A. L.; Yu S.; Thompson T.; Siegel D. J.; Dasgupta N. P.; Sakamoto J. Surface Chemistry Mechanism of Ultra-Low Interfacial Resistance in the Solid-State Electrolyte Li7La3Zr2O12. Chem. Mater. 2017, 29 (18), 7961–7968. 10.1021/acs.chemmater.7b03002. [DOI] [Google Scholar]
- Zheng H.; Wu S.; Tian R.; Xu Z.; Zhu H.; Duan H.; Liu H. Intrinsic Lithiophilicity of Li–Garnet Electrolytes Enabling High-Rate Lithium Cycling. Adv. Funct Mater. 2020, 30 (6), 1906189. 10.1002/adfm.201906189. [DOI] [Google Scholar]
- Huo H.; Luo J.; Thangadurai V.; Guo X.; Nan C. W.; Sun X. Li2CO3: A Critical Issue for Developing Solid Garnet Batteries. ACS Energy Lett. 2020, 5 (1), 252–262. 10.1021/acsenergylett.9b02401. [DOI] [Google Scholar]
- Cheng L.; Liu M.; Mehta A.; Xin H.; Lin F.; Persson K.; Chen G.; Crumlin E. J.; Doeff M. Garnet Electrolyte Surface Degradation and Recovery. ACS Appl. Energy Mater. 2018, 1 (12), 7244–7252. 10.1021/acsaem.8b01723. [DOI] [Google Scholar]
- Zhu Y.; Connell J. G.; Tepavcevic S.; Zapol P.; Garcia-Mendez R.; Taylor N. J.; Sakamoto J.; Ingram B. J.; Curtiss L. A.; Freeland J. W.; Fong D. D.; Markovic N. M. Dopant-Dependent Stability of Garnet Solid Electrolyte Interfaces with Lithium Metal. Adv. Energy Mater. 2019, 9 (12), 1803440. 10.1002/aenm.201803440. [DOI] [Google Scholar]
- Brugge R. H.; Pesci F. M.; Cavallaro A.; Sole C.; Isaacs M. A.; Kerherve G.; Weatherup R. S.; Aguadero A. The Origin of Chemical Inhomogeneity in Garnet Electrolytes and Its Impact on the Electrochemical Performance. J. Mater. Chem. A Mater. 2020, 8 (28), 14265–14276. 10.1039/D0TA04974C. [DOI] [Google Scholar]
- McConohy G.; Xu X.; Cui T.; Barks E.; Wang S.; Kaeli E.; Melamed C.; Gu X. W.; Chueh W. C. Mechanical Regulation of Lithium Intrusion Probability in Garnet Solid Electrolytes. Nat. Energy 2023, 8 (3), 241–250. 10.1038/s41560-022-01186-4. [DOI] [Google Scholar]
- Grissa R.; Payandeh S.; Heinz M.; Battaglia C. Impact of Protonation on the Electrochemical Performance of Li7La3Zr2O12 Garnets. ACS Appl. Mater. Interfaces 2021, 13 (12), 14700–14709. 10.1021/acsami.0c23144. [DOI] [PubMed] [Google Scholar]
- Cai J.; Polzin B.; Fan L.; Yin L.; Liang Y.; Li X.; Liu Q.; Trask S. E.; Liu Y.; Ren Y.; Meng X.; Chen Z. Stoichiometric Irreversibility of Aged Garnet Electrolytes. Mater. Today Energy 2021, 20, 100669. 10.1016/j.mtener.2021.100669. [DOI] [Google Scholar]
- Xia W.; Xu B.; Duan H.; Tang X.; Guo Y.; Kang H.; Li H.; Liu H. Reaction Mechanisms of Lithium Garnet Pellets in Ambient Air: The Effect of Humidity and CO2. J. Am. Ceram. Soc. 2017, 100 (7), 2832–2839. 10.1111/jace.14865. [DOI] [Google Scholar]
- Wang Y.; Lai W. Phase Transition in Lithium Garnet Oxide Ionic Conductors Li7La3Zr2O12: The Role of Ta Substitution and H2O/CO2 Exposure. J. Power Sources 2015, 275, 612–620. 10.1016/j.jpowsour.2014.11.062. [DOI] [Google Scholar]
- Dunstan M. T.; Griffin J. M.; Blanc F.; Leskes M.; Grey C. P. Ion Dynamics in Li2CO3 Studied by Solid-State NMR and First-Principles Calculations. J. Phys. Chem. C 2015, 119 (43), 24255–24264. 10.1021/acs.jpcc.5b06647. [DOI] [Google Scholar]
- Tetenbaum M.; Johnson C. E. Vaporization Behavior of Lithium Oxide: Effect of Water Vapor in Helium Carrier Gas. J. Nucl. Mater. 1984, 120 (2–3), 213–216. 10.1016/0022-3115(84)90058-8. [DOI] [Google Scholar]
- Shchukarev A.; Korolkov D. XPS Study of Group IA Carbonates. Open Chem. 2004, 2 (2), 347–362. 10.2478/BF02475578. [DOI] [Google Scholar]
- Shao Y.; Wang H.; Gong Z.; Wang D.; Zheng B.; Zhu J.; Lu Y.; Hu Y. S.; Guo X.; Li H.; Huang X.; Yang Y.; Nan C. W.; Chen L. Drawing a Soft Interface: An Effective Interfacial Modification Strategy for Garnet-Type Solid-State Li Batteries. ACS Energy Lett. 2018, 3 (6), 1212–1218. 10.1021/acsenergylett.8b00453. [DOI] [Google Scholar]
- Trotochaud L.; Head A. R.; Pletincx S.; Karslloǧlu O.; Yu Y.; Waldner A.; Kyhl L.; Hauffman T.; Terryn H.; Eichhorn B.; Bluhm H. Water Adsorption and Dissociation on Polycrystalline Copper Oxides: Effects of Environmental Contamination and Experimental Protocol. J. Phys. Chem. B 2018, 122 (2), 1000–1008. 10.1021/acs.jpcb.7b10732. [DOI] [PubMed] [Google Scholar]
- Arinicheva Y.; Guo X.; Gerhards M. T.; Tietz F.; Fattakhova-Rohlfing D.; Finsterbusch M.; Navrotsky A.; Guillon O. Competing Effects in the Hydration Mechanism of a Garnet-Type Li7La3Zr2O12 Electrolyte. Chem. Mater. 2022, 34 (4), 1473–1480. 10.1021/acs.chemmater.1c02581. [DOI] [Google Scholar]
- Bakiz B.; Guinneton F.; Arab M.; Benlhachemi A.; Villain S.; Satre P.; Gavarri J.-R. Carbonatation and Decarbonatation Kinetics in the La2O3 -La2O2CO3 System under CO2 Gas Flows. Adv. Mater. Sci. Eng. 2010, 2010, 1–6. 10.1155/2010/360597. [DOI] [Google Scholar]
- Vardar G.; Bowman W. J.; Lu Q.; Wang J.; Chater R. J.; Aguadero A.; Seibert R.; Terry J.; Hunt A.; Waluyo I.; Fong D. D.; Jarry A.; Crumlin E. J.; Hellstrom S. L.; Chiang Y.-M.; Yildiz B. Structure, Chemistry, and Charge Transfer Resistance of the Interface between Li7 La3 Zr2 O12 Electrolyte and LiCoO2 Cathode. Chem. Mater. 2018, 30 (18), 6259–6276. 10.1021/acs.chemmater.8b01713. [DOI] [Google Scholar]
- Kim Y.; Kim D.; Bliem R.; Vardar G.; Waluyo I.; Hunt A.; Wright J. T.; Katsoudas J. P.; Yildiz B. Thermally Driven Interfacial Degradation between Li7La3Zr2O12 Electrolyte and LiNi0.6Mn0.2Co0.2O2 Cathode. Chem. Mater. 2020, 32 (22), 9531–9541. 10.1021/acs.chemmater.0c02261. [DOI] [Google Scholar]
- Kim Y.; Waluyo I.; Hunt A.; Yildiz B. Avoiding CO2 Improves Thermal Stability at the Interface of Li7La3Zr2O12 Electrolyte with Layered Oxide Cathodes. Adv. Energy Mater. 2022, 12 (13), 2102741. 10.1002/aenm.202102741. [DOI] [Google Scholar]
- Flatscher F.; Philipp M.; Ganschow S.; Wilkening H. M. R.; Rettenwander D. The Natural Critical Current Density Limit for Li7La3Zr2O12 garnets. J. Mater. Chem. A Mater. 2020, 8 (31), 15782–15788. 10.1039/C9TA14177D. [DOI] [Google Scholar]
- Lu Y.; Zhao C.; Yuan H.; Cheng X.; Huang J.; Zhang Q. Critical Current Density in Solid-State Lithium Metal Batteries: Mechanism, Influences, and Strategies. Adv. Funct Mater. 2021, 31 (18), 2009925. 10.1002/adfm.202009925. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.