

Abstract

Leishmaniasis is a collection of diseases caused by more than 20 Leishmania parasite species that manifest as either visceral, cutaneous, or mucocutaneous leishmaniasis. Despite the significant mortality and morbidity associated with leishmaniasis, it remains a neglected tropical disease. Existing treatments have variable efficacy, significant toxicity, rising resistance, and limited oral bioavailability, which necessitates the development of novel and affordable therapeutics. Here, we report on the continued optimization of a series of imidazopyridines for visceral leishmaniasis and a scaffold hop to a series of substituted 2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazoles with improved absorption, distribution, metabolism, and elimination properties.
Keywords: leishmaniasis, neglected tropical diseases, structure−property optimization
Neglected tropical diseases (NTDs) are a collection of 20 infectious diseases identified by the World Health Organization (WHO), for which over 1 billion people worldwide received chemotherapy between 2015 and 2019.1 These communicable diseases are often found in tropical and subtropical regions of the world. The most vulnerable populations are those living in poverty, often with inadequate means of sanitation in densely populated regions proximal to disease vectors.2 These diseases are designated as NTDs due to the lack of dedicated funding by the for-profit sector for the discovery and development of new drugs, further diluting the limited financial resources available.
Leishmaniasis is caused by a variety of Leishmania species and subspecies and is the third most common3 parasitic disease worldwide. The disease commonly presents in three forms, visceral (VL), which is the most serious and often fatal if left untreated, cutaneous which is the most common and causes disfiguring skin lesions, and mucocutaneous, which destroys mucous membranes. Existing treatments for VL have variable efficacy and significant toxicity, and there are limited oral medications that exist (miltefosine),4 which necessitates the development of novel, affordable, and orally available drugs.
Although large compound libraries have been screened against Leishmania spp., the discovery of quality leads remains challenging. As a consequence, the modest hit selection criteria of pEC50 > 5.3 against intracellular L. donovani has been described.5 The Drugs for Neglected Diseases initiative (DNDi) developed a program that utilizes extensive proprietary chemical libraries of partner pharmaceutical companies in an iterative, noncompetitive environment. A hit identified in this effort, referred to as the “Booster Program”, was typified by the imidazo[1,2-a]pyridine (DNDI0003363576),6 which was progressed to a pharmacokinetic (PK) study to assess the translation based on a series of in vitro absorption, distribution, metabolism, and elimination (ADME) parameters and in vivo bioavailability. Insufficient exposure of the compound was observed to warrant advancement into an in vivo proof-of-concept infection study. We report the continued optimization of this hit, with a focus on improving oral bioavailability. To inform the optimization campaign and to ensure the future success of a pre-clinical candidate, more stringent target candidate properties (TCP) were identified (Table 1). The full suite of ADME was not obtained for DNDI0003363576; however, the series suffered from poor solubility, selectivity, and low metabolic stability. Furthermore, improvements that positively modified the structure–property relationships (SPR) detrimentally affected the structure–activity relationships (SAR).
Table 1. TCP Summary of Desired Potency, Toxicity, ADME, and Physicochemical Properties.
| Desired criteria | DNDI0003363576 | ||
|---|---|---|---|
| Potency | pEC50 Leish spp | >5.3 | 5.7 |
| Leish LLEa | ≥4 | 2.1 | |
| Toxicity | PMMb pCC50 | >10 | 4.2 |
| ADME and physicochemical properties | MW | ≤360 | 350 |
| cLogP | ≤3 | 3.6 | |
| LogD7.4 | ≤2 | 3.6 | |
| MLMc Clint | ≤60 | 62 | |
| revised ADME assessment criteria | |||
| RHd Clint | ≤27 | ||
| HLMe Clint | ≤47 | ||
| Aq. Sol.f | >10 | ||
| PK exposure | free plasma concentration > EC90 | ||
Ligand lipophilicity efficiency pEC50 – LogD7.4.
Primary mouse macrophage.
Mouse liver microsomes μL/min/mg protein.
Rat hepatocyte μL/min/106 cells; 5.1 ≤ moderate ≤27.
Human liver microsomes μL/min/mg protein; 8.6 ≤ moderate ≤47.
Thermodynamic aqueous solubility μM.
Chemistry
Synthesis of the hetericyclic core was achieved via a three-step synthesis represented in Scheme 1. Amines 1a–e were converted to the corresponding isocyanides 2a–e using the Hoffmann isocyanide synthesis,7 followed by the Groebke–Blackburn–Bienaymé three component reaction,8 which provided rapid access to the desired imidazo[1,2-a]-heterocycles 3a–ai.8N-alkyl derivatives 4a–n were synthesized using the corresponding alkyl halide.
Scheme 1. Synthesis of Diarylimidazole Derivatives 3a–ai and 4a–n.

Reagents and reaction conditions: (i) NaOH (50% v/v), TEAC, CH3Cl, CH2Cl2, 40 °C, 3–8 h; (ii) appropriate pyridin-2-amine or 2-aminothiazole, aldehyde, Yb(OTf)3, 120 °C, microwave, 40 min; (iii) alkyl halide, Cs2CO3, DMF, 55 °C, 24 h; (iv) alkyl halide, pyridine, CH2Cl2, room temperature, 17 h.
There were some examples where the isocyanide synthesis was not achievable due to instability of the reactants (Scheme 2). The synthesis could be achieved starting from differently substituted pyridine amines which were converted to the corresponding derivatives 6a,b. The removal of the tert-butyl group with HBr provided amines 7a,b, followed by Buchwald–Hartwig coupling to achieve the desired imidazo[1,2-a]-heterocycle 8.9 Alternatively, bromination of ketone 9 provided 10 as a HBr salt which was cyclized with the corresponding 2-aminoaryl to provide 11a-b. Treatment of 11b with NCS provided 12; the Vilsmeier–Haack reaction provided aldehyde 13, and subsequent reductive amination in the presence of NaCNBH3 provided 14.
Scheme 2. Synthesis of Imidazo[1,2-a] Heterocycles 8, 11a,b, 12, and 14.
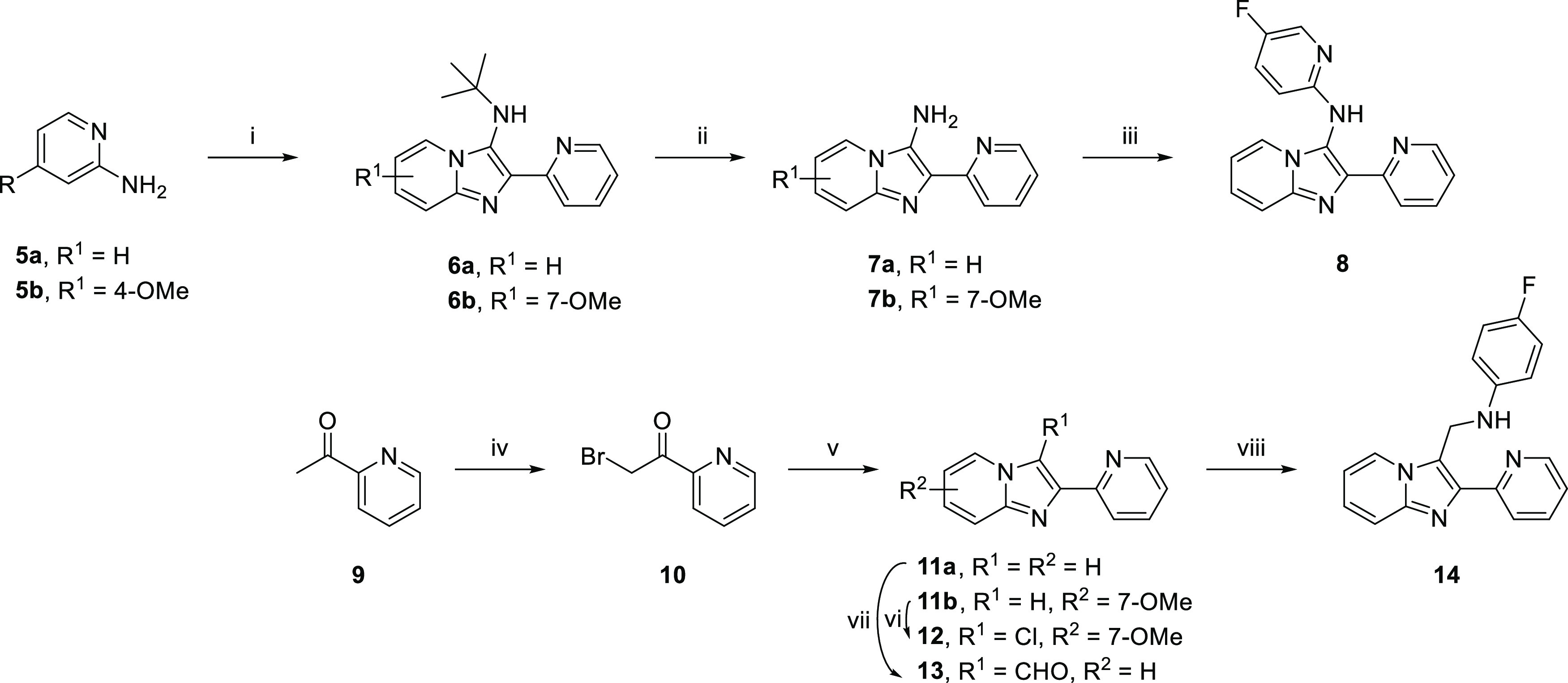
Reagents and reaction conditions: (i) pyridine-2-carboxaldehyde, tert-butyl isocyanide, Yb(OTf)3, 120 °C, microwave, 40 min; (ii) HBr, H2O, 110 °C, 4 h; (iii) 2-bromo-5-fluoropyridine, NaOtBu, XPhos Pd G2, t-BuOH, 75 °C, 2 h; (iv) HBr, Br2, room temperature, overnight; (v) 2-aminoaryl, Na2CO3, DMF, 85 °C, 18 h; (vi) NCS, ACN, room temperature, 1 h; (vii) POCl3, 1,2-DCA, DMF, 70 °C, 2 h; (viii) NaCNBH3, MeOH, AcOH, room temperature, 1 h.
An alternative scaffold could be synthesized with condensation of dihydropyrrolamine and α-bromoketone 10 providing intermediate 15a. Subsequent bromination with NBS afforded 15b. A final Suzuki coupling provides the desired substituted 6,7-dihydro-5H-pyrrolo[1,2-a]imidazoles 16a–ad (Scheme 3).
Scheme 3. Synthesis of Substituted 6,7-Dihydro-5H-pyrrolo[1,2-a]imidazoles 16a–ad.

Reagents and reaction conditions: (i) dihydropyrrolamine, Na2CO3, DMF, 50 °C, overnight; (ii) NBS, CH2Cl2, room temperature, 1 h; (iii) appropriate aryl boronic acid or pinacol boronic ester, Pd(PPh3)4, toluene/MeOH (3:1 v/v, 0.2 M), Na2CO3 (aq. 2 M), 120 °C 18 h or microwave, 40 min.
Results and Discussion
To better understand the SPR and the pharmacophore required for potent activity, we synthesized a series of truncated analogues (Table 2, 7b, 11a–b, 12). The introduction of a 7-methoxy group (11a vs 11b) resulted in a modest boost in parasite selectivity versus primary mouse macrophages (PMM), although this was at the expense of microsomal stability, potentially due to o-demethylation. Introduction of the amine in the 3-position (7b vs 11b) was tolerated with a significant increase in the lipophilic ligand efficiency (LLE) and provided a handle for future modifications. However, the aqueous solubility of these analogues was reduced, suggesting that hydrogen bonding with the adjacent pyridyl may increase the planarity of the series. Compounds without 3-arylamine had rapid human microsomal and low rat hepatocyte intrinsic clearance, necessitating the re-introduction of the arylamine functionality.10
Table 2. Biological Activity and Physicochemical Properties of Truncated Analoguesa.
| ID | R | 7 | L.inf pEC50 | PMM pCC50 | SI | aq. sol.b | HLM clintc | RH clintd | LLEe |
|---|---|---|---|---|---|---|---|---|---|
| 7b | NH2 | OMe | 4.5 | 4.2 | 2 | 290 | 120 | 7.1 | 4.1 |
| 11a | H | H | 4.3 | 4.2 | 1 | 828 | 130 | 140 | 2.6 |
| 11b | H | OMe | 4.6 | 4.2 | 3 | 723 | >300 | 53 | 2.4 |
| 12 | Cl | OMe | 4.8 | 4.5 | 2 | 562 | >300 | 17 | 2.2 |
nt, not tested.
Thermodynamic aqueous solubility μM.
μL/min/mg protein.
μL/min/106 cells.
LLE = pEC50 – LogD7.4.
We first synthesized the unadorned imidazopyridine 3a to act as a reference point to the exploration of the core. This compound had a reasonable selectivity index [SI, expressed as the ratio CC50 μM (cell line)/EC50 μM(L.inf), rounded to the nearest integer] (16-fold), although aqueous solubility was poor and metabolism was high. We hypothesized that there was a correlation between selectivity and the electronics and, to understand this, a fluorine and methoxy scan was conducted (Table 3, 3b, 3c, 3e, 3f, 3h, 3i, 3k, 3l). Fluorine afforded minimal preference to substitution, with no position affording SI greater than ten-fold. Conversely, a strong correlation with SI was observed for the methoxy substitutions, with the best SI observed for the 6-methoxy (3f) derivative and no selectivity for the 5- or 7-methoxy substitutions (3c and 3i). Given this, we sought to further explore the substituent at the 6-position (3n–q) of the imidazopyridine and explore alternate scaffolds that incorporated additional heteroatoms to modulate the ADME and physicochemical properties. The introduction of an additional nitrogen into any position (3d, 3g, 3j, 3m) or thiazole (3r) failed to improve the SI with only small gains in aqueous solubility.
Table 3. Biological Activity and Physicochemical Properties of the Imidazo[1,2-a]-Pyridine.
| ID | 5 | 6 | 7 | 8 | L.inf pEC50 | PMM pCC50 | SI | aq. sol.a | HLM clintb | RH clintc | LLEd |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3a | H | H | H | H | 5.4 | 4.2 | 16 | 8 | 137 | >300 | 2 |
| 3b | F | H | H | H | 5.1 | 4.2 | 8 | 26 | 60 | 190 | 1.6 |
| 3c | OMe | H | H | H | 5.1 | 5.1 | 1 | 62 | 28 | 30 | 0.6 |
| 3d | [N] | H | H | H | 4.8 | 4.5 | 2 | 68 | 212 | >300 | 3.2 |
| 3e | H | F | H | H | 5.1 | 4.3 | 6 | 25 | >300 | >300 | |
| 3f | H | OMe | H | H | 5.7 | 4.5 | 16 | ||||
| 3g | H | [N] | H | H | 4.3 | 4.2 | 1 | 64 | |||
| 3h | H | H | F | H | 4.6 | 4.2 | 3 | 0.1 | >300 | 146 | |
| 3i | H | H | OMe | H | 5.1 | 5.1 | 1 | ||||
| 3j | H | H | [N] | H | 4.2 | 4.2 | 1 | 61 | 202 | 64.2 | 2.7 |
| 3k | H | H | H | F | 4.4 | 4.2 | 2 | 12 | 220 | 101 | 1.4 |
| 3l | H | H | H | OMe | 5.1 | 4.2 | 8 | 2 | 143 | 29 | 1.7 |
| 3m | H | H | H | [N] | 4.3 | 4.2 | 1 | 180 | 42.4 | 64.8 | 1.7 |
| 3n | H | SMe | H | H | 5.1 | 5.1 | 1 | 0.5 | 247 | >300 | 1.8 |
| 3o | H | SO2Me | H | H | 4.5 | 4.5 | 1 | 5 | 92 | >300 | 2.1 |
| 3p | H | cyclopropyl | H | H | 5.2 | 5.1 | 1 | 0.7 | 91.6 | 148 | 1.1 |
| 3q | H | 4-isoxazole | H | H | 4.6 | 4.2 | 3 | 3 | 75 | 128 | 0.7 |
| 3r | 5.1 | 4.5 | 4 | 76 | 270 | >300 | 0.9 |
Thermodynamic aqueous solubility μM.
μL/min/mg protein.
μL/min/106 cells.
LLE = pEC50 – LogD7.4.
Attention was directed toward optimizing the 3-position substituent (Table 4). This region was previously explored and identified as having the potential to positively affect leishmanicidal activity.6 Small lipophilic alkyl groups demonstrated moderate antiparasitic activities as well as N-phenyl derivatives adorned with polar heteroatoms and halogens. When compared to 3a, difluoro and trifluoromethyl compounds (3s, 3t, and 3u) had reduced selectivity and only 3s demonstrated improved solubility. Compounds 3v and 8 had improved metabolic stability, although it was still classified as moderate in human liver microsomes for both compounds and high and moderate, respectively, in rat hepatocytes; however, this came at the expense of antiparasitic activity. The presence of a short methylene linker between the imidazopyridine core and aniline portion was found to be detrimental to antiparasitic potency (14).
Table 4. Biological Activity and Physicochemical Properties of the 3-Position Substituenta.

| ID | R | L.inf pEC50 | PMM pCC50 | SI | aq. sol.b | HLM clintc | RH clintd | LLEe |
|---|---|---|---|---|---|---|---|---|
| 3a | A | 5.4 | 4.2 | 16 | 8 | 140 | >300 | 2 |
| 3s | B | 5.3 | 4.5 | 6 | 32 | 64 | 131 | 1.9 |
| 3t | C | 5.1 | 4.5 | 4 | 0.8 | 96 | 194 | 3.9 |
| 3u | D | 5.2 | 4.8 | 3 | 2 | 59 | >300 | 1.9 |
| 3v | E | 4.8 | 4.5 | 2 | 8 | 25 | 88 | 2.9 |
| 8 | F | 4.2 | 4.2 | 1 | 220 | 44 | 18 | 1.6 |
| 14 | G | 4.9 | 4.3 | 4 | nd | nd | nd | NA |
NA, not applicable; nd, not determined.
Thermodynamic aqueous solubility μM.
μL/min/mg protein.
μL/min/106 cells.
LLE = pEC50 – LogD7.4.
Due to the lipophilic nature of the binding sites of metabolizing enzymes, reduction in lipophilicity has been demonstrated to be an effective method to improve metabolic stability.11−13 Hence, we next explored modifications to the 2-pyridyl moiety utilizing an unadorned core to allow an improved understanding of the SPR (Table 5). We replaced the pyridyl with more polar nitrogen-containing heterocycles, and while 3y exhibited the highest LLE to date, the pyrazole detrimental to selectivity. Improved solubility and metabolic stability were obtained with several other compounds, although this was at the expense of selectivity. X-ray crystallography of 3a and 3ac indicated that the improved aqueous solubility of the ortho-methoxy-substituted compound likely resulted from increased dihedral rotation to reduce steric interaction, reducing the planarity of the compound (Table S1).
Table 5. Biological Activity and Physicochemical Properties of the 2-Position Substituenta.

| ID | R | L.inf pEC50 | PMM pCC50 | SI | aq. sol.b | HLM clintc | RH clintd | logD7.4 | LLEe |
|---|---|---|---|---|---|---|---|---|---|
| 3a | D | 5.4 | 4.2 | 16 | 8 | 140 | >300 | 3.4 | 2 |
| 3w | A | 4.5 | 4.2 | 2 | 190 | 64 | 290 | 1.6 | 2.9 |
| 3x | B | 5.1 | 4.8 | 2 | 4 | 17 | 14 | nd | NA |
| 3y | C | 4.2 | 4.2 | 1 | 410 | 45 | 51 | –0.1 | 4.3 |
| 3z | E | 4.2 | 4.2 | 1 | 4 | 137 | >300 | 4.5 | –0.3 |
| 3aa | F | 5.3 | 4.7 | 4 | 3 | 35 | 88 | 3.8 | 1.5 |
| 3ab | G | 5.3 | 4.8 | 3 | 0.2 | 120 | 190 | 4.0 | 1.3 |
| 3ac | H | 4.5 | 4.2 | 2 | 690 | 19 | 29 | 1.7 | 2.8 |
| 3ad | I | 4.8 | 4.2 | 4 | 13 | 130 | >300 | 4.5 | 0.3 |
| 3ae | J | 4.9 | 4.2 | 5 | 2 | 89 | >300 | 3.9 | 1 |
| 3af | K | 5.0 | 4.8 | 2 | 2 | 160 | >300 | nd | NA |
| 3ag | L | 4.7 | 4.2 | 3 | 49 | 86 | 100 | 3.5 | 1.2 |
NA, not applicable; nd, not determined.
Thermodynamic aqueous solubility μM.
μL/min/mg protein.
μL/min/106 cells.
LLE = pEC50 – LogD7.4.
The final area of exploration was alkylation of the exocyclic amine (Table 6). Improvement to the solubility was observed for the N-methyl derivative (4a) when compared to the N-H derivative (3a), presumably via interruption of the planarity reinforced by hydrogen bonding. However, this was accompanied by a reduction in selectivity. This selectivity could be restored by replacing the methyl with the deuterium-labeled isostere (4b) with no loss of potency. Incorporation of deuterium isosteres has been shown to positively modulate pharmacokinetics due to the stronger deuterium-carbon bond;14 however, 4b failed to improve metabolic stability. Increasing the alkyl-chain length resulted in a lipophilicity-driven reduction in aqueous solubility and selectivity (4a vs 4c vs 4d). In general, the improved antiparasitic activity of the N-alkyl compounds was coupled with erosion of metabolic stability.
Table 6. Biological Activity and Physicochemical Properties of N-Alkyl Substituents.


Thermodynamic aqueous solubility μM.
μL/min/mg protein.
μL/min/106 cells.
LLE = pEC50 – LogD7.4.
The synthesis of compounds that encompassed the optimal modifications from each of the explored regions was done with the goal of achieving additive improvements to SAR and SPR (Table 7). Re-introduction of the 6-methoxy group into the core with methoxy-substituted pyridyl 3ah and 3ai improved the SI relative to their unadorned analogues (Table 4, 3aa and 3ab). The introduction of the ionizable N-methyl-linked substituted pyridyl (4m) improved aqueous solubility with comparable SI to 3ah. N-methylated derivatives 4k and 4l did provide some SI improvement, although metabolism was still high.
Table 7. Hybrid Analogues That Combine the Most Promising Modifications for L. infantum Activity and Physicochemical Property Modulation.

| ID | R1 | R2 | R3 | L.inf pEC50 | PMM pCC50 | SI | aq. sol.a | HLM clintb | RH clintc | logD7.4 | LLEd |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3ah | OMe | H | A | 5.9 | 5.1 | 6 | 0.4 | 240 | >300 | 5.2 | 0.7 |
| 3ai | OMe | H | B | 5.6 | 4.8 | 6 | 1 | 44 | 76 | 4.8 | 0.8 |
| 4m | OMe | Me | B | 6.1 | 5.4 | 5 | 27 | 260 | >300 | 3.7 | 2.4 |
| 4k | H | Me | A | 6.3 | 4.5 | 63 | 26 | 210 | >300 | 3.4 | 2.9 |
| 4l | H | Me | B | 6.3 | 5.1 | 16 | 10 | 93 | >300 | 3.7 | 2.6 |
| 4a | H | Me | C | 6.0 | 5.4 | 4 | 120 | 110 | >300 | 3.7 | 2.3 |
| 4n | OMe | Me | C | 6.1 | 5.1 | 10 | 17 | 250 | >300 | 2.9 | 3.2 |
Thermodynamic aqueous solubility μM.
μL/min/mg protein.
μL/min/106 cells.
LLE = pEC50 – LogD7.4.
Acknowledging that the presence of basic nitrogens and high lipophilicity can correlate to potential liabilities,15,16 we sought to get a better understanding of the limitations of this series. Representative compounds of the series were selected for further in vitro biological screening. We investigated blockage of the potassium ion channel coded by the human ether-a-go-go related gene (hERG) which has been associated with long QT syndrome and has led to the withdrawal of several marketed drugs, such as sertindole.17 The series exhibits micromolar activity against hERG (Table 8); however, the SAR shows that strategies to mitigate this activity, such as reduction of lipophilicity and modulation of basic nitrogen pKa via steric and electronic alteration, are tolerated, suggesting that this liability may be mitigated. Inhibition of cytochrome P450 (CYP450) enzymes blocks the metabolic activity of the enzyme which can result in clinically significant drug–drug interactions that can cause adverse reactions or therapeutic failures. The most clinically significant CYP450 isoforms are CYP3A4 and CYP2D6 as they account for 23.6 and 13.8% of known CYP450-drug interactions, respectively.18 Only moderate inhibition of these CYPs was observed for 3p (Table 8), with strong inhibition for 3ak (Table S6), indicating that structural modifications of the series can impact this liability.
Table 8. Further Biological Profiling of Compound 3p.
| L. inf pEC50 | 5.2 |
| PMM pCC50 | 5.1 |
| hERG pIC50a | 4.9 |
| MDCK Papp A-Bb | 16.7 × 10–6 cm/s |
| kinetic aq. sol. | 3 μM |
| CYP isoform | inhibition |
| CYP1A2 | 0.59 μM, strong inhibitor |
| CYP2C19 | 1.8 μM, moderate inhibitor |
| CYP2C8 | 4.9 μM, moderate inhibitor |
| CYP2C9 | 0.76 μM, strong inhibitor |
| CYP2D6 | 1.5 μM, moderate inhibitor |
| CYP3A4 | 1.6 μM, moderate inhibitor |
Human ether-a-go-go.
Madin-Darby canine kidney Apparent permeability.
We assessed the bioavailability of the series by progressing 4n (Table 7) into PK studies, as it had the best balance of potency (L. infantum pEC50: 6.1), SI (10-fold), and LLE (3.2) for the series, with moderate aqueous solubility (17 μM) (Figure 1). However, this compound did not possess the desired metabolic stability outlined in the TCP (Table 1) and it was hypothesized that co-dosing with the pan-CYP450 inhibitor, 1-aminobenzotriazole (ABT), would increase exposure. Two studies were run in parallel: the first was 4n only, and the second included a 50 mg/kg dose of ABT 2 h prior to dosing with 4n. In both studies, 4n was dosed at 50 mg/kg P.O. in fasted male CD1 mice. When accounting for the low (2%) free plasma concentration, the criterion for progression into the in vivo proof-of-concept efficacy study was not met (criteria: free plasma concentration > EC90) in either study.
Figure 1.
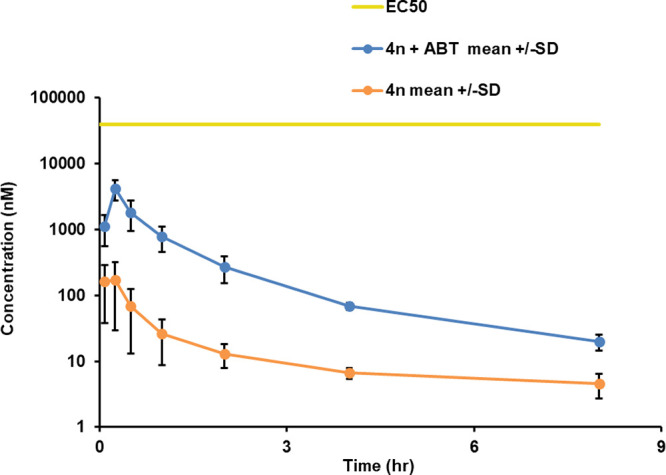
Mean plasma levels of 4n in fasted male CD1 mice, with and without ABT. 4n was dosed at 50 mg/kg P.O. (formulation: 5% DMSO, 45% PEG400, 50% MilliQ water; pH 4.0–5.0), and ABT was dosed 2 h before compound (P.O., in saline).
At this point, we connected with the Open Science Antibiotics (OSA) consortium who were working on a related chemotype that came from the Published Kinase Inhibitor Set19 (SB-400868) for Methicillin-resistantStaphylococcus aureus (MRSA). Compound SB-400868 was originally generated in an effort targeting transforming growth factor beta receptor I (TGFBR1/ALK5)20 and identified at University of North Carolina, Chapel Hill, as a hit against Mycobacterium tuberculosis. Preliminary optimization leading to compounds with activity against MRSA was carried out to yield OSA-822 (Figure 2).
Figure 2.

Origin of the OSA series from SB-400868.
The LogD was generally improved for this series, and we tested compounds for the in vitro L. infantum potency, host cell toxicity, and ADME profile. From this set, we identified several compounds that met or exceeded the aqueous solubility threshold as well as demonstrated improved metabolic stability. Substituent exploration of the northern aromatic region revealed that introduction of a pyridyl nitrogen (Table 9, 16h,i and 16k,l) significantly improved the ADME profile, with millimolar solubility and clearance in the medium to low range for both HLM and RH, although this was also accompanied by abrogation of potency. The para substituent could also be modified to improve the ADME profile, although this was similarly accompanied by loss of potency (cf. 16u–16w). The best compounds from this series, when considering potency, selectivity, and ADME, were 16f, 16g, and 16t. Nevertheless, the narrow therapeutic window of 16f and 16t, and the high clearance of 16g advocated against advancing these compounds further.
Table 9. Derivatives of SB-400868 and their Activity against L. infantum and Associated ADME Dataa.
| ID | R | L.inf pEC50 | PMM pCC50 | SI | aq. sol.b | HLM clintc | RH clintd | logD7.4 | LLEe |
|---|---|---|---|---|---|---|---|---|---|
| 16a | 5.7 | 5.1 | 4 | 180 | 64 | 26 | 2.2 | 3.5 | |
| 16b | 2-Me | 6.0 | 5.7 | 2 | nt | nt | nt | nt | NA |
| 16c | 3,4-Cl | 6.2 | 5.1 | 13 | 3 | 100 | 38 | 3.4 | 2.8 |
| 16d | 3,4-OMe | 5.7 | 4.5 | 16 | nt | nt | nt | nt | NA |
| 16e | 3,5-OMe | 5.9 | 5.1 | 6 | 97 | 53 | >300 | 2.4 | 3.5 |
| 16f | 3-Me | 6.2 | 5.1 | 13 | 560 | 76 | 63 | 2.8 | 2.4 |
| 16g | 3-Cl | 6.1 | 4.6 | 32 | 51 | 140 | 51 | 3.0 | 3.1 |
| 16h | 3-[N] | 4.2 | 4.2 | 1 | >1000 | 24 | 2.4 | 1.1 | 3.1 |
| 16i | 3-[N], 4-CN | 4.2 | 4.2 | 1 | nt | nt | nt | nt | NA |
| 16j | 3-CN | 5.0 | 4.2 | 6 | 37 | 44 | 23 | 2.0 | 3.0 |
| 16k | 4-[N] | 4.2 | 4.2 | 1 | >1000 | 26 | 4.8 | 1.2 | 3.0 |
| 16l | 3-F, 4[N] | 4.6 | 4.5 | 1 | nt | nt | nt | nt | NA |
| 16m | 4-CN | 4.5 | 4.2 | 2 | 38 | 99 | 8.0 | 1.9 | 2.6 |
| 16n | 3-OMe, 4-CN | 4.6 | 4.2 | 3 | nt | nt | nt | nt | NA |
| 16o | 3-CF3, 4-CN | 4.6 | 4.2 | 3 | nt | nt | nt | nt | NA |
| 16p | 4-Cl | 5.8 | 5.1 | 5 | 4 | 170 | 22 | 3.0 | 2.8 |
| 16q | 4-F | 5.5 | 4.5 | 10 | nt | nt | nt | nt | NA |
| 16r | 4-Me | 6.3 | 5.7 | 4 | 32 | 82 | 49 | 2.7 | 3.6 |
| 16s | 4-CF3 | 5.7 | 4.5 | 16 | nt | nt | nt | nt | NA |
| 16t | 4-OCF3 | 6.2 | 5.1 | 13 | 38 | 62 | 54 | 2.9 | 3.3 |
| 16u | 4-SOMe | 4.7 | 4.2 | 3 | 760 | <3 | <1 | 0.9 | 3.8 |
| 16v | 4-SO2Me | 4.2 | 4.2 | 1 | 320 | <3 | <1 | 1.2 | 3.0 |
| 16w | 4-CON(Me)2 | 4.4 | 4.2 | 2 | 780 | <3 | <1 | 1.1 | 3.3 |
| 16x | 4-COMe | 5.2 | 4.5 | 5 | nt | nt | nt | nt | NA |
| 16y | 4-NH2 | 5.6 | 5.1 | 3 | nt | nt | nt | nt | NA |
| 16z | 5.5 | 4.3 | 16 | 10 | 34 | 49 | 2.6 | 2.9 | |
| 16aa | 5.4 | 4.5 | 8 | nt | nt | nt | nt | NA | |
| 16ab | 6.5 | 5.7 | 6 | 3 | 43 | 150 | 4.1 | 2.4 | |
| 16ac | 6.5 | 5.7 | 6 | 3 | 62 | >300 | 2.9 | 3.6 | |
| 16ad | 5.9 | 5.1 | 6 | nt | nt | nt | nt | NA |
NA, not applicable; nt, not tested.
Thermodynamic aqueous solubility μM.
μL/min/mg protein.
μL/min/106 cells.
LLE = pEC50 – LogD7.4.
While we were unable to achieve our original goal of increasing in vivo exposure for the imidazopyridines, we did identify a modification that led to a boost in potency (N-alkylation), and we were able to modulate the aqueous solubility by lowering LogD7.4. We serendipitously uncovered a novel scaffold for Leishmania, the 6,7-dihydro-5H-pyrrolo[1,2-a]imidazoles, with superior LogD7.4 and improved aqueous solubility and metabolic stability. While 16g is currently untested in an in vivo PK study to determine its exposure, this latter series serves as a potential starting point for continued optimization as a therapeutic for leishmaniasis, given the dearth of leads for this NTD.
Methods
General Procedures for Biological Evaluation
Test compounds were formulated in 100% dimethyl sulfoxide (DMSO) at 20 mM and were 4-fold serially diluted to obtain 10 concentrations. Following an intermediate dilution in MilliQ water, the highest in-test concentration of 64 μM was obtained while assuring a DMSO concentration of <1%. Compounds were evaluated against intramacrophage amastigotes. Briefly, L. infantum (MHOM/MA(BE)/67/ITMAP263) amastigotes were derived from the spleens of heavily infected Syrian golden hamsters (Mesocricetus auratus) and used to infect primary peritoneal mouse macrophages (PMM) of Swiss mice. PMM (3 × 104) were infected with 4.5 × 105 parasites/well in 190 μL. Compound dilutions (10 μL) were added after 2 h of infection. Cells were maintained at 37 °C and 5% CO2 atmosphere in RPMI-1640 medium, supplemented with 2 mM l-glutamine, 16.5 mM NaHCO3, and 5% inactivated fetal calf serum. After 5 days of incubation, parasite burdens (mean number of amastigotes/PMM) were determined microscopically after staining with a 10% Giemsa solution. Percentage (%) reduction in growth compared to untreated controls was used to calculate pEC50 values. Cytotoxicity toward PMM was scored semi-quantitatively by observation of compound-induced cell detachment, lysis and granulation of at least 500 cells. The percentage reduction in cell viability compared to untreated controls was used to calculate pCC50 values.
Pharmacokinetic Study
The study was performed under the protocol approved by the Institutional Animal Ethical Committee (IAEC) at TCGLS as per guidelines of Committee for Purpose of Control and Supervision of Experiments on Animals (CPCSEA). IAEC protocol number: TCGLS/M-PK/21-20.
General Chemistry
Reagents purchased were used as received, unless otherwise noted. Purification of intermediates and final compounds was performed using silica gel or reverse phase chromatography using the Biotage Isolera, or Selekt flash purification systems. LCMS analysis was performed using either of the following:
-
a.
Waters Alliance reverse phase HPLC (columns Waters SunFire C18 4.6 × 50 mm, 3.5 μm, or Waters SunFire C8 4.6 × 50 mm, 3.5 μm) using a multiwavelength photodiode array detector from 210 to 600 nm and either a Waters Micromass ZQ detector (electrospray ionization) or Waters Micromass QDA detector.
-
b.
Waters Alliance reverse phase HPLC (2695; Xbridge C18 4.6 × 50 mm, 3.5 μm), using a multi-wavelength photodiode array detector from 210 to 600 nm and Waters Micromass QDA detector.
-
c.
Agilent 1260 Infinity HPLC (Agilent SB-C18 column (1.8 μm, 2.1 × 50 mm)), using a multiwavelength photodiode array detector monitoring 210, 230, 254, and 280 nm coupled to an Agilent 6120 single quad MS (ESI source).
All compounds tested had a purity of >95% as measured by LCMS, unless otherwise noted. 1H NMR spectra were obtained with Bruker NMR systems, operating at either 400 or 500 MHz at room temperature. Chemical shifts (δ, ppm) are reported relative to the solvent peak (CDCl3: 7.26 [1H]; DMSO-d6: 2.50 [1H]; or CD3OD: 3.31 [1H]). Data for 1H NMR spectra are reported as follows: chemical shift (ppm), multiplicity (s for singlet, d for doublet, t for triplet, dd for doublet of doublet, m for multiplet), coupling constant (Hz), and integration.
General Procedure a for the Synthesis of Isocyanides
To a vigorously stirred solution of a 50% NaOH in water (36 mL), TEAC (0.18 equiv) was added. The reaction mixture was efficiently stirred and heated at 40 °C; then, a solution of the selected amine (1 equiv) and CHCl3 (1.05 equiv) in CH2Cl2 (36 mL) was added dropwise over 50 min. The reaction was stirred and heated at 40 °C for 3–8 h. After completion, the reaction mixture was allowed to cool to room temperature and cold water (150 mL) was added. The aqueous phase was extracted with CH2Cl2. The organic layers were combined and washed with brine, dried over Na2SO4, and evaporated to give an oil which was then purified via a bulb-to-bulb distillation condensing with reaction dewar acetone dry ice (55 °C, 0.2 mbar).
General Procedure B for the Synthesis of Imidazopyridines
Amine (1 equiv), aldehyde (1.2 equiv), appropriate fluorophenyl isocyanide (1.2 equiv), and ytterbium(III) trifluoromethanesulfonate (0.01 equiv) were combined in a microwave vial and irradiated at 120 °C for 40 min. The reaction was cooled to room temperature, dissolved in a minimal amount of DMSO, and purified via a 30 g Biotage SH-C18 reverse phase column. The purified fractions were partially concentrated until a precipitate formed which was collected on a Hirsch funnel. The precipitate was then dried overnight with a high vacuum pump to provide the titled compound.
General Procedure C for N-Alkylation
To a solution of the corresponding imidazopyridine (1 equiv) in DMF (1–2 mL), cesium carbonate (2 equiv) and appropriate alkyl halide (1.2–1.5 equiv) were added. The resultant mixture was stirred at 55 °C for 24 h. After cooling to room temperature, the reaction mixture was partitioned between EtOAc and water. The organic layer was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue that was dissolved in a minimal amount of DMSO and purified via a 30 g Biotage SH-C18 reverse chromatography. The purified fractions were partially concentrated until a precipitate formed which was collected on a Hirsch funnel. The precipitate was then dried overnight with a high vacuum pump to provide the title compound.
General Procedure D for the Synthesis of 3-Aryl-2-(pyridyl)imidazoles
A reaction vial was charged with the corresponding 3-bromo-2-(pyridyl)imidazole (1 equiv), the appropriate aryl boronic acid or pinacol boronic ester (1.3 equiv), and Pd(PPh3)4 (0.12 equiv). The vial was sealed with a Teflon septum, evacuated, and backfilled with nitrogen (this sequence was carried out three times). Under an inert atmosphere, a mixture of toluene and methanol (3:1, v/v, 0.2 M) was added via a syringe, followed by the addition of 2 M aqueous Na2CO3 (4 equiv). The mixture was heated at 120 °C for 18 h or in the microwave at 120 °C for 30 min. After cooling to room temperature, the mixture was either diluted with CH2Cl2 and the organic layer was washed with brine, dried over Na2SO4, filtered, and concentrated under reduced pressure; or alternatively, the reaction mixture was diluted with MeOH, filtered through celite, and concentrated. The crude residue was purified by flash chromatography on silica and the product triturated to afford a fine powder.
General Procedure E for the Synthesis of 2-(Pyridyl)imidazoles
2-Bromo-1-(pyridyl)ethan-1-one hydrobromide (1 equiv), heterocycloalkenyl, or heteroaryl amine (1 equiv), and Na2CO3 (2 equiv) were stirred in EtOH (15–20 mL) at 85 °C for 1 h. After cooling to room temperature, the solvent was evaporated under reduced pressure and the crude reaction mixture was diluted with water and neutralized using 2 N HCl, then extracted twice with CH2Cl2. The organic layer was separated, dried over Na2SO4, filtered, and concentrated under reduced pressure to give a residue that was purified by flash chromatography on silica.
5-(Isoxazol-4-yl)pyridin-2-amine (1f)
A reaction vial was charged with 5-bromopyridin-2-amine (100 mg, 0.58 mmol), 4-isoxazoleboronic acid pinacol ester (135 mg, 0.69 mmol), potassium fluoride (101 mg, 1.73 mmol), and PdCl2dppf complex (47 mg, 0.58 mmol). The vial was sealed with a Teflon septum, evacuated, and backfilled with N2 three times. Under an inert atmosphere, a mixture of DMF and H2O (2:1, v/v) was added via a syringe and stirred at 50 °C for 4 h. After cooling to room temperature, the reaction mixture was filtered through celite, washed with MeOH, and concentrated under reduced pressure to give a residue that was purified by column chromatography (20/80 EtOAc:Hex) to afford a light brown solid (46 mg, 49%).
1-Fluoro-4-isocyanobenzene (2a)
Compound 2a was synthesized using 4-fluoroaniline (2.1 g, 18.90 mmol) according to General Procedure A. The crude product was purified by distillation to yield the title compound as a faint green oil (yield: 60%). 1H NMR (500 MHz, DMSO-d6) δ 7.68 (dd, J = 4.9, 8.8 Hz, 2H), 7.37 (t, J = 8.8 Hz, 2H).
1,3-Difluoro-5-isocyanobenzene (2b)
Compound 2b was synthesized using 1,3-difluoroaniline (1.4 g, 10.84 mmol) according to General Procedure A. The crude product was used as is without further purification.
2,4-Difluoro-1-isocyanobenzene (2c)
Compound 2c was synthesized using 3,5-difluoroaniline (100 mg, 0.79 mmol) according to General Procedure A. The crude product was used as is without further purification.
1-Isocyano-3-(trifluoromethyl)benzene (2d)
Compound 2d was synthesized using 3-trifluoromethylaniline (150 mg, 0.12 mmol) according to General Procedure A. The crude product was used as is without further purification.
4-Isocyanobenzo[d][1,3]dioxole (2e)
Compound 2e was synthesized using benzo[d][1,3]dioxol-4-amine (100 mg, 0.73 mmol) according to General Procedure A. The crude product was used as is without further purification.
N-(4-Fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3a)
Compound 3a was synthesized using 2-aminopyridine (400 mg, 4.25 mmol), picolinaldehyde (546 mg, 5.10 mmol), and 2a (882 mg, 5.10 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a beige solid (996 mg, 77%). LCMS [M + H]+ 305.3 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.57 (d, J = 4.5 Hz, 1H), 8.34 (s, 1H), 8.22 (d, J = 8.4 Hz, 1H), 7.79 (t, J = 7.9 Hz, 1H), 7.66 (d, J = 9.3 Hz, 1H), 7.60 (d, J = 6.8 Hz, 1H), 7.13–7.23 (m, 2H), 6.95 (t, J = 8.4 Hz, 2H), 6.78 (t, J = 6.8 Hz, 1H), 6.64 (dd, J = 4.5, 7.9 Hz, 2H).
5-Fluoro-N-(4-fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3b)
Compound 3b was synthesized using 6-fluoropyridin-2-amine (60 mg, 0.54 mmol), picolinaldehyde (69 mg, 0.64 mmol), and 2a (78 mg, 0.64 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a beige solid (116 mg, 67%). LCMS [M + H]+ 323.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.58 (d, J = 4.4 Hz, 1H), 8.14–8.27 (m, 2H), 7.74–7.84 (m, 1H), 7.43 (d, J = 9.3 Hz, 1H), 7.10–7.23 (m, 2H), 6.93 (t, J = 8.6 Hz, 2H), 6.69 (dd, J = 4.4, 8.6 Hz, 2H), 6.34 (t, J = 6.8 Hz, 1H).
N-(4-Fluorophenyl)-5-methoxy-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3c)
Compound 3c was synthesized using 6-methoxypyridin-2-amine (150 mg, 0.54 mmol), picolinaldehyde (155 mg, 1.45 mmol), and 2a (176 mg, 1.45 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a beige solid (309 mg, 76%). LCMS [M + H]+ 335.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.57 (d, J = 4.9 Hz, 1H), 8.35 (br. s., 1H), 8.22 (d, J = 7.3 Hz, 1H), 7.76 (t, J = 7.8 Hz, 1H), 7.27–7.31 (m, 1H), 7.10–7.20 (m, 2H), 6.90 (t, J = 8.5 Hz, 2H), 6.62–6.72 (m, 2H), 5.95 (d, J = 7.3 Hz, 1H), 3.63 (s, 3H).
N-(4-Fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-b]pyridazin-3-amine (3d)
Compound 3d was synthesized using 3-aminopyridazine (100 mg, 1.05 mmol), picolinaldehyde (135 mg, 1.26 mmol), and 2a (153 mg, 1.26 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a brown solid (158 mg, 50%). LCMS [M + H]+ 306.0 m/z.b 1H NMR (400 MHz, CDCl3) δ 8.82 (s, 1H), 8.61 (d, J = 4.7 Hz, 1H), 8.31 (d, J = 4.7 Hz, 1H), 8.24 (d, J = 8.2 Hz, 1H), 7.95 (d, J = 9.4 Hz, 1H), 7.81 (t, J = 8.2 Hz, 1H), 7.17–7.25 (m, 1H), 6.90–7.04 (m, 3H), 6.75 (dd, J = 4.7, 8.2 Hz, 2H).
6-Fluoro-N-(4-fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3e)
Compound 3e was synthesized using 5-fluoropyridin-2-amine (50 mg, 0.45 mmol), picolinaldehyde (96 mg, 0.89 mmol), and 2a (108 mg, 0.89 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (40–80% MeOH:H2O) to yield the title compound as a white solid (60 mg, 42%). LCMS [M + H]+ 323.1 m/z.a 1H NMR (500 MHz, CD3OD) δ 8.62–8.56 (m, 1H), 8.01 (dt, J = 8.0, 1.2 Hz, 1H), 7.88–7.85 (m, 1H), 7.83 (td, J = 7.6, 1.8 Hz, 1H), 7.66 (dd, J = 9.9, 4.8 Hz, 1H), 7.35 (ddd, J = 10.1, 8.2, 2.4 Hz, 1H), 7.28 (ddd, J = 7.6, 4.8, 1.2 Hz, 1H), 6.96–6.87 (m, 2H), 6.60–6.52 (m, 2H).
N-(4-Fluorophenyl)-6-methoxy-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3f)
Compound has been synthesized as previously reported in the literature.6
N-(4-Fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-c]pyrimidin-3-amine (3g)
Compound 3g was synthesized using 4-aminopyrimidine (200 mg, 2.10 mmol), picolinaldehyde (270 mg, 2.52 mmol), and 2a (270 mg, 2.52 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as an orange solid (95 mg, 18%). LCMS [M + H]+ 306.2 m/z.a 1H NMR (500 MHz, CDCl3) δ 9.76 (s, 1H), 9.55 (d, J = 1.0 Hz, 1H), 8.72 (d, J = 4.4 Hz, 1H), 8.00 (d, J = 5.9 Hz, 1H), 7.79–7.88 (m, 1H), 7.61–7.69 (m, 2H), 7.42–7.49 (m, 1H), 7.12–7.19 (m, 1H), 7.01–7.09 (m, 2H).
7-Fluoro-N-(4-fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3h)
Compound 3h was synthesized using 4-fluoropyridin-2-amine (150 mg, 1.34 mmol), picolinaldehyde (172 mg, 1.61 mmol), and 2a (194 mg, 1.61 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (40–70% MeOH:H2O) to yield the title compound as a beige solid (105 mg, 24%). LCMS [M + H]+ 323.1 m/z.a 1H NMR (500 MHz, CD3OD) δ 8.59 (ddd, J = 4.9, 1.8, 1.0 Hz, 1H), 7.99 (dt, J = 7.9, 1.0 Hz, 1H), 7.95 (dd, J = 7.5, 5.6 Hz, 1H), 7.82 (td, J = 7.9, 1.8 Hz, 1H), 7.34–7.24 (m, 2H), 6.95–6.88 (m, 3H), 6.60–6.53 (m, 2H).
N-(4-Fluorophenyl)-7-methoxy-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3i)
Compound 3i has been synthesized as previously reported in the literature.6
N-(4-Fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyrazin-3-amine (3j)
Compound 3j was synthesized using 2-aminopyrazine (100 mg, 1.05 mmol), picolinaldehyde (135 mg, 1.26 mmol), and 2a (153 mg, 1.26 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a brown solid (51 mg, 16%). LCMS [M + H]+ 306.0 m/z.b 1H NMR (400 MHz, CDCl3) δ 9.08 (s, 1H), 8.79 (s, 1H), 8.59 (d, J = 4.3 Hz, 1H), 8.28 (d, J = 7.8 Hz, 1H), 7.83 (t, J = 7.8 Hz, 1H), 7.78 (d, J = 4.7 Hz, 1H), 7.39 (d, J = 4.7 Hz, 1H), 7.17–7.26 (m, 1H), 7.00 (t, J = 8.4 Hz, 2H), 6.68 (dd, J = 4.3, 8.4 Hz, 2H).
8-Fluoro-N-(4-fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3k)
Compound 3k was synthesized using 3-fluoropyridin-2-amine (150 mg, 1.34 mmol), picolinaldehyde (172 mg, 1.61 mmol), and 2a (194 mg, 1.61 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a beige solid (182 mg, 42%). LCMS [M + H]+ 323.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.56 (d, J = 5.1 Hz, 1H), 8.49 (br. s., 1H), 8.31 (d, J = 8.4 Hz, 1H), 7.80 (dt, J = 1.5, 7.7 Hz, 1H), 7.40 (d, J = 6.7 Hz, 1H), 7.19 (dd, J = 5.1, 6.7 Hz, 1H), 6.95 (t, J = 8.4 Hz, 2H), 6.89 (dd, J = 7.7, 10.2 Hz, 1H), 6.61–6.71 (m, 3H).
N-(4-Fluorophenyl)-8-methoxy-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3l)
Compound 3l was synthesized using 3-methoxypyridin-2-amine (150 mg, 1.21 mmol), picolinaldehyde (155 mg, 1.45 mmol), and 2a according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a beige solid (350 mg, 87%). LCMS [M + H]+ 335.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.53 (d, J = 4.9 Hz, 1H), 8.40 (s, 1H), 8.34 (d, J = 8.3 Hz, 1H), 7.76 (t, J = 7.8 Hz, 1H), 7.26 (d, J = 6.8 Hz, 1H), 7.14 (dd, J = 5.9, 6.8 Hz, 1H), 6.93 (t, J = 8.8 Hz, 2H), 6.59–6.72 (m, 3H), 6.48 (d, J = 7.8 Hz, 1H), 4.08 (s, 3H).
N-(4-Fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyrimidin-3-amine (3m)
Compound 3m was synthesized using 2-aminopyrimidine (100 mg, 1.05 mmol), picolinaldehyde (135 mg, 1.26 mmol), and 2a (153 mg, 1.26 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a beige solid (55 mg, 17%). LCMS [M + H]+ 306.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.50–8.60 (m, 3H), 8.38 (d, J = 8.5 Hz, 1H), 7.78–7.87 (m, 2H), 7.17–7.23 (m, 1H), 6.98 (t, J = 8.5 Hz, 2H), 6.82 (dd, J = 3.9, 6.8 Hz, 1H), 6.68 (dd, J = 4.4, 8.5 Hz, 2H).
N-(4-Fluorophenyl)-6-(methylsulfanyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3n)
Compound 3n was synthesized using 5-(methylthio)pyridin-2-amine (150 mg, 1.07 mmol), 4-methoxypicolinaldehyde (138 mg, 1.28 mmol), and 2a (156 mg, 1.28 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a beige solid (320 mg, 85%). LCMS [M + H]+ 351.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.56 (d, J = 4.4 Hz, 1H), 8.39 (br. s., 1H), 8.22 (d, J = 7.7 Hz, 1H), 7.80 (t, J = 7.7 Hz, 1H), 7.60 (d, J = 9.3 Hz, 1H), 7.46 (s, 1H), 7.14–7.23 (m, 2H), 6.96 (t, J = 8.6 Hz, 2H), 6.64 (dd, J = 4.4, 8.6 Hz, 2H), 2.38 (s, 3H).
N-(4-Fluorophenyl)-6-methanesulfonyl-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3o)
Compound 3o was synthesized using 5-(methylsulfonyl)pyridin-2-amine (140 mg, 0.81 mmol),21 picolinaldehyde (105 mg, 0.96 mmol), and 2a (118 mg, 0.96 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to provide a yellow solid (206 mg, 66%). LCMS [M + H]+ 383.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.59 (d, J = 4.4 Hz, 1H), 8.44 (s, 1H), 8.28 (s, 1H), 8.23 (d, J = 7.8 Hz, 1H), 7.82 (dt, J = 1.5, 7.8 Hz, 1H), 7.76 (d, J = 9.3 Hz, 1H), 7.53 (dd, J = 1.7, 9.5 Hz, 1H), 7.22 (dd, J = 5.1, 6.6 Hz, 1H), 6.97 (t, J = 8.5 Hz, 2H), 6.61–6.70 (m, 2H), 3.10 (s, 3H).
6-Cyclopropyl-N-(4-fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3p)
Compound 3p was synthesized using 5-cyclopropylpyridin-2-amine (40 mg, 0.30 mmol),22 picolinaldehyde (64 mg, 0.60 mmol), and 2a (72 mg, 0.60 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (50–70% MeOH:H2O) to provide a beige solid (33 mg, 32%). LCMS [M + H]+ 345.2 m/z.a 1H NMR (500 MHz, CDOD3) δ 8.59 (d, J = 4.9 Hz, 1H), 8.00 (d, J = 8.3 Hz, 1H), 7.82 (d, J = 1.5 Hz, 1H), 7.65 (s, 1H), 7.53 (d, J = 9.3 Hz, 1H), 7.24–7.29 (m, 1H), 7.13 (dd, J = 1.5, 9.3 Hz, 1H), 6.91 (t, J = 8.8 Hz, 2H), 6.52–6.58 (m, 2H), 1.93 (s, 1H), 0.96 (dd, J = 1.5, 8.3 Hz, 2H), 0.66 (d, J = 6.4 Hz, 2H).
N-(4-Fluorophenyl)-6-(1,2-oxazol-4-yl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3q)
Compound 3q was synthesized using 1f (45 mg, 0.28 mmol), picolinaldehyde (60 mg, 0.56 mmol), and 2a (68 mg, 0.56 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (30–60% MeOH:H2O) to provide a beige solid (39 mg, 38%). LCMS [M + H]+ 372.2 m/z.a 1H NMR (500 MHz, CDOD3) δ 9.13 (s, 1H), 8.83 (s, 1H), 8.60 (d, J = 4.9 Hz, 1H), 8.23 (s, 1H), 8.02 (d, J = 7.8 Hz, 1H), 7.84 (s, 1H), 7.68–7.73 (m, 1H), 7.63–7.67 (m, 1H), 7.29 (d, J = 4.9 Hz, 1H), 6.91 (t, J = 8.8 Hz, 2H), 6.59 (dd, J = 4.4, 8.8 Hz, 2H).
N-(4-Fluorophenyl)-6-(pyridin-2-yl)imidazo[2,1-b][1,3]thiazol-5-amine (3r)
Compound 3r was synthesized using 2-aminothiazole (50 mg, 0.50 mmol), picolinaldehyde (107 mg, 1.00 mmol), and 2a (121 mg, 1.00 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as an orange solid (27 mg, 17%). LCMS [M + H]+ 311.1 m/z.a 1H NMR (500 MHz, DMSO-d6) δ 8.48 (d, J = 4.5 Hz, 1H), 8.29 (s, 1H), 7.93–7.86 (m, 1H), 7.78 (td, J = 7.8, 1.8 Hz, 1H), 7.37 (d, J = 4.5 Hz, 1H), 7.26 (d, J = 4.5 Hz, 1H), 7.17 (ddd, J = 7.4, 4.8, 1.2 Hz, 1H), 6.99 (t, J = 8.8 Hz, 2H), 6.67–6.59 (m, 2H).
N-(3,5-Difluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3s)
Compound 3s was synthesized using pyridin-2-amine (90 mg, 0.96 mmol), picolinaldehyde (123 mg, 1.15 mmol), and 2b (160 mg, 1.15 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a brown solid (243 mg, 78%). LCMS [M + H]+ 323.2 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.55–8.62 (m, 1H), 8.18–8.25 (m, 2H), 7.79 (dt, J = 1.7, 7.7 Hz, 1H), 7.65–7.72 (m, 2H), 7.23–7.27 (m, 1H), 7.19 (ddd, J = 1.0, 5.5, 6.7 Hz, 1H), 6.86 (t, J = 6.7 Hz, 1H), 6.36 (tt, J = 2.2, 9.0 Hz, 1H), 6.07–6.22 (m, 2H).
N-(2,4-Difluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3t)
Compound 3t was synthesized using pyridin-2-amine (50 mg, 0.53 mmol), picolinaldehyde (114 mg, 1.06 mmol), and 2c (148 mg, 1.06 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (5–70% MeOH:H2O) to yield the title compound as a brown solid (53 mg, 31%). LCMS [M + H]+ 323.1 m/z.a 1H NMR (500 MHz, CDOD3) δ 8.58 (ddd, J = 4.9, 1.8, 1.1 Hz, 1H), 8.03 (dt, J = 7.8, 1.1 Hz, 1H), 7.99 (dt, J = 6.9, 1.1 Hz, 1H), 7.84 (td, J = 7.8, 1.8 Hz, 1H), 7.64 (dt, J = 9.2, 1.1 Hz, 1H), 7.40 (ddd, J = 9.2, 6.9, 1.3 Hz, 1H), 7.28 (ddd, J = 7.8, 4.9, 1.1 Hz, 1H), 7.05–6.97 (m, 2H), 6.67–6.60 (m, 1H), 6.14 (td, J = 9.4, 5.4 Hz, 1H).
2-(Pyridin-2-yl)-N-[3-(trifluoromethyl)phenyl]imidazo[1,2-a]pyridin-3-amine (3u)
Compound 3u was synthesized using pyridin-2-amine (50 mg, 0.53 mmol), picolinaldehyde (114 mg, 1.06 mmol), and 2d (182 mg, 1.06 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (30–70% MeOH:H2O) to yield the title compound as a yellow solid (80 mg, 42%). LCMS [M + H]+ 355.1 m/z.a 1H NMR (500 MHz, CDOD3) δ 8.59 (ddd, J = 4.9, 1.9, 1.1 Hz, 1H), 8.00 (ddt, J = 14.4, 6.8, 1.1 Hz, 2H), 7.83 (td, J = 7.8, 1.8 Hz, 1H), 7.66 (dt, J = 9.1, 1.1 Hz, 1H), 7.42 (ddd, J = 9.1, 6.8, 1.1 Hz, 1H), 7.32–7.25 (m, 2H), 7.05 (d, J = 7.8 Hz, 1H), 7.00 (td, J = 6.8, 1.1 Hz, 1H), 6.86 (t, J = 1.9 Hz, 1H), 6.71–6.64 (m, 1H).
N-(Benzo[d][1,3]dioxol-4-yl)-6-methoxy-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3v)
Compound 3v was synthesized using pyridin-2-amine (20 mg, 0.21 mmol), picolinaldehyde (45 mg, 0.43 mmol), and 2e (47 mg, 0.32 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (40–65% MeOH:H2O) to yield the title compound as a beige solid (14 mg, 20%). LCMS [M + H]+ 331.1 m/z; 1H NMR (500 MHz, Methanol-d4) δ 8.60 (d, J = 4.9 Hz, 1H), 8.02 (d, J = 8.0 Hz, 1H), 7.97 (dt, J = 6.8, 1.2 Hz, 1H), 7.84 (td, J = 7.7, 1.7 Hz, 1H), 7.61 (dt, J = 9.1, 1.2 Hz, 1H), 7.37 (ddd, J = 9.2, 6.7, 1.3 Hz, 1H), 7.28 (dd, J = 7.4, 5.1 Hz, 1H), 6.96 (td, J = 6.8, 1.1 Hz, 1H), 6.59 (t, J = 8.1 Hz, 1H), 6.39 (dd, J = 7.9, 1.0 Hz, 1H), 5.89 (s, 2H), 5.87 (dd, J = 8.3, 1.1 Hz, 1H).
N-(4-Fluorophenyl)-2-(pyridazin-3-yl)imidazo[1,2-a]pyridin-3-amine (3w)
Compound 3w was synthesized using pyridin-2-amine (90 mg, 0.96 mmol), pyridazine-3-carbaldehyde (124 mg, 1.15 mmol), and 2a (174 mg, 1.15 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (20–70% MeOH:H2O) to yield the title compound as a dark brown solid (80 mg, 27%). LCMS [M + H]+ 306.6 m/z.a 1H NMR (500 MHz, CDCl3) δ 9.04 (dd, J = 1.4, 4.9 Hz, 1H), 8.38 (dd, J = 1.4, 8.5 Hz, 1H), 8.23 (s, 1H), 7.59–7.69 (m, 2H), 7.57 (dd, J = 4.9, 8.8 Hz, 1H), 7.20–7.25 (m, 1H), 6.94 (t, J = 8.5 Hz, 2H), 6.80 (t, J = 6.6 Hz, 1H), 6.60–6.68 (m, 2H).
N-(4-Fluorophenyl)-2-(1-methyl-1H-imidazol-4-yl)imidazo[1,2-a]pyridin-3-amine (3x)
Compound 3x was synthesized using pyridin-2-amine (90 mg, 0.96 mmol), 1-methyl-1H-imidazole-4-carbaldehyde (172 mg, 1.15 mmol), and 2a (154 mg, 1.15 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (20–70% MeOH:H2O) to yield the title compound as a beige solid (115 mg, 40%). LCMS [M + H]+ 308.2 m/z.a 1H NMR (500 MHz, CDCl3) δ 7.68 (d, J = 6.8 Hz, 1H), 7.58 (d, J = 9.3 Hz, 1H), 7.42 (s, 2H), 7.13–7.20 (m, 1H), 7.06 (s, 1H), 6.89 (t, J = 8.8 Hz, 2H), 6.75 (t, J = 6.3 Hz, 1H), 6.49–6.59 (m, 2H), 3.70 (s, 3H).
N-(4-Fluorophenyl)-2-[(1H-pyrazol-1-yl)methyl]imidazo[1,2-a]pyridin-3-amine (3y)
Compound 3y was synthesized using pyridin-2-amine (45 mg, 0.48 mmol), 2-(1H-pyrazol-1-yl)acetaldehyde (105 mg, 0.96 mmol), and 2a (116 mg, 0.96 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (30–60% MeOH:H2O) to yield the title compound as a beige solid (9 mg, 6%). LCMS [M + H]+ 308.1 m/z.a 1H NMR (500 MHz, CDOD3) δ 7.94 (dt, J = 6.9, 1.3 Hz, 1H), 7.63 (d, J = 2.3 Hz, 1H), 7.53 (d, J = 9.0 Hz, 1H), 7.43 (d, J = 1.9 Hz, 1H), 7.36 (ddd, J = 9.0, 6.9, 1.3 Hz, 1H), 6.94 (td, J = 6.9, 1.3 Hz, 1H), 6.90–6.83 (m, 2H), 6.48–6.42 (m, 2H), 6.24 (t, J = 2.3 Hz, 1H), 5.39 (s, 2H).
N-(4-Fluorophenyl)-2-(6-methoxypyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3z)
Compound 3z was synthesized using pyridin-2-amine (90 mg, 0.96 mmol), 6-methoxypicolinaldehyde (158 mg, 1.15 mmol), and 2a (174 mg, 1.15 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (30–80% MeOH:H2O) to yield the title compound as a yellow solid (50 mg, 16%). LCMS [M + H]+ 335.6 m/z.a 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 8.12–8.18 (m, 1H), 7.95–8.03 (m, 1H), 7.62 (s, 2H), 7.16–7.22 (m, 2H), 6.89–6.97 (m, 2H), 6.74–6.81 (m, 1H), 6.61 (d, J = 4.3 Hz, 2H), 3.89 (s, 3H).
N-(4-Fluorophenyl)-2-(5-methoxypyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3aa)
Compound 3aa was synthesized using 2-aminopyridine (200 mg, 2.13 mmol), 5-methoxypicolinaldehyde (350 mg, 2.55 mmol), and 2a (309 mg, 2.55 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a colorless powder (49 mg, 31%). LCMS [M + H]+ 335.5 m/z.a 1H NMR (400 MHz, CDCl3) δ 8.28 (d, J = 3.1 Hz, 1H), 8.15 (d, J = 9.0 Hz, 1H), 7.97–8.02 (m, 1H), 7.57–7.66 (m, 1H), 7.29–7.33 (m, 2H), 7.15–7.22 (m, 1H), 6.90–6.96 (m, 2H), 6.74–6.80 (m, 1H), 6.60 (dd, J = 4.3, 9.0 Hz, 2H), 3.89 (s, 3H).
11,111 (3ab)
Compound 3ab was synthesized using 2-aminopyridine (45 mg, 0.48 mmol), 4-methoxypicolinaldehyde (79 mg, 0.57 mmol), and 2a (87 mg, 0.57 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a beige solid (55 mg, 35%). LCMS [M + H]+ 335.5 m/z.a 1H NMR (400 MHz, CDCl3) δ 8.55 (s, 1H), 8.36 (d, J = 5.5 Hz, 1H), 7.75 (d, J = 2.4 Hz, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.56 (d, J = 7.0 Hz, 1H), 7.14–7.23 (m, 1H), 6.94 (t, J = 8.6 Hz, 2H), 6.76 (t, J = 7.0 Hz, 1H), 6.70 (dd, J = 2.4, 5.5 Hz, 1H), 6.63 (dd, J = 4.5, 8.6 Hz, 2H), 3.96 (s, 3H).
N-(4-Fluorophenyl)-2-(3-methoxypyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3ac)
Compound 3 ac was synthesized using 2-aminopyridine (45 mg, 0.48 mmol), 3-methoxypicolinaldehyde (45 mg, 0.48 mmol), and 2a (87 mg, 0.57 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a faint brown solid (35 mg, 22%). LCMS [M + H]+ 335.5 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.32 (d, J = 3.9 Hz, 1H), 7.64–7.74 (m, 2H), 7.33 (s, 1H), 7.31 (d, J = 8.7 Hz, 1H), 7.14–7.25 (m, 2H), 6.86 (t, J = 8.7 Hz, 2H), 6.77 (t, J = 6.8 Hz, 1H), 6.44–6.54 (m, 2H), 3.90 (s, 3H).
N-(4-Fluorophenyl)-2-(6-fluoropyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3ad)
Compound 3ad was synthesized using 2-aminopyridine (45 mg, 0.48 mmol), 6-fluoropicolinaldehyde (72 mg, 0.57 mmol), and 2a (87 mg, 0.57 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a brown solid (68 mg, 44%). LCMS [M + H]+ 323.5 m/z.a 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 5.8 Hz, 1H), 7.87 (q, J = 8.2 Hz, 1H), 7.72 (s, 1H), 7.64 (d, J = 8.6 Hz, 2H), 7.18–7.26 (m, 1H), 6.95 (t, J = 8.6 Hz, 2H), 6.74–6.84 (m, 2H), 6.61 (dd, J = 4.3, 8.9 Hz, 2H).
N-(4-Fluorophenyl)-2-(5-fluoropyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3ae)
Compound 3ae was synthesized using 2-aminopyridine (45 mg, 0.48 mmol), 5-fluoropicolinaldehyde (72 mg, 0.57 mmol) and 2a (87 mg, 0.57 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a brown solid (38 mg, 25%). LCMS [M + H]+ 323.5 m/z.a 1H NMR (400 MHz, CDCl3) δ 8.43 (d, J = 2.7 Hz, 1H), 8.23 (dd, J = 4.4, 8.9 Hz, 1H), 7.88 (s, 1H), 7.59–7.67 (m, 2H), 7.50 (dt, J = 2.7, 8.6 Hz, 1H), 7.17–7.25 (m, 1H), 6.94 (t, J = 8.6 Hz, 2H), 6.75–6.82 (m, J = 6.4, 6.4 Hz, 1H), 6.60 (dd, J = 4.4, 8.9 Hz, 2H).
2-(4-Chloropyridin-2-yl)-N-(4-fluorophenyl)imidazo[1,2-a]pyridin-3-amine (3af)
Compound 3af was synthesized using 2-aminopyridine (45 mg, 0.48 mmol), 4-chloropicolinaldehyde (81 mg, 0.57 mmol), and 2a (87 mg, 0.57 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a brown solid (52 mg, 32%). LCMS [M + H]+ 339.5 m/z.a 1H NMR (400 MHz, CDCl3) δ 8.44 (d, J = 5.5 Hz, 1H), 8.27 (s, 1H), 8.18 (s, 1H), 7.64 (d, J = 9.4 Hz, 1H), 7.58 (d, J = 6.5 Hz, 1H), 7.13–7.24 (m, 2H), 6.95 (t, J = 8.4 Hz, 2H), 6.77 (t, J = 6.5 Hz, 1H), 6.64 (d, J = 4.5 Hz, 2H).
N-(4-Fluorophenyl)-2-(3-fluoropyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3ag)
Compound 3ag was synthesized using 2-aminopyridine (90 mg, 0.96 mmol), 3-fluoropicolinaldehyde (144 mg, 1.15 mmol), and 2a (174 mg, 1.15 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a faint beige solid (123 mg, 40%). LCMS [M + H]+ 323.5 m/z.a 1H NMR (400 MHz, CDCl3) δ 8.46 (d, J = 4.7 Hz, 1H), 7.79 (s, 1H), 7.73 (d, J = 8.9 Hz, 1H), 7.67 (d, J = 7.0 Hz, 1H), 7.49–7.58 (m, 1H), 7.17–7.26 (m, 2H), 6.91 (t, J = 8.9 Hz, 2H), 6.80 (t, J = 7.0 Hz, 1H), 6.56 (dd, J = 4.7, 8.9 Hz, 2H).
N-(4-Fluorophenyl)-6-methoxy-2-(4-methoxypyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3ah)
Compound 3ah was synthesized using 5-methoxypyridin-2-amine (90 mg, 0.72 mmol), 4-methoxypicolinaldehyde (119 mg, 0.87 mmol) and 2a (132 mg, 0.87 mmol), according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a white solid (110 mg, 42%). LCMS [M + H]+ 365.7 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.54 (br. s., 1H), 8.34 (d, J = 5.8 Hz, 1H), 7.70 (d, J = 2.4 Hz, 1H), 7.54 (d, J = 9.3 Hz, 1H), 6.92–7.02 (m, 4H), 6.69 (dd, J = 2.4, 5.8 Hz, 1H), 6.59–6.66 (m, 2H), 3.95 (s, 3H), 3.68 (s, 3H).
N-(4-Fluorophenyl)-6-methoxy-2-(5-methoxypyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (3ai)
Compound 3ai was synthesized using 5-methoxypyridin-2-amine (150 mg, 1.21 mmol), 5-methoxypicolinaldehyde (199 mg, 1.45 mmol) and 2a (176 mg, 1.45 mmol) according to General Procedure B. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a yellow solid (135 mg, 31%). LCMS [M + H]+ 365.1 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.27 (d, J = 2.9 Hz, 1H), 8.12 (d, J = 8.8 Hz, 1H), 7.99 (s, 1H), 7.54 (d, J = 9.8 Hz, 1H), 7.30 (dd, J = 2.9, 8.8 Hz, 1H), 7.07 (d, J = 1.5 Hz, 1H), 6.99 (d, J = 9.8 Hz, 1H), 6.94 (t, J = 8.5 Hz, 2H), 6.60 (dd, J = 4.4, 8.8 Hz, 2H), 3.88 (s, 3H), 3.70 (s, 3H).
N-(4-Fluorophenyl)-N-methyl-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (4a)
Compound 4a was synthesized using 3a (100 mg, 0.33 mmol) and iodomethane (21.5 μL, 0.34 mmol) according to General Procedure C. The crude product was then purified by Rilas technologies to afford the title compound as a yellow solid (66 mg, 64%). LCMS [M + H]+ 319.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.62 (d, J = 4.4 Hz, 1H), 7.93 (d, J = 7.8 Hz, 1H), 7.76 (d, J = 6.8 Hz, 1H), 7.63–7.73 (m, 2H), 7.25 (d, J = 8.7 Hz, 1H), 7.14 (dd, J = 5.4, 6.8 Hz, 1H), 6.90 (t, J = 8.7 Hz, 2H), 6.80 (t, J = 6.8 Hz, 1H), 6.47–6.58 (m, 2H), 3.41 (s, 3H).
N-(4-Fluorophenyl)-N-(methyl-d3)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (4b)
Compound 4b was synthesized using 3a (56 mg, 0.18 mmol) and iodomethane-d3 (13 μL, 0.19 mmol) according to General Procedure C. The crude product was then purified by Rilas technologies to afford the title compound as a yellow solid (34 mg, 58%). LCMS [M + H]+ 322.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.63 (d, J = 4.4 Hz, 1H), 7.94 (d, J = 7.8 Hz, 1H), 7.78 (d, J = 6.8 Hz, 1H), 7.66–7.74 (m, 2H), 7.24–7.28 (m, 1H), 7.16 (dd, J = 5.4, 6.8 Hz, 1H), 6.91 (t, J = 9.0 Hz, 2H), 6.81 (t, J = 6.8 Hz, 1H), 6.54 (dd, J = 4.0, 9.0 Hz, 2H).
N-Ethyl-N-(4-fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (4c)
Compound 4c was synthesized from 3a (50 mg, 0.16 mmol) and iodoethane (0.16 μL, 0.20 mmol) according to General Procedure C. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a light orange solid (26 mg, 49%). LCMS [M + H]+ 333.2 m/z.a 1H NMR (500 MHz, DMSO-d6) δ 8.48 (s, 1H), 8.12 (d, J = 8.0 Hz, 1H), 8.01 (d, J = 6.8 Hz, 1H), 7.82 (t, J = 7.6 Hz, 1H), 7.66 (d, J = 9.2 Hz, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.23 (s, 1H), 6.94 (t, J = 8.8 Hz, 3H), 6.44 (d, J = 5.0 Hz, 2H), 3.82 (d, J = 36.1 Hz, 2H), 1.11–0.92 (m, 3H).
N-(4-Fluorophenyl)-N-propyl-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (4d)
Compound 4d was prepared from 3a (50 mg, 0.16 mmol) and 1-iodopropane (33.5 μL, 0.20 mmol) according to General Procedure C. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a yellow solid (26 mg, 46%). LCMS [M + H]+ 347.1 m/z.b 1H NMR (500 MHz, CD3OD) δ 8.63–8.54 (m, 1H), 7.96 (dt, J = 6.7, 1.3 Hz, 1H), 7.88–7.83 (m, 1H), 7.79 (td, J = 7.7, 1.8 Hz, 1H), 7.66 (dt, J = 9.0, 1.1 Hz, 1H), 7.41 (ddd, J = 9.0, 6.7, 1.3 Hz, 1H), 7.28 (ddd, J = 7.5, 4.9, 1.3 Hz, 1H), 6.97 (td, J = 6.7, 1.3 Hz, 1H), 6.94–6.89 (m, 2H), 6.59–6.54 (m, 2H), 3.71 (br. s, 2H), 1.56 (br. s, 2H), 0.85 (t, J = 7.4 Hz, 3H).
N-(Cyclopropylmethyl)-N-(4-fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (4e)
Compound 4e was synthesized from 3a (50 mg, 0.16 mmol) and (iodomethyl)cyclopropane (18.4 μL, 19.72 mmol) according to General Procedure C. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a beige solid (13 mg, 22%). LCMS [M + H]+ 359.2 m/z.a 1H NMR (500 MHz, DMSO-d6) δ 8.51–8.44 (m, 1H), 8.13 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 6.9 Hz, 1H), 7.81 (td, J = 7.8, 1.8 Hz, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.34 (dd, J = 9.0, 6.9 Hz, 1H), 7.22 (dd, J = 7.4, 4.9 Hz, 1H), 6.94 (td, J = 8.8, 7.8, 4.3 Hz, 3H), 6.45 (dd, J = 9.2, 4.3 Hz, 2H), 3.90–3.74 (m, 1H), 3.61–3.41 (m, 1H), 0.95–0.81 (m, 1H), 0.13 (d, J = 13.3 Hz, 3H), −0.21 (s, 1H).
N-(4-Fluorophenyl)-N-isobutyl-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (4f)
Compound 4f was synthesized using 3a (50 mg, 0.16 mmol) and 1-iodo-2-methylpropane (28.3 μL, 0.24 mmol) according to General Procedure C. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a colorless solid (18 mg, 31%). LCMS [M + H]+ 361.6 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.66 (d, J = 4.4 Hz, 1H), 7.91 (d, J = 8.4 Hz, 1H), 7.78 (d, J = 6.8 Hz, 1H), 7.65–7.73 (m, 2H), 7.24–7.26 (m, 1H), 7.17 (ddd, J = 1.0, 5.5, 6.8 Hz, 1H), 6.90 (t, J = 8.4 Hz, 2H), 6.81 (dt, J = 1.0, 6.8 Hz, 1H), 6.54 (td, J = 3.9, 8.4 Hz, 2H), 3.39–3.75 (m, 2H), 1.89 (spt, J = 6.6 Hz, 1H), 0.70–1.02 (m, 6H).
N-(4-Fluorophenyl)-N-(3-methylbutyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (4g)
Compound 4 g was synthesized from 3a (50 mg, 0.16 mmol) and 1-bromo-3-methylbutane (30 μL, 0.25 mmol) according to General Procedure C. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a brown oil (10 mg, 16%). LCMS [M + H]+ 375.8 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.65 (d, J = 3.9 Hz, 1H), 7.90 (d, J = 8.3 Hz, 1H), 7.75 (d, J = 6.8 Hz, 1H), 7.61–7.72 (m, 2H), 7.24 (ddd, J = 1.2, 6.8, 9.0 Hz, 1H), 7.11–7.18 (m, 1H), 6.84–6.93 (m, 2H), 6.78 (dt, J = 1.0, 6.8 Hz, 1H), 6.49–6.55 (m, 2H), 3.74 (br. s., 2H), 1.30–1.64 (m, 3H), 0.75–0.90 (m, 6H).
N-{3-[(tert-Butyldimethylsilyl)oxy]propyl}-N-(4-fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (4h)
Compound 4 h was synthesized from 3a (50 mg, 0.16 mmol) and (3-bromopropoxy)(tert-butyl)dimethylsilane (57 μL, 0.25 mmol) according to General Procedure C. The crude product was purified by Rilas technologies to afford the title compound as a brown solid (20 mg, 26%). LCMS [M + H]+ 477.9 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.65 (d, J = 4.9 Hz, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.75 (d, J = 6.8 Hz, 1H), 7.64–7.73 (m, 2H), 7.26 (d, J = 1.5 Hz, 1H), 7.10–7.18 (m, 1H), 6.89 (t, J = 8.4 Hz, 2H), 6.79 (s, 1H), 6.55–6.65 (m, 2H), 3.76–4.14 (m, 2H), 3.59 (br. s., 2H), 1.61–1.96 (m, 2H), 0.87 (s, 9H), −0.01 (s, 6H).
N-{2-[(tert-Butyldimethylsilyl)oxy]ethyl}-N-(4-fluorophenyl)-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (4i)
Compound 4i was synthesized from 3a (100 mg, 0.33 mmol) and (2-bromoethoxy)(tert-butyl)dimethylsilane (94 mg, 0.39 mmol) according to General Procedure C. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a yellow solid (10 mg, 6%). LCMS [M + H] + 463.2 m/z.c 1H NMR (500 MHz, CDCl3) δ 8.58–8.69 (m, 1H), 8.00 (d, J = 7.8 Hz, 1H), 7.91 (d, J = 6.8 Hz, 1H), 7.68 (d, J = 10.3 Hz, 2H), 7.22–7.26 (m, 1H), 7.13–7.19 (m, 1H), 6.87 (t, J = 8.8 Hz, 2H), 6.75 (s, 1H), 6.62 (dd, J = 4.4, 9.3 Hz, 2H), 3.45–4.18 (m, 4H), 0.80 (s, 9H), −0.27–0.06 (m, 6H).
N-(4-Fluorophenyl)-N-(2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-yl)acetamide (4j)
An oven-dried 8 mL vial had added 3a (200 mg, 0.66 mmol), CH2Cl2 (3.3 mL) and pyridine (58 μL, 0.72 mmol). The reaction was then cooled with an external ice bath before the dropwise addition of acetyl chloride (50 μL, 0.69 mmol). The reaction was then slowly warmed to room temperature and stirred for 17 h. To the solution was added CH2Cl2 (15 mL) and then diluted HCl till pH was adjusted to ∼5–6. The two phases are separated, and combined organic washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was dissolved in a minimal amount of DMSO and purified by reverse phase chromatography (40–100% MeOH:H2O) to yield the title compound as a yellow solid (55 mg, 24%). LCMS [M + H]+ 379.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.65 (d, J = 4.9 Hz, 1H), 8.29 (d, J = 8.3 Hz, 1H), 7.99 (d, J = 6.8 Hz, 1H), 7.82 (t, J = 7.8 Hz, 1H), 7.69 (d, J = 9.3 Hz, 1H), 7.39 (br. s., 2H), 7.32 (t, J = 7.8 Hz, 1H), 7.24–7.27 (m, 1H), 6.88–7.01 (m, 3H), 2.02 (br. s., 3H).
N-(4-Fluorophenyl)-2-(4-methoxypyridin-2-yl)-N-methylimidazo[1,2-a]pyridin-3-amine (4k)
Compound 4 k was synthesized from 3ab (100 mg, 0.30 mmol) and iodomethane (20 μL, 0.31 mmol) according to General Procedure C. The crude product was then purified by Rilas technologies to afford the title compound as a yellow solid (55 mg, 53%). LCMS [M + H]+ 349.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.42 (d, J = 5.4 Hz, 1H), 7.79 (d, J = 6.6 Hz, 1H), 7.70 (d, J = 9.3 Hz, 1H), 7.49 (d, J = 2.4 Hz, 1H), 7.22–7.31 (m, 1H), 6.90 (t, J = 8.8 Hz, 2H), 6.81 (t, J = 6.6 Hz, 1H), 6.70 (dd, J = 2.4, 5.4 Hz, 1H), 6.52 (dd, J = 4.4, 8.8 Hz, 2H), 3.81 (s, 3H), 3.42 (s, 3H).
N-(4-Fluorophenyl)-2-(5-methoxypyridin-2-yl)-N-methylimidazo[1,2-a]pyridin-3-amine (4l)
Compound 4l was synthesized from 3aa (100 mg, 0.30 mmol) and iodomethane (20 μL, 0.31 mmol) according to General Procedure C. The crude product was then purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the afford the title compound as a brown solid (10 mg, 10%). LCMS [M + H]+ 349.1 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.32 (br. s., 1H), 7.85 (d, J = 8.5 Hz, 1H), 7.75 (t, J = 8.5 Hz, 2H), 7.24–7.32 (m, 1H), 7.20 (td, J = 1.5, 8.5 Hz, 1H), 6.86–6.93 (m, 2H), 6.82 (t, J = 6.6 Hz, 1H), 6.45–6.57 (m, 2H), 3.86 (d, J = 1.5 Hz, 3H), 3.39 (s, 3H).
N-(4-Fluorophenyl)-6-methoxy-2-(5-methoxypyridin-2-yl)-N-methylimidazo[1,2-a]pyridin-3-amine (4m)
Compound 4 m was synthesized using 3ai (115 mg, 0.32 mmol) and iodomethane (29.5 μL, 0.47 mmol) according to General Procedure C. The crude product was then purified by Rilas technologies to afford the title compound as a yellow solid (25 mg, 24%). LCMS [M + H]+ 379.0 m/z.c 1H NMR (500 MHz, CDCl3) δ 8.38 (d, J = 5.4 Hz, 1H), 7.59 (d, J = 9.8 Hz, 1H), 7.43 (d, J = 2.4 Hz, 1H), 7.27–7.29 (m, 1H), 7.05 (dd, J = 2.2, 9.5 Hz, 1H), 6.91 (t, J = 8.5 Hz, 2H), 6.67 (dd, J = 2.4, 5.9 Hz, 1H), 6.51–6.58 (m, 2H), 3.80 (s, 3H), 3.75 (s, 3H), 3.41 (s, 3H).
N-(4-Fluorophenyl)-6-methoxy-N-methyl-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (4n)
Compound 4n was prepared using 3f (50 mg, 0.15 mmol) and iodomethane (11 μL, 0.18 mmol) according to General Procedure C. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a light yellow solid (14 mg, 27%). LCMS [M + H]+ 349.2 m/z.a 1H NMR (500 MHz, DMSO-d6) δ 8.44–8.38 (m, 1H), 8.08–8.03 (m, 1H), 7.78 (td, J = 7.8, 1.8 Hz, 1H), 7.60 (d, J = 9.6 Hz, 1H), 7.49 (d, J = 2.3 Hz, 1H), 7.18 (ddd, J = 7.4, 4.8, 1.2 Hz, 1H), 7.14 (dd, J = 9.6, 2.3 Hz, 1H), 6.94 (t, J = 8.8 Hz, 2H), 6.48–6.38 (m, 2H), 3.71 (s, 3H), 3.35 (s, 3H).
N-(tert-Butyl)-7-methoxy-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (6b)
Compound 6b was synthesized using 4-methoxypyridin-2-amine 5b (200 mg, 1.61 mmol), pyridine-2-carboxaldehyde (305 μL, 3.22 mmol), and tert-butyl isocyanide (364.4 μL, 3.22 mmol) according to General Procedure B. The crude product was purified by flash chromatography (5:95 EtOAc/Hex) to yield the title compound as a solid (318 mg, 71%). LCMS [M + H]+ 297.1 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.54 (ddd, J = 4.9, 1.9, 1.0 Hz, 1H), 8.12–8.08 (m, 2H), 7.73 (td, J = 7.8, 1.8 Hz, 1H), 7.12 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H), 6.78 (d, J = 2.3 Hz, 1H), 6.46 (dd, J = 7.6, 2.4 Hz, 1H), 5.36 (s, 1H), 3.85 (s, 3H), 1.13 (s, 9H).
2-(Pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (7a)
A suspension of N-(tert-butyl)-amine 6a (660 mg, 2.48 mmol)8 in water (30 mL) was treated with HBr (9.8 mL, ∼8.95 M in water), and stirred at 110 °C for 4 h. After the reaction was complete, the pH has been raised to ∼12, and the was precipitate collected by filtration (430 mg, 83%). 1H NMR (500 MHz, CDCl3) δ 8.57 (d, J = 3.9 Hz, 1H), 8.20 (d, J = 7.8 Hz, 1H), 7.81 (d, J = 6.5 Hz, 1H), 7.71–7.79 (m, 1H), 7.56 (d, J = 9.3 Hz, 1H), 7.03–7.16 (m, 2H), 6.81 (t, J = 6.5 Hz, 1H), 5.41 (br. s., 2H).
7-Methoxy-2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-amine (7b)
A suspension of 6b (340 mg, 1.15 mmol) in 3 M HBr (20 mL) was stirred at 110 °C for 3 h. After the reaction was complete, the pH has been raised to ∼12, and the precipitate was collected by filtration (220 mg, 80%). LCMS [M + H]+ 241.1 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.53 (d, J = 4.8 Hz, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.77–7.68 (m, 1H), 7.63 (d, J = 7.5 Hz, 1H), 7.06 (t, J = 6.2 Hz, 1H), 6.83–6.74 (m, 1H), 6.51 (dd, J = 7.6, 2.3 Hz, 1H), 5.22 (s, 2H), 3.85 (s, 3H).
5-Fluoro-N-[2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-yl]pyridin-2-amine (8)
Compound 8 was synthesized using amine 7a (100 mg, 0.48 mmol) and 2-bromo-5-fluoropyridine (100 mg, 0.57 mmol), in the presence of NaOtBu and XPhos Pd G2 (19 mg, 0.24 mmol) in t-BuOH (2 mL). The reaction was stirred at 75 °C for 2 h, then quenched with brine (30 mL) and extracted with CH2Cl2 (3 × 40 mL), dried over Na2SO4, filtered, and concentrated under vacuum. The crude product was purified by reverse phase chromatography (20–80% MeOH:H2O) to yield the title compound as a yellow solid (141 mg, 97%). LCMS [M + H]+ 306.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.71 (s, 1H), 8.59 (d, J = 3.9 Hz, 1H), 8.21 (d, J = 8.4 Hz, 1H), 8.04 (d, J = 2.4 Hz, 1H), 7.72–7.81 (m, 2H), 7.64 (d, J = 9.3 Hz, 1H), 7.27–7.32 (m, 1H), 7.22 (ddd, J = 1.2, 7.3, 8.4 Hz, 1H), 7.16 (ddd, J = 1.0, 5.0, 7.7 Hz, 1H), 6.75–6.83 (m, 1H), 6.55 (dd, J = 3.4, 8.8 Hz, 1H).
2-(Pyridin-2-yl)imidazo[1,2-a]pyridine (11a)
Compound 11a was synthesized using pyridin-2-amine (258 mg, 2.74 mmol) according to General Procedure E. The crude product was then purified by flash chromatography (35–100% MeOH in CH2Cl2) to yield the title compound as a brown solid (10 mg, 10%). LCMS [M + H]+ 196.0 m/z.a 1H NMR (500 MHz, CDOD3) δ 8.58 (d, J = 4.9 Hz, 1H), 8.49 (d, J = 6.8 Hz, 1H), 8.38 (s, 1H), 8.12 (d, J = 8.0 Hz, 1H), 7.92 (td, J = 7.7, 1.8 Hz, 1H), 7.61–7.56 (m, 1H), 7.36 (tdd, J = 8.0, 4.2, 1.4 Hz, 2H), 6.95 (td, J = 6.8, 1.1 Hz, 1H).
7-Methoxy-2-(pyridin-2-yl)imidazo[1,2-a]pyridine (11b)
Compound 11b was synthesized using 4-methoxypyridin-2-amine (127 mg, 1.02 mmol) according to General Procedure E. The crude product was then purified by flash chromatography (30:70 EtOAc/Hex) to yield the title compound as a white solid (136 mg, 67%). LCMS [M + H]+ 226.0 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.60 (ddd, J = 4.8, 1.8, 0.9 Hz, 1H), 8.15 (d, J = 7.9 Hz, 1H), 8.08 (s, 1H), 7.96 (d, J = 7.4 Hz, 1H), 7.76 (td, J = 7.8, 1.8 Hz, 1H), 7.20 (ddd, J = 7.5, 4.8, 1.2 Hz, 1H), 6.92 (d, J = 2.4 Hz, 1H), 6.54 (dd, J = 7.4, 2.4 Hz, 1H), 3.87 (s, 3H).
3-Chloro-7-methoxy-2-(pyridin-2-yl)imidazo[1,2-a]pyridine (12)
To a solution of 11b (100 mg, 0.44 mmol) in ACN (5 mL) was added N-chlorosuccinimide (59.28 mg, 0.44 mmol), and the mixture was stirred at room temperature for 4 h. After the reaction was complete, all the volatiles were removed under vacuum. The residue was diluted with CH2Cl2 and washed with a saturated solution of NaCl. The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by RP chromatography (40–60% MeOH:H2O) to give the title compound as light orange solid (57 mg, 50%). LCMS [M + H]+ 260.1 m/z.a 1H NMR (500 MHz, CDOD3) δ 8.67 (d, J = 4.9 Hz, 1H), 8.23 (d, J = 7.5 Hz, 1H), 8.05 (d, J = 7.9 Hz, 1H), 7.93 (td, J = 7.8, 1.8 Hz, 1H), 7.39 (dd, J = 7.5, 5.0 Hz, 1H), 6.95 (d, J = 2.4 Hz, 1H), 6.84 (dd, J = 7.5, 2.4 Hz, 1H), 3.93 (s, 3H).
2-(Pyridin-2-yl)imidazo[1,2-a]pyridine-3-carbaldehyde (13)
A mixture of DMF (7.5 mL) and POCl3 (7.5 mL) was stirred in an ice bath for 15 min under argon. After being warmed to room temperature, it was stirred for 30 min. Then, 10 (490 mg, 2.5 mmol) in CH2Cl2 (60 mL) was added. Then the temperature was raised to 70 °C, and the mixture was stirred for additional 2 h. The reaction mixture was cooled to room temperature, and slowly poured into NH3 H2O under ice-cold conditions. After being warmed to room temperature, the reaction mixture was further stirred for 30 min and extracted with CH2Cl2. The organic layers were combined, dried over anhydrous Na2SO4, and evaporated in vacuo. The pure product was obtained as a white solid (300 mg, 70%). 1H NMR (400 MHz, CDCl3) δ 11.04 (s, 1H), 9.75 (d, J = 7.0 Hz, 1H), 8.72 (d, J = 4.3 Hz, 1H), 8.36 (d, J = 8.2 Hz, 1H), 7.87 (t, J = 7.8 Hz, 1H), 7.81 (d, J = 9.0 Hz, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.33–7.42 (m, 1H), 7.13 (t, J = 6.8 Hz, 1H).
4-Fluoro-N-{[2-(pyridin-2-yl)imidazo[1,2-a]pyridin-3-yl]methyl}aniline (14)
To a suspension of previously prepared aldehyde 13 (110 mg, 0.49 mmol)23 in MeOH (2 mL), acetic acid (141 μL, 2.46 mmol) is added, followed by 4-fluoroaniline (109 mg, 0.93 mmol) and NaCNBH3 (124 mg, 1.97 mmol). After stirring for 1 h, the reaction was quenched with NaHCO3 solution and extracted with EtOAc, dried over Na2SO4, filtered, and concentrated under reduced pressure to yield the title compound as a faint beige powder (560 mg, quantitative). The residue was used in the subsequent reaction without purification. LCMS [M + H]+ 319.1 m/z.b 1H NMR (400 MHz, CDCl3) δ 8.63 (d, J = 4.7 Hz, 1H), 8.26 (d, J = 7.8 Hz, 1H), 8.20 (d, J = 6.6 Hz, 1H), 7.81 (t, J = 7.8 Hz, 1H), 7.65 (d, J = 9.0 Hz, 1H), 7.23 (d, J = 7.0 Hz, 2H), 6.80–6.99 (m, 3H), 6.67–6.80 (m, 2H), 5.02 (s, 2H), 4.52 (br. s., 1H).
2-(Pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (15a)
Compound 15a was synthesized using 3,4-dihydro-2H-pyrrol-5-amine hydrochloride (521 mg, 4.32 mmol) according to General Procedure E. The crude product was then purified by flash chromatography (0–10% MeOH in CH2Cl2) to yield the title compound as an orange solid (242 mg, 45%). 1H NMR (400 MHz, CDCl3) δ 8.89–8.82 (m, 1H), 8.38–8.30 (m, 1H), 7.94 (dt, J = 7.9, 2.0 Hz, 1H), 7.19–7.14 (m, 1H), 7.13 (s, 1H), 3.88 (t, J = 7.0 Hz, 2H), 2.80 (t, J = 7.6 Hz, 2H), 2.57–2.43 (m, 2H).
3-Bromo-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (15b)
To a solution of the 15a (150 mg, 0.81 mmol) in CH2Cl2 (4 mL) was added N-bromosuccinimide (151 mg, 0.85 mmol) and the mixture was stirred at 25 °C for 1 h. On completion, the volatiles were evaporated. The residue was diluted with EtOAc and washed with a saturated solution of NaHCO3 and brine. The organic phase was dried over Na2SO4, filtered, and concentrated under reduced pressure to afford the desired product as a brown solid, which was used without further purification (205 mg, 96%). 1H NMR (400 MHz, CDCl3) δ 8.61 (ddd, J = 4.9, 1.8, 0.9 Hz, 1H), 7.93 (dt, J = 8.0, 1.1 Hz, 1H), 7.66 (ddd, J = 8.1, 7.5, 1.9 Hz, 1H), 7.11 (ddd, J = 7.5, 4.9, 1.2 Hz, 1H), 3.98–3.90 (m, 2H), 3.00–2.90 (m, 2H), 2.64–2.50 (m, 2H).
3-Phenyl-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16a)
Synthesized as reported in a previous study.24
2-(Pyridin-2-yl)-3-(o-tolyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16b)
Synthesized as reported in a previous study.24
3-(3,4-Dichlorophenyl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16c)
Synthesized as reported in a previous study.24
3-(3,4-Dimethoxyphenyl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16d)
Synthesized as reported in a previous study.24
3-(3,5-Dimethoxyphenyl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16e)
Compound 16e was prepared using 15 (75 mg, 0.28 mmol) and 2-(3,5-dimethoxyphenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (98 mg, 0.37 mmol) according to General Procedure D. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a faint beige solid (21 mg, 22%). LCMS [M + H]+ 322.0 m/z.b 1H NMR (500 MHz, CDCl3) δ 8.52 (d, J = 4.4 Hz, 1H), 7.52–7.61 (m, 2H), 7.05 (ddd, J = 1.2, 5.4, 6.6 Hz, 1H), 6.59 (d, J = 2.0 Hz, 2H), 6.44 (t, J = 2.0 Hz, 1H), 3.98 (t, J = 7.3 Hz, 2H), 3.74 (s, 6H), 2.98 (t, J = 7.3 Hz, 2H), 2.55–2.66 (m, 2H).
2-(Pyridin-2-yl)-3-(m-tolyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16f)
Synthesized as reported in a previous study.24
3-(3-Chlorophenyl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16g)
Compound 16g was prepared using 15 (75 mg, 0.28 mmol) and (3-chlorophenyl)boronic acid (58 mg, 0.37 mmol) according to General Procedure D. The crude product was purified by reverse phase chromatography (35–100% MeOH–H2O) to yield the title compound as a colorless solid (10 mg, 12%). LCMS [M + H]+ 296.2 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.46 (d, J = 3.9 Hz, 1H), 7.70 (d, J = 8.3 Hz, 1H), 7.61 (dt, J = 1.6, 7.8 Hz, 1H), 7.47 (d, J = 1.5 Hz, 1H), 7.28–7.37 (m, 3H), 7.05–7.10 (m, 1H), 3.99 (t, J = 7.3 Hz, 2H), 3.01 (t, J = 7.3 Hz, 2H), 2.65 (quin, J = 7.3 Hz, 2H).
2-(Pyridin-2-yl)-3-(pyridin-3-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16h)
Compound 16h was prepared using 15 (75 mg, 0.28 mmol) and pyridin-3-ylboronic acid (45 mg, 0.37 mmol) according to General Procedure D. The crude product was purified by reverse phase chromatography (35–100% MeOH:H2O) to yield the title compound as a colorless solid (10 mg, 11%). LCMS [M + H]+ 263.1 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.74 (d, J = 1.5 Hz, 1H), 8.57 (dd, J = 1.5, 4.9 Hz, 1H), 8.38–8.43 (m, 1H), 7.85 (td, J = 1.9, 7.8 Hz, 1H), 7.81 (d, J = 7.8 Hz, 1H), 7.64 (dt, J = 1.9, 7.8 Hz, 1H), 7.30–7.37 (m, 1H), 7.08 (ddd, J = 1.5, 4.9, 7.3 Hz, 1H), 4.01 (t, J = 7.1 Hz, 2H), 3.03 (t, J = 7.6 Hz, 2H), 2.68 (quin, J = 7.3 Hz, 2H).
5-(2-(Pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl)picolinonitrile (16i)
Synthesized as reported in a previous study.24
3-(2-(Pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl)benzonitrile (16j)
Synthesized as reported in a previous study.24
2-(Pyridin-2-yl)-3-(pyridin-4-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16k)
Compound 16k was prepared using 15 (75 mg, 0.28 mmol) and 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine (76 mg, 0.37 mmol) according to General Procedure D. The crude product was purified by reverse phase chromatography (35–100% MeOH–H2O) to yield the title compound as a colorless solid (12 mg, 16%). LCMS [M + H]+ 263.1 m/z.a 1H NMR (500 MHz, CDCl3) δ 8.61 (d, J = 6.3 Hz, 2H), 8.45 (d, J = 4.9 Hz, 1H), 7.79 (d, J = 7.8 Hz, 1H), 7.67 (dt, J = 1.5, 7.8 Hz, 1H), 7.34–7.41 (m, 2H), 7.13 (dd, J = 5.4, 6.8 Hz, 1H), 4.06 (t, J = 7.1 Hz, 2H), 3.03 (t, J = 7.6 Hz, 2H), 2.69 (quin, J = 7.3 Hz, 2H).
3-(2-Fluoropyridin-4-yl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16l)
Synthesized as reported in a previous study.24
4-(2-(Pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl)benzonitrile (16m)
Synthesized as reported in a previous study.24
2-Methoxy-4-(2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl)benzonitrile (16n)
Synthesized as reported in a previous study.24
4-(2-(Pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl)-2-(trifluoromethyl)benzonitrile (16o)
Synthesized as reported in a previous study.24
3-(4-Chlorophenyl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16p)
Synthesized as reported in a previous study.24
3-(4-Fluorophenyl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16q)
Synthesized as reported in a previous study.24
2-(Pyridin-2-yl)-3-(p-tolyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16r)
Synthesized as reported in a previous study.24
2-(Pyridin-2-yl)-3-(4-(trifluoromethyl)phenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16s)
Synthesized as reported in a previous study.24
2-(Pyridin-2-yl)-3-(4-(trifluoromethoxy)phenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16t)
Synthesized as reported in a previous study.24
3-(4-(Methylsulfinyl)phenyl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16u)
Synthesized as reported in a previous study.24
3-(4-(Methylsulfonyl)phenyl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16v)
Synthesized as reported in a previous study.24
N,N-Dimethyl-4-(2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl)benzamide (16w)
Synthesized as reported in a previous study.24
1-(4-(2-Pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl)phenyl)ethan-1-one (16x)
Synthesized as reported in a previous study.24
4-(2-(Pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl)aniline (16y)
Synthesized as reported in a previous study.24
2-Methyl-5-(2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl)benzo[d]thiazole (16z)
Synthesized as reported in a previous study.24
7-(2-(Pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazol-3-yl)quinoline (16aa)
Synthesized as reported in a previous study.24
3-(Dibenzo[b,d]thiophen-2-yl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16ab)
Synthesized as reported in a previous study.24
3-(Benzo[b]thiophen-5-yl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16ac)
Synthesized as reported in a previous study.24
3-(Benzo[d][1,3]dioxol-5-yl)-2-(pyridin-2-yl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (16ad)
Synthesized as reported in a previous study.20
Acknowledgments
The authors acknowledge funding from the National Institute of Allergy and Infectious Diseases (M.P.P. R33AI141227) and Sao Paulo Research Foundation (grant # 2021/11899-0 and grant #2017/21146-4). We thank AstraZeneca, and Charles River Laboratories for the provision of the in vitro ADME and physicochemical properties data included in the manuscript. We would like to acknowledge Professor Roman Manetsch’s lab at Northeastern University for allowing us ad hoc access to their LCMS system. Use of JChem/ChemAxon software (Product version 20.11.0, ChemAxon (https://www.chemaxon.com)) is acknowledged. We acknowledge support from the Major Research Instrumentation (MRI) Program of the National Science Foundation under NSF award no. 2216066 for the X-ray facility. DNDi thanks the following donors for funding the Drug Discovery Booster: Federal Ministry of Education and Research (BMBF) through KfW, Germany; Global Health Innovative Technology Fund (GHIT Fund), Japan; Dutch Ministry of Foreign Affairs (DGIS), the Netherlands. DNDi also thanks UK aid, UK; Médecins Sans Frontières International and the Swiss Agency for Development and Cooperation (SDC), Switzerland, for funding its overall mission.
Glossary
Abbreviations Used
- ABT
1-aminobenzotriazole
- ADME
absorption distribution metabolism excretion
- Clint
intrinsic clearance
- HLM
human liver microsomes
- HTS
high-throughput screen
- PO
Per os
- LLE
lipophilic ligand efficiency
- MLM
mouse liver microsomes
- NBS
N-bromosuccinimide
- NTD
neglected tropical disease
- PK
pharmacokinetic
- PPB
plasma protein binding
- RH
rat hepatocytes
- TCP
target candidate profile
- TEAC
tetraethylammonium chloride
- TPSA
topological polar surface area
- VL
visceral leishmaniasis
- WHO
World Health Organization
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.3c00040.
Author Contributions
○ M.D. and Q.J.S. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Organization, W. H. https://www.who.int/data/gho/data/themes/neglected-tropical-diseases (1st September 2021).
- Hotez P. J.; Fenwick A.; Savioli L.; Molyneux D. H. Rescuing the bottom billion through control of neglected tropical diseases. Lancet 2009, 373, 1570–1575. 10.1016/S0140-6736(09)60233-6. [DOI] [PubMed] [Google Scholar]
- Pisarski K. The Global Burden of Disease of Zoonotic Parasitic Diseases: Top 5 Contenders for Priority Consideration. Trop. Med. Infect. Dis. 2019, 4, 44. 10.3390/tropicalmed4010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore E.; Lockwood D. Treatment of visceral leishmaniasis. J. Glob. Infect. Dis. 2010, 2, 151–158. 10.4103/0974-777X.62883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno K.; Burrows J. N.; Duncan K.; van Huijsduijnen R. H.; Kaneko T.; Kita K.; Mowbray C. E.; Schmatz D.; Warner P.; Slingsby B. T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015, 14, 751–758. 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- Akao Y.; Canan S.; Cao Y.; Condroski K.; Engkvist O.; Itono S.; Kaki R.; Kimura C.; Kogej T.; Nagaoka K.; Naito A.; Nakai H.; Pairaudeau G.; Radu C.; Roberts I.; Shimada M.; Shum D.; Watanabe N.-A.; Xie H.; Yonezawa S.; Yoshida O.; Yoshida R.; Mowbray C.; Perry B. Collaborative virtual screening to elaborate an imidazo[1,2-a]pyridine hit series for visceral leishmaniasis. RSC Med. Chem. 2021, 12, 384–393. 10.1039/D0MD00353K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokel G. W.; Widera R. P.; Weber W. P. Phase-Transfer Hofmann Carbylamine Reaction: tert-butyl isocyanide. Org. Synth. 2003, 55, 96–99. 10.1002/0471264180.os055.25. [DOI] [Google Scholar]
- Ansari A. J.; Sharma S.; Pathare R. S.; Gopal K.; Sawant D. M.; Pardasani R. T. Solvent–free Multicomponent Synthesis of Biologically–active Fused–imidazo Heterocycles Catalyzed by Reusable Yb(OTf)3 Under Microwave Irradiation. ChemistrySelect 2016, 1, 1016–1021. 10.1002/slct.201600241. [DOI] [Google Scholar]
- Yin J.; Buchwald S. L. Pd-Catalyzed Intermolecular Amidation of Aryl Halides: The Discovery that Xantphos Can Be Trans-Chelating in a Palladium Complex. J. Am. Chem. Soc. 2002, 124, 6043–6048. 10.1021/ja012610k. [DOI] [PubMed] [Google Scholar]
- Di L.; Keefer C.; Scott D. O.; Strelevitz T. J.; Chang G.; Bi Y.-A.; Lai Y.; Duckworth J.; Fenner K.; Troutman M. D.; Obach R. S. Mechanistic insights from comparing intrinsic clearance values between human liver microsomes and hepatocytes to guide drug design. Eur. J. Med. Chem. 2012, 57, 441–448. 10.1016/j.ejmech.2012.06.043. [DOI] [PubMed] [Google Scholar]
- van de Waterbeemd H.; Smith D. A.; Beaumont K.; Walker D. K. Property-Based Design: Optimization of Drug Absorption and Pharmacokinetics. J. Med. Chem. 2001, 44, 1313–1333. 10.1021/jm000407e. [DOI] [PubMed] [Google Scholar]
- Di L.; Kerns E. H.. Metabolic Stability. In Drug-Like Properties (Second Edition), Di L.; Kerns E. H., Eds.; Academic Press: Boston, 2016, 161–194. [Google Scholar]
- Nassar A.-E. F.; Kamel A. M.; Clarimont C. Improving the decision-making process in the structural modification of drug candidates: enhancing metabolic stability. Drug Discov. Today 2004, 9, 1020–1028. 10.1016/S1359-6446(04)03280-5. [DOI] [PubMed] [Google Scholar]
- Timmins G. S. Deuterated drugs: where are we now?. Expert Opin. Ther. Pat. 2014, 24, 1067–1075. 10.1517/13543776.2014.943184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalgutkar A. S. Designing around Structural Alerts in Drug Discovery. J. Med. Chem. 2020, 63, 6276–6302. 10.1021/acs.jmedchem.9b00917. [DOI] [PubMed] [Google Scholar]
- Morgenthaler M.; Schweizer E.; Hoffmann-Röder A.; Benini F.; Martin R. E.; Jaeschke G.; Wagner B.; Fischer H.; Bendels S.; Zimmerli D.; Schneider J.; Diederich F.; Kansy M.; Müller K. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amine Basicities. ChemMedChem 2007, 2, 1100–1115. 10.1002/cmdc.200700059. [DOI] [PubMed] [Google Scholar]
- Nielsen J.; Wang F.; Graff C.; Kanters J. K. QT dynamics during treatment with sertindole. Ther. Adv. Psychopharmacol. 2015, 5, 26–31. 10.1177/2045125314560738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissner S.; Kroll K.; Dunkel M.; Senger C.; Goldsobel G.; Kuzman D.; Guenther S.; Winnenburg R.; Schroeder M.; Preissner R. SuperCYP: a comprehensive database on Cytochrome P450 enzymes including a tool for analysis of CYP-drug interactions. Nucleic Acids Res. 2010, 38, D237–D243. 10.1093/nar/gkp970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins J. M.; Fedele V.; Szklarz M.; Abdul Azeez K. R.; Salah E.; Mikolajczyk J.; Romanov S.; Sepetov N.; Huang X.-P.; Roth B. L.; Al Haj Zen A.; Fourches D.; Muratov E.; Tropsha A.; Morris J.; Teicher B. A.; Kunkel M.; Polley E.; Lackey K. E.; Atkinson F. L.; Overington J. P.; Bamborough P.; Müller S.; Price D. J.; Willson T. M.; Drewry D. H.; Knapp S.; Zuercher W. J. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol. 2016, 34, 95–103. 10.1038/nbt.3374. [DOI] [PubMed] [Google Scholar]
- Callahan J. F.; Burgess J. L.; Fornwald J. A.; Gaster L. M.; Harling J. D.; Harrington F. P.; Heer J.; Kwon C.; Lehr R.; Mathur A.; Olson B. A.; Weinstock J.; Laping N. J. Identification of Novel Inhibitors of the Transforming Growth Factor β1 (TGF-β1) Type 1 Receptor (ALK5). J. Med. Chem. 2002, 45, 999–1001. 10.1021/jm010493y. [DOI] [PubMed] [Google Scholar]
- Kana Kon-I, C.-g. M. M., Chita-gun; Akiko Shima, Chita-gun . Sulfonyl Benzimidazole Derivatives. US 7,598,393 B2, 2009.
- Striela R.; Urbelis G.; Su̅džius J.; Stončius S.; Sadzevičienė R.; Labanauskas L. Synthesis of bromocyclopropylpyridines via the Sandmeyer reaction. Tetrahedron Lett. 2017, 58, 1681–1683. 10.1016/j.tetlet.2017.03.029. [DOI] [Google Scholar]
- Zhang S.; Yuan W.; Qin Y.; Zhang J.; Lu N.; Liu W.; Li H.; Wang Y.; Li Y. Bidentate BODIPY-appended 2-pyridylimidazo[1,2-a]pyridine ligand and fabrication of luminescent transition metal complexes. Polyhedron 2018, 148, 22–31. 10.1016/j.poly.2018.03.023. [DOI] [Google Scholar]
- Klug D. M.; Tse E. G.; Silva D. G.; Cao Y.; Chauhan J.; Dichiara M.; Drewry D.; Emery F. D. S.; Evans L.; Simpson Q.; Graves L.; Lorente-Macías Á.; Charman S. A.; Perry B.; Quiroga B.; Quotadamo A.; Sabatino G.; Sama A.; Schätzlein A.; Shanu-Wilson J.; Ferrins L.; Sjö P.; Stapleton P.; Swain C.; Vaideanu A.; Xie H.; Zuercher B.; Todd M. H.. Open Source Antibiotics - Simple Diarylimidazoles are Potent Against Methicillin Resistant Staphylococcus Aureus. 2022, Submitted, 10.1121/10.0015246. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.