

Abstract
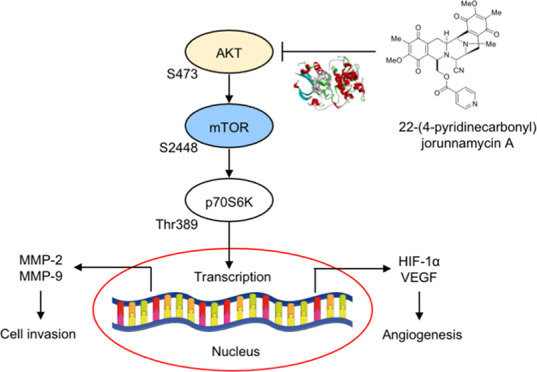
Non-small-cell lung cancer (NSCLC), the most prevalent form of lung cancer, is associated with an unfavorable prognosis owing to its high rate of metastasis. Thus, the identification of new drugs with potent anticancer activities is essential to improve the clinical outcome of this disease. Marine organisms exhibit a diverse source of biologically active compounds with anticancer effects. The anticancer effects of jorunnamycin A (JA) derived from the Thai blue sponge (Xestospongia sp.) and 22-(4′-pyridinecarbonyl) jorunnamycin A (22-(4′-py)-JA), the semisynthetic derivative of JA, have been reported. The present study aimed to investigate the impact of 22-(4′-py)-JA on NSCLC metastasis using in vitro, in vivo, and in silico approaches. The JA derivative inhibited tumor cell invasion and tube formation in human umbilical vein endothelial cells (HUVECs). The computational analysis demonstrated strong and stable interactions between 22-(4′-py)-JA and the AKT protein. Further examinations into the molecular mechanisms revealed the suppression of AKT/mTOR/p70S6K signaling by 22-(4′-py)-JA, leading to the downregulation of matrix metalloproteinases (MMP-2 and MMP-9), hypoxia-inducible factor-1α (HIF-1α), and vascular endothelial growth factor (VEGF). Furthermore, 22-(4′-py)-JA suppressed in vivo metastasis by decreasing the number of colonies in the lung. These findings indicated the antimetastasis activity of 22-(4′-py)-JA, which might prove useful for further clinical applications.
Keywords: 22-(4′-py)-JA, invasion, angiogenesis, metastasis, non-small-cell lung cancer
Lung cancer exhibits a leading cause of cancer-related mortality worldwide, and non-small-cell lung cancer (NSCLC) is the predominant subtype observed in lung cancer patients.1 Surgical resection serves as the primary treatment for early-stage NSCLC, and chemotherapy and radiotherapy play important roles as adjuvant therapies administered before and after surgery.2 However, the clinical outcome of advanced NSCLC remains poorly understood. Despite significant advances in the treatment of this disease, several severe undesired effects have been reported.3,4 The five-year mortality rate of patients with NSCLC exceeds 80% primarily due to the high metastatic potential of this tumor.1 Therefore, studies on developing a new anticancer drug are crucial to overcome and treat these exceedingly metastatic tumors.
The process of cancer metastasis is a highly coordinated and sequential series of events, beginning with the dissemination of cells from the primary site, followed by the local degradation of the surrounding basement membrane through proteolytic activity. The detached cells consequently intravasate and circulate within the blood and lymphatic systems, eventually extravasating and establishing secondary tumors at distant sites.5 Cancer invasion requires a group of proteolytic enzymes known as matrix metalloproteinases (MMPs), which degrade the components of the extracellular matrix (ECM).6 Accumulative studies have demonstrated a strong association between the overexpression of MMPs, specifically MMP-2 and MMP-9, and cancer metastasis.6,7 Enlargement of the primary tumor triggers the formation of new blood vessels, a process known as angiogenesis. Cancer cells respond to a hypoxic microenvironment by releasing angiogenic factors, particularly vascular endothelial growth factor (VEGF). These factors mediate the proliferation and differentiation of endothelial cells.8 In lung cancer patients, increased VEGF expression has been reported and is correlated with an unfavorable prognosis.9,10 The transcription of VEGF is regulated by HIF-1α, a protein frequently dysregulated and overexpressed in cancers, particularly in metastatic cells.11,12 HIF-1α is controlled by the protein kinase B (AKT)/mammalian target of the rapamycin (mTOR) signaling,13,14 which serves as an essential pathway that regulates cancer metastasis.15 Activation of AKT stimulates the phosphorylation of downstream effectors, such as mTOR (serine 2448) and p70S6K (threonine 389), which induce the transcription of several genes involved with metastasis regulation, including HIF-1α and MMPs.13,15,16 Inhibition of the AKT/mTOR signaling pathway was able to decrease the metastatic potential of the cancer cells by downregulating MMPs.17 Consequently, targeting the AKT/mTOR/p70S6K signaling pathway represents a promising therapeutic strategy for attenuating cancer metastasis.
A significant number of marine derivatives have demonstrated notable antitumor activity.18−20 Trabectedin, cytarabine, and lurbinectedin obtained from marine natural products have been approved as novel anticancer agents.21−23 Additionally, renieramycin M, renieramycin T, and jorunnamycin A (JA) from Xetospongia sp. were reported to display significant cytotoxicity, attenuation of tumor growth and metastasis, and inhibition of stemness phenotypes in previous studies.19,20,24−26 Due to highly potent cytotoxicity, JA was further structurally modified. Derivatives of JA with fluorobenzoyl and trifluoromethyl benzoyl substitutions were synthesized, which resulted in a significant ten-fold increase in cytotoxicity compared to the original compound.27 However, in contrast, the leaving group at the C-22 position of JA is crucial for its cytotoxic activity. According to the structure of trabectedin, the incorporation of a 4′-pyridine carbonyl ester moiety remarkably led to the most substantial improvement in cytotoxic activity among all of the derivatives tested.27 The incorporation of the 4′-pyridine carbonyl group at the C-22 position led to the formation of the 22-(4′-py)-JA derivative (Figure 1A), which displayed a strong cytotoxic activity relative to the other derivatives27 and induced apoptosis through an ERK/Bcl-2-dependent mechanism.28 However, the impact of 22-(4′-py)-JA on cancer metastasis remains unknown. The objective of this study was to evaluate the antimetastatic activity of 22-(4′-py)-JA, specifically its effect on the invasiveness and angiogenesis using in vitro NSCLC culture and in vivo metastasis model. Furthermore, the underlying mechanisms, particularly the involvement with AKT/mTOR signaling, were investigated through biochemical analysis and an in silico molecular docking experiment.
Figure 1.

Cytotoxicity of 22-(4′-pyridinecarbonyl) jorunnamycin A (22-(4′-py)-JA) and jorunnamycin A (JA) in the H460 and A549 cells. (A) Chemical structures of 22-(4′-py)-JA and JA. (B) H460 and (C) A549 cells were treated with various concentrations of 22-(4′-py)-JA or JA for 24, 48, and 72 h and cell viability was measured using the MTT assay and presented as a percentage of the cell viability. (D) IC50 value of 22-(4′-py)-JA and JA in H460 and A549 cells. (E) H460 and (F) A549 cells were incubated with nontoxic concentrations of 22-(4′-py)-JA for 24 and 48 h and cell proliferation was examined using the MTT assay and presented as a percentage of cell proliferation. Data are presented as the mean ± SEM (n = 3). *p < 0.05 vs untreated control cells. All data were analyzed by using analysis of variance (ANOVA) followed by the Tukey post hoc test.
Results
Cytotoxicity of 22-(4′-py)-JA on NSCLC Cells
The cytotoxicity of 22-(4′-py)-JA and JA on NSCLCs was evaluated using the MTT assay. H460 and A549 cells were treated with varying concentrations (0–1 μM) of the compounds for 24, 48, and 72 h. The results demonstrated significant reductions in the number of viable cells for both compounds at all time points. Notably, 22-(4′-py)-JA exhibited significant cytotoxicity at lower concentrations (5 and 10 nM) in H460 and A549 cells after 24 h, with 50% inhibitory concentrations (IC50) of 810 ± 33 and 835 ± 30 nM, respectively, while IC50 of JA was more than 1000 nM for both cells (Figure 1B–D). At 48 and 72 h, the reduction in cell viability following treatment with 22-(4′-py)-JA was more pronounced than that seen after treatment with a similar concentration of JA, thus indicating the greater potency of the JA derivative. The antiproliferative effect of 22-(4′-py)-JA was investigated using nontoxic doses (≤1 nM) to minimize the interference of the cytotoxic effect of the compound. However, 22-(4′-py)-JA did not significantly affect NSCLC proliferation in the H460 and A549 cells (Figure 1E,F).
22-(4′-py)-JA Represses the Invasiveness of NSCLC Cells by Downregulating the MMPs
The effect of 22-(4′-py)-JA on NSCLC cell invasion was evaluated using the transwell invasion assay, wherein H460 and A549 cells were incubated with a nontoxic concentration range of 0–1 nM of 22-(4′-py)-JA. The results demonstrated that at a dose of ≥0.5 nM, 22-(4′-py)-JA significantly reduced the number of invading cells (Figure 2A), whereas the same concentration of JA did not demonstrate any significant suppressive activity (Figure 2B).
Figure 2.

Fluorescent images and bar graphs showing the effects of JA and 22-(4′-py)-JA on NSCLC cell invasion. Following the transwell invasion assay, the invaded cells were identified by staining with DAPI and imaged by fluorescence microscopy. (A) 22-(4′-py)-JA (dose, ≥0.5 nM) decreased the number of invading H460 and A549 cells. (B) Same concentration of JA did not demonstrate any significant suppressive activity. Scale bar, 10 μm. (C) Cells were incubated with nontoxic doses of 22-(4′-py)-JA for 24 h, and MMP-2 and MMP-9 mRNA expressions were evaluated by qRT-PCR. (D) MMP-2 and MMP-9 activities assessed by gelatin zymography analysis revealed a marked reduction in the gelatinase activities of both enzymes. Data are presented as mean ± SEM (n = 3). *p < 0.05 vs untreated control group. All data were analyzed by using analysis of variance (ANOVA) followed by the Tukey post hoc test.
MMPs are required for degrading the extracellular matrix surrounding cancer cells, particularly MMP-2 and MMP-9 associated with an invasiveness of cancer cells;6,7 the impact of 22-(4′-py)-JA on MMP-2 and MMP-9 mRNA expression was investigated. The present study showed that 22-(4′-py)-JA significantly downregulated the levels of MMP-2 and MMP-9 mRNA expression, which corresponded with their anti-invasive activities (Figure 2C). Furthermore, zymography experiments revealed a marked decrease in the gelatinase activities of MMP-2 and MMP-9 (Figure 2D), indicating that 22-(4′-py)-JA inhibits cancer cell invasion by suppressing MMP-2 and MMP-9 expression.
22-(4′-py)-JA Inhibits Angiogenesis in Human Endothelial Cells by Downregulating the Hif-1α and Vegf Expressions in NSCLC
Next, the effect of 22-(4′-py)-JA on angiogenesis was investigated. Human umbilical vein endothelial cells (HUVECs) were exposed in conditioned media obtained from H460 and A549 cells treated with or without 22-(4′-py)-JA. The tube formation parameters, i.e., the number of nodes and fragments and the tube length, were analyzed (Figure 3A). The results revealed that 22-(4′-py)-JA attenuated the tube formation ability of HUVECs compared to that of the controls (Figure 3B,C), suggesting that 22-(4′-py)-JA could attenuate angiogenesis. Since VEGF secreted by cancer cells in response to a low oxygen environment was required for mediating new vascularization in endothelial cells, the expression levels of Hif-1α and Vegf in NSCLCs was examined. The data indicated that treatment with 22-(4′-py)-JA led to a gradual decrease in the mRNA expression levels of Hif-1α and Vegf in H460 and A549 cells (Figure 3D,E), indicating that 22-(4′-py)-JA attenuated Hif-1α and Vegf expressions in cancer cells, thereby suppressing an angiogenic activity in endothelial cells.
Figure 3.

Effects of 22-(4′-py)-JA on tube formation in HUVECs. (A) Schematic diagram demonstrates the parameters for tube formation analysis (i.e., the number of nodes and fragments and their length). HUVECs were treated with the medium obtained from (B) H460 cells and (C) A549 cells. After 24 h, tube formation was imaged, and its parameters were quantified using ImageJ software. Plots present the fold change of node number, fragment number, and fragment length. Scale bar, 100 μm. Bar graphs showing the mRNA expression levels of (D) Hif-1α and (E) Vegf in H460 and A549 cells incubated with or without a nontoxic dose of of 22-(4′-py)-JA for 24 h. Data are presented as mean ± SEM (n = 3). *p < 0.05 vs untreated control group. All data were analyzed by using analysis of variance (ANOVA) followed by the Tukey post hoc test.
22-(4′-py)-JA Inhibits the AKT/mTOR Pathway
The results demonstrated that treatment with 22-(4′-py)-JA gradually downregulated phosphorylated AKT (p-AKTS473), p-mTORS2448, and p-p70S6KT389 in both H460 and A549 cells, whereas their total forms remained unchanged (Figure 4A,B). However, 22-(4′-py)-JA did not have a suppressive effect on p-PI3K and p-ERK1/2 (Figure S1). These findings suggested that 22-(4′-py)-JA particularly targets AKT signaling. Furthermore, the same concentration of JA showed no effects on p-AKTS473, p-mTORS2448, and p-p70S6KT389 levels (Figure S2), thus indicating the greater efficacy of the JA derivative compared to its parent compound.
Figure 4.

Effects of of 22-(4′-py)-JA on the AKT/mTOR/p70S6K signaling pathway. Western blot images and bar graphs showing the levels of p-AKT, AKT, p-mTOR, mTOR, and p-p70S6K relative to the control in (A) H460 and (B) A549 cells treated with or without nontoxic concentrations of 22-(4′-py)-JA for 24 h. Western blot images and bar graphs showing the levels of p-AKT, AKT, p-mTOR, mTOR, and p-p70S6K relative to the control in (C) H460 and (D) A549 cells pretreated with either EGF (100 ng/mL) or LY294002 (10 μM) for 1 h and incubated with 1 nM 22-(4′-py)-JA for 24 h. The upregulation of p-AKTS473, p-mTOR S2448, and p-p70S6KT389 mediated by EGF was reversely declined in response to 22-(4′-py)-JA in both cell types. Data are expressed as mean ± SEM (n = 3). *p < 0.05 vs untreated control group. #p < 0.05 vs indicated group. All data were analyzed by using analysis of variance (ANOVA) followed by the Tukey post hoc test.
To confirm the underlying mechanism involved in this phenomenon, the H460 and A549 cells were incubated with 22-(4′-py)-JA after pretreatment with the epidermal growth factor (EGF), an upstream activator of the receptor tyrosine kinase, or LY294002, an AKT inhibitor. Western blot analysis demonstrated that the upregulation of p-AKTS473, p-mTORS2448, and p-p70S6KT389 mediated by EGF was reversely declined in response to 22-(4′-py)-JA (Figure 4C,D). In contrast, the inhibitory effect of LY294002 on these phosphorylated molecules was notably pronounced when cotreated with 22-(4′-py)-JA. These findings suggest that 22-(4′-py)-JA mediated its antimetastasis effects by suppressing AKT/mTOR signaling in NSCLC.
22-(4′-py)-JA Interacts with AKT
We next hypothesized that the inhibition of AKT phosphorylation by 22-(4′-py)-JA was attributed to its binding with AKT. To confirm this, an in silico molecular docking experiment was conducted between 22-(4′-py)-JA and AKT (PDB: 3MVH). The docking score of 22-(4′-py)-JA with AKT at the ATP-binding site was −9.3 kcal/mol and the ligand efficiency was −0.211 kcal/mol/heavy atom (Figure 5A,B), while the binding energy and ligand efficacy of JA were −8.1 and −0.225 kcal/mol/heavy atom, respectively (Figure S3). Both 22-(4′-py)-JA and JA bound to AKT through hydrogen bonding and van der Waals, hydrophobic, and electrostatic interactions (Table S1), with 22-(4′-py)-JA having a higher number of bonding to AKT in correspondence to the binding energy. The interaction of 22-(4′-py)-JA and AKT was then subjected to molecular dynamic analysis. The complex between 22-(4′-py)-JA and AKT remained stable during the equilibrium phase for 100 ns, and the average root-mean-square deviation (RMSD) of the ligand conformation was 1.49 ± 0.01 Å. These findings indicated that 22-(4′-py)-JA interacted with AKT and interfered with AKT phosphorylation.
Figure 5.

Molecular docking analysis and dynamic interaction of 22-(4′-py)-JA with AKT1. Interactions between 22-(4′-py)-JA and AKT1 (PDB ID: 3MVH) are shown in (A) 3D and (B) 2D images. Molecular docking was performed using PyRx Autodock Vina, and schematization of the interaction between 22-(4′-py)-JA and AKT1 was obtained using the BIOVIA Discovery Studio Visualizer. (C) RMSD ligand conformation represents the stable interaction between 22-(4′-py)-JA and AKT1.
22-(4′-py)-JA Inhibits Lung Cancer Metastasis In Vivo
To confirm the antimetastatic effect of 22-(4′-py)-JA, an in vivo tail vein metastasis assay was conducted using A549 cells injected into the tail vein of nude mice. The next day, mice were administrated 22-(4′-py)-JA (1 mg/kg body weight), erlotinib (100 mg/kg body weight), or a vehicle (5% DMSO in phosphate-buffered saline (PBS)) (Figure 6A). No significant differences in body weights were observed among the treatment groups (Figure 6B). The serum profiles of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) at the end of the experiment were found to be within the normal ranges (Figure 6C), thus indicating that 22-(4′-py)-JA had less hepatotoxicity. Interestingly, the number of metastatic nodules was significantly reduced in the 22-(4′-py)-JA and erlotinib (a positive control) groups compared to those in the vehicle control group (Figure 6D,E). Microscopic examination using hematoxylin and eosin staining confirmed a high density of metastatic tumor cells in the lung tissue of the vehicle group, which was remarkably lower in the 22-(4′-py)-JA and erlotinib groups (Figure 6F). These findings confirmed the potent antimetastatic activity of 22-(4′-py)-JA in NSCLC.
Figure 6.

Effects of 22-(4′-py)-JA on metastasis in vivo. (A) Timeline overview of the in vivo metastasis experiment. The tail veins of female BALB/C nude mice were intravenously injected with A549 cells (5 × 106 cells), followed by the intraperitoneal injection of either 22-(4′-py)-JA or a vehicle (5% DMSO in PBS) the next day (twice a week), or erlotinib administered orally (once per week). After 6 weeks, the lungs and blood were collected for biochemical analyses. (B) Body weights of the mice throughout the experiment showed no significant differences. (C) Serum profiles of AST and ALT at the end of the experiment were found to be within the normal ranges. (D) The number of metastatic nodules was significantly decreased in the 22-(4′-py)-JA and erlotinib groups compared to those in the vehicle control group. Data are presented as a percentage of mean ± SEM (n = 6). *p < 0.05 vs the vehicle group. (E) Macroscopic images showing the reduction of lung nodules in the 22-(4′-py)-JA and erlotinib groups compared to those in the vehicle control group. (F) Hematoxylin and eosin staining demonstrated a high density of metastatic tumor cells in the lung tissue in the vehicle group, which was significantly lower than those in the 22-(4′-py)-JA and erlotinib groups. Arrowheads indicate the tumors formed on the lungs. Scale bar, 200 μm for the upper panel, and 100 μm for the lower panel.
Discussion
The discovery of an effective therapeutic for NSCLC is urgently needed due to the rapid progression of the disease, especially cancer metastasis. The natural compound 22-(4′-py)-JA semisynthesized from the marine sponge Xestospongia sp. exhibits a promising anticancer activity.19 Evidence from in vitro studies shows that 22-(4′-py)-JA induces apoptosis in multiple NSCLC cell lines via an ERK/Bcl-2-dependent mechanism.28 The present study explores an antimetastasis activity and elucidates its underlying molecular mechanism. The results indicate that 22-(4′-py)-JA effectively inhibits in vitro invasion and angiogenesis and in vivo metastasis through the AKT/mTOR pathway. Its efficacy was more potent than erlotinib with the effective dose in a micromolar range (Figure S4). This EGFR inhibitor is commonly employed in the therapeutic management of NSCLC. However, this drug does not exert direct inhibitory effects on AKT and mTOR, and the suppression of EGFR activity can effectively attenuate downstream signaling cascades involving AKT and mTOR.29 This finding emphasized the potential of 22-(4′-py)-JA for further exploration in drug research and development with clinical implications.
Numerous studies have consistently reported the significant involvement of MMPs in tumorigenesis, contributing the establishment of a complex microenvironment that supports malignant transformation within lung tissue.30,31 Consequently, the inhibition of MMP expression or activity has emerged as an interesting strategy employed by several anticancer agents.17,32,33 Erianthridin effectively suppressed both in vitro and in vivo lung cancer metastasis by downregulating MMP-2 and MMP-9, together with their respective activities.17 The reduction of both MMP-2 and MMP-9 expressions and gelatinolytic activities by sinulariolide and stellettin B successfully impeded cancer cell migration and invasion in bladder cancer and hepatocellular carcinoma cells.32,33 Furthermore, this study also substantiates the ability of 22-(4′-py)-JA to downregulate MMP-2 and -9, consequently inhibiting cancer cell invasion.
Angiogenesis plays an essential role in facilitating cancer metastasis by providing necessary nutrients and prosurvival factors to the tumor as well as establishing a pathway for tumor cells to metastasize to distant sites.34 In response to the hypoxic microenvironment, primary tumors release growth factors and cytokines to stimulate endothelial growth and differentiation within the tumor mass. HIF-1α, a member of the transcription factor family, is activated under low oxygen conditions, which participated in the regulation of angiogenesis by inducing the transcription of Vegf in cancer cells.11,12 VEGF, a crucial growth factor, binds to its receptor on endothelial cells, promoting their proliferation and differentiation and leading to new blood vessel formation.9 Elevated levels of VEGF have been observed in the serum of patients with various cancer types, including NSCLC,10,11 and it is well recognized that HIF-1α plays a pivotal role in regulating this phenomenon.12 Inhibition of HIF-1α/VEGF expression has been shown to reduce vascularization in cancer,35−37 suggesting a potential antiangiogenic activity of 22-(4′-py)-JA through an HIF-1α/VEGF mechanism.
Several studies have highlighted the role of AKT/mTOR/p70S6K signaling pathway in the regulation of cancer metastasis, with its overactivation being observed in lung cancer.15,38 Activation of this signaling pathway enhances the phosphorylation of mTOR and its downstream target, p70S6K, initiating cascades that upregulate various oncogenes, including MMPs, HIF-1α, and VEGF. The upregulation of these proteins is associated with increased invasiveness and angiogenesis in cancers.39,40 Inhibition of AKT activity has been shown to result in the downregulation of MMP-2 and MMP-9, consequently impeding cancer cell metastasis.17,32 Additionally, AKT inhibition has been reported to attenuate the EGFR-mediated increase in VEGF and the endothelial tube-forming process.41,42 In the context of this study, 22-(4′-py)-JA exhibits molecular signaling in lung cancer metastasis through the AKT/mTOR mechanism, effectively inhibiting EGF-mediating AKT/mTOR activation. It is worth noting that there is a crosstalk between AKT and ERK signaling,43 and a recent study demonstrated that 22-(4′-py)-JA suppressed ERK activity, contributing to apoptosis induction.28 However, in the present study, the phosphorylated ERK level was unaffected by a low dose of 22-(4′-py)-JA in suppressing cancer invasion and angiogenesis. This discrepancy may be potentially attributed to the concentration-dependent efficacy of the compound. In pharmacology, it is widely recognized that compounds can selectively target specific signaling pathways or molecular targets at lower doses, resulting in the suppression of specific cellular processes. As the dose increases, the compound’s effects may extend to additional signaling pathways or targets, exhibiting a more widespread impact. This phenomenon, known as a dose–response relationship, is commonly observed.44 Furthermore, it is important to consider that ERK signaling has an upstream regulator that may be regulated differently.45 At low doses, 22-(4′-py)-JA might not be sufficiently potent to influence this upstream regulator, thereby not affecting the ERK pathway in the present study.
The concept of molecular docking can be used to accurately anticipate the optimal orientation of a ligand on its target at the atomic level, facilitating a detailed understanding of the ligand’s behavior at the binding site of the target protein.44 This approach provides valuable insights into the underlying biochemical processes and aids in the development of new drugs with improved selectivity and effectivity.46 Our finding demonstrated that 22-(4′-py)-JA exhibits stable interaction with AKT, characterized by more negative energy values and stronger bonds compared to JA. A great negative energy value indicates better ligand-binding properties.17,47 This suggests that 22-(4′-py)-JA may exert a more pronounced suppressive effect on AKT activation. Furthermore, the complex formed by 22-(4′-py)-JA and AKT demonstrated less fluctuation in the RMSD value throughout the 100 ns stimulation, indicating a stable interaction between the compound and protein target.48,49 The investigation of the interaction between AKT and 22-(4′-py)-JA in an in vitro setting would indeed provide further support for our findings. Collectively, the present study provided crucial preclinical evidence of the antimetastatic activity and molecular mechanism of 22-(4′-py)-JA in NSCLC. These findings suggest that 22-(4′-py)-JA might be considered as a significant candidate for further drug research and development.
In relation to the pharmacokinetic parameters encompassing absorption, distribution, metabolism, excretion, and toxicity, it is worth noting that both compounds fall within the acceptable range as indicated in Table S2. These compounds demonstrate the potential to serve as substrates for CYP3A4 and undergo metabolism in the liver. Therefore, it is crucial to identify their respective metabolites in order to determine whether they may play a role in mediating the anticancer effects of the compounds. Although both compounds may induce hepatotoxicity, 22-(4′-py)-JA exhibits a lower level of oral rat chronic toxicity. Taking into account its high molecular weight and low water solubility, further study aims to develop a formulation of the compound as a lyophilized powder suitable for intravenous injection. This approach will enhance compound stability, minimize the impact of first-pass metabolism, and improve drug bioavailability. Nevertheless, further investigation is required to obtain a more comprehensive understanding of the pharmacokinetic and physiochemical characteristics associated with these compounds.
Conclusions
The current study highlights the remarkable antimetastatic properties of 22-(4′-py)-JA in NSCLC through in vitro, in vivo, and in silico approaches. With potent efficacy demonstrated at the nanoscale level, 22-(4′-py)-JA effectively inhibited cell invasion in NSCLC and angiogenesis in HUVECs. These effects were attributed to the compound’s interaction with AKT and subsequently suppression of the AKT/mTOR/p70S6K signaling pathway. Consequently, the downstream targets including MMPs, Hif-1α, and Vegf were downregulated. These findings provide valuable insights into the potential of 22-(4′-py)-JA as a novel compound, which can be further explored for its clinical application in the management of metastasis in NSCLC.
Material and Methods
Preparation of 22-(4′-py)-JA
22-(4′-py)-JA (Figure 1A) was semisynthesized from JA (25 mg) with slight modification by mixing with 4-dimethylaminopyridine (DMAP) (30.9 mg), 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI·HCl) (48.56 mg), and isonicotinoyl chloride (45.08 mg). The stock solution was prepared by solubilizing 22-(4′-py)-JA in dimethyl sulfoxide (DMSO; Merck Millipore, Billerica, MA, USA), which was further diluted with cell culture medium to achieve the desired working concentration for experimental use. The concentration of DMSO was less than 0.5% and was nontoxic to the cells.
Cell Culture
Human NSCLC cell lines, H460 and A549, as well as HUVEC cells were obtained from the American Type Culture Collection (Manassas, VA). H460 cells were cultured in Roswell Park Memorial Institute (RPMI) 1640, A549 cells were maintained in Dulbecco’s modified Eagle medium, and the HUVECs were cultivated in M199. All culture media were supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, and 100 U/mL penicillin/streptomycin. The media and supplements were purchased from GIBCO (Grand Island, NY). All cells were incubated at 37 °C in a humidified atmosphere with 5% CO2.
Cell Viability Assay
Cell viability was assessed using the MTT (Invitrogen, Carlsbad, CA) colorimetric assay. A549 and H460 cells (5 × 103 cells/well) were seeded onto 96-well plates and allowed to adhere overnight. Subsequently, cells were treated with various concentrations of 22-(4′-py)-JA (ranging from 0 to 1 μM) for 24, 48, and 72 h. After the treatment period, the cells were incubated with MTT solution (0.5 mg/mL) at 37 °C for 4 h. The formed purple formazan crystal was dissolved in DMSO (100 μL/well), and the intensity was measured at 570 nm using a microplate reader (Victor, PerkinElmer). The mean optical density in each experimental group was calculated as a percentage of the cell viability, with the control group set as 100%.
Cell Proliferation Assay
The A549 and H460 cells (2 × 103 cells/well) were seeded onto 96-well microplates and allowed to attach to the plate overnight. Then, the cells were treated with 22-(4′-py)-JA (0–1 nM) for 24 and 48 h, following which MTT solution (0.5 mg/mL) was added to each well and incubated for another 4 h at 37 °C. The formazan crystals were dissolved by DMSO, and the intensity was measured at 570 nm using a microplate reader (Victor, PerkinElmer) as described above. The mean optical density was calculated as a percentage of cell proliferation.
Transwell Invasion Assay
The A549 and H460 cells (5 × 104 cells/well) were cultured in the upper chamber of a 24-well transwell plate consisting of a membrane insert coated with 10 μg/mL of Matrigel (BD Biosciences, Bedford, MA), and subsequently treated with 22-(4′-py)-JA (ranging from 0 to 1 nM) in serum-free medium, while the lower chamber of the transwell plate contained cell culture medium supplemented with 10% FBS. After 24 h of incubation, the cells that remained in the upper chamber were removed. The cells that had invaded through the Matrigel-coated membrane and reached the lower side of the insert were fixed and stained with DAPI (Sigma-Aldrich Corporation, St. Louis, MO) for 30 min. Images of the invaded cells were captured using a fluorescence microscope (Nikon Inverted Microscope Eclipse Ti-U Ti-U/B, NY) from at least five different fields in each group.
Gelatin Zymography Assay
The effects of 22-(4′-py)-JA on the activities of MMP-2 and -9 were assessed by gelatin zymography. A549 and H460 cells (1 × 106 cells/well) were seeded onto a 60 mm culture dish. The cells were treated with various concentrations of 22-(4′-py)-JA (ranging from 0 to 1 nM) for 24 h. After the treatment period, the medium was collected and centrifuged at 2000g for 5 min at 4 °C to remove any cellular debris. The supernatant was transferred to a gel and subjected to gelatin-polyacrylamide gel electrophoresis under nonreducing conditions. The gels were subsequently washed with a solution containing 50 mM Tris-HCl (pH 7.5) and 2.5% Triton X-100, followed by rinsing with 10 mM Tris-HCl (pH 6.8). The gels were then incubated in a gelatinase buffer at 37 °C for 72 h to facilitate gelatin digestion. After incubation, the gels were stained with Coomassie Blue staining solution until clear bands representing gelatinolytic activity by MMPs appeared against a blue background. The clear bands indicating MMP activity were visualized and measured using the ImageJ (NIH) software to quantify the gelatinolytic activity.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
To assess the mRNA expression levels of MMP-2 and -9, qRT-PCR was performed. A549 and H460 cells (3 × 105 cells/well) were seeded onto a 6-well plate. The cells were treated with different concentrations of 22-(4′-py)-JA (ranging from 0 to 1 nM) for 24 h. RNA isolation was performed using the GENEzol reagent (Geneaid Biotech, Shijr, New Taipei, Taiwan), following the manufacturer’s protocol. RNA was reverse transcribed to cDNA using ProtoScript II Reverse Transcriptase (Carlsbad, CA), following which the cDNA was mixed with a specific primer (Table S3) using the SensiFAST SYBR No-ROX Kit (Bioline, Taunton, MA). Amplification of the target genes was conducted using a T100 thermal cycler (Bio-Rad, California) under the following conditions: (1) denaturation at 95 °C for 30 s, (2) extension for 40 cycles at 95 °C for 5 s, and (3) annealing at a temperature based on the specific primer sequences indicated in Table S3 for 10 s. The relative expression levels of the genes were analyzed using the ΔΔCt method.
Tube Formation Assay
HUVECs (1.5 × 104 cells/100 μL) were seeded onto a 96-well plate coated with 80 μL of Corning Matrigel (Fischer Scientific, Loughborough, U.K.) and incubated with conditioned medium obtained from lung cancer cells treated with 22-(4′-py)-JA (0–1 nM) at 37 °C for 24 h. Images of the tube formation were randomly taken from at least three different fields/group using an inverted light microscope (Nikon Inverted Microscope Eclipse Ti-U Ti-U/B, NY). The number of nodes and fragments and their lengths were quantified using the ImageJ (NIH) software.
Western Blot Analysis
Cells at a density of 3 × 105 cells/well were seeded onto 6-well plates and treated with 22-(4′-py)-JA for 24 h. After the treatment, cells were lysed using TMEM lysis buffer supplemented with a protease inhibitor cocktail (Roche Diagnostics). The protein content in the lysates was determined using the bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL). After being separated by sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel electrophoresis, proteins were transferred onto a poly(vinylidene difluoride) (PVDF) membrane (Bio-Rad, California). To block nonspecific binding, the membranes were incubated with 5% non-fat milk, subsequently incubating with specific primary antibodies at 4 °C overnight. After washing, the membranes were incubated with the appropriate secondary antibody at room temperature for 2 h. The following antibodies were used in this study: anti-p-PI3K (no. 4228), anti-PI3K (no. 4257), anti-p-AKT (no. 9271), anti-AKT (no. 9272), anti-p-mTOR (no. 5536), anti-mTOR (no. 2938), anti-p-p70S6K (no. 9234), anti-GAPDH (no. 97166), anti-rabbit horseradish peroxidase (HRP; no. 7074), and anti-mouse HRP (no. 7076), which were obtained from Cell Signaling Technology (Massachusetts). The protein bands were visualized using an enhanced chemiluminescence kit (EMD Millipore, Darmstadt, Germany) and quantified using the ImageJ software (NIH) to analyze the protein expression levels.
Computational Molecular Docking and Molecular Dynamic
The X-ray crystal structure of AKT1 (3MVH) was obtained from the Protein Data Bank (PDB). The two-dimensional (2D) structure of 22-(4′-py)-JA was drawn using ChemDraw Ultra 15.0 (PerkinElmer, Waltham, MA). To convert the 2D structure into a three-dimensional (3D) structure, ChemDraw 3D (PerkinElmer, Waltham, MA) was utilized. The molecular docking of 22-(4′-py)-JA with AKT1 was performed using the PyRx Virtual Screening Tool (version 0.8). The docking process generated multiple ligand conformations, and the conformations with the highest number of clusters were analyzed for their free binding energy (ΔG), which indicates the strength of the ligand–protein interaction. The interactions between the ligand and protein targets were visualized and analyzed using PyMOL (Schrödinger, New York, NY) and BIOVIA Discovery Studio Visualizer 2022 (Biovia, San Diego, CA). The best ligand-target trajectory of docked complexes was analyzed through 100 ns of molecular dynamic simulation by using YASARA dynamics with the AMBER14 plugin.
In Vivo Tail Vein Metastasis
The animal studies conducted in this research were approved by the Institutional Animal Care and Use Committee of Chulalongkorn University (Approval No: 21-33-003). All procedures were carried out following the guidelines and regulations set by the committee. Eighteen female BALB/c nude mice were obtained from Nomura Siam International Co. Ltd (Bangkok, Thailand) and were housed under specific pathogen-free conditions throughout the study. To establish the metastasis model, A549 cells (5 × 106 cells/100 μL PBS) were injected into the tail vein of mice. The following day, mice were randomly divided into three groups (n = 6/group). The groups received either a vehicle (5% DMSO in PBS) via an intraperitoneal (i.p.) injection twice a week, 22-(4′-py)-JA at a dose of 1 mg/kg BW via i.p. injection twice a week, or erlotinib at a dose of 100 mg/kg BW via oral administration once a week. After 6 weeks of treatment, the mice were anesthetized, and the lungs were excised. The lung nodules or metastatic colonies were counted using the dissecting microscope. Subsequently, the lung tissues were fixed, sectioned, and stained with hematoxylin and eosin. Histological analyses were performed on randomly selected sections using an inverted light microscope (Nikon Inverted Microscope Eclipse Ti-U Ti-U/B, NY).
Statistical Analysis
All experimental data were analyzed and presented as means ± standard error of the mean (SEM) derived from at least three independent experiments. Statistical analysis was performed using Prism 9 (San Diego, CA). One-way analysis of variance (ANOVA) followed by a post hoc test was conducted to analyze the obtained results. A p-value of less than 0.05 was considered statistically significant, indicating a significant difference between groups or conditions.
Acknowledgments
This research was supported by Fundamental Fund, Thailand Science Research and Innovation Fund, Chulalongkorn University (HEA663300005, to V.P.) and The Second Century Fund, Chulalongkorn University (C2F to I.I.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.3c00046.
Effect of 22-(4′-py)-JA on PI3K and ERK signaling (Figure S1); effect of JA on AKT/mTOR/p70S6K signaling (Figure S2); molecular docking of 22-(4′-py)-JA and AKT1(Figure S3); anticancer activities of erlotinib on NSCLC cells (Figure S4); interactions between AKT1 and 22-(4′-py)-JA (Table S1); pharmacokinetic parameters of 22-(4′-py)-JA and JA obtained from pkCSM tools (Table S2); and list of primers used for qRT-PCR (Table S3) (PDF)
Author Contributions
V.P. conceived and designed the experiments, contributed reagents/materials/analysis tools, analyzed the data, and wrote the manuscript. I.I. performed the experiments, analyzed the data, and wrote the manuscript. S.Se., V.L, S.Si., K.B., and S.C. performed the experiments. All authors read and approved the final manuscript
The authors declare no competing financial interest.
Supplementary Material
References
- Thai A. A.; Solomon B. J.; Sequist L. V.; Gainor J. F.; Heist R. S. Lung cancer. Lancet 2021, 398, 535–554. 10.1016/S0140-6736(21)00312-3. [DOI] [PubMed] [Google Scholar]
- Duma N.; Santana-Davila R.; Molina J. R. Non-small cell lung cancer: Epidemiology, screening, diagnosis, and treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. 10.1016/j.mayocp.2019.01.013. [DOI] [PubMed] [Google Scholar]
- Alexander M.; Kim S. Y.; Cheng H. Update 2020: Management of non-small cell lung cancer. Lung 2020, 198, 897–907. 10.1007/s00408-020-00407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler E.; Arondekar B.; Aguilar K. M.; Zhou J.; Chang J.; Zhang X.; Pawar V. Treatment patterns and clinical outcomes in patients with advanced non-small cell lung cancer initiating first-line treatment in the US community oncology setting: a real-world retrospective observational study. J. Cancer Res. Clin. Oncol. 2021, 147, 671–690. 10.1007/s00432-020-03414-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majidpoor J.; Mortezaee K. Steps in metastasis: an updated review. Med. Oncol. 2021, 38, 3 10.1007/s12032-020-01447-w. [DOI] [PubMed] [Google Scholar]
- Song Z.; Wang J.; Su Q.; Luan M.; Chen X.; Xu X. The role of MMP-2 and MMP-9 in the metastasis and development of hypopharyngeal carcinoma. Braz. J. Otorhinolaryngol. 2021, 87, 521–528. 10.1016/j.bjorl.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobin E.; Bagwell K.; Wagner J.; Mysona D.; Sandirasegarane S.; Smith N.; Bai S.; Sharma A.; Schleifer R.; She J. X. A pan-cancer perspective of matrix metalloproteases (MMP) gene expression profile and their diagnostic/prognostic potential. BMC Cancer 2019, 19, 581 10.1186/s12885-019-5768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alevizakos M.; Kaltsas S.; Syrigos K. N. The VEGF pathway in lung cancer. Cancer Chemother. Pharmacol. 2013, 72, 1169–1181. 10.1007/s00280-013-2298-3. [DOI] [PubMed] [Google Scholar]
- Zang J.; Hu Y.; Xu X.; Ni J.; Yan D.; Liu S.; He J.; Xue J.; Wu J.; Feng J. Elevated serum levels of vascular endothelial growth factor predict a poor prognosis of platinum-based chemotherapy in non-small cell lung cancer. OncoTargets Ther. 2017, 10, 409–415. 10.2147/OTT.S124124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Mathy N. W.; Lu H. The role of VEGF in the diagnosis and treatment of malignant pleural effusion in patients with non-small cell lung cancer. Mol. Med. Rep. 2018, 17, 8019–8030. 10.3892/mmr.2018.8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv X.; Li J.; Zhang C.; Hu T.; Li S.; He S.; Yan H.; Tan Y.; Lei M.; Wen M.; Zuo J. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017, 4, 19–24. 10.1016/j.gendis.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid M.; Zadeh L. R.; Baradaran B.; Molavi O.; Ghesmati Z.; Sabzichi M.; Ramezani F. Up-down regulation of HIF-1α in cancer progression. Gene 2021, 798, 145796 10.1016/j.gene.2021.145796. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Xu J.; Dong Y.; Huang B. Down-regulation of HIF-1α inhibits the proliferation, migration, and invasion of gastric cancer by inhibiting PI3K/AKT pathway and VEGF expression. Biosci. Rep. 2018, 38, BSR20180741 10.1042/BSR20180741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Zhang C.; Xu L.; Zang K.; Ning Z.; Jiang F.; Chi H.; Zhu X.; Meng Z. Bufalin suppresses hepatocellular carcinoma invasion and metastasis by targeting HIF-1α via the PI3K/AKT/mTOR pathway. Oncotarget 2016, 7, 20193–20208. 10.18632/oncotarget.7935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iksen; Pothongsrisit S.; Pongrakhananon V. Targeting the PI3K/AKT/mTOR signaling pathway in lung cancer: an update regarding potential drugs and natural products. Molecules 2021, 26, 4100 10.3390/molecules26134100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. Y.; Wong A. S. Activation of p70S6K induces expression of matrix metalloproteinase 9 associated with hepatocyte growth factor-mediated invasion in human ovarian cancer cells. Endocrinology 2006, 147, 2557–2566. 10.1210/en.2005-1404. [DOI] [PubMed] [Google Scholar]
- Pothongsrisit S.; Arunrungvichian K.; Hayakawa Y.; Sritularak B.; Mangmool S.; Pongrakhananon V. Erianthridin suppresses non-small-cell lung cancer cell metastasis through inhibition of Akt/mTOR/p70S6K signaling pathway. Sci. Rep. 2021, 11, 6618 10.1038/s41598-021-85675-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oo Y.; Nealiga J. Q. L.; Suwanborirux K.; Chamni S.; Ecoy G. A. U.; Pongrakhananon V.; Chanvorachote P.; Chaotham C. 22-O-(N-Boc-L-glycine) ester of renieramycin M inhibits migratory activity and suppresses epithelial-mesenchymal transition in human lung cancer cells. J. Nat. Med. 2021, 75, 949–966. 10.1007/s11418-021-01549-3. [DOI] [PubMed] [Google Scholar]
- Sirimangkalakitti N.; Chamni S.; Suwanborirux K.; Chanvorachote P. Renieramycin M attenuates cancer stem cell-like phenotypes in H460 lung cancer cells. Anticancer Res. 2017, 37, 615–621. 10.21873/anticanres.11355. [DOI] [PubMed] [Google Scholar]
- Ecoy G. A. U.; Chamni S.; Suwanborirux K.; Chanvorachote P.; Chaotham C. Jorunnamycin A from Xestospongia sp. suppresses epithelial to mesenchymal transition and sensitizes anoikis in human lung cancer cells. J. Nat. Prod. 2019, 82, 1861–1873. 10.1021/acs.jnatprod.9b00102. [DOI] [PubMed] [Google Scholar]
- Barone A.; Chi D. C.; Theoret M. R.; Chen H.; He K.; Kufrin D.; Helms W. S.; Subramaniam S.; Zhao H.; Patel A.; Goldberg K. B.; Keegan P.; Pazdur R. FDA approval summary: Trabectedin for unresectable or metastatic liposarcoma or leiomyosarcoma following an anthracycline-containing regimen. Clin. Cancer Res. 2017, 23, 7448–7453. 10.1158/1078-0432.CCR-17-0898. [DOI] [PubMed] [Google Scholar]
- Krauss A. C.; Gao X.; Li L.; Manning M. L.; Patel P.; Fu W.; Janoria K. G.; Gieser G.; Bateman D. A.; Przepiorka D.; Shen Y. L.; Shord S. S.; Sheth C. M.; Banerjee A.; Liu J.; Goldberg K. B.; Farrell A. T.; Blumenthal G. M.; Pazdur R. FDA approval summary: (Daunorubicin and cytarabine) liposome for injection for the treatment of adults with high-risk acute myeloid leukemia. Clin. Cancer Res. 2019, 25, 2685–2690. 10.1158/1078-0432.CCR-18-2990. [DOI] [PubMed] [Google Scholar]
- Trigo J.; Subbiah V.; Besse B.; Moreno V.; López R.; Sala M. A.; Peters S.; Ponce S.; Fernández C.; Alfaro V.; Gómez J.; Kahatt C.; Zeaiter A.; Zaman K.; Boni V.; Arrondeau J.; Martínez M.; Delord J. P.; Awada A.; Kristeleit R.; Olmedo M. E.; Wannesson L.; Valdivia J.; Rubio M. J.; Anton A.; Sarantopoulos J.; Chawla S. P.; Mosquera-Martinez J.; D’Arcangelo M.; Santoro A.; Villalobos V. M.; Sands J.; Paz-Ares L. Lurbinectedin as second-line treatment for patients with small-cell lung cancer: a single-arm, open-label, phase 2 basket trial. Lancet Oncol. 2020, 21, 645–654. 10.1016/S1470-2045(20)30068-1. [DOI] [PubMed] [Google Scholar]
- Sumkhemthong S.; Chamni S.; Ecoy G. U.; Taweecheep P.; Suwanborirux K.; Prompetchara E.; Chanvorachote P.; Chaotham C. Jorunnamycin A suppresses stem-like phenotypes and sensitizes cisplatin-induced apoptosis in cancer stem-like cell-enriched spheroids of human lung cancer cells. Mar. Drugs 2021, 19, 261 10.3390/md19050261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirimangkalakitti N.; Chamni S.; Suwanborirux K.; Chanvorachote P. Renieramycin M sensitizes anoikis-resistant H460 lung cancer cells to anoikis. Anticancer Res. 2016, 36, 1665–1671. [PubMed] [Google Scholar]
- Petsri K.; Chamni S.; Suwanborirux K.; Saito N.; Chanvorachote P. Renieramycin T induces lung cancer cell apoptosis by targeting Mcl-1 degradation: a new insight in the mechanism of action. Mar. Drugs 2019, 17, 301 10.3390/md17050301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirimangkalakitti N.; Chamni S.; Charupant K.; Chanvorachote P.; Mori N.; Saito N.; Suwanborirux K. Chemistry of renieramycins. 15. Synthesis of 22-O-ester derivatives of jorunnamycin A and their cytotoxicity against non-small-cell lung cancer cells. J. Nat. Prod. 2016, 79, 2089–2093. 10.1021/acs.jnatprod.6b00433. [DOI] [PubMed] [Google Scholar]
- Iksen I.; Sinsook S.; Wattanathamsan O.; Buaban K.; Chamni S.; Pongrakhananon V. Target identification of 22-(4-pyridinecarbonyl) jorunnamycin A, a tetrahydroisoquinoline derivative from the sponge Xestospongia sp., in mediating non-small-cell lung cancer cell apoptosis. Molecules 2022, 27, 8948 10.3390/molecules27248948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliesath J.; Huser N.; Omori M.; Bunag D.; Proffitt C.; Streiner N.; Ho C.; Siddiqui-Jain A.; O’Brien S. E.; Lim J. K.; Ryckman D. M.; Anderes K.; Rice W. G.; Drygin D. Combined inhibition of EGFR and CK2 augments the attenuation of PI3K-Akt-mTOR signaling and the killing of cancer cells. Cancer Lett. 2012, 322, 113–118. 10.1016/j.canlet.2012.02.032. [DOI] [PubMed] [Google Scholar]
- El-Badrawy M. K.; Yousef A. M.; Shaalan D.; Elsamanoudy A. Z. Matrix metalloproteinase-9 expression in lung cancer patients and its relation to serum mmp-9 activity, pathologic type, and prognosis. J. Bronchol. Interventional Pulmonol. 2014, 21, 327–334. 10.1097/LBR.0000000000000094. [DOI] [PubMed] [Google Scholar]
- Han L.; Sheng B.; Zeng Q.; Yao W.; Jiang Q. Correlation between MMP2 expression in lung cancer tissues and clinical parameters: a retrospective clinical analysis. BMC Pulm. Med. 2020, 20, 283 10.1186/s12890-020-01317-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T. C.; Din Z. H.; Su J. H.; Wu Y. J.; Liu C. I. Sinulariolide suppresses cell migration and invasion by inhibiting matrix metalloproteinase-2/-9 and urokinase through the PI3K/AKT/mTOR signaling pathway in human bladder cancer cells. Mar. Drugs 2017, 15, 238 10.3390/md15080238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai T. C.; Wu W. T.; Lin J. J.; Su J. H.; Wu Y. J. Stellettin B isolated from Stelletta Sp. reduces migration and invasion of hepatocellular carcinoma cells through reducing activation of the MAPKs and FAK/PI3K/AKT/mTOR signaling pathways. Int. J. Cell Biol. 2022, 2022, 1–9. 10.1155/2022/4416611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugano R.; Ramachandran M.; Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 77, 1745–1770. 10.1007/s00018-019-03351-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M. H.; Jeong Y. J.; Cho H. J.; Hoe H. S.; Park K. K.; Park Y. Y.; Choi Y. H.; Kim C. H.; Chang H. W.; Park Y. J.; Chung I. K.; Chang Y. C. Delphinidin inhibits angiogenesis through the suppression of HIF-1α and VEGF expression in A549 lung cancer cells. Oncol. Rep. 2017, 37, 777–784. 10.3892/or.2016.5296. [DOI] [PubMed] [Google Scholar]
- Li G.; Shan C.; Liu L.; Zhou T.; Zhou J.; Hu X.; Chen Y.; Cui H.; Gao N. Tanshinone IIA inhibits HIF-1α and VEGF expression in breast cancer cells via mTOR/p70S6K/RPS6/4E-BP1 signaling pathway. PloS One 2015, 10, e0117440 10.1371/journal.pone.0117440. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Shenoy S. K.; Han S.; Zhao Y. L.; Hara M. R.; Oliver T.; Cao Y.; Dewhirst M. W. β-arrestin1 mediates metastatic growth of breast cancer cells by facilitating HIF-1-dependent VEGF expression. Oncogene 2012, 31, 282–292. 10.1038/onc.2011.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tungsukruthai S.; Reamtong O.; Roytrakul S.; Sukrong S.; Vinayanwattikun C.; Chanvorachote P. Targeting AKT/mTOR and Bcl-2 for autophagic and apoptosis cell death in lung cancer: novel activity of a polyphenol compound. Antioxidants 2021, 10, 534 10.3390/antiox10040534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng J.; Li X.; Zhou Z.; Wu C. L.; Dai M.; Bai X. EZH2 promotes tumor progression via regulating VEGF-A/AKT signaling in non-small cell lung cancer. Cancer Lett. 2015, 359, 275–287. 10.1016/j.canlet.2015.01.031. [DOI] [PubMed] [Google Scholar]
- Yang H.; Geng Y. H.; Wang P.; Zhou Y. T.; Yang H.; Huo Y. F.; Zhang H. Q.; Li Y.; He H. Y.; Tian X. X.; Fang W. G. Extracellular ATP promotes breast cancer invasion and epithelial-mesenchymal transition via hypoxia-inducible factor 2α signaling. Cancer Sci. 2019, 110, 2456–2470. 10.1111/cas.14086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W.; Tian W.; Ijaz M.; Wang F. Inhibition of EGF-induced migration and invasion by sulfated polysaccharide of Sepiella maindroni ink via the suppression of EGFR/Akt/p38 MAPK/MMP-2 signaling pathway in KB cells. Biomed. Pharmacother. 2017, 95, 95–102. 10.1016/j.biopha.2017.08.050. [DOI] [PubMed] [Google Scholar]
- Jeong Y. J.; Cho H. J.; Magae J.; Lee I. K.; Park K. G.; Chang Y. C. Ascofuranone suppresses EGF-induced HIF-1α protein synthesis by inhibition of the Akt/mTOR/p70S6K pathway in MDA-MB-231 breast cancer cells. Toxicol. Appl. Pharmacol. 2013, 273, 542–550. 10.1016/j.taap.2013.09.027. [DOI] [PubMed] [Google Scholar]
- Mendoza M. C.; Er E. E.; Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. 10.1016/j.tibs.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallarida R. J.; Jacob L. S.. The Dose-Response Relation. In The Dose-Response Relation in Pharmacology, 1st ed.; Tallarida R. J., Ed.; Springer: New York, 1979; pp 1–16. [Google Scholar]
- Guo Y. J.; Pan W. W.; Liu S. B.; Shen Z. F.; Xu Y.; Hu L. L. ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 2020, 19, 1997–2007. 10.3892/etm.2020.8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X. Y.; Zhang H. X.; Mezei M.; Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr. Comput.-Aided Drug Des. 2011, 7, 146–157. 10.2174/157340911795677602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanzione F.; Giangreco I.; Cole J. C. Use of molecular docking computational tools in drug discovery. Prog. Med. Chem. 2021, 60, 273–343. 10.1016/bs.pmch.2021.01.004. [DOI] [PubMed] [Google Scholar]
- Iksen I.; Witayateeraporn W.; Wirojwongchai T.; Suraphan C.; Pornputtapong N.; Singharajkomron N.; Nguyen H. M.; Pongrakhananon V. Identifying molecular targets of aspiletrein-derived steroidal saponins in lung cancer using network pharmacology and molecular docking-based assessments. Sci. Rep. 2023, 13, 1545 10.1038/s41598-023-28821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusani L.; Palmer D. S.; Somers D. O.; Wall I. D. Exploring ligand stability in protein crystal structures using binding pose metadynamics. J. Chem. Inf. Model. 2020, 60, 1528–1539. 10.1021/acs.jcim.9b00843. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.