

Abstract
Objectives
Leucine-rich glioma-inactivated 1 (LGI1) encephalitis and IgG4-related disease (IgG4RD) have traditionally been regarded as 2 distinct disease entities.
Methods
We detail the presentation, investigations, and management of a patient who showed typical signs and symptoms of LGI1 encephalitis and also found to possess pancreatic changes and a serum profile in keeping with IgG4RD.
Results
Serum and CSF analyses at presentation showed a significant hyponatraemia (117 mmol/L), elevated IgG4 concentration (1.73 g/L), and the presence of LGI1 antibodies. MRI revealed symmetrical diffuse T2-weighted hyperintensity and mild swelling throughout both medial temporal lobes. CT of the chest, abdomen and pelvis revealed an edematous, bulky pancreas with loss of lobulation, typical for IgG4RD. A glucocorticoid weaning regimen was commenced, facilitated by 2 rituximab infusions, with the patient showing an effective treatment response. HLA testing confirmed the presence of HLA DRB1 and HLA DQB1 risk alleles.
Discussion
This case suggests that there may be shared mechanisms between LGI1 encephalitis and IgG4RD, supported by common risk HLA associations and treatment strategies/responses. To our knowledge, this represents the first instance that LGI1 encephalitis and IgG4RD have been reported in the same patient and emphasizes the continued development of our understanding of the wide range of IgG4-mediated conditions.
Clinical Presentation
An 82-year-old woman presented with an 8-month history of progressive confusion, unsteady gait with falls, and speech difficulties. She struggled to remember her daily routine and retain new information, and her sleep-wake cycle was disrupted. Furthermore, she often suffered episodes of 'fidgeting' with tonic posturing of her right hand, associated with vacancy. These episodes were increasing in frequency and duration, now occurring hourly and lasting up to 15 seconds with no clear trigger. She otherwise reported no weight loss or gastrointestinal symptoms.
Her medical history included bilateral knee replacements complicated by a deep vein thrombosis and an episode of speech disturbance 9 months previously, diagnosed as an ischemic stroke. Her medications included clopidogrel 75 mg, atorvastatin 40 mg, and lansoprazole 30 mg once daily. There was no significant family medical history.
Diagnostic Process
Physical examination of the neurologic system was unremarkable besides mild ataxia. However, during cognitive testing (in which clock drawing and memory impairment was evident), she experienced a brief episode of vacancy in which she was unresponsive and experienced tonic posturing of the left hand. After a few seconds, her right arm lifted and became rigid. This self-terminated after 10 seconds when she subsequently became reoriented.
Routine blood tests were unremarkable besides significant hyponatraemia (117 mmol/L). Paired serum and urine osmolalities supported a diagnosis of syndrome of inappropriate antidiuretic hormone secretion (SIADH), which normalized to 135 mmol/L with fluid restriction. MRI revealed relatively symmetrical diffuse T2-weighted hyperintensity and mild swelling throughout both medial temporal lobes (Figure 1). Distinctively unilateral high T1-weighted and T2-weighted signal in the ventral right putamen was also noted. Appearances were suggestive of limbic encephalitis, and owing to the clinical presentation and putaminal high T1-weighted signal, leucine-rich glioma-inactivated 1 (LGI1) encephalitis was felt most likely. An electroencephalogram demonstrated slow wave activity and no epileptiform features despite the patient experiencing 2 episodes of flexion of the right arm and contraction of the right side of the face lasting a few seconds during testing.
Figure 1. MRI Brain Imaging.

Axial fluid-attenuated inversion recovery (FLAIR) MRI sequences (A, B) at presentation demonstrating T2-weighted hyperintensity in the ventral right putamen (A, arrow) and in both medial temporal lobes (B, arrows). Coronal T1-weighted image displays corresponding mildly increased T1-weighted signal in the ventral right putamen (C, with magnified bottom panel). Axial postcontrast T1-weighted image shows asymmetric abnormal enhancement of the right hippocampus (D, with magnified panel). Repeat FLAIR (E, F) and susceptibility-weighted sequences (G) 6 months after presentation and therapy shows regression of prior abnormal high T2-weighted signal (E, F) with new mild volume loss in the affected regions and new mineralization of the ventral right putamen (G, arrow). FDG-PET/CT performed 3 weeks into presentation (and after commencement of corticosteroids) fused with T2-weighted axial MRI (H) demonstrates corresponding asymmetric hypometabolism in the affected ventral right putamen (arrow).
A paraneoplastic, autoimmune and infection screen confirmed the presence of serum LGI1 antibodies in blood and CSF, further indicating a diagnosis of LGI1 encephalitis (CSF glucose = 3.7 mmol/L, lactate = 1.8 mmol/L and protein = 0.58g/L). A CT of the chest, abdomen, and pelvis was undertaken to exclude a secondary cause, which revealed an edematous, bulky appearance to her pancreas with loss of lobulation (Figure 2), typical in appearance of IgG4-related disease (IgG4RD). Serum IgG4 concentration was elevated at 1.73 g/L (normal <1.3 g/L). Whole-body FDG PET/CT (3 weeks after commencement of corticosteroids) confirmed no avid regions or evidence of malignancy.
Figure 2. CT of the Chest, Abdomen, and Pelvis Imaging.
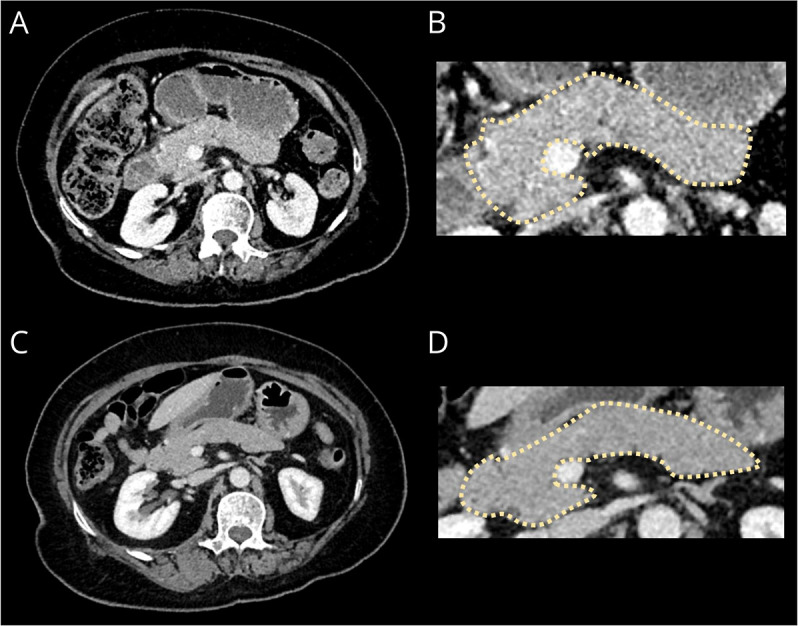
Axial contrast-enhanced CT portovenous phase imaging of the abdomen at presentation (A) with a magnified panel of the pancreas (B) demonstrates minimal peripancreatic fat stranding and diffuse enlargement of the pancreas with loss of the definition of the pancreatic clefts causing a “sausage-shape” morphology, typical of IgG4-related disease.6 Repeat CT (C) with a magnified panel (D) 5 months later demonstrates resolution of the fat stranding and reduced pancreatic swelling.
Management and Follow-up
The patient was commenced on IV methylprednisolone 1 g through 3 daily pulses, followed by oral prednisolone 60 mg daily with a weaning reduction of 10 mg per week. Sodium valproate MR 300 mg twice daily was commenced for seizure prevention.
This led to a rapid, significant improvement in cognitive symptoms over a matter of days. The patient was discharged from hospital 3 weeks after admission and 9 days after commencing corticosteroids. Outpatient reviews confirmed continued improvement in cognitive function toward her premorbid baseline. Three months after admission, she received rituximab treatment (two 1g IV infusions, a fortnight apart) to facilitate corticosteroid weaning and maintain disease control. Repeat CT of the pancreas and MR brain imaging at 6 months after commencement of treatment showed resolution of the pancreatic swelling and regression of the prior medial temporal and right putaminal abnormal signal with mild interval volume loss in the involved regions (Figures 1 and 2). Serum IgG4 levels had normalized (0.97 g/L). At the time of last follow-up 9 months after starting treatment, the patient remained well and off corticosteroids, and serum LGI1 autoantibodies were no longer detectable. Serum samples were sent for HLA testing revealing HLA DRB1 and HLA DQB1 alleles (Table).
Table.
Full HLA Genotyping (High Resolution) Results

Discussion
This novel case represents an intriguing overlap of 2 rare conditions relating to IgG4 antibodies: LGI1 antibody encephalitis and IgG4RD. LGI1 encephalitis is characterized by the presence of IgG4 antibodies against the LGI1 neuronal protein, which is a component of a transsynaptic complex that interacts with voltage gated-potassium channels found on the cell surface of neurons.1 The condition typically affects older men (median age 60 years), manifesting as an autoimmune limbic encephalitis. Presenting features subsequently include a subacute onset of confusion, prominent amnesia with autobiographical memory impairment, behavioral abnormalities, and seizures (normally preceding the memory impairment with faciobrachial dystonic seizures being particularly characteristic).1 Hyponatraemia reportedly occurs in 60% of patients and is thought to be driven by SIADH, potentially secondary to inflammatory processes occurring within the hypothalamus. MRI may demonstrate abnormal signal in the medial temporal lobe and approximately 60% of cases possess lesions in the basal ganglia which, as in this case, are typically unilateral in the putamen or caudate (usually contralateral to faciobrachial dystonic seizures) and distinctively high on T1-weighted imaging.1,2 Serum IgG4 is shown to be marginally elevated in up to 20% of patients with LGI1 encephalitis, although no extracranial inflammatory manifestations were reported in these cases.3
IgG4 antibodies evolve as an anti-inflammatory response to chronic antigenic stimulation and are normally the least common IgG subclass found within healthy individuals.4 Conversely, IgG4 neurologic disorders are characterized by the direct action of IgG4 antibodies on specific enzymatic or protein-protein interactions to disrupt their usual downstream signal transduction pathways.1 Unlike other autoimmune neurologic disorders, which primarily involve the IgG1 subclass, IgG4 antibodies are unable to activate complement-mediated immune responses and are less effective in crosslinking and internalizing their target antigen.4
Encephalitis is not a typical feature of the distinct and broader disease entity, IgG4RD, which is characterized by mass-forming lesions containing IgG4+ plasma cell infiltrates that can cause permanent organ injury.4 IgG4RD can mimic neoplastic, infectious, and other inflammatory diseases in typical organ patterns affecting salivary glands and lymph nodes of the head and neck, the pancreas, biliary tree, kidneys, and retroperitoneum. It is recognized in our presented case that the absence of histopathologic confirmation does not allow a definitive diagnosis of IgG4RD; however, the typical pancreas appearances (with no other obvious pancreatitis cause) and elevated serum IgG4 concentration in the absence of any other causal neoplastic/inflammatory disease partly satisfy the diagnostic criteria.5,6 Pancreatic biopsy was not pursued given the potential risks involved and the need to treat LGI1 encephalitis with corticosteroids regardless. Treating and closely observing therapeutic response with surveillance imaging, therefore, seemed most appropriate.
HLA association studies confirm common gene loci and risk alleles to both LGI1 encephalitis and IgG4RD. Studies of the Japanese population have identified susceptibility loci in regions of the HLA-DRB1 and DQB1 genes toward the development of IgG4RD.7,8 Similarly, studies involving patients with LGI1 patients have also revealed susceptibility loci on regions of HLA-DRB1.9 In our patient's HLA typing, the following risk alleles were confirmed: DQB1* 03:03, HLA-DRB1*14:04 (pertaining to IgG4RD susceptibility) with DRB1* 07:01 (pertaining to LGI1 encephalitis).
Good responses to corticosteroid and B-cell depleting therapies, such as rituximab, are described in both LGI1 encephalitis4,10 and IgG4RD.11 The use of rituximab in this case allowed corticosteroid weaning once disease was controlled, helping to reduce the risks of corticosteroid side effects and disease relapse. Long-term outcome reports indicate that only 35% of patients with LGI1 encephalitis return to their baseline cognitive function, associated with greater disease severity and delays to immunotherapy.12 However, relapses of LGI1 encephalitis are uncommon after the use of rituximab. This compares with IgG4RD, which frequently encounters relapses after B-cell reconstitution.13 For our patient, longer-term surveillance of disease activity will, therefore, likely be required with further dosing of rituximab according to B-cell counts to maintain effective disease control.
Acknowledgment
The authors would like to thank our patient for kindly giving us the opportunity to present and learn from their interesting case. Infrastructure support for this research was provided by the NIHR Imperial Biomedical Research Centre (BRC).
Appendix. Authors

Study Funding
The authors report no targeted funding.
Disclosure
The authors report no relevant disclosures. Go to Neurology.org/NN for full disclosures.
References
- 1.Dalmau J, Geis C, Graus F. Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the central nervous system. Physiol Rev. 2017;97(2):839-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flanagan EP, Kotsenas AL, Britton JW, et al. Basal ganglia T1 hyperintensity in LGI1-autoantibody faciobrachial dystonic seizures. Neurol Neuroimmunol Neuroinflamm. 2015;2(6):e161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Endmayr V, Tunc C, Ergin L, et al. Anti-neuronal IgG4 autoimmune diseases and IgG4-Related diseases may not be part of the same spectrum: a comparative study. Front Immunol. 2021;12:785247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dalakas MC. IgG4-mediated neurologic autoimmunities: understanding the pathogenicity of IgG4, ineffectiveness of IVIg, and long-lasting benefits of anti-B cell therapies. Neurol Neuroimmunol Neuroinflamm. 2022;9(1):e1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Culver EL, Sadler R, Simpson D, et al. Elevated serum IgG4 levels in diagnosis, treatment response, organ involvement, and relapse in a prospective IgG4-related disease UK cohort. Am J Gastroenterol. 2016;111(5):733-743. [DOI] [PubMed] [Google Scholar]
- 6.Wallace ZS, Naden RP, Chari S, et al. The 2019 American College of Rheumatology/European League against rheumatism classification criteria for IgG4-related disease. Arthritis Rheumatol. 2020;72(1):7-19. [DOI] [PubMed] [Google Scholar]
- 7.Terao C, Ota M, Iwasaki T, et al. IgG4-related disease in the Japanese population: a genome-wide association study. Lancet Rheumatol. 2019;1(1):e14-e22. [DOI] [PubMed] [Google Scholar]
- 8.Kawa S, Ota M, Yoshizawa K, et al. HLA DRB10405-DQB10401 haplotype is associated with autoimmune pancreatitis in the Japanese population. Gastroenterology. 2002;122(5):1264-1269. [DOI] [PubMed] [Google Scholar]
- 9.Binks S, Varley J, Lee W, et al. Distinct HLA associations of LGI1 and CASPR2-antibody diseases. Brain. 2018;141(8):2263-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez A, Klein CJ, Sechi E, et al. LGI1 antibody encephalitis: acute treatment comparisons and outcome. J Neurol Neurosurg Psychiatry. 2022;93(3):309-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamisawa T, Okazaki K. Diagnosis and treatment of IgG4-related disease. Curr Top Microbiol Immunol. 2017;401:19-33. [DOI] [PubMed] [Google Scholar]
- 12.Ariño H, Armangué T, Petit-Pedrol M, et al. Anti-LGI1–associated cognitive impairment. Neurology. 2016;87(8):759-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebbo M, Grados A, Samson M, et al. Long-term efficacy and safety of rituximab in IgG4-related disease: data from a French nationwide study of thirty-three patients. PLoS One. 2017;12(9):e0183844. [DOI] [PMC free article] [PubMed] [Google Scholar]