

Abstract
Background and Objectives
The clinical criteria for autoimmune encephalitis (AE) were proposed by Graus et al. in 2016. In this study, the AE criteria were validated in the real world, and common AE mimics were described. In addition, criteria for probable anti-LGI1 encephalitis were proposed and validated.
Methods
In this retrospective cohort study, patients referred to our national referral center with suspicion of AE and specific neuroinflammatory disorders with similar clinical presentations were included from July 2016 to December 2019. Exclusion criteria were pure cerebellar or peripheral nerve system disorders. All patients were evaluated according to the AE criteria.
Results
In total, 239 patients were included (56% female; median age 42 years, range 1–85). AE was diagnosed in 104 patients (44%) and AE mimics in 109 patients (46%). The most common AE mimics and misdiagnoses were neuroinflammatory CNS disorders (26%), psychiatric disorders (19%), epilepsy with a noninflammatory cause (13%), CNS infections (7%), neurodegenerative diseases (7%), and CNS neoplasms (6%). Common confounding factors were mesiotemporal lesions on brain MRI (17%) and false-positive antibodies in serum (12%). Additional mesiotemporal features (involvement extralimbic structures, enhancement, diffusion restriction) were observed more frequently in AE mimics compared with AE (61% vs 24%; p = 0.005). AE criteria showed the following sensitivity and specificity: possible AE, 83% (95% CI 74–89) and 27% (95% CI 20–36); definite autoimmune limbic encephalitis (LE), 10% (95% CI 5–17) and 98% (95% CI 94–100); and probable anti-NMDAR encephalitis, 50% (95% CI 26–74) and 96% (95% CI 92–98), respectively. Specificity of the criteria for probable seronegative AE was 99% (95% CI 96–100). The newly proposed criteria for probable anti-LGI1 encephalitis showed a sensitivity of 66% (95% CI 47–81) and specificity of 96% (95% CI 93–98).
Discussion
AE mimics occur frequently. Common pitfalls in AE misdiagnosis are mesiotemporal lesions (predominantly with atypical features) and false-positive serum antibodies. As expected, the specificity of the criteria for possible AE is low because these criteria represent the minimal requirements for entry in the diagnostic algorithm for AE. Criteria for probable AE (-LGI1, -NMDAR, seronegative) and definite autoimmune LE are applicable for decisions on immunotherapy in early disease stage, as specificity is high.
Introduction
The discovery of anti-NMDAR encephalitis (anti-NMDARE) in 2007 is regarded as a major breakthrough by introducing autoimmune encephalitis (AE) as a new disease entity with unique characteristics.1,2 In contrast to classical paraneoplastic neurologic syndromes (PNSs),3 AE is associated with neuronal autoantibodies against extracellular antigens, which are directly pathogenic.4 Prompt diagnosis is essential in AE because early administration of immunotherapy improves outcome in most patients with AE.5-7 The diagnosis of AE strongly relies on the identification of neuronal autoantibodies in serum and CSF.4,5 However, it has been stated that former clinical criteria for AE were too reliant on neuronal autoantibody status8 because comprehensive antibody testing can be time consuming and can result in diagnostic and therapeutic delay.5 In addition, the absence of neuronal autoantibodies does not exclude AE5,9 while on the other hand false-positive results may produce an incorrect diagnosis of AE.5,10-12 An important improvement in the clinical approach of patients with suspicion of AE was the publication of the clinical AE criteria in 2016 by Graus et al.5 based on conventional clinical neurologic assessment and standard diagnostic tests (MRI, EEG, and CSF studies). The 2016 AE criteria allow preliminary treatment with immunotherapy by establishment of an early diagnosis of probable or definite AE awaiting neuronal autoantibody status.5 In addition, a novel diagnosis of autoantibody-negative but probable AE and criteria for probable anti-NMDAR encephalitis (anti-NMDARE) were introduced (Figure 1).5 However, because many diseases can resemble AE and immunotherapy may induce serious adverse events or delay of alternative diagnoses,11,13 the diagnostic accuracy of the 2016 AE criteria, particularly specificity, is highly relevant for clinical practice. In this study, we validate the 2016 clinical AE criteria and describe frequently recognized mimics of AE and the red flags to prevent misdiagnosis. In addition, we propose criteria for probable anti-LGI1 encephalitis, to improve outcome by early diagnosis and treatment in this relatively common subtype of AE.7
Figure 1. 2016 Clinical Criteria for Autoimmune Encephalitis by Graus et al.5.

Reprinted with permission from Elsevier.
Methods
Patient Selection and Diagnostics
In this retrospective cohort study, we included children and adults referred to the Erasmus MC University Medical Center with suspicion of AE in the period of July 2016 until December 2019. This study was performed according to the Strengthening the Reporting of Observational Studies in Epidemiology reporting guideline for observational research. The Erasmus MC University Medical Center is the Dutch national referral center for neuroinflammation and accredited European Reference Network site (ERA-RITA). All disorders presented in the diagnostic algorithm for AE proposed by Graus et al.5 were included. Patients with autoimmune cerebellopathies, opsoclonus-myoclonus syndrome, and disorders exclusively affecting the peripheral nerve system were excluded because the 2016 clinical AE criteria focus on patients presenting with a subacute onset of memory deficits or altered mental status.5 Ancillary testing included blood analysis, lumbar puncture, EEG, MRI, and cerebral biopsy, if necessary. All patients underwent extensive neuronal autoantibody testing in serum and CSF, if available, using a combination of tests, including in-house immunohistochemistry (IHC) on rat brain slices.14 Specific cell surface autoantibodies were tested using commercial cell-based assays (CBAs; Euroimmun, Lübeck, Germany) or in-house CBAs. Only those samples with positive CBAs were considered positive that could be confirmed by IHC or, if necessary, live hippocampal cell cultures (LN).14-16 GlyR,17 KLHL-11,18 GFAP,19 IgLON5,20 mGluR1, and mGluR5 were tested by in-house CBAs.21,22 Anti-MOG antibodies were tested using CBA, as described elsewhere.23 Anti-GAD65 was tested by ELISA (Medizym anti-GAD 96, Medipan, Berlin, Germany) and considered clinically relevant if serum concentration was >10.000 IU/mL or CSF concentration was >100 IU/mL (high titer) and IHC showed a compatible staining pattern.24 Antibodies against paraneoplastic neurologic (‘onconeural’) antigens amphiphysin, CV2, Ma1/2, Ri, Yo, Hu, and Tr (DNER) were detected by the combined use of line immunoassay (EUROLINE Paraneoplastic neurological Syndrome 12 Ag (IgG), Euroimmun, Lubeck, Germany), and when positive, it is confirmed using a second antigen-specific line immunoassay (PNS-Blot, Ravo Diagnostika, Freiburg, Germany) and indirect immunofluorescence (Cerebellum (Primate) Slide, Inova Diagnostics, San Diego, CA).
Definitions
All patients were physically seen by the authors (R.W.V.S, A.L.B., Y.S.C., A.E.M.B., J.M.D.V, R.F.N., and M.J.T.). Medical records were reviewed, and definite diagnoses were made by consensus. Patients with positive neuronal autoantibody status and a compatible clinical syndrome, including PNS, were classified as definite AE. An AE mimic was defined as a patient with initial strong suspicion of AE and an alternative final diagnosis. All patients were evaluated according to the 2016 AE criteria,5 including our proposed criteria for probable anti-LGI1 encephalitis (Figure 2). Patients were classified as probable seronegative AE (SN-AE), if the 2016 criteria were satisfied.5 Established criteria were used to define acute disseminated encephalomyelitis (ADEM) and Hashimoto encephalopathy (HE).5 ADEM and HE were classified as separate inflammatory categories (i.e., not as inflammatory AE mimic) because these disorders were also separately included in the 2016 AE criteria.5 Patients with strong evidence of a neuroinflammatory disorder, who did not fulfill the criteria for probable SN-AE or any other specific inflammatory CNS disorder, were classified in this study as probable neuroinflammatory disorder (PNID), which was considered as an inflammatory subcategory of the mimics. In this study, strong evidence of a neuroinflammatory disorder was defined as the presence of ≥2 of the following characteristics: brain MRI suggestive of AE, CSF pleocytosis, specific oligoclonal bands in CSF, repeated steroid responsiveness, or similar staining pattern on IHC in serum and CSF in the absence of a known neuronal autoantibody. Patients exclusively demonstrating a pleocytosis and an altered mental status, new-onset seizures, or focal deficits without other specific evidence of an infectious or inflammatory cause were labeled as encephalitis with unknown cause.
Figure 2. Proposed Criteria for Probable Anti-LGI1 Encephalitis.

Standard Protocol Approvals, Registrations, and Patient Consents
IRB approval was waivered, but informed consent for usage of medical information for research purposes was obtained from all patients or proxies that could be reached.
Statistics
We used IBM SPSS 25.0 (SPSS Inc) and Prism 8.4.3 (GraphPad) for statistical analysis. The Pearson χ2 test or the Fisher-Freeman-Halton test, when appropriate, was used for patient characteristics analysis and group comparisons, encompassing categorical data. p-values were two-sided and considered statistically significant when below 0.05. Sensitivity and specificity of the 2016 clinical AE criteria were calculated. Sensitivity was defined as the percentage of definite AE and probable SN-AE of all patients fulfilling a specific category of the clinical AE criteria (i.e., true positive), whereas specificity was defined as the percentage of diagnoses other than AE, including other neuroinflammatory disorders, of all patients not fulfilling these criteria (i.e., true negative). No sensitivity was determined for probable SN-AE, in view of the low expected number of patients and absence of a gold standard for this particular diagnosis. Criteria for probable anti-LGI1 encephalitis were also applied to a previous nonoverlapping cohort from our center, described earlier.25 Similarly, criteria for probable anti-NMDARE were calculated in the whole national cohort (that includes the patients from this cohort with anti-NMDARE) to account for potential bias.26
Data Availability
The data of this study, coded to adhere to legal privacy regulations, are available on request from any qualified investigator.
Results
Patient Characteristics
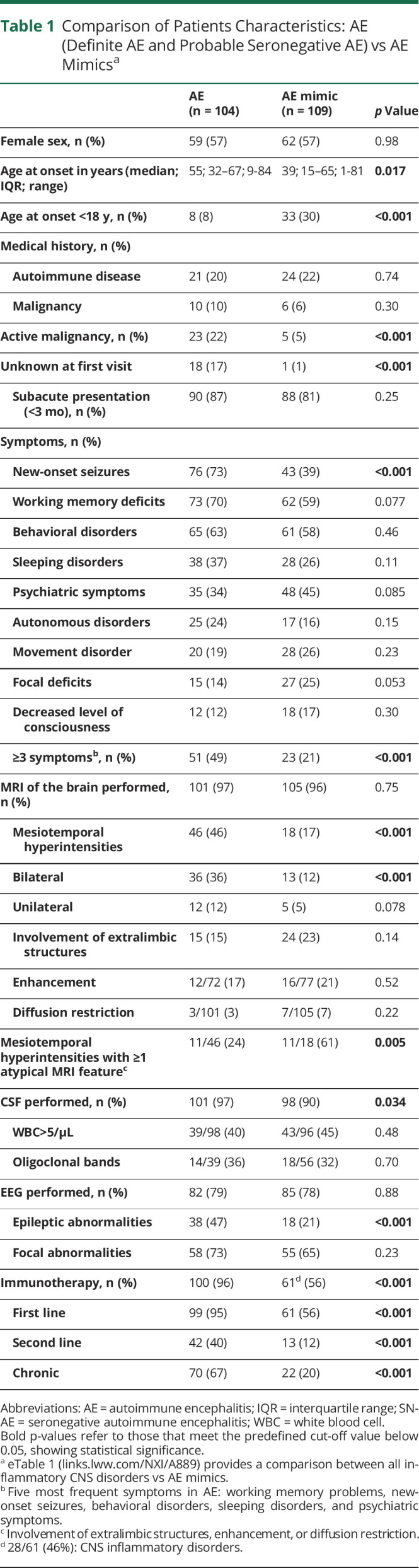
Over a three-and-a-half year period, 310 patients were assessed for eligibility (eFigure 1, links.lww.com/NXI/A889). A total of 239 patients with a suspicion of AE were included, of whom 134 (56%) were female. The median follow-up was 11.0 months (interquartile range [IQR] 1–24.5, range 0–277). Sixty patients (25%) were children (younger than 18 years) at the onset of symptoms. The median age was 42 years (IQR 18–65, range 1–85). Definite AE was diagnosed in 96 patients (40%) and probable SN-AE in 8 patients (3%; Figure 3). Other inflammatory categories included ADEM (9%) and HE (2%). A total of 109 patients (46%) were ultimately classified as AE mimic. In adult patients (age at onset of symptoms 18 years or older), definite AE was diagnosed more frequently (49% vs 13%; p < 0.001) while ADEM was observed more often in children (32% vs 1%; p < 0.001). Patient characteristics and comparison of AE (definite AE and probable SN-AE) vs AE mimics are summarized in Table 1, eTable 1, and eFigure 2. New-onset seizures were observed more frequently in AE than in AE mimics (73% vs 39%; p < 0.001). In addition, patients with AE presented more often (49% vs 21%, p < 0.001) with ≥3 of the following symptoms: working memory deficits, new-onset seizures, behavioral disorders, psychiatric symptoms, and sleeping disorders. Regarding ancillary testing, bilateral mesiotemporal hyperintensities on brain MRI (36% vs 12%; p < 0.001) and epileptic abnormalities on EEG (47% vs 21%; p < 0.001) were described more frequently in AE. A newly diagnosed malignancy was observed more often in patients with AE than in AE mimics (17% vs 1%; p < 0.001).
Figure 3. Overview of Diagnostic Categories Specified by Age Category (Total Group vs Mimics).

The pie charts on the left represent the specific inflammatory categories (depicted in blue and green) and AE mimics (depicted in red) in the total group (n = 239), adults (n = 179), and children (n = 60). Diagnostic subcategories of AE mimics are demonstrated per age category on the right side. ADEM = acute disseminated encephalomyelitis; AE = autoimmune encephalitis; HE = Hashimoto encephalitis; PNID = probable neuroinflammatory disorder; SN-AE = seronegative autoimmune encephalitis.
Table 1.
Comparison of Patients Characteristics: AE (Definite AE and Probable Seronegative AE) vs AE Mimicsa
AE Mimics and Confounders
The most frequent AE mimics were CNS inflammatory disorders (26%: PNID 14% and other CNS inflammatory diseases 12%), primary psychiatric disorders (19%), epilepsy with a noninflammatory cause (13%), CNS infectious diseases (7%), encephalitis with unknown cause (7%), neurodegenerative diseases (7%), and primary CNS neoplasms (6%) (Figure 3, specific diagnoses per subcategory are provided in eTable 2, links.lww.com/NXI/A889). In children, primary psychiatric disorders were observed more frequently (36% vs 12%; p = 0.016). Overall, the most frequent confounding factor for AE misdiagnosis was MRI T2/FLAIR hyperintensities involving the mesiotemporal lobe(s), observed in 18 of 109 (17%) AE mimics (Table 1). The presence of ≥1 atypical radiologic feature (involvement of extralimbic structures, enhancement, diffusion restriction), in addition to mesiotemporal T2/FLAIR hyperintensities, was observed more frequently in AE mimics compared with AE (61% vs 24%; p = 0.005). Brain biopsy was performed in 5 of 18 patients with mesiotemporal lobe abnormalities and provided a definite diagnosis in all 5 patients, including GBM (n = 3), B-cell lymphoma, and CNS Whipple disease. The second most common confounder was false-positive or clinically irrelevant antibodies in serum, which was observed in 13 of 109 (12%) AE mimics, including thyroid peroxidase antibodies (anti-TPO; n = 4), NMDAR antibodies (n = 5), a positive VGKC antibody test in the absence of LGI1 and CASPR2 antibodies (n = 2), and Hu and Ma2 antibodies (both n = 1). Sixty-one AE mimics (56%) were treated with immunotherapy, of whom 13 had an CNS inflammatory disease and 15 were classified as PNID (eTable 3, links.lww.com/NXI/A889).
Validation Clinical AE Criteria
An evaluation using the diagnostic algorithm for AE is provided for all patients in Figure 4 and for the adult population (age 18 years and older) in eFigure 3 (links.lww.com/NXI/A889).5 Fifty-five of all 239 patients (23%) with suspicion of AE did not meet the criteria for possible AE, of whom 35 of 55 (63%) were AE mimics and 18 of 55 (33%) definite AE. AE mimics not fulfilling criteria for possible AE included predominantly primary psychiatric disorders (19/35; 54%). Patients with probable and definite AE not fulfilling criteria for possible AE had more frequently high titer anti-GAD65 antibodies (15/18, 83%; p < 0.001), were female (17/18, 94%; p < 0.001), and had lower median age at symptom onset (31 years, p < 0.001, eTable 4). A total of 184 of all 239 patients (77%) fulfilled the criteria for possible AE, and 78/184 (42%) of patients satisfying these criteria were diagnosed with definite AE and 8/184 (4%) with probable SN-AE. Thirteen of 184 patients (7%) fulfilling the criteria for possible AE also met the criteria for definite LE, of whom 8 patients (62%) had a diagnosis of definite AE and 2 patients (15%) were finally diagnosed as probable SN-AE. Three patients fulfilling the criteria for definite LE (23%) were classified as AE mimic (Figure 5). Nine of 18 (50%) patients meeting the criteria for probable anti-NMDARE were diagnosed as definite anti-NMDARE while 3 of 9 remaining patients had an alternative neuroinflammatory disorder and 3 patients were classified as PNID. Nine of 184 patients with possible AE (5%) fulfilled the criteria for probable SN-AE. One of these patients was ultimately diagnosed with a GBM (Figure 5). Twenty four of 184 (13%) patients had another specific CNS inflammatory disorder (ADEM 11%, HE 2%). In total, 74 of 184 (40%) patients with possible AE were ultimately classified as AE mimic. The accuracy of the 2016 AE criteria is provided in Table 2, eTable 5, and eTable 6.
Figure 4. Evaluation of Patients According to Diagnostic Algorithm for Autoimmune Encephalitis.

Adapted from Graus et al.,5 reprinted with permission from Elsevier. ♦ Anti-MOG was tested in all (n = 21) patients with ADEM; in 10/21 (48%) antibodies were present. *Two patients ultimately diagnosed as probable SN-AE. Three patients fulfilling criteria for definite autoimmune limbic encephalitis were diagnosed as AE mimic after applying diagnostic AE algorithm. † Anti-NMDARE (1), PACNS (1), PML (1), MS (1), and neuro-Sjögren (1). ¥ Encephalitis with unknown cause (3), PNID (3), definite AE (2), and HE (1). Ψ Caspr2 (6), Ma2 (1), and NMDAR (1). Abs = antibodies; ADEM = acute disseminated encephalomyelitis; AE = autoimmune encephalitis; BBE = Bickerstaff brainstem encephalitis; HE = Hashimoto encephalopathy; MOG = myelin oligodendrocyte glycoprotein; NMDARE = anti-NMDA receptor encephalitis; PNID = probable neuroinflammatory disorder; SN-AE = seronegative autoimmune encephalitis.
Figure 5. AE Mimic Examples (Brain MRI).

(A–D) Glioblastoma multiforme (GBM): 47-year-old female patient with subacute working memory deficits and new-onset focal seizures. Left temporal hyperintensities on T2/FLAIR images (A) with subtle left temporal leptomeningeal enhancement (B). Brain MRI after 6 months showed progression of T2/FLAIR hyperintense left temporal lesion (C) and enhancement (D). (E–F) CNS Whipple disease: 69-year-old male patient with rapidly progressive dementia and diarrhea. Bilateral mesiotemporal hyperintensities on T2/FLAIR images (E) and parenchymal enhancement in corresponding regions. (This patient was also published elsewhere by Kloek et al.48). (G) Neurofibromatosis type 1 (NF-1): 52-year-old male patient with a chronic course focal epilepsy and cognitive decline. Bilateral mesiotemporal T2/FLAIR hyperintensities, showing no progression for approximately 10 years, regarded as CNS lesion due to NF-1.49 (H) 3,4-Methyl enedioxy methamphetamine (MDMA) intoxication: 27-year-old male patient with acute amnestic syndrome. No seizures were observed, and hippocampal damage was probably causes by direct MDMA-neurotoxicity, as described earlier.50
Table 2.
Accuracy of the 2016 AE Criteriaa

Criteria for Probable Anti-LGI1 Encephalitis
In total, 32 patients were diagnosed as definite anti-LGI1 encephalitis, of whom 13 (41%) demonstrated faciobrachial dystonic seizures (FBDS) and 12 (38%) demonstrated frequent (>5 per day) stereoytypical focal seizures. In all patients, focal seizures had a nonmotor onset, predominantly dyscognitive and autonomous. In the diagnostic algorithm for AE, one patient presented with isolated faciobrachial dystonic and did not fulfill the criteria for possible AE (Figure 4, eFigure 4, links.lww.com/NXI/A889). Three patients with anti-LGI1 encephalitis met the criteria for definite autoimmune LE. Twenty five of 171 (15%) remaining patients with possible AE not fulfilling criteria for definite autoimmune LE met the proposed criteria for probable anti-LGI1 encephalitis. Eighteen of these 25 (72%) patients could be confirmed by antibody testing, whereas the other 7 patients were diagnosed as definite AE with another antibody (eFigure 4). FBDS was exclusively observed in anti-LGI1 encephalitis. Overall, the criteria for probable anti-LGI1 encephalitis showed a sensitivity of 66% (95% CI 47–81) and specificity of 96% (95% CI 93–98; Table 2). It allowed earlier treatment in 25 of 171 (15%) without treating noninflammatory AE mimics erroneously. The criteria for probable anti-LGI1 encephalitis were validated on an earlier described cohort, of whom one overlapping patient was excluded (n = 37),25 showing a comparable sensitivity (65%; 95% CI 47–80; p = 0.9).
Discussion
In this retrospective cohort study, we describe common AE mimics and validate the 2016 AE criteria using real-world data.5 In addition, we propose criteria for probable anti-LGI1 encephalitis. We demonstrate that the specificity for probable AE (NMDAR, SN-AE, and LGI1) and definite autoimmune LE criteria is reassuringly high (>95%). Furthermore, we show that AE mimics occur frequently and are diverse. The most common diagnostic categories are primary psychiatric disorders, CNS inflammatory disorders, epilepsy with a noninflammatory cause, CNS infections, neurodegenerative diseases, and primary CNS neoplasms. The sensitivity of the criteria for possible AE was relatively high (83%), which was comparable with previous studies by Li et al.27 and Costa et al.28 This implicates that most patients with AE can be identified by these criteria. However, a substantial part of AE did not fulfill the criteria for possible AE, of whom the majority had anti-GAD65 antibodies with a chronic course. This demonstrates that these criteria focus on patients with a subacute presentation, and sensitivity for neuronal autoantibodies associated with a chronic course (i.e., Caspr2, IgLON5, GAD65) is only moderate. Ninety percent of patients with a primary psychiatric disorder did not meet the criteria for possible AE, indicating high specificity in this category. However, the overall specificity of the criteria for possible AE was markedly lower (27%), indicating a relatively high rate of false-positive cases and potentially erroneous treatment with immunotherapy. Previous studies reported higher specificities (72%–94%),11,27,28 which is probably explained by differences in AE mimic population. Compared with an earlier study performed by Flanagan et al.,11 we observed a higher frequency of CNS inflammatory and CNS infectious disorders in our study, of whom the majority fulfilled the criteria for possible AE, whereas the occurrence of neurodegenerative diseases was lower. Our findings emphasize that the criteria for possible AE are useful as entry criteria for the diagnostic algorithm of AE. However, possible AE should not be regarded as an established diagnosis and requires ancillary testing because specificity is (too) low. The criteria for probable AE (NMDAR, SN-AE) and definite autoimmune LE were highly specific (>95%), indicating a very low risk of false-positive cases. Li et al.27 reported comparable specificities for probable anti-NMDARE and definite autoimmune LE. We deliberately chose to include other inflammatory CNS disorders in the control group (i.e., patients with a diagnosis other than AE) to obtain optimal specificity for AE. The sensitivity of probable anti-NMDARE criteria in this study (50%) was lower compared with earlier research (81%–90%),29-31 which is probably explained by an underrepresentation of severely affected anti-NMDARE patients in our study. This is supported by a higher sensitivity (58%) if we applied these criteria to the cohort of all anti-NMDARE patients from the Netherlands in the period of 2007–2019 (n = 126, including the 18 anti-NMDARE patients in this study) with relatively more ill patients.26 Previous studies showed anti-NMDARE patients not identified by the clinical criteria were mainly post-HSV1 encephalitis, milder affected, or showed atypical demyelinating syndromes.31,32 Although the moderate sensitivity of the 2016 AE criteria for anti-NMDARE could be problematic, these patients were identified shortly afterward by neuronal autoantibody studies. Our findings confirm that the criteria for probable and definite AE can be applied for decisions on immunotherapy in early disease stage because the risk of false-positive cases is low. We show that the newly proposed criteria for probable anti-LGI1 encephalitis are highly specific (96%). We intentionally designed highly specific criteria because the prevention of false-positive cases and potential erroneous immunotherapy was considered highly important. Similar sensitivity of the criteria of probable anti-LGI encephalitis was found in the cohort of this study and an earlier cohort (66% and 65%). This was considered acceptable because patients who did not fulfill these criteria were mostly mildly affected and identified by neuronal autoantibody testing shortly afterward. We emphasize that the criteria for probable anti-LGI1 encephalitis should be applied in addition to the 2016 clinical AE criteria (i.e., as part of the diagnostic algorithm) and that these criteria are only applicable for patients who also fulfill the criteria for possible AE. FBDS was exclusively observed in anti-LGI1 encephalitis and can be regarded as pathognomonic for this disorder.33 Therefore, when recognizing FBDS, patients should always be tested for LGI1 antibodies, also in those without cognitive or additional symptoms and not fulfilling criteria for possible AE. Although focal epilepsy has a broad differential diagnosis, we found that frequent stereotypical focal seizures (>5 seizures per day) were also specific for anti-LGI1 encephalitis. All anti-LGI1 encephalitis patients with focal seizures had a nonmotor onset in this study. Particularly, high frequent focal dyscognitive and autonomic seizures should raise suspicion for anti-LGI1 encephalitis, as described earlier.25 In this study, the most important differential diagnosis of subacute cognitive decline and frequent focal seizures was anti-Caspr2 encephalitis, requiring similar treatment regimens. Three of 32 (10%) patients with anti-LGI1 encephalitis fulfilled the criteria for definite autoimmune LE because bilateral mesiotemporal hyperintensities were observed only in a minority of patients, as described earlier.25 By including the criteria for probable anti-LGI1 encephalitis to the diagnostic algorithm for AE proposed by Graus et al.,5 a substantial part of anti-LGI1 encephalitis patients can be identified and treated earlier (e.g., prior to antibody test results), which improves outcome.7 In this study, the criteria for probable anti-LGI1 encephalitis were validated in another cohort from our center without overlap of patients. The LGI1 criteria would profit from validation in cohorts from other countries. A considerable part (46%) of patients were classified as AE mimic. We found that more than half of AE mimics were treated with steroids, supporting the importance of early AE mimic identification because steroids may induce various adverse effects, including a reduction of the diagnostic yield of brain biopsy in CNS lymphoma,34 deterioration of symptoms in psychiatric disorders, or exacerbation of CNS infections.35,36 We described various diagnostic pitfalls that had an important contribution to AE misdiagnosis. First, bilateral mesiotemporal hyperintensities on brain MRI were highly specific for AE, as reported earlier.5 However, mesiotemporal lesions were also the most important pitfall in AE misdiagnosis (17%) and should therefore be interpreted with caution and rigor. Notably, most AE mimics with mesiotemporal lesions demonstrated additional radiologic features, including enhancement, diffusion restriction, or involvement of extralimbic regions. These features were also observed in a minority of patients with AE but were usually mild and transient, as described earlier.37 Earlier research showed that diffusion restriction can distinguish HSV1 encephalitis from AE in early stages,38 whereas mass effect, involvement of extralimbic regions, and enhancement can be seen in gliomas.39 Furthermore, bilateral mesiotemporal enhancement has been reported in neurosyphilis and CNS Whipple disease.40,41 We suggest that mesiotemporal lesions on brain MRI with pronounced additional radiologic features should raise suspicion of an AE mimic, necessitating ancillary testing, including follow-up MRI and brain biopsy in selected cases, particularly if a CNS tumor is suspected. The second most common confounding factor in AE misdiagnosis was false-positive or clinically irrelevant antibody test results (12%). This percentage was notably lower compared with an earlier study by Flanagan et al.,11 reporting positive serum antibodies in 50% of AE misdiagnoses. The testing of extensive antibody panels, with the adjoining risks of false-positive or clinically irrelevant results, which is not advocated nor commonly used within our country, might explain this difference. These findings emphasize the importance of adequate patient selection for antibody studies, as stated earlier,12 and the relevance of adequate neuronal antibody test methodologies, by using confirmatory test modalities and inclusion of CSF in antibody studies.5,15,16 In addition to diagnostic characteristics, we identified various clinical characteristics that may aid to discriminate between AE and AE mimics. First, the occurrence of seizures was higher in AE and should raise suspicion for AE in patients presenting with subacute cognitive impairment. In particular anti-LGI1,25 anti-NMDAR,42 anti-GABABR,43 anti-GABAAR,44 and anti-GAD6524 antibodies are associated with seizures. Second, we show that patients with AE had more frequently a polysymptomatic presentation, as described earlier in various AE subtypes, probably reflecting diffuse or multifocal brain inflammation.42,45 Third, a systemic tumor was more common in AE and might suggest a PNS. However, tumor status is frequently unknown at the onset of neurologic symptoms (78% in our study) and therefore less useful. In addition, it is essential to establish causality between type and neurologic syndrome by using the updated PNS-Care Score because comorbid tumors may be detected.46 In this study, 8 patients (3%) were diagnosed with SN-AE, which was markedly lower compared with earlier studies.9 This might be partially explained by extensive testing for relevant antibodies, also in research setting, as well as a very rigorous application of the criteria for SN-AE in our study. However, we observed that a substantial part of patients (n = 15, 8%) could not be classified as SN-AE nor a specific neuroinflammatory disorder, despite a high suspicion of an inflammatory etiology (e.g., suggestive brain MRI or inflammatory CSF profile). In this study, we classified these patients as probable neuroinflammatory disorder (PNID), after thorough exclusion of other diseases, particularly CNS infections and malignancies. This category should be interpreted with caution because heterogeneity is high, and some patients may be diagnosed with another disease at a later stage. Therefore, PNID was classified as AE mimic, to prevent overinterpretation. However, it should be noted that AE mimics also include inflammatory disorders and some patients with PNID might have an inflammatory disorder requiring immunotherapy, although formal criteria are not satisfied. Further research is needed to characterize this heterogeneous patient category, clarify underlying pathogenic mechanisms, and identify new biomarkers. This study has some limitations. First, selection bias probably occurred in this cohort because it was a single-center study from a specialized institution with a relatively high occurrence of neuroinflammatory disorders. However, because we are a national referral center for AE and related disorders, patients from many other (e.g., nonspecialized) institutions were included. Similarly, selection bias mostly influences positive value and negative predictive values, whereas sensitivity and specificity should remain the same. Second, only a small number of pediatric patients with AE were included in our study, in line with the low incidence of AE in children.32 Consequently, results for this specific patient category should be interpreted carefully. Although the 2016 AE criteria were also considered to allow for inclusion of children, additional pediatric AE criteria were proposed in 2020, that require validation in future research.47 In summary, criteria for probable and definite AE, including newly proposed criteria for anti-LGI1 encephalitis, are applicable for early decisions on immunotherapy because specificity is high. Specificity of possible AE criteria is low and should, therefore, be regarded as an entry criterion for more extensive investigations, instead of established diagnosis. Various disorders can present as an AE mimic and cause misdiagnosis. Particularly, early identification of CNS infections and CNS tumors is essential because treatment strategies differ substantially.
Acknowledgment
The authors thank all patients for their participation and all referring physicians. M.W.J. Schreurs, P.A. Sillevis Smitt, J.M. de Vries, R.F. Neuteboom, and M.J. Titulaer of this publication are members of the European Reference Network for Rare Immunodeficiency, Autoinflammatory and Autoimmune Diseases-Project ID No 739543 (ERN-RITA; HCP Erasmus MC).
Glossary
- ADEM
acute disseminated encephalomyelitis
- AE
autoimmune encephalitis
- CBA
cell-based assay
- FBDS
faciobrachial dystonic seizures
- HE
Hashimoto encephalopathy
- IHC
immunohistochemistry
- IQR
interquartile range
- PNID
probable neuroinflammatory disorder
- PNS
paraneoplastic neurologic syndromes
- SN-AE
seronegative AE
Appendix. Authors
Study Funding
Dr Titulaer was supported by an Erasmus MC fellowship, has received funding from the Netherlands Organization for Scientific Research (NWO, Veni incentive), ZonMw (Memorabel program), the Dutch Epilepsy Foundation (NEF 14-19 & 19-08), Dioraphte (2001 0403), and E-RARE JTC 2018 (UltraAIE, 90030376505).
Disclosure
P.A. Sillevis Smitt holds a patent for the detection of anti-DNER, he received research support from Euroimmun. R.F. Neuteboom reports participates in pediatric MS studies with Novartis, Roche, and Sanofi-Genzyme; he received consultancy fees from Novartis, Sanofi-Genzyme, and Zogenix; he received research grants from the Dutch MS research foundation, DreaMS foundation, Postcode Loterij, Vrienden Loterij, Stichting Vrienden van het Sophia. M.J. Titulaer has filed a patent, on behalf of the Erasmus MC, for methods for typing neurologic disorders and cancer, and devices for use therein; has received research funds for serving on a scientific advisory board of Horizon Therapeutics, for consultation at Guidepoint Global LLC, for consultation at UCB, for teaching colleagues at Novartis; and has received an unrestricted research grant from Euroimmun AG and from CSL Behring. The other authors report no relevant disclosures. Go to Neurology.org/NN for full disclosures.
References
- 1.Dalmau J, Tuzun E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol. 2007;61(1):25-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dalmau J, Gleichman AJ, Hughes EG, et al. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7(12):1091-1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349(16):1543-1554. [DOI] [PubMed] [Google Scholar]
- 4.Dalmau J, Graus F. Antibody-mediated encephalitis. N Engl J Med. 2018;378(9):840-851. [DOI] [PubMed] [Google Scholar]
- 5.Graus F, Titulaer MJ, Balu R, et al. A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol. 2016;15(4):391-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrne S, Walsh C, Hacohen Y, et al. Earlier treatment of NMDAR antibody encephalitis in children results in a better outcome. Neurol Neuroimmunol Neuroinflamm. 2015;2(4):e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson J, Bi M, Murchison AG, et al. The importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain. 2018;141(2):348-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venkatesan A, Tunkel AR, Bloch KC, et al. Case definitions, diagnostic algorithms, and priorities in encephalitis: consensus statement of the international encephalitis consortium. Clin Infect Dis. 2013;57(8):1114-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee WJ, Lee HS, Kim DY, et al. Seronegative autoimmune encephalitis: clinical characteristics and factors associated with outcomes. Brain. 2022;145(10):3509-3521. [DOI] [PubMed] [Google Scholar]
- 10.van Sonderen A, Schreurs MW, de Bruijn MA, et al. The relevance of VGKC positivity in the absence of LGI1 and Caspr2 antibodies. Neurology. 2016;86(18):1692-1699. [DOI] [PubMed] [Google Scholar]
- 11.Flanagan EP, Geschwind MD, Lopez-Chiriboga AS, et al. Autoimmune encephalitis misdiagnosis in adults. JAMA Neurol. 2023;80(1):30-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalmau J, Graus F. Autoimmune encephalitis-misdiagnosis, misconceptions, and how to avoid them. JAMA Neurol. 2023;80(1):12-14. [DOI] [PubMed] [Google Scholar]
- 13.Thomas C, Lehrich C, Gross CC, Wiendl H, Meuth SG, Melzer N. Primary B cell Lymphoma of the CNS mimicking anti-LGI1 limbic encephalitis. Front Neurol. 2018;9:658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Coevorden-Hameete MH, Titulaer MJ, Schreurs MW, de Graaff E, Sillevis Smitt PA, Hoogenraad CC. Detection and characterization of autoantibodies to neuronal cell-surface antigens in the central nervous system. Front Mol Neurosci. 2016;9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain. 2005;128(Pt 8):1764-1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gresa-Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2014;13(2):167-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hutchinson M, Waters P, McHugh J, et al. Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody. Neurology. 2008;71(16):1291-1292. [DOI] [PubMed] [Google Scholar]
- 18.Mandel-Brehm C, Dubey D, Kryzer TJ, et al. Kelch-like protein 11 antibodies in seminoma-associated paraneoplastic encephalitis. N Engl J Med. 2019;381(1):47-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang B, McKeon A, Hinson SR, et al. Autoimmune glial fibrillary acidic protein astrocytopathy: a novel meningoencephalomyelitis. JAMA Neurol. 2016;73(11):1297-1307. [DOI] [PubMed] [Google Scholar]
- 20.Sabater L, Gaig C, Gelpi E, et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: a case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014;13(6):575-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spatola M, Sabater L, Planaguma J, et al. Encephalitis with mGluR5 antibodies: symptoms and antibody effects. Neurology. 2018;90(22):e1964-e1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sillevis Smitt P, Kinoshita A, De Leeuw B, et al. Paraneoplastic cerebellar ataxia due to autoantibodies against a glutamate receptor. N Engl J Med. 2000;342(1):21-27. [DOI] [PubMed] [Google Scholar]
- 23.Ketelslegers IA, Van Pelt DE, Bryde S, et al. Anti-MOG antibodies plead against MS diagnosis in an acquired demyelinating syndromes cohort. Mult Scler. 2015;21(12):1513-1520. [DOI] [PubMed] [Google Scholar]
- 24.Munoz-Lopetegi A, de Bruijn M, Boukhrissi S, et al. Neurologic syndromes related to anti-GAD65: clinical and serologic response to treatment. Neurol Neuroimmunol Neuroinflamm. 2020;7(3):e696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Sonderen A, Thijs RD, Coenders EC, et al. Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology. 2016;87(14):1449-1456. [DOI] [PubMed] [Google Scholar]
- 26.Bastiaansen AEM, de Bruijn M, Schuller SL, et al. Anti-NMDAR encephalitis in The Netherlands, focusing on late-onset patients and antibody test accuracy. Neurol Neuroimmunol Neuroinflamm. 2022;9(2):e1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Sun L, Du R, et al. Application of the 2016 diagnostic approach for autoimmune encephalitis from Lancet Neurology to Chinese patients. BMC Neurol. 2017;17(1):195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costa D, Sardoeira A, Carneiro P, et al. Autoimmune encephalitis: suspicion in clinical practice and mimics. J Neuroimmunol. 2022;365:577824. [DOI] [PubMed] [Google Scholar]
- 29.Nishida H, Kohyama K, Kumada S, et al. Evaluation of the diagnostic criteria for anti-NMDA receptor encephalitis in Japanese children. Neurology. 2021;96(16):e2070-e2077. [DOI] [PubMed] [Google Scholar]
- 30.Ho ACC, Mohammad SS, Pillai SC, et al. High sensitivity and specificity in proposed clinical diagnostic criteria for anti-N-methyl-D-aspartate receptor encephalitis. Dev Med Child Neurol. 2017;59(12):1256-1260. [DOI] [PubMed] [Google Scholar]
- 31.Kaneko A, Kaneko J, Tominaga N, et al. Pitfalls in clinical diagnosis of anti-NMDA receptor encephalitis. J Neurol. 2018;265(3):586-596. [DOI] [PubMed] [Google Scholar]
- 32.de Bruijn M, Bruijstens AL, Bastiaansen AEM, et al. Pediatric autoimmune encephalitis: recognition and diagnosis. Neurol Neuroimmunol Neuroinflamm. 2020;7(3):e682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Irani SR, Michell AW, Lang B, et al. Faciobrachial dystonic seizures precede Lgi1 antibody limbic encephalitis. Ann Neurol. 2011;69(5):892-900. [DOI] [PubMed] [Google Scholar]
- 34.Scheichel F, Marhold F, Pinggera D, et al. Influence of preoperative corticosteroid treatment on rate of diagnostic surgeries in primary central nervous system lymphoma: a multicenter retrospective study. BMC Cancer. 2021;21(1):754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pulivarthi S, Reshi RA, McGary CT, Gurram MK. Cerebral toxoplasmosis in a patient on methotrexate and infliximab for rheumatoid arthritis. Intern Med. 2015;54(11):1433-1436. [DOI] [PubMed] [Google Scholar]
- 36.Warrington TP, Bostwick JM. Psychiatric adverse effects of corticosteroids. Mayo Clin Proc. 2006;81(10):1361-1367. [DOI] [PubMed] [Google Scholar]
- 37.Kelley BP, Patel SC, Marin HL, Corrigan JJ, Mitsias PD, Griffith B. Autoimmune encephalitis: pathophysiology and imaging review of an overlooked diagnosis. AJNR Am J Neuroradiol. 2017;38(6):1070-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bani-Sadr A, Ruitton-Allinieu MC, Brisset JC, et al. Contribution of diffusion-weighted imaging to distinguish herpetic encephalitis from auto-immune encephalitis at an early stage. J Neuroradiol. 2023;50(3):288-292. [DOI] [PubMed] [Google Scholar]
- 39.Zoccarato M, Valeggia S, Zuliani L, et al. Conventional brain MRI features distinguishing limbic encephalitis from mesial temporal glioma. Neuroradiology. 2019;61(8):853-860. [DOI] [PubMed] [Google Scholar]
- 40.Scheid R, Voltz R, Vetter T, Sabri O, von Cramon DY. Neurosyphilis and paraneoplastic limbic encephalitis: important differential diagnoses. J Neurol. 2005;252(9):1129-1132. [DOI] [PubMed] [Google Scholar]
- 41.Blanc F, Ben Abdelghani K, Schramm F, et al. Whipple limbic encephalitis. Arch Neurol. 2011;68(11):1471-1473. [DOI] [PubMed] [Google Scholar]
- 42.Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013;12(2):157-165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Coevorden-Hameete MH, de Bruijn M, de Graaff E, et al. The expanded clinical spectrum of anti-GABABR encephalitis and added value of KCTD16 autoantibodies. Brain. 2019;142(6):1631-1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spatola M, Petit-Pedrol M, Simabukuro MM, et al. Investigations in GABAA receptor antibody-associated encephalitis. Neurology. 2017;88(11):1012-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gruter T, Mollers FE, Tietz A, et al. Clinical, serological and genetic predictors of response to immunotherapy in anti-IgLON5 disease. Brain. 2023;146(2):600-661. [DOI] [PubMed] [Google Scholar]
- 46.Graus F, Vogrig A, Muniz-Castrillo S, et al. Updated diagnostic criteria for paraneoplastic neurologic syndromes. Neurol Neuroimmunol Neuroinflamm. 2021;8(4):e1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cellucci T, Van Mater H, Graus F, et al. Clinical approach to the diagnosis of autoimmune encephalitis in the pediatric patient. Neurol Neuroimmunol Neuroinflamm. 2020;7(2):e663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kloek AT, Piet JR, Adriani KS. Pearls & Oy-sters: a rare presentation of Whipple disease: still waters run deep. Neurology. 2020;94(7):e758-e761. [DOI] [PubMed] [Google Scholar]
- 49.Gill DS, Hyman SL, Steinberg A, North KN. Age-related findings on MRI in neurofibromatosis type 1. Pediatr Radiol. 2006;36(10):1048-1056. [DOI] [PubMed] [Google Scholar]
- 50.Bruggemann N, Heldmann M, Sprenger A, Repenthin J, Munte TF. Acute amnestic syndrome due to MDMA exposure. J Neurol. 2016;263(5):1022-1023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data of this study, coded to adhere to legal privacy regulations, are available on request from any qualified investigator.