

Abstract
This study aimed to develop a physiologically‐based pharmacokinetic pharmacodynamic (PBPK/PD) parent‐metabolite model of edoxaban, an oral anticoagulant with a narrow therapeutic index, and to predict pharmacokinetic (PK)/PD profiles and potential drug–drug‐disease interactions (DDDIs) in patients with renal impairment. A whole‐body PBPK model with a linear additive PD model of edoxaban and its active metabolite M4 was developed and validated in SimCYP for healthy adults with or without interacting drugs. The model was extrapolated to situations including renal impairment and drug‐drug interactions (DDIs). Observed PK and PD data in adults were compared with predicted data. The effect of several model parameters on the PK/PD response of edoxaban and M4 was investigated in sensitivity analysis. The PBPK/PD model successfully predicted PK profiles of edoxaban and M4 as well as anticoagulation PD responses with or without the influence of interacting drugs. For patients with renal impairment, the PBPK model successfully predicted the fold change in each impairment group. Inhibitory DDI and renal impairment had a synergistic effect on the increased exposure of edoxaban and M4, and their downstream anticoagulation PD effect. Sensitivity analysis and DDDI simulation show that renal clearance, intestinal P‐glycoprotein activity, and hepatic OATP1B1 activity are the major factors affecting edoxaban‐M4 PK profiles and PD responses. Anticoagulation effect induced by M4 cannot be ignored when OATP1B1 is inhibited or downregulated. Our study provides a reasonable approach to adjust the dose of edoxaban in several complicated scenarios especially when M4 cannot be ignored due to decreased OATP1B1 activity.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
A physiologically‐based pharmacokinetic (PBPK) model of edoxaban has been previously developed and applied for the prediction of its pharmacokinetic (PK) properties. However, the pharmacodynamic (PD) effect of its active metabolites and drug–drug‐disease interactions (DDDIs) were not implemented in the model.
WHAT QUESTION DID THIS STUDY ADDRESS?
A PBPK parent‐metabolite model with a linear additive PD model was developed for predicting edoxaban and its active metabolite M4 anticoagulation responses in DDDIs. Factors affecting PK/PD responses were further investigated by sensitivity analysis.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Inhibitory drug‐drug interactions and renal impairment had a synergistic effect on the increased exposure of edoxaban and M4, and their anticoagulation effect. Renal clearance, intestinal P‐glycoprotein, and hepatic OATP1B1 are the major factors affecting the anticoagulation effect of edoxaban. Despite of a very low abundance generally, M4 cannot be ignored when OATP1B1 is inhibited or downregulated.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
This PBPK/PD model could be applied for dose adjustment of edoxaban in complicated scenarios especially when M4 cannot be ignored due to decreased OATP1B1 activity.
INTRODUCTION
Edoxaban, a direct factor Xa inhibitor, is the latest of the direct‐acting oral anticoagulants (DOACs) with a narrow therapeutic index. Edoxaban given at a dose of 30 or 60 mg once daily was approved for the stroke and systemic embolism prevention in patients with nonvalvular atrial fibrillation and the treatment and secondary prevention of venous thromboembolism. 1 Although compared with warfarin, edoxaban has a rapid onset and offset of action, few drug and food interactions, and predictable pharmacokinetics (PKs) with no need of routine monitoring of anticoagulation activity. Analysis of clinical PK/pharmacodynamic (PK/PD) studies implies the need for dose adjustment in patients with impaired renal function or under the influence of drug–drug interactions (DDIs). 2 , 3
Edoxaban is rapidly absorbed after oral administration with a time to peak plasma concentration of 1–2 h and an absolute oral bioavailability of 61.8%. Its absorption is not affected by food. 3 Approximately 50% of edoxaban is excreted by the kidneys and the remaining 50% via the hepatobiliary system. Edoxaban is metabolized in the liver by carboxylesterase 1 (CES1; major) and cytochrome P450 3A4 (CYP3A4; minor). In healthy subjects, the major metabolite M4, formed by CES1, is present in plasma at less than 10% of the total edoxaban exposure. 4 , 5 Previous in vitro studies confirmed that edoxaban is a substrate of intestinal P‐glycoprotein (P‐gp), whereas M4 is a substrate of hepatic organic anion transporting polypeptide 1B1 (OATP1B1). 6 , 7 Different from other members of DOACs (e.g., rivaroxaban and apixaban), edoxaban's major metabolite M4 is biologically active. The half‐maximal inhibitory concentration (IC50) on factor Xa is 3 and 1.8 nmol/L for edoxaban and M4, respectively. 4 Although M4 is generally not expected to contribute to the pharmacological effects of edoxaban due to its low abundance and high protein binding, in cases such as severe renal dysfunction and DDIs, 8 , 9 plasma exposure of M4 as well as the ratio of M4 to edoxaban is significantly elevated and the anticoagulation effect of M4 might not be ignored.
Edoxaban is generally well‐tolerated; however, accumulative evidence also suggested that DDIs and renal impairment could significantly increase edoxaban exposure and subsequently lead to severe adverse events, such as major bleeding. 10 , 11 Although the approved edoxaban label recommended reduction to half the approved dose in the above situations, it is still challenging to reasonably adjust the dose under the circumstances when DDIs and renal impairment co‐exist, especially if anticoagulation effect of M4 needs to be considered.
The physiologically‐based pharmacokinetic (PBPK) model can be used to predict potential DDIs and parent‐metabolite PK profiles in special populations especially when several complicated scenarios co‐exist, which made drug dosing more challenging. 12 Several in vitro in vivo extrapolation approaches have been proposed to predict the in vivo drug metabolism and transport and DDIs using data from the in vitro systems, 13 such as pooled human liver microsomes, 14 P‐gp/OATP overexpressing cell lines, and caco‐2 cell monolayers. 6 With the advancement of these methods, prediction of parent‐metabolite PKs, DDIs, and other complicated scenarios using mechanism‐based PBPK modeling was made much more feasible and accurate.
Prediction of the PD profiles of edoxaban considering its active metabolite M4 in special clinical scenarios using PBPK/PD modeling was not reported previously, to the best of our knowledge. The present study aimed to develop a PBPK/PD parent‐metabolite model for edoxaban, and investigate the impact of renal impairment and DDIs on edoxaban‐M4 PKs and the downstream anticoagulation effect. The whole‐body PBPK/PD model for healthy subjects was first established in SimCYP and extrapolated to renal impairment and DDI situations. Sensitivity analysis was further conducted to investigate the primary factors affecting the PKs and PDs of edoxaban. Finally, the contribution of M4 to the PD response of edoxaban in special scenarios can be estimated.
METHODS
PBPK model development
The SimCYP (version 21; SimCYP Limited) population‐based absorption, distribution, metabolism, and excretion (ADME) simulator platform was used to develop the PBPK models for edoxaban and M4 and run the subsequent simulations. The PBPK model was first developed by using drug‐dependent and population‐dependent parameters, followed by verification through the comparison of the predicted exposures and the observed data from healthy subjects after intravenous and oral administrations. The validated model was used to predict exposure of edoxaban and M4 under the influence of renal impairment and DDIs. Each scenario was simulated with 10 trials containing 10 subjects (10 × 10) randomly selected by the simulator. Interindividual variabilities of model parameters were incorporated in this PBPK model using the SimCYP simulator default values and the proportion of female subjects was controlled to match the observed data. All simulations after oral administrations were performed under fast conditions as the way PK studies were conducted.
PBPK modeling in healthy subjects
The mechanistic PBPK model was first constructed to describe the PK profiles of edoxaban in healthy subjects using the default SimCYP virtual Sim‐Healthy Population (Sim‐Chinese Healthy Volunteers was chosen for simulation in the Chinese population). The model was built based on physicochemical properties, in vitro experiments, and the clinical PK parameters of edoxaban obtained from literature and in silico prediction (Table 1). The oral absorption of edoxaban was modeled mechanistically using the Advanced Dissolution, Absorption and Metabolism model in SimCYP, which divides the gastrointestinal tract into nine heterogeneous compartments considering several physiological factors, such as size, gut wall permeation, transit time, and transporter (P‐gp) efflux of edoxaban. 15 The effective permeability (P eff,man) was estimated using data obtained from an in vitro permeability study in Caco‐2 cells. 6 P‐gp kinetic parameters maximal rate of metabolism and kinetic metabolite were calculated using the three‐compartment model 16 by fitting Caco‐2 data. 6
TABLE 1.
Parameter‐values used for PBPK/PD models of edoxaban and M4 in SimCYP.
| Parameter | Edoxaban | M4 |
|---|---|---|
| Physiochemical and blood binding | ||
| Molecular weight (g/mol) | 548.6 (DrugBank) | 521.0 |
| logP | 1.61 (DrugBank) | 1.68 (ACD) |
| Compound type | Monoprotic base | Monoprotic acid |
| pKa | 6.7 (DrugBank) | 4.09 (ACD) |
| Blood‐to‐plasma ratio | 0.96 17 | 0.96 (assumed to be the same as edoxaban) |
| f u | 0.45 (Drug label) | 0.2 4 |
| Absorption model | ADAM |
|---|---|
| f u, gut predicted | 0.24 |
| P eff,man type | Regional |
| P Caco‐2 (10−6 cm/s) | 10.3 6 |
| Input form | Solid formulation |
| Formulation | Immediate release (IR) |
| Total solubility in segment (mg/mL) | |
| Stomach | 4.40 17 |
| Duodenum | 1.80 17 |
| Jejunum | 0.54 17 |
| Ileum | 0.14 17 |
| Colon | 0.6 17 |
| Distribution model | Full PBPK model | Full PBPK model |
|---|---|---|
| V ss input type | Predicted using Method 1 | Predicted using Method 2 |
| V ss predicted (L/kg) | 1.04 | 0.16 |
| Tissue to plasma partition coefficients | ||
| Adipose | 1.30 | 0.06 |
| Skin | 1.08 | 0.35 |
| Gut | 1.47 | 0.22 |
| Liver | 1.30 | Permeability limited |
| Lungs | 0.68 | 0.28 |
| Heart | 0.84 | 0.22 |
| Kidneys | 1.02 | 0.20 |
| Spleen | 1.03 | 0.17 |
| Muscle | 1.01 | 0.10 |
| Bone | 1.64 | 0.13 |
| Brain | 1.78 | 0.11 |
| Elimination | ||
| CYP3A4 CLint (μL/min/mg protein) | 1.1 (See text) | |
| CES1 CLint (μL/min/mg protein) | 2.2, forms metabolite M4 | |
| Biliary CLint (μL/min/106) | 1.1 (See text) | 0.4 (Optimized) |
| Renal clearance (L/h) | 10.7 18 | 1 (Optimized) |
| Transporter kinetics | ||
| Intestines | ||
| P‐gp | ||
| J max (pmol/min) | 108 (See text) | |
| K m (μM) | 9.1 (See text) | |
| K i value of ketoconazole (μM) | 0.17 29 | |
| K i value of erythromycin (μM) | 1.1 (Optimized) | |
| Indmax value of rifampicin | 4.01 30 | |
| IndC50 value of rifampicin (μM) | 0.0639 30 | |
| Liver | ||
| CLPD (mL/min/106 cells) | 0.001 (See text) | |
| OATP1B1 | ||
| CLint,T (μL/min/106 cells) | 4.6 (See text) | |
| Scaling factor | 22 19 | |
| K i value of cyclosporine (μM) | 0.014 34 | |
| Indmax value of rifampicin | 2.23 31 | |
| IndC50 value of rifampicin (μM) | 0.0639 31 | |
| Pharmacodynamic | ||
| PD input type | Free concentration | Free concentration |
| PK/PD input compartment | Plasma | Plasma |
| Effect model | Linear | Linear |
| Slope for PT | 20 | 33 |
| Slope for aPTT | 56 | 93 |
| Baseline model | Additive | Additive |
Abbreviations: ADAM, Advanced Dissolution, Absorption and Metabolism; CLint, intrinsic clearance; f u, fraction of unbound drug in plasma; Indmax, maximal fold induction; J max, maximal flux value; K i, inhibition constant; K m, kinetic metabolite; PBPK, physiologically‐based pharmacokinetic; PD, pharmacodynamic; Peff, effective permeability; PK, pharmacokinetic; Vss, volume of distribution at steady state.
The distribution of edoxaban in the body was described with a full PBPK model. The tissue‐plasma partition (k p) coefficients were predicted using the Poulin and Theil method (Method 1 in SimCYP). The k p scalar parameter was set to 1 as the default value. The fraction of unbound drug in plasma (f u) was 0.45. The intrinsic hepatic metabolic clearance was divided into two parts, clearance through CES1 (CLint,CES1) to form M4 and clearance through CYP3A4 (CLint,CYP3A4). CLint,CES1 was assumed to be two times of CLint,CYP3A4 according to mass balance study. 5 Because the involvement of biliary excretion of edoxaban was calculated to be 10% (the calculation was based on 49.1% − [100% − 61.8%]) = 10.9%, where 49% represents the amount of drug in feces, and [100% − 61.8%] represents the amount of drug not absorbed and directly excreted in feces) 17 and about 10% of the total dose was metabolized in the liver, 4 , 5 we assumed that the hepatic biliary clearance was the same as the metabolic clearance. Renal clearance of edoxaban was based on the results of a single intravenous infusion dose of 30 mg edoxaban in a clinical study. 18
The disposition characteristics of M4 were integrated into the above developed PBPK model. The formation of M4 was described as mediated by CES1. Table 1 also presents the physicochemical, biopharmaceutical, and PK parameters of M4 obtained from literature and in silico prediction. A full PBPK distribution was modeled for M4; volume of distribution at steady state and k p coefficients were predicted using the Rodgers and Rowland method (Method 2 in SimCYP) and f u was 0.2. The elimination properties of M4 were not well‐studied. Clearance of hepatic passive diffusion and active uptake mediated by OATP1B1 were calculated from in vitro studies and the in vitro‐in vivo scaling factor of OATP1B1 clearance was set to be 22 according to previous reports. 19 Mass balance studies indicated that M4 can be detected in both feces and urine, 5 therefore we assumed that M4 was further eliminated by biliary and renal pathway. The renal clearance (CLR,M4) and biliary clearance of M4 (CLB,M4) were optimized by fitting the observed PK profiles of M4 after p.o. administration of 60 mg edoxaban.
Scaling to the renal impairment
The physiological properties related to renal impairment were described using the SimCYP population library files Sim‐Renal Impaired_Mild, Sim‐Renal Impaired_Moderate, and Sim‐Renal Impaired_Severe for mild, moderate, and severe renal impairment, respectively. No modifications were made to these model files. According to the latest review about the effect of chronic kidney disease (CKD) on OATPs activity, expression and activity of hepatic OATPs were decreased at least 20% in CKD rats, and clearance of OATP substrates was also decreased as kidney function declines in clinical trials. Thus, the scaling factor of OATP1B1 clearance was set to be 0.7, 0.6, and 0.5 for mild, moderate, and severe renal impairment, respectively, based on the median values of ratios of clearance with and without CKD for OATP substrates observed in the clinic. 20 , 21 , 22 , 23 Therefore, the effect of renal impairment on hepatic OATP1B1 clearance was integrated in the model for M4 prediction. The model was also validated in renal impairment patients using observed data. 24
DDI models
The PKs of ketoconazole, erythromycin, verapamil, quinidine, cyclosporine (with cyclosporine metabolite AM1), and rifampicin after oral administration were described using the SimCYP compound library files Sim‐ketoconazole‐400 mg q.d., Sim‐Erythromycin, SV‐Verapamil, Sim‐Quinidine, SV‐cyclosporine_Neoral (Wsp‐Cyclosporine_AM1), and Sim‐rifampicin‐MD, respectively. Ketoconazole, erythromycin, and quinidine are potent inhibitors of CYP3A4/P‐gp. Verapamil is a potent inhibitor of P‐gp. Cyclosporine is a strong inhibitor for multiple transporters, including P‐gp and OATP1B1; its metabolite AM1 can also significantly inhibit OATP1B1. Rifampicin is a potent inducer of CYP3A4/P‐gp/OATP1B1/CES1 as well as an inhibitor of OATP1B1. 8 , 25 No modification was made to these model files, except for the inhibition constant (K i) values of transporters listed in Table 1. The K i value of ketoconazole for P‐gp inhibition and that of cyclosporine for OATP1B1 inhibition were based on previous reports. 26 , 27 , 28 , 29 The K i value of erythromycin for P‐gp inhibition was optimized by clinical DDI data, as shown in Table S3. The induction of rifampicin on intestinal P‐gp and hepatic OATP1B1 was described using the maximum effect model, as previously reported. 30 , 31 The induction parameters in SimCYP including Indmax and IndC50 are listed in Table 1. The induction profile of rifampicin on hepatic CES1 was not well‐quantified: previous studies only reported a moderate induction of human CES‐1 gene expression in human hepatocytes by rifampicin at 10 micromolar 25 , 32 ; therefore a conservative assumption of 1.5‐fold induction of hepatic CES1 activity was introduced when co‐administered with multiple doses of rifampicin.
Pharmacokinetic pharmacodynamic model development
Exposure‐response analysis of edoxaban concentration versus PT and aPTT suggested linear relationship of edoxaban concentration against these PD measurements. 33 In the present study, PT and aPTT were selected as the PD markers and the effect of edoxaban and its M4 metabolite on both terms were modeled using a linear additive model (Equation 1). The response of the drug was calculated as the difference between the levels of PD markers after drug administration and those at the baseline (Equation 2).
| (1) |
| (2) |
where E 0 is the baseline of the PD marker before edoxaban administration. E edo and E M4 are the anticoagulant effect of edoxaban and M4, respectively. Slopeedo and SlopeM4 represent the anticoagulant activity of edoxaban and M4, respectively. Cu is the plasma free concentration which is used to link PKs and PDs. The relative value of Slopeedo and SlopeM4 was fixed the same as the ratio of IC50,edo to IC50,M4 according to literature reports, 4 and Slopeedo and SlopeM4 values were further estimated using previous clinical data. 33 All PD model parameters are listed in Table 1.
Data source
PK data for edoxaban and its metabolite M4 in healthy subjects and patients with renal impairment after single drug administration or combined with DDI drugs were collected from literature reports. These data were used to support the development and qualification of the PBPK model. Reports were selected based on the following criteria: (i) edoxaban was orally or intravenously administered in single dose or multiple doses; and (ii) plasma concentration‐time profiles and/or PK parameters are available in the reports.
Analysis of model predictability
The compatibility between the observed mean values determined from clinical trials and the predicted values calculated from the model and their 95% confidence interval (CI) ranges was examined. The observed data were digitized from the literature using the Plot Digitizer software. The accuracy of the prediction was graphically evaluated by examining whether the observed mean value fell within the 95% CI of the predicted value. In addition, the ratio of the predicted mean of PK parameters to the observed one was also compared. Predicted PK parameters (area under the curve [AUC] and maximum plasma concentration [C max]) and PD parameter (AUC of response [AUCR]) were calculated from the simulated plasma concentration‐time profiles and response‐time profiles by noncompartmental analysis. AUC was calculated using the linear trapezoidal rule, with extrapolation to infinity. C max was obtained directly from the plasma concentration‐time profiles. The extent of DDI and renal impairment influence was assessed as the ratio of AUC, C max, or AUCR with or without DDI or renal impairment. The predictions were considered successful if the ratio of predication to observation fell between 0.5 and 2.0. 34
Sensitivity analysis
Sensitivity analysis was conducted in the SimCYP global sensitivity analysis module using the Morries method. The analyzed independent parameters are listed in Table 1, which might affect edoxaban and M4 absorption and elimination. Each independent parameter was varied from 1/5 to five times of the estimated value. The dependent variables included both AUC and C max of edoxaban and M4, and the combined PD effect from edoxaban and M4, which is evaluated as AUC over a dosing interval.
RESULTS
Prediction of PK profiles of edoxaban and M4 in healthy adults
The established PBPK model was first used to simulate the concentration‐time profiles in healthy subjects after both i.v. and p.o. doses of edoxaban at different dose levels. For i.v. dose simulation, healthy subjects were given 30 mg edoxaban. For oral dose simulation, healthy subjects in each dose group were given edoxaban at a single dose of 10, 30, 60, 90, 120, and 150 mg or multiple doses of 60 mg for 4 days. 4 , 33 As shown in Figure 1, the model described the observed PK profile of edoxaban and M4 well across the regimens investigated. The terminal elimination after a single dose and the accumulation after multiple doses can also be well predicted. These results indicate a reasonable assumption of the absorption and elimination mechanism of edoxaban and M4.
FIGURE 1.

Prediction of the pharmacokinetic (PK) profiles for edoxaban and M4 at a series of doses in healthy subjects using physiologically‐based PK modeling. Simulation (mean predictions in black lines and 5th–95th percentiles of predictions in gray shade) of PK profiles was conducted for a single i.v. dose of 30 mg, a single oral dose of 10, 30, 60, 90, 120, and 150 mg and multiple oral doses of 60 mg edoxaban (log scale was on the right top in each dose panel).
Prediction of PK profiles of edoxaban and M4 combined with DDI drugs in healthy subjects and in patients with renal impairment
The mechanistic PBPK model was then applied to simulate the plasma concentration‐time profiles of edoxaban with or without DDI drugs (ketoconazole, erythromycin, cyclosporine, and rifampicin). For ketoconazole DDI prediction, healthy subjects received a single oral dose of 60 mg edoxaban on day 4. In addition, from days 1–7, subjects received 400 mg once daily of ketoconazole. For erythromycin DDI prediction, healthy subjects received a single oral dose of 60 mg edoxaban on day 7, and, from days 1–8, subjects received 500 mg erythromycin four times a day. For cyclosporine DDI prediction, subjects were administered a single oral dose of edoxaban 60 mg concomitant with a single oral dose of cyclosporine 500 mg. 8 For rifampicin DDI prediction, healthy subjects received a single oral dose of 60 mg edoxaban on day 7, and, from days 1–7, subjects received 600 mg rifampicin once daily. 25 As shown in Figure 2, the model‐predicted PK profiles of edoxaban and M4 were comparable with the observed data for both edoxaban alone and in combination with the DDI drugs. Additionally, the effect of DDI drugs (ketoconazole, erythromycin, cyclosporine, rifampicin, quinidine, and verapamil) and renal impairment on fold changes of C max and AUC of edoxaban and M4 were predicted and all the predictions fell within 0.5–2.0 folds of clinic observations, as shown in Table 2 and Table S1. 8 , 24 , 25 , 35 Overall, the present model captures the impact of DDI drugs and renal impairment on the PK profiles of both edoxaban and its metabolite M4.
FIGURE 2.
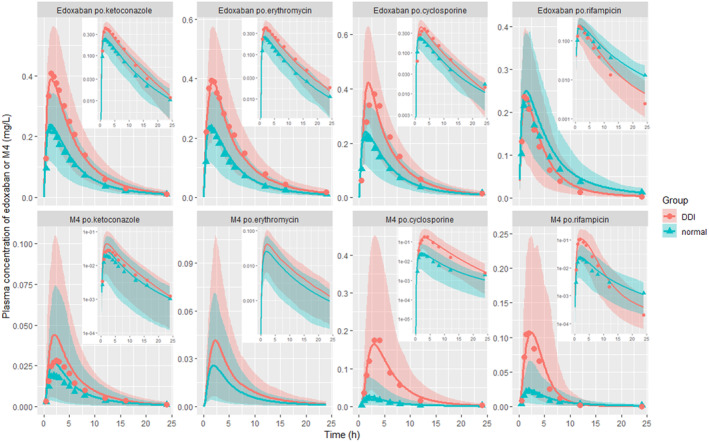
Prediction of the drug–drug interaction (DDI) of edoxaban and M4. The line represents the prediction, the dot represents the observed value, and the shade represents the 5th–95th percentiles of predictions with log scale on the right top in each dose panel (the observed pharmacokinetic profile data of M4 in erythromycin DDI scenario were missing in the cited original literature).
TABLE 2.
Fold change of Edo and M4 exposure (AUC and C max) and PD response (AUCR) in adults with impaired renal function (±DDI) compared with normal renal function without DDI.
| Renal impairment | Observed C max ratio a (Edo/M4) | Observed AUC ratio a (Edo/M4) | Predicted C max ratio a (Edo/M4) | Predicted AUC ratio a (Edo/M4) | Predicted AUCR ratio a |
|---|---|---|---|---|---|
| Normal | NA/NA | NA/NA | 1/1 | 1/1 | 1 |
| Normal + ketoconazole | 1.9/1.6 | 1.9/1.5 | 1.7/1.7 | 1.7/1.7 | 1.7 |
| Normal + erythromycin | 1.6/1.7 | 1.8/1.7 | 1.7/1.7 | 1.7/1.7 | 1.7 |
| Normal + cyclosporine | 1.7/8.7 | 1.7/6.9 | 1.9/7.6 | 1.6/6.7 | 2.0 |
| Mild | 1.1/1.8 | 1.3/2.2 | 1.2/1.7 | 1.5/2.4 | 1.6 |
| Mild + ketoconazole | 1.8/2.6 | 2.5/3.4 | 2.6 | ||
| Mild + erythromycin | 2.0/2.5 | 2.5/3.0 | 2.6 | ||
| Mild + cyclosporine | 2.0/10.3 | 2.2/11.9 | 3.0 | ||
| Moderate | 1.2/2.7 | 1.8/3.7 | 1.2/2.1 | 1.9/3.4 | 2.1 |
| Moderate + ketoconazole | 2.0/3.6 | 2.8/5.8 | 2.8 | ||
| Moderate + erythromycin | 2.0/3.3 | 2.9/5.5 | 3.0 | ||
| Moderate + cyclosporine | 2.1/12.6 | 2.7/17.3 | 3.7 | ||
| Severe | 1.0/2.3 | 1.8/3.9 | 1.3/2.1 | 2.5/4.3 | 2.7 |
| Severe + ketoconazole | 2.2/3.5 | 4.2/6.9 | 4.4 | ||
| Severe + erythromycin | 2.2/3.4 | 4.4/6.0 | 4.7 | ||
| Severe + cyclosporine | 2.3/14.8 | 3.8/22.0 | 5.7 |
Abbreviations: AUC, area under the curve; AUCR, area under the curve ratio; C max, maximum plasma concentration; DDI, drug‐drug interaction; Edo, edoxaban; NA, not analysis; PD, pharmacodynamic.
The ratio represents impaired renal function (±DDI)/normal renal function without DDI.
Prediction of the PD profiles in healthy subjects and patients
After the validation of the PK model, the PK/PD model was further applied to simulate the PD profiles when healthy subjects were orally administered a series of a single dose (30, 60, and 90 mg) of edoxaban 33 as well as administered edoxaban with or without rifampicin. 25 The model‐predicted PT and aPTT were comparable with the observed data for both edoxaban alone and in combination with rifampicin (Figure 3).
FIGURE 3.
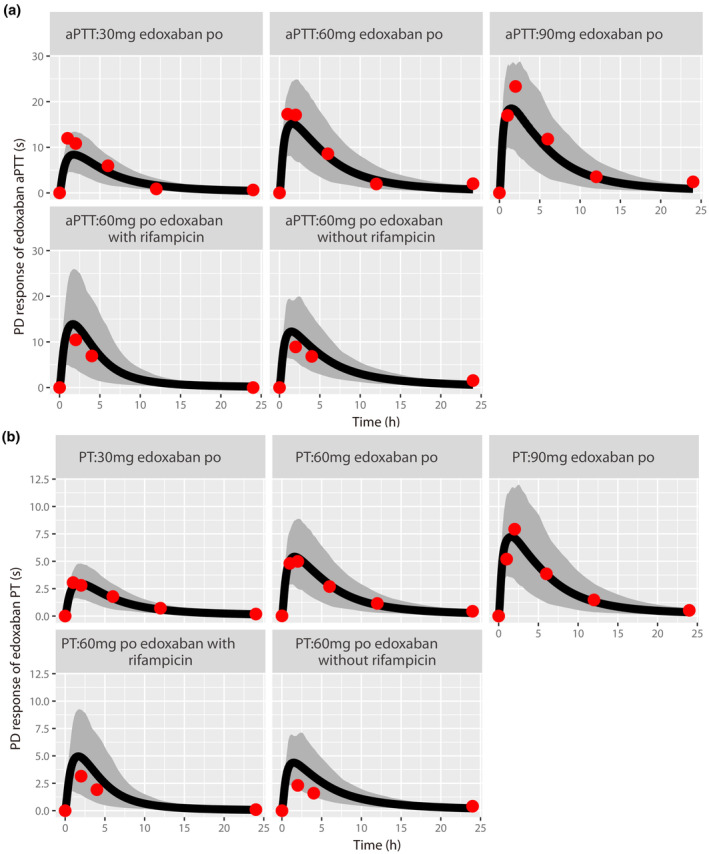
Prediction of pharmacodynamic (PD) profiles for aPTT (a) and PT (b) in healthy subjects using pharmacokinetic PD modeling. Simulation (mean predictions in black lines and 5th–95th percentiles of predictions in gray shade) of PD profiles was conducted for a series single oral dose of 30, 60, and 90 mg edoxaban or a single oral dose of 60 mg edoxaban with or without rifampicin. In the edoxaban + rifampicin group, 600 mg rifampicin was orally administered once daily for 7 days, and a single dose of edoxaban was combined administered on day 7.
DDI combined with renal impairment was further predicted, as shown in Table 2. The results showed that renal impairment combined with CYP3A4/P‐gp inhibitors (ketoconazole and erythromycin) or P‐gp/OATP inhibitor cyclosporine would have a synergistic effect on the increase of edoxaban and M4 exposure as well as the anticoagulation PD response. For renal impairment combined with ketoconazole or erythromycin, the fold change of PD response was the same as the fold change of edoxaban or M4 exposure, and the PD change combined with these drugs can be estimated by edoxaban monitoring. However, for renal impairment combined with cyclosporine, the fold change of PD response was much higher than that of edoxaban exposure due to the significant increase of M4 to edoxaban ratio. In this case, not only edoxaban but also M4 exposure should be considered to estimate the PD response.
Sensitivity analysis
Sensitivity analysis of the solubility of edoxaban in each gastrointestinal tract segment and ADME parameters of edoxaban and M4 on the PK and PD are shown in Figures 4 and 5, respectively. The rank order of the effect of the absorption site solubility of edoxaban on AUC at higher dose was jejunum > duodenum > ileum > colon > stomach. The rank order of the contribution of ADME factors to the AUC of edoxaban was CLR,edo > Jmax,P‐gp,edo > CLint,CES,edo > other factors, and to the AUC of M4 was CLint,OATP1B1,M4 > CLB,M4 > CLint,CES,edo > other factors. The identified rank order of ADME factors on PD effect is CLR,edo > Jmax,P‐gp,edo > CLint,OATP1B1,M4 > other factors, which largely follows that of the parent drug edoxaban with the exception of CLint,OATP1B1,M4 having a stronger effect than CLint,CES,edo. This is not surprising considering the abundance of edoxaban over M4.
FIGURE 4.

Sensitivity analysis of the solubility in gastrointestinal tract segment (a: stomach; b: duodenum; c: jejunum; d: ileum; e: colon) on the area under the curve (AUC) over dose of edoxaban.
FIGURE 5.

Global sensitivity analysis of absorption, distribution, metabolism, and excretion parameters of edoxaban and M4 on AUC of edoxaban (a), area under the curve (AUC) of M4 (b) and AUC of anticoagulation pharmacodynamic response (c). μ*: The average of absolute elementary effects. CLint, intrinsic clearance; J max, maximal flux value; Peff, effective permeability.
DISCUSSION
Edoxaban is the latest approved oral direct factor Xa inhibitor for the treatment of venous thromboembolism. Edoxaban 7 , 19 has a rapid absorption, a linear PKs, and a once daily dosing regimen. Its maximal plasma concentrations are achieved at 1.5 h after oral administration, and has a half‐life of 10–14 h. CYP‐mediated edoxaban metabolism is limited; therefore, a CYP‐related DDI usually is not a concern for clinical use. However, edoxaban is a substrate of P‐glycoprotein, and increased edoxaban exposure was reported when edoxaban was co‐administered with transporter inhibitors. Due to its good PK/PD properties, there are increasing needs of edoxaban used in circumstances such as DDIs and special populations.
The fact that both the parent drug and the metabolite M4 have anticoagulation activity is one of the major differences between edoxaban and other DOACs. Usually, M4 is present at less than 10% of the total edoxaban exposure and not expected to contribute significantly to the overall pharmacological activity of edoxaban, notwithstanding, this is actually not true. In comparison with subjects with normal renal function, the total exposure to M4 in cases of severe renal impairment and cyclosporine interaction were 4.3‐ and 6.7‐fold higher, whereas the fold change for edoxaban was only predicted to be 2.5‐ and 1.6‐fold higher, respectively. Therefore, knowledge of the exposure of edoxaban active metabolite is usually necessary for estimation of its overall pharmacology effect, especially in circumstances such as DDIs and special populations.
To our knowledge, this is the first study reporting a PBPK/PD model of edoxaban and its active metabolite M4, and the first model exploring the pharmacological effect of both edoxaban and M4 in complicated scenarios. Therefore, the model provides a useful quantitative tool with which to evaluate alternative dosing strategies under circumstances where the PKs of both edoxaban and M4 are altered.
This study confirms some findings from previous PK studies and provides new insights. The present model can successfully predict the slightly less‐than‐dose‐proportional dose‐exposure relationship, which is consistent with previous reports. 36 Further sensitivity analysis indicates that edoxaban solubility in jejunum is the major factor that results in the decrease of the bioavailability at higher doses. The present model can also predict the DDI scenarios and renal impairment well across the regimens investigated. Although there are subtle differences between the observed scenarios and the simulation settings for renal impairment, this would make a minimal influence on the accuracy of prediction. In the observed clinical study, renal impairment was categorized according to GFR values as greater than 80 (normal), 50–80 (mild), 30–50 (moderate), and less than 30 with no endstage renal disease (severe) compared with greater than 90 (normal), 60–90 (mild), 30–60 (moderate), and 15–30 (severe) in the SimCYP setting.
Contribution of P‐gp and OATP to edoxaban/M4 PK/PD properties cannot be estimated due to lack of experimental data. As such, sensitivity analysis was conducted to investigate its impact. The results indicated that CLR and intestinal P‐gp transport activity were the most important factors affecting edoxaban exposure; hepatic OATP1B1 was the most important factor affecting M4 exposure. When considering the efficacy of both parent drug and metabolite, CLR of edoxaban, intestinal P‐gp transport of edoxaban, hepatic CES metabolism of edoxaban, and hepatic OATP1B1 uptake of M4 were the most important factors affecting PD markers. Therefore, P‐gp might be the major factor causing significant elevation of edoxaban plasma concentration when combined with CYP3A4/P‐gp inhibitors, whereas OATP1B1 might be the key factor causing disproportional increase of M4 compared with parent drug in the cases of rifampin and cyclosporine DDIs. In addition, the anticoagulation effect of M4 cannot be ignored when OATP1B1 is inhibited or downregulated, especially combined with renal impairment.
According to the sensitivity analysis, renal impairment might significantly increase the edoxaban and M4 exposure as well as the PD response. Generally, half the dose of edoxaban is recommended to be used in DDI scenarios or patients with renal impairment who have creatinine clearance higher than 15 mL/min. However, when a DDI drug has to be dosed together with edoxaban to patients with renal impairment, PBPK simulation in the current analysis shows that the exposure of both parent drug edoxaban and metabolite M4 would be significantly increased and further dose adjustment might be needed in this complex dosing situation. For non‐OATP1B1 inhibitors combined with renal impairment, the dose adjustment could be estimated according to the exposure of edoxaban. However, for OATP1B1 inhibitors combined with renal impairment, not only edoxaban but also metabolite M4 should be considered for the dosage estimation due to the disproportional increase of M4 compared with parent drug. Although mainly supported by modeling and simulation, these results suggested the important role of metabolite M4 in the pharmacological effect of edoxaban in some special cases. Further study of these scenarios in clinical trials or collection of real‐world data from clinical practice is warranted further to understand the extent of the impact and improve rationalized dosing in patients.
In the present study, we developed the first PBPK/PD parent‐metabolite model of edoxaban successfully describing PKs of edoxaban and its active metabolite M4 as well as the PDs after administration of edoxaban in several scenarios, such as DDIs and renal impairment. CLR, intestinal P‐gp activity, and hepatic OATP1B1 activity were the major factors affecting edoxaban and M4 PK and PD profiles. The simulated results indicated that the M4 effect might not be ignored in the cases in which OATP1B1 was inhibited or downregulated, which should be especially considered in clinical practice.
AUTHOR CONTRIBUTIONS
R.X. and W.L. wrote the manuscript. R.X., H.H., and Q.J. designed the research. R.X., W.L., and W.G. performed the research. H.H. and Q.J. analyzed the data.
FUNDING INFORMATION
This work was supported by research grants from National Natural Science Foundation of China (No. 81603184), Natural Science Foundation of Jiangsu Province of China (No. BK20160124), Key Project supported by Medical Science and Technology Development Foundation, Nanjing Department of Health (No. YKK17080), and funding for Clinical Trials from the Affiliated Drum Tower Hospital, Medical School of Nanjing University (No. 2022‐LCYJ‐PY‐41).
CONFLICT OF INTEREST STATEMENT
The authors declared no competing interests for this work.
Supporting information
Tables S1–S3
Data S1
ACKNOWLEDGMENTS
Certara UK Limited (SimCYP Division) granted access to the SimCYP simulators through a sponsored academic license (subject to conditions).
Xu R, Liu W, Ge W, He H, Jiang Q. Physiologically‐based pharmacokinetic pharmacodynamic parent‐metabolite model of edoxaban to predict drug–drug‐disease interactions: M4 contribution. CPT Pharmacometrics Syst Pharmacol. 2023;12:1093‐1106. doi: 10.1002/psp4.12977
Contributor Information
Hua He, Email: huahe_cpupk@cpu.edu.cn.
Qing Jiang, Email: qingj@nju.edu.cn.
REFERENCES
- 1. Stacy ZA, Call WB, Hartmann AP, Peters GL, Richter SK. Edoxaban: a comprehensive review of the pharmacology and clinical data for the Management of Atrial Fibrillation and Venous Thromboembolism. Cardiol Ther. 2016;5(1):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bhatt MD, Portman MA, Berger F, et al. ENNOBLE‐ATE trial: an open‐label, randomised, multi‐Centre, observational study of edoxaban for children with cardiac diseases at risk of thromboembolism. Cardiol Young. 2021;31(8):1213‐1219. [DOI] [PubMed] [Google Scholar]
- 3. Corsini A, Ferri N, Proietti M, Boriani G. Edoxaban and the issue of drug–drug interactions: from pharmacology to clinical practice. Drugs. 2020;80(11):1065‐1083. [DOI] [PubMed] [Google Scholar]
- 4. Parasrampuria DA, Truitt KE. Pharmacokinetics and pharmacodynamics of edoxaban, a non‐vitamin K antagonist Oral anticoagulant that inhibits clotting factor Xa. Clin Pharmacokinet. 2016;55(6):641‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bathala MS, Masumoto H, Oguma T, He L, Lowrie C, Mendell J. Pharmacokinetics, biotransformation, and mass balance of edoxaban, a selective, direct factor Xa inhibitor, in humans. Drug Metab Dispos. 2012;40(12):2250‐2255. [DOI] [PubMed] [Google Scholar]
- 6. Mikkaichi T, Yoshigae Y, Masumoto H, et al. Edoxaban transport via P‐glycoprotein is a key factor for the drug's disposition. Drug Metab Dispos. 2014;42(4):520‐528. [DOI] [PubMed] [Google Scholar]
- 7. Vandell AG, Lee J, Shi M, Rubets I, Brown KS, Walker JR. An integrated pharmacokinetic/pharmacogenomic analysis of ABCB1 and SLCO1B1 polymorphisms on edoxaban exposure. Pharmacogenomics J. 2018;18(1):153‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Parasrampuria DA, Mendell J, Shi M, Matsushima N, Zahir H, Truitt K. Edoxaban drug–drug interactions with ketoconazole, erythromycin, and cyclosporine. Br J Clin Pharmacol. 2016;82(6):1591‐1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jonsson S, Simonsson U, Miller R, Karlsson M. Population pharmacokinetics of edoxaban and its main metabolite in a dedicated renal impairment study. J Clin Pharmacol. 2015;55(11):1268‐1279. [DOI] [PubMed] [Google Scholar]
- 10. Ferri N, Colombo E, Tenconi M, Baldessin L, Corsini A. Drug–drug interactions of direct Oral anticoagulants (DOACs): from pharmacological to clinical practice. Pharmaceutics. 2022;14(6):1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shroff GR, Stoecker R, Hart A. Non‐vitamin K‐dependent Oral anticoagulants for nonvalvular atrial fibrillation in patients with CKD: pragmatic considerations for the clinician. Am J Kidney Dis. 2018;72(5):717‐727. [DOI] [PubMed] [Google Scholar]
- 12. Hartmanshenn C, Scherholz M, Androulakis IP. Physiologically‐based pharmacokinetic models: approaches for enabling personalized medicine. J Pharmacokinet Pharmacodyn. 2016;43(5):481‐504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Storelli F, Yin M, Kumar AR, et al. The next frontier in ADME science: predicting transporter‐based drug disposition, tissue concentrations and drug–drug interactions in humans. Pharmacol Ther. 2022;238:108271. [DOI] [PubMed] [Google Scholar]
- 14. Zhou S, Yung Chan S, Cher Goh B, et al. Mechanism‐based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin Pharmacokinet. 2005;44(3):279‐304. [DOI] [PubMed] [Google Scholar]
- 15. Willemin ME, Zannikos P, Mannens G, de Zwart L, Snoeys J. Prediction of drug–drug interactions after Esketamine intranasal administration using a physiologically based pharmacokinetic model. Clin Pharmacokinet. 2022;61(8):1115‐1128. [DOI] [PubMed] [Google Scholar]
- 16. Tachibana T, Kitamura S, Kato M, et al. Model analysis of the concentration‐dependent permeability of P‐gp substrates. Pharm Res. 2010;27(3):442‐446. [DOI] [PubMed] [Google Scholar]
- 17. Kato T, Mikkaichi T, Yoshigae Y, et al. Quantitative analysis of an impact of P‐glycoprotein on edoxaban's disposition using a human physiologically based pharmacokinetic (PBPK) model. Int J Pharm. 2021;597:120349. [DOI] [PubMed] [Google Scholar]
- 18. Matsushima N, Lee F, Sato T, Weiss D, Mendell J. Bioavailability and safety of the factor Xa inhibitor edoxaban and the effects of quinidine in healthy subjects. Clin Pharmacol Drug Dev. 2013;2(4):358‐366. [DOI] [PubMed] [Google Scholar]
- 19. Chothe PP, Wu SP, Ye Z, Hariparsad N. Assessment of transporter‐mediated and passive hepatic uptake clearance using rifamycin‐SV as a pan‐inhibitor of active uptake. Mol Pharm. 2018;15(10):4677‐4688. [DOI] [PubMed] [Google Scholar]
- 20. Drozdzik M, Oswald S, Drozdzik A. Impact of kidney dysfunction on hepatic and intestinal drug transporters. Biomed Pharmacother. 2021;143:112125. [DOI] [PubMed] [Google Scholar]
- 21. Miners JO, Yang X, Knights KM, Zhang L. The role of the kidney in drug elimination: transport, metabolism, and the impact of kidney disease on drug clearance. Clin Pharmacol Ther. 2017;102(3):436‐449. [DOI] [PubMed] [Google Scholar]
- 22. Nolin TD, Naud J, Leblond FA, Pichette V. Emerging evidence of the impact of kidney disease on drug metabolism and transport. Clin Pharmacol Ther. 2008;83(6):898‐903. [DOI] [PubMed] [Google Scholar]
- 23. Tan ML, Yoshida K, Zhao P, et al. Effect of chronic kidney disease on nonrenal elimination pathways: a systematic assessment of CYP1A2, CYP2C8, CYP2C9, CYP2C19, and OATP. Clin Pharmacol Ther. 2018;103(5):854‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. FDA . Clinical Pharmacology and Biopharmaceutics Review(s). 2014; Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206316Orig1Orig2s000ClinPharmR.pdf
- 25. Mendell J, Chen S, He L, Desai M, Parasramupria DA. The effect of rifampin on the pharmacokinetics of edoxaban in healthy adults. Clin Drug Investig. 2015;35(7):447‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kiso S, Cai SH, Kitaichi K, et al. Inhibitory effect of erythromycin on P‐glycoprotein‐mediated biliary excretion of doxorubicin in rats. Anticancer Res. 2000;20(5A):2827‐2834. [PubMed] [Google Scholar]
- 27. Amundsen R, Christensen H, Zabihyan B, Åsberg A. Cyclosporine a, but not tacrolimus, shows relevant inhibition of organic anion‐transporting protein 1B1‐mediated transport of atorvastatin. Drug Metab Dispos. 2010;38(9):1499‐1504. [DOI] [PubMed] [Google Scholar]
- 28. Varma MV, Bi YA, Kimoto E, Lin J. Quantitative prediction of transporter‐ and enzyme‐mediated clinical drug–drug interactions of organic anion‐transporting polypeptide 1B1 substrates using a mechanistic net‐effect model. J Pharmacol Exp Ther. 2014;351(1):214‐223. [DOI] [PubMed] [Google Scholar]
- 29. Cheong EJY, Teo DWX, Chua DXY, Chan ECY. Systematic development and verification of a physiologically based pharmacokinetic model of rivaroxaban. Drug Metab Dispos. 2019;47(11):1291‐1306. [DOI] [PubMed] [Google Scholar]
- 30. Asaumi R, Nunoya KI, Yamaura Y, Taskar KS, Sugiyama Y. Robust physiologically based pharmacokinetic model of rifampicin for predicting drug–drug interactions via P‐glycoprotein induction and inhibition in the intestine, liver, and kidney. CPT Pharmacometrics Syst Pharmacol. 2022;11(7):919‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Asaumi R, Menzel K, Lee W, et al. Expanded physiologically‐based pharmacokinetic model of rifampicin for predicting interactions with drugs and an endogenous biomarker via complex mechanisms including organic anion transporting polypeptide 1B induction. CPT Pharmacometrics Syst Pharmacol. 2019;8(11):845‐857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Staudinger JL, Xu C, Cui YJ, Klaassen CD. Nuclear receptor‐mediated regulation of carboxylesterase expression and activity. Expert Opin Drug Metab Toxicol. 2010;6(3):261‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen X, Liu D, Wu Y, et al. A single‐dose study investigating the pharmacokinetics and pharmacodynamics of edoxaban at 30–90 mg in healthy Chinese volunteers. Xenobiotica. 2017;47(7):592‐599. [DOI] [PubMed] [Google Scholar]
- 34. Yang Y, Li P, Zhang Z, Wang Z, Liu L, Liu X. Prediction of cyclosporin‐mediated drug interaction using physiologically based pharmacokinetic model characterizing interplay of drug transporters and enzymes. Int J Mol Sci. 2020;21(19):7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mendell J, Zahir H, Matsushima N, et al. Drug–drug interaction studies of cardiovascular drugs involving P‐glycoprotein, an efflux transporter, on the pharmacokinetics of edoxaban, an oral factor Xa inhibitor. Am J Cardiovasc Drugs. 2013;13(5):331‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yin OQ, Miller R. Population pharmacokinetics and dose‐exposure proportionality of edoxaban in healthy volunteers. Clin Drug Investig. 2014;34(10):743‐752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Data S1