

Abstract
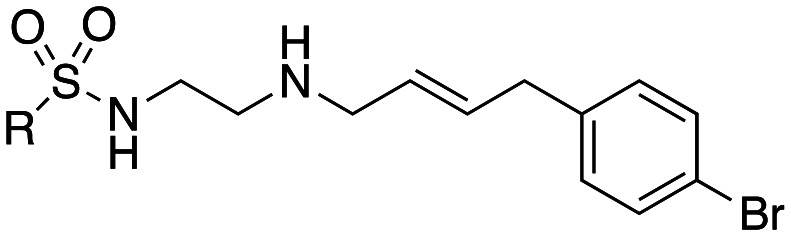
We show that dansylcadaverine (1) a known in-cell inhibitor of clathrin mediated endocytosis (CME), moderately inhibits dynamin I (dynI) GTPase activity (IC50 45 μM) and transferrin (Tfn) endocytosis in U2OS cells (IC50 205 μM). Synthesis gave a new class of GTP-competitive dynamin inhibitors, the Sulfonadyns™. The introduction of a terminal cinnamyl moiety greatly enhanced dynI inhibition. Rigid diamine or amide links between the dansyl and cinnamyl moieties were detrimental to dynI inhibition. Compounds with in vitro inhibition of dynI activity <10 μM were tested in-cell for inhibition of CME. These data unveiled a number of compounds, e.g. analogues 33 ((E)-N-(6-{[(3-(4-bromophenyl)-2-propen-1-yl]amino}hexyl)-5-isoquinolinesulfonamide)) and 47 ((E)-N-(3-{[3-(4-bromophenyl)-2-propen-1-yl]amino}propyl)-1-naphthalenesulfonamide)isomers that showed dyn IC50 <4 μM, IC50(CME) <30 μM and IC50(SVE) from 12–265 μM. Both analogues (33 and 47) are at least 10 times more potent that the initial lead, dansylcadaverine (1). Enzyme kinetics revealed these sulfonamide analogues as being GTP competitive inhibitors of dynI. Sulfonadyn-47, the most potent SVE inhibitor observed (IC50(SVE) = 12.3 μM), significantly increased seizure threshold in a 6 Hz mouse psychomotor seizure test at 30 (p = 0.003) and 100 mg kg−1 ip (p < 0.0001), with similar anti-seizure efficacy to the established anti-seizure medication, sodium valproate (400 mg kg−1). The Sulfonadyn™ class of drugs target dynamin and show promise as novel leads for future anti-seizure medications.
Modification of the known clathrin mediated endocytosis inhibitor dansylcadaverine to the sulfonadyns, dynamin GTPase inhibitors active in the 6 Hz psychomotor animal model of seizures.
Introduction
The epilepsies are a common group of neurological conditions affecting ∼1% of the world's population that impose a significant clinical and economic burden on society.1 Current epilepsy therapies provide inadequate seizure control in about one third of patients.1,2 The most common drug-resistant epilepsies in adults are focal epilepsies, including mesial temporal lobe epilepsy and post-traumatic epilepsy.2 Although the epilepsies are heterogeneous conditions, a uniting principle is that all seizures are associated with a massive burst of synaptic transmission, which requires the release of neurotransmitter through synaptic vesicle (SV) exocytosis.3 Correspondingly, synaptic transmission cannot be sustained for more than a few seconds without compensation by synaptic vesicle endocytosis (SVE) to generate new SVs, maintaining the vesicle supply and hence maintaining continuous neurotransmission.3–8 Given the involvement of pathologically large bursts of excitatory neurotransmitter release, distinct from conventional neurotransmission, it follows that an inhibitor of SVE could potentially interrupt seizure activity without impacting normal, physiological neuronal firing.
A common principle of most currently prescribed anti-seizure medications is their ability to inhibit synaptic transmission, generally by inhibiting neuronal excitability. Most anti-seizure medications appear to achieve their therapeutic action by this principle, e.g. inhibiting voltage gated sodium channels at excitatory synapses or enhancing inhibitory GABAergic activity at inhibitory synapses.3,9–12 There are rare exceptions to this approach, such as levetiracetam, which targets the synaptic vesicle protein 2A (SV2A).3,7,9,10,13 However, an underexplored mechanism for anti-seizure therapy is to reduce synaptic transmission via inhibition of SVE.14,15
There is growing evidence for the therapeutic potential of targeting dynamin and inhibitors of endocytosis in a number of clinically relevant diseases, opening the gateway to potential therapeutic interventions involving dynamin and endocytosis modulation.16,17 Mutations in dynI cause epileptic encephalopathy in humans18 and mutations in dynII cause leukemia,19 Charcot–Marie–Tooth disease20 and centronuclear myopathy.21–24 Pharmacological or antisense oligonucleotides dynamin modulators show promise in different animal models of these disorders and in other diseases that may not involve dynamin mutation. In disorders involving mutant dynII, its reduction with antisense oligonucleotides can alleviate severe forms of centronuclear myopathy.25 There are examples of diseases that do not involve mutant dynamin. The anti-seizure medication potential of dynamin inhibitors was demonstrated in a pilocarpine rat epilepsy model by the anti-seizure effects of stereotactically injected dynasore,26,27 a dynamin inhibitor with an IC50 of 15 μM for both dynI GTPase activity and CME.28 Endocytosis inhibition with prochlorperazine is effective in combination with immunotherapy for head and neck cancer in mice,29 while dynole-34-2 is effective in animal models of leukemia30 and glioblastoma.31 The dynamin inhibitor Dyngo®-4a is effective in botulism,32 while both Dyngo®-4a and antisense oligonucleotides are effective in models of neuropathic pain.33 Dynamin inhibition by Dyngo®-4a and MiTMAB causes death of leukemia and lymphoma cancer cells.34 We have demonstrated that clinically used antipsychotic medications in the phenothiazine class, including the archetypical inhibitor chlorpromazine, are dynamin inhibitors.35 In a human study the phenothiazine prochlorperazine reduces endocytosis in humans demonstrating that endocytosis inhibitor therapy is not inherently unsafe.29
The best known druggable targets in the CME and SVE pathways are clathrin and dynamin.17 DynI is a GTPase enzyme that plays a key role in maintaining SV supply in presynaptic terminals. DynI is able to assemble as a helix at the neck of a newly forming SV, and brings about fission by twisting the helix.36 There are three classical dynamins, dynamin I, II and III each with the same domain organization and 80% overall amino acid sequence homology. They have overlapping biological roles,30,38,39 but distinct tissue expression patterns.37,38 DynI is found predominately in neurons and is crucial for SVE. However it also plays a role in at least two non-clathrin modes of neuronal endocytosis called activity-dependent bulk endocytosis27 and ultrafast endocytosis.39
Herein we explore the non-specific CME inhibitor, dansylcadaverine (1),40 and describe the development and dynamin inhibition of a new series of compounds we call the Sulfonadyns™. We started with the observation that the autofluorescent compound dansylcadaverine (1) (also called monodansylcadaverine (MDC), Fig. 1) is a known low potency in-cell inhibitor of CME (IC50 is typically in the 200–500 μM range) with an unidentified molecular target, and is also an inhibitor of tissue transglutaminase enzymes.41,42 Its CME inhibitory activity was initially attributed to the stabilization of clathrin-coated pits, but this has not been validated.43 We explored dynamin as its possible molecular target to potentially explain the phenotype and we uncovered modest potency. We then used this sulfonamide as a platform to improve dynamin and CME potency in concert, supporting that dynamin remains the potential molecular target of its CME (and SVE) action. We then tested the resultant compounds as potentially novel dynamin-targeting anti-seizure agents.
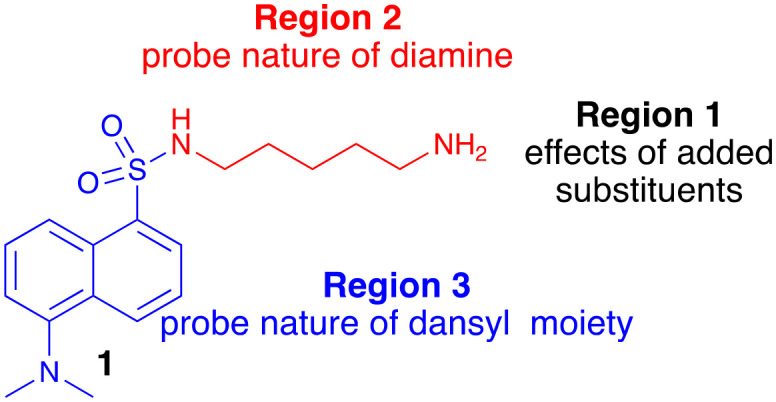
Fig. 1. Chemical structure of dansylcadaverine (1) and possible sites for modification and focused library development.
Results and discussion
Focussed libraries based on dansylcadaverine lead to potent inhibitors of dynamin GTPase activity
Dansylcadaverine, N-(5-aminopentyl)-5-dimethylamino-1-naphthalenesulfonamide (1) is known as a poor inhibitor of endocytosis, operating via an unknown mechanism. Based on its structural relationship to H89 (ref. 44 and 45) we rationalised that a potential mode of action might be via inhibition of dynamin's GTP binding. Given the low numbers of GTP domain targeting dynamin inhibitors, we viewed this as a potential opportunity to reveal the CME mode of action of 1, and develop a novel series of potentially GTP competitive dynamin inhibitors. Thus, dansylcadaverine (1) was first examined for inhibition of two forms of endocytosis: SVE in synaptosomes (mediated by dynI) and CME of transferrin in non-neuronal cells (mediated by dynII). Dansylcadaverine (1) inhibited CME relatively poorly (IC50(CME) = 205 ± 67 μM, Table 1), and was not an effective inhibitor of SVE (IC50(SVE) >300 μM).49 The compound also showed moderate inhibition of dynamin I (dynI) GTPase activity in vitro (IC50(dynI) = 45.1 ± 2.6 μM). With this preliminary evidence for dynamin inhibition, we next explored the SAR associated with 1.
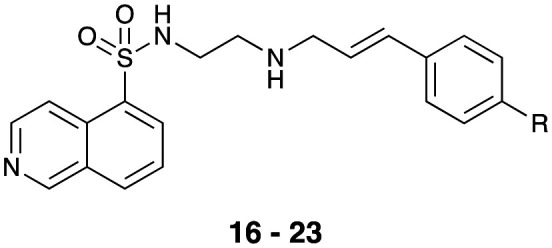
Inhibition of dynamin I lipid-stimulated GTPase activity (IC50a (μM)) by analogues 1, 7, and 16–23.
| Compound | DynI IC50a (μM) | |
|---|---|---|
| 1 | Dansylcadaverine | 45.1 ± 2.6 |
|
R |
|
| 7 | — | Not active |
| 16 | –H | 38.2 ± 6.0 |
| 17 | –CH3 | 22.4 ± 3.0 |
| 18 | –C(CH3)3 | 6.6 ± 0.8 |
| 19 | –Br | 7.0 ± 0.8 |
| 20 | –Cl | 9.2 ± 1.1 |
| 21 | –F | 20.3 ± 1.6 |
| 22 | –CF3 | 12.4 ± 1.5 |
| 23 | –N(CH3)2 | 19.9 ± 3.5 |
IC50 and 95% CI of triplicates, n = 1.
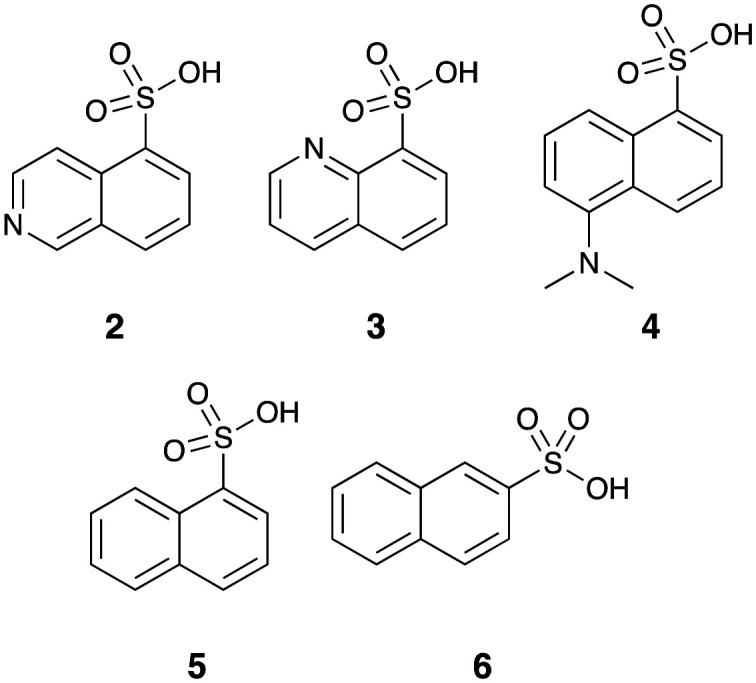
We had access to a number of aromatic sulfonic acids (2–6) which offered the potential for rapid synthesis of analogues with specific modifications to the dansyl moiety (Fig. 2). We also have experience in development of molecules with a diamine linker. Together, these suggested that a full SAR evaluation of 1 would be best approached by a pseudo de novo approach.
Fig. 2. Starting sulfonic acids (2–6) for the development of focused libraries.
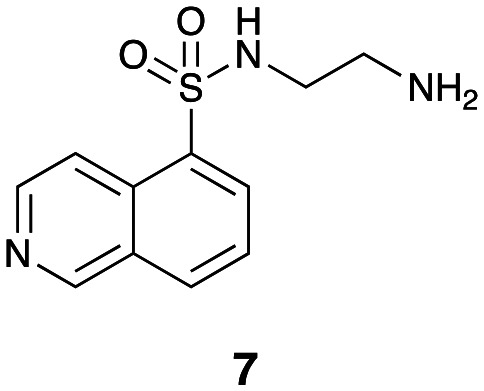
Our SAR evaluation commenced with sulfonic acid (2) which was readily converted to the corresponding sulfonyl chloride on treatment with thionyl chloride, and the aminosulfonamide (7) following addition of 1,2-diaminoethane.46 Further elaboration of the –NH2 moiety was achieved by Na(OAc)3BH mediated reductive amination with selected aromatic aldehydes (Scheme 1 and Table 1). Our initial efforts amounted to a rapid survey of the type of substituent tolerated in region 1 (Fig. 1), and these data (not shown) indicated a notable preference for the introduction of substituted 4-cinnamyl moieties (Scheme 1 and Table 1).
Scheme 1. Reagents and conditions: i) SOCl2, DMF (cat.) reflux, 8 h; ii) ethane-1,2-diamine, CH2Cl2, 0–25 °C, 18 h; iii) Ph3P CHCO2Et, EtOH, 25 °C, 18 h, chromatography; iv) DIBAL-H, −78 °C, 1 h; v) MnO2, CH2Cl2, 25 °C, 18 h; vi) Na(OAc)3BH, THF, 25 °C, 18 h.
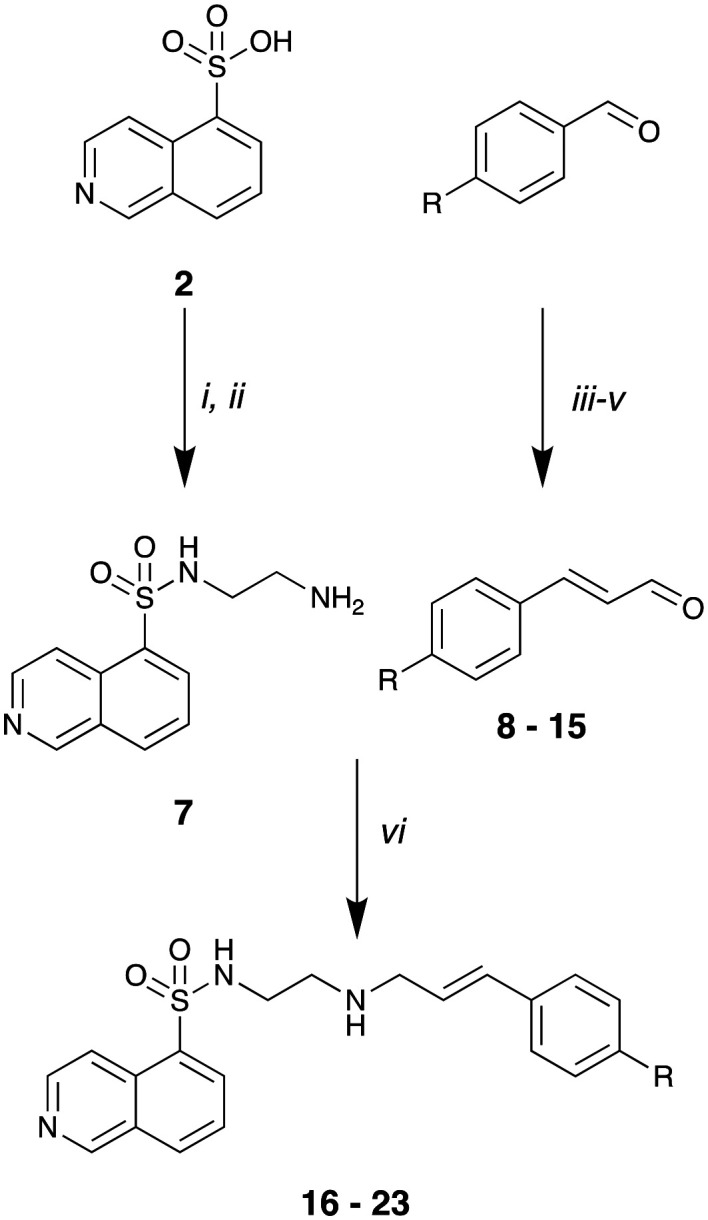
As the commercial availability of substituted cinnamaldehydes was limited, we prepared a small library of 4-substituted cinnamaldehydes. Wittig reaction of a series of 4-substituted benzaldehydes (see Table 1 for details) and the phosphorane ylid, Ph3P CHCOOEt, after flash chromatography allowed isolation, of the desired (E)-ethyl cinnamates in good yields (62–75%). Further treatment with 1 M DIBAl-H gave the corresponding cinnamoyl alcohols, which on MnO2 oxidation gave cinnamaldehydes (9–15) in good to excellent yields (73–93%).47–53 Subsequent reductive amination with 7 and Na(OAc)3BH gave analogues 16–23 in moderate to good (54–74%) yields (Scheme 1). These compounds were then evaluated for their ability to inhibit dynI GTPase activity (Table 1).
With the exception of primary amine 7, they were moderate to good dynI inhibitors (Table 1). The parent cinnamyl analogue (16; R = H, IC50 = 38.2 ± 6.0 μM) was the poorest dynamin inhibitor, but was more potent than 1, demonstrating the importance of the cinnamoyl moiety. The introduction of alkyl and simple aromatic substituents analogues to the cinnamoyl moiety improved potency, with IC50 values of 22.4 ± 3.3 (17; R = CH3), and 6.6 ± 0.8 (18; R = C(CH3)3). Bioisosteric replacement of 16's 4-H moiety with F, Cl and Br resulted in a steady increase in potency, with these analogues returning IC50 values of 20.3 ± 1.6 (21; R = F), 9.2 ± 1.1 (20; R = Cl) and 7.0 ± 0.8 μM (19; R = Br, 19 is also known as H89). Introduction of a 4-CF3 moiety increased potency 2-fold relative to 16 (22; IC50 = 12.4 ± 1.5 μM). However, the introduction of electron donating substituents only had a modest effect on dynamin inhibition with IC50 value of 19.9 ± 3.5 (23; R = N(CH3)2) noted. These data suggest that inhibitor potency is dependent upon both the size (bulkier groups favoured) and electronic properties (electronegative groups favoured) of the cinnamoyl group substituents. The equipotent nature of electron donating 23 with the weakly donating 17 (R = CH3; IC50 = 22.4 ± 3.0 μM) suggests that steric bulk is the primary driver of increased activity. This is consistent with the 4-cinnamoyl substituent interaction with active site hydrophobic residues or via π–π or π-electrostatic. The Br–Cl interchange had little effect on activity (19; R = Br, IC50 = 7.0 ± 0.8 vs.20; R = Cl, 9.2 ± 1.1 μM). It is possible that hydrophobic interactions counterbalance any competing electronic effects or that the observed effects are a combination including or via π–π or π-electrostatic effects, resulting in the observed potency increase. This is consistent with our modelling data (below).
Region 2 modifications were explored by altering the distance and orientation between the two aryl moieties of these compounds (Fig. 1). To this end, reductive amination of 7 in the presence of Na(OAc)3BH with phenylacetaldehyde and 3-phenylpropionaldehyde gave 24 and 25 in good yields (73% and 75%, respectively). The amide analogue of 19 (26) was prepared as per Scheme 2 to evaluate the potential role of the amine moiety.
Scheme 2. Reagents and conditions: i) Na(OAc)3BH, 2-phenylacetaldehyde or 3-phenylpropanal, THF, 25 °C, 18 h; ii) (E)-3-(4-bromophenyl)acryloyl chloride, CH2Cl2, TEA, 0 to 25 °C, 18 h.
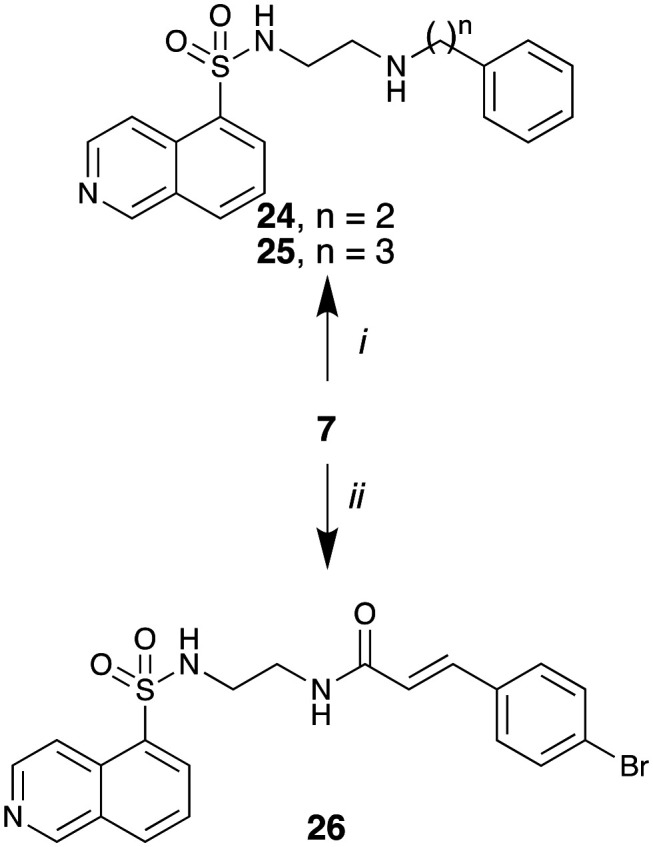
The dynI inhibition data in Table 2 shows that the removal of double bond had little effect on analogue potency (16; IC50 = 38.2 ± 6.0 μM vs.25; IC50 = 30.1 ± 9.7 μM). However, double bond removal concurrent with a one carbon chain length reduction resulted in a 3-fold potency reduction (24; IC50 = 96.8 ± 13.1 μM). While this does not confirm the lack of importance of a double bond, its retention increases compound rigidity, combined with chain length allows appropriate aryl ring localization in the binding pocket. Introduction of an amide (vs. amine) moiety resulted in complete loss of activity (19; IC50 = 7.0 ± 0.8 μM vs.26; IC50 > 300 μM). The amine is, most likely, involved in hydrogen bonding interactions with Ser45, or the conformational restriction arising from the amide prevents optimal positioning of the aromatic ring within the active site.
Inhibition of dynamin I lipid-stimulated GTPase activity (IC50a (μM)) by analogues 16, 19 and 24–26.
| Compound | Structure | Dynamin I IC50a (μM) |
|---|---|---|
| 16 |
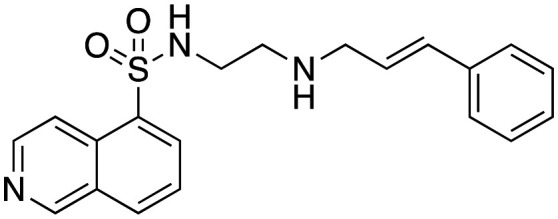
|
38.2 ± 6.0 |
| 19 |
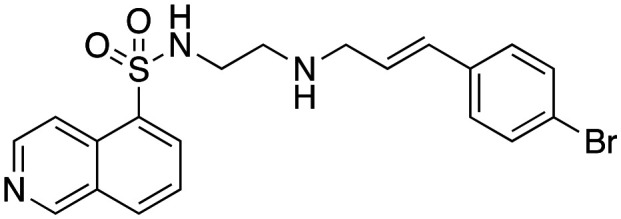
|
7.0 ± 0.8 |
| 24 |
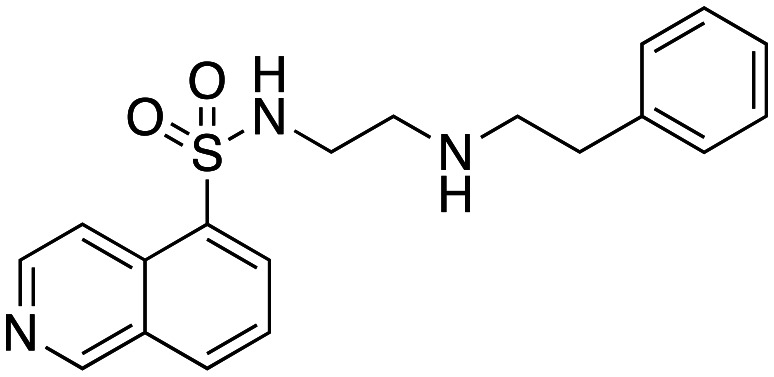
|
96.8 ± 13.1 |
| 25 |
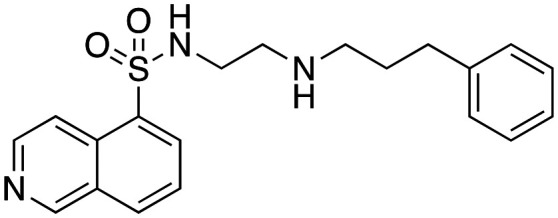
|
30.1 ± 9.7 |
| 26 |
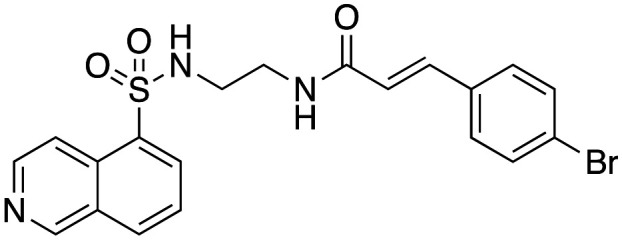
|
>300b |
IC50 and 95% CI of triplicates, n = 1;
Less than 50% inhibition at 300 μM drug concentration.
Given our region 1 findings we developed sub-library of compounds to examine the nature of the diamine linker, region 2 (Fig. 1). The 4-Br 20 was chosen as the lead for this library due to more favourable physicochemical properties and potential ease of further synthetic modifications. This sub-library was synthesized as per Scheme 3 to afford analogues 31–34 in low to moderate yields (11–65%).
Scheme 3. Reagents and conditions: i) SOCl2, DMF (cat.), reflux, 8 h; then diamine, CH2Cl2, 0 to 25 °C, 18 h; ii) Na(OAc)3BH, 12 THF, 25 °C, 18 h.

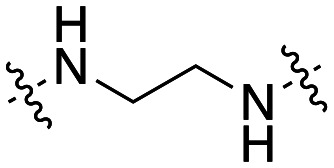
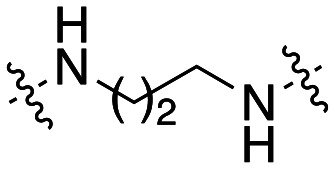
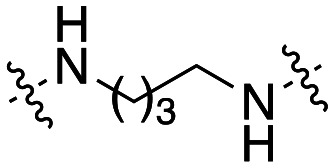
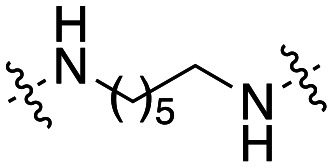
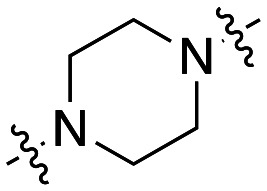
The dynI inhibition data in Table 3 highlights no detriment to dynI activity with ethyl (19; IC50 = 7.0 ± 0.8 μM) to propyl (31; IC50 = 7.1 ± 1.1 μM) to butyl (32; IC50 = 8.1 ± 1.1 μM) to hexyl (33; IC50 = 3.7 ± 1.7 μM) linker modifications. Linker conformational restriction via a piperazine moiety results in a significant decrease in activity (34; IC50 = 58.2 ± 8.6 μM). The similarity in overall size between piperazine 34 and the ethyl linked 19, the effect noted with the amido analogue 26, and the effects noted with analogues 24 and 25, together strongly support both a length and conformational requirement of the diamine linker moiety.
Inhibition of dynamin I lipid-stimulated GTPase activity (IC50a (μM)) by analogues 19, and 31–34.
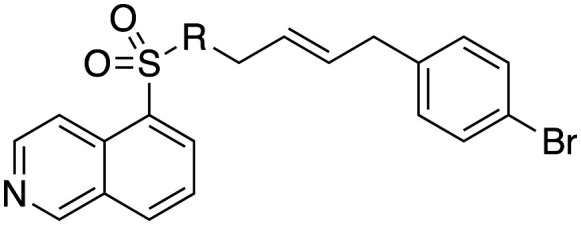
IC50 and 95% CI of triplicates, n = 1;
Mean and SEM of 3 independent experiments each performed in triplicate.
The 5-isoquinoline moiety of region 3 was next examined with 19 as the lead. Treatment of sulfonic acids 3–6 with SOCl2 gave the anticipated sulfonyl chlorides, which on treatment with ethane-1,2-diamine yielded sulfonamides 35–38. Reductive amination with 4-bromocinnamaldehyde and Na(OAc)3BH, gave the target analogues, 39–42, in moderate yields (51–59%) (Scheme 4).
Scheme 4. Reagents and conditions: i) SOCl2, DMF (cat.) reflux; 8 h; ii) ethane-1,2-diamine, CH2Cl2, 0 to 25 °C, 18 h; iii) 11, Na(OAc)3BH, THF, 25 °C, 18 h.

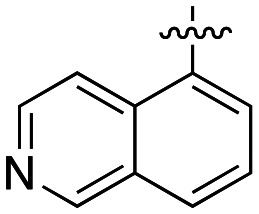
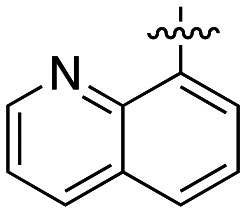
The dynI inhibition data in Table 4 shows that the position of the nitrogen within the aromatic ring had no effect on potency (19; IC50 = 7.0 ± 0.8 μM vs.39; IC50 = 9.5 ± 1.8 μM). However, removal or exocyclic positioning of the nitrogen resulted in a slight increase in potency (40; IC50 = 3.9 ± 0.4 μM, 41; IC50 = 4.4 ± 0.4 μM and 42; IC50 = 3.6 ± 0.7 μM). Analogue 40 differing only in the addition of the 4-bromocinnamyl substituent (relative to lead 1) which resulted in a >10 fold increase in dynamin inhibition activity.
Inhibition of dynamin I lipid—stimulated GTPase activity (IC50a (μM)) by analogues 19 and 39–42.
| ||
|---|---|---|
| Compound | R | Dynamin I IC50a (μM) |
| 19 |
|
7.0 ± 0.8 |
| 39 |
|
9.5 ± 1.8 |
| 40 |
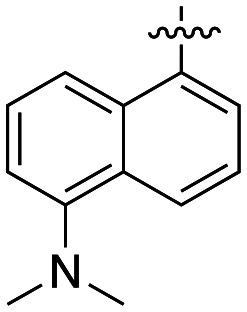
|
3.9 ± 0.4 |
| 41 |
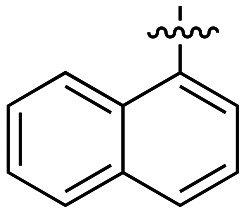
|
4.4 ± 0.4 |
| 42 |
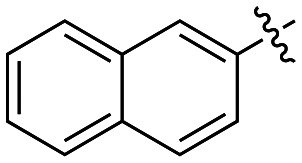
|
3.6 ± 0.7 |
IC50 and 95% CI of triplicates, n = 1.
In our final focused library, we sought to confirm the effects that we had noted in discrete libraries, by the combination of selected features for the analogue series developed thus far. We also chose to examine the role of the sulfonamide and amino NH moieties within the diamine linker. Synthesis of our target analogues was carried out as per Scheme 5 and commenced with the treatment of 1-napthalenesulfonic acid (5) with SOCl2 and the appropriate diamine. The resulting sulfonamides were reductively aminated with 4-bromocinnamaldehyde (1) in the presence of Na(OAc)3BH, to afford the desired compounds 47–50 in low (11%) to excellent (93%) yields. The N-methylated adduct 51 was prepared via the treatment of 42 with formaldehyde and Na(OAc)3BH.
Scheme 5. Reagents and conditions: i) SOCl2, DMF (cat.), reflux, 8 h; ii) diamine, CH2Cl2, 0–25 °C, 18 h; iii) Na(OAc)3BH, aldehyde, THF, 25 °C, 18 h.

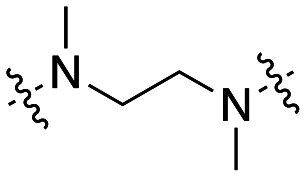
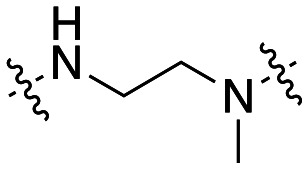
The dynI inhibition data in Table 5 shows that increasing chain length had little effect on the potency of the naphthalene sulfonamide analogues. Elongation of the linker from two to six carbons results in a no significant change in potency (41; IC50 = 4.4 ± 0.4 μM vs.47; IC50 = 3.5 ± 1.3 μM vs.48; IC50 = 3.0 ± 0.6 μM vs.49; IC50 = 5.2 ± 0.7 μM), in agreement with our findings with the equivalent 5-isoquinoline sulfonamide analogues 31–34 (Table 3). Methylation of the amino nitrogen reduced potency 2-fold (50; IC50 = 8.7 ± 1.0 μM) while methylation of both nitrogen atoms had no further effect on activity (51; IC50 = 10.5 ± 1.2 μM). This highlights the delicate balance that linker elongation may have in reaching additional hydrophobic or van der Waals interactions between the naphthalene ring and in facilitating optimal spacing between the two aromatic rings, whilst ensuring the correct placement of other key moieties within the active site. Additionally, these results again suggest that the amine hydrogen is important for inhibition, while the sulfonamide hydrogen does not appear to interact with the active site. The above data suggests that conformational flexibility in the linker may be required for inhibition, as the introduction of a rigid piperazine ring reduced activity 5-fold (19; IC50 = 7.0 ± 0.8 μM vs.34; IC50 = 58.2 ± 8.6 μM).
Inhibition of dynamin I lipid-stimulated GTPase activity (IC50a (μM)) by analogues 41 and 47–51.
| ||
|---|---|---|
| Compound | R | Dynamin I IC50 (μM) |
| 41 |
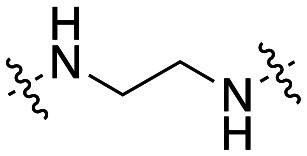
|
4.4 ± 0.4 |
| 47 |
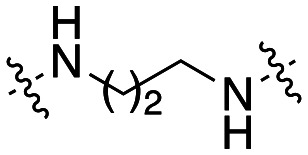
|
3.5 ± 1.3 |
| 48 |
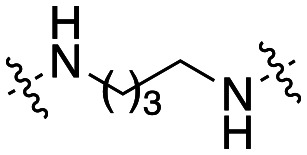
|
3.0 ± 0.6 |
| 49 |
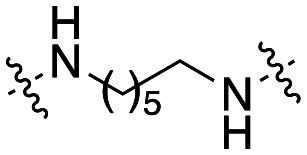
|
5.2 ± 0.7 |
| 50 |
|
8.7 ± 1.0 |
| 51 |
|
10.5 ± 1.2 |
IC50 and 95% confidence interval of triplicates, n = 1.
In-cell inhibition of dynamin reduces CME and SVE
Given that dynamin is essential for endocytosis we next determined the ability of selected sulfonamides (33, 40–42, 47 and 48) to block in-cell CME using our previously established quantitative method for transferrin (Tfn-A594) uptake in human bone osteosarcoma epithelial (U2OS) cells (Table 6).54
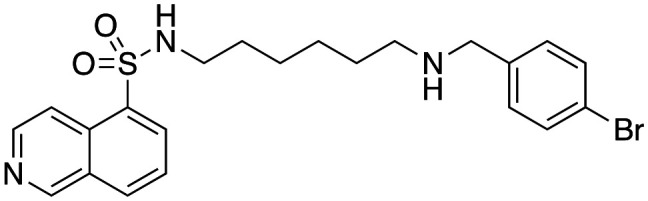
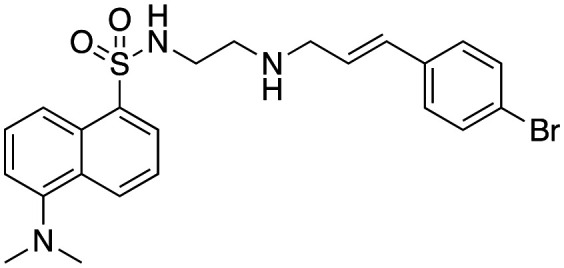
Effect of analogues 33, 40–42, 47 and 48 on purified dyn I lipid-stimulated GTPase activity (IC50a (μM)), in-cell endocytosis of Alexa labelled Tfn in U2OS cells (CME) or depolarisation-stimulated uptake of the styryl dye FM4-64 in synaptosomes (SVE).
| Compound | Structure | Dynamin I IC50a (μM) | CME IC50 (μM) | SVE IC50 (μM) |
|---|---|---|---|---|
| 33 |
|
3.7 ± 1.7 | 15.9 ± 1.6 | 264.9b |
| 40 |
|
3.9 ± 0.4 | 29 ± 4.3 | 126.8b |
| 41 |
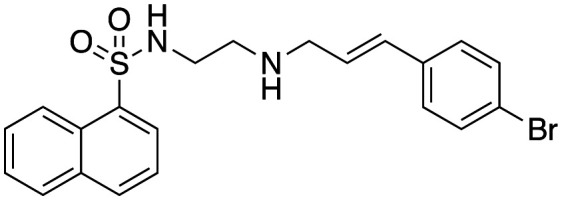
|
4.4 ± 0.4 | 12.4 ± 4 | Not active |
| 42 |
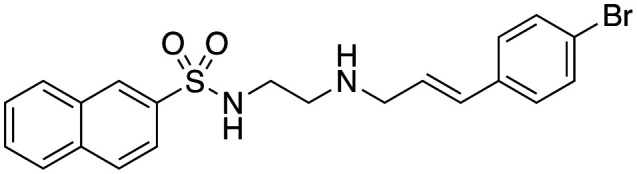
|
3.6 ± 0.7 | 9.3 ± 2.5 | Not active |
| 47 |
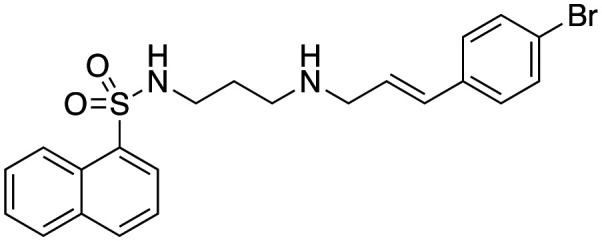
|
3.5 ± 1.3 | 27.3 ± 2.0 | 12.3c |
| 48 |
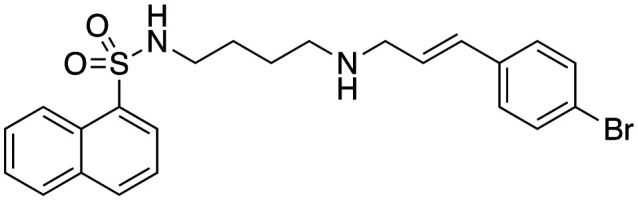
|
3.0 ± 0.6 | 11.3 ± 2.4 | Not active |
| 49 |
|
5.2 ± 0.7 | 27.1 ± 0.8 | Not active |
IC50 and 95% CI of triplicates, n = 1, except
and
. Mean and SEM of 3 and 2 independent experiments, each in triplicate respectively.
All the compounds tested showed significant inhibition of CME (9.3–27.3 μM), which suggests that the mechanism of CME inhibition is through inhibition of dynamin. While all seven compounds had very similar dynI potency, there were discernible differences in CME inhibition. Isoquinoline 33 retained modest CME inhibition (IC50(CME) = 15.9 ± 1.6 μM) as did the dansyl analogue 40. Incorporation of the dimethylamino moiety incurred a two-fold loss in CME inhibition (40; IC50(CME) = 29.0 ± 4.3 μM vs.41; IC50(CME) = 12.4 ± 4.0 μM). The most active inhibitor of CME was the constitutional isomer of 41, the 2-naphthyl sulfonamide 42 (IC50(CME) = 9.3 ± 2.5 μM). Chain elongation was well tolerated in the dynI inhibition assay, however there was no clear trend in CME inhibition between the ethyl-diamine linker of 41 (IC50(CME) = 12.4 ± 4 μM) and the longer propyl (47; IC50(CME) = 27.3 ± 2.0 μM), butyl (48; IC50(CME) = 11.3 ± 2.4 μM) and hexyl (50; IC50(CME) = 27.1 ± 0.8 μM) linkers. Compound 42 is among the more potent inhibitors of CME reported to date, with only the Dyngo®-4a (IC50(CME) 5.7 μM),55,56 Rhodadyn57 (IC50(CME) ~6 μM), Dynole 2-24 (ref. 58) (IC50(CME) ∼1.9 μM) and Iminodyn59 (IC50(CME) ∼11 μM) series of dynamin inhibitors being slightly more active.
SVE inhibition by the Sulfonadyns™ did not show the same level of correlation with the dynI IC50 values and significant inhibitory activity was only noted with sulfonadyn-47. This was unexpected as no major differences in calculated physicochemical characteristics such as clogP or polar surface area (PSA) that might have been expected to play a role in modulation of compound distribution were observed (ESI†). These differences could be associated with the nuances that modify membrane permeability and compound uptake or efflux in biological systems.
Since endocytosis and exocytosis are balanced in nerve terminals, it was possible that the apparent decrease in SVE by 47 was a consequence of reduced exocytosis. Given that SVE is induced to compensate for the loss of SVs through exocytosis, we reasoned that an inhibition of exocytosis could result in a secondary non-specific reduction in SVE. To rule out the possibility of exocytosis inhibition, we next examined whether 47 had any effect on glutamate release, evoked by 4-aminopyridine (4-AP) in a well-characterized assay.60 The presence of 80 μM 47 had no effect on glutamate release (Fig. 3), consistent with 47 blocking endocytosis without direct inhibition of exocytosis.
Fig. 3. Treatment of rat brain synaptosomes with 47 (80 μM) had no effect on Ca2+-dependent exocytosis of glutamate over time, compared to vehicle control (1% DMSO). Data are from n = 3 independent experiments. The curves represent the data mean while vertical lines represent SEM.

Mechanism of dynamin in vitro inhibition
To explore the mechanism of inhibition of the sulfonadyns on dynI, we used enzyme kinetics on two of the series leads. Michaelis–Menten kinetic experiments with sulfonadyns-33 and -47 (3–7 μM) and varying concentrations of the Mg2+·GTP substrate (80–160 mM) were performed to ask if they were competitive inhibitors of dynI with respect to GTP. The Lineweaver–Burk plots for both compounds (Fig. 4 shows data for compound 33) reveals that both are GTP-competitive inhibitors of dynI, acting at the active site in the GTPase domain of dynamin.
Fig. 4. Kinetics of sulfonadyn-33 with respect to GTP. Double-reciprocal plots of various sulfonadyn-33 concentrations at varying substrate (GTP at 80–160 μM, S) and reaction velocity (V). The concentrations were 3 (▼) 5 (◆), 6 (■), and 7 (●) μM sulfonadyn-33. Error bars are the mean ± SEM of three independent experiments each conducted in triplicate. Kinetic constants for GTP were Km 20.8 ± 3.8 μM and Vmax 590 ± 67 nmol mg−1 min−1.

Modelling analysis of selected compounds
We next examined the docking poses of selected sulfonadyn analogues, 1, 19, 41 and 47 in the dynamin I GTP binding site (Fig. 5). The terminal NH2 moiety of 1 interacts with Ser-41 and the Mg2+ within the active site, with additional predicted interactions with the diamine side chain and Gly50, and arene–H interactions between the naphthalene moiety and Lys205 (Fig. 5A and B). Structure modification to isoquinoline 19 saw additional interactions between the quinoline-N and Asn236 and an aromatic interaction with Asp208. The sulfonamide NH engages with Arg237, one of the S O engages with Lys-06, with the terminal 4-BrPh moiety engaging in an aromatic interaction with Ser64 (Fig. 5C and D). Removal of the quinoline-N with 41 results in a loss of the Asn235 interaction, retention of the Lys-206 and Arg-237 interactions with an additional H-bonding interaction from the ethylamine NH to Gly60. With the final analogue examined, sulfonadyn-47, the key interactions were predicted to be a naphthyl-Lys206 and 4-BrPh to Lys44 aromatic engagements. In all instances, no major change in binding orientation were observed and the nature of the binding poses were consistent with the general trend in observed dynamin inhibition activity noted herein.
Fig. 5. Docked poses and molecular interaction descriptors as determined on docking of selected sulfonadyn analogues into the GTPase domain of dynI (pdb: 3ZYC) using the Molecular Operating Environment (MOE) software. A and B, dansylcadaverine 1; C and D, sulfonadyn-19; E and F, sulfonadyn-41; and G and H, sulfonadyn-47.

Anti-seizure effects of sulfonadyn-47
Finally we tested the effect of these inhibitors on seizures after i.p. administration of the compounds in mice. We employed the 6 Hz psychomotor seizure test in mice to assess sulfonadyn-47. Seizures are provoked in otherwise healthy mice by electrical stimulation of the cornea.61 We first tested the widely clinically used anti-seizure medication, sodium valproate, demonstrating that at a dose of 400 mg kg−1 ip, the current required to provoke seizures in 50% of mice (the EC50 value) was significantly higher compared to vehicle treated mice (p < 0.0001; Fig. 6A). We then examined sulfonadyn-47 in this model, and observed it significantly increased seizure threshold at doses of 30 (p < 0.01) and 60 mg kg−1 (p < 0.05), with the threshold for the 60 mg kg−1 dose being equivalent to that to induce seizures in 50% of mice given 400 mg kg−1 of valproate. The lower dose of 47, i.e. 10 mg kg−1, was without effect (p = 0.9), indicating a dose-dependent antiseizure effect of the dynamin inhibitor (Fig. 6B–D).
Fig. 6. A. Significant anticonvulsant effects were observed in the 6 Hz acute mouse model of seizures by valproate treatment (400 mg kg−1) as evidenced by increased EC50 value (indicative of the amount of current required to induce a seizure in 50% of mice), compared to vehicle. Sulfonadyn-47 showed dose-dependent anticonvulsant activity. B. 10 mg kg−1 was ineffective. C and D. 30 and 60 mg kg−1 significantly inhibited seizures. Four different cohorts of mice (n = 30/cohort) were used for these experiments. Data represent group mean ± SEM. *p < 0.05; **p < 0.01; ****p < 0.0001 indicate significant difference between the treatment groups.

Conclusions
We show that from a previously known compound with weak CME activity we were able to design a series of potent GTP-competitive dynamin inhibitors with anti-seizure activity in mice. Dansylcadaverine (1) is a widely-used, low-potency inhibitor of CME in multiple cell types. At 200 μM the compound blocked vesicular stomatitis virus uptake in cells, consistent with our CME IC50 determination.59 Similarly, 1 inhibits the internalization of alpha2 adrenergic and dopamine D2 receptors, but not in cells lacking clathrin heavy chain, also at similar potency to our study with transferrin uptake.62 This broad body of data is consistent with our observation that 1 inhibited transferrin uptake via dynamin-dependent CME. One known molecular target for 1 is the family of transglutaminases which are involved in protein cross-linking.63 Here we identify a second dansylcadaverine target as dynamin GTPase activity. Its weak dynamin potency is consistent with the possibility that its weak inhibition of CME may be explained by targeting dynamin. This action mechanism is consistent with previous suggestions that its CME inhibitory activity could be attributed to the stabilization of clathrin-coated pits.64
Through the design, synthesis and subsequent screening of a series of small-targeted compound libraries, based on the lead compound dansylcadaverine (1),40,65,66 we have developed a novel class of dynamin inhibitors and generated a preliminary SAR profile for this class. We demonstrated that the Sulfonadyns™ are a new class of GTP-competitive dynamin GTPase inhibitors, joining the pthaladyns,67 quinodyns68 and naphthaladyns69 as the only compounds targeting the active site, rather than allosteric sites on dynamin. In our initial library, region 1 was probed through exploration of the binding role of the cinnamyl moiety. By altering this substituent, we discovered that electron withdrawing groups and bulky substituents were optimal for activity. Removal of the double bond showed no decrease in dynI inhibition, however the incorporation of an amide was detrimental to activity. We are currently investigating the effects of different substituent positions, multiple aryl substituents and the introduction of different ring structures. Replacement of the amine with an amide was detrimental to compound potency suggesting that the nitrogen lone pair is involved in a crucial interaction with the active site.
Exploration of region 2 revealed the importance of diamine linker length and flexibility. Linker elongation increased dynamin potency 1.8-fold for the isoquinoline series (19; IC50 = 7.0 ± 0.8 μM vs.33; IC50 = 3.7 ± 1.7 μM), however our naphthalene analogues (Table 5) were unaffected by this alteration. Rigidification of region 2 in the form of a piperazine linker caused an 8-fold decrease in dynI inhibition (19; IC50 = 7.0 ± 0.8 μM vs.34; IC50 = 58.2 ± 8.6 μM). Amine methylation was shown to reduce potency and methylation of both nitrogen atoms had no further effect on potency (50; IC50 = 8.7 ± 1.0 μM vs.51; IC50 = 10.5 ± 1.2 μM). This suggests that the amine hydrogen is involved in mediating active site binding, however the sulfonamide hydrogen does not interact with the binding site. Thus, further sulfonamide substitution may allow the exploration of additional chemical space within the binding pocket.
Region 3 was probed to establish the importance of the dansyl moiety of lead 1. Positioning of the nitrogen within the aromatic ring led to decreased activity (19; IC50 = 7.0 ± 0.8 μM, 39; IC50 = 9.5 ± 1.8 μM) relative to the dansyl substituent (40; IC50 = 3.9 ± 0.4 μM). Complete removal of the nitrogen resulted in improved activity in the naphthyl analogues (41 and 42), with the 2-naphthalenesulfonamide 42 resulting in the most active modification to region 3 (42; IC50 = 3.6 ± 0.7 μM).
With a preliminary SAR established, the most potent dynI inhibitors were screened for in-cell CME inhibition. All compounds that were tested showed good CME inhibition, with 41 (IC50(CME) = 12.4 ± 4.0 μM), 42 (IC50(CME) = 9.3 ± 2.5 μM) and 48 (IC50(CME) = 11.3 ± 2.4 μM) proving to be the most active inhibitors. Sulfonadyn-33 and -47 were shown to be competitive inhibitors of dynamin with respect to GTP, revealing that this class of dynamin inhibitors targets the active site of dynamin. Enzyme kinetics support the sulfonadyns inhibiting dynamin activity through binding at dynamin's GTP binding site. This, and the observed SAR, data was supported by a molecular docking study which revealed key interactions between the motifs in the sulfonamides and Asn236 (quinoline N), Asp208 (aromatic interaction), Arg237 (sulfonamide NH), Lys206 (sulfonamide S O) and Ser64 (4-BrPh aromatic interaction). The Sulfonadyn™ compound class represents the most drug-like of the dynamin inhibitor series to date and act by a distinct mechanism to Dyngo®56 or dynasore inhibitors, which instead target unidentified allosteric sites that affect dynamin helical assembly.28,56
In seizure models and in brain tissue extracted from epilepsy patients, elevated levels of dynI have been noted,26 suggesting that overexpression may be involved in the pathogenesis of seizures and thus epilepsy. In addition, mutations in dynI cause epileptic encephalopathy in humans18,70 and also mice,71 and dynamin dephosphorylation state has been linked to neuronal seizures.15,27 Examination of the most SVE active analogue, sulfonadyn-47, in the 6 Hz psychomotor model of focal seizures in mice revealed efficacy in raising the threshold current required to elicit seizures, with significant anti-seizure activity noted at 30 mg kg−1 which compares favourably with the clinically used valproate (400 mg kg−1). Importantly, no dose limiting toxicity was observed for these compounds, even though the parent dansylcadaverine (1) is cytotoxic at about 300 μM.62 The compounds described herein display enhanced potency, drug character, relative to the lead dansylcadaverine, and are functionally active in animal models of epilepsy. Thus, the sulfonadyns represent a promising class of new dynI inhibitors with clinical potential for the future treatment of epilepsy.
Experimental
Biology materials
Phosphatidylserine (PS), phenylmethylsulfonylfluoride (PMSF), Tween 80, fibronectin and DAPI were from Sigma-Aldrich (St Louis, CA). GTP was from Roche Applied Science (Germany), leupeptin was from Bachem (Bubendorf, Switzerland). Gel electrophoresis reagents, equipment and protein molecular weight markers were from Bio-Rad (Hercules, CA). Paraformaldehyde (PFA) was from Merck Pty Ltd (Kilsyth, Australia). Penicillin/streptomycin, phosphate buffered salts, foetal bovine serum (FBS) and Dulbecco's modified Eagle's media (DMEM) were from Invitrogen (Mount Waverley, Victoria, Australia). Alexa-594 conjugated Tfn (Tfn-A594) was from Molecular Probes (Oregon, USA). All other reagents were of analytical reagent grade or better.
Compounds
Small molecule compounds were synthesized in-house and made up as 30 mM stock solutions in 100% DMSO and diluted in 50% v/v DMSO/20 mM Tris/HCl pH 7.4 or cell media prior to further dilution in the assay. The final DMSO concentration in the GTPase or endocytosis assays was at most 3.3% or 1% respectively. The GTPase assay for dynamin I was unaffected by DMSO up to 3.3%. Stocks were stored at −20 °C for several months.
Protein production
Native dynamin I was purified from sheep brain by extraction from the peripheral membrane fraction of whole brain72 and affinity purification on GST-Amph2-SH3-sepharose as described,73 yielding 8–10 mg protein from 250 g sheep brain.
Malachite green GTPase assay
The malachite green method was used for the sensitive colorimetric detection of orthophosphate (Pi) release from GTP as previously described.56,67 Dynamin I activity was stimulated by sonicated phosphatidylserine (PS) liposomes. Data analysis and enzyme kinetics using non-linear regression was performed using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA). IC50 values are determined as described by the software program manufacturer using the model: Y = bottom + (top − bottom)/(1 + 10((log IC50 − X) × hillslope)).54,74,75 The curves were generated using the Michaelis–Menten equation v = Vmax[S]/(Km + [S]) where S = PS activator or GTP substrate. After the Vmax and Km values were determined, the data was transformed using the Lineweaver–Burk equation 1/v = 1/Vmax + (Km/Vmax) × (1/[S]).
CME assay
Human bone osteosarcoma epithelial (U2OS) cells were cultured in DMEM supplemented with 10% FBS at 37 °C and in 5% CO2 in a humidified incubator. Tfn uptake was analyzed as previously described.54 Briefly, cells were grown in fibronectin-coated (5 μg mL−1) 96-well plates. The cells were serum-starved overnight (16 h) in DMEM minus FBS. Cells were then incubated with the sulfonamide (usually at 6 concentrations per drug from 1 to 300 μM) or vehicle for 30 min prior to addition of 4 μg mL−1 Tfn-A594 for 8 min at 37 °C. Cell surface-bound Tfn was removed by incubating the cells in an ice-cold acid wash solution (0.2 M acetic acid + 0.5 M NaCl, pH 2.8) for 10 min and washed with ice-cold PBS for 5 min. Cells were immediately fixed with 4% PFA for 10 min at room temperature. Nuclei were stained using DAPI. Quantitative analysis of the inhibition of Tfn-A594 endocytosis in U2OS cells was performed on large numbers of cells by an automated acquisition and analysis system (Image Xpress Micro, Molecular Devices, Sunnyvale, CA). Nine images were collected from each well, averaging 40–50 cells per image. The average integrated intensity of Tfn-A594 signal per cell was calculated for each well using IXM system, and the data expressed as a percentage of control cells (vehicle treated). The average number of cells for each data point was ∼1200. IC50 values were calculated using Graphpad Prism v7 and data was expressed as mean ±95% confidence interval (CI) for 3 wells and ∼1200 cells.
Synaptic vesicle endocytosis and exocytosis in synaptosomes
Highly purified synaptosomes were prepared from the cerebrum of adult male Sprague–Dawley rats.76 The animals (6–8 weeks old, 250 g body weight) were euthanised by cervical dislocation with approval from the Animal Care and Ethics Committee for the Children's Medical Research Institute, Sydney, Australia (approval number C116). Synaptosomes were attached to 96 well glass-bottom plates by low-speed centrifugation. Synaptosomes were maintained at 30 °C in modified Kreb's buffer (143 mM NaCl, 4.7 mM KCl, 19 mM HEPES, 1.2 mM MgSO4, 0.1 mM CaCl2 pH 7.4). Once attached, synaptosomes were incubated in buffer containing either DMSO alone (control) or drug dissolved in DMSO. DMSO was kept at a concentration of 1% in all samples. Compound exposure was carried out for 30 min. The synaptosomes were then exposed to 1 μM FM 4-64 in the presence of compound for 2 min. Synaptosomes were then depolarised in 80 mM KCl for a further 2 min to induce exocytosis/endocytosis, thereby facilitating uptake of FM 4-64. After depolarisation, synaptosomes were returned to standard Hepes-buffered Kreb's buffer. Fluorescent images of synaptosomes were acquired using an ImageXpress Micro system with a 20× air objective at excitation 476–524 nm and 608–742 nm emission. Images were analysed using MetaXpress software and fluorescence intensity values were normalised to control uptake of dye (set at 1.0).77
Glutamate exocytosis was performed using enzyme-linked fluorescent detection of released glutamate.60 In briefly, the synaptosomes (0.6 mg in 2 ml) were resuspended in calcium-containing (1.2 mM CaCl2) or calcium-free (1 mM EGTA) Krebs'-like buffer (in mM: 118.5 NaCl, 4.7 KCl, 1.18 MgCl2, 0.1 Na2HPO4, 20 HEPES, and 10 glucose, pH 7.4) at 37 °C. Experiments were started after addition of 1 mM NADP and, after 1 min further, 50 U of glutamate dehydrogenase was added and the synaptosomes stimulated after 4 min with 4-aminopyridine (4-AP). Increases in fluorescence caused by production of NADPH were monitored in a Perkin-Elmer (Emeryville, CA) LS-50B spectrofluorimeter at 340 nm excitation and 460 nm emission. Standard curves were produced by addition of 4 nmol of glutamate. Data are presented as Ca2+-dependent glutamate release, being the difference between release in presence or absence of Ca2+.
Animal model of seizure
To test the influence of sulfonadyn-47 on seizures, we used the 6 Hz psychomotor seizure model.61 Adult male c57Bl/6 mice (n = 120) purchased from the Australian Resource Centre, Perth, Australia. All procedures were approved by the University of Melbourne Animal Ethics Committee, and were conducted in the Department of Medicine (RMH), University of Melbourne. Corneal stimulations were administered to mice using an ECT Unit from Ugo Basile, Varese, Italy (6 Hz, 0.2 ms rectangular pulse for 3 s using varying current amplitude) as previously described.78 The protocol was designed to determine the current intensity required to elicit seizures in 50% of mice (i.e. the EC50 current). Animals were manually restrained and released immediately following the stimulation and observed for the presence or absence of seizure, which were characterized by a stunned or fixed posture, rearing, forelimb clonus or twitching. The experiments compared anti-seizure efficacy of different doses of sulfonadyn-47 (10, 30 or 60 mg kg−1 ip), or sodium valproate (400 mg kg−1 ip). The vehicle group for sulfonadyn-47 was treated with 20% dimethylacetate (DMA), 5% Tween-80 and 75% PEG300, whereas for sodium valproate (purchased from Sigma), the vehicle was 0.9% saline. Injection volumes were 0.1 mL/10 g. Drugs were injected 30 min before stimulation, and each mouse was used only once. To determine the EC50 current, varying intensities of stimulation were delivered to different mice using the ‘up and down’ method using 2 mA steps.78 Data were analyzed using Mann–Whitney U-test for each dose of drug compared to its vehicle control.
Molecular docking
Molecular modelling studies were performed using the docking engine of Molecular Operating Environment (MOE) software, using the crystal structure of dyn1 GTPase domain bound to GMPPCP ligand (PDB code: 3ZYC).79 Docking was performed using MOE's default setting using the “Triangle Matcher” as the placement method in combination with the “London dG” scoring function for the initial placement of the ligand, followed by a refinement of the top 30 poses with rigid receptor setting and the “GBVI/WSA” scoring function. Poses for each compound were relaxed in the binding pocket using LigX energy minimisation. Analysis and visualization of the docking output were performed in MOE.69,80,81
Chemistry
General methods
THF was freshly distilled from sodium–benzophenone. Flash chromatography was carried out using silica gel 200–400 mesh (60 Å). Thin layer chromatography (TLC) was performed on Merck silica gel 60 F254 precoated aluminium plates with a thickness of 0.2 mm. Column chromatography was performed under “flash” conditions on Merck silica gel 60 (230–400 mesh). All NMR spectra were recorded using a Bruker Avance 300 (1H NMR at 300 MHz and 13C NMR at 75 MHz) or Bruker Ascend 400 (1H NMR at 400 MHz and 13C NMR at 100 MHz) spectrometer in CDCl3 or CD3OD. Chemical shifts (δ) are reported in parts per million (ppm) measured relative to the internal standards, and coupling constants (J) are expressed in hertz (Hz). Melting points were recorded on a Stuart Scientific melting point apparatus (UK) and are uncorrected. GCMS was performed using a Shimadzu GCMS-QP2100. The instrument uses a quadrupole mass spectrometer and detects samples via electron impact ionization (EI). Compound purity was confirmed by a combination of LC-MS (HPLC) and NMR analysis. All analogues are ≥95% purity.
General procedure 1 – sulfonyl chloride generation and sulfonamide synthesis
The requisite sulfonic acid (10 mmol) was treated with SOCl2 (150 mmol) and 2 drops of DMF. The reaction mixed was heated at reflux for 8 h, cooled and the excess SOCl2 removed in vacuo. The crude sulfonyl chloride was taken up in dry CH2Cl2 (50 mL) and added dropwise to a cooled (0 °C) solution of the diamine (50 mmol) in CH2Cl2 (100 mL). The reaction mixture was stirred at 25 °C for 18 h and 10% NaHCO3 added (50 mL). The organic layer was separated, washed with brine (4 × 50 mL), dried (MgSO4) and concentrated in vacuo to afford the desired amino sulfonamide analogue.
General procedure 2 – synthesis of cinnamaldehydes
To a solution of the requisite aldehyde (10.8 mmol) in EtOH (20 mL) was added dropwise a solution of ylid (Ph3P CHCO2Et) (13.5 mmol) in EtOH (10 mL). The reaction mixture was stirred at 25 °C for 18 h and the solvent removed in vacuo. The residue was suspended in petroleum spirit (50 mL), filtered and evaporated to yield a mixture of E/Z isomers. The desired E isomer was isolated by flash column chromatography eluting with petroleum spirit/EtOAc (100 : 5).
The ethyl cinnamate analogue (15.66 mmol) was taken up in dry toluene (100 mL) and cooled to −78 °C under nitrogen. DIBAl-H (1 M in toluene, 34.5 mL, 34.5 mmol) was added dropwise and the reaction mixture stirred at −78 °C for 1 h before being quenched by the addition of 1 M HCl (70 mL). The resulting mixture was warmed to room temperature and ethyl acetate (100 mL) added. The organic layer was washed with 1 M HCl (2 × 50 mL) and brine (2 × 50 mL), dried (MgSO4) and the solvent removed in vacuo to afford the corresponding crude alcohol that was used directly in the next step.
The alcohol (7 mmol) was taken up in CH2Cl2 (100 mL) and treated with activated MnO2 (56 mmol). The reaction mixture was stirred at 25 °C for 18 h, filtered and the solvent removed in vacuo. The residue was purified by flash column chromatography, eluting with petroleum spirit/EtOAc (100 : 5) to afford the desired cinnamaldehyde.
General procedure 3 – reductive amination
To a stirred solution of the requisite amine (0.59 mmol) and aldehyde (0.44 mmol) in THF (20 mL) was added Na(OAc)3BH (1.29 mmol). The reaction mixture was stirred at 25 °C for 18 h, quenched with 1 M NaOH (5 mL) and EtOAc added (40 mL). The organic layer was separated, washed with 10% NaHCO3 (25 mL) and brine (25 mL), dried (MgSO4) and the solvent removed in vacuo. The residue was purified by flash column chromatography eluting with EtOAc followed by EtOAc/MeOH/TEA (20 : 1 : 1) to give the desired amines.
N-(2-Aminoethyl)-5-isoquinolinesulfonamide (7)46
Compound 7 was synthesized using general procedure 1 from isoquinoline-5-sulfonic acid (2) and ethane-1,2-diamine to afford the title compound (1.17 g, 47%) as an orange oil.
1H NMR (300 MHz, CDCl3): δ 9.34 (s, 1H), 8.60 (d, J = 6.1 Hz, 1H), 8.51 (d, J = 6.1 Hz, 1H), 8.42 (d, J = 7.3 Hz, 1H), 8.18 (d, J = 8.1 Hz, 1H), 7.67 (t, J = 7.8 Hz, 1H), 3.02 (t, J = 5.7 Hz, 2H), 2.77 (t, J = 5.7 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ 152.9, 144.4, 134.5, 133.1, 132.7, 130.9, 128.7, 125.8, 117.2, 45.3, 41.0.
3-(4-Methylphenyl)-(2E)-propenal (9)50
Compound 9 was synthesized using general procedure 2 from 4-methylbenzaldehyde to afford 9 (471 mg, 83% over 3 steps) as a pale yellow oil.
1H NMR (300 MHz, CDCl3): δ 9.71 (d, J = 7.7 Hz, 1H), 7.50–7.43 (m, 3H), 7.26 (d, J = 8.1 Hz, 2H), 6.70 (dd, J = 15.9, 7.7 Hz, 1H), 2.42 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 193.7, 152.9, 142.0, 131.4, 129.9, 128.6, 127.7, 21.6.
3-(4-tert-Butylphenyl)-(2E)-propenal (10)82
Compound 10 was synthesized using general procedure 2 from 4-tert-butylbenzaldehyde to afford 10 (498 mg, 60% over 3 steps) as an off-white solid, mp 52–54 °C (lit. 57–59 °C).82
1H NMR (300 MHz, CDCl3): δ 9.60 (d, J = 7.7 Hz, 1H), 7.44–7.34 (m, 5H), 6.61 (dd, J = 15.9, 7.7 Hz, 1H), 1.25 (s, 9H); 13C NMR (75 MHz, CDCl3): δ 193.7, 155.1, 152.7, 131.4, 128.4, 127.9, 126.1, 35.0, 31.1.
3-(4-Bromophenyl)-(2E)-propenal (11)83
Compound 11 was synthesized using general procedure 2 from 4-bromobenzaldehyde to afford 11 (1.28 g, 53% over 3 steps) as a yellow solid, mp 74–76 °C (lit. 78–79 °C).83
1H NMR (400 MHz, CDCl3): δ 9.66 (d, J = 7.6 Hz, 1H), 7.51 (d, J = 8.4 Hz, 2H), 7.43–7.34 (m, 3H), 6.65 (dd, J = 16.0, 7.6 Hz, 1H); 13C NMR (101 MHz, CDCl3): δ 193.3, 151.0, 132.9, 132.3, 129.8, 129.0, 125.6.
3-(4-Chlorophenyl)-(2E)-propenal (12)84
Compound 12 was synthesized using general procedure 2 from 4-chlorobenzaldehyde to afford 12 (1.12 g, 65% over 3 steps) a light yellow solid, mp 66–68 °C (lit. 62–63 °C).84
1H NMR (300 MHz, CDCl3): δ 9.70 (d, J = 7.6 Hz, 1H), 7.52–7.37 (m, 5H), 6.68 (dd, J = 16.0, 7.6 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ 193.4, 151.1, 137.2, 132.5, 129.7, 129.4, 129.0.
3-(4-Fluorophenyl)-(2E)-propenal (13)49
Compound 13 was synthesized using general procedure 2 from 4-fluorobenzaldehyde to afford 13 (547 mg, 57% over 3 steps) as a pale yellow oil.
1H NMR (300 MHz, CDCl3): δ 9.70 (d, J = 7.6 Hz, 1H), 7.62–7.56 (m, 2H), 7.47 (d, J = 16.0 Hz, 1H), 7.13 (t, J = 8.7 Hz, 2H), 6.66 (dd, J = 16.0, 7.6 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ 193.2, 164.2 (d, 1JCF = 252.8 Hz), 151.1, 130.4 (d, 3JCF = 8.7 Hz), 130.3 (d, 4JCF = 3.6 Hz), 128.1 (d, 5JCF = 2.3 Hz), 116.1 (d, 2JCF = 22.0 Hz).
3-(4-Trifluoromethylphenyl)-(2E)-propenal (14)53
Compound 14 was synthesized using general procedure 2 from 4-trifluoromethylbenzaldehyde to afford 14 (459 mg, 59% over 3 steps) as a pale yellow oil.
1H NMR (300 MHz, CDCl3): δ 9.75 (d, J = 7.5 Hz, 1H), 7.70–7.63 (m, 4H), 7.51 (d, J = 16.1 Hz, 1H), 6.77 (dd, J = 16.0, 7.5 Hz, 1H); 13C NMR (75 MHz, CDCl3): δ 193.1, 150.2, 137.4 (q, 4JCF = 1.2 Hz), 132.4 (q, 2JCF = 32.6 Hz), 130.5, 128.6, 125.9 (q, 3JCF = 3.8 Hz), 123.7 (q, 1JCF = 272.3 Hz).
3-(4-Dimethylaminophenyl)-(2E)-propenal (15)85
Compound 15 was synthesized using general procedure 2 from 4-N,N-dimethylaminobenzaldehyde. However, the DIBAl-H reduction was quenched by the addition of EtOH (10 mL) rather than 1 M HCl, to afforded 15 (1.27 g, 69% over 3 steps) as a reddish solid, mp 134–136 °C (lit. 140–141 °C).85
1H NMR (300 MHz, CDCl3): δ 9.59 (d, J = 7.9 Hz, 1H), 7.45 (d, J = 8.7 Hz, 2H), 7.37 (d, J = 15.6 Hz, 1H), 6.68 (d, J = 9.0 Hz, 2H), 6.54 (dd, J = 15.6, 7.9 Hz, 1H), 3.05 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 193.8, 154.0, 152.5, 130.6, 123.9, 121.9, 111.9, 40.1.
(E)-N-{2-[(3-Phenyl-2-propen-1-yl)amino]ethyl}-5-isoquinolinesulfonamide (16)86
Compound 16 was synthesized using general procedure 3 from amine 7 and commercially available cinnamaldehyde (8) to afford 17 (870 mg, 60%) as a light yellow oil.
1H NMR (400 MHz, CDCl3): δ 9.34 (s, 1H), 8.70 (d, J = 6.1 Hz, 1H), 8.50–8.38 (m, 2H), 8.18 (d, J = 8.2 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.36–7.24 (m, 5H), 6.37 (d, J = 15.9 Hz, 1H), 6.04 (dt, J = 15.8, 6.3 Hz, 1H), 3.16 (d, J = 5.6 Hz, 2H), 3.04–2.96 (m, 2H), 2.72–2.64 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 153.5, 145.5, 136.8, 134.3, 133.7, 133.5, 132.1, 131.4, 129.2, 128.8, 127.8, 127.2, 126.4, 126.0, 117.3, 51.0, 47.1, 42.4.
(E)-N-(2-{[3-(4-Methylphenyl)-2-propen-1-yl]amino}ethyl)-5-isoquinolinesulfonamide (17)
Compound 17 was synthesized using general procedure 3 from amine 7 and cinnamaldehyde 9 to afford 17 (680 mg, 72%) as a light yellow solid, mp 114–116 °C.
1H NMR (300 MHz, CDCl3): δ 9.30 (s, 1H), 8.63 (d, J = 6.1 Hz, 1H), 8.47–8.40 (m, 2H), 8.13 (d, J = 8.2 Hz, 1H), 7.66–7.60 (m, 1H), 7.16 (d, J = 8.1 Hz, 2H), 7.07 (d, J = 7.9 Hz, 2H), 6.35 (d, J = 15.9 Hz, 1H), 6.00 (dt, J = 15.8, 6.5 Hz, 1H), 3.23 (d, J = 6.5 Hz, 2H), 3.09–3.03 (m, 2H), 2.74 (t, J = 5.6 Hz, 2H), 2.30 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 153.3, 145.2, 137.7, 134.5, 133.7, 133.6, 133.3, 133.0, 131.4, 129.4, 129.1, 126.4, 126.0, 124.7, 117.4, 50.8, 47.3, 42.0, 21.3.
(E)-N-(2-{[3-(4-tert-Butylphenyl)-2-propen-1-yl]amino}ethyl)-5-isoquinolinesulfonamide (18)
Compound 18 was synthesized using general procedure 3 from amine 7 and cinnamaldehyde 10 to afford 18 (683 mg, 72%) as a light yellow solid, mp 52–54 °C.
1H NMR (300 MHz, CDCl3): δ 9.31 (s, 1H), 8.65 (d, J = 6.1 Hz, 1H), 8.48–8.41 (m, 2H), 8.15–8.11 (m, 1H), 7.64 (dd, J = 8.1, 7.4 Hz, 1H), 7.31 (d, J = 8.5 Hz, 2H), 7.22 (d, J = 8.4 Hz, 2H), 6.38 (d, J = 15.9 Hz, 1H), 6.02 (dt, J = 15.8, 6.5 Hz, 1H), 3.23 (dd, J = 6.5, 1.1 Hz, 2H), 3.08–3.03 (m, 2H), 2.77–2.71 (m, 2H), 1.30 (s, 9H); 13C NMR (75 MHz, CDCl3): δ 153.4, 151.0, 145.3, 134.6, 133.8, 133.6, 133.3, 132.8, 131.4, 129.1, 126.2, 126.0, 125.6, 125.1, 117.4, 50.8, 47.2, 42.1, 34.7, 31.4.
(E)-N-(2-{[3-(4-Bromophenyl)-2-propen-1-yl]amino}ethyl)-5-isoquinolinesulfonamide (19)
Compound 19 was synthesized using general procedure 3 from amine 7 and cinnamaldehyde 11 to afford 19 (124 mg, 63%) as a light yellow oil.
1H NMR (300 MHz, CDCl3): δ 9.28 (s, 1H), 8.60 (d, J = 6.1 Hz, 1H), 8.46–8.38 (m, 2H), 8.12 (d, J = 8.2 Hz, 1H), 7.62 (dd, J = 8.2, 7.4 Hz, 1H), 7.34 (d, J = 8.5 Hz, 2H), 7.09 (d, J = 8.5 Hz, 2H), 6.27 (d, J = 15.9 Hz, 1H), 6.01 (dt, J = 15.9, 6.3 Hz, 1H), 3.16 (dd, J = 6.3, 1.1 Hz, 2H), 3.06–3.00 (m, 2H), 2.75–2.62 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 153.3, 145.0, 135.5, 134.3, 133.6, 133.3, 131.7, 131.2, 131.0, 129.0, 127.9, 127.5, 126.0, 121.4, 117.4, 50.7, 47.4, 42.2.
(E)-N-(2-{[3-(4-Chlorophenyl)-2-propen-1-yl]amino}ethyl)-5-isoquinolinesulfonamide (20)86
Compound 20 was synthesized using general procedure 3 from amine 7 and cinnamaldehyde 12 to afford 20 (764 mg, 64%) as a light yellow oil.
1H NMR (300 MHz, CDCl3): δ 9.30 (s, 1H), 8.64 (d, J = 6.1 Hz, 1H), 8.49–8.39 (m, 2H), 8.14 (d, J = 8.2 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.24–7.16 (m, 4H), 6.33 (d, J = 15.9 Hz, 1H), 6.03 (dt, J = 15.8, 6.4 Hz, 1H), 3.22 (d, J = 6.3 Hz, 2H), 3.08–3.01 (m, 2H), 2.80–2.65 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 153.4, 145.2, 135.1, 134.3, 133.7, 133.4, 131.5, 131.3, 129.1, 128.8, 127.6, 126.8, 126.0, 117.4, 50.7, 47.4, 42.1.
(E)-N-(2-{[3-(4-Fluorophenyl)-2-propen-1-yl]amino}ethyl)-5-isoquinolinesulfonamide (21)86
Compound 21 was synthesized using general procedure 3 from amine 7 and cinnamaldehyde 13 to afford 21 (624 mg, 62%) a light yellow oil.
1H NMR (300 MHz, CDCl3): δ 9.29 (s, 1H), 8.63 (d, J = 6.1 Hz, 1H), 8.47–8.39 (m, 2H), 8.13 (d, J = 8.2 Hz, 1H), 7.67–7.61 (m, 1H), 7.22 (m, 2H), 6.94 (t, J = 8.7 Hz, 2H), 6.34 (d, J = 15.9 Hz, 1H), 5.97 (dt, J = 15.8, 6.4 Hz, 1H), 3.22 (d, J = 6.4 Hz, 2H), 3.09–3.03 (m, 2H), 2.78–2.70 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 162.4 (d, 1JCF = 245.6 Hz), 153.3, 145.2, 134.4, 133.6, 133.3, 132.8 (d, 4JCF = 3.3 Hz), 131.7, 131.3, 129.1, 128.0 (d, 3JCF = 8.0 Hz), 126.0, 125.8 (d, 5JCF = 2.2 Hz), 117.4, 115.6 (d, 2JCF = 21.4 Hz), 50.7, 47.4, 42.1.
(E)-N-(2-{[3-(4-Trifluoromethylphenyl)-2-propen-1-yl]amino}ethyl)-5-isoquinolinesulfonamide (22)
Compound 22 was synthesized using general procedure 3 from amine 7 and cinnamaldehyde 14 to afford 22 (678 mg, 68%) as a light yellow oil.
1H NMR (300 MHz, CDCl3): δ 9.32 (s, 1H), 8.66 (d, J = 6.1 Hz, 1H), 8.47–8.41 (m, 2H), 8.16 (d, J = 8.2 Hz, 1H), 7.69–7.63 (m, 1H), 7.52 (d, J = 8.2 Hz, 2H), 7.37 (d, J = 8.2 Hz, 2H), 6.45 (d, J = 15.9 Hz, 1H), 6.18 (dt, J = 15.8, 6.3 Hz, 1H), 3.29 (d, J = 6.2 Hz, 2H), 3.09–3.04 (m, 2H), 2.81–2.72 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 153.4, 145.3, 140.1 (q, 4JCF = 1.4 Hz), 134.4, 133.7, 133.4, 131.5, 131.4129.2 (q, 2JCF = 32.2 Hz), 129.2, 126.6, 126.0, 125.7 (q, 3JCF = 3.8 Hz), 123.0 (q, 1JCF = 242.4 Hz), 117.4, 50.7, 47.4, 42.1.
(E)-N-(2-{[3-(4-Dimethylaminophenyl)-2-propen-1-yl]amino}ethyl)-5-isoquinolinesulfonamide (23)
Compound 23 was synthesized using general procedure 3 from amine 7 and cinnamaldehyde 15 to afford 23 (754 mg, 74%) as an orange oil.
1H NMR (300 MHz, CDCl3): δ 9.29 (s, 1H), 8.63 (d, J = 6.1 Hz, 1H), 8.47 (d, J = 6.0 Hz, 1H), 8.40 (d, J = 7.3 Hz, 1H), 8.11 (d, J = 8.2 Hz, 1H), 7.62 (t, J = 7.8 Hz, 1H), 7.16 (d, J = 8.7 Hz, 2H), 6.61 (d, J = 8.8 Hz, 2H), 6.31 (d, J = 15.7 Hz, 1H), 5.84 (dt, J = 15.7, 6.7 Hz, 1H), 3.25 (d, J = 6.7 Hz, 2H), 3.12–3.05 (m, 2H), 2.92 (s, 6H), 2.81–2.75 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 153.3, 150.4, 145.2, 134.5, 134.1, 133.5, 133.2, 131.3, 129.1, 127.5, 126.0, 124.6, 119.8, 117.5, 112.4, 50.8, 47.0, 41.6, 40.5.
N-{2-[(2-Phenylethyl)amino]ethyl}-5-isoquinolinesulfonamide (24)86
Compound 24 was synthesized using general procedure 3 from amine 7 and commercially available phenylacetaldehyde to afford 24 (666 mg, 75%) as a light yellow oil.
1H NMR (300 MHz, CDCl3): δ 9.34 (s, 1H), 8.66 (d, J = 6.1 Hz, 1H), 8.47–8.39 (m, 2H), 8.18 (d, J = 8.2 Hz, 1H), 7.67 (t, J = 7.8 Hz, 1H), 7.29–7.16 (m, 3H), 7.12–7.04 (m, 2H), 3.03–2.95 (m, 2H), 2.73–2.60 (m, 6H); 13C NMR (75 MHz, CDCl3): δ 153.3, 145.2, 139.3, 134.4, 133.6, 133.3, 131.4, 129.1, 128.7, 128.6, 126.4, 126.0, 117.4, 50.0, 47.7, 42.1, 35.8.
N-{2-[(3-Phenylpropyl)amino]ethyl}-5-isoquinolinesulfonamide (25)86
Compound 25 was synthesized using general procedure 3 from amine 7 and commercially available 3-phenylpropionaldehyde to afford 25 (884 mg, 73%) a light yellow oil.
1H NMR (300 MHz, CDCl3): δ 9.31 (s, 1H), 8.64 (d, J = 6.1 Hz, 1H), 8.48–8.39 (m, 2H), 8.15 (d, J = 8.2 Hz, 1H), 7.65 (t, J = 7.8 Hz, 1H), 7.27–7.12 (m, 3H), 7.08 (d, J = 7.0 Hz, 2H), 3.10–3.02 (m, 2H), 2.79–2.69 (m, 2H), 2.52 (t, J = 7.5 Hz, 4H), 1.80–1.65 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 153.3, 145.2, 141.3, 134.4, 133.6, 133.3, 131.3, 129.1, 128.5, 128.4, 126.1, 126.0, 117.5, 48.4, 48.0, 41.6, 33.3, 30.4.
(E)-3-(4-bromophenyl)-N-{2-[(5-isoquinolinylsulfonyl)amino]ethyl}-2-propenamide (26)
4-Bromocinnamic acid (0.10 g, 0.44 mmol) was taken up in dry CHCl3 (15 mL) containing two drops of DMF and treated with SOCl2 (0.47 g, 3.96 mmol). The resulting solution was heated at refluxed for 4 h, cooled and the solvent and excess SOCl2 removed in vacuo. The crude acid chloride was taken up in dry CH2Cl2 (10 mL) and added dropwise to a cooled (0 °C) solution of 7 (0.1 g, 0.4 mmol) and TEA (0.060 g, 0.6 mmol) in CH2Cl2 (10 mL). The reaction mixture was stirred at 25 °C for 18 h and diluted with EtOAc (30 mL) and 10% NaHCO3 (20 mL). The organic layer was separated, washed with 10% NaHCO3 (20 mL) and brine (20 mL), dried (MgSO4) and concentrated in vacuo. The desired product was subjected to flash column chromatography eluting with EtOAc/petroleum spirit (7 : 3) affording 26 (981 mg, 51%) as an off-white solid, mp 175–178 °C.
1H NMR (300 MHz, CDCl3) δ 9.24 (s, 1H), 8.58 (d, J = 6.1 Hz, 1H), 8.44 (d, J = 6.1 Hz, 1H), 8.39 (dd, J = 7.4, 1.1 Hz, 1H), 8.11 (d, J = 8.2 Hz, 1H), 7.63 (dd, J = 8.1, 7.5 Hz, 1H), 7.38–7.31 (m, 3H), 7.15 (d, J = 8.4 Hz, 2H), 7.08 (bt, J = 4.9 Hz, 1H), 6.84 (t, J = 5.7 Hz, 1H), 6.21 (d, J = 15.7 Hz, 1H), 3.51–3.40 (m, 2H), 3.16–3.10 (m, 2H).13C NMR (75 MHz, CDCl3): δ 166.9, 153.2, 144.9, 140.2, 134.2, 133.8, 133.4, 133.3, 132.1, 131.3, 129.6, 129.3, 126.2, 124.1, 120.6, 117.5, 43.1, 39.8.
N-(3-Aminopropyl)-5-isoquinolinesulfonamide (27)
Compound 27 was synthesized using general procedure 1 from sulfonic acid 2 and commercially available propane-1,3-diamine to afford 27 (870 mg, 57%) as an orange oil.
1H NMR (400 MHz, CDCl3): δ 9.36 (s, 1H), 8.68 (d, J = 6.1 Hz, 1H), 8.48–8.40 (m, 2H), 8.20 (d, J = 8.2 Hz, 1H), 7.70 (t, J = 7.8 Hz, 1H), 3.06 (t, J = 5.9 Hz, 2H), 2.73 (t, J = 5.7 Hz, 2H), 1.54–1.47 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 153.5, 145.1, 134.7, 133.4, 133.4, 131.4, 129.2, 126.0, 117.6, 43.9, 41.5, 30.1.
N-(4-Aminobutyl)-5-isoquinolinesulfonamide (28)
Compound 28 was synthesized using general procedure 1 from sulfonic acid 2 and butane-1,4-diamine to afford 28 (376 mg, 19%) as an orange oil.
1H NMR (300 MHz, CDCl3): δ 9.59 (d, J = 0.7 Hz, 1H), 8.81 (d, J = 6.1 Hz, 1H), 8.58 (d, J = 6.1 Hz, 1H), 8.53 (d, J = 8.3 Hz, 1H), 8.46 (dd, J = 7.4, 1.1 Hz, 1H), 7.97–7.91 (m, 1H), 2.89 (t, J = 6.8 Hz, 2H), 2.47 (t, J = 6.7 Hz, 2H), 1.46–1.28 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 153.2, 144.7, 135.1, 133.2, 133.0, 131.3, 129.1, 126.0, 117.6, 43.0, 41.2, 30.2, 27.6.
N-(6-Aminohexyl)-5-isoquinolinesulfonamide (29)
Compound 29 was synthesized using general procedure 1 from sulfonic acid 2 and hexane-1,6-diamine to afford 29 (1.04 g, 77%) as light yellow oil.
1H NMR (400 MHz, CDCl3): δ 9.35 (s, 1H), 8.65 (d, J = 6.1 Hz, 1H), 8.51–8.37 (m, 2H), 8.19 (d, J = 8.2 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 2.92 (t, J = 6.9 Hz, 2H), 2.57 (t, J = 6.7 Hz, 2H), 1.43–1.33 (m, 2H), 1.31–1.22 (m, 2H), 1.19–1.09 (m, 4H); 13C NMR (101 MHz, CDCl3): δ 153.4, 145.1, 134.8, 133.5, 133.3, 131.4, 129.2, 126.1, 117.4, 43.1, 41.9, 33.2, 29.6, 26.2, 26.2.
1-(5-Isoquinolinesulfonyl)piperazine (30)87
Compound 30 was synthesized using general procedure 1 from sulfonic acid 2 and piperazine to afford 30 (679 mg, 34%) as an orange oil.
1H NMR (400 MHz, CDCl3): δ 9.33 (s, 1H), 8.66 (d, J = 6.2 Hz, 1H), 8.52 (d, J = 6.2 Hz, 1H), 8.35 (dd, J = 7.4, 1.1 Hz, 1H), 8.21 (d, J = 8.2 Hz, 1H), 7.70 (t, J = 7.8 Hz, 1H), 3.17–3.07 (m, 4H), 2.94–2.79 (m, 4H); 13C NMR (101 MHz, CDCl3): δ 153.4, 145.2, 134.4, 134.0, 132.2, 132.0, 129.2, 126.0, 117.8, 46.5, 45.5.
(E)-N-(3-{[3-(4-Bromophenyl)-2-propen-1-yl]amino}propyl)-5-isoquinolinesulfonamide (31)
Compound 31 was synthesized using general procedure 3 from amine 31 and cinnamaldehyde 11 to afford the title compound (73 mg, 65%) as a yellow oil.
1H NMR (300 MHz, CDCl3): δ 9.31 (s, 1H), 8.58 (d, J = 6.1 Hz, 1H), 8.45–8.38 (m, 2H), 8.14 (d, J = 8.2 Hz, 1H), 7.64 (dd, J = 8.1, 7.4 Hz, 1H), 7.38 (d, J = 8.5 Hz, 2H), 7.18 (d, J = 8.5 Hz, 2H), 6.42 (d, J = 15.9 Hz, 1H), 6.16 (dt, J = 15.9, 6.2 Hz, 1H), 3.26 (dd, J = 6.2, 1.2 Hz, 2H), 3.02 (t, J = 6.0 Hz, 2H), 2.64 (t, J = 5.9 Hz, 2H), 1.65–1.54 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 153.3, 144.9, 135.7, 134.6, 133.3, 133.2, 131.7, 131.3, 131.0, 129.1, 127.9, 127.7, 126.0, 121.4, 117.4, 51.3, 48.1, 43.5, 27.7.
(E)-N-(4-{[3-(4-Bromophenyl)-2-propen-1-yl]amino}butyl)-5-isoquinolinesulfonamide (32)
Compound 32 was synthesized using general procedure 3 from amine 28 and cinnamaldehyde 11 to afford 32 (12 mg, 11%) as a yellow oil.
1H NMR (300 MHz, CDCl3): δ 9.32 (s, 1H), 8.56 (d, J = 6.0 Hz, 1H), 8.48 (d, J = 6.2 Hz, 1H), 8.41 (d, J = 7.3 Hz, 1H), 8.15 (d, J = 8.2 Hz, 1H), 7.64 (t, J = 7.8 Hz, 1H), 7.41 (d, J = 8.4 Hz, 2H), 7.25 (d, J = 8.4 Hz, 2H), 6.52 (d, J = 15.9 Hz, 1H), 6.39 (dt, J = 15.9, 6.1 Hz, 1H), 3.44 (d, J = 6.2 Hz, 2H), 2.93–2.86 (m, 2H), 2.65 (t, J = 5.4 Hz, 2H), 1.60–1.50 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 153.4, 145.0, 135.7, 135.2, 133.2, 132.2, 131.9, 131.8, 131.5, 129.2, 128.1, 127.4, 126.0, 121.6, 117.6, 51.5, 48.7, 43.1, 28.6, 27.8.
(E)-N-(6-{[3-(4-Bromophenyl)-2-propen-1-yl]amino}hexyl)-5-isoquinolinesulfonamide (33)
Compound 33 was synthesized using general procedure 3 from amine 29 and cinnamaldehyde 12 to afford 33 (73 mg, 51%) as a yellow solid, mp 80–82 °C.
1H NMR (300 MHz, CDCl3): δ 9.32 (s, 1H), 8.60 (d, J = 6.2 Hz, 1H), 8.50 (d, J = 6.1 Hz, 1H), 8.40 (dd, J = 7.3, 1.1 Hz, 1H), 8.15 (d, J = 7.8 Hz, 1H), 7.63 (t, J = 7.5 Hz, 1H), 7.35 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.5 Hz, 2H), 6.47 (d, J = 15.9 Hz, 1H), 6.27 (dt, J = 15.8, 6.5 Hz, 1H), 3.45 (d, J = 6.4 Hz, 2H), 2.89 (t, J = 6.8 Hz, 2H), 2.63 (t, J = 7.2 Hz, 2H), 1.50–1.34 (m, 4H), 1.19–1.13 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 153.3, 145.0, 135.5, 134.9, 133.4, 133.2, 132.5, 131.7, 131.4, 129.1, 128.1, 126.1, 126.0, 121.6, 117.6, 51.0, 48.3, 43.0, 29.4, 28.4, 26.3, 26.0.
(E)-5-({4-[3-(4-Bromophenyl)-2-propen-1-yl]-1-piperazinyl}sulfonyl)Isoquinoline (34)
Compound 34 was synthesized using general procedure 3 from amine 30 and cinnamaldehyde 11 to afford 34 (112 mg, 62%) as a yellow oil.
1H NMR (300 MHz, CDCl3) δ 9.32 (s, 1H), 8.65 (d, J = 5.3 Hz, 1H), 8.52 (d, J = 5.9 Hz, 1H), 8.34 (dd, J = 7.4, 1.0 Hz, 1H), 8.18 (d, J = 8.2 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.36 (d, J = 8.4 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 6.38 (d, J = 15.9 Hz, 1H), 6.07 (dt, J = 15.8, 6.7 Hz, 1H), 3.21–3.15 (m, 4H), 3.07 (d, J = 6.6 Hz, 2H), 2.53–2.48 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 153.3, 145.1, 135.6, 134.3, 133.9, 132.4, 132.1, 131.9, 131.7, 129.1, 127.8, 126.5, 125.9, 121.4, 117.8, 60.3, 52.2, 45.8.
N-(2-Aminoethyl)-8-quinolinesulfonamide (35)
Compound 35 was synthesized using general procedure 1 from sulfonic acid 3 and ethane-1,2-diamine to afford 35 (1.73 g, 83%) as a yellow oil.
1H NMR (300 MHz, CDCl3): δ 8.98 (dd, J = 4.2, 1.4 Hz, 1H), 8.38 (d, J = 7.3 Hz, 1H), 8.24 (d, J = 8.3 Hz, 1H), 8.01 (d, J = 8.2 Hz, 1H), 7.59 (t, J = 7.8 Hz, 1H), 7.50 (dd, J = 8.4, 4.3 Hz, 1H), 2.87 (t, J = 5.7 Hz, 2H), 2.71 (t, J = 5.7 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ 151.3, 143.4, 137.0, 136.3, 133.3, 131.1, 128.9, 125.7, 122.3, 46.4, 41.5.
N-(2-Aminoethyl)-5-(dimethylamino)-1-naphthalenesulfonamide (36)88
Compound 36 was synthesized using general procedure 1 from sulfonic acid 4 and ethane-1,2-diamine to afford 36 (1.23 g, 83%) as a yellow solid, mp 117–119 °C (lit. 145–147 °C).88
1H NMR (300 MHz, CDCl3): δ 8.52 (d, J = 8.5 Hz, 1H), 8.31 (d, J = 8.7 Hz, 1H), 8.22 (dd, J = 7.3, 1.2 Hz, 1H), 7.58–7.45 (m, 2H), 7.16 (d, J = 7.6 Hz, 1H), 2.93–2.85 (m, 8H), 2.70–2.66 (m, 2H).
13C NMR (75 MHz, CDCl3): δ 152.1, 135.0, 130.5, 130.0, 129.7, 129.6, 128.4, 123.3, 118.9, 115.3, 45.6, 45.5, 41.1.
N-(2-Aminoethyl)-1-naphthalenesulfonamide (37)
Compound 37 was synthesized using general procedure 1 from sulfonic acid 5 and ethane-1,2-diamine to afford 37 (1.33 g, 80%) as a clear oil.
1H NMR (400 MHz, MeOD): δ 8.71 (d, J = 8.3 Hz, 1H), 8.20 (dd, J = 7.3, 1.1 Hz, 1H), 8.07 (d, J = 8.3 Hz, 1H), 7.95 (d, J = 8.1 Hz, 1H), 7.69–7.62 (m, 1H), 7.59–7.49 (m, 2H), 2.87 (t, J = 6.2 Hz, 2H), 2.57 (t, J = 6.2 Hz, 2H); 13C NMR (101 MHz, MeOD): δ 136.4, 135.7, 135.1, 130.2, 130.1, 129.2, 129.1, 127.9, 125.6, 125.3, 46.2, 42.1.
N-(2-Aminoethyl)-2-naphthalenesulfonamide (38)
Compound 38 was synthesized using general procedure 1 from sulfonic acid 6 and ethane-1,2-diamine to afford 38 (1.92 g, 64%) as a white solid, mp 124–126 °C.
1H NMR (300 MHz, CDCl3): δ 8.43 (s, 1H), 7.98–7.80 (m, 4H), 7.67–7.53 (m, 2H), 3.05–2.96 (m, 2H), 2.85–2.73 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 136.9, 134.9, 132.3, 129.6, 129.3, 128.9, 128.5, 128.0, 127.7, 122.4, 45.4, 41.1.
(E)-N-(2-{[3-(4-Bromophenyl)-2-propen-1-yl]amino}ethyl)-8-quinolinesulfonamide (39)
Compound 39 was synthesized using general procedure 3 from amine 35 and cinnamaldehyde 11 to afford 39 (132 mg, 67%) a light yellow oil.
1H NMR (300 MHz, CDCl3): δ 8.98 (dd, J = 4.3, 1.7 Hz, 1H), 8.40 (dd, J = 7.3, 1.4 Hz, 1H), 8.21 (dd, J = 8.4, 1.7 Hz, 1H), 8.00 (dd, J = 8.3, 1.4 Hz, 1H), 7.59 (dd, J = 8.2, 7.3 Hz, 1H), 7.47 (dd, J = 8.3, 4.3 Hz, 1H), 7.37 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H), 6.32 (d, J = 15.9 Hz, 1H), 6.05 (dt, J = 15.9, 6.2 Hz, 1H), 3.20 (dd, J = 6.2, 1.3 Hz, 2H), 3.04–2.99 (m, 2H), 2.77–2.69 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 151.3, 143.3, 137.0, 135.9, 133.4, 131.6, 131.2, 130.6, 128.8, 128.2, 127.9, 125.7, 122.3, 121.2, 50.8, 47.8, 42.9.
(E)-N-(2-{[3-(4-Bromophenyl)-2-propen-1-yl]amino}ethyl)-5-dimethylamino-1-naphthalenesulfonamide (40)
Compound 40 was synthesized using general procedure 3 from amine 36 and cinnamaldehyde 11 to afford 40 (104 mg, 58%) as an orange solid, mp 42–44 °C.
1H NMR (300 MHz, CDCl3): δ 8.52 (dt, J = 8.5, 0.9 Hz, 1H), 8.33 (d, J = 8.7 Hz, 1H), 8.25 (dd, J = 7.3, 1.3 Hz, 1H), 7.57–7.45 (m, 2H), 7.39 (d, J = 8.4 Hz, 2H), 7.16–7.07 (m, 3H), 6.25 (d, J = 15.9 Hz, 1H), 5.97 (dt, J = 15.9, 6.2 Hz, 1H), 3.04 (dd, J = 6.2, 1.3 Hz, 2H), 3.00–2.94 (m, 2H), 2.84 (s, 6H), 2.64–2.57 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 152.1, 135.9, 134.7, 131.7, 130.5, 130.2, 129.9, 129.7, 128.7, 128.5, 127.9, 123.2, 121.2, 118.8, 115.2, 50.8, 47.4, 45.4, 42.6.
(E)-N-(2-{[3-(4-Bromophenyl)-2-propen-1-yl]amino}ethyl)-1-naphthalenesulfonamide (41)
Compound 41 was synthesized using general procedure 3 from amine 37 and cinnamaldehyde 11 to afford 42 (121 mg, 57%) as a light yellow oil.
1H NMR (300 MHz, CDCl3): δ 8.69 (d, J = 8.4 Hz, 1H), 8.25 (d, J = 7.3 Hz, 1H), 8.01 (d, J = 8.2 Hz, 1H), 7.89 (d, J = 8.1 Hz, 1H), 7.64–7.47 (m, 3H), 7.37 (d, J = 8.2 Hz, 2H), 7.09 (d, J = 8.4 Hz, 2H), 6.22 (d, J = 15.9 Hz, 1H), 5.95 (dt, J = 15.9, 6.2 Hz, 1H), 3.05–2.96 (m, 4H), 2.59 (t, J = 5.6 Hz, 2H); 13C NMR (75 MHz, CDCl3): δ 135.9, 134.6, 134.3, 134.2, 131.6, 130.2, 129.7, 129.1, 128.6, 128.4, 128.2, 127.8, 126.9, 124.4, 124.2, 121.1, 50.7, 47.4, 42.5.
(E)-N-(2-{[3-(4-Bromophenyl)-2-propen-1-yl]amino}ethyl)-2-naphthalenesulfonamide (42)
Compound 42 was synthesized using general procedure 3 from amine 38 and cinnamaldehyde 11 to afford 42 (108 mg, 51%) as a light yellow oil.
1H NMR (300 MHz, MeOD + CDCl3): δ 8.38 (d, J = 1.2 Hz, 1H), 7.90 (d, J = 8.2 Hz, 2H), 7.82 (m, 2H), 7.62–7.52 (m, 2H), 7.34 (d, J = 8.5 Hz, 2H), 7.12 (d, J = 8.5 Hz, 2H), 6.34 (d, J = 15.9 Hz, 1H), 6.10 (dt, J = 15.9, 6.4 Hz, 1H), 3.24 (dd, J = 6.4, 1.1 Hz, 2H), 3.03 (t, J = 5.9 Hz, 2H), 2.68 (t, J = 5.9 Hz, 2H); 13C NMR (75 MHz, MeOD + CDCl3): δ 136.5, 135.5, 134.5, 131.9, 131.3, 130.8, 129.2, 128.8, 128.5, 127.8, 127.6, 127.5, 127.4, 127.3, 121.8, 120.9, 50.4, 47.5, 41.9.
N-(3-Aminopropyl)-1-naphthalenesulfonamide (43)
Compound 43 was synthesized using general procedure 1 from sulfonic acid 5 and propane-1,3-diamine to afford 43 (2.75 g, 74%) as an orange oil.
1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 8.4 Hz, 1H), 8.23 (dd, J = 7.3, 0.9 Hz, 1H), 8.01 (d, J = 8.3 Hz, 1H), 7.90 (d, J = 8.2 Hz, 1H), 7.68–7.44 (m, 3H), 2.97 (t, J = 6.3 Hz, 2H), 2.66 (t, J = 6.5 Hz, 2H), 1.56–1.50 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 134.8, 134.1, 133.8, 129.0, 128.8, 127.9, 127.9, 126.7, 124.4, 124.0, 41.5, 39.2, 30.9.
N-(4-Aminobutyl)-1-naphthalenesulfonamide (44)
Compound 44 was synthesized using general procedure 1 from sulfonic acid 5 and butane-1,4-diamine to afford 44 (835 mg, 68%) as a yellow oil.
1H NMR (300 MHz, CDCl3): δ 8.67 (d, J = 8.6 Hz, 1H), 8.14 (d, J = 7.3 Hz, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.51–7.36 (m, 3H), 2.88–2.64 (m, 2H), 2.56–2.40 (m, 2H), 1.36–1.26 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 135.2, 134.1, 133.7, 128.9, 128.8, 128.1, 127.9, 126.7, 124.6, 124.1, 42.7, 40.5, 28.7, 26.9.
N-(6-Aminohexyl)-1-naphthalenesulfonamide (45)
Compound 45 was synthesized using general procedure 1 from sulfonic acid 5 and hexane-1,4-diamine to afford 45 (102 mg, 77%) as a yellow oil.
1H NMR (300 MHz, CDCl3): δ 8.67 (d, J = 8.6 Hz, 1H), 8.15 (d, J = 7.3 Hz, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.57–7.33 (m, 4H), 2.79 (t, J = 6.9 Hz, 2H), 2.44 (t, J = 7.0 Hz, 2H), 1.26–1.10 (m, 4H), 1.02–0.93 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 135.2, 134.0, 133.7, 128.9, 128.7, 128.0, 127.9, 126.6, 124.5, 123.9, 42.7, 41.1, 32.0, 29.1, 25.8, 25.7.
N-Methyl-N-(2-methylaminoethyl)-1-naphthalenesulfonamide (46)
Compound 46 was synthesized using general procedure 1 from sulfonic acid 5 and N,N′-dimethylethane-1,2-diamine to afford 46 (554 mg, 88%) as a light yellow oil; 88%.
1H NMR (300 MHz, CDCl3): δ 8.70 (d, J = 8.6 Hz, 1H), 8.16 (dd, J = 7.4, 1.1 Hz, 1H), 8.03 (d, J = 8.2 Hz, 1H), 7.90 (d, J = 8.6 Hz, 1H), 7.66–7.48 (m, 3H), 3.27 (t, J = 6.2 Hz, 2H), 2.84 (s, 3H), 2.72 (t, J = 6.2 Hz, 2H), 2.32 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 134.5, 134.3, 133.9, 130.0, 129.0, 128.9, 128.1, 126.9, 125.1, 124.2, 49.2, 49.2, 36.0, 34.8.
(E)-N-(3-{[3-(4-Bromophenyl)-2-propen-1-yl]amino}propyl)-1-naphthalenesulfonamide (47)
Compound 47 was synthesized using general procedure 3 from amine 43 and cinnamaldehyde 11 to afford 48 (137 mg, 63%) as a light yellow solid, mp 123–125 °C.
1H NMR (300 MHz, CDCl3): δ 8.69 (d, J = 8.2 Hz, 1H), 8.23 (dd, J = 7.3, 1.1 Hz, 1H), 8.03 (d, J = 8.3 Hz, 1H), 7.95–7.87 (m, 1H), 7.62–7.47 (m, 3H), 7.39 (d, J = 8.5 Hz, 2H), 7.18 (d, J = 8.4 Hz, 2H), 6.47 (d, J = 15.9 Hz, 1H), 6.19 (dt, J = 15.9, 6.5 Hz, 1H), 3.36 (d, J = 5.9 Hz, 2H), 3.01 (t, J = 6.0 Hz, 2H), 2.72 (t, J = 6.1 Hz, 2H), 1.75–1.61 (m, 2H); 13C NMR (75 MHz, CDCl3): δ 135.5, 134.9, 134.4, 134.1, 132.7, 131.8, 131.8, 129.6, 129.2, 128.3, 128.2, 127.0, 125.8, 124.7, 124.3, 121.8, 51.0, 47.3, 42.8, 27.5.
(E)-N-(4-{[3-(4-Bromophenyl)-2-propen-1-yl]amino}butyl)-1-naphthalenesulfonamide (48)
Compound 48 was synthesized using general procedure 1 from amine 44 and cinnamaldehyde 11 to afford 48 (102 mg, 49%) as a light yellow oil.
1H NMR (300 MHz, CDCl3): δ 8.72 (d, J = 7.1 Hz, 1H), 8.23 (d, J = 7.3 Hz, 1H), 8.01 (d, J = 8.2 Hz, 1H), 7.93–7.88 (m, 1H), 7.57–7.47 (m, 3H), 7.39 (d, J = 8.5 Hz, 2H), 7.21 (d, J = 8.4 Hz, 2H), 6.46 (d, J = 16.0 Hz, 1H), 6.31 (dt, J = 15.9, 6.2 Hz, 1H), 3.34 (d, J = 6.1 Hz, 2H), 2.89 (t, J = 5.8 Hz, 2H), 2.54 (t, J = 6.0 Hz, 2H), 1.51–1.42 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 136.0, 135.4, 134.4, 133.9, 131.7, 130.9, 129.4, 129.1, 128.5, 128.4, 128.0, 128.0, 126.8, 124.8, 124.3, 121.3, 51.6, 48.7, 43.2, 28.2, 27.6.
(E)-N-(6-{[3-(4-Bromophenyl)-2-propen-1-yl]amino}hexyl)-1-naphthalenesulfonamide (49)
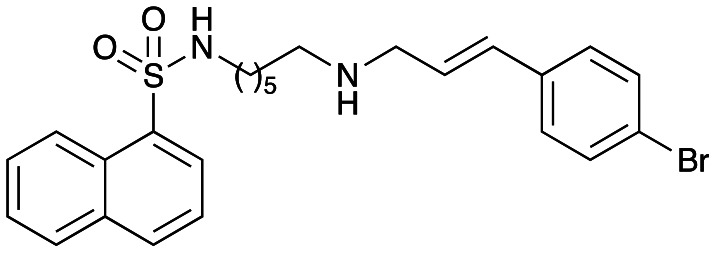
Compound 49 was synthesized using general procedure 1 from amine 45 and cinnamaldehyde 11 to afford 49 (95 mg, 48%) as a light yellow oil.
1H NMR (300 MHz, CDCl3): δ 8.68 (d, J = 8.4 Hz, 1H), 8.24 (dd, J = 7.3, 1.0 Hz, 1H), 8.03 (d, J = 8.2 Hz, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.64–7.48 (m, 3H), 7.39 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.4 Hz, 2H), 6.42 (d, J = 15.9 Hz, 1H), 6.24 (dt, J = 15.9, 6.2 Hz, 1H), 3.33 (d, J = 6.1 Hz, 2H), 2.90 (t, J = 6.9 Hz, 2H), 2.51 (t, J = 7.1 Hz, 2H), 1.39–1.29 (m, 4H), 1.18–1.09 (m, 4H); 13C NMR (75 MHz, CDCl3): δ 136.2, 135.1, 134.4, 134.2, 131.7, 130.1, 129.6, 129.5, 129.1, 128.4, 128.3, 127.9, 126.9, 124.6, 124.2, 121.1, 51.8, 49.2, 43.2, 29.8, 29.5, 26.6, 26.3.
(E)-N-(2-{Methyl[3-(4-bromophenyl)-2-propen-1-yl]amino}ethyl)-1-naphthalenesulfonamide (50)
Compound 50 was synthesized using general procedure 3 from sulfonamide 41 and 40% formaldehyde (26 eq.) to afford 50 (86 mg, 59%) as a light yellow solid, mp 47–49 °C.
1H NMR (300 MHz, CDCl3): δ 8.69 (d, J = 8.6 Hz, 1H), 8.27 (dd, J = 7.3, 1.2 Hz, 1H), 8.05 (d, J = 8.3 Hz, 1H), 7.92 (d, J = 8.1 Hz, 1H), 7.68–7.52 (m, 3H), 7.43 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.6 Hz, 2H), 6.28 (d, J = 15.9 Hz, 1H), 5.94 (dt, J = 15.9, 6.6 Hz, 1H), 2.97–2.92 (m, 2H), 2.85 (d, J = 6.6 Hz, 2H), 2.35–2.29 (m, 2H), 1.88 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 135.8, 134.5, 134.4, 134.3, 134.3, 131.8, 131.8, 129.9, 129.2, 128.4, 128.0, 127.3, 127.0, 124.6, 124.3, 121.4, 59.6, 54.8, 41.1, 40.3.
(E)-N-Methyl-N-(2-{methyl[3-(4-bromophenyl)-2-propen-1-yl]amino}ethyl)-1-naphthalenesulfonamide (51)
Compound 50 was synthesized using general procedure 3 from amine 46 and cinnamaldehyde 11 to afford 50 (209 mg, 94%) as a light yellow oil.
1H NMR (300 MHz, CDCl3): δ 8.76 (d, J = 8.4 Hz, 1H), 8.20 (d, J = 7.3 Hz, 1H), 8.02 (d, J = 8.1 Hz, 1H), 7.90 (d, J = 7.8 Hz, 1H), 7.66–7.47 (m, 3H), 7.42 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.4 Hz, 2H), 6.41 (d, J = 15.9 Hz, 1H), 6.14 (dt, J = 15.9, 6.5 Hz, 1H), 3.37 (t, J = 6.9 Hz, 2H), 3.11 (d, J = 6.5 Hz, 2H), 2.91 (s, 3H), 2.59 (t, J = 6.9 Hz, 2H), 2.24 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 135.9, 134.4, 134.3, 134.1, 131.6, 131.5, 129.6, 128.8, 128.8, 128.0, 127.9, 127.8, 126.8, 125.2, 124.1, 121.1, 60.0, 54.9, 47.5, 42.2, 34.9.
Ethical statement
Experimental and animal care practices were conducted with approval from the University of Melbourne Animal Ethics Committee (#0707408) in accordance with the Australian code for the care of animals for scientific purposes, 8th edition. This work was conducted according to the approved ethical standards of the Australian National Medical and Health Research Council for the use of live animals.
Author contributions
AM, PJR and TJO'B conceived the study and obtained the necessary funding. NCJ conducted the animal models of epilepsy experiments; NC, AW, JX conducted the in vitro and in cell biological analysis (dynamin and CME IC50); NNG and JAD conducted the exocytosis and kinetics studies; LRO, MJR, JIA, FMD and KAY were responsible for compound synthesis, characterisation and purification. KP was responsible for compound modelling and docking.
Conflicts of interest
The authors declare the following competing financial interest(s): we have entered into a commercial agreement with Abcam (Cambridge, UK) for the supply of some of our lead dynamin and clathrin inhibitors. This includes some of the compounds listed in this paper.
Supplementary Material
Acknowledgments
We are grateful for financial support from the National Health & Medical Research Council (Australia), the Australia Research Council, the Children's Medical Research Institute (CMRI) and the University of Newcastle (UON) and for equipment from the Australian Cancer Research Foundation, the Ramaciotti Foundation and the Cancer Institute NSW. The National Institute for Neurological Disorders and Stroke is thanked for early-stage seizure testing.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2md00371f
References
- Singh G. Sander J. W. Epilepsy Behav. 2020;105:106949. doi: 10.1016/j.yebeh.2020.106949. [DOI] [PubMed] [Google Scholar]
- Becker A. J. Neuropathol. Appl. Neurobiol. 2018;44:112–129. doi: 10.1111/nan.12451. [DOI] [PubMed] [Google Scholar]
- Bialer M. White H. S. Nat. Rev. Drug Discovery. 2010;9:68–82. doi: 10.1038/nrd2997. [DOI] [PubMed] [Google Scholar]
- Leo A. Citraro R. Constanti A. Sarro G. D. Russo E. Expert Opin. Ther. Targets. 2015;19:911–926. doi: 10.1517/14728222.2015.1026258. [DOI] [PubMed] [Google Scholar]
- N'Gouemo P. Expert Opin. Ther. Targets. 2011;15:1283–1295. doi: 10.1517/14728222.2011.620607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S. S. Buckmaster P. S. J. Neurosci. 2006;26:4613–4623. doi: 10.1523/JNEUROSCI.0064-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twyman R. E. Neurochem. Res. 2017;42:2099–2115. doi: 10.1007/s11064-017-2300-2. [DOI] [PubMed] [Google Scholar]
- Binotti B. Jahn R. Pérez-Lara Á. Arch. Biochem. Biophys. 2021;709:108966. doi: 10.1016/j.abb.2021.108966. [DOI] [PubMed] [Google Scholar]
- Kuzmanova R. Stefanova I. Acta Med. Bulg. 2017;44:52–58. [Google Scholar]
- Özbek O. Gürdere M. B. Med. Chem. Res. 2020;29:1553–1578. doi: 10.1007/s00044-020-02595-4. [DOI] [Google Scholar]
- Pal R. Kumar B. Akhtar Md. J. Chawla P. A. Bioorg. Chem. 2021;115:105230. doi: 10.1016/j.bioorg.2021.105230. [DOI] [PubMed] [Google Scholar]
- Hinckley C. A. Kuryshev Y. Sers A. Barre A. Buisson B. Naik H. Hajos M. Mol. Pharmacol. 2021;99:49–59. doi: 10.1124/molpharm.120.000079. [DOI] [PubMed] [Google Scholar]
- Meldrum B. S. Rogawski M. A. Neurotherapeutics. 2007;4:18–61. doi: 10.1016/j.nurt.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casillas-Espinosa P. M. Powell K. L. O'Brien T. J. Epilepsia. 2012;53:41–58. doi: 10.1111/epi.12034. [DOI] [PubMed] [Google Scholar]
- McCabe M. P. Shore A. N. Frankel W. N. Weston M. C. eNeuro. 2021;8:ENEURO.0269-20.2020. doi: 10.1523/ENEURO.0269-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper C. B. Popoff M. R. McCluskey A. Robinson P. J. Meunier F. A. Trends Cell Biol. 2013;23:90–101. doi: 10.1016/j.tcb.2012.10.007. [DOI] [PubMed] [Google Scholar]
- Eschenburg S. Reubold T. F. Biol. Chem. 2018;399:1421–1432. doi: 10.1515/hsz-2018-0257. [DOI] [PubMed] [Google Scholar]
- Appenzeller S. Balling R. Barisic N. Baulac S. Caglayan H. Craiu D. Jonghe P. D. Depienne C. Dimova P. Djémié T. Gormley P. Guerrini R. Helbig I. Hjalgrim H. Hoffman-Zacharska D. Jähn J. Klein K. M. Koeleman B. Komarek V. Krause R. Kuhlenbäumer G. Leguern E. Lehesjoki A.-E. Lemke J. R. Lerche H. Linnankivi T. Marini C. May P. Møller R. S. Muhle H. Pal D. Palotie A. Pendziwiat M. Robbiano A. Roelens F. Rosenow F. Selmer K. Serratosa J. M. Sisodiya S. Stephani U. Sterbova K. Striano P. Suls A. Talvik T. von Spiczak S. Weber Y. Weckhuysen S. Zara F. Abou-Khalil B. Alldredge B. K. Andermann E. Andermann F. Amrom D. Bautista J. F. Berkovic S. F. Bluvstein J. Boro A. Cascino G. Consalvo D. Crumrine P. Devinsky O. Dlugos D. Epstein M. P. Fiol M. Fountain N. B. French J. Friedman D. Geller E. B. Glauser T. Glynn S. Haas K. Haut S. R. Hayward J. Helmers S. L. Joshi S. Kanner A. Kirsch H. E. Knowlton R. C. Kossoff E. H. Kuperman R. Kuzniecky R. Lowenstein D. H. McGuire S. M. Motika P. V. Novotny E. J. Ottman R. Paolicchi J. M. Parent J. Park K. Poduri A. Sadleir L. Scheffer I. E. Shellhaas R. A. Sherr E. Shih J. J. Singh R. Sirven J. Smith M. C. Sullivan J. Thio L. L. Venkat A. Vining E. P. G. Allmen G. K. V. Weisenberg J. L. Widdess-Walsh P. Winawer M. R. Allen A. S. Berkovic S. F. Cossette P. Delanty N. Dlugos D. Eichler E. E. Epstein M. P. Glauser T. Goldstein D. B. Han Y. Heinzen E. L. Johnson M. R. Kuzniecky R. Lowenstein D. H. Marson A. G. Mefford H. C. Nieh S. E. O'Brien T. J. Ottman R. Petrou S. Petrovski S. Poduri A. Ruzzo E. K. Scheffer I. E. Sherr E. Am. J. Hum. Genet. 2017;100:179. doi: 10.1016/j.ajhg.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. Ding L. Holmfeldt L. Wu G. Heatley S. L. Payne-Turner D. Easton J. Chen X. Wang J. Rusch M. Lu C. Chen S.-C. Wei L. Collins-Underwood J. R. Ma J. Roberts K. G. Pounds S. B. Ulyanov A. Becksfort J. Gupta P. Huether R. Kriwacki R. W. Parker M. McGoldrick D. J. Zhao D. Alford D. Espy S. Bobba K. C. Song G. Pei D. Cheng C. Roberts S. Barbato M. I. Campana D. Coustan-Smith E. Shurtleff S. A. Raimondi S. C. Kleppe M. Cools J. Shimano K. A. Hermiston M. L. Doulatov S. Eppert K. Laurenti E. Notta F. Dick J. E. Basso G. Hunger S. P. Loh M. L. Devidas M. Wood B. Winter S. Dunsmore K. P. Fulton R. S. Fulton L. L. Hong X. Harris C. C. Dooling D. J. Ochoa K. Johnson K. J. Obenauer J. C. Evans W. E. Pui C.-H. Naeve C. W. Ley T. J. Mardis E. R. Wilson R. K. Downing J. R. Mullighan C. G. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züchner S. Noureddine M. Kennerson M. Verhoeven K. Claeys K. Jonghe P. D. Merory J. Oliveira S. A. Speer M. C. Stenger J. E. Walizada G. Zhu D. Pericak-Vance M. A. Nicholson G. Timmerman V. Vance J. M. Nat. Genet. 2005;37:289–294. doi: 10.1038/ng1514. [DOI] [PubMed] [Google Scholar]
- Buono S. Ross J. A. Tasfaout H. Levy Y. Kretz C. Tayefeh L. Matson J. Guo S. Kessler P. Monia B. P. Bitoun M. Ochala J. Laporte J. Cowling B. S. Proc. Natl. Acad. Sci. U. S. A. 2018;115:201808170. doi: 10.1073/pnas.1808170115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catteruccia M. Fattori F. Codemo V. Ruggiero L. Maggi L. Tasca G. Fiorillo C. Pane M. Berardinelli A. Verardo M. Bragato C. Mora M. Morandi L. Bruno C. Santoro L. Pegoraro E. Mercuri E. Bertini E. D'Amico A. Neuromuscular Disord. 2013;23:229–238. doi: 10.1016/j.nmd.2012.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujise K. Noguchi S. Takeda T. Int. J. Mol. Sci. 2022;23:6274. doi: 10.3390/ijms23116274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Oca R. Cowling B. S. Laporte J. Int. J. Mol. Sci. 2021;22:11377. doi: 10.3390/ijms222111377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz X. M. Kretz C. Silva-Rojas R. Ochala J. Menuet A. Romero N. B. Cowling B. S. Laporte J. JCI Insight. 2020;5:e137899. doi: 10.1172/jci.insight.137899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. Chen X. Fan X. Zhang Y. Gu J. Fu X. Wang Z. Wang X. Xiao Z. Synapse. 2015;69:67–77. doi: 10.1002/syn.21788. [DOI] [PubMed] [Google Scholar]
- Li Y.-Y. Zhou J.-X. Fu X.-W. Bao Y. Xiao Z. Epilepsy Res. 2022;182:106915. doi: 10.1016/j.eplepsyres.2022.106915. [DOI] [PubMed] [Google Scholar]
- Macia E. Ehrlich M. Massol R. Boucrot E. Brunner C. Kirchhausen T. Dev. Cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Chew H. Y. Lima P. O. D. Cruz J. L. G. Banushi B. Echejoh G. Hu L. Joseph S. R. Lum B. Rae J. O'Donnell J. S. de Long L. M. Okano S. King B. Barry R. Moi D. Mazzieri R. Thomas R. Souza-Fonseca-Guimaraes F. Foote M. McCluskey A. Robinson P. J. Frazer I. H. Saunders N. A. Parton R. G. Dolcetti R. Cuff K. Martin J. H. Panizza B. Walpole E. Wells J. W. Simpson F. Cell. 2020;180:895–914.e27. doi: 10.1016/j.cell.2020.02.019. [DOI] [PubMed] [Google Scholar]
- Tremblay C. S. Chiu S. K. Saw J. McCalmont H. Litalien V. Boyle J. Sonderegger S. E. Chau N. Evans K. Cerruti L. Salmon J. M. McCluskey A. Lock R. B. Robinson P. J. Jane S. M. Curtis D. J. Nat. Commun. 2020;11:6211. doi: 10.1038/s41467-020-20091-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luwor R. Morokoff A. P. Amiridis S. D'Abaco G. Paradiso L. Stylli S. S. Nguyen H. P. T. Tarleton M. Young K. A. O'Brien T. J. Robinson P. J. Chircop M. McCluskey A. Jones N. C. Cancer Invest. 2019;37:1–12. doi: 10.1080/07357907.2019.1582060. [DOI] [PubMed] [Google Scholar]
- Harper C. B. Martin S. Nguyen T. H. Daniels S. J. Lavidis N. A. Popoff M. R. Hadzic G. Mariana A. Chau N. McCluskey A. Robinson P. J. Meunier F. A. J. Biol. Chem. 2011;286:35966–35976. doi: 10.1074/jbc.M111.283879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen D. D. Lieu T. Halls M. L. Veldhuis N. A. Imlach W. L. Mai Q. N. Poole D. P. Quach T. Aurelio L. Conner J. Herenbrink C. K. Barlow N. Simpson J. S. Scanlon M. J. Graham B. McCluskey A. Robinson P. J. Escriou V. Nassini R. Materazzi S. Geppetti P. Hicks G. A. Christie M. J. Porter C. J. H. Canals M. Bunnett N. W. Sci. Transl. Med. 2017;9:eaal3447. doi: 10.1126/scitranslmed.aal3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Beek C. Alriksson L. Palle J. Gustafson A.-M. Grujic M. Melo F. R. Sellin M. E. Pejler G. PLoS One. 2021;16:e0256708. doi: 10.1371/journal.pone.0256708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel J. A. Chau N. Abdel-Hamid M. K. Hu L. von Kleist L. Whiting A. Krishnan S. Maamary P. Joseph S. R. Simpson F. Haucke V. McCluskey A. Robinson P. J. Traffic. 2015;16:635–654. doi: 10.1111/tra.12272. [DOI] [PubMed] [Google Scholar]
- Kaksonen M. Roux A. Nat. Rev. Mol. Cell Biol. 2018;19:313–326. doi: 10.1038/nrm.2017.132. [DOI] [PubMed] [Google Scholar]
- Ferguson S. M. Camilli P. D. Nat. Rev. Mol. Cell Biol. 2012;13:75–88. doi: 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonny B. Burd C. Camilli P. D. Chen E. Daumke O. Faelber K. Ford M. Frolov V. A. Frost A. Hinshaw J. E. Kirchhausen T. Kozlov M. M. Lenz M. Low H. H. McMahon H. Merrifield C. Pollard T. D. Robinson P. J. Roux A. Schmid S. EMBO J. 2016;35:2270–2284. doi: 10.15252/embj.201694613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imoto Y. Raychaudhuri S. Ma Y. Fenske P. Sandoval E. Itoh K. Blumrich E.-M. Matsubayashi H. T. Mamer L. Zarebidaki F. Söhl-Kielczynski B. Trimbuch T. Nayak S. Iwasa J. H. Liu J. Wu B. Ha T. Inoue T. Jorgensen E. M. Cousin M. A. Rosenmund C. Watanabe S. Neuron. 2022;110:2815–2835.e13. doi: 10.1016/j.neuron.2022.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A. I. Methods Mol. Biol. 2008;440:15–33. doi: 10.1007/978-1-59745-178-9_2. [DOI] [PubMed] [Google Scholar]
- Leuven F. V. Cassiman J.-J. Berghe H. V. D. Cell. 1980;20:37–43. doi: 10.1016/0092-8674(80)90232-9. [DOI] [PubMed] [Google Scholar]
- Levitzki A. Willingham M. Pastan I. Proc. Natl. Acad. Sci. U. S. A. 1980;77:2706–2710. doi: 10.1073/pnas.77.5.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi P. K. Jaarsveld P. P. V. Lippoldt R. E. Edelhoch H. Biochemistry. 1981;20:6706–6710. doi: 10.1021/bi00526a028. [DOI] [PubMed] [Google Scholar]
- Marunaka Y. Niisato N. Biochem. Pharmacol. 2003;66:1083–1089. doi: 10.1016/S0006-2952(03)00456-8. [DOI] [PubMed] [Google Scholar]
- Engh R. A. Girod A. Kinzel V. Huber R. Bossemeyer D. J. Biol. Chem. 1996;271:26157–26164. doi: 10.1074/jbc.271.42.26157. [DOI] [PubMed] [Google Scholar]
- Morikawa A. Sone T. Asano T. J. Med. Chem. 1989;32:46–50. doi: 10.1021/jm00121a010. [DOI] [PubMed] [Google Scholar]
- Considine W. J. J. Org. Chem. 1962;27:647–649. doi: 10.1021/jo01049a519. [DOI] [Google Scholar]
- Nordqvist A. Björkelid C. Andaloussi M. Jansson A. M. Mowbray S. L. Karlén A. Larhed M. J. Org. Chem. 2011;76:8986–8998. doi: 10.1021/jo201715x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranovsky A. Schmitt B. Fowler D. J. Schneider B. Synth. Commun. 2003;33:1019–1045. doi: 10.1081/SCC-120016367. [DOI] [Google Scholar]
- Chuzel O. Piva O. Synth. Commun. 2003;33:393–402. doi: 10.1081/SCC-120015768. [DOI] [Google Scholar]
- Dasgupta R. Ghatak U. R. Tetrahedron Lett. 1985;26:1581–1584. doi: 10.1016/S0040-4039(00)98557-5. [DOI] [Google Scholar]
- Olsen D. K. Torian B. E. Morgan C. D. Braun L. L. J. Org. Chem. 1980;45:4049–4052. doi: 10.1021/jo01308a022. [DOI] [Google Scholar]
- Wu Z. Minhas G. S. Wen D. Jiang H. Chen K. Zimniak P. Zheng J. J. Med. Chem. 2004;47:3282–3294. doi: 10.1021/jm0499615. [DOI] [PubMed] [Google Scholar]
- Cardoso D. A. Chau N. Robinson P. J. Methods Mol. Biol. 2021;2233:71–91. doi: 10.1007/978-1-0716-1044-2_5. [DOI] [PubMed] [Google Scholar]
- Robertson M. J. Deane F. M. Robinson P. J. McCluskey A. Nat. Protoc. 2014;9:851–870. doi: 10.1038/nprot.2014.046. [DOI] [PubMed] [Google Scholar]
- McCluskey A. Daniel J. A. Hadzic G. Chau N. Clayton E. L. Mariana A. Whiting A. Gorgani N. N. Lloyd J. Quan A. Moshkanbaryans L. Krishnan S. Perera S. Chircop M. von Kleist L. McGeachie A. B. Howes M. T. Parton R. G. Campbell M. Sakoff J. A. Wang X. Sun J. Robertson M. J. Deane F. M. Nguyen T. H. Meunier F. A. Cousin M. A. Robinson P. J. Traffic. 2013;14:1272–1289. doi: 10.1111/tra.12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson M. J. Hadzic G. Ambrus J. Pomè D. Y. Hyde E. Whiting A. Mariana A. von Kleist L. Chau N. Haucke V. Robinson P. J. McCluskey A. ACS Med. Chem. Lett. 2012;3:352–356. doi: 10.1021/ml200284s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon C. P. Venn-Brown B. Robertson M. J. Young K. A. Chau N. Mariana A. Whiting A. Chircop M. Robinson P. J. McCluskey A. J. Med. Chem. 2012;56:46–59. doi: 10.1021/jm300844m. [DOI] [PubMed] [Google Scholar]
- Hill T. A. Mariana A. Gordon C. P. Odell L. R. Robertson M. J. McGeachie A. B. Chau N. Daniel J. A. Gorgani N. N. Robinson P. J. McCluskey A. J. Med. Chem. 2010;53:4094–4102. doi: 10.1021/jm100119c. [DOI] [PubMed] [Google Scholar]
- Cousin M. A. Robinson P. J. Neurosci. Lett. 1998;253:1–4. doi: 10.1016/S0304-3940(98)00610-7. [DOI] [PubMed] [Google Scholar]
- Jones N. C. Nguyen T. Corcoran N. M. Velakoulis D. Chen T. Grundy R. O'Brien T. J. Hovens C. M. Neurobiol. Dis. 2012;45:897–901. doi: 10.1016/j.nbd.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Guo S. Zhang X. Zheng M. Zhang X. Min C. Wang Z. Cheon S. H. Oak M.-H. Nah S.-Y. Kim K.-M. Biochim. Biophys. Acta, Biomembr. 2015;1848:2101–2110. doi: 10.1016/j.bbamem.2015.05.024. [DOI] [PubMed] [Google Scholar]
- Davies P. J. A. Davies D. R. Levitzki A. Maxfield F. R. Milhaud P. Willingham M. C. Pastan I. H. Nature. 1980;283:162–167. doi: 10.1038/283162a0. [DOI] [PubMed] [Google Scholar]
- Ivanov A. I. Methods Mol. Biol. 2008;440:15–33. doi: 10.1007/978-1-59745-178-9_2. [DOI] [PubMed] [Google Scholar]
- Davies P. J. Cornwell M. M. Johnson J. D. Reggianni A. Myers M. Murtaugh M. P. Diabetes Care. 1984;7(Suppl 1):35–41. [PubMed] [Google Scholar]
- Schlegel R. Dickson R. B. Willingham M. C. Pastan I. H. Proc. Natl. Acad. Sci. U. S. A. 1982;79:2291–2295. doi: 10.1073/pnas.79.7.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odell L. R. Howan D. Gordon C. P. Robertson M. J. Chau N. Mariana A. Whiting A. E. Abagyan R. Daniel J. A. Gorgani N. N. Robinson P. J. McCluskey A. J. Med. Chem. 2010;53:5267–5280. doi: 10.1021/jm100442u. [DOI] [PubMed] [Google Scholar]
- MacGregor K. A. Abdel-Hamid M. K. Odell L. R. Chau N. Whiting A. Robinson P. J. McCluskey A. Eur. J. Med. Chem. 2014;85:191–206. doi: 10.1016/j.ejmech.2014.06.070. [DOI] [PubMed] [Google Scholar]
- Abdel-Hamid M. K. Macgregor K. A. Odell L. R. Chau N. Mariana A. Whiting A. Robinson P. J. McCluskey A. Org. Biomol. Chem. 2015;13:8016–8028. doi: 10.1039/C5OB00751H. [DOI] [PubMed] [Google Scholar]
- Appenzeller S. Balling R. Barisic N. Baulac S. Caglayan H. Craiu D. De Jonghe P. Depienne C. Dimova P. Djémié T. Gormley P. Guerrini R. Helbig I. Hjalgrim H. Hoffman-Zacharska D. Jähn J. Klein K. M. Koeleman B. Komarek V. Krause R. Kuhlenbäumer G. Leguern E. Lehesjoki A.-E. Lemke J. R. Lerche H. Linnankivi T. Marini C. May P. Møller R. S. Muhle H. Pal D. Palotie A. Pendziwiat M. Robbiano A. Roelens F. Rosenow F. Selmer K. Serratosa J. M. Sisodiya S. Stephani U. Sterbova K. Striano P. Suls A. Talvik T. von Spiczak S. Weber Y. Weckhuysen S. Zara F. Abou-Khalil B. Alldredge B. K. Andermann E. Andermann F. Amron D. Bautista J. F. Berkovic S. F. Bluvstein J. Boro A. Cascino G. Consalvo D. Crumrine P. Devinsky O. Dlugos D. Epstein M. P. Fiol M. Fountain N. B. French J. Friedman D. Geller E. B. Glauser T. Glynn S. Haas K. Haut S. R. Hayward J. Helmers S. L. Joshi S. Kanner A. Kirsch H. E. Knowlton R. C. Kossoff E. H. Kuperman R. Kuzniecky R. Lowenstein D. H. McGuire S. M. Motika P. V. Novotny E. J. Ottman R. Paolicchi J. M. Parent J. Park K. Poduri A. Sadleir L. Scheffer I. E. Shellhaas R. A. Sherr E. Shih J. J. Singh R. Sirven J. Smith M. C. Sullivan J. Thio L. L. Venkat A. Vining E. P. G. Von Allmen G. K. Weisenberg J. L. Widdess-Walsh P. Winawer M. R. Allen A. S. Berkovic S. F. Cossette P. Delanty N. Dlugos D. Eichler E. E. Epstein M. P. Glauser T. Goldstein D. B. Han Y. Heinzen E. L. Johnson M. R. Kuzniecky R. Lowenstein D. H. Marson A. G. Mefford H. C. Nieh S. E. O'Brien T. J. Ottman R. Petrou S. Petrovski S. Poduri A. Ruzzo E. K. Scheffer I. E. Sherr E. Am. J. Hum. Genet. 2014;95:360–370. doi: 10.1016/j.ajhg.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boumil R. M. Letts V. A. Roberts M. C. Lenz C. Mahaffey C. L. Zhang Z. Moser T. Frankel W. N. PLoS Genet. 2010;6:e1001046. doi: 10.1371/journal.pgen.1001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson P. J. Sontag J.-M. Liu J.-P. Fykse E. M. Slaughter C. McMahontt H. Südhof T. C. Nature. 1993;365:163–166. doi: 10.1038/365163a0. [DOI] [PubMed] [Google Scholar]
- Quan A. Robinson P. J. Methods Enzymol. 2005;404:556–569. doi: 10.1016/S0076-6879(05)04049-8. [DOI] [PubMed] [Google Scholar]
- Cossar P. J. Cardoso D. Mathwin D. Russell C. C. Chiew B. Hamilton M. P. Baker J. R. Young K. A. Chau N. Robinson P. J. McCluskey A. Eur. J. Med. Chem. 2023;247:115001. doi: 10.1016/j.ejmech.2022.115001. [DOI] [PubMed] [Google Scholar]
- Odell L. R. Chau N. Russell C. C. Young K. A. Gilbert J. Robinson P. J. Sakoff J. A. McCluskey A. ChemMedChem. 2022;17:e202100560. doi: 10.1002/cmdc.202100560. [DOI] [PubMed] [Google Scholar]
- Dunkley P. R. Jarvie P. E. Robinson P. J. Nat. Protoc. 2008;3:1718–1728. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- Daniel J. A. Malladi C. S. Kettle E. McCluskey A. Robinson P. J. Nat. Protoc. 2012;7:1439–1455. doi: 10.1038/nprot.2012.070. [DOI] [PubMed] [Google Scholar]
- Samala R. Willis S. Borges K. Epilepsy Res. 2008;81:119–127. doi: 10.1016/j.eplepsyres.2008.05.001. [DOI] [PubMed] [Google Scholar]
- Chappie J. S. Mears J. A. Fang S. Leonard M. Schmid S. L. Milligan R. A. Hinshaw J. E. Dyda F. Cell. 2011;147:209–222. doi: 10.1016/j.cell.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker J. R. Russell C. C. Gilbert J. Sakoff J. A. McCluskey A. ChemMedChem. 2020;15:490–505. doi: 10.1002/cmdc.201900643. [DOI] [PubMed] [Google Scholar]
- Abdel-Hamid M. K. McCluskey A. Molecules. 2014;19:6609–6622. doi: 10.3390/molecules19056609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyi S. Erman M. Novikov N. A. Aul'chenko I. Vol'pin M. E. Zh. Org. Khim. 1983;19:808–818. [Google Scholar]
- Maltsev O. V. Kucherenko A. S. Beletskaya I. P. Tartakovsky V. A. Zlotin S. G. Eur. J. Org. Chem. 2010;2010:2927–2933. doi: 10.1002/ejoc.201000239. [DOI] [Google Scholar]
- Tichotová L. Matoušová E. Špulák M. Kuneš J. Votruba I. Buchta V. Pour M. Bioorg. Med. Chem. Lett. 2011;21:6062–6066. doi: 10.1016/j.bmcl.2011.08.059. [DOI] [PubMed] [Google Scholar]
- Ullrich F. Breitmaier E. Synthesis. 1983:641–645. doi: 10.1055/s-1983-30457. [DOI] [Google Scholar]
- Kuijl C. Savage N. D. L. Marsman M. Tuin A. W. Janssen L. Egan D. A. Ketema M. van den Nieuwendijk R. van den Eeden S. J. F. Geluk A. Poot A. van der Marel G. Beijersbergen R. L. Overkleeft H. Ottenhoff T. H. M. Neefjes J. Nature. 2007;450:725–730. doi: 10.1038/nature06345. [DOI] [PubMed] [Google Scholar]
- Ricouart A. Gesquiere J. C. Tartar A. Sergheraert C. J. Med. Chem. 1991;34:73–78. doi: 10.1021/jm00105a012. [DOI] [PubMed] [Google Scholar]
- Hoshino Y. Haberaecker W. W. H. Kodama T. Zeng Z. Okahata Y. Shea K. J. J. Am. Chem. Soc. 2010;132:13648–13650. doi: 10.1021/ja1058982. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.