

Abstract

Macrocyclic kinase inhibitors (MKIs) are gaining attention due to their favorable selectivity and potential to overcome drug resistance, yet they remain challenging to design because of their novel structures. To facilitate the design and discovery of MKIs, we investigate MKI rational design starting from initial acyclic compounds by performing microsecond-scale atomistic simulations for multiple MKIs, constructing an MKI database, and analyzing MKIs using hierarchical cluster analysis. Our studies demonstrate that the binding modes of MKIs are like those of their corresponding acyclic counterparts against the same kinase targets. Importantly, within the respective binding sites, the MKI scaffolds retain the same conformations as their corresponding acyclic counterparts, demonstrating the rigidity of scaffolds before and after molecular cyclization. The MKI database includes 641 nanomole-level MKIs from 56 human kinases elucidating the features of rigid scaffolds and the core structures of MKIs. Collectively these results and resources can facilitate MKI development.
Keywords: kinase, macrocyclic kinase inhibitors, binding mechanisms, drug discovery, scaffolds, database
1. Introduction
The human kinome represents one of the largest gene families, consisting of over 500 protein kinases that regulate almost all aspects of cellular function.1 Thus, alterations of gene expression, dysregulation of signaling pathways, or gene mutation of kinases can cause a wide variety of cancers and other diseases.2,3 Consequently, kinases have been considered primary drug targets.4 Kinases typically share a conserved catalytic domain that contains a highly similar ATP binding site.5,6 As such, it is a daunting challenge to achieve the desired selectivity, where inhibitors bind to the desired kinase but not to the others.7,8 Nevertheless, over the last 30 years, a great variety of kinase-targeted inhibitors or degraders have been successfully developed,9 such as type I inhibitors that occupy the ATP binding site, type II inhibitors that not only occupy the ATP binding site but also extend into nearby allosteric space, allosteric inhibitors, covalent inhibitors, macrocyclic inhibitors, PROTAC degraders, and molecular glues.10−14 To date, more than 70 small-molecule kinase inhibitors have been approved by the FDA since the first kinase drug Imatinib was approved by the FDA in 2001.15 Clinically, these drugs have substantially alleviated patients’ anguish and prolonged their lives.16,17 However, unexpected side effects and acquired drug resistance also mean that designing more innovative, efficient kinase drugs is warranted.1,16,18
Macrocyclic (at least 12-membered ring) kinase inhibitors (MKIs) have attracted more attention as aids in developing innovative, efficient kinase inhibitors because of their unique cyclic structure and potential to overcome drug resistance.9,19,20 Here we concentrate on small-molecule MKIs that bind to the ATP-binding pocket. Thus, MKIs that do not bind to the ATP-binding site or comprise peptide macrocycles (namely, Rapamycin and its derivatives binding to the FKBP—Rapamycin binding domain of mTOR kinase21 and DNA-template macrocyclic Src inhibitors that were discovered based on a DNA-templated synthetic small-molecule macrocycle library,22−24Figure 1) are not discussed.25,26 Hitherto, nine small-molecule MKIs have been in clinical trials (https://www.clinicaltrials.gov/), including two FDA-approved drugs Lorlatinib and Pacritinib (Figure 2).27,28
Figure 1.

Excluded macromolecular MKIs.
Figure 2.

Small-molecule MKIs approved by the FDA or in clinical trials.
Lorlatinib, approved in November 2018, is the first third-generation ALK-targeted drug to overcome multiple recalcitrant resistance mutations, such as G1202R or the most common gatekeeper mutation L1196M, during the treatment of ALK-positive non-small cell lung cancer (NSCLC) after a year or two of first- or second-generation ALK drug therapy (i.e., Crizotinib, Ceritinib, Alectinib, and Brigatinib).28−30 Pacritinib, approved in February 2022, is a JAK2/FLT3 inhibitor for the treatment of high-risk myelofibrosis with severe thrombocytopenia.27,31 Except for E6201 (Figure 2),32 which was inspired by a natural product called resorcylic acid lactone f152A1,33 all other eight macrocyclic inhibitors (Figure 2) were rationally designed starting from generic acyclic active compounds (called “counterparts”).30,31,34−37 For example, Lorlatinib was designed based on the acyclic molecule, Crizotinib (Figure 3).38 Macrocyclization in Lorlatinib not only improves selectivity and inhibitory potency for the primary targets ROS1/ALK30,39 but also provides excellent central nervous system (CNS) penetration compared to Crizotinib.40 Pacritinib was designed based on an acyclic, multi-targeted kinase inhibitor, compound-1 (Figure 3). Pacritinib not only inhibits JAK2 and FLTS (IC50 = 23, 19, and 22 nM for JAK2WT, JAK2V617F, and FLT3, respectively) but is also selective for JAK1 and JAK3 (IC50 = 1.28 and 0.52 μM, respectively).31 In the clinic, Pacritinib is more effective in patients with myelofibrosis than other available therapies.41 Inhibitor BI-4020 is a fourth-generation EGFR-targeted inhibitor designed to overcome drug resistance acquired from EGFR del19/T790M/C797M mutation.42 BI-4020 was designed based on an acyclic, broad kinase inhibitor (here, called ligand-1, Figure 3). EGFR mutations like del19 or L858R are related to NSCLC.43 Clinical evidence shows that the first-, second-, and third-generation EGFRmutated inhibitors, including Afatinib, Dacomitinib, Erlotinib, Gefitinib, and Osimertinib, can shrink lung cancers with different response rates.1,44 However, the response is not durable given the presence of acquired T790M and/or C797S mutations. In contrast, BI-4020 shows high potency on EGFR variants (IC50 = 0.6 and 1.0 nM for EGFRdel19/T790M/C797S variant and EGFRdel19, respectively) with limited activity against EGFRwt (IC50 = 190 nM).45 These successes demonstrate that designing MKIs starting from acyclic structures is an effective pathway.46,47 However, current drug-like chemical space (at least 1060 compounds) is too large and diverse to screen for potent acyclic counterparts that can be used as the starting point in designing MKIs.48 Narrowing chemical space and determining which acyclic structures are promising for macrocyclic kinase drug design is a prerequisite.
Figure 3.

Chemical structures of three MKIs (lower row) and their corresponding acyclic counterparts (upper row).
To this end, we sought to explore the molecular characteristics and binding modes of MKIs. We performed microsecond-scale atomistic simulations for three pairwise systems (Lorlatinib and Crizotinib,30 Pacritinib and compound-1,31 and BI-4020 and ligand-1,45Figure 3) to identify the different binding characteristics between MKIs and their corresponding acyclic counterparts within the kinase binding pockets. Based on these simulations, a systematic analysis of the binding characteristics of MKIs before and after their cyclization was performed using a functional–site interaction fingerprint (Fs-IFP) method.49−51 Subsequently, we manually constructed an MKI database from the published MKI literature. Thus far, a total of 641 nanomolar MKIs, covering 56 human kinases, have been curated. The data can be accessed and downloaded freely from (https://zhengzhster.github.io/MKIs). Harnessing the MKI database, we obtained an overview of the properties of MKIs, paying particular attention to the characteristics of MKI scaffolds. In addition, the core structures of MKIs that interact with the hinge regions are discussed, and design strategies are proposed. Together the binding modes, database, and design strategies for MKIs provide a resource for advancing MKIs’ design and discovery.
2. Results
2.1. Binding Modes of MKIs and Their Corresponding Acyclic Counterparts
We first analyzed the binding modes of MKIs and their corresponding acyclic counterparts within the kinase binding site using microsecond-scale all-atom MD simulation (see the Experimental Section).49,52
2.1.1. Binding Modes of Crizotinib and Lorlatinib
The binding modes of the two ALK inhibitors are illustrated in Figure 4a,b. The common aminopyridine cores of Crizotinib and Lorlatinib form stable interactions with residues E1197, L1198, M1199, and L1256 (the probabilities of interactions >0.8, Figure 4b). Of these, E1197, L1198, and M1199 are at the hinge. L1256 is beneath the aminopyridine core. Thus, the aminopyridine scaffold nestles in the ATP-binding site against the hinge. E1197 and M1199 interact with the aminopyridine fragment through two H-bond interactions (the probability of hydrogen bond formation is close to 100%, Figure 4b).30,38 Linking to the aminopyridine core, the common fluorophenyl groups of Crizotinib and Lorlatinib lies between the roof (β3) and the DFG peptide with similar interactions with V1130, K1150, N1256, and D1270. Another common pyrazole moiety, connecting the aminopyridine core, is located between L1122 and G1202. However, in Crizotinib, the piperidine-substituted pyrazole moiety is exposed to the solvent while interacting with G1123, A1200, and S1206 in the front-pocket area (Figure 4a,b). By contrast, in Lorlatinib the pyrazole was optimized forming 2-methylpyrazole-3-carbonitrile, in which the nitrile group interacts with R1120 and E1132.30 In summary, the core scaffolds of Crizotinib and Lorlatinib present similar binding modes with differences in the substructures.
Figure 4.

(a–f) Binding modes of each pairwise system (MKIs and their corresponding acyclic counterparts; PDB templates: 5aa9, 7ree, 7kxz for (a,c,e) respectively; GK gatekeeper; FP front-pocket; αC α-helix). (e) Blue highlights the hydrophobic sub-pocket at the back cleft of the binding pocket.
2.1.2. Binding Modes of Compound-1 and Pacritinib
Pacritinib, a JAK2-targeted drug, was designed based upon an acyclic counterpart, compound-1, which was then optimized by cyclization and R-group modification.31 Thus, Pacritinib and compound-1 share the same scaffold, which is composed of an aminopyrimidine group coplanar with two phenyl groups (Figures 3 and 4c). In compound-1, the aminopyrimidine core structure, located at the adenine binding area, has stable hydrogen-bond interactions with residues M929 (gatekeeper) and L932 at the hinge (probability of interaction >0.9, Figure 4c,d). The 2-methoxyphenol fragment, connecting to the aminopyrimidine core, forms stable hydrophobic interactions with residues V863 at the β2 and L983 at the β6 strand. The terminal dimethylphenylamine moiety makes stable hydrophobic interactions with residue L855 at β1 (probability of interaction >0.9, Figure 4d). Compared to compound-1, the two open ends between the two phenyl groups in Pacritinib are bridged. After cyclization, the binding patterns between the Pacritinib scaffold and residues L855, V863, M929, L932, and L983 are similar to that of compound-1 (Figure 4d). Apart from the common scaffolds Pacritinib was further optimized through a terminal pyrrolidine displacement on the 4-aminophenol fragment. The pyrrolidine extends into the solvent front-pocket interacting with residues D939, Y940, and K943, accounting for the improved selectivity against JAK2.27
2.1.3. Binding Modes of Ligand-1 and BI-4020
BI-4020 is a potential fourth-generation EGFR inhibitor that overcomes del19/T790M/C797S mutation-induced drug resistance.45 Based on the scaffold of ligand-1, BI-4020 was rationally designed. The common scaffold of the two share a similar binding mode of Type-I kinase inhibitors (Figure 4e). Specifically, the common aminobenzimidazole core has stable interactions with residues Q791, L792, or M793 at the hinge, residue L718 at β1, and residue L844 at β6 (probability > 0.9, Figure 4f). The “head” groups of both, attached to the aminobenzimidazole group, extend into the kinase hydrophobic sub-pocket, which is located at the back cleft adjacent to the adenine binding site and consists of αC residues M766 and A743 on β3,53 forming key interactions with residues A743, M766, C775, and T854 (Figure 4f). In contrast, the terminal piperazine group of BI-4020 reaches the solvent, forming interactions with residue D800 at the front pocket. Moreover, the bridging linker of BI-4020 also provides unique interactions with residues R841 and D855 to achieve selectivity among mutant variants (Figure 4f).
Overall, MKIs and their corresponding acyclic counterparts have the same type-I binding modes, forming the typical hydrogen-bond interactions with the hinge.53 The same scaffolds of MKIs and their corresponding acyclic counterparts show similar binding patterns, meaning the binding modes of the scaffolds are not affected upon macrocyclization. We investigated the fluctuation of ligands within the binding sites by analyzing the MD trajectories of MKIs and their corresponding acyclic counterparts separately. The analyses showed that the scaffolds remain more rigid than other fragments of the ligands either in MKIs or in the acyclic counterparts (Figure 5). Considering the consistency of scaffolds before and after cyclization, we further studied the properties of MKIs to explore promising new scaffolds for developing MKIs.
Figure 5.

(a–f) Rigidity of Crizotinib, Lorlatinib, compound-1, Pacritinib, ligand-1, and BI-4020, respectively, while interacting with the hinge (gray). Rigid scaffolds to flexible fragments are colored from blue to red.
2.2. Characteristics of MKIs and Scaffolds
The consistent rigid properties of the scaffolds before and after the aforementioned cyclization inspired us to establish the characteristics of such scaffolds so that they may be used to screen MKIs from across acyclic chemical space. We collected all of the released MKIs as found in the literature, constructing an MKI database containing 641 MKIs and 56 human kinases with nanomolar affinity (Figure 6a, Tables S1 and S2). The 56 kinase targets belonged to different kinase groups (TK, CMGC, CAMK, AGC, STE, TKL, Other, Lipid, and Atypical) excluding the CK1 group; 20 out of the 56 belonged to the TK group. The two kinase targets (ALK and JAK2) with the approved macrocyclic kinase drugs, Lorlatinib and Pacritinib, belong to the TK group. We obtained the scaffold of every MKI by using the tool strip-it.54 After deleting the redundant scaffolds, a total of 95 unique scaffolds were obtained (Table S3) and then clustered using an “average linkage” hierarchical clustering algorithm.55 Ten scaffold clusters were identified using a dissimilarity distance >0.8 as the threshold (Figure 6b, Table S4). The 10 clusters provide an available conformational space for designing MKIs with diverse properties (Figure S1: one compound representative per cluster). The details reveal the important conformations and properties.
Figure 6.

(a) Distribution of kinase targets with released MKIs across the human kinome (red). Two kinases with approved macrocyclic kinase drugs are highlighted in blue. (b) Distributions of ten clustered scaffolds.
For every scaffold and MKI, molecular weight (MW), number of H-bond donors (HbD), number of H-bond acceptors (HbA), log P, number of aromatic rings (AR), and molecular flexibility (MF) have been analyzed (Figure 7, Tables S5 and S6). For MKIs, the MW ranges from 296 to 720 g mol–1, the number of HbDs from 0 to 6, the number of HbAs from 3 to 10, log P has a range of [−6.01, 8.74], the number of ARs from 1 to 5, and the MF from 3.62 to 15.85. The averages for MWs, HbDs, HbAs, log Ps, ARs, and MFs of the MKIs are 457.4, 2.3, 6.3, 1.9, 3.0, and 7.7, respectively. According to Lipinski’s rule of five (Ro5, Table S7), 99.4% of MKIs have HbD values of ≤5, 100% of MKIs have HbA values of ≤10, and 96.1% of MKIs have log P values of ≤5. Notably, only 74.5% of MKIs have a MW of ≤500, which is in agreement with the trend in MW increase for FDA-approved drugs over the last 20 years.56
Figure 7.

(a–f) Properties of MKIs and the corresponding scaffolds: molecular weight, number of H-bond donors, number of H-bond acceptors, log P, number of aromatic rings, and molecular flexibility, respectively. The ordinate is probability density. The dashed line indicates the maximum probability density and the corresponding abscissa.
The maximum probability densities (MPDs) for MW, number of HbD, and number of HbA of the scaffolds are 274.5, 1.0, and 3.0, respectively, smaller than those for MKIs (477.4, 3.0, and 6.0) (Figure 7a–c). Not surprising since the ring_with_link scaffolds are obtained by excising the bridging linkers and deleting the R-groups of MKIs.54 The log Ps of scaffolds and MKIs have similar MPDs (2.1 and 2.0), which means that both have similar hydrophobicity profiles.57 From this point of view, it may be necessary to consider hydrophobicity when designing MKIs starting with the choice of scaffold. Likewise, the number of ARs for scaffolds and MKIs are similar with MPDs of 3.0 (Figure 7e), suggesting that ARs are part of scaffolds but no ARs are in bridging linkers for MKIs. The scaffolds have much low flexibility (3.2) than MKIs (7.7) (Figure 7f). Not surprising since on trimming the R-groups of MKIs to obtain scaffolds, the MFs of the scaffolds would decrease. Scaffolds consist of a few rigid ARs (Figure 7e), consistent with the aforementioned rigid binding modes. More specifically, since the number of ARs and MFs of scaffolds have similar MPDs (3.0 vs 3.2), it means scaffolds are mostly composed of 3 ARs directly connected to one another. This suggests that ARs-composed scaffolds with strong rigidity should be considered for rational MKI design. The majority of MKIs obey the Ro5, and when combined with the characteristics of MKIs, it suggests that multiple-AR, rigid scaffolds are practicable in developing MKIs.
2.3. Strategies to Design MKIs from Core Structures That Interact with the Hinges
To date, all published MKIs are type I or type II kinase inhibitors that occupy the ATP-binding site and form hydrogen bond interactions with the hinge.7 While the hinge-MKI interaction appears to be conserved, the types and positions of the atoms involved in hydrogen bonding can vary significantly. Accordingly, understanding what chemical structures can serve as the core structures for recognizing and binding into the adenine sub-pocket is essential to choreograph scaffolds for MKI design. Hence, we investigated all MKIs based on differences in core structures interacting with the hinge, establishing 73 different core structures (Table S8). The 73 core structures were further categorized into eight groups (Figure 8). In the dendrogram, each node represents a core structure, and the edges connect nodes that share the same chemical fragments interacting with the hinge (Figure 8).
Figure 8.

Eight MKI core structure categories. One representative per node (the full list is in Table S8).
Specifically, the eight groups are (I) indole derivatives; (II) benzomorpholine derivatives; (III) thienopyrimidine derivatives; (IV) 2-amino-pyrimidine derivatives; (V) 2-amino pyridine derivatives; (VI) phenol derivatives; (VII) quinoline derivatives; and (VIII) benzamide derivatives. On the innermost layer, the nodes represent common fragments interacting with the hinge for every group. The outer-layer nodes show examples of structural derivatives in groups I–VIII. For example, in group V, the innermost layer is 2-amino pyridine, in which the amino and the nitrogen atom of the heterocyclic pyridine provide the hydrogen-bond interactions with the hinge. Moreover, the amino group is located on the side close to the gatekeeper.53,58 In the middle layer, the derivatives were first divided into two prongs: the first is a 5-substituted 2-aminopyrimidine derivative (MKI-242), and the second branch is a 3,5-substituted 2-aminopyridine derivative. Then, based on the 5-substituted difference, the second branch was further derived into three prongs in the outer layer with representatives MKI-1, MKI-14, and MKI-18.
Similarly, the other seven groups illustrate different categories of core structures interacting with the hinge. In the largest group, group I, the indole nitrogen atom provides the hydrogen bond interaction with the hinge. Given heterocycles with more nitrogen atoms, such as in the 1, 2, 3, 4, 5, or 6 position, the group diverges into 9 branches: pyrazolo[1,5-a]pyrimidine, 1H-indazole, 5H-pyrrolo[2,3-b]pyrazine, 7-azaindole, 7H-pyrrolo[2,3-d]pyrimidine, imidazo[1,2-a]pyridine, imidazo[1,2-b]pyridazine, 7-azaindazole, and purine, as shown on the middle layer. Notably, they share the same planar ring structures akin to adenine, but the different heterocycles provide opportunities for scaffold hopping for drug design.59 It is not surprising that adenine-like structures tend to appear in adenine pockets, and this suggests that adenine-like core structures are practical MKI scaffolds.
3. Conclusions
Here we focus on MKIs, a class of emerging kinase inhibitors with novel molecular scaffolds, that provide potential opportunities for establishing new chemical entities through studying the binding modes of MKIs in kinase pockets, curating an MKI database, and determining the characteristics of MKIs’ scaffolds.
We first explored the binding modes of MKIs against kinase targets and compared them to their corresponding acyclic counterparts by performing three pairs of μs-scale MD simulations. Our simulations show that the binding modes of MKIs and their acyclic counterparts are similar. The binding patterns of scaffold fragments before and after cyclization retain higher rigidity and consistency, inspiring us to pay more attention to acyclic compounds, especially those that can be used for MKI design. To this end, we investigated MKI’s characteristics by creating an MKI database, containing 641 MKIs covering 56 kinases. Based on analysis of the MKI database, we found 95 diverse scaffolds where a large proportion obeys the Ro5. Compared to MKIs, the 95 scaffolds have the same log P profiles, which suggests that we should emphasize solubility and lipophilicity when designing MKIs. The scaffolds are generally composed of 3 ARs connected by one bond, which makes for rigid MKIs’ scaffolds. Further, according to typical hinge–ligand interactions of type-I/II kinase inhibitors, we investigated the core structures of MKIs and demonstrated that adenine-like core structures tend to appear in the adenine pockets.
In summary, this work systematically studied MKIs, revealing the promise of rigid scaffolds for designing MKIs and a preference of adenine-like core structures for developing scaffolds. The MKI database used in this study is freely available at https://zhengzhster.github.io/MKIs. The resultant database and understanding of the chemical characteristics can go a long way in reducing the vast chemical space needed for MKI screening.
4. Experimental Section
4.1. MD Simulations
All-atom MD simulations have been widely used for exploring drug binding mechanisms.60,61 Here, the goal is to explore the binding modes of macrocyclic inhibitors before and after cyclization using microsecond-scale MD simulations. To do so, three pairs of MD systems for both the macrocyclic inhibitors and their corresponding counterparts (i.e., the initial acyclic hit compounds for starting to design the MKIs) were prepared. Lorlatinib was rationally designed based on an acyclic drug Crizotinib.30 Thus, the MD simulations of Crizotinib and Lorlatinib bound to ALK kinase were set up, and their starting conformations were taken from Protein Data Bank (PDB)62 (PDB ids: 2xp2 and 5aa9, respectively). Likewise, the starting conformations of BI-4020 and its corresponding counterpart (named ligand-1)45 bound to EGFR kinase were taken from the PDB (PDB ids: 7kxz and 6s9b, respectively). Because the cocrystal structures of the macrocyclic drug Pacritinib and its counterpart (named compound-1)31 bound to JAK2 kinase were not available in any protein structure database, we docked the two molecules Pacritinib and compound-1 into JAK2 ATP binding pockets (PDB id 7ree as the kinase template) using the AutoDock4.2 software.63 From the docked lists of complexes, the top scoring complexes were selected as the starting conformations of the Pacritinib- and compound-1-bound JAK2 systems.
All six kinase-ligand complexes were processed using the VMD software,64 with missing residues added based to the corresponding kinase sequence. Redundant structures were deleted based on the amino acid sequence of each kinase domain (ALK residues 1116–1392, EGFR residues 712–979, and JAK2 residues 849–1124). Mutations at E746_A750del (representing the del19 mutations65), T790M, and C797S were set for the EGFR kinase system as BI-4020 is sensitive to the triple mutation while sparing wildtype EGFR.45 These 6 systems were solvated in a rectangular water box with margins at an 18.0 Å buffer distance from any solute atom. The protonation states of all charged amino acids were automatically assigned assuming a PH of 7.0. The counterions (Na+ and Cl–) were added to reach establish an ion concentration of 0.20 M and electroneutrality. The CHARMM36 all-atom protein force field,66 CHARMM general force field,67,68 and TIP3P water model were used to describe kinases, ligands, and water molecules, respectively. The parameter files and the topology files of all ligand molecules (i.e., Crizotinib, Lorlatinib, compound-1, Pacritinib, ligand-1, and BI-4020) were prepared using the online server (https://cgenff.umaryland.edu/).69
For MD simulations, every system was first optimized using an ACEMD MD protocol: 500 steps of minimization and a 5 × 10 ns restraining MD simulation with gradual reduced restraining force constants (i.e., 10.0, 5.0, 2.5, 1.0, and 0.5 kcal mol–1 Å–2, respectively).70 A 1.2 μs MD simulation was carried out (Figure S2), and the last 0.8 μs equilibrated MD trajectory was used to analyze the ligand-binding details for each system. All MD simulations were performed using the ACEMD software package.70 During the MD simulations, the integration time step was 4 fs, and the SHAKE method was used as the bond constraint.71 The temperature and the pressure were maintained using a Langevin thermostat at 298.15 K and a Berendsen barostat at 1 atm, respectively. All trajectories and ligand fluctuations were analyzed using a Wordom program.72
4.2. Kinase-Ligand Interaction Fingerprint Analysis
As stated, all conformations were derived from the last 0.8 μs equilibrated MD trajectories for each system in order to analyze the binding characteristics. Specifically, we encoded every conformation into a one-dimension array as a bit string,49 representing the kinase-ligand atom–scale interaction details, by using an Fs-IFP method with predefined geometric rules.50−52,73 In the Fs-IFP method, first all kinase-ligand conformations were aligned using a 3D binding pocket alignment tool SMAP.74 Second, in the ligand-binding binding site, every residue–ligand interaction was described using 7 kinds of interaction fingerprints (IFPs): van der Waals, aromatic interaction (face-to-face or face-to-edge), hydrogen bond (protein as acceptor or donor), and electrostatic interactions (protein positively charged or negatively charged).75 Thus, the interactions between every residue within the binding site and the ligand were encoded into a 7 bit substring, such as “1000000”, where “1” indicates the interaction exists and “0” means no interaction detected between the given residue and ligand. The residue-based bit string was encoded for every kinase-ligand conformation. Finally, based on the aligned binding pockets, all Fs-IFPs were aligned for analysis. The IChem software package was used for encoding IFPs.75
The probability of interaction between
the ligand and every residue comprising the binding sites was calculated.
From MD simulation trajectories, the probability of interaction of
every residue is obtained using the equation 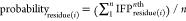 , where i is the index
of amino acids comprising the binding site and n is
the number of conformations extracted from the corresponding MD trajectory.
In the nth conformation, if a 7 bit substring between
the residue(i) and the ligand is “0000000”,
it means no interactions, i.e., IFPresidue(i)nth =
0, otherwise IFPresidue(i) = 1.
, where i is the index
of amino acids comprising the binding site and n is
the number of conformations extracted from the corresponding MD trajectory.
In the nth conformation, if a 7 bit substring between
the residue(i) and the ligand is “0000000”,
it means no interactions, i.e., IFPresidue(i)nth =
0, otherwise IFPresidue(i) = 1.
4.3. MKI Database
The available MKIs were collected from review papers25,46 and scientific databases, including PubMed76 and PDB.62 Databases were searched using the query keywords “macrocyclic”, “kinase”, and “inhibitors”. The search results were manually checked, and all MKIs with nanomolar-level inhibition were collated, including SMILES format, primary target(s), assay data (IC50, Ki, or Kd), clinical states, PDB structures, and references. MKI scaffolds were obtained by manually cutting off the bridge linkers and then calculating the Rings_with_Linkers (RWL) scaffold using the tool strip-it.54 For every MKI and its corresponding RWL scaffold, the molecular properties were calculated using JChem77 and Mold2 for calculating Kier flexibility indices for MF.78,79 The larger the flexibility index, the more flexible the molecule. The pairwise similarity of the RWL scaffolds was calculated using JChem with the molecular descriptor ECFP and Tanimoto distance. The RWL scaffolds were clustered using the average-linkage algorithm (Table S4) and shown using a circular dendrogram in RStudio (version 1.4.1106).
Based on the types and positions of atoms of the core structures interacting with the hinge, we sorted and clustered them into eight different types (Table S8). The Reingold-Tilford Tree network diagram was created using the networkD3 package in RStudio (version 1.4.1106) showing the 8 types of core structures. All compounds were illustrated with professional ChemDraw (version 20.0.0.38).
Acknowledgments
The authors acknowledge Research Computing at The University of Virginia for providing computational resources and technical support. The work described here was supported by the University of Virginia (P.E.B.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.3c00078.
MKI database, 95 nonredundant scaffolds, distributions of 10 clusters of scaffolds, physicochemical properties of MKIs, physicochemical properties of scaffolds, and physicochemical properties of MKIs and Ro5 (XLSX)
73 core structures, one compound representative per cluster; and RMSDs of all six MD systems for validating the MD processes (PDF)
Author Contributions
The manuscript was written with contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
The online MKI database is at https://zhengzhster.github.io/MKIs.
Special Issue
Published as part of the ACS Pharmacology & Translational Science virtual special issue “New Drug Modalities in Medicinal Chemistry, Pharmacology, and Translational Science”.
Supplementary Material
References
- Cohen P.; Cross D.; Janne P. A. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat. Rev. Drug Discovery 2021, 20, 551–569. 10.1038/s41573-021-00195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P.; Hunter T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- Lahiry P.; Torkamani A.; Schork N. J.; Hegele R. A. Kinase mutations in human disease: interpreting genotype-phenotype relationships. Nat. Rev. Genet. 2010, 11, 60–74. 10.1038/nrg2707. [DOI] [PubMed] [Google Scholar]
- Vogelstein B.; Papadopoulos N.; Velculescu V. E.; Zhou S.; Diaz L. A. Jr.; Kinzler K. W. Cancer genome landscapes. Science 2013, 339, 1546–1558. 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubersax J. A.; Ferrell J. E. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- Kannan N.; Taylor S. S.; Zhai Y.; Venter J. C.; Manning G. Structural and functional diversity of the microbial kinome. PLoS Biol. 2007, 5, e17 10.1371/journal.pbio.0050017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhullar K. S.; Lagaron N. O.; McGowan E. M.; Parmar I.; Jha A.; Hubbard B. P.; Rupasinghe H. P. V. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol. Cancer 2018, 17, 48. 10.1186/s12943-018-0804-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwood M. M.; Fabbro D.; Sokolov A. V.; Knapp S.; Schioth H. B. Trends in kinase drug discovery: targets, indications and inhibitor design. Nat. Rev. Drug Discovery 2021, 20, 839–861. 10.1038/s41573-021-00252-y. [DOI] [PubMed] [Google Scholar]
- Lu X.; Smaill J. B.; Ding K. New Promise and Opportunities for Allosteric Kinase Inhibitors. Angew. Chem., Int. Ed. 2020, 59, 13764–13776. 10.1002/anie.201914525. [DOI] [PubMed] [Google Scholar]
- Rana S.; Mallareddy J. R.; Singh S.; Boghean L.; Natarajan A. Inhibitors, PROTACs and Molecular Glues as Diverse Therapeutic Modalities to Target Cyclin-Dependent Kinase. Cancers 2021, 13, 5506. 10.3390/cancers13215506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.; Xie L.; Bourne P. E. Insights into the binding mode of MEK type-III inhibitors. A step towards discovering and designing allosteric kinase inhibitors across the human kinome. PLoS One 2017, 12, e0179936 10.1371/journal.pone.0179936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.; Liu Q.; Bliven S.; Xie L.; Bourne P. E. Determining Cysteines Available for Covalent Inhibition Across the Human Kinome. J. Med. Chem. 2017, 60, 2879–2889. 10.1021/acs.jmedchem.6b01815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.; Bourne P. E.. Overview of Current Type I/II Kinase Inhibitors. Next Generation Kinase Inhibitors, 1st ed.; Springer International Publishing, 2020; pp 13–28. [Google Scholar]
- Roskoski R. Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2023 update. Pharmacol. Res. 2023, 187, 106552. 10.1016/j.phrs.2022.106552. [DOI] [PubMed] [Google Scholar]
- Gharwan H.; Groninger H. Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nat. Rev. Clin. Oncol. 2016, 13, 209–227. 10.1038/nrclinonc.2015.213. [DOI] [PubMed] [Google Scholar]
- Chaft J. E.; Rimner A.; Weder W.; Azzoli C. G.; Kris M. G.; Cascone T. Evolution of systemic therapy for stages I-III non-metastatic non-small-cell lung cancer. Nat. Rev. Clin. Oncol. 2021, 18, 547–557. 10.1038/s41571-021-00501-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman J. A.; Settleman J. Acquired resistance to tyrosine kinase inhibitors during cancer therapy. Curr. Opin. Genet. Dev. 2008, 18, 73–79. 10.1016/j.gde.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Marsault E.; Peterson M. L. Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J. Med. Chem. 2011, 54, 1961–2004. 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- Ferguson F. M.; Gray N. S. Kinase inhibitors: the road ahead. Nat. Rev. Drug Discovery 2018, 17, 353–377. 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- Abraham R. T.; Gibbons J. J.; Graziani E. I.. Chemistry and Pharmacology of Rapamycin and Its Derivatives; The Enzymes; Academic Press, 2010; pp 329–366. [Google Scholar]
- Georghiou G.; Kleiner R. E.; Pulkoski-Gross M.; Liu D. R.; Seeliger M. A. Highly specific, bisubstrate-competitive Src inhibitors from DNA-templated macrocycles. Nat. Chem. Biol. 2012, 8, 366–374. 10.1038/nchembio.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner R. E.; Dumelin C. E.; Tiu G. C.; Sakurai K.; Liu D. R. In vitro selection of a DNA-templated small-molecule library reveals a class of macrocyclic kinase inhibitors. J. Am. Chem. Soc. 2010, 132, 11779–11791. 10.1021/ja104903x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S.; Lerner R. A. Encoded combinatorial chemistry. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 5381–5383. 10.1073/pnas.89.12.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen A.; Dymock B.. CHAPTER 5. Small Molecule Macrocyclic Kinase Inhibitors. Kinase Drug Discovery; Drug Discovery, 2018; pp 97–127. [Google Scholar]
- Amrhein J. A.; Knapp S.; Hanke T. Synthetic Opportunities and Challenges for Macrocyclic Kinase Inhibitors. J. Med. Chem. 2021, 64, 7991–8009. 10.1021/acs.jmedchem.1c00217. [DOI] [PubMed] [Google Scholar]
- Hart S.; Goh K. C.; Novotny-Diermayr V.; Tan Y. C.; Madan B.; Amalini C.; Ong L. C.; Kheng B.; Cheong A.; Zhou J.; Chng W. J.; Wood J. M. Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer J. 2011, 1, e44 10.1038/bcj.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw A. T.; Bauer T. M.; de Marinis F.; Felip E.; Goto Y.; Liu G.; Mazieres J.; Kim D. W.; Mok T.; Polli A.; Thurm H.; Calella A. M.; Peltz G.; Solomon B. J. First-Line Lorlatinib or Crizotinib in Advanced ALK-Positive Lung Cancer. N. Engl. J. Med. 2020, 383, 2018–2029. 10.1056/NEJMoa2027187. [DOI] [PubMed] [Google Scholar]
- Song X.; Zhong H.; Qu X.; Yang L.; Jiang B. Two novel strategies to overcome the resistance to ALK tyrosine kinase inhibitor drugs: Macrocyclic inhibitors and proteolysis-targeting chimeras. MedComm 2021, 2, 341–350. 10.1002/mco2.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T. W.; Richardson P. F.; Bailey S.; Brooun A.; Burke B. J.; Collins M. R.; Cui J. J.; Deal J. G.; Deng Y. L.; Dinh D.; Engstrom L. D.; He M.; Hoffman J.; Hoffman R. L.; Huang Q.; Kania R. S.; Kath J. C.; Lam H.; Lam J. L.; Le P. T.; Lingardo L.; Liu W.; McTigue M.; Palmer C. L.; Sach N. W.; Smeal T.; Smith G. L.; Stewart A. E.; Timofeevski S.; Zhu H.; Zhu J.; Zou H. Y.; Edwards M. P. Discovery of (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-8,4-(m etheno)pyrazolo[4,3-h] [2,5,11]-benzoxadiazacyclotetradecine-3-carbonitrile (PF-06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c-ros oncogene 1 (ROS1) with preclinical brain exposure and broad-spectrum potency against ALK-resistant mutations. J. Med. Chem. 2014, 57, 4720–4744. 10.1021/jm500261q. [DOI] [PubMed] [Google Scholar]
- William A. D.; Lee A. C.; Blanchard S.; Poulsen A.; Teo E. L.; Nagaraj H.; Tan E.; Chen D.; Williams M.; Sun E. T.; Goh K. C.; Ong W. C.; Goh S. K.; Hart S.; Jayaraman R.; Pasha M. K.; Ethirajulu K.; Wood J. M.; Dymock B. W. Discovery of the Macrocycle 11-(2-Pyrrolidin-1-yl-ethoxy)-14,19-dioxa-5,7,26-triaza-tetracyclo[19.3.1.1(2,6).1(8,12)]heptacosa-1(25),2(26),3,5,8,10,12(27),16,21,23-decaene (SB1518), a Potent Janus Kinase 2/Fms-Like Tyrosine Kinase-3 (JAK2/FLT3) Inhibitor for the Treatment of Myelofibrosis and Lymphoma. J. Med. Chem. 2011, 54, 4638–4658. 10.1021/jm200326p. [DOI] [PubMed] [Google Scholar]
- Goto M.; Chow J.; Muramoto K.; Chiba K.; Yamamoto S.; Fujita M.; Obaishi H.; Tai K.; Mizui Y.; Tanaka I.; Young D.; Yang H.; Wang Y. J.; Shirota H.; Gusovsky F. E6201 [(3S,4R,5Z,8S,9S,11E)-14-(ethylamino)-8, 9,16-trihydroxy-3,4-dimethyl-3,4,9,19-tetrahydro-1H-2-benzoxacyclotetradecine-1,7 (8H)-dione], a novel kinase inhibitor of mitogen-activated protein kinase/extracellular signal-regulated kinase kinase (MEK)-1 and MEK kinase-1: in vitro characterization of its anti-inflammatory and antihyperproliferative activities. J. Pharmacol. Exp. Ther. 2009, 331, 485–495. 10.1124/jpet.109.156554. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Boivin R.; Yoneda N.; Du H.; Schiller S.; Matsushima T.; Goto M.; Shirota H.; Gusovsky F.; Lemelin C.; Jiang Y.; Zhang Z.; Pelletier R.; Ikemori-Kawada M.; Kawakami Y.; Inoue A.; Schnaderbeck M.; Wang Y. Discovery of anti-inflammatory clinical candidate E6201, inspired from resorcylic lactone LL-Z1640-2, III. Bioorg. Med. Chem. Lett. 2010, 20, 3155–3157. 10.1016/j.bmcl.2010.03.087. [DOI] [PubMed] [Google Scholar]
- William A. D.; Lee A. C.; Goh K. C.; Blanchard S.; Poulsen A.; Teo E. L.; Nagaraj H.; Lee C. P.; Wang H.; Williams M.; Sun E. T.; Hu C.; Jayaraman R.; Pasha M. K.; Ethirajulu K.; Wood J. M.; Dymock B. W. Discovery of kinase spectrum selective macrocycle (16E)-14-methyl-20-oxa-5,7,14,26-tetraazatetracyclo[19.3.1.1(2,6).1(8,12)]heptaco sa-1(25),2(26),3,5,8(27),9,11,16,21,23-decaene (SB1317/TG02), a potent inhibitor of cyclin dependent kinases (CDKs), Janus kinase 2 (JAK2), and fms-like tyrosine kinase-3 (FLT3) for the treatment of cancer. J. Med. Chem. 2012, 55, 169–196. 10.1021/jm201112g. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Yu P.; Dong L.; Wang W.; Duan S.; Wang B.; Gong X.; Ye L.; Wang H.; Tian J. Discovery of the Next-Generation Pan-TRK Kinase Inhibitors for the Treatment of Cancer. J. Med. Chem. 2021, 64, 10286–10296. 10.1021/acs.jmedchem.1c00712. [DOI] [PubMed] [Google Scholar]
- William A. D.; Lee A. C.; Poulsen A.; Goh K. C.; Madan B.; Hart S.; Tan E.; Wang H.; Nagaraj H.; Chen D.; Lee C. P.; Sun E. T.; Jayaraman R.; Pasha M. K.; Ethirajulu K.; Wood J. M.; Dymock B. W. Discovery of the Macrocycle (9E)-15-(2-(Pyrrolidin-1-yl)ethoxy)-7,12,25-trioxa-19,21,24-triaza-tetracyclo[18.3.1.1(2,5).1(14,18)]hexacosa-1(24),2,4,9,14(26),15,17,20,22-nonaene (SB1578), a Potent Inhibitor of Janus Kinase 2/Fms-LikeTyrosine Kinase-3 (JAK2/FLT3) for the Treatment of Rheumatoid Arthritis. J. Med. Chem. 2012, 55, 2623–2640. 10.1021/jm201454n. [DOI] [PubMed] [Google Scholar]
- Gijsen M.; King P.; Perera T.; Parker P. J.; Harris A. L.; Larijani B.; Kong A. HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer. PLoS Biol. 2010, 8, e1000563 10.1371/journal.pbio.1000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. J.; Tran-Dube M.; Shen H.; Nambu M.; Kung P. P.; Pairish M.; Jia L.; Meng J.; Funk L.; Botrous I.; McTigue M.; Grodsky N.; Ryan K.; Padrique E.; Alton G.; Timofeevski S.; Yamazaki S.; Li Q.; Zou H.; Christensen J.; Mroczkowski B.; Bender S.; Kania R. S.; Edwards M. P. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- Basit S.; Ashraf Z.; Lee K.; Latif M. First macrocyclic 3(rd)-generation ALK inhibitor for treatment of ALK/ROS1 cancer: Clinical and designing strategy update of lorlatinib. Eur. J. Med. Chem. 2017, 134, 348–356. 10.1016/j.ejmech.2017.04.032. [DOI] [PubMed] [Google Scholar]
- Bauer T. M.; Shaw A. T.; Johnson M. L.; Navarro A.; Gainor J. F.; Thurm H.; Pithavala Y. K.; Abbattista A.; Peltz G.; Felip E. Brain Penetration of Lorlatinib: Cumulative Incidences of CNS and Non-CNS Progression with Lorlatinib in Patients with Previously Treated ALK-Positive Non-Small-Cell Lung Cancer. Target. Oncol. 2020, 15, 55–65. 10.1007/s11523-020-00702-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascarenhas J.; Hoffman R.; Talpaz M.; Gerds A. T.; Stein B.; Gupta V.; Szoke A.; Drummond M.; Pristupa A.; Granston T.; Daly R.; Al-Fayoumi S.; Callahan J. A.; Singer J. W.; Gotlib J.; Jamieson C.; Harrison C.; Mesa R.; Verstovsek S. Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients With Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2018, 4, 652–659. 10.1001/jamaoncol.2017.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X.; Smaill J. B.; Ding K. Medicinal Chemistry Strategies for the Development of Kinase Inhibitors Targeting Point Mutations. J. Med. Chem. 2020, 63, 10726–10741. 10.1021/acs.jmedchem.0c00507. [DOI] [PubMed] [Google Scholar]
- da Cunha Santos G.; Shepherd F. A.; Tsao M. S. EGFR mutations and lung cancer. Annu. Rev. Pathol. 2011, 6, 49–69. 10.1146/annurev-pathol-011110-130206. [DOI] [PubMed] [Google Scholar]
- Morgillo F.; Della Corte C. M.; Fasano M.; Ciardiello F. Mechanisms of resistance to EGFR-targeted drugs: lung cancer. ESMO Open 2016, 1, e000060 10.1136/esmoopen-2016-000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt H.; Bose D.; Petronczki M.; Scharn D.; Bader G.; Baum A.; Bergner A.; Chong E.; Dobel S.; Egger G.; Engelhardt C.; Ettmayer P.; Fuchs J. E.; Gerstberger T.; Gonnella N.; Grimm A.; Grondal E.; Haddad N.; Hopfgartner B.; Kousek R.; Krawiec M.; Kriz M.; Lamarre L.; Leung J.; Mayer M.; Patel N. D.; Simov B. P.; Reeves J. T.; Schnitzer R.; Schrenk A.; Sharps B.; Solca F.; Stadtmuller H.; Tan Z.; Wunberg T.; Zoephel A.; McConnell D. B. Start Selective and Rigidify: The Discovery Path toward a Next Generation of EGFR Tyrosine Kinase Inhibitors. J. Med. Chem. 2019, 62, 10272–10293. 10.1021/acs.jmedchem.9b01169. [DOI] [PubMed] [Google Scholar]
- Alihodzic S.; Bukvic M.; Elenkov I. J.; Hutinec A.; Kostrun S.; Pesic D.; Saxty G.; Tomaskovic L.; Ziher D. Current Trends in Macrocyclic Drug Discovery and beyond-Ro5. Prog. Med. Chem. 2018, 57, 113–233. 10.1016/bs.pmch.2018.01.002. [DOI] [PubMed] [Google Scholar]
- Cummings M. D.; Sekharan S. Structure-Based Macrocycle Design in Small-Molecule Drug Discovery and Simple Metrics To Identify Opportunities for Macrocyclization of Small-Molecule Ligands. J. Med. Chem. 2019, 62, 6843–6853. 10.1021/acs.jmedchem.8b01985. [DOI] [PubMed] [Google Scholar]
- Reymond J. L. The chemical space project. Acc. Chem. Res. 2015, 48, 722–730. 10.1021/ar500432k. [DOI] [PubMed] [Google Scholar]
- Deng Z.; Chuaqui C.; Singh J. Structural interaction fingerprint (SIFt): a novel method for analyzing three-dimensional protein-ligand binding interactions. J. Med. Chem. 2004, 47, 337–344. 10.1021/jm030331x. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Xie L.; Xie L.; Bourne P. E. Delineation of Polypharmacology across the Human Structural Kinome Using a Functional Site Interaction Fingerprint Approach. J. Med. Chem. 2016, 59, 4326–4341. 10.1021/acs.jmedchem.5b02041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.; Bourne P. E. Harnessing systematic protein-ligand interaction fingerprints for drug discovery. Drug Discov. Today 2022, 27, 103319. 10.1016/j.drudis.2022.07.004. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Bourne P. E. Revealing Acquired Resistance Mechanisms of Kinase-Targeted Drugs Using an on-the-Fly, Function-Site Interaction Fingerprint Approach. J. Chem. Theory Comput. 2020, 16, 3152–3161. 10.1021/acs.jctc.9b01134. [DOI] [PubMed] [Google Scholar]
- Liao J. J. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J. Med. Chem. 2007, 50, 409–424. 10.1021/jm0608107. [DOI] [PubMed] [Google Scholar]
- Strip-itTM is a tool to extract predefined scaffolds from input molecules, Strip-it 1.0.2; Silicos-it, 2012. (acccessed Nov/27/2017).
- Murtagh F.; Contreras P. Algorithms for hierarchical clustering: an overview. Wiley Interdiscip. Rev. Data Min. Knowl. Discov. 2011, 2, 86–97. 10.1002/widm.53. [DOI] [Google Scholar]
- Shultz M. D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem. 2019, 62, 1701–1714. 10.1021/acs.jmedchem.8b00686. [DOI] [PubMed] [Google Scholar]
- Viswanadhan V. N.; Ghose A. K.; Revankar G. R.; Robins R. K. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships. 4. Additional parameters for hydrophobic and dispersive interactions and their application for an automated superposition of certain naturally occurring nucleoside antibiotics. J. Chem. Inf. Model. 1989, 29, 163–172. 10.1021/ci00063a006. [DOI] [Google Scholar]
- McIver A. L.; Zhang W.; Liu Q.; Jiang X.; Stashko M. A.; Nichols J.; Miley M. J.; Norris-Drouin J.; Machius M.; DeRyckere D.; Wood E.; Graham D. K.; Earp H. S.; Kireev D.; Frye S. V.; Wang X. Discovery of Macrocyclic Pyrimidines as MerTK-Specific Inhibitors. ChemMedChem 2017, 12, 207–213. 10.1002/cmdc.201600589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eduful B. J.; O’Byrne S. N.; Temme L.; Asquith C. R. M.; Liang Y.; Picado A.; Pilotte J. R.; Hossain M. A.; Wells C. I.; Zuercher W. J.; Catta-Preta C. M. C.; Zonzini Ramos P.; Santiago A. D. S.; Counago R. M.; Langendorf C. G.; Nay K.; Oakhill J. S.; Pulliam T. L.; Lin C.; Awad D.; Willson T. M.; Frigo D. E.; Scott J. W.; Drewry D. H. Hinge Binder Scaffold Hopping Identifies Potent Calcium/Calmodulin-Dependent Protein Kinase Kinase 2 (CAMKK2) Inhibitor Chemotypes. J. Med. Chem. 2021, 64, 10849–10877. 10.1021/acs.jmedchem.0c02274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal J.; Tiwary P.; Berne B. J. How a Kinase Inhibitor Withstands Gatekeeper Residue Mutations. J. Am. Chem. Soc. 2016, 138, 4608–4615. 10.1021/jacs.6b01232. [DOI] [PubMed] [Google Scholar]
- Sztain T.; Ahn S. H.; Bogetti A. T.; Casalino L.; Goldsmith J. A.; Seitz E.; McCool R. S.; Kearns F. L.; Acosta-Reyes F.; Maji S.; Mashayekhi G.; McCammon J. A.; Ourmazd A.; Frank J.; McLellan J. S.; Chong L. T.; Amaro R. E. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat. Chem. 2021, 13, 963–968. 10.1038/s41557-021-00758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris G. M.; Huey R.; Lindstrom W.; Sanner M. F.; Belew R. K.; Goodsell D. S.; Olson A. J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zheng R.; Hu P.; Zhang Z.; Shen S.; Li X. Patients harboring uncommon EGFR exon 19 deletion-insertion mutations respond well to first-generation EGFR inhibitors and osimeritinib upon acquisition of T790M. BMC Cancer 2021, 21, 1215. 10.1186/s12885-021-08942-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best R. B.; Zhu X.; Shim J.; Lopes P. E.; Mittal J.; Feig M.; Mackerell A. D. Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; Mackerell A. D. Jr. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.; He X.; Vanommeslaeghe K.; MacKerell A. D. Jr. Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. 10.1002/jcc.23067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; MacKerell A. D. Jr. Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. 10.1021/ci300363c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey M. J.; Giupponi G.; Fabritiis G. D. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- Kräutler V.; van Gunsteren W. F.; Hünenberger P. H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. . [DOI] [Google Scholar]
- Seeber M.; Cecchini M.; Rao F.; Settanni G.; Caflisch A. Wordom: a program for efficient analysis of molecular dynamics simulations. Bioinformatics 2007, 23, 2625–2627. 10.1093/bioinformatics/btm378. [DOI] [PubMed] [Google Scholar]
- Marcou G.; Rognan D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J. Chem. Inf. Model. 2007, 47, 195–207. 10.1021/ci600342e. [DOI] [PubMed] [Google Scholar]
- Xie L.; Bourne P. E. Detecting evolutionary relationships across existing fold space, using sequence order-independent profile-profile alignments. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 5441–5446. 10.1073/pnas.0704422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desaphy J.; Raimbaud E.; Ducrot P.; Rognan D. Encoding protein-ligand interaction patterns in fingerprints and graphs. J. Chem. Inf. Model. 2013, 53, 623–637. 10.1021/ci300566n. [DOI] [PubMed] [Google Scholar]
- Roberts R. J. PubMed Central: The GenBank of the published literature. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 381–382. 10.1073/pnas.98.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JChem Base was used for structure searching and chemical database access and management, JChem 22.22.0; ChemAxon, 2022. (http://www.chemaxon.com).
- Kier L. B. Structural Information from Molecular Connectivity 4Xpc index. J. Pharm. Sci. 1980, 69, 1034–1039. 10.1002/jps.2600690914. [DOI] [PubMed] [Google Scholar]
- Hong H.; Xie Q.; Ge W.; Qian F.; Fang H.; Shi L.; Su Z.; Perkins R.; Tong W. Mold(2), molecular descriptors from 2D structures for chemoinformatics and toxicoinformatics. J. Chem. Inf. Model. 2008, 48, 1337–1344. 10.1021/ci800038f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.