

Abstract
Background and Objectives
Glial fibrillary acidic protein (GFAP) antibodies can associate with an astrocytopathy often presenting as a meningoencephalitis. Visual involvement has been reported but scarcely defined. We describe 2 cases of GFAP astrocytopathy with predominant visual symptoms and present a systematic review of the literature.
Methods
We describe 2 patients with GFAP astrocytopathy from our neurology department. We performed a systematic review of the literature according to PRISMA guidelines, including all patients with this disease and available clinical data, focusing on visual involvement.
Results
Patient 1 presented with bilateral optic disc edema and severe sudden bilateral loss of vision poorly responsive to therapy. Patient 2 showed bilateral optic disc edema, headache, and mild visual loss with complete recovery after steroids. We screened 275 records and included 84 articles (62 case reports and 22 case series) for a total of 592 patients. Visual involvement was reported in 149/592 (25%), with either clinical symptoms or paraclinical test-restricted abnormalities. Bilateral optic disc edema was found in 80/159 (50%) of patients investigated with fundoscopy, among which 49/80 (61%) were asymptomatic. One hundred (100/592, 17%) reported visual symptoms, often described as blurred vision or transient visual obscurations. Optic neuritis was rare and diagnosed in only 6% of all patients with GFAP astrocytopathy, often without consistent clinical and paraclinical evidence to support the diagnosis. Four patients (including patient 1) manifested a severe, bilateral optic neuritis with poor treatment response. In patients with follow-up information, a relapsing disease course was more frequently observed in those with vs without visual involvement (35% vs 11%, p = 0.0035, OR 3.6 [CI 1.44–8.88]).
Discussion
Visual system involvement in GFAP astrocytopathy is common and heterogeneous, ranging from asymptomatic bilateral optic disc edema to severe bilateral loss of vision, but optic neuritis is rare. GFAP CSF antibody testing should be considered in patients with encephalitis/meningoencephalitis or myelitis and bilateral optic disc edema, even without visual symptoms, and in patients with severe bilateral optic neuritis, especially when AQP4 antibodies are negative. Visual symptoms might associate with a higher relapse risk and help to identify patients who may require chronic immunosuppression.
Introduction
Glial fibrillary acidic protein (GFAP) antibody–associated astrocytopathy is an autoimmune, inflammatory CNS disorder characterized by a corticosteroid responsive encephalitis or meningoencephalitis commonly associated with myelitis, seizures, brainstem involvement, or psychiatric symptoms.1,2 Visual abnormalities are a common manifestation of GFAP astrocytopathy and may include optic neuritis, bilateral optic disc edema, or both.3 However, comprehensive descriptions of visual involvement in this condition are lacking.
In GFAP astrocytopathy, both sexes can be affected, with only a slight female predominance.2 Recent case series describing pediatric patients,4-7 who account for around 10% of total cases,1 have shown similar presentations and outcomes compared with adults. A concomitant neoplasm is a common finding, observed in just under one-third of patients, and most frequently reported as ovarian teratomas in young women.1,2
Many patients show brain MRI lesions with a linear, radial perivascular pattern of contrast enhancement, radiating from the periventricular regions through white matter in the centra semiovale, which is believed to be an expression of perivenular inflammation.8 Moreover, some patients show T2 hyperintense white matter lesions that involve the periventricular regions, centrum semiovale, deep brain structures, brainstem, and the spinal cord, often with the features of longitudinally extensive transverse myelitis (LETM), as seen in neuromyelitis optica spectrum disorder (NMOSD).2,9-12
Response to treatment is generally good in the acute phase, but up to 20% of patients with GFAP astrocytopathy experience a relapsing course.1
We present 2 cases of GFAP astrocytopathy with visual involvement, illustrating 2 different clinical scenarios: one, more common, characterized by mild symptoms, while the other is notable for its severe outcome. We also integrated the findings of our cases into those arising from a systematic review of the literature, aiming to describe in detail the characteristics of visual involvement in this disease.
Methods
Case Descriptions
We collected information from clinic consultations and discharge letters on 2 GFAP antibody-positive patients with visual involvement observed in our institution. Both patients provided written informed consent to disclose for data publication. Optic neuritis was defined according to recently published criteria.13
Data Retrieval
The review process followed published PRISMA guidelines for systematic reviews.14 Articles were retrieved using a systematic search strategy performed independently by 2 authors (G.G., P.B.) from the PubMed/MEDLINE database with the following search strings: ‘GFAP’ OR ‘Glial fibrillary acidic protein’ AND ‘autoantibodies’ OR ‘antibodies OR ‘autoantibody’ OR ‘antibody’ OR ‘IgG’ associated with common presentations: AND ‘meningoencephalitis’ OR ‘meningitis’ OR ‘meningoencephalomyelitis’ OR ‘encephalomyelitis’ OR ‘optic neuritis (ON)’ OR ‘optic disc edema’. Relevant studies not included in the search were added manually after reference screening. The search was last updated on February 15th, 2023. The present review was unregistered.
Inclusion Criteria
We included patients with (1) GFAP-IgG antibodies in serum and/or CSF and (2) sufficient clinical data to assess neurologic signs and symptoms. Three neurologists independently evaluated studies (M.G., S.M., and P.B.) before inclusion. Consensus was reached on all cases by mutual agreement.
Data Extraction and Outcomes
Two authors (G.G. and P.B.) independently gathered information on visual involvement, i.e., reported visual symptoms and optic system objective findings. Moreover, for all patients, we collected information on sex, ethnicity, age group, and evidence of concomitant cancer. In patients with visual abnormalities, a more thorough evaluation included visual involvement severity, symptoms lateralization, attack recovery, MRI evidence of optic system involvement, neurophysiology studies, acute and chronic phase treatments, disease course (monophasic vs relapsing at last follow up), disease severity at acme and outcome, classified using the modified Rankin Scale [mRS]15 as low disability/good outcome [mRS 2, or less] or serious disability/severe outcome [mRS 3 or more]. To compare the differences between patients with or without visual symptoms, when data were available from the studies selected through the review, the Chi-square or Fisher exact test were performed when appropriate. The measure of odds ratio (OR) was reported to quantify the risk between groups when opportune. p values of <0.05 were considered significant.
Visual function impairment was assessed using subcategories derived from the Visual Functional System Score (FSS) in the Expanded Disability Status Scale and divided in mild/asymptomatic, moderate, and severe. For further detail on categories definition, see supplementary eTable 1 (links.lww.com/NXI/A883).
Statistical analyses were performed with GraphPad Prism (version 9.0, GraphPad Software, La Jolla, CA) and Stata/IC 14.0 for Mac (64-bit, StataCorp, College Station, TX).
Quality and Bias Assessment
Two authors (G.G. and P.B.) independently assessed methodological strength and quality of the included studies using a specific tool proposed to evaluate case reports and case series,16 which examines 8 items categorized into 4 domains: selection, ascertainment, causality, and reporting. The scores of the 8 binary responses were summed into an aggregate score, and each study was rated as carrying low (score 0–2), moderate (score 3–5), or high risk of bias (score 6–8), supplementary eTable 2 (links.lww.com/NXI/A883). Disagreement among authors was solved by mutual consensus.
Patient Consents
Written informed consent to disclose was obtained from the 2 patients presented in the study in the form of case reports.
Data Availability
The final data set is available in Zenodo (doi:10.5281/zenodo.8052535). Anonymized data not published within this article will be made available by request from any qualified investigator.
Case Reports
Case 1
In 2017, a 33-year-old, previously healthy woman presented to the emergency department with a 4-day history of fever and cough. X-ray scans showed paracardiac pneumonia that was treated with oral antibiotics.
She came back 3 days later reporting headache and blurred vision. A course of diuretics and oral steroids was empirically started in the suspicion of idiopathic intracranial hypertension (IIH).
Two days later she experienced a dramatic drop in visual acuity, leading to severe bilateral loss of vision. Ophthalmic examination showed marked loss of vision (no light perception in the right eye; 6/36 in the left eye) with bilateral optic disc edema and an unremarkable fluorangiography. Neurologic examination was normal except for a bilateral mydriasis unresponsive to light. Brain, optic nerve, and spinal cord MRI was negative at admission and subsequent controls. Visual-evoked potentials (VEPs) showed absent cortical responses in the right eye and reduced amplitudes with preserved latencies in the left, indicating a severe bilateral axonal impairment. The combination of clinical and paraclinical evidence allows for the definition of a “possible ON” according to the recently proposed diagnostic criteria.13 CSF examination showed 96 lymphomonocytes/mm3, with normal blood-CSF barrier permeability. Isoelectric focusing showed a mirror pattern of oligoclonal IgG bands (type 4). Autoantibodies [including a standard panel for neuronal surface antibodies (NMDAR, LGI1, CASPR2, GABABR and AMPAR), aquaporin-4 (AQP4), myelin oligodendrocyte glycoprotein (MOG), ANA, ENA, and ANCA] were negative. Angiotensin-converting enzyme, thyroid function, and immunophenotyping of peripheral blood lymphocytes were within reference ranges. Immunohistochemistry on rat brain revealed an uncharacterized antibody in serum and CSF, which bound to the cytoplasm of astrocytes (Figure 1, panel A). This antibody reactivity completely disappeared in the serum samples collected after plasma exchange (PLEX) (Figure 1, panel B). A few years later, when the first reports of GFAP astrocytopathy were published, we established an in-house cell-based assay (CBA)8 that revealed the presence of GFAP antibodies in the patient's stored serum and CSF samples (Figure 1, panel C). At the time of clinical presentation, the patient had been initially treated with a course of IV methylprednisolone (1 g/die for 5 days), with no improvement. Subsequently, she performed 5 PLEX sessions associated with oral steroids, followed by IvIg (24 g/d for 5 days) and a 6-month oral steroid tapering. Repetition of the lumbar puncture after PLEX showed reduced cell number (30 lymphomonocytoids/mm3) and negativization of the antibody reactivity to astrocytes (Figure 1, panel D). The patient experienced limited therapeutic response during the acute phase. In the following months, she partially recovered her vision in the left eye (best visual acuity, 6/9) but not in her right eye (light perception). Visual acuity measured repeatedly over the following years showed stable findings. She did not experience any relapses after a 5-year follow-up.
Figure 1. GFAP Antibodies Detection in Patient 1.
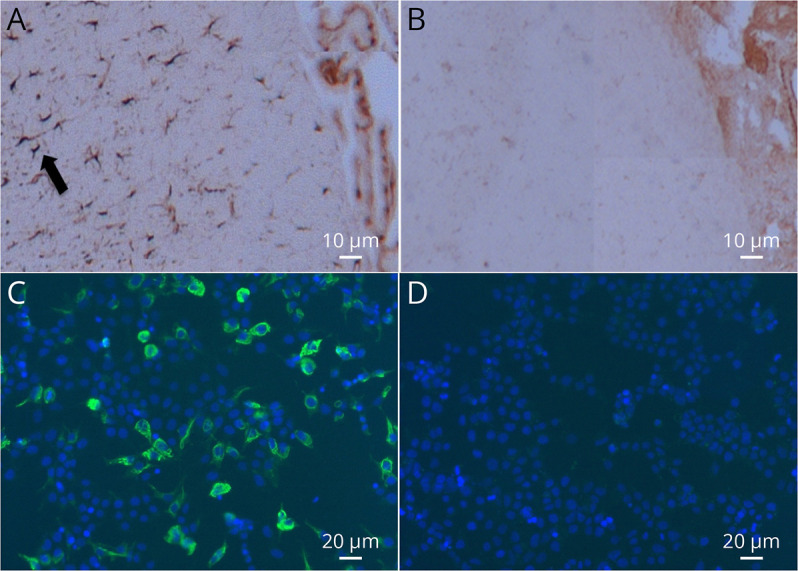
(A, B) Immunohistochemistry on rat brain and (C, D) in-house GFAP cell-based assay for the detection of GFAP antibodies. (A) CSF of patient 1 stains cytoplasm of astrocytes in periventricular areas (arrow). (B) This staining completely disappears after plasma exchange. (C) Cell-based assay for GFAP antibodies. The CSF IgG bind GFAP-transfected HEK293T cells. (D) Negative control (serum from a healthy subject; green: anti-human IgG; blue: 4',6-diamidino-2-phenylindole).
Case 2
A 19-year-old, previously healthy woman presented with fever, bilateral blurred vision, and headache developed acutely after a transient rash on her arms and torso. She was admitted to the Infectious Disease division, where a full blood panel for inflammatory markers, blood cultures, autoimmune and infectious screening (including Chlamydia pneumoniae, Mycoplasma pneumoniae, human Cytomegalovirus, Borrelia burgdorferi, and Rickettsiae) were negative. A full-body CT scan was also unremarkable. CSF examination showed pleocytosis (45 lymphomonocytoids/mm3), slightly elevated total proteins (64 mg/dL), and negative results on all culture and PCR tests for infectious agents.
Brain MRI showed diffuse, bilateral, T2-hyperintense lesions in the supratentorial white matter. After contrast administration, those lesions did not show pathologic enhancement, but there was a substantial increase of fine vascular images at many cortical sulci suggestive of diffuse venular congestion. MRI images from this admission are shown in Figure 2 (panels A and B).
Figure 2. MRI and OCT Features in Patient 2.
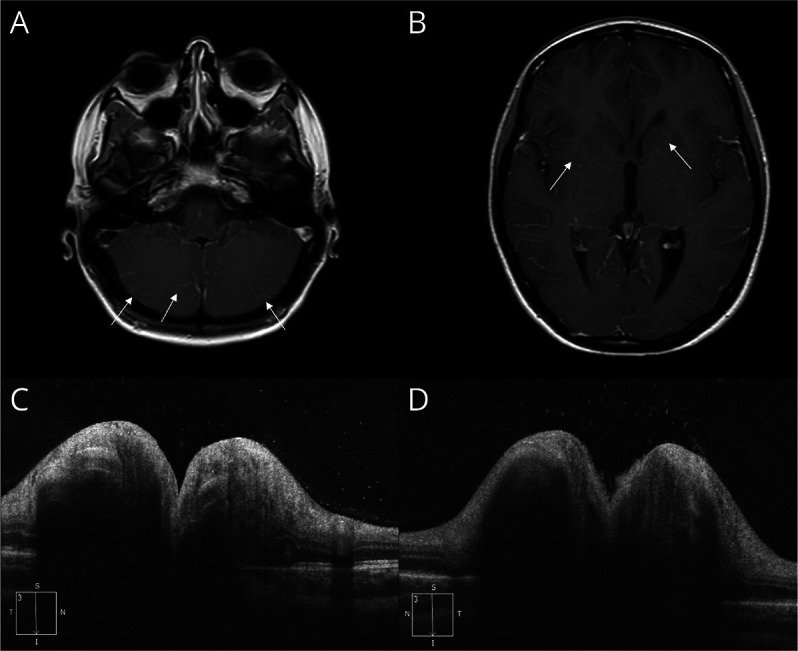
Postcontrast axial T1 MRI shows a very subtle radial enhancement and a diffuse venular congestion, both supratentorial and infratentorial, indicated by the white arrows (panels A, B).
Fundus oculi showed a bilateral optic disc edema, with focal hemorrhages and peripapillary exudates.
She was treated with IV antibiotics, without clinical improvement, and fever persistence. She was then transferred to our department.
On admission, she still suffered from headache and blurred vision, and the rest of the neurologic examination was normal. OCT confirmed the bilateral optic disc edema (Figure 2, panels C and D), although VEPs were normal. We performed an extended autoimmunity panel (as described for patient 1) that revealed the presence of CSF GFAP antibodies. The patient was treated with high-dose IV steroids (methylprednisolone 1 g for 5 days, followed by a 3-month oral tapering), with prompt and full regression of all her symptoms. At 12-month follow-up, the patient showed complete regression of symptoms, without any relapses.
Results of the Systematic Review
After identifying 275 records, we included 84 studies, consisting of 62 case reports and 22 case series (i.e., at least 3 patients described). The detailed search algorithm and flow chart are reported in Figure 3.
Figure 3. Flowchart Presenting the Search Process Based on the PRISMA Systematic Review of the Literature.

Quality control of the included studies indicated a low-to-moderate risk of bias. The risk of reporting bias cannot be excluded because some patients from larger cohorts might have been subject to subanalysis and could, therefore, be repeated in our database.
Total Cohort and Visual System Involvement
The overall number of GFAP antibody-positive patients identified was 649 (301 females; 46%). When age was reported, adult patients were 347 of 424 (82%). In 124 patients, the ethnic group was specified: 80 of 124 were Asians and 44 of 124 were White. A concomitant neoplasm, either found during diagnostic workup, or known in patient history, was present in 105 of 649 patients (16%).
Among the total 649 patients, only 592 had sufficient information regarding possible visual involvement and were, therefore, considered for further analysis.
Patients with visual system involvement were 149 of 592 (25%), of whom 31 of 149 (21%) had both clinical symptoms and paraclinical test confirmation (bilateral optic disc edema at fundus examination), 69 of 149 (46%) visual symptoms only, and 49 of 149 (33%) exclusive paraclinical involvement (asymptomatic disc edema). A diagnosis of ON was made in 36 cases, 6% of all GFAP astrocytopathy presentations. However, no detailed clinical information to support ON diagnosis was available for most of these patients. In all the included patients, visual involvement was caused by optic nerve dysfunction except for 2 patients with uveitis and one patient with large occipital lesions.17,18
Among the 149 patients with visual involvement, detailed information regarding the clinicoradiological phenotype were available in only 47. The most common extravisual presentation in this group was encephalomyelitis, found in 29 of 47 (62%), followed by meningoencephalitis/encephalitis in 15 of 47 (32%) and myelitis in 3 of 47 (6%). Visual involvement, when reported, was described as concomitant with the other neurologic manifestations in 40 patients (85%). On the other hand, isolated visual system involvement was reported for only 6 patients (6 of 149; 4%).
Patients With Symptomatic Visual Involvement
A total of 100 of 592 patients (17%) had visual symptoms. These were always classified as visual loss and mainly described as blurred vision or transient visual obscurations. No patient (except our patient 2) reported positive visual symptoms, such as flashing lights or myodesopsias.
Visual impairment was bilateral (including the description of “blurred vision”) in 97 of 100 patients (97%). We did not identify any significant differences in clinicodemographic features for patients with or without visual symptoms (Table 1).
Table.
Demographic, Disease Course, and Outcome Features of Patients With vs Without Visual Symptoms and Optic Disc Edema

Fundoscopic Alterations
Optic disc edema, reported in 80 of 159 (50%) patients who underwent fundoscopic examination, was always bilateral and symmetric. Among these patients, 49 of 80 (61%) did not report visual symptoms.
On the other hand, within the group of symptomatic patients, information on fundus examination was available for 34, and nearly all (32/34; 95%) showed bilateral optic disc edema (Figure 4).
Figure 4. Proportion of Patients With vs Without Visual Symptoms, and Optic Disc Edema.

Pie graphs on the left describe all included patients. Graphs on the right indicate the proportion of symptomatic patients with optic disc edema (when assessed; top right) and the proportion of those with optic disc edema who did or did not report visual symptoms (bottom right).
In the whole cohort of patients with GFAP astrocytopathy, clinicodemographic features of those with vs without optic disc involvement did not significantly differ (Table 1).
Neurophysiology and Optic Nerve Imaging
VEPs results were reported for only 15 patients with visual symptoms. Fourteen (93%) had pathologic findings (mainly reduced amplitudes or absent cortical responses, suggestive of axonal damage). In one patient, VEPs showed a W-shaped pattern, an aberrant response rarely seen in normal examinations that indicates a demyelinating optic pathway disturbance.19
Optic nerve MRI findings were reported in 88 patients and were abnormal in only 12 of 88 (19%), all showing monolateral optic nerve T2-hyperintense lesions.
Concomitant Antibodies
Concomitant antibodies were described in 14 patients with visual symptoms; 10 in CSF only (AQP4, n = 5; NMDAR, n = 2; unknown neural antibody, n = 3),20 1 in serum only (AQP4),21 and one in both CSF and serum (MOG).22 The patient with MOG antibodies presented with severe bilateral ON with poor response to steroid therapy. Four of 6 patients with AQP4 antibodies presented with visual disturbances described as “blurred vision” and diagnosed as ON, although no clinical or paraclinical tests were reported. One patient had a known seropositivity for serum AChR antibodies.23
Treatment, Disability, and Outcome
Nearly all patients with GFAP antibodies and visual involvement were treated in the acute phase with high-dose IV steroids (137/149; 93%), sometimes associated with IvIg (81/149; 55%) and/or PLEX (15/149; 10%). No specific association between treatment and outcome could be performed because of the heterogeneity of aggregate data.
Visual impairment was classified as mild in 144 of 149 of patients (97%), all showing good recovery. Conversely, the remaining 5 cases18,22,24,25 developed a severe visual impairment (including our patient 1). In one of them, loss of vision was caused by vast occipital lesions18 while the remaining 4 patients had severe, subacute, painless bilateral vision loss. All 4 patients fulfilled the criteria for a diagnosis of “possible ON” because of a definite visual acuity drop and varying degrees of paraclinical evidence. All patients received high-dose IV methylprednisolone, 2 were also treated with both IvIg and PLEX. Despite treatment, the final visual outcome was extremely poor, with mild or no signs of visual recovery in all 4 patients. One of those patients had an isolated visual system involvement (our case 1); one presented with a mild ataxic gait but no MRI abnormalities,25 one with multiple supra and subtentorial lesions, extraocular movement impairment, and gait disturbances,22 and one with large, tumefactive white matter lesions and flaccid tetraplegia.24 None of the patients had a concomitant cancer. One patient had coexisting MOG-IgG, although the disease course and scarce treatment response were atypical for MOG-IgG associated disease.
Relapses (both inside and outside the visual system) occurred in 24 of 141 (17%) patients with GFAP astrocytopathy with available follow-up. Considering only those with sufficient clinical information to ascertain visual involvement, relapses were more common in patients with visual symptoms (15/42, 35%) than in those without (11/83, 13%, p = 0.0035). The presence of visual symptoms was, therefore, associated with a higher risk of relapse (OR 3.6 [CI 1.44–8.88], not shown) (Table 1). There was no difference in sex, age group, concomitant malignancy, disability at acme, or severe outcome among patients with vs without visual symptoms or optic disc edema (Table 1).
Discussion
This systematic review highlights often overlooked features of visual abnormalities in GFAP astrocytopathy, including their clinical and paraclinical correlates and prognostic implications.
GFAP astrocytopathy is a relatively novel disease entity, sharing a common differential diagnostic ground with more frequent inflammatory disorders, such as NMOSD, or MOG antibodies–associated disease (MOGAD). For instance, NMOSD, MOGAD, or GFAP astrocytopathy can present with LETM.26 However, visual involvement characteristics could help differential diagnosis.
The typical visual manifestation in NMOSD and MOGAD is ON, which presents distinctive features. In NMOSD, ON is often severe, can be bilateral, simultaneous, or rapidly sequential (20% of cases), with very limited treatment response and visual recovery.3,27 In MOGAD, on the other hand, ON can present as a perineuritis with extensive lesions, prominent pain, and substantial recovery after steroid therapy; bilateral involvement is much more common in up to 50% of presentations.3,27 Optic disc edema occurs in around 5–33% patients with ON and NMOSD and in up to 80% of patients with MOGAD.28,29 In GFAP astrocytopathy, the occurrence of ON is rare, and many reported cases are based on nondetailed assessments. Notably, recently published peer-reviewed diagnostic criteria for ON13 require strict clinical or paraclinical features to reach a conclusive diagnosis, especially when vision loss is binocular, and further highlight the rarity of a definite ON phenotype in patients with GFAP astrocytopathy.
This suggests different mechanisms could underlie the pathogenesis of visual involvement. Fluorescein angiography studies in GFAP astrocytopathy have shown prominent retinal venular leakage, highly suggestive of a primary venous inflammation.30 This is consistent with the peculiar radial perivascular enhancement often found on brain MRI, therefore potentially suggesting a CNS vasculitic, venulocentric pathogenic process in GFAP astrocytopathy.30 ON without abnormalities of the optic disc orients the diagnosis toward NMOSD and MS, rather than GFAP astrocytopathy.
Although visual manifestations in GFAP astrocytopathy are generally mild, a severe bilateral optic neuropathy, unresponsive to immunotherapies, can rarely occur. Common features of such cases include subacute onset, unrelenting course until visual nadir, and limited response to high-dose steroid therapy and second-line acute-phase treatments. Neurophysiologic data show an axonal optic neuropathy with irreversible damage, similar to the optic neuropathy observed in NMOSD. CSF testing of GFAP antibodies should be considered in patients with this severe clinical presentation, especially when MOG and AQP4 antibodies are negative, and future studies should carefully evaluate therapeutic strategies.
Data from this review show that more than 60% of patients with GFAP astrocytopathy and optic disc edema did not report any visual symptoms, implying that this pathologic feature could be underestimated. Performing fundoscopic examination and identifying optic disc edema, even in the absence of visual symptoms, could thus be an important diagnostic clue to prompt GFAP antibody testing in patients with other clinical features of GFAP astrocytopathy, such as transverse myelitis, encephalitis, or meningoencephalitis. Notably, meningoencephalitis signs and symptoms can be subtle, as in our patient 2.
GFAP astrocytopathy should also be considered in the differential diagnosis of IIH. Visual symptoms of IIH could resemble the mild, fluctuant manifestations described in GFAP astrocytopathy in our review.31 An important difference between the 2 pathologies is lumbar puncture opening pressure that is abnormally high in IIH but normal in most patients with GFAP astrocytopathy, including those with optic disc edema.17
CSF is the preferred specimen for GFAP antibody testing because available data indicate that GFAP-IgG detection in CSF is highly specific for an inflammatory meningoencephalomyelitis phenotype when tested with a CBA.8 While serum positivity for GFAP antibody has been observed by some investigators in various disorders (including traumatic brain injury, brain tumors, autism, lead-exposed workers, and diabetes), such positivity is often transient and less specific than CSF-based testing.8 Pleocytosis and blood-CSF barrier damage is frequent,17,26more so than other inflammatory disorders such as MS, MOGAD, and NMOSD, probably owing to the frequent inflammatory involvement of meninges in GFAP astrocytopathy.
Finally, another important finding of our review indicates that patients with visual symptoms can have up to a three-fold higher risk of relapses. Twenty to 50% of patients with GFAP astrocytopathy experience relapses, but predictors of a relapsing disease course have not been reported so far. Visual symptoms could help clinicians to stratify patients and identify those who could benefit from long-term immunosuppression. However, this warrants prospective, multicenter studies for confirmation.
Our study has limitations. First, we cannot exclude a reporting bias because some of the described patients come from large cohorts and could have been subject to further studies in later studies. However, large cohorts in our review were infrequent, and this issue likely affected only a small fraction of patients. Second, despite the large numbers of patients, detailed information was frequently not available for many of those with visual involvement. This has led to a consistent reduction in number from the original pool of cases. Third, some patients, although less than 10%, presented with concomitant autoantibodies, including AQP4 and MOG, which are frequently associated with visual involvement, and this might have contributed to heterogeneity of clinical manifestations.
Visual system involvement in GFAP astrocytopathy is common and heterogeneous, ranging from asymptomatic bilateral optic disc edema to severe bilateral loss of vision, although ON is rare.
CSF GFAP antibody testing should be considered in patients with encephalitis/meningoencephalitis or myelitis and bilateral optic disc edema, even without visual symptoms, and in patients with severe bilateral ON, especially when AQP4 antibodies are negative.
Patients with visual abnormalities could be at higher risk of relapse and addressed to more aggressive treatments or chronic immunosuppression. Systematic studies of the optic pathways in these patients are needed to better understand disease pathology and guide treatment.
Glossary
- AQP4
aquaporin-4
- CBA
cell-based assay
- EDSS
Expanded Disability Status Scale
- FSS
Functional System Score
- GFAP
glial fibrillary acidic protein
- IIH
idiopathic intracranial hypertension
- LETM
longitudinally extensive transverse myelitis
- MOG
myelin oligodendrocyte glycoprotein
- MOGAD
MOG antibodies–associated disease
- NMOSD
neuromyelitis optica spectrum disorder
- OR
odds ratio
- VEPs
visual-evoked potentials
Appendix. Authors

Study Funding
The present study was supported by the Italian Ministry of Health, 'Ricerca Corrente 2022-2024.
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NN for full disclosures.
References
- 1.Kunchok A, Zekeridou A, McKeon A. Autoimmune glial fibrillary acidic protein astrocytopathy. Curr Opin Neurol. 2019;32(3):452-458. doi: 10.1097/WCO.0000000000000676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shan F, Long Y, Qiu W. Autoimmune glial fibrillary acidic protein astrocytopathy: a review of the literature. Front Immunol. 2018;9:2802. doi: 10.3389/fimmu.2018.02802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett JL, Costello F, Chen JJ, et al. Optic neuritis and autoimmune optic neuropathies: advances in diagnosis and treatment. Lancet Neurol. 2023;22(1):89-100. doi: 10.1016/S1474-4422(22)00187-9 [DOI] [PubMed] [Google Scholar]
- 4.Huang H, Bai K, Fu Y, et al. Glial fibrillary acidic protein astrocytopathy in pediatric patients: a retrospective study. Front Pediatr. 2021;8:626564. doi: 10.3389/fped.2020.626564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Francisco C, Meddles K, Waubant E. Pediatric glial fibrillary acidic protein meningoencephalomyelitis: a case report and review of the literature. Mult Scler Relat Disord. 2019;29:148-152. doi: 10.1016/j.msard.2018.12.008 [DOI] [PubMed] [Google Scholar]
- 6.Fang H, Hu W, Jiang Z, et al. Autoimmune glial fibrillary acidic protein astrocytopathy in children: a retrospective analysis of 35 cases. Front Immunol. 2021;12:761354. doi: 10.3389/fimmu.2021.761354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhuang X, Jin K, Li X, Li J. Autoimmune glial fibrillary acidic protein astrocytopathy in children: a retrospective study. Eur J Med Res. 2022;27(1):11. doi: 10.1186/s40001-022-00641-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flanagan EP, Hinson SR, Lennon VA, et al. Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients. Ann Neurol. 2017;81(2):298-309. doi: 10.1002/ana.24881 [DOI] [PubMed] [Google Scholar]
- 9.Fang B, McKeon A, Hinson SR, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. 2016;73(11):1297-1307. doi: 10.1001/jamaneurol.2016.2549 [DOI] [PubMed] [Google Scholar]
- 10.Long Y, Liang J, Xu H, et al. Autoimmune glial fibrillary acidic protein astrocytopathy in Chinese patients: a retrospective study. Eur J Neurol. 2018;25(3):477-483. doi: 10.1111/ene.13531 [DOI] [PubMed] [Google Scholar]
- 11.Iorio R, Damato V, Evoli A, et al. Clinical and immunological characteristics of the spectrum of GFAP autoimmunity: a case series of 22 patients. J Neurol Neurosurg Psychiatry. 2018;89(2):138-146. doi: 10.1136/jnnp-2017-316583 [DOI] [PubMed] [Google Scholar]
- 12.Dubey D, Hinson SR, Jolliffe EA, et al. Autoimmune GFAP astrocytopathy: prospective evaluation of 90 patients in 1 year. J Neuroimmunol. 2018;321:157-163. doi: 10.1016/j.jneuroim.2018.04.016 [DOI] [PubMed] [Google Scholar]
- 13.Petzold A, Fraser CL, Abegg M, et al. Diagnosis and classification of optic neuritis. Lancet Neurol. 2022;21(12):1120-1134. doi: 10.1016/S1474-4422(22)00200-9 [DOI] [PubMed] [Google Scholar]
- 14.Page MJ, Moher D, Bossuyt PM, et al. PRISMA 2020 explanation and elaboration: updated guidance and exemplars for reporting systematic reviews. BMJ. 2021;372:n160. doi: 10.1136/bmj.n160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saver JL, Filip B, Hamilton S, et al. Improving the reliability of stroke disability grading in clinical trials and clinical practice: the Rankin Focused Assessment (RFA). Stroke. 2011;41(5):992-995. doi: 10.1161/STROKEAHA.109.571364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murad MH, Sultan S, Haffar S, Bazerbachi F. Methodological quality and synthesis of case series and case reports. Evid Based Med. 2018;23(2):60-63. doi: 10.1136/bmjebm-2017-110853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gravier-Dumonceau A, Ameli R, Rogemond V, et al. Glial fibrillary acidic protein autoimmunity: a French Cohort Study. Neurology. 2022;98(6):E653-E668. doi: 10.1212/WNL.0000000000013087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimura A, Kato S, Takekoshi A, et al. Autoimmune glial fibrillary acidic protein astrocytopathy resembling isolated central nervous system lymphomatoid granulomatosis. J Neuroimmunol. 2021:361:577748. doi: 10.1016/j.jneuroim.2021.577748 [DOI] [PubMed] [Google Scholar]
- 19.Tokimura R, Matsuda N, Kobayashi S, Kimura A, Kanai K. Abnormal evoked potentials in autoimmune glial fibrillary acidic protein astrocytopathy. eNeurologicalSci. 2020;18:100229. doi: 10.1016/j.ensci.2020.100229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang X, Xu H, Ding M, et al. Overlapping autoimmune syndromes in patients with glial fibrillary acidic protein antibodies. Front Neurol 2018;9:251. doi: 10.3389/fneur.2018.00251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin H, Huang Y, Zeng H, et al. Overlapping clinical syndromes in patients with glial fibrillary acidic protein IgG. Neuroimmunomodulation. 2020;27(1):69-74. doi: 10.1159/000505730 [DOI] [PubMed] [Google Scholar]
- 22.Ding J, Ren K, Wu J, et al. Overlapping syndrome of MOG-IgG-associated disease and autoimmune GFAP astrocytopathy. J Neurol. 2020;267(9):2589-2593. doi: 10.1007/s00415-020-09869-2 [DOI] [PubMed] [Google Scholar]
- 23.White D, Mollan SP, Ramalingam S, Nagaraju S, Hayton T, Jacob S. Enlarged and enhancing optic nerves in advanced glial fibrillary acidic protein meningoencephalomyelitis. J Neuroophthalmol. 2019;39(3):411-415. doi: 10.1097/WNO.0000000000000842 [DOI] [PubMed] [Google Scholar]
- 24.Mabrouki FZ, Aziouaz F, Sekhsoukh R, Yassine M. Subacute blindness revealing an autoimmune glial fibrillary acidic protein astrocytopathy. Cureus. 2021;13(8):e17588. doi: 10.7759/cureus.17588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Utley WJ, El-Dieb A, Chancellor AM. Anti-GFAP neuroinflammation with synchronous bilateral papillitis and characteristic imaging. Pract Neurol. 2021;21(2):171-172. doi: 10.1136/practneurol-2020-002805 [DOI] [PubMed] [Google Scholar]
- 26.Cacciaguerra L, Sechi E, Rocca MA, Filippi M, Pittock SJ, Flanagan EP. Neuroimaging features in inflammatory myelopathies: a review. Front Neurol. 2022;13. doi: 10.3389/fneur.2022.993645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moheb N, Chen JJ. The neuro-ophthalmological manifestations of NMOSD and MOGAD—a comprehensive review. Eye (Lond). 2023. doi: 10.1038/s41433-023-02477-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banwell B, Bennett JL, Marignier R, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD panel proposed criteria. Lancet Neurol. 2023;4422(22):1-15. doi: 10.1016/s1474-4422(22)00431-8 [DOI] [PubMed] [Google Scholar]
- 29.Marignier R, Hacohen Y, Cobo-Calvo A, et al. Myelin-oligodendrocyte glycoprotein antibody-associated disease. Lancet Neurol. 2021;20(9):762-772. doi: 10.1016/S1474-4422(21)00218-0 [DOI] [PubMed] [Google Scholar]
- 30.Chen JJ, Aksamit AJ, McKeon A, et al. Optic disc edema in glial fibrillary acidic protein autoantibody-positive meningoencephalitis. J Neuroophthalmol. 2018;38(3):276-281. doi: 10.1097/WNO.0000000000000593 [DOI] [PubMed] [Google Scholar]
- 31.Yetimler B, Tzartos J, Şengül B, et al. Serum glial fibrillary acidic protein (GFAP)-antibody in idiopathic intracranial hypertension. Int J Neurosci. 2021;131(8):775-779. doi: 10.1080/00207454.2020.1758084 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The final data set is available in Zenodo (doi:10.5281/zenodo.8052535). Anonymized data not published within this article will be made available by request from any qualified investigator.