

Abstract
The Telomere-to-Telomere consortium recently assembled the first truly complete sequence of a human genome. To resolve the most complex repeats, this project relied on manual integration of ultra-long Oxford Nanopore sequencing reads with a high-resolution assembly graph built from long, accurate PacBio HiFi reads. We have improved and automated this strategy in Verkko, an iterative, graph-based pipeline for assembling complete, diploid genomes. Verkko begins with a multiplex de Bruijn graph built from long, accurate reads and progressively simplifies this graph by integrating ultra-long reads and haplotype-specific markers. The result is a phased, diploid assembly of both haplotypes, with many chromosomes automatically assembled from telomere to telomere. Running Verkko on the HG002 human genome resulted in 20 of 46 diploid chromosomes assembled without gaps at 99.9997% accuracy. The complete assembly of diploid genomes is a critical step towards the construction of comprehensive pangenome databases and chromosome-scale comparative genomics.
Ed summary:
Integration of long and ultra-long reads results in improved phased, diploid assemblies.
Recent advances in sequencing technologies have greatly increased the accuracy and length of sequencing reads 1. Pacific Biosciences’ high-fidelity (HiFi) reads can achieve accuracies of over 99.9% with read lengths of 18–25 kilobases (kb) 2 and Oxford Nanopore Technologies (ONT) reads routinely reach median lengths of 50–150 kb with accuracies around 95% 3,4. Recently, ONT has demonstrated the ability to generate relatively shorter reads (median 25–35 kb) at 99.9% accuracy. For convenience when not referring to a specific technology, we will refer to “long, accurate reads” (LA reads) as those with lengths greater than 10 kb and accuracy greater than 99.9%, and “ultra-long reads” (UL reads) as those with lengths over 100 kb and accuracies over 90%.
These technological advances have greatly simplified the process of reconstructing a genome from overlapping sequencing reads 5,6, yielding highly continuous genome assemblies 4,7–11. With the recent completion of the CHM13 human reference genome, the Telomere-to-Telomere (T2T) consortium demonstrated that gapless and accurate assembly of human genomes is now possible 12–14. However, this consortium effort required considerable resources, and the complete assembly of human chromosomes is not yet routine. While careful assembly of LA reads can automatically complete a handful of chromosomes in a haploid human genome 15, the majority of chromosomes remain unresolved and require manual intervention 11–13. Chromosome-scale scaffolding of LA-based assemblies requires additional technologies such as Bionano 16, Strand-seq 17–19, or Hi-C 20–22, which can be error-prone 11 and require careful curation and validation 23.
Genome assembly is complicated due to the presence of large, highly similar repeats. The resolution of repeats during assembly requires either UL reads, to span repeat instances, or LA reads, to identify repeat-specific variants. UL reads can span exact repeats but lack the necessary accuracy to resolve long, very similar repeats. In contrast, LA reads excel at diverged repeats but fail on exact repeats, such as recent tandem duplications. Current methods for sequencing LA reads, such as PacBio HiFi, also suffer from known systematic sequencing errors 8, leading to regions of the genome with no coverage. On diploid genomes, LA reads can be used to build high-resolution haplotype-resolved graphs 9,11, where variants between haplotypes are represented as separate graph nodes (unitigs) with high accuracy and not collapsed into a mosaic assembly 8,9,11. However, their relatively short read lengths limit phasing to a few hundred kb in human genomes. As a result, most LA assemblers output pseudo-haplotypes, a random walk combining the short, but haplotype-resolved, unitigs into long, but haplotype-mixed, contigs. In contrast, UL reads can phase over megabases 24 but have lacked the accuracy to separate haplotypes within the assembly graph or generate a high-quality consensus sequence 4. Thus, LA and UL sequencing reads are complementary for genome assembly but no tools exist for co-assembling them.
Here, we present our new assembler, Verkko, which combines the best features of LA and UL reads into one workflow. Previous hybrid assembly approaches have relied on short reads to build the initial graph and were limited to haploid genomes or the unphased assembly of diploid genomes 25–27. Verkko was specifically designed for large, eukaryotic genomes and incorporates LA reads, UL reads, and haplotype information from familial trios 28, Hi-C 20,21, or Strand-seq 29,30 for the complete assembly of diploid chromosomes.
Results
Verkko overview
Verkko builds on lessons learned from the completion of the CHM13 human reference genome and adopts a similar graph-first approach. However, the methods developed for CHM13 were semi-manual and not directly applicable to heterozygous genomes 14. Verkko extends this strategy to diploid genomes and fully automates the process. Conceptually, Verkko constructs a high-resolution assembly graph from LA reads that resolves diverged repeats and captures small variants between the repeats and haplotypes. This graph is then progressively simplified by the integration of UL reads and haplotype markers to bridge exact repeats, fill coverage gaps, and phase haplotypes (Fig. 1) (Online Methods).
Figure 1. Verkko assembly workflow.
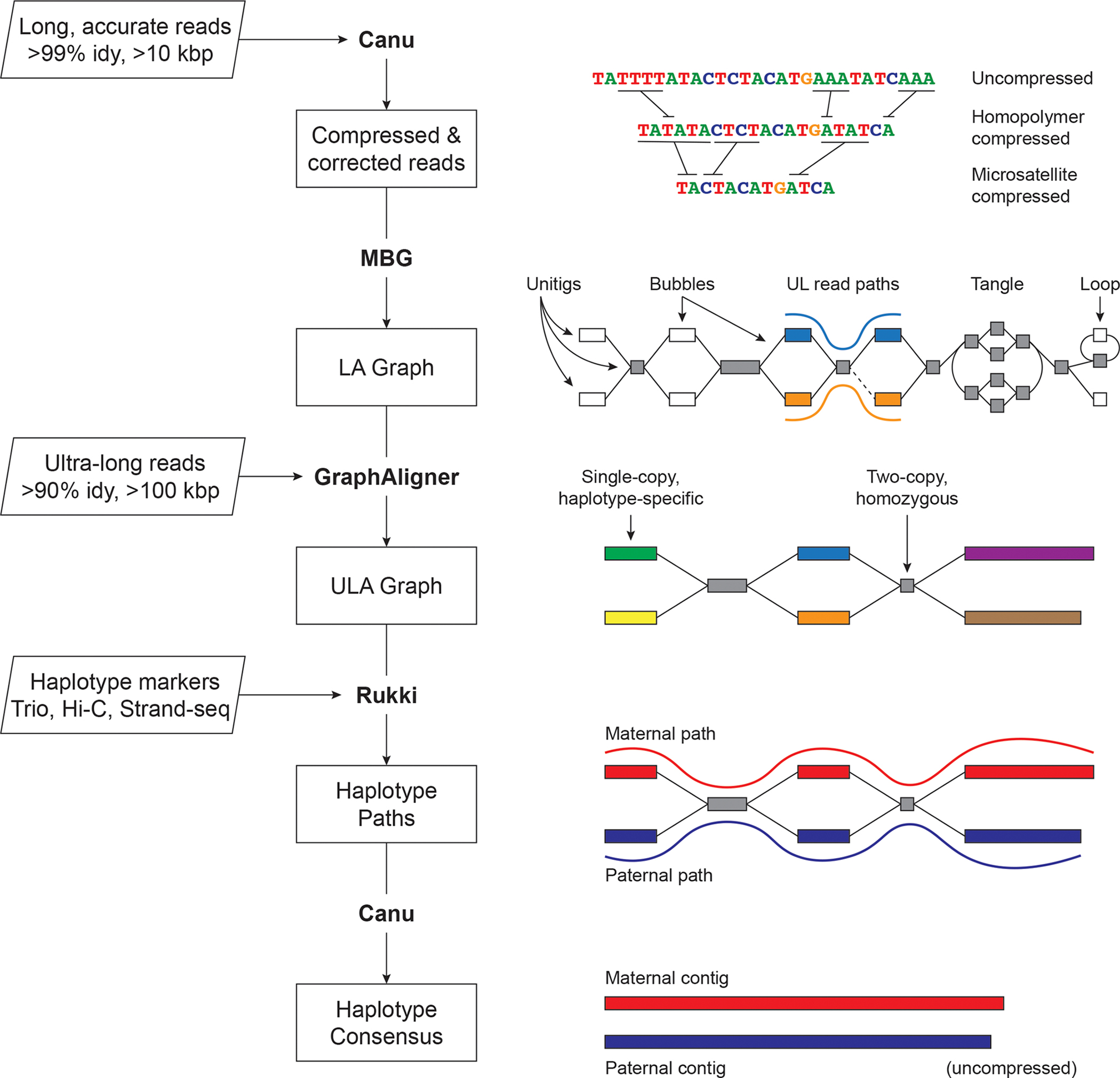
Inputs are on the left, with program outputs listed in rectangles. Key components of the pipeline are highlighted, including Canu 8, MBG 60, GraphAligner 61, and Rukki (Online Methods). Using the outputs of these tools, Verkko performs successive rounds of processing and graph resolution. Input reads are first homopolymer compressed and error corrected, and remain so throughout the process. MBG also compresses microsatellites to aid graph construction, but only internally. The first graph output by Verkko is an accurate, high-resolution de Bruijn graph built from the long, accurate reads (LA Graph). This is a node-labeled graph comprising unitigs (nodes) and their adjacency relationships (edges). Ultra-long reads are then aligned to the LA graph to identify read paths (blue curve, orange curve), and Verkko uses them for phasing bubbles, filling gaps (dotted edge), and resolving loops and tangles. The resulting simplified graph (ULA Graph) is typically composed of single-copy, haplotype specific unitigs, separated by two-copy, homozygous unitigs that could not be phased from the read data alone. Haplotype-specific markers are then used to label nodes in the ULA graph and identify haplotype paths through the graph (maternal, red; paternal, blue). These paths are converted to haplotype-specific contigs using a consensus algorithm that recovers the homopolymers.
Homopolymer insertions and deletions are one of the primary sources of error in long-read sequencing, and compressing them (e.g. A1…An becomes A1 for all n > 1) simplifies the assembly process 8,14,31. The entire Verkko pipeline operates on homopolymer-compressed sequences, which are recovered during the final consensus phase. After compression, the LA reads are error-corrected and used to build a multiplex de Bruijn graph 15,32. UL reads are then aligned to this graph to patch coverage gaps and further resolve repeats and haplotypes. The graph is finally cleaned, and haplotype paths are identified using haplotype-specific markers from additional parental or long-range sequencing information. To restore homopolymers, LA read paths from the initial graph are lifted to the final graph and a consensus sequence is computed for all nodes and haplotype paths.
Verkko’s final output is a phased, diploid assembly of both paternal and maternal haplotypes, as well as a highly accurate and resolved assembly graph that can assist with additional genome finishing. In cases where the available data could not fully resolve a chromosome as a single contig, Verkko leverages the assembly graph structure to generate telomere-to-telomere, haplotype-resolved scaffolds, thus removing the need for a separate, error-prone scaffolding step 11,23.
Complete haploid genome assembly
We tested Verkko (version beta2) (Code availability) on a recently published Arabidopsis thaliana genome 33 (Data availability), and benchmarked it against state-of-the art assemblers designed for HiFi 9,15,34 and ONT 7 data (Supplementary Note 1, Supplementary Table 1). We compared the assemblers to the published A. thaliana reference 33 using Quast 35 (Supplementary Note 2). The errors were filtered to exclude those arising from likely mis-assemblies in the reference. Using only the HiFi data, Verkko was comparable to other HiFi-only assemblers, failing to resolve any chromosomes end-to-end. However, when combining HiFi and ONT data, Verkko assembled 4 of 5 chromosomes into single unitigs spanning >99.5% of the reference genome, with 2 of the 4 having canonical telomeric repeats on both ends (Supplementary Table 2). The only exception was Chr4, which had a single heterozygous bubble and unresolved 45S rDNA tangle. Verkko also had the lowest error count and comparable base accuracy to other HiFi-based assemblers (Supplementary Table 2). All assemblies had a lower number of differences when compared to the Verkko assembly than the published reference, suggesting that the Verkko assembly was more correct (Supplementary Table 3). This finding was further supported by a reference-free evaluation using VerityMap 36 (Extended Data Figure 1).
The T2T consortium relied on an almost fully homozygous cell line for completion of the human genome 14. Due to its completeness, extensive validation, and cataloged variants 37, the CHM13 cell line provides an excellent test case for benchmarking the assembly of a single haplotype. We ran Verkko with the same HiFi and ONT data that was originally used to assemble the CHM13 genome (Data availability, Supplementary Note 1), and compared to HiFi-only and ONT-only assemblers (Table 1, Fig. 2, Supplementary Table 4). The assemblies were validated with QUAST 35 and VerityMap 36 while filtering for known heterozygous variants. Verkko correctly assembled 12 chromosomes from telomere to telomere, with 5 additional chromosomes assembled into a single unitig containing >95% of the expected sequence. This is double that of any assembly based on a single technology, with the next best result coming from LJA 15, which completely assembled 6 chromosomes from HiFi data alone.
Table 1.
Validation of CHM13 assemblies.
| Asm | NG50 (Mb) | #Errors Quast / VerityMap | %Comp | Mismatch /100 kb |
Indel /100 kb |
Resolved BACs | T2T | >95% |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Verkko | 135.13 | 21 / 20 | 99.75 | 2.46 | 0.61 | 644 / 644 | 12 | 17 |
| Verkko (HiFi) | 70.13 | 17 / 20 | 99.42 | 2.17 | 0.55 | 629 / 644 | 1 | 3 |
| LJA | 96.69 | 12 / 29 | 99.60 | 2.14 | 0.85 | 639 / 644 | 6 | 9 |
| Flye | 69.53 | 114 / 30 | 93.58 | 6.03 | 110.92 | 511 / 644 | 0 | 0 |
| Hifiasm | 90.22 | 32 / 39 | 99.54 | 2.55 | 0.70 | 643 / 644 | 2 | 5 |
We used the published reference of CHM13v1.1 14 to evaluate the assemblies with QUAST and ran VerityMap as a reference-free alternative. The relative assembler ranking between QUAST and VerityMap shows good agreement, only switching LJA and Verkko in terms of fewest errors. %Comp: chromosome completeness as measured by QUAST alignments. Number of mismatches and Indels reported by QUAST are given per 100 kb. T2T: reports complete chromosomes with a single unitig covering >99% of the reference bases and having canonical telomeres on each end. >95%: reports chromosomes with a single unitig covering >95% of the reference with no requirement for telomeres. QUAST errors intersecting known heterozygous variants or errors 37, as well as those within the core rDNA arrays, were excluded using a filter script from Shafin et al. 4. CHM13 reference BACs were evaluated as in Nurk et al. 8. Canonical telomeres were identified using the VGP pipeline 10. The best result for each coverage level and category is highlighted in bold.
Figure 2. Continuity of assembled CHM13 chromosomes.
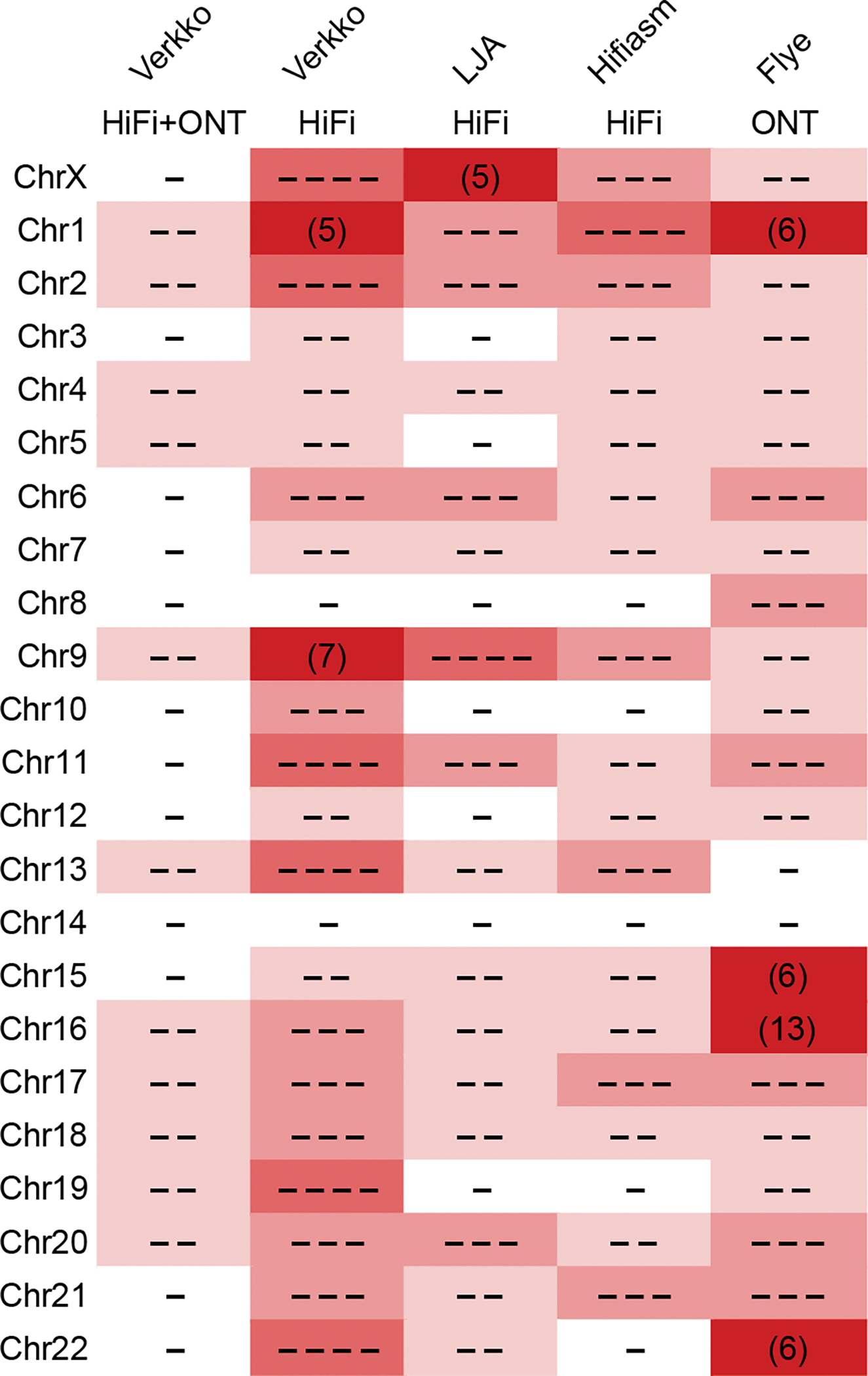
The minimum number of continuous alignments needed to cover 85% of each chromosome (LGA85) is represented by the number of dashes, or plain numbers when ≥5. Higher numbers are shaded progressively darker. Alignments were taken from QUAST output versus the CHM13v1.1 reference, after filtering errors at heterozygous sites and potential erroneous regions in the reference as in Table 1. Verkko using HiFi data alone is comparable to other HiFi-based assemblies 9,15, but Verkko combining HiFi and ONT data has the most complete and correct chromosomes (12).
Even in cases where Verkko did not assemble a complete chromosome, the assemblies were close to complete and the correct resolution was often clear from the final graph (Extended Data Figure 2A). Unresolved regions were limited to the highly repetitive rDNA arrays and centromeric human satellite arrays on Chr9 and Chr16. However, even the rDNA regions were partially resolved, correctly separating Chr15 and Chr22 into their own connected components. The component containing Chr13, Chr14, and Chr21 had three separate rDNA tangles, with the distal and proximal regions of each chromosome connected to the corresponding rDNA tangle (Extended Data Figure 2B). It was previously noted that the these chromosomes shared the highest degree of inter-chromosomal similarity in the human genome 14, which may explain why they are the only CHM13 chromosomes not separated into distinct components of the graph.
Verkko’s results show the advantage of combining LA and UL data types. For example, Chr8 was >90% complete in LJA 15, Hifiasm 9, and HiFi-only Verkko. However, there is a coverage gap at the end of this chromosome due to a GA-rich microsatellite that requires ONT-based gap-filling to resolve 8,13. Verkko correctly identified the missing component and patched this gap. Other chromosomes, such as ChrX, are specifically enriched for HiFi coverage gaps and large near-perfect repeats 8. No HiFi-only assembly recovered more than 40% of ChrX in a single contig, and at least 7 contigs were needed to cover >99% of it. Flye’s 7 ONT-based assembly was more continuous but failed to resolve the centromeric repeats. In contrast, a single unitig in Verkko contained >99.98% of ChrX, missing only the last 50 kb on the q-arm due to a retained heterozygous bubble. We manually inspected the VerityMap output for the Verkko assembly and found that some reported QUAST errors may be false positives or errors in the CHM13 reference, as there is better agreement with the Verkko assembly in these regions (Extended Data Figure 3A,B for Chr4 and Chr17, respectively).
Complete diploid genome assembly
To evaluate Verkko on a highly heterozygous sample, we chose the Darwin Tree of Life insect genome Harmonia axyridis 38 (Data availability). Since Verkko outputs phased unitigs, we compared its output to fully phased equivalents from other assemblers. Despite relatively short ONT data (<2× sequence read coverage ≥100 kb), Verkko’s unitig NG50 was 14.53 Mb, similar to the pseudo-haplotype reference NG50 of 15.11 Mb and much larger than the phased unitigs produced by HiFi-only assemblers: Verkko (HiFi) (6.17 Mb), LJA (5.24 Mb), or Hifiasm (7.08 Mb). We did not evaluate the quality of the assemblies with QUAST as the reference likely contains errors, but Verkko had a low number of errors (18) identified by VerityMap 36 versus 21 for Hifiasm and 289 for the reference. Thus, Verkko’s combination of HiFi and ONT sequencing not only produces highly accurate and complete assemblies but can produce phased unitigs rivaling the best unphased pseudo-haplotypes for diverse genomes. When incorporating Hi-C data 39, Verkko’s contig NG50 increased to 25.31 Mb, far exceeding the continuity of the pseudo-haplotype reference.
We ran Verkko on the benchmark HG002 human sample from Genome in a Bottle (GIAB) 40 and the Human Pangenome Reference Consortium (HPRC) 41 (Data availability). We used the downsampled 35× dataset base-called with DeepConsensus, which has been shown to improve coverage and assembly quality 42, along with 60× ONT ultra-long reads. We compared the results to the recently finished HG002 ChrX and ChrY 14 using QUAST and evaluated assembly quality and accuracy using reference-free methods (Supplementary Note 2). We layered Hi-C and trio information (Supplementary Note 1) onto the Verkko graph and compared it to other state-of-the-art phased assembly results 9,34 (Table 2, Supplementary Tables 5, 6).
Table 2.
Quality and completeness of HG002 diploid assemblies.
| Asm | Contig NG50 (Mb) |
Scaffold NG50 (Mb) |
Hamming error |
QV | #Errors (Chr X & Y) |
|---|---|---|---|---|---|
|
| |||||
| Downsampled (35× HiFi / 60× ONT-UL) | |||||
|
| |||||
| Verkko | 12.90 | 0.13% | 52.18 | 10 | |
| Verkko + trio | 80.77 | 102.55 | 0.13% | 52.40 | 10 |
| Verkko + Hi-C | 58.24 | 82.42 | 0.16% | 52.48 | 10 |
| LJA | 0.39 | 0.14% | 55.75 | 7 | |
| Hifiasm (unitigs) | 0.35 | 0.23% | 61.05 | 2 | |
| Hifiasm + trio | 64.50 | 0.06% | 60.86 | 26 | |
| Hifiasm + Hi-C | 66.34 | 0.79% | 60.57 | 37 | |
|
| |||||
| Full-coverage (>100× HiFi / 85× ONT-UL) | |||||
|
| |||||
| Verkko | 17.35 | 0.05% | 54.91 | 5 | |
| Verkko + trio | 134.00 | 135.80 | 0.05% | 55.77 | 5 |
| Verkko + Hi-C | 68.32 | 85.97 | 0.05% | 55.57 | 5 |
| HPRC curated | 72.70 | 146.75 | 0.13% | 61.35 | 26 |
We evaluated contig NG50 and phase accuracy for all assemblies using Merqury 58. With a goal of phased assemblies, pseudo-haplotype outputs from Flye and Hifiasm were not considered. We evaluated hamming error rate (the fraction of non-dominant allele variants in a unitig) and Phred QV 59 using Merqury. We measured errors versus the recently finished HG002 chromosomes X and Y 14 using QUAST. Results are shown for DeepConsensus HiFi; results from the original HiFi reads are in Supplementary Table 6. LJA and Hifiasm do not output scaffolds, so no scaffold NG50 values are reported. Verkko assemblies used 105× HiFi coverage (mean 17.5 kbp, Data availability) while the HPRC curated assembly used 130× HiFi coverage (mean 14.8 kbp) 11. The best result for each coverage level and category is highlighted in bold.
Verkko produced megabase-scale phase blocks, similar to our observation on non-human genomes. We found that DeepConsensus HiFi increased NG50 without introducing errors and decreased the switch rate in most cases (Hifiasm + Hi-C has a higher switch rate). Verkko has a low switch rate and a low error count with a QV over 50 (1 error per 100 kb, Extended Data Figure 4). When adding Hi-C or trio information, the Verkko assembly had fewer errors when compared to Hifiasm (3.7-fold less with Hi-C and 2.6-fold less with trios). Verkko filled 48 HiFi gaps in this assembly using ONT sequences, totaling less than 0.25 Mbp of final assembled sequence. Verkko has a higher switch error rate than Hifiasm using trios but a lower switch rate when using Hi-C. Our current integration of Hi-C or trio data does not correct switch errors in the initial assembly and, thus, cannot be below the 0.13% switch rate of the Verkko unitigs.
Both Verkko and Hifiasm assemblies are highly complete, with Verkko recovering slightly more multi-copy genes but at the expense of a slightly higher false-duplication rate within individual haplotypes (Supplementary Table 7). This effect was most evident when using Hi-C data due to the higher rate of haplotype misassignment or lack of assignment compared to trios. Post-processing tools, such as purge_dups 43, or improved Hi-C handling by Verkko can potentially address these duplicated sequences when trio information is unavailable.
Uniquely, Verkko is able to output accurate scaffolds using only the connectivity information contained within the assembly graph. For the downsampled HG002 dataset, this feature produced chromosome-scale scaffolds for 7 and 4 chromosomes with trio and Hi-C information, respectively. Verkko is able to generate T2T scaffolds with even lower HiFi and ONT coverage (Supplementary Table 5). Previously, such continuity would have required a separate and error-prone scaffolding step 11.
Finally, we used the full-coverage HG002 dataset (105× HiFi, 85× ONT-UL) to produce the most continuous assembly of this genome to date (Fig. 3). We compared the Verkko assembly to a Hifiasm trio assembly (Supplementary Table 6) and a recently published benchmark assembly which used high-coverage HiFi data for contig assembly, ONT assembly for gap filling, trio information for phasing, BioNano as well as Hi-C data for scaffolding, and manual curation. Both the Verkko trio and Hi-C assemblies have fewer errors and more accurate phasing 11, and the trio-based Verkko assembly has nearly double the contig NG50 size (Table 2). Verkko filled 102 HiFi gaps with ONT data, despite the higher starting HiFi coverage. However, the total bases added was still a small fraction (< 1 Mbp) of the assembly. The improvement in accuracy is partially due to the avoidance of large-scale switches within complex regions of the centromeric repeat arrays (Extended Data Figure 5). In contrast to Hifiasm, Verkko was able to resolve these regions using the complementary ONT data, enabling more accurate phasing.
Figure 3. Verkko assembly graph of the HG002 diploid genome.
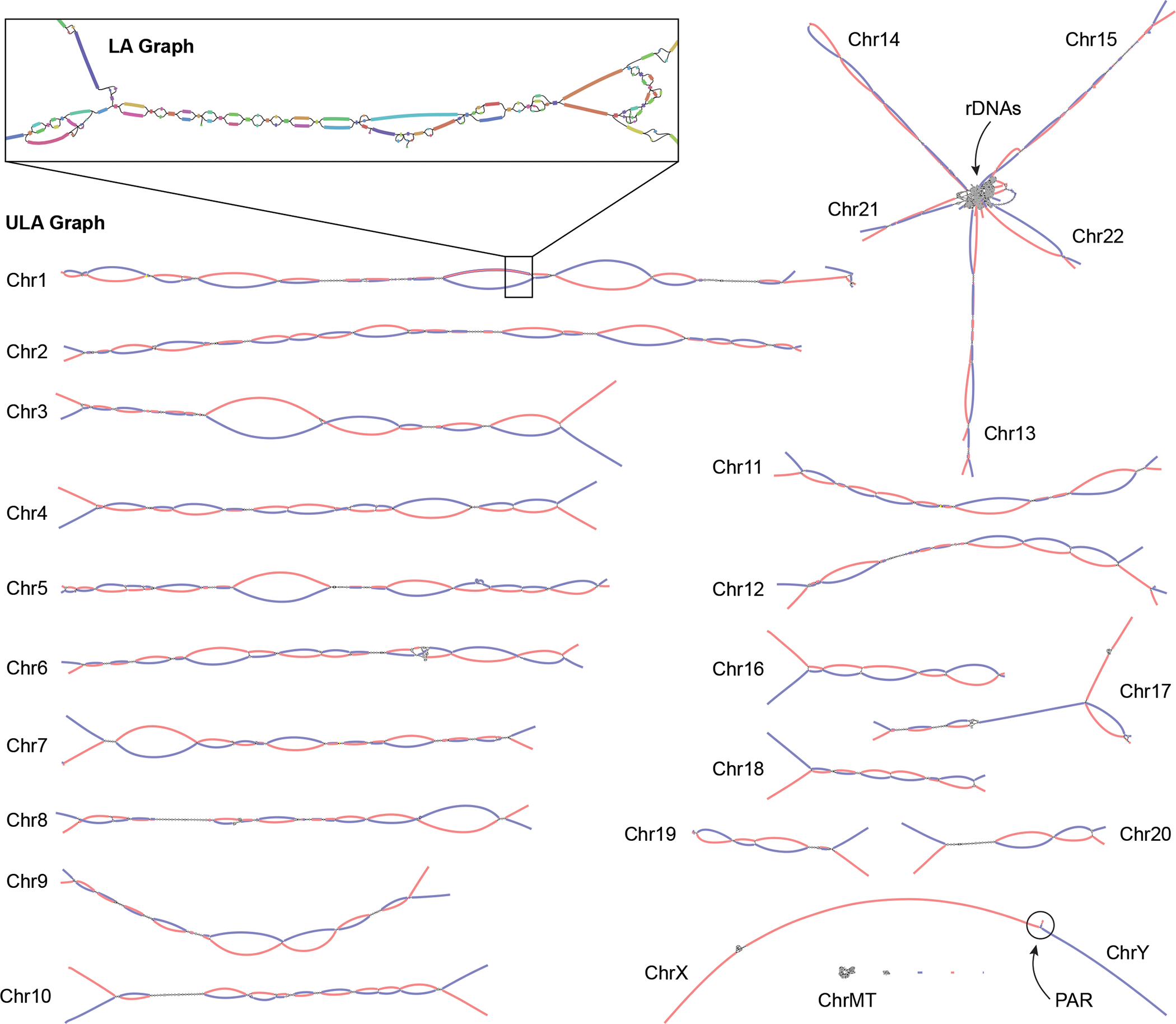
ULA graph integrating both PacBio HiFi and ONT ultra-long reads visualized using Bandage62. Inset shows a portion of the LA graph with multiple bubbles, tangles, and tips prior to resolution using the ultra-long reads. This particular region contains stretches of homozygosity and exact repeats that could not be resolved by HiFi alone. After integration of the ONT data, the corresponding region in the ULA graph is resolved into two linear haplotypes. Each chromosome in HG002 is mostly resolved as a single connected component, with the exception of the acrocentric (13, 14, 15, 21, and 22) and sex chromosomes (X and Y), which are joined by the highly similar rDNA arrays and pseudoautosomal regions (PAR), respectively. The final unitigs in the ULA graph have been colored after assembly by trio-derived haplotype markers (maternal, red; paternal, blue). Using this information to determine haplotype paths through the graph, Verkko was able to completely assemble 20 chromosomal haplotypes in HG002 from telomere to telomere without gaps.
Of the 46 chromosomes in HG002, Verkko resolves 27 of them into single scaffolds using the trio information. Of these, 20 are completely assembled from telomere to telomere without gaps, which is a dramatic improvement over the previous HG002 benchmark assembly 11 that includes 19 complete scaffolds and only one complete contig. Using the Hi-C data alone, Verkko was able to resolve 9 chromosomes into single scaffolds, 7 of which are gapless.
We evaluated the full-coverage Verkko trio assembly using orthogonal Strand-seq 29,44 data from the same sample, which allows for the detection of inversions and translocations in the assembly. We found that the Verkko trio assembly had a higher fraction of contigs and bases correctly assigned to chromosomes than the benchmark assembly 11 (Extended Data Figure 6). The majority of chromosomes were covered by a single Verkko scaffold for both the maternal and paternal haplotypes, and there were no mis-orientations or regions which failed to resolve a homozygous inversion, again an improvement on the previous result. There were a handful of regions, accounting for approximately 30 Mb per haplotype, where there was an ambiguity, either due to true heterozygous inversions between the haplotypes or due to low mappability of Strand-seq data (Extended Data Figure 7A,B). After filtering ambiguous regions, we confirmed 18 heterozygous inversions ranging from 6 kb to 4.1 Mb (median 237 kb) and accounting for 9 Mb per haplotype (Supplementary Table 8, Extended Data Figure 7C). This includes three medically relevant regions which were not correctly resolved in previous HG002 assemblies 11. The largest of these is a known, medically relevant, recurrent inversion on Chr8 13,45. As observed with CHM13, the Verkko reconstruction improves on the manually curated reference and was used to confirm an inversion error in the T2T assembly of ChrY 46 (Extended Data Figure 7D).
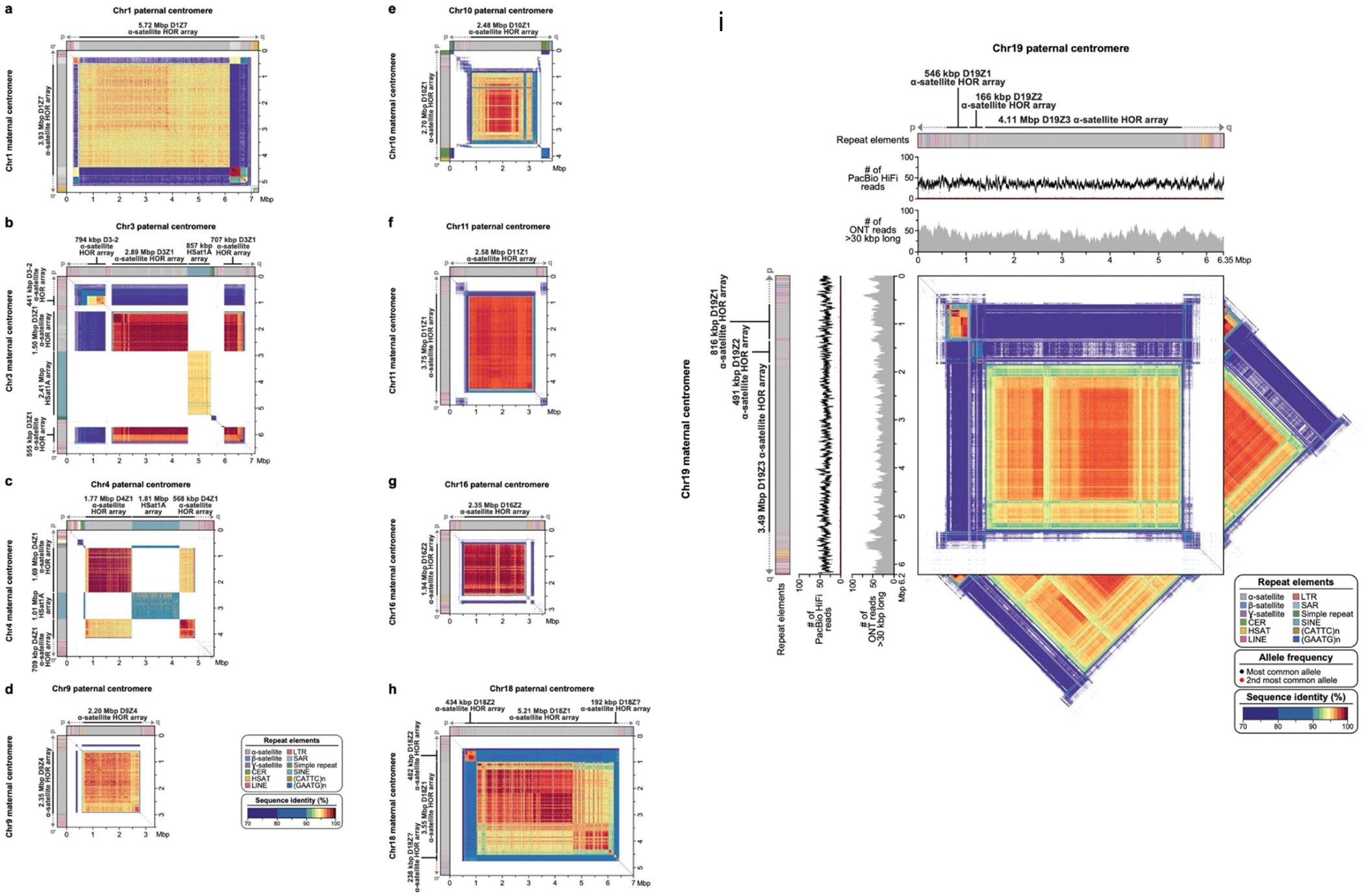
Verkko was able to completely resolve both haplotypes of a single chromosome in several cases (Fig. 4, Extended Data Figure 8). This enabled us to assess variability between centromeric regions in a diploid human genome. Consistent with prior studies 47–54, we found that centromeric ɑ-satellite higher-order repeat (HOR) arrays often vary in length by tens to hundreds of kilobases. For example, Figure 4 shows the centromeric HOR arrays from Chr19. Both Chr19 haplotypes are well-supported by read alignments, indicating no large-scale mis-assemblies. The arrays differ in length with respect to different individual HOR units, suggesting distinct patterns of HOR expansion between the maternal and paternal haplotypes. The paternal haplotype’s D19Z3 array is approximately 620 kb larger than the maternal. The D19Z3 array also differs in HOR structure between the haplotypes. While both arrays are primarily composed of a 2-mer, the paternal haplotype is flanked by 4-mer and 8-mer HORs and the maternal haplotype is dominated by the 2-mer HOR. The HOR arrays show evidence of layered expansions 13, with more recently evolved HOR repeats that have expanded within the core of the arrays (shown in dark orange) and, consequently, pushing more divergent HOR arrays to the sides (shown in green to yellow). The other 8 pairs of completely assembled centromeric satellite arrays (chromosomes 1, 3, 4, 9, 10, 11, 16, and 18) show similar patterns of differential expansion, contraction, and homogenization, underscoring the ubiquity of centromere HOR array size variation between haplotypes (Extended Data Figure 9).
Figure 4. Comparison of the maternal (a) and paternal (b) Chr19 centromeric regions in HG002.
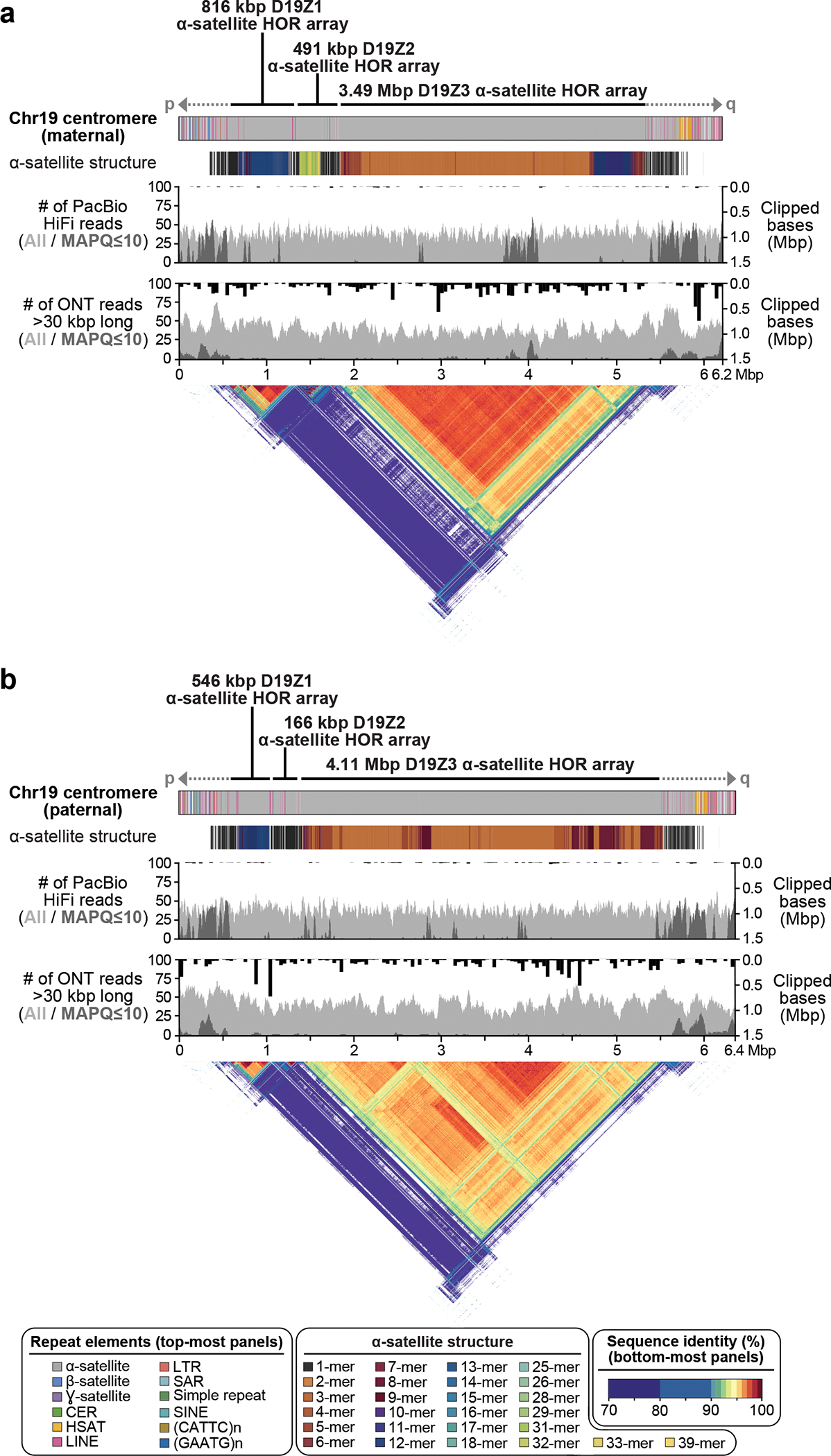
Each haplotype is annotated with its repeat structure (top-most track), HOR structure, PacBio HiFi coverage, and ONT coverage. Dark gray indicates reads with low mapping quality, which may be due to either mis-assembly or repetitive regions in the genome. Bars indicate the sum of clipped bases within each window along the assembly (Supplementary Table 9). Last, the triangle, corresponding to the upper-triangular portion of a dot-plot rotated so the diagonal is the x-axis and colored by identity, shows sequence similarity within each haplotype 63. Both haplotypes reveal the presence of three higher-order repeat (HOR) arrays (D19Z1, D19Z2, and D19Z3) that vary in size and structure.
Discussion
Verkko is a new assembler that leverages the complementarity of long, accurate reads and ultra-long reads to produce assemblies that are more continuous and accurate than when using either technology alone. Verkko relies on high-accuracy reads to build an initial assembly graph which is further resolved with long, noisy reads. Haplotype markers can then be mapped to identify haplotype paths through the graph. Verkko is not currently compatible with only ONT “simplex” sequencing. While Verkko can generate HiFi-only assemblies, we note that other tools perform better at this task based on our evaluation. Verkko’s runtime is also longer than these HiFi-only assemblers but recent improvements make it comparable to ONT-only assemblers. For complete, diploid genome assembly, we currently recommend sequencing approximately 50× genomic coverage of LA reads, 50× in UL reads >100 kb, and 50× of parental short reads. High-quality draft assemblies are possible with lower coverage, and Hi-C or Strand-seq can be used for chromosome-scale phasing in the absence of trios.
Vekko is a modular pipeline that is adaptable to different technologies or the substitution of specific components. For example, Verkko should be compatible with other LA read technologies, such as ONT “duplex” sequencing 55. Additionally, the graphs produced by Verkko are similar in spirit to those from LJA 15, so LJA graphs could potentially be used as a basis instead of MBG graphs. However, Verkko’s methods for UL resolution and haplotype walking were developed using HiFi-based MBG graphs and would likely require tuning for the idiosyncrasies of different data types and tools. Verkko heuristics also assume uniform coverage so metagenomic assemblies would be sub-optimal without further development.
On haploid genomes, Verkko automates telomere-to-telomere assembly for the majority of human chromosomes. On diploid genomes, Verkko generates megabase-scale phase blocks, rivaling the continuity of pseudo-haplotypes single-technology assemblers on this genome. Verkko initially treated long accurate and ultra-long reads identically during consensus which led to lower QV than HiFi-only tools. Recent improvements correct this and increase QV (Supplementary Table 5). In combination with Hi-C or trio information, Verkko can generate complete, haplotype-resolved scaffolds for a subset of chromosomes given sufficient sequencing coverage.
Verkko’s current Hi-C integration requires pre-binned haplotype reads or assemblies, such as those generated by DipAsm 39 or Hifiasm 34. However, it would be possible and likely more accurate to infer haplotype walks from Hi-C reads aligned directly to the graph 34. Another limitation of our current approach is that trio or Hi-C information is used after LA and UL read resolution. As a result, switch errors introduced during graph construction are not corrected by later stages. To address this, future versions could incorporate haplotype information in tandem with graph construction, or could break erroneous graph nodes based on haplotype markers. Despite this limitation, we found haplotype switches to be rare in practice, with <1% of the assembly in nodes with ≥5% hamming error in the downsampled HG002 dataset, and <0.3% in the full-coverage dataset.
With accurate and phased assemblies of complete human chromosomes, we observed large-scale variation between the most repetitive regions of human haplotypes, with the centromeres having an elevated level of heterozygosity compared to the rest of the genome (Extended Data Figure 8). The autosomal centromeric arrays show differentiated expansion of HOR elements and differ in their HOR copy age. This is in contrast to the high similarity previously observed between ChrX centromeric arrays 56 but agrees with a previous analysis of Chr8 13. Our observation is consistent with a low rate of recombination within the centromeric arrays and supports the idea of centromere haplogroups 57.
With its ability to resolve complete haplotypes, Verkko ushers in a new era of comprehensive genomic analysis. The complete assembly of vertebrate haplotypes has direct application for the construction of new reference genomes, and ultimately, to better understanding of the relationships between large, complex structural variation, phenotype, and disease.
Methods
Verkko (“network” or “graph” in Finnish) includes a core set of tools, including HiCanu 8, MBG 60, and GraphAligner 61, that have been extended and integrated together for the assembly of complete haplotypes. All steps of the pipeline are described below along with the details of any improvements made to the existing tools since their published versions.
Although “sequencing coverage” traditionally means the total bases sequenced divided by the haploid genome size, this section considers the diploid genome. Thus, 50× genomic coverage equates to roughly 25× coverage per haplotype. Similarly, when discussing “single-copy” or “unique” nodes within the assembly graph, this is again in regard to the diploid genome. A typical node-labeled diploid assembly graph comprises single-copy nodes that are haplotype-specific and multi-copy nodes that represent homozygous sequences between the two haplotypes (typically two-copy) or genomic repeats (two or more copies).
Verkko’s goal is to resolve each chromosome into one single-copy node spanning from telomere to telomere. However, early iterations of the assembly graph usually contain a number of “bubbles” and “tangles”. A bubble is an acyclic subgraph between source and sink nodes v and w, where only v and w have connections to nodes outside the bubble 64. Note that while simple paths satisfy the above definition, bubble detection is expected to run after unitigification of the graph (removal of simple paths). Heterozygous variants are a common cause of bubbles, where nodes v and w are both two-copy, homozygous nodes connected by exactly two single-copy, disjoint paths representing the distinct haplotypes. Somatic variants and uncorrected sequencing errors are another source of bubbles, and can be identified by their low coverage. Lastly, a tangle is a subgraph containing multi-copy nodes and whose nodes only connect to other non-unique nodes within the tangle or to unique nodes outside the tangle. Tangles are caused by long, exact or near-exact repeats either within or between haplotypes.
Error correction and homopolymer compression
Correction of the LA reads follows the procedure described in HiCanu 8 with incremental improvements to the accuracy. First, to mask a primary source of errors in LA reads such as PacBio HiFi, all homopolymers are compressed to a single base. The reads are then aligned in an all-vs-all manner and compared. If a read has a position which is covered by multiple aligned reads and most other reads agree on a difference at that position, the position in the read is considered to be erroneous and corrected. If at least two other reads support the base, it is left unchanged. The corrected, homopolymer-compressed LA reads are used for all downstream stages of the pipeline, and only reverted to their original form during the final consensus stage.
Microsatellite compression
Although homopolymer errors are the most common systematic error type in HiFi reads 2,8, they are not the only error type. Microsatellite errors happen when a read contains a short, exact, tandem repeat with some copies incorrectly deleted or inserted. We define a unit as a single sequence 2–6bp that is repeated in the sequence. For example, the genomic sequence ACGACGACG, composed of the unit ACG repeating three times, could be miscalled as ACGACGACGACG, containing one extra unit copy. We have extended MBG to perform microsatellite compression to mask these errors.
The microsatellite compression used by MBG works analogously to, and follows directly after, homopolymer compression. Each microsatellite repeat unit is represented by a unique character, and any tandemly repeating characters are then merged together as with homopolymers. MBG transforms the input read into a special alphabet where each possible microsatellite repeat with a unit size of up to 6 bp gets its own character. Microsatellites are detected in the reads whenever a unit repeats at least twice and are encoded by three properties: the unit sequence, unit length, and overhang length. We define the overhang as the incomplete unit prefix at the end of the microsatellite repeat. The overhang length is always strictly less than the unit length and can be zero. The overhang sequence does not need to be encoded since it is always a prefix of the unit sequence. This encoding considers repeats with different overhangs to be distinct characters and does not include any information regarding copy number. Given a unit length n, there are n4n possible microsatellite characters. Considering all possible units of size 1 to 6 nucleotides, there are less than 216 characters in the alphabet and so each can be represented by a 16-bit integer.
Reverse complements are handled by computing the reverse complement of the unit sequence and rotating it according to the overhang length. Here we represent the encoding with the notation (x)y, where x is the unit sequence and y is the overhang sequence. For example, the repeat ACGACGACGAC is written as (ACG)AC, with a unit length 3, unit sequence ACG, and an overhang length of 2. The reverse complement of a character can be found in this notation by reverse complementing the string and keeping the parenthesis in place. In this example, the reverse complement of (ACG)AC is (GTC)GT.
Some microsatellite repeats can be encoded in multiple ways. For example, the sequence ATATATATATATAT could be encoded as (AT), (ATAT)AT, or (ATATAT)AT. If there are multiple possible encodings, we pick the one with the shortest unit length. Also, any microsatellite repeat contained within another microsatellite repeat is discarded. For example, in the sequence CGTGTCGTGT the two-copy repeat (CGTGT) is retained and the (GT) repeats are discarded. Microsatellite repeats can also overlap. For example, the sequence ACGACGACGTCGTCGT contains two microsatellite repeats, (ACG) and (CGT), which overlap by two characters, CG. In this case, the repeat boundaries are trimmed to avoid the overlap, but the repeat overhangs are not updated and the coded repeat character is kept the same. That is, the sequence ACGACGACGTCGTCGT will be represented by a repeat of type (ACG) coding for the sequence ACGACGA, then the non-repeating characters CG, followed by a repeat of type (CGT) coding for the sequence TCGTCGT. The overlap between two microsatellite repeats is always shorter than the shorter of the two motifs, and so trimming always leaves at least one nucleotide in the repeat and, therefore, cannot eliminate a microsatellite repeat. However, the final collapsed character may represent less than one copy of the repeat unit.
After graph construction, the reads are once again accessed to expand the microsatellite compressed strings by computing a consensus for each repeat. Reads are encoded as the original nucleotide string consisting of the nucleotides {A, C, T, G}, a stream of 16-bit integers representing the string in the microsatellite collapsed alphabet, and a character mapping that describes which substrings in the original string are represented by each character in the microsatellite collapsed string. The support for each encoded character is collected from all reads, and the most frequent nucleotide string is chosen as the consensus for each microsatellite collapsed character. If there is a tie, MBG selects the lexicographically highest one according to C++ string ordering. Note that after expansion of the microsatellites, the strings remain homopolymer compressed.
In practice, this compression had a large effect on the initial MBG graph. On CHM13, the MBG graph with compression had 10-fold fewer nodes (19,981 vs 206,180) and a 20-fold increase in N50 (591,485 vs 28,081).
Multiplex de Bruijn graph
MBG is a tool for building sparse de Bruijn graphs from long, accurate reads. Originally based on a minimizer 65 de Bruijn graph, the updated version of MBG uses closed syncmers 66 to sparsify the k-mer space and implements the multiplex de Bruijn graph strategy of Bankevich et al. 15 to simplify the graph. In summary, once the sparse de Bruijn graph is built for an initial size k (k=1001 by default), the reads are threaded through the graph according to exact k-mer matches. The threaded reads are then used to resolve the graph by locally increasing k. Bankevich et al. describe an exact de Bruijn graph with sequences stored on the edges. A detailed description of read threading and graph resolution is provided in their manuscript. Here, we focus only on how MBG adapts the multiplex de Bruijn graph algorithm to a sparse de Bruijn graph with sequences stored on the nodes.
Given a k-mer size k, all nodes with length k are potentially resolvable and taken under consideration. Paths which cross through a node are used to find spanning triplets for the potentially resolvable nodes. A spanning triplet for a node n is a subpath of length 3 where the middle node is n. The number of reads supporting each spanning triplet is found for all potentially resolvable nodes. Given a resolving coverage threshold t, if the read support for a spanning triplet is at least t, it is considered a solid triplet. By default a potentially resolvable node is marked unresolvable if any of its edges are not covered by a solid triplet, since an edge not covered by a solid triplet would be removed, introducing a gap in the graph. However, we disable this check for small k (empirically, k < 4,000) because these cases are usually caused by sequencing errors. Note that an edge is allowed to be covered by multiple solid triplets. After this, any solid triplets whose first and third nodes are of length k and marked unresolvable are removed. This process of marking nodes unresolvable and removing solid triplets is repeated until the set of potentially resolvable nodes and solid triplets has converged, and the final set of potentially resolvable nodes are marked resolvable.
Next, all resolvable nodes are removed and a set of edge-nodes, representing k+1-mers, is created. An edge-node is created for each edge touched by a resolvable node. If the edge touches one resolvable node, the edge-node has length k+1, containing the entire sequence of the resolvable node and one base pair from the successor node, and an edge is added between the newly created edge-node and the unresolvable node. If the edge connects two resolvable nodes, the sequence of the edge-node is the sequence of the path containing the two nodes. The edge-nodes are connected according to the solid triplets. Given a solid triplet (n1, n2, n3), an edge is added between the edge-nodes (n1, n2) and (n2, n3). The read paths are then rerouted to use these new edge-nodes, and non-branching paths are collapsed into a single node.
During the local k increase, MBG also performs graph cleaning. After each resolution step, tips and crosslinks are removed. A tip is a node with connections only on one end. Tips are removed if they are short (≤10 kb by default), low coverage (≤3 by default), and removing them would not create a new tip. Crosslinks are nodes which falsely connect two different genomic regions. We consider low coverage nodes with exactly one edge on each side as possible crosslink nodes and remove them if the tip removal condition applies to both ends of the node. After this, non-branching paths are again collapsed.
This process of increasing k and cleaning the graph is repeated until there are no more resolvable nodes or k=15,000, by default. MBG first resolves the graph with a coverage threshold t=2, and afterwards again with t=1 to resolve areas with lower coverage. Gaps in the graph caused by errors in the reads are then patched using the read paths, resulting in the final LA graph.
Ultra-long read to graph alignment
After construction of the LA graph, we use GraphAligner61 to align UL reads to the graph for further resolution. Since the graph sequences remain homopolymer-compressed, all UL reads are compressed prior to alignment. During development, we noticed a low rate of systematic misalignments in GraphAligner and implemented changes which improved both accuracy and runtime compared with the previously published version.
First, to reduce memory requirements, we switched to maximal exact match seeding using an FM-index 67 instead of the default minimizer-based seeding. Second, the dynamic programming alignment formulation was systematically causing the alignment at the start of the read to be less accurate than the end of the read when aligning to bubbles in the graph. This was because the alignment was forced to pass through all the seeds, and when a seed was selected from the incorrect haplotype in a bubble, the previous implementation would not recover the optimal alignment. To address this, we adopted a Smith-Waterman formulation; however, to avoid computing the entire dynamic programming matrix, we implemented a sparse Smith-Waterman which restricts alignments from starting anywhere except for the start position of a seed match.
The sparse Smith-Waterman allows for the extension of seed hits from both sides of a bubble, without the runtime penalty of a full Smith-Waterman alignment. The runtime and memory requirements are further improved by banding. If there is a correct seed at the start of the read, the result of the sparse Smith-Waterman alignment is guaranteed to be optimal, regardless of the other seeds. The presence of correct seed hits near the beginning, but not at the start of the read will also have a high probability of recovering the optimal alignment. To counter the problem of spurious seeds at the beginning of a read, we also run the algorithm backwards, starting from the end of the read and extending towards the beginning, and the better of two alignments is taken.
In order to use a Smith-Waterman recurrence, the scoring matrix must have positive scores for good alignments and negative scores for bad alignments. However, GraphAligner uses Myers’ bit vector algorithm 68 which is based on edit distance, where positive scores correspond to bad alignments. We formulated a connection between edit distances and a scoring matrix suitable for Smith-Waterman. Given a desired identity threshold p and a dynamic programming matrix with the read represented by rows and the graph represented by columns, we define a match score m = 1, mismatch and deletion score d = −p/(1−p), and insertion score i = −p/(1−p) − 1 (with deletions and insertions defined relative to the graph sequence). Given this scoring scheme, an alignment consisting only of matches and mismatches with an identity of p will have an alignment score of 0, while higher identity alignments will have positive scores and lower identity negative scores. Given an alignment at row n in the dynamic programming matrix with an edit distance e, the edit distance can be used to compute the alignment score s = n − (d+1)e up to an additive factor. We use this equality to set the edit distance of alignment starting cells to a value which approximates an alignment score of 0. The identity threshold p also allows GraphAligner to discard alignments which are below the expected identity of highly accurate input reads. Alignments are also clipped based on alignment identity, something that cannot be easily done with an edit distance.
Graph resolution with ultra-long reads
After the UL reads have been aligned to the LA graph, they are used to fill gaps and resolve repeats. The first step of the process is to connect nodes in the LA graph that were left disconnected due to coverage gaps or errors. Next, unique nodes are identified within the graph and connected based on the UL read paths. After this, the multiplex de Bruijn graph algorithm, identical to the one used by MBG, is run using the UL read paths to further resolve the graph.
When filling gaps in the LA graph, both the alignments of the UL reads and the topology of the graph are considered. First, tips are detected in the graph (i.e. nodes which don’t have an edge on both sides). If a UL read has an alignment ending at a tip, and another alignment starting at a tip, the UL read supports a gap fill between those two tips. If a pair of tips has a gap fill supported by a minimum number of UL reads (3 by default), a new gap node is inserted into the graph and connected to the tips. Each UL read supporting a gap fill has a gap length, estimated by the positions of the alignments within the read. Due to the higher error of the UL reads, there is no requirement that all gap fills have the same length. The length of the gap is estimated by taking the median gap length of all UL reads supporting the gap fill. If the gap has a positive length, the sequence of the gap node is represented by a corresponding number of “N”s. If it has a negative length, the sequences are copied from the tip nodes, and the edges are marked to have an appropriate overlap.
Unique nodes are those with a sequence that appears exactly once in the diploid genome. They are useful for identifying the correct genomic traversal of the graph, and Verkko uses multiple heuristics to discover unique nodes. The average genomic coverage is first estimated as the average LA read coverage observed for nodes ≥100 kb. All nodes are then compared to this average coverage, and any node which is both long and close to the average coverage is marked as unique. Next, any node close to the average coverage, regardless of length, is marked as unique if it is path consistent. A node is considered path consistent if at least 80% of the UL read paths touching it are either identical, prefixes of each other, or suffixes of each other. The topology of the graph is then used to discover chains of bubbles, as defined previously. Chains of bubbles are classified as 1-copy, 2-copy, or multi-copy based on the coverage of the core nodes in the chain (i.e. the nodes separating the bubbles). The core nodes of 1-copy chains are marked unique. For 2-copy chains, if a bubble has two paths with roughly equal coverage close to half the chain coverage, then the bubble nodes are marked unique. Multi-copy chains are ignored.
Once unique nodes are marked, the UL reads are used to find bridging paths between them. A bridge connects two unique nodes, with no unique nodes in between. The subpaths of UL alignments are collected as bridges and inconsistent bridges are resolved. Two bridges are considered inconsistent if they share one, but not both, unique node endpoints. At this stage, only the endpoints of the bridges are considered and not the specific paths. If an inconsistent bridge has less than half the read support of another, it is assumed to be erroneous and removed. If neither of the bridges has twice the coverage of the other, they are both retained. Once the bridges connecting the unique nodes are found, Verkko looks at the paths connecting the endpoints. The path with the most read support between each pair of unique nodes is taken as the consensus bridge path. All paths with at least half the coverage of the consensus path are kept, and all other lower coverage paths are discarded.
Lastly, tangles are marked as resolvable if all unique nodes touching the tangle have a bridge, and all high-coverage nodes and edges in the tangle are covered by a bridge (node coverage is measured from both the LA graph and aligned UL reads, and edge coverage from the aligned UL reads). These tangles are resolved by connecting the unique nodes based on the bridges. Nodes in a resolvable tangle that are not covered by a path are assumed to be erroneous and removed, while non-unique nodes covered by multiple paths are duplicated onto the corresponding paths. Unresolvable tangles for which there are no unique nodes, or those where the unique nodes were misidentified by the coverage checks, are left unchanged.
After the unique nodes have been connected, the multiplex de Bruijn graph algorithm, identical to the one implemented in MBG, is applied to the graph using the UL read paths. This can further resolve tangles which were not resolved by the unique node connection, either completely or partially.
Graph cleaning
Once UL resolution is complete, tips are clipped from the graph. If there is at least one long path starting at a fork, then other shorter paths starting at the same fork may be removed. By default, long paths are ≥35 kb, and the clipped paths are either <35 kb or <10% of the longer path’s length, whichever is shorter.
Each step of resolution outputs a node mapping which relates the nodes in the previous graph to the simplified graph. This node mapping can be used recursively to discover which nodes in the original MBG graph are contained in a node at any point during resolution. This information is used to generate coverage estimates for each node in the subsequent graphs. The coverage estimate of a node is defined as the weighted average of the coverages of the MBG nodes contained in it, considering only MBG nodes contained in a single node in the final graph.
These coverage estimates are used for resolving bubbles caused by sequencing errors. If a chain of bubbles appears to be single-copy, any bubbles comprising it must be caused by sequencing errors. The highest coverage path through a bubble is kept and the nodes and edges outside of this path are discarded. The final graph combining both the LA and UL reads is named the ULA graph.
Haplotype reconstruction
Rukki (“spinning wheel” in Finnish) is a companion tool for extracting haplotypes from a labeled assembly graph. Its main purpose is to facilitate haplotype-resolved assembly of diploid genomes by analysis of haplotype-specific markers within the graph nodes. For now, we have primarily targeted trio-based haplotype reconstruction, using parent-specific k-mers identified from parental Illumina reads as haplotype markers (Supplementary Note 1).
Rukki first labels graph nodes as maternal or paternal based on the prevalence of the corresponding haplotype markers, or leaves them unlabeled if the markers are ambiguous. This step considers an absolute number of markers attributed to the node, their average density, as well as the ratio between the marker counts of the two haplotypes. Rukki also detects nodes with a high abundance of markers from both haplotypes, as these nodes are likely to be misassembled or represent spurious cross-haplotype connections.
We call a graph node “long” if its length exceeds a certain threshold (>500 kb). This threshold is empirically chosen so that long nodes are common, but are very likely to be single-copy and haplotype-specific. Next, Rukki labels homozygous nodes by identifying neighborhoods where differently labeled regions converge. Due to minor flaws in the graph, nodes representing homozygous regions may contain a few parent-specific markers, which can lead to their initial mislabeling, so both unlabeled and previously labeled nodes with elevated coverage (as compared to the weighted average across all long nodes) are examined. The remaining long nodes are used to seed heuristic extension of the haplotype paths. Naturally, paternal nodes are considered incompatible with any path derived from a maternal node and vice versa, while homozygous and unlabeled nodes can potentially be incorporated in any path.
To make the extension procedure more robust to spurious cross-haplotype nodes, every time a long node s is incorporated into the path, Rukki analyzes its neighborhood to try and identify the next long candidate t. To do so, Rukki considers a subgraph bounded by long nodes, with s being one of the sources. If all sources and sinks in the subgraph are labeled, and there exists only a single source and sink compatible with the current path’s haplotype (with s being the source), the corresponding sink is marked at the next long candidate t. If found, Rukki further constrains the extension heuristic to prioritize nodes along any paths connecting s to t. Note that we only need to consider extensions in the forward direction, since a backward extension can be expressed via forward extension of the corresponding reverse complement path. The haplotype path extension can infer some missing haplotype labels, so the entire process is repeated a second time to generate a final set of haplotype paths. Extended Data Figure 10 provides some examples of haplotype paths produced by Rukki.
A unique feature of Rukki is its ability to “scaffold” across local ambiguities and breaks in the assembly graph, without the use of any additional scaffolding technologies. For example, when the extension procedure reaches a bubble it will either completely traverse it (Extended Data Figure 10A) or introduce a gap, depending on the bubble characteristics and provided options. Another example is scaffolding across a “loop tangle”, typically caused by near-identical tandem repeats and defined as a strongly connected subgraph with one source and one sink (Extended Data Figure 10B). In this case the gap size is estimated based on the total length of the nodes in the tangle and their overlaps. Rukki can also scaffold across a coverage break in one haplotype when the other haplotype is intact (Extended Data Figure 10C). In this case, the complete haplotype is used to estimate the size of the gap in the broken haplotype.
Rukki is an independent module taking as input only an assembly graph in GFA 69 format and the counts of haplotype-specific markers assigned to each node. Thus, it should be compatible with alternative methods of graph construction and haplotype assignment (e.g. based on Hi-C and Strand-seq). However, Rukki’s current heuristics have been optimized for Verkko’s ULA graphs, and would likely need further tuning for other graphs and contexts. Regarding alternative sources of phasing information, note that Rukki expects exactly two haplotype labels, which in the case of trios correspond to the paternal and maternal haplotypes. Fortunately, most phasing methods produce exactly two sets of contigs or read bins, typically labeled H1 and H2. The preliminary Hi-C integration results presented here are based on markers extracted from reads binned by DipAsm 39, but one could also use phased contigs from Hifiasm (Hi-C) or PGAS 18 (Strand-seq). A more sophisticated incorporation of phasing data is left as future work, and would likely involve mapping the Hi-C or Strand-seq reads directly to the Verkko assembly graph. Phased polyploid assembly is also left as future work and would require modifications to both Verkko and Rukki.
Consensus
Verkko’s primary data structure is a homopolymer-compressed assembly graph so a consensus stage is required to recover the genomic sequence of individual nodes or haplotype paths. Verkko’s node mapping is used to translate each node or path in the final graph into a sequence of MBG nodes. The sequences of MBG nodes and the paths of the LA reads in the MBG graph are used to build a layout of reads for each node in the final graph. Each LA read is assigned to the location with the longest exact match through the MBG graph. If there are multiple equally good locations, the read is assigned to all of them. Due to their lower quality, UL reads are only included in the layout where they were used to fill a gap in the initial LA graph. A consensus sequence is then called for the reads in the layout using a module from Canu70. By default, the ends of the consensus sequence supported by only a single read are trimmed.
Data availability
No new data was generated for this study. All assemblies generated in this paper are archived at zenodo 71 and we have provided convenience links to download both data and assemblies 72. The data is also hosted in public databases: A thaliana PRJCA005809, H axyridis PRJEB45202, CHM13 PRJNA559484, HG002 SAMN03283347 and the HPRC AWS bucket 73.
Code availability
Verkko code is available from GitHub 74 and all code used for the paper is archived at zenodo 71.
Extended Data
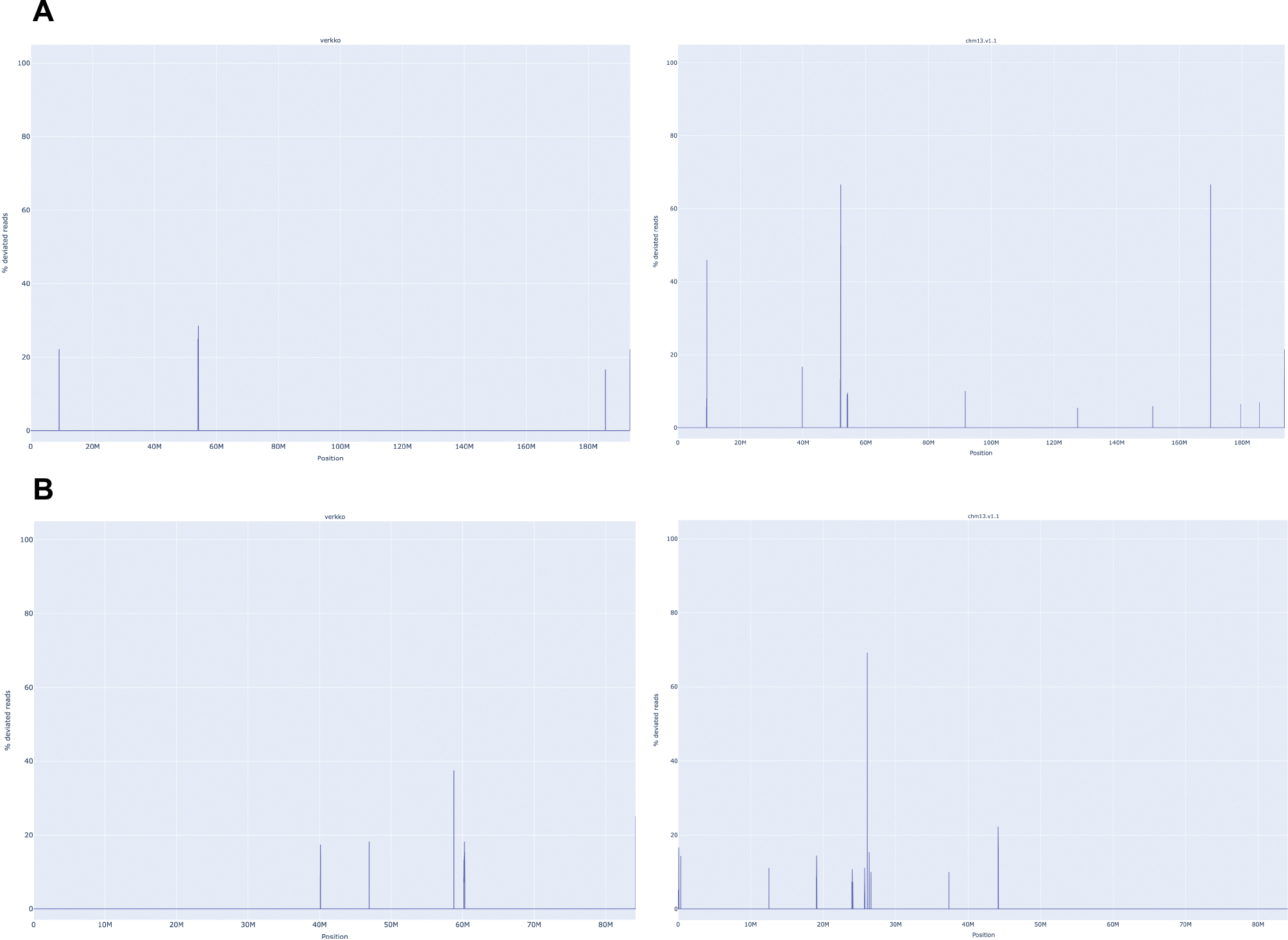
Extended Data Figure 1. A. thaliana chromosome unitigs in Verkko (left) vs published assembly chromosomes evaluated by VerityMap (right).
From top to bottom, Chr1, Chr2, Chr3, Chr4, and Chr5. VerityMap compares the spacing of unique k-mers within the HiFi reads to the spacing observed in the assembly. Whenever there is a disagreement, the plot shows a spike at the discrepant location. The x-axis indicates the coordinates along the assembly contig or scaffold while the y-axis shows the fraction of disagreeing reads (0–100%). A disagreement greater than 50% is likely not a heterozygous variant but a true error in the assembly. The BED file produced by VerityMap also indicates the size of the discrepancy, estimated from the difference in k-mer spacing between the reads and the assembly.
Extended Data Figure 2. Verkko CHM13 assembly sub-graphs.
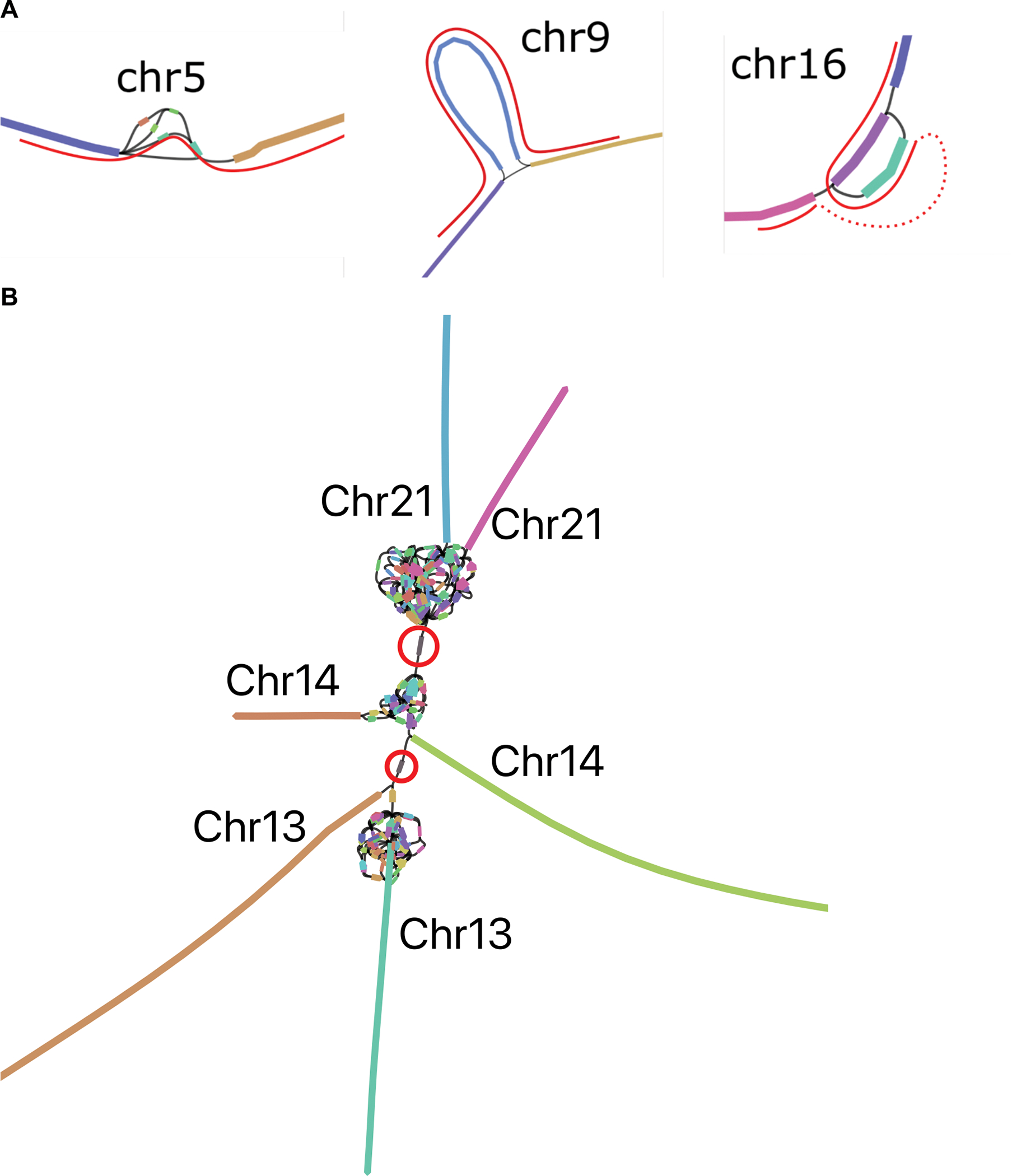
A. The remaining unresolved regions in CHM13 chromosomes 5, 9 and 16, visualized using Bandage62, with the correct resolution marked in red paths. Left: Chr5 has a spurious edge causing a cycle, and three spurious low-coverage nodes which were not removed by bubble popping since they are a part of the cycle. Middle: Chr9 has a spurious edge. Right: Chr16 has two spurious edges, and one missing edge (dashed red curve). The spurious non-genomic edges are caused by noisy ONT alignments switching between highly similar repeats in the LA graph, while the missing edge is caused by low HiFi coverage. B. rDNA cluster mixing in CHM13 chromosomes 13, 14, and 21, visualized using Bandage62. Each chromosome has a separate rDNA tangle. There are two cross-chromosomal connections by erroneous low coverage (<4x) nodes circled in red. For all three chromosomes, the remainder of the p and q arms are contained in the long unitigs shown.
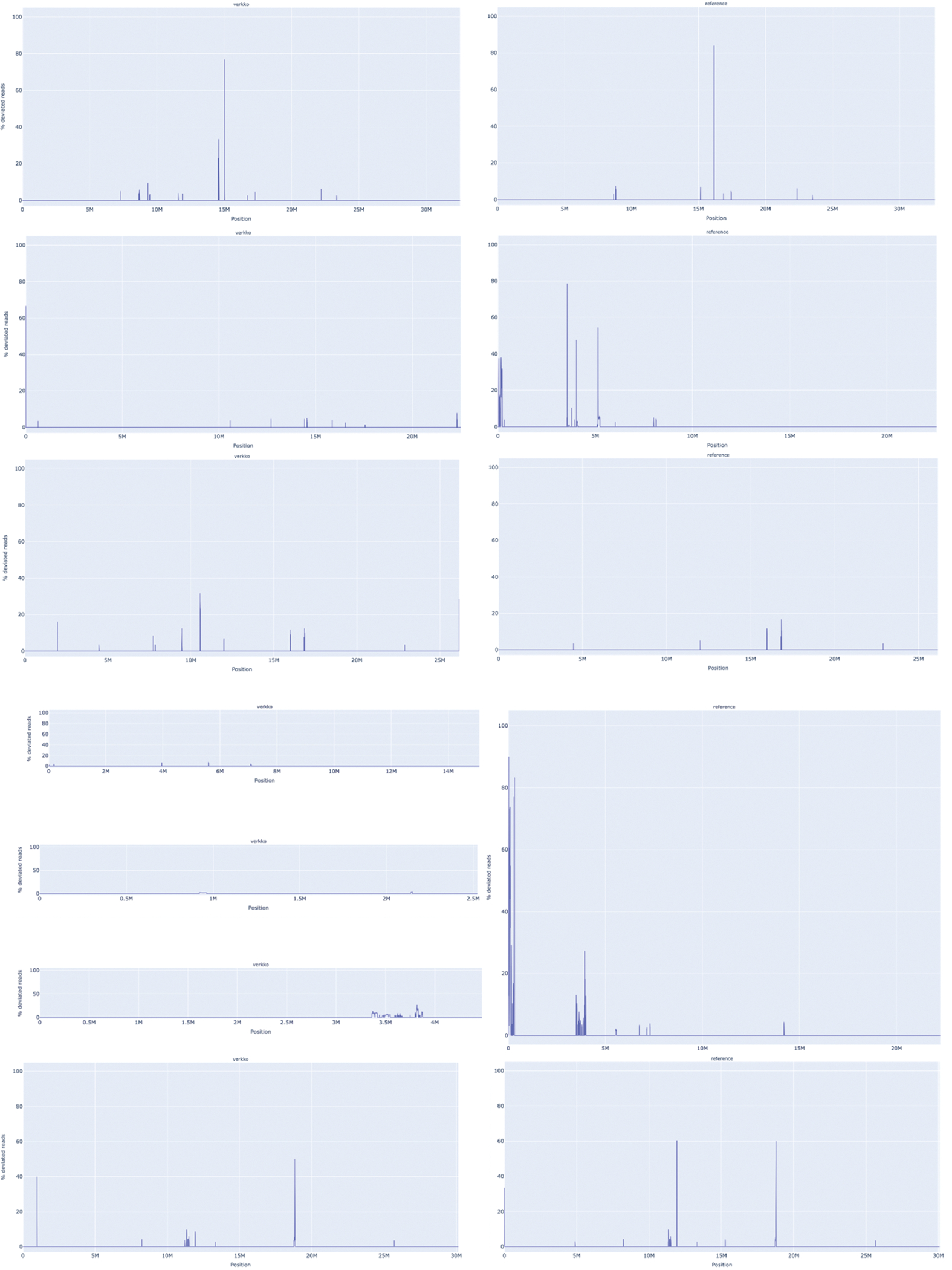
Extended Data Figure 3. VerityMap discrepant reads plot for CHM13 HiFi and ONT unitigs assembled by Verkko (left) and CHM13 v1.1 14 (right).
A. The assemblies for Chromosome 4. The Verkko assembly has no regions where a large fraction of reads are deviated even though QUAST marks an error at approximately 52 Mb. This corresponds to a position in the reference with a large fraction of deviated reads and an estimated 19 kb discrepancy. B. same for Chromosome 17. There are no regions with a large fraction (>50%) of discrepant reads in the Verkko assembly despite QUAST reporting an error at approximately 25 Mb on the reference. This corresponds to an approximately 3 kb discrepancy identified by VerityMap in CHM13 v1.1.
Extended Data Figure 4. Merqury 58 haplotype blob plots.
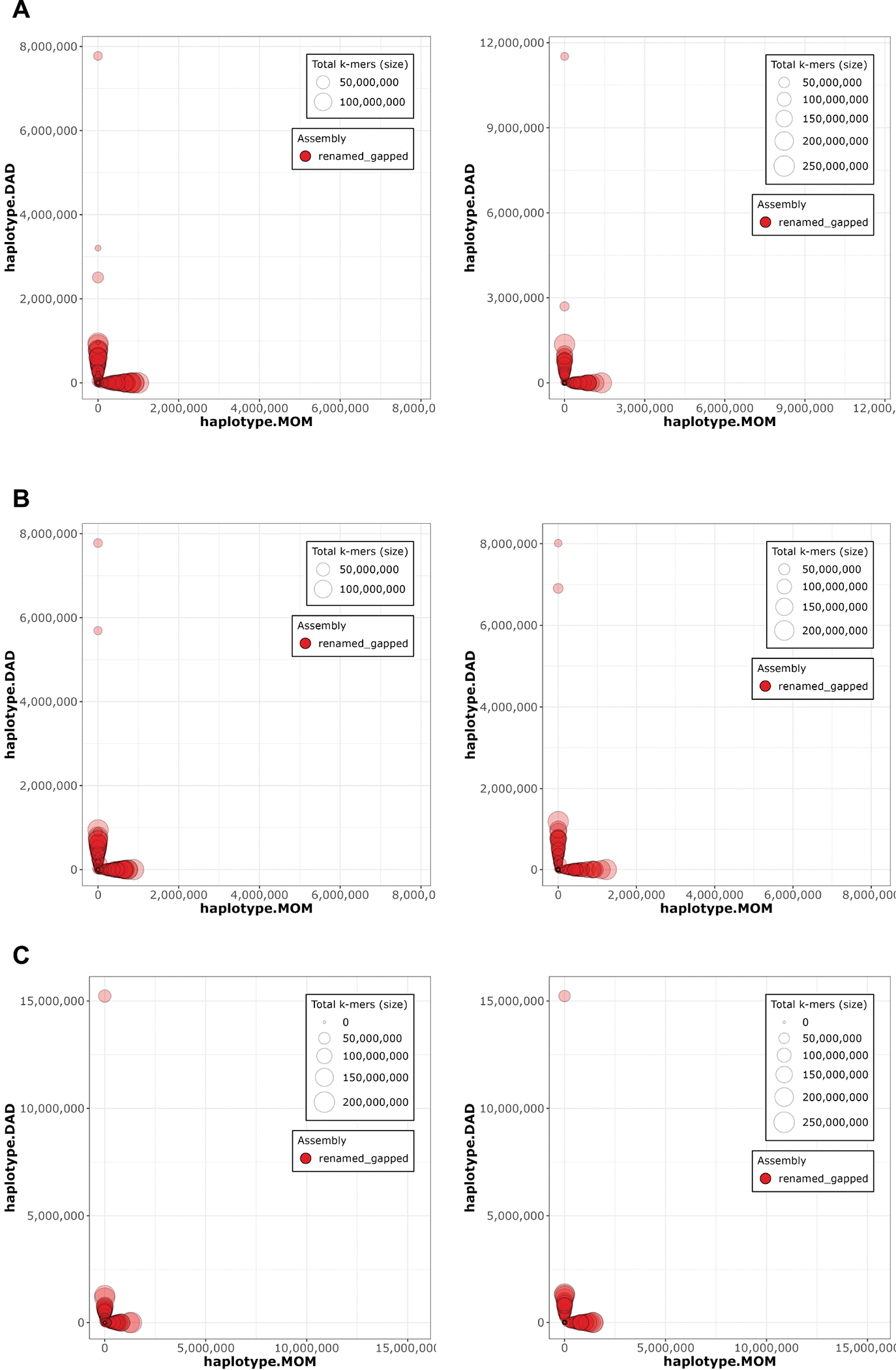
A. HG002 downsampled Verkko B. HG002 downsampled DeepConsensus HiFi Verkko and C. HG002 full-coverage Verkko assemblies. The Hi-C phased assembly is on the left and the trio-phased assembly is on the right. Each contig/scaffold is a circle on the plot, with the size scaled based on contig/scaffold length. The x-axis shows the number of maternal markers while the y-axis shows the number of paternal markers. Contigs which lie along either the x-axis or y-axis show no haplotype errors and are consistently maternal or paternal. Contigs which mixed haplotypes would appear along the diagonal but are not observed in these plots, indicating an accurately phased assembly.
Extended Data Figure 5. IGV 82 views of a recently published HG002 diploid assembly of paternal Chromosome 10 11 (top) and the Verkko full-coverage trio assembly of the same chromosome (bottom).
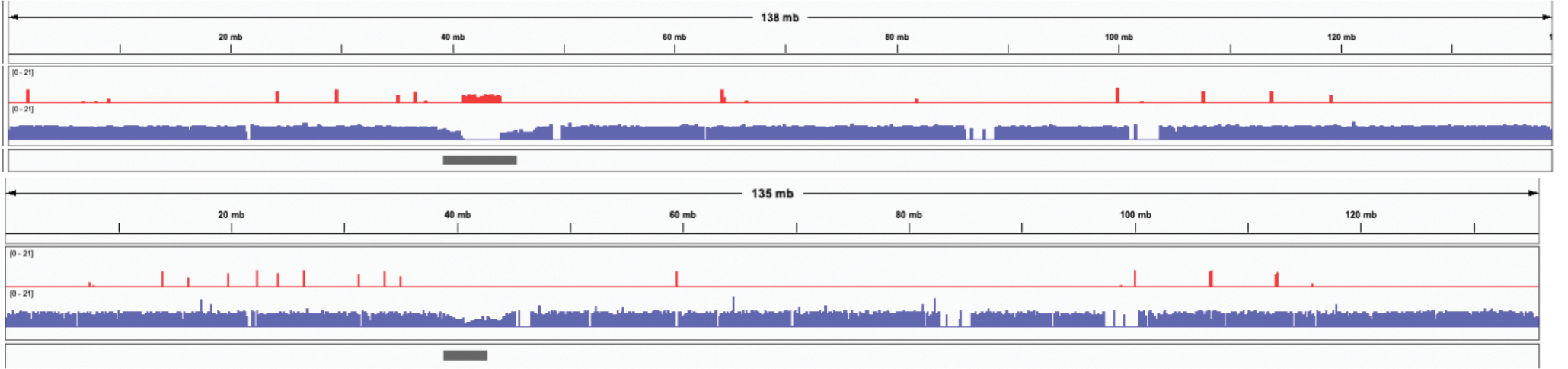
The tracks show the maternal (red) and paternal (blue) markers. The centromere location is shown in gray. The published assembly has extensive switching within the centromere array, indicated by the presence of maternal markers and the absence of paternal markers. In contrast, the Verkko assembly centromere shows only paternal markers. The Verkko paternal centromere array is shorter but shows no signs of mis-assembly (Extended Data Figure 8) indicating the larger array in the published assembly is likely due to the incorrect insertion of maternal sequence. Overall, the Verkko assembly is more continuous, with 0 gaps vs 4, and a lower hamming error rate, 0.03%, versus 1.98% compared to the published assembly.
Extended Data Figure 6. Strand-seq validation of the full-coverage Verkko trio assembly and HPRC manually curated assembly 11.
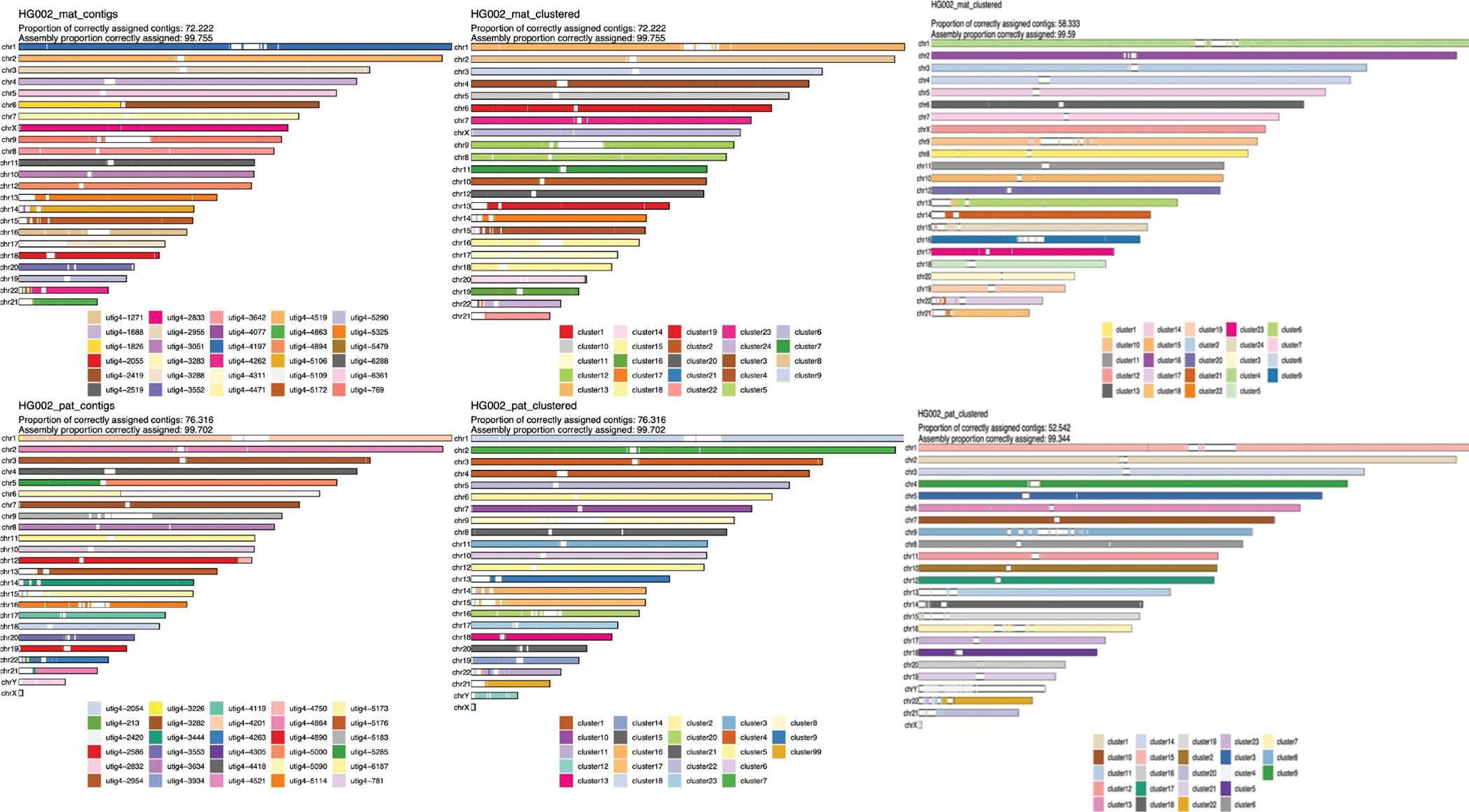
The maternal haplotype is shown along the top row and the paternal along the bottom row. Leftmost: alignment-based scaffold assignment to the maternal haplotype (top) and paternal haplotype (bottom) for the full-coverage Verkko assembly. Almost all chromosomes are a single color, indicating that Verkko scaffolds resolved most chromosomes end-to-end. The only exceptions are in the acrocentrics, where some of the scaffolds could not be assigned due to low mappability and maternal Chromosome 6 and paternal Chromosomes 5 which are each composed of two large scaffolds. Over 99.7% of the scaffold bases could be assigned to chromosomes. Middle: the cluster assignment for the maternal haplotype (top) and paternal haplotype (bottom) based on Strand-seq data for the full-coverage Verkko assembly. Here, cluster ID is assigned to each 200 kb window in a scaffold. In case of large scale chromosomal mis-joins, we expect to see multiple colors in a chromosome. The Verkko assembly is consistent with scaffolds all representing a single chromosome bin. Once again, >99.7% of the scaffold bases can be assigned using Strand-seq. Only 2 and 4 Mb of sequence not scaffolded by Verkko could be assigned to the maternal and paternal haplotypes, respectively. Right: The cluster assignment for the maternal haplotype (top) and the paternal haplotype (bottom) based on Strand-seq data for the HPRC manually curated assembly. Here, cluster ID is assigned to each 200 kb window in a scaffold. In case of large scale chromosomal mis-joins, we expect to see multiple colors in a chromosome. A smaller fraction of contigs (and a slightly lower fraction of bases) was assigned than for the Verkko assembly, despite the combination of technologies and manual curation. This may be due to shorter contigs from unresolved repeats which are resolved through Verkko’s ONT integration. There is also visible chromosome mixing within the acrocentric chromosomes unlike in the Verkko result.
Extended Data Figure 7. Strand-seq structural variant analysis for Verkko full-coverage assembly.
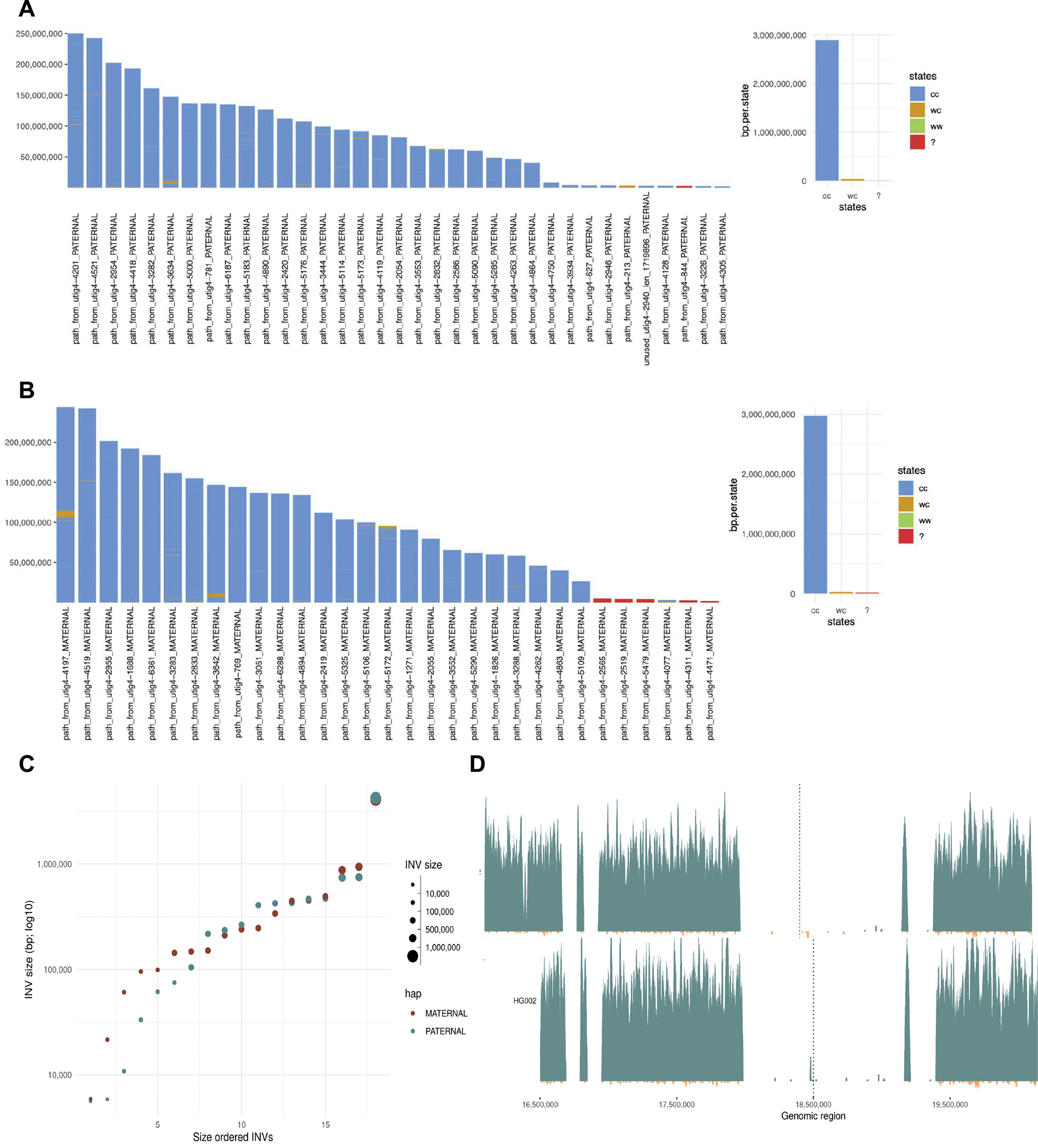
The states assigned to each scaffold in the paternal (A) and maternal (B) for the full-coverage Verkko trio assembly. Strand-seq reads aligned to each assembly are genotype based on their directionality into three possible strand states. Crick-Crick (‘cc’) state in which both homologs in Strand-seq data map in direct orientation and thus such regions are consistent with Strand-seq directional information. Watson-Watson (‘ww’) state in which both homologs in Strand-seq data map in inverted orientation and are indicative of assembly misorientation or unresolved homozygous inversion. Lastly, there are a few (<1% of bases) Watson-Crick (‘wc’) where there is a mixture of Watson and Crick reads and such regions are indicative of heterozygous inversions between haplotypes or might also be cause by low-mappability regions for short Strand-seq reads. C. The size of the heterozygous inversion versus the count of inversions of that size in the maternal and paternal haplotypes of the full-coverage Verkko trio assembly. These regions have confident Strand-seq alignments and normal copy number so these regions indicate potential true heterozygous variation between the haplotypes. D. Strand-seq alignments to the reference Chromosome Y before it was corrected (top) and full-coverage Verkko trio Chromosome Y assembly (bottom). Each plot shows Strand-seq directional read coverage reported as binned (bin size: 10,000, step size: 1,000) read counts represented as vertical bars above (teal; Crick read counts) and below (orange; Watson read counts) the midline. The top plot shows an inversion (dashed line) where directly oriented reads (Crick; teal) switch to inversely oriented reads (Watson, orange) and then back to directly oriented reads. The Verkko assembly in contrast is consistent with only Crick reads present in the same location (dashed line).
Extended Data Figure 8. Full-coverage Verkko trio assemblies of chromosome 1 (a), 3 (b), 4 (c), 11 (d), 9 (e), 10 (f), 16 (g), and 18 (h) centromeric regions in the HG002 genome.
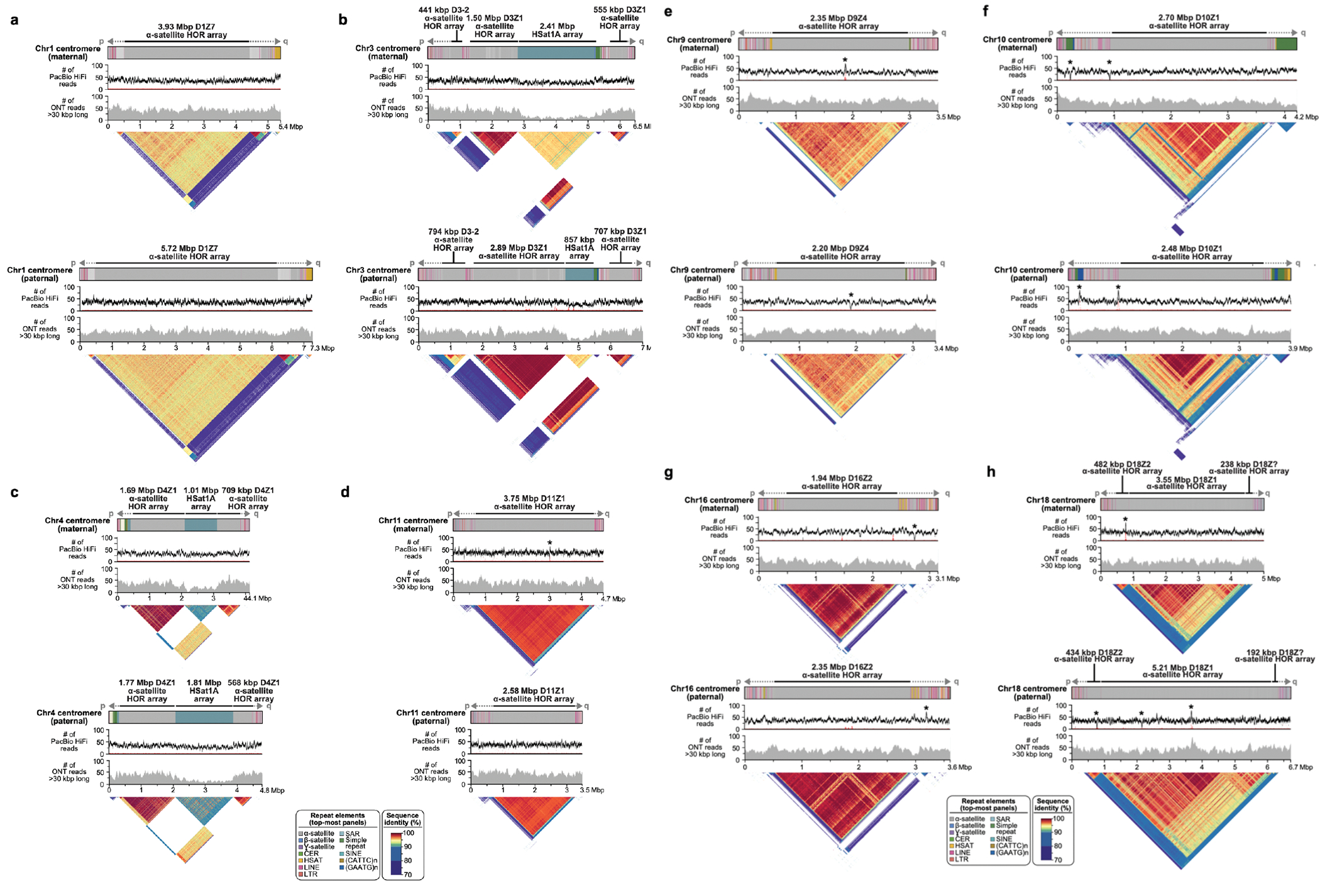
Both maternal and paternal haplotypes are shown, with repeat element annotation shown on top, followed by PacBio HiFi coverage, ONT coverage, and StainedGlass 63 plots. As with the Chromosome 19 centromeres (Fig. 4), the maternal and paternal haplotypes show large-scale structural variation, with alpha-satellite HOR arrays sizes varying by tens to hundreds of kb. Sites with discrepant HiFi mappings (low coverage or high coverage) are marked with an asterisk. There are few sites in the centromeres, and the artifacts are localized and often inconsistent between ONT and HiFi alignments, indicating the assembly is overall of high quality. To further validate assembly accuracy, we intersected centromere array locations with VerityMap errors and found that in all but four cases (two on the Chr1 paternal centromere, Chr9 paternal centromere, and Chr10 maternal centromere), the errors were short (≤1 kb) or lower frequency (≤50% of the reads). VerityMap also identified one issue, with ≥50% of reads deviating in the Chr4 maternal centromere. However, this was not visible in the NucFreq 37,83 plots above, and the region only had a total of three mapped reads.
Extended Data Figure 9. Comparison of the HG002 maternal and paternal full-coverage Verkko trio assemblies for the centromeric regions of chromosomes 1 (a), 3 (b), 4 (c), 9 (d), 10 (e), 11 (f), 16 (g), 18 (h), and 19 (i) in the HG002 genome.
The plots show the similarity between the two haplotypes, with the maternal haplotype on the y-axis and the paternal on the x-axis. The centromeric regions show varying ɑ-satellite HOR array sizes and sequence identity between the two haplotypes, consistent with earlier reports that indicate that centromeric HOR arrays often expand and contract due to their repetitive nature and their propensity for unequal crossing over 84–86 and gene conversion 87 events. For Chromosome 19, as in Figure 4, the tracks show the repeat annotations and read coverages. The triangles show the self-similarity within each haplotype for comparison.
Extended Data Figure 10. Examples of haplotype scaffolding by Rukki in the HG002 genome.
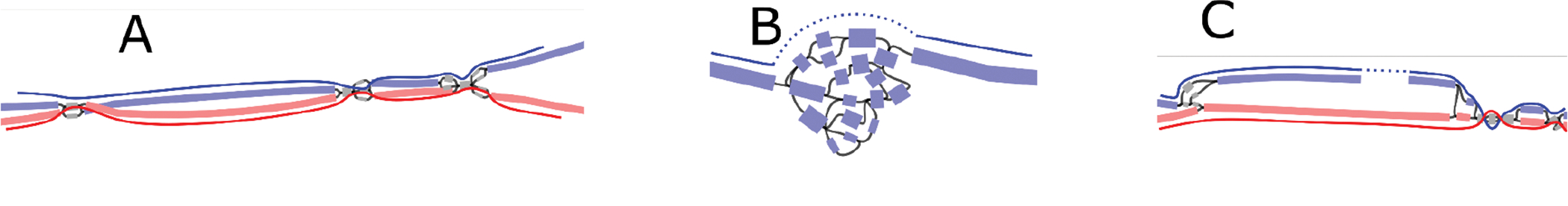
The nodes are colored according to their haplotype assignments. Nodes with at least 100 total markers where 90% of the markers agree are colored: red for maternal, blue for paternal. Nodes with less than 100 markers are colored gray for unassigned. The haplotype paths are marked with solid curves with dotted curves for gaps. (A) A well behaved genomic region consisting of phased heterozygous bubbles, homozygous nodes, and spurious nodes caused by sequencing errors. Where possible, Rukki connects the nodes attributed to the same haplotype across the homozygous regions, producing two phased unitigs without gaps. (B) A tangle within one haplotype. Rukki scaffolds across the tangle (dotted line), reporting an estimated size of the tangled region. (C) A gap in the paternal haplotype. Rukki uses haplotype assignments and the topology of the graph to scaffold across the gap (dotted line), and estimates the size of the gap based on the size of the paired haplotype.
Supplementary Material
Acknowledgements
This work was supported, in part, by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health (MR, SN, AR, BPW, AMP, and SK) as well as grants from the U.S. National Institutes of Health (NIH HG010169 and HG002385 to EEE) and National Institute of General Medical Sciences (NIGMS 1F32GM134558 to GAL). EEE is an investigator of the Howard Hughes Medical Institute. This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov).
Footnotes
Competing interest declaration
EEE is on the scientific advisory board of DNAnexus, Inc. SK has received travel funds to speak at events hosted by Oxford Nanopore Technologies. SN is an employee of Oxford Nanopore Technologies. The remaining authors declare no competing interests.
References
- 1.Logsdon GA, Vollger MR & Eichler EE Long-read human genome sequencing and its applications. Nat. Rev. Genet. 21, 597–614 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wenger AM et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nature Biotechnology vol. 37 1155–1162 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain M et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shafin K et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat. Biotechnol. (2020) doi: 10.1038/s41587-020-0503-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagarajan N & Pop M Sequencing and genome assembly using next-generation technologies. Methods in molecular biology (Clifton, N.J.) vol. 673 1–17 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Koren S & Phillippy AM One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr Opin Microbiol vol. 23C 110–120 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Kolmogorov M, Yuan J, Lin Y & Pevzner PA Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Nurk S et al. HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. Genome Res. (2020) doi: 10.1101/gr.263566.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng H, Concepcion GT, Feng X, Zhang H & Li H Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rhie A et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature 592, 737–746 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jarvis ED et al. Automated assembly of high-quality diploid human reference genomes. bioRxiv 2022.03.06.483034 (2022) doi: 10.1101/2022.03.06.483034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miga KH et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature (2020) doi: 10.1038/s41586-020-2547-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Logsdon GA et al. The structure, function and evolution of a complete human chromosome 8. Nature 593, 101–107 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nurk S et al. The complete sequence of a human genome. Science 2021.05.26.445798 (2022) doi: 10.1126/science.abj6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bankevich A, Bzikadze AV, Kolmogorov M, Antipov D & Pevzner PA Multiplex de Bruijn graphs enable genome assembly from long, high-fidelity reads. Nat. Biotechnol. (2022) doi: 10.1038/s41587-022-01220-6. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz DC et al. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science vol. 262 110–4 (1993). [DOI] [PubMed] [Google Scholar]
- 17.Ghareghani M et al. Strand-seq enables reliable separation of long reads by chromosome via expectation maximization. Bioinformatics 34, i115–i123 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Porubsky D et al. Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads. Nat. Biotechnol. 39, 302–308 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.O’Neill K et al. Assembling draft genomes using contiBAIT. Bioinforma. Oxf. Engl. 33, 2737–2739 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burton JN et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nature Biotechnology vol. 31 1119–25 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olga Dudchenko et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghurye J et al. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLOS Comput. Biol. 15, e1007273 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howe K et al. Significantly improving the quality of genome assemblies through curation. GigaScience 10, giaa153 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edge P, Bafna V & Bansal V HapCUT2: robust and accurate haplotype assembly for diverse sequencing technologies. Genome Res vol. 27 801–812 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antipov D, Korobeynikov A, McLean JS & Pevzner PA hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics vol. 32 1009–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wick RR, Judd LM, Gorrie CL & Holt KE Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput. Biol. 13, e1005595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Genova A, Buena-Atienza E, Ossowski S & Sagot M-F Efficient hybrid de novo assembly of human genomes with WENGAN. Nat. Biotechnol. 39, 422–430 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koren S et al. De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol. (2018) doi: 10.1038/nbt.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Falconer E et al. DNA template strand sequencing of single-cells maps genomic rearrangements at high resolution. Nat. Methods 9, 1107–1112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanders AD, Falconer E, Hills M, Spierings DCJ & Lansdorp PM Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat. Protoc. 12, 1151–1176 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Miller JR et al. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics vol. 24 2818–2824 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Idury RM & Waterman MS A new algorithm for DNA sequence assembly. Journal of computational biology : a journal of computational molecular cell biology vol. 2 291–306 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Wang B et al. High-quality Arabidopsis thaliana Genome Assembly with Nanopore and HiFi Long Reads. Genomics Proteomics Bioinformatics (2021) doi: 10.1016/j.gpb.2021.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheng H et al. Haplotype-resolved assembly of diploid genomes without parental data. Nat. Biotechnol. (2022) doi: 10.1038/s41587-022-01261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gurevich A, Saveliev V, Vyahhi N & Tesler G QUAST: quality assessment tool for genome assemblies. Bioinformatics vol. 29 1072–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mikheenko A, Bzikadze AV, Gurevich A, Miga KH & Pevzner PA TandemTools: mapping long reads and assessing/improving assembly quality in extra-long tandem repeats. Bioinformatics 36, i75–i83 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mc Cartney AM et al. Chasing perfection: validation and polishing strategies for telomere-to-telomere genome assemblies. Nat. Methods (2022) doi: 10.1038/s41592-022-01440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boyes D et al. The genome sequence of the harlequin ladybird, Harmonia axyridis (Pallas, 1773). (2021) doi: 10.12688/wellcomeopenres.17349.1. [DOI] [Google Scholar]
- 39.Garg S et al. Chromosome-scale, haplotype-resolved assembly of human genomes. Nat. Biotechnol. 39, 309–312 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zook JM et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. Sci. Data 3, 1–26 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang T et al. The Human Pangenome Project: a global resource to map genomic diversity. Nature 604, 437–446 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baid G et al. DeepConsensus: Gap-Aware Sequence Transformers for Sequence Correction. bioRxiv 2021.08.31.458403 (2021) doi: 10.1101/2021.08.31.458403. [DOI] [Google Scholar]
- 43.Guan D et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics (2020) doi: 10.1093/bioinformatics/btaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Porubsky D et al. Recurrent inversion polymorphisms in humans associate with genetic instability and genomic disorders. Cell 185, 1986–2005.e26 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohajeri K et al. Interchromosomal core duplicons drive both evolutionary instability and disease susceptibility of the Chromosome 8p23.1 region. Genome Res. 26, 1453–1467 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rhie A et al. The complete sequence of a human Y chromosome. bioRxiv 2022.12.01.518724 (2022) doi: 10.1101/2022.12.01.518724. [DOI] [Google Scholar]
- 47.McNulty SM & Sullivan BA Alpha satellite DNA biology: finding function in the recesses of the genome. Chromosome Res. 26, 115–138 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mahtani MM & Willard HF Pulsed-field gel analysis of alpha-satellite DNA at the human X chromosome centromere: high-frequency polymorphisms and array size estimate. Genomics 7, 607–613 (1990). [DOI] [PubMed] [Google Scholar]
- 49.Wevrick R & Willard HF Physical map of the centromeric region of human chromosome 7: relationship between two distinct alpha satellite arrays. Nucleic Acids Res. 19, 2295–2301 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waye JS & Willard HF Chromosome specificity of satellite DNAs: short- and long-range organization of a diverged dimeric subset of human alpha satellite from chromosome 3. Chromosoma 97, 475–480 (1989). [DOI] [PubMed] [Google Scholar]
- 51.Waye JS et al. Chromosome-specific alpha satellite DNA from human chromosome 1: hierarchical structure and genomic organization of a polymorphic domain spanning several hundred kilobase pairs of centromeric DNA. Genomics 1, 43–51 (1987). [DOI] [PubMed] [Google Scholar]
- 52.Willard HF et al. Detection of restriction fragment length polymorphisms at the centromeres of human chromosomes by using chromosome-specific alpha satellite DNA probes: implications for development of centromere-based genetic linkage maps. Proc. Natl. Acad. Sci. U. S. A. 83, 5611–5615 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wevrick R & Willard HF Long-range organization of tandem arrays of alpha satellite DNA at the centromeres of human chromosomes: high-frequency array-length polymorphism and meiotic stability. Proc. Natl. Acad. Sci. U. S. A. 86, 9394–9398 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Lima LG et al. PCR amplicons identify widespread copy number variation in human centromeric arrays and instability in cancer. Cell Genomics 1, 100064 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maize B73 Oxford Nanopore duplex sequence data release. https://www.keygene.com/news-events/maize-b73-oxford-nanopore-duplex-sequence-data-release/ (2022).
- 56.Altemose N et al. Complete genomic and epigenetic maps of human centromeres. Science (2022) doi: 10.1126/science.abl4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Langley SA, Miga KH, Karpen GH & Langley CH Haplotypes spanning centromeric regions reveal persistence of large blocks of archaic DNA. eLife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rhie A, Walenz BP, Koren S & Phillippy AM Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 21, 245 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ewing B & Green P Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Research vol. 8 186–94 (1998). [PubMed] [Google Scholar]
- 60.Rautiainen M & Marschall T MBG: Minimizer-based sparse de Bruijn Graph construction. Bioinformatics (2021) doi: 10.1093/bioinformatics/btab004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rautiainen M & Marschall T GraphAligner: rapid and versatile sequence-to-graph alignment. Genome Biol. 21, 253 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wick RR, Schultz MB, Zobel J & Holt KE Bandage: interactive visualization of de novo genome assemblies. Bioinformatics vol. 31 3350–2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vollger MR, Kerpedjiev P, Phillippy AM & Eichler EE StainedGlass: interactive visualization of massive tandem repeat structures with identity heatmaps. Bioinformatics 38, 2049–2051 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Onodera T, Sadakane K & Shibuya T Detecting Superbubbles in Assembly Graphs. in Algorithms in Bioinformatics (eds. Darling A & Stoye J) 338–348 (Springer; Berlin Heidelberg, 2013). [Google Scholar]
- 65.Roberts M, Hayes W, Hunt BR, Mount SM & Yorke JA Reducing storage requirements for biological sequence comparison. Bioinformatics vol. 20 3363–9 (2004). [DOI] [PubMed] [Google Scholar]
- 66.Edgar R Syncmers are more sensitive than minimizers for selecting conserved k-mers in biological sequences. PeerJ 9, e10805 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferragina P & Manzini G Indexing compressed text. J. ACM vol. 52 552–581 (2005). [Google Scholar]
- 68.Myers G A fast bit-vector algorithm for approximate string matching based on dynamic programming. J. ACM vol. 46 395–415 (1999). [Google Scholar]
- 69.Li H Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics vol. 32 2103–10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koren S et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koren S Zenodo verkko publication data. 10.5281/zenodo.6618379. [DOI]
- 72.Koren S GitHub verkko publication readme. https://github.com/marbl/verkko/blob/master/paper/README.md.
- 73.HPRC HG002 public data. https://s3-us-west-2.amazonaws.com/human-pangenomics/index.html?prefix=.
- 74.Koren S GitHub verkko repository. https://github.com/marbl/verkko/.
- 75.Chin C-S & Khalak A Human Genome Assembly in 100 Minutes. bioRxiv (2019) doi: 10.1101/705616. [DOI] [Google Scholar]
- 76.Li H New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jain C, Koren S, Dilthey A, Phillippy AM & Aluru S A fast adaptive algorithm for computing whole-genome homology maps. Bioinformatics 34, i748–i756 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics vol. 25 1754–60 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics vol. 25 2078–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Porubsky D et al. breakpointR: an R/Bioconductor package to localize strand state changes in Strand-seq data. Bioinformatics 36, 1260–1261 (2020). [DOI] [PubMed] [Google Scholar]
- 82.Robinson JT et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vollger MR et al. Long-read sequence and assembly of segmental duplications. Nat. Methods 16, 88–94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smith George P Evolution of Repeated DNA Sequences by Unequal Crossover. Science 191, 528–535 (1976). [DOI] [PubMed] [Google Scholar]
- 85.Alkan C, Eichler EE, Bailey JA, Sahinalp SC & Tüzün E The role of unequal crossover in alpha-satellite DNA evolution: a computational analysis. J. Comput. Biol. J. Comput. Mol. Cell Biol. 11, 933–944 (2004). [DOI] [PubMed] [Google Scholar]
- 86.Alkan C, Bailey JA, Eichler EE, Sahinalp SC & Tuzun E An algorithmic analysis of the role of unequal crossover in alpha-satellite DNA evolution. Genome Inform. Int. Conf. Genome Inform. 13, 93–102 (2002). [PubMed] [Google Scholar]
- 87.Schindelhauer D & Schwarz T Evidence for a fast, intrachromosomal conversion mechanism from mapping of nucleotide variants within a homogeneous alpha-satellite DNA array. Genome Res. 12, 1815–1826 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new data was generated for this study. All assemblies generated in this paper are archived at zenodo 71 and we have provided convenience links to download both data and assemblies 72. The data is also hosted in public databases: A thaliana PRJCA005809, H axyridis PRJEB45202, CHM13 PRJNA559484, HG002 SAMN03283347 and the HPRC AWS bucket 73.