

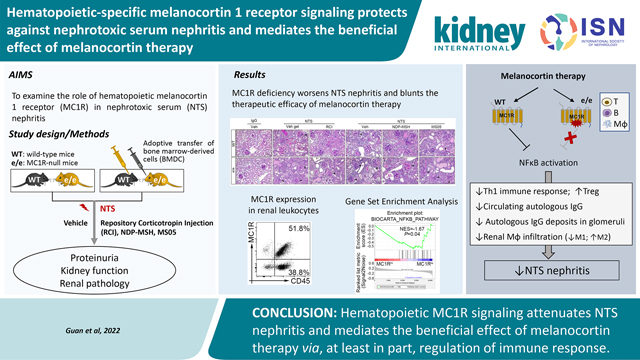
Abstract
The melanocortin hormone system has emerged as a novel therapeutic target for treating refractory glomerular diseases. However, the role of hematopoietic melanocortin 1 receptor (MC1R) signaling remains unknown. Upon insult by rabbit nephrotoxic serum, MC1R null-mutant mice developed more severe crescentic glomerulonephritis than wild-type mice, marked by aggravated proteinuria, kidney dysfunction and histologic lesions. Melanocortin therapy, using Repository Corticotropin Injection (Acthar Gel), the pan-melanocortin receptor agonist NDP-MSH, or the MC1R agonist MS05, ameliorated experimental nephritis in wild-type mice but this effect was blunted in null mice. Exacerbated experimental nephritis in null mice was associated with increased glomerular deposition of autologous IgG and C5b-9, in parallel with higher circulating levels of autologous IgG2c and IgG3. Additionally, the Th1 immune response was potentiated in null mice with experimental nephritis, accompanied by diminished kidney FoxP3+ regulatory T cells. Kidney infiltration of macrophages was also augmented by MC1R deficiency with an enhanced M1 polarization. Moreover, adoptive transfer of syngeneic bone marrow-derived cells from wild-type mice mitigated experimental nephritis in null mice and restored the beneficial efficacy of melanocortins. Mechanistically, MC1R was expressed by diverse subsets of kidney leukocytes, including macrophages, T and B lymphocytes, and inversely associated with the NFκB pathway, a key player in immune responses. MS05 attenuated the production of rabbit IgG-specific IgG2c and IgG3 in cultured wild-type splenocytes, and promoted M2 polarization in M1-primed wild-type macrophages, associated with NFκB inhibition. In contrast in null splenocytes or macrophages, this effect of MS05 was barely detectable, but was mimicked by an NFκB inhibitor. Thus, hematopoietic MC1R signaling attenuates experimental nephritis and mediates the beneficial effect of melanocortin therapy via, in part, by regulating the immune response.
Keywords: crescentic glomerulonephritis, macrophage, lymphocytes, ACTH, immune response, inflammation
Graphical Abstract
Introduction
Glomerular diseases are the third leading cause of end stage renal failure in the US, with no definitive therapy available yet.1 Adrenocorticotropic hormone (ACTH) was the first treatment approved by the US Food and Drug Administration to induce remission of proteinuria due to idiopathic nephrotic syndrome or lupus erythematosus, and was commonly used in the 1950s.2–5 At that time, the mechanism of action was assumed to be mediated indirectly via steroidogenesis.6 Subsequently, with the introduction of synthetic glucocorticoids such as prednisone in the 1960s, clinical use of ACTH became uncommon.7–9 Two different types of ACTH are currently available for clinical use, synthetic ACTH, which contains ACTH1–24, and natural ACTH, known as repository corticotropin injection (RCI) or Acthar gel, which contains a complex mixture of melanocortin peptides, including ACTH1–39.5 In recent years, a growing body of evidence has suggested that ACTH is effective in a variety of glomerular diseases, including minimal change disease,10–12 focal segmental glomerulosclerosis,13, 14 idiopathic membranous nephropathy,15–17 and proliferative glomerulonephritis.18, 19 Moreover, the beneficial action of ACTH is unlikely to result from steroidogenesis, because ACTH therapy is effective in patients resistant to steroids.12, 13 Furthermore, in addition to regulating adrenal production of corticosteroids, ACTH also functions as a major ligand of the melanocortin system, a neuroimmunoendocrine hormone system that is involved in a diverse array of physiological functions including melanogenesis, immunomodulation, energy homeostasis, sexual function and exocrine secretion.20
In agreement with the above clinical findings,20 pre-clinical studies reveal that non-steroidogenic melanocortins, such as α-melanocyte stimulating hormone (MSH) and its peptidomimetics, exert a unique salutary effect in rodent models of glomerular disease, including lipopolysaccharide - or adriamycin-elicited podocytopathy21, 22 and passive Heymann nephritis.23 The mechanism underlying this renoprotective action is not fully understood. Evidence suggests that melanocortin 1 receptor (MC1R) is expressed by glomerular cells, such as podocytes, and that MC1R-specific agonists are able to lessen proteinuria and glomerular injury in experimental membranous nephropathy, implying that they play a protective role via glomerulus-specific MC1R signaling.23 In contrast, selective MC1R agonists barely triggered the cyclic-adenosine monophosphate (cAMP) response downstream of MC1R signaling in cultured wild-type podocytes.24 Moreover, the selective MC1R agonist had minimal effects on proteinuria and glomerular injury in murine models of acute podocytopathy elicited by adriamycin,25 suggesting that glomerular-specific MC1R signaling alone, may not be sufficient for glomerular protection. Also potentially relevant is the fact that most human glomerular diseases involve not only glomerular cell injury but also systemic immune dysregulation.26 Despite extensive investigation, the extent to which hematopoietic MC1R signaling contributes to the renoprotective action of melanocortin therapy remains uncertain. This study aims to address this issue by harnessing MC1Re/e mice (e/e mice; homozygous for the mutant recessive Mc1re allele) with the naturally occurring loss-of-function null mutation of MC1R. Because many murine models of nephrotic glomerulopathy are produced by administration of direct podocytoxic agents or by podocyte depletion with only minimal involvement of immunopathogenic mechanisms,27 this study employed the nephrotoxic serum (NTS) nephritis model, which is characterized by heavy proteinuria and diffuse proliferative crescentic glomerulonephritis, and shares pathologic features of human lupus nephritis.
Methods
More detailed information on methods is provided in the Supplementary Methods.
Animal experimental design
Animal studies were approved by the animal care and use committee of the University of Toledo, and they conformed to US Department of Agriculture regulations and the National Institutes of Health guidelines for humane care and use of laboratory animals. Mice carrying the recessive yellow Mc1re null allele on a C57BL/6 genetic background were acquired from the Jackson Laboratory. They were bred to generate the MC1Re/e (e/e) mice that were homozygous for the mutant recessive Mc1re allele and were identified based on the yellow coat color. Mice carrying the dominant Mc1rE allele had the wild-type (WT) trait of black coat color, and were generated from the same crossings as MC1Re/e mice and they served as WT controls. The murine model of NTS nephritis was established in male WT or e/e mice as previously described, with some modifications,28 followed by treatment with vehicles or melanocortins, including the RCI (Acthar Gel, Mallinckrodt ARD), the pan-melanocortin receptor agonist [Nle, DPhe]-α-MSH (NDP-MSH), and the MC1R agonist MS05 (custom-made peptide, GL Biochem).
Adoptive transfer of bone marrow-derived cells
Bone marrow-derived cells (BMDCs) were freshly prepared from syngeneic donor mice by flushing the dissected femurs and tibiae, and labelled as previously reported with PKH26 (Sigma-Aldrich),29 a red fluorescent cell-labeling dye for in vivo live cell imaging and tracking. WT or e/e mice received adoptive transfer of 1×106 syngeneic BMDCs every 10 days, for a total of 3 times via tail vein infusion, followed by NTS injury and treatment with melanocortins or vehicles.
Statistical analyses
The integrated pixel density of immunoblot bands and mean fluorescence intensity of fluorescence staining were determined using the ImageJ program (National Institutes of Health, Bethesda, MD). All in vitro studies were repeated at least 3 times. Power analysis was performed based on pilot data, using the G*power software to determine the sample size needed to detect a 25% difference in proteinuria between WT and e/e mice upon NTS injury with 80% power and a 2-sided P-value of 0.05. GraphPad Prism 8.0 was used for statistical analysis. Data are expressed as mean± SD (or as mean difference, with 95% confidence intervals [95%CI] for the mean differences). One-way analysis of variance tests were performed to compare means of normally distributed data across several groups, and when these differed significantly, the Student’s t test for equal variance was used for pairwise comparisons. Data from 2 groups were analyzed using an unpaired t test. P<0.05 was considered statistically significant.
Results
MC1R deficiency exacerbates NTS nephritis and lessens the protective efficacy of melanocortin therapy
After 14 days of NTS injury (Figure 1a), vehicle-treated WT mice demonstrated prominent proteinuria, as quantitated by urine albumin-to-creatinine ratios (uACR; Figure 1b), and kidney dysfunction, as evidenced by elevated blood urea nitrogen (BUN) levels (Figure 1c). These functional impairments were associated with histologic evidence of renal injury, including glomerular hypercellularity, crescent formation, mesangial expansion, protein casts, inflammation and fibrosis, as shown by periodic acid–Schiff (PAS) staining (Figure 1d) and evaluated by semi-quantitative morphometric analysis of glomerulonephritis scores, tubulointerstitial nephritis scores, and percentage of crescentic glomeruli (Figure 1e–g). As expected in animals with massive proteinuria, vehicle-treated WT mice displayed prominent podocyte injury, characterized by podocytopenia, marked by loss of glomerular staining for podocyte markers such as wilms’ tumor-1 and podocin, and by de novo expression of podocyte injury marker desmin, as shown by dual-color fluorescent immunostaining for desmin and synaptopodin (Figure 2a). These morphologic findings were further corroborated by immunoblot analysis of isolated glomeruli (Figure 2b) in conjunction with densitometry (Figure 2c and d). The above signs of NTS nephritis were exacerbated in vehicle-treated e/e mice, as compared with vehicle-treated WT mice, with significant differences (P<0.05) in uACR (9.416, 95%CI: 6.521, 12.31), BUN (14.76, 95%CI: 3.668, 25.84), glomerulonephritis score (0.8444, 95%CI: 0.2926, 1.396), tubulointerstitial nephritis score (0.9167, 95%CI: 0.5225, 1.311), and percentage of crescentic glomeruli (17.23, 95%CI: 10.44, 24.03). RCI or NDP-MSH treatment significantly reduced uACRs (Figure 1b) and BUN levels (Figure 1c), improved kidney pathology (Figure 1d–g), and ameliorated podocyte injury (Figure 2) in WT mice after NTS injury. Similar beneficial effects were also achieved, though to a lesser extent, by MS05 (a selective MC1R agonist) in NTS-injured WT mice (Supplementary Figure S1). In contrast, in NTS-injured e/e mice, MS05 and vehicle treatment resulted in comparable effects with regard to uACR (−0.9999, 95%CI: −3.895, 1.896), BUN (0.5617, 95%CI: −10.52, 11.65), glomerulonephritis score (−0.3945, 95%CI: −0.9463, 0.1573), tubulointerstitial nephritis score (−0.100, 95%CI: −0.4942, 0.2942), and percentage of crescentic glomeruli (−1.700, 95%CI: −8.497, 5.097). Given that the CIs for mean differences contain the null value zero, the evidence based on this study was insufficient to conclude that a difference was present between MS05 and vehicle treatment in NTS-injured e/e mice (Figure 1). This finding warrants validation in future larger studies. Likewise, the protective efficacy of RCI and NDP-MSH was blunted in NTS-injured e/e mice, as evidenced by the lower percentage of reduction in uACR and BUN levels (Supplementary Figure S1a and b), and a lesser-fold change in wilms’ tumor-1 and podocin expressions in e/e mice (Supplementary Figure S1c and d).
Figure 1. The e/e loss-of-function mutation of MC1R aggravates proteinuria, kidney injury and dysfunction in murine models of nephrotoxic serum (NTS) nephritis, and blunts the protective efficacy of melanocortin therapy.
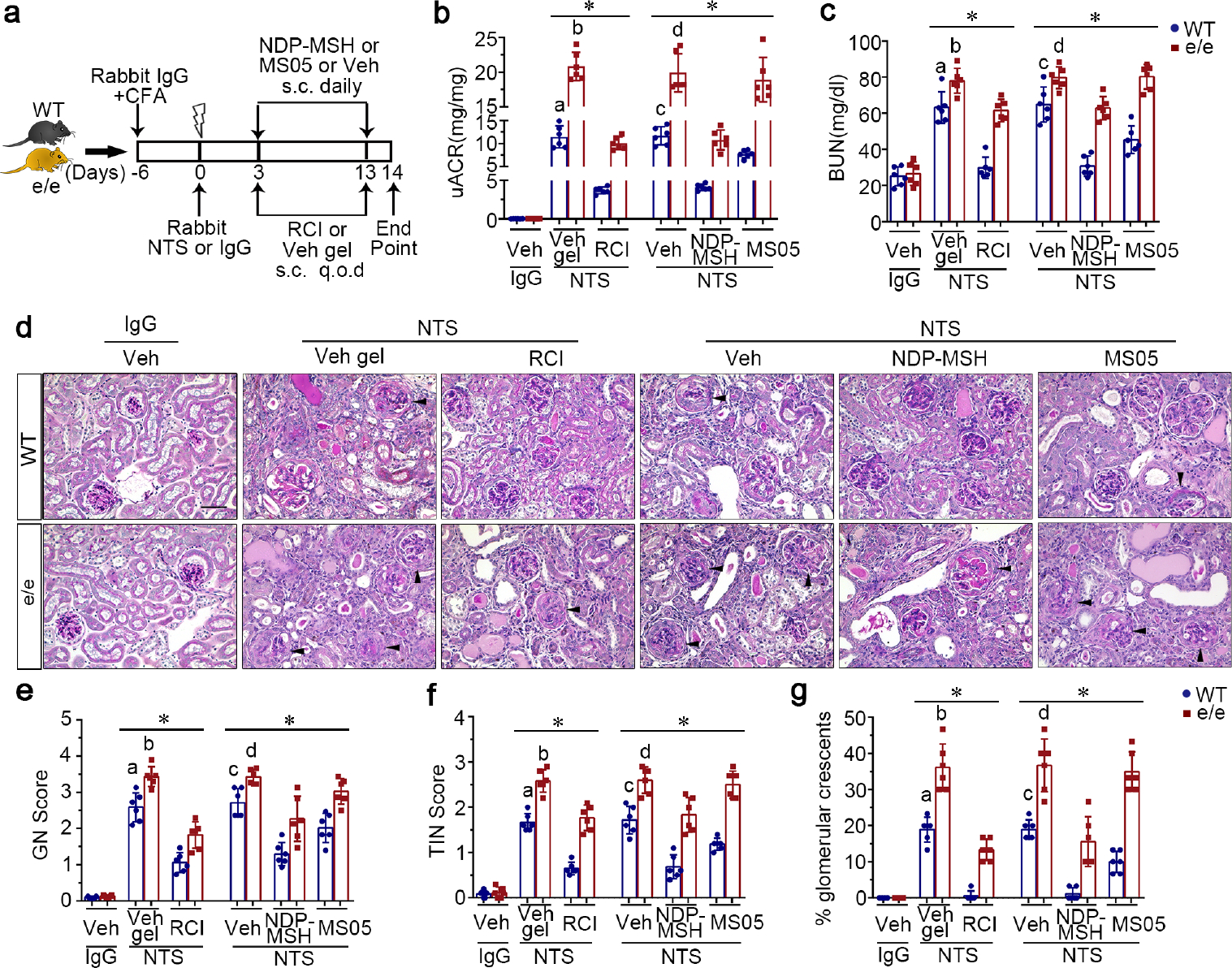
(a) Schematic diagram depicts the animal study design. Wild-type (WT) and MC1R-null (e/e) mice received an intraperitoneal injection of 250μl rabbit IgG emulsified in complete Freund’s adjuvant (CFA) 6 days before a tail vein injection of either rabbit NTS or nonimmune IgG, followed by treatment with repository corticotropin injection (RCI), vehicle gel (Veh gel), the pan-melanocortin receptor agonist [Nle, DPhe]-α-MSH (NDP-MSH), MC1R agonist MS05 (custom-made peptide, GL Biochem), or vehicle (Veh) till day 14. (b) Proteinuria was estimated using the urinary albumin-to-creatinine ratios (uACR). (c) Kidney function was assessed by measuring blood urea nitrogen (BUN) levels in sera. (d) Representative micrographs of periodic acid-Schiff staining of mouse kidneys are shown. Arrowheads indicate glomeruli with crescents (Scale bar = 50 μm). (e~g) Semi-quantitative morphometric analysis of (e) glomerulonephritis (GN) score, (f) tubulointerstitial nephritis (TIN) score, and (g) the percentage of glomeruli with crescents based on periodic acid-Schiff staining of mouse kidneys. *P<0.05 by analysis of variance. aP<0.05 versus e/e group treated with NTS and Veh gel or WT group with NTS and RCI treatment; bP<0.05 versus e/e mice treated with NTS and RCI; cP<0.05 versus e/e mice treated with NTS and Veh or WT mice treated with NTS and NDP-MSH or MS05; dP<0.05 versus e/e mice treated with NTS and NDP-MSH; (n=6). q.o.d., every other day; s.c., subcutaneous. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Figure 2. Melanocortin 1 receptor (MC1R) deficiency exacerbates podocyte injury in murine models of nephrotoxic serum (NTS) nephritis and blunts the protective efficacy of melanocortins.
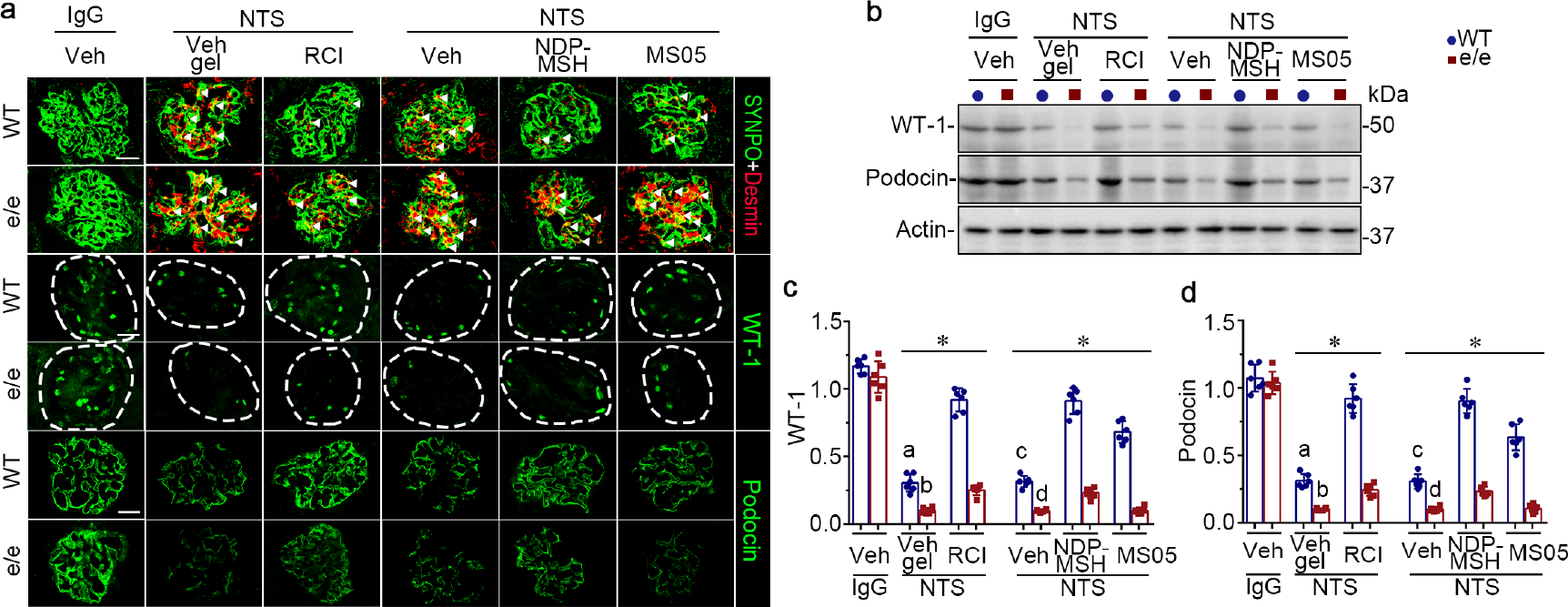
(a) Animals were treated as shown in Figure 1. Representative micrographs show fluorescent immunohistochemistry staining of kidney specimens for indicated proteins, with glomeruli outlined by dashed circles. (bar=20 μm). White arrowheads indicate desmin expression in synaptopodin (SYNPO)-positive podocytes. (b) Representative blots show immunoblot analysis of isolated glomeruli for indicated proteins. (c, d) Estimation of the abundance of wilms’ tumor-1 (WT-1) and podocin expression by densitometric analyses of immunoblots, presented as relative levels normalized to actin. *P<0.05 by analysis of variance. aP<0.01 versus e/e (MC1R-null) group treated with NTS and Vehicle (Veh) gel or wild-type (WT) group with NTS and repository corticotropin injection (RCI) treatment; bP<0.01 versus e/e mice treated with NTS and RCI; cP<0.05 versus e/e mice treated with NTS and Veh or WT mice treated with NTS and pan-melanocortin receptor agonist [Nle, DPhe]-α-MSH (NDP-MSH) or MC1R agonist MS05 (custom-made peptide, GL Biochem); dP<;0.05 versus e/e mice treated with NTS and NDP-MSH; (n=6). To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
MC1R signaling is involved in the humoral immune response of NTS nephritis and contributes to the protective effect of melanocortin therapy
Evidence suggests that podocyte-specific MC1R signaling may protect against glomerular injury.23, 24, 30 To test whether glomerular MC1R signaling is present in NTS nephritis, WT and e/e mice were examined 1 day after NTS injury (Supplementary Figure S2a), an early stage of NTS nephritis when adaptive immunity is unlikely to be prominent. Shown in Supplementary Figures S2 and S3, e/e and WT mice presented a comparable-magnitude albuminuria (0.02507, 95%CI: −0.03803, 0.08816) (Figure S2b), glomerular damage (Figure S2c and d), and podocyte injury (Figure S3a–e) in response to similar NTS insults, as shown by linear deposition to a similar extent of the glomerular basement membrane (GBM)-reactive NTS IgG, together with the terminal complement complex C5b-9 along glomerular capillary loops (Supplementary Figure S3f–h). The negligible effect of MC1R deficiency on proteinuria or glomerular injury at this very early stage of NTS injury may suggest that the aggravated NTS nephritis that develops later in e/e mice is unlikely to be due to a sensitized response of the e/e kidney cells to injury. However, because other possible confounding factors also exist including the early and premature immune response, and the short time to renal injury, this finding needs to be validated in future studies in a purely heterologous model of NTS nephritis.
To determine whether alterations of an extrarenal, MC1R-mediated process contribute to the aggravated NTS nephritis in e/e mice on day 14, kidney specimens were processed for immunofluorescence staining for mouse IgG and C5b-9 (Figure 3a), followed by morphometric measurements of mean fluorescence intensity (Figure 3b and c). As shown in Figure 3, deposition of mouse IgG, together with C5b-9 along glomerular capillary loops, was evident in vehicle-treated WT mice after NTS injury, consistent with an autologous immune nephritis. This effect was markedly enhanced in vehicle-treated e/e mice, suggesting that the humoral immune response to the xenogeneic NTS may be amplified in e/e mice. Melanocortin treatment markedly reduced glomerular deposition of mouse IgG and C5b-9 in WT mice (Figure 3), whereas MC1R deficiency blunted this beneficial effect and largely abrogated the protective action of MS05 (Supplementary Figure S4a and b).
Figure 3. Melanocortin 1 receptor (MC1R) deficiency promotes glomerular deposition of mouse IgG and the terminal complement complex C5b-9 along glomerular capillary loops in murine models of NTS nephritis, and lessens the protective efficacy of melanocortin therapy.
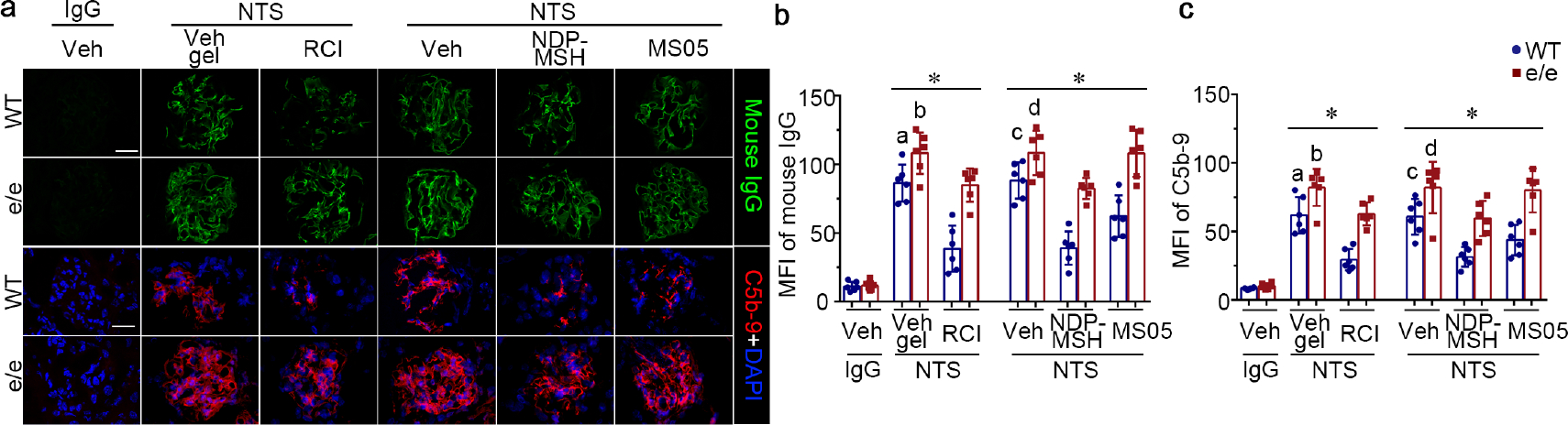
(a) Representative micrographs show immunofluorescence staining of kidney cryosections for mouse IgG or the complement membrane attack complex C5b-9 (bar=20 μm). (b) Arbitrary levels of mean fluorescence intensity (MFI) of mouse IgG staining or (c) C5b-9 staining in glomeruli, as estimated by computerized morphometric analysis. *P<0.05 by analysis of variance. aP<0.05 versus e/e group treated with NTS and Vehicle (Veh) gel or wild-type (WT) group with NTS and repository corticotropin injection (RCI) treatment; bP<;0.05 versus e/e (MC1R-null) mice treated with NTS and RCI; cP<0.05 versus e/e mice treated with NTS and Veh or WT mice treated with NTS and pan-melanocortin receptor agonist [Nle, DPhe]-α-MSH (NDP-MSH) or MC1R agonist MS05 (custom-made peptide, GL Biochem); dP<0.05 versus e/e mice treated with NTS and NDP-MSH; (n=6). To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Different IgG subclasses have variable complement-fixing and opsonizing activities.31 For instance, IgG3 has potent complement-fixing activity, and IgG2a/c has strong Fc gamma receptor (FcγR) affinity. To determine whether the differences in glomerular injury and glomerular accumulation of autologous IgG between WT and e/e mice result from deposition of different IgG subclasses, kidney sections were processed for immunofluorescent staining for mouse IgG1, IgG2c, and IgG3. Shown in Figure 4a, glomerular deposition of IgG2c and IgG3 was more intense in e/e mice than in WT mice after NTS injury. In contrast, glomerular deposition of IgG1 was similar in both the WT and e/e groups (data not shown). To further examine whether a difference in systemic humoral immune responses also contributes, serum levels of mouse anti-rabbit IgG and IgG subclasses were next assayed. As shown in Figure 4b, compared with WT mice, e/e mice had significantly (P<0.05) higher circulating levels of IgG2c (0.8678, 95%CI: 0.1083, 1.627) and IgG3 (0.79, 95%CI: 0.1477, 1.432) after NTS injury, despite similar levels of IgG1 in the 2 groups (0.479, 95%CI: −0.766, 1.724).
Figure 4. Melanocortin 1 receptor (MC1R) deficiency augments glomerular deposition and circulating levels of mouse autologous IgG2c and IgG3 in nephrotoxic serum (NTS) nephritis.
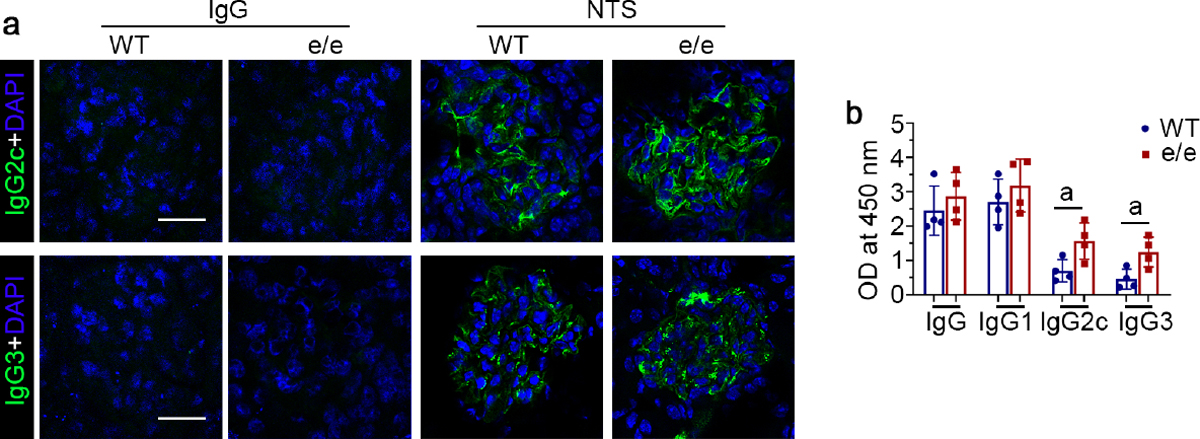
(a) Representative micrographs show immunofluorescence staining of kidney specimens procured from IgG or NTS-injured and vehicle-treated mice for mouse IgG2c and IgG3. (bar=25 μm). (b) Arbitrary levels of rabbit IgG-specific mouse IgG, IgG1, IgG2c and IgG3 in sera collected from vehicle-treated wild-type (WT) or e/e (MC1R-null) mice with NTS nephritis, as determined by enzyme-linked immunosorbent assay of diluted sera. aP<0.05 by unpaired t test (n=4). OD, optical density. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
MC1R deficiency promotes T helper type 1 (Th1) immune response in NTS nephritis
Not only humoral immunity, but also cell-mediated immune response contributes to glomerular injury in NTS nephritis.32 To examine whether MC1R signaling modulates T helper (Th) immunity, signature cytokines of Th immunity were assayed in kidney homogenates. As shown in Figure 5a, compared with WT mice, e/e mice with NTS nephritis displayed higher renal expression of interferon- γ (IFN-γ), and a trend toward increased expression of tumor necrosis factor-α, consistent with an enhanced Th1 immune response. In parallel, the number of regulatory T cells (Treg) in the nephritic kidneys was diminished in e/e mice, compared with that in WT mice, as shown by fluorescent immunohistochemistry staining for the Treg marker FoxP3 (Figure 5b).
Figure 5. Melanocortin 1 receptor (MC1R) deficiency potentiates T helper type 1 (Th1) immune response and diminishes renal regulatory T cells in nephrotoxic serum (NTS) nephritis.
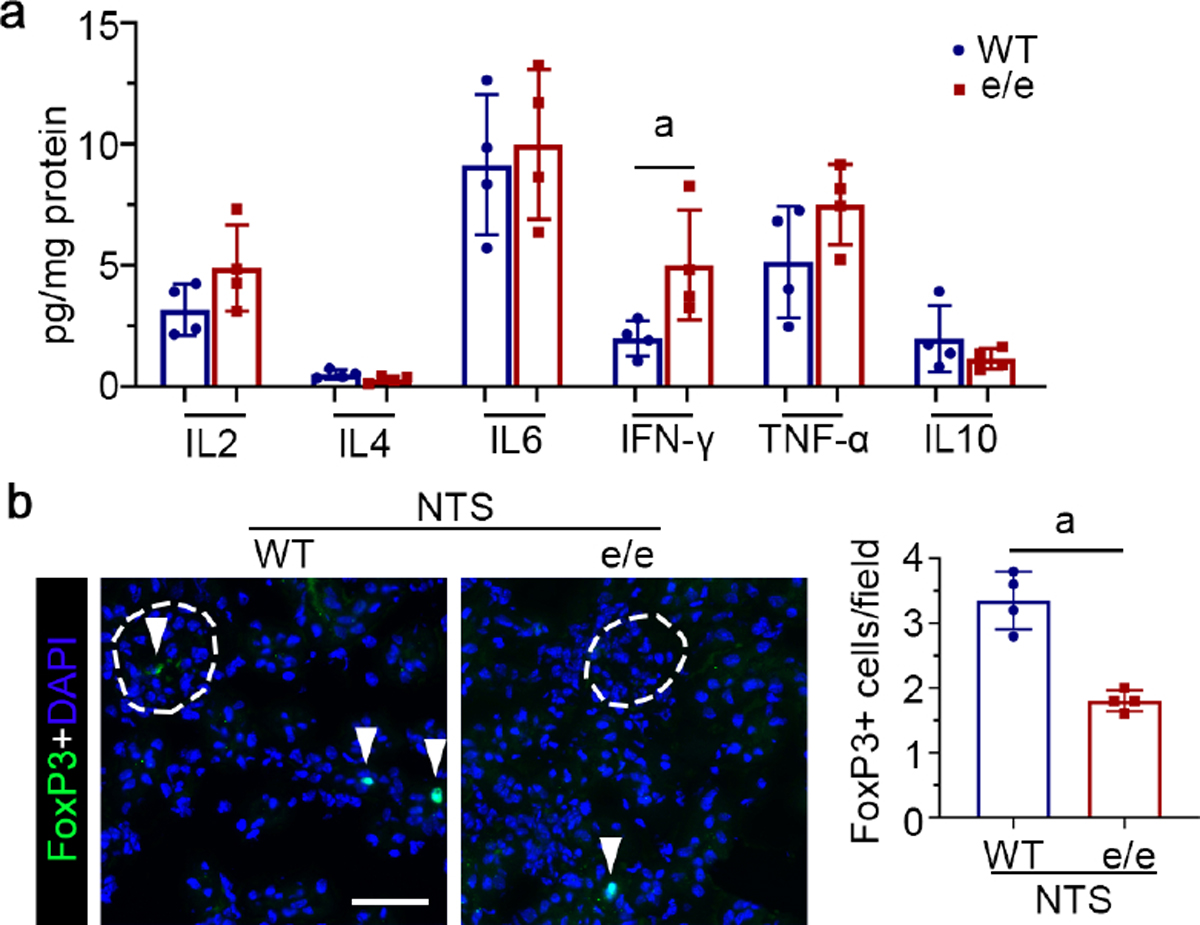
(a) Kidney homogenates derived from NTS-injured vehicle-treated wild-type (WT) or e/e mice (MC1R-null) were processed for profiling of signature cytokines of Th immunity. aP<0.05 by unpaired t test (n=4). (b) Representative micrographs show immunofluorescence staining of kidney cryosections for forkhead box protein P3 (FoxP3), with glomeruli outlined by dashed circles. Arrowheads indicate the positive staining of FoxP3. (bar=50 μm). Absolute counting of FoxP3+ cells per microscopic field. aP<0.05 by unpaired t test (n=4). IFN, interferon; TNF, tumor necrosis factor. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
MC1R signaling mitigates glomerular infiltration of macrophages in NTS nephritis and favors M2 polarization
Macrophage infiltration also plays a key role in NTS nephritis.32 To determine whether MC1R regulation of NTS nephritis was associated with changes in macrophage infiltration, kidney specimens were processed for dual-color fluorescent immunohistochemistry staining for the macrophage marker F4/80 and the M1 macrophage-specific marker inducible nitric oxide synthase (iNOS) or the M2 macrophage marker mannose receptor (MR; Figure 6a), which were barely detectable in normal kidney tissues. NTS injury resulted in evident renal infiltration of F4/80-positive macrophages expressing iNOS or MR in WT mice (Figure 6a). In contrast, MC1R deficiency in e/e mice augmented renal infiltration of F4/80-positive macrophages mostly positive for iNOS upon NTS injury, in association with diminished MR-positive macrophages, suggesting an enhanced proinflammatory M1 polarization (Figure 6a). The morphologic findings were further verified by immunoblot analysis of kidney specimens (Figure 6b and c). Conversely, MC1R activation in NTS-injured WT mice by MS05, RCI, or NDP-MSH mitigated kidney infiltration of macrophages, concomitant with a macrophage polarization skewed toward the anti-inflammatory M2 phenotype, as evidenced by the reduced expression of F4/80, iNOS and interleukin (IL-1β), but elevated expression of MR and arginase-1 on immunoblot analysis (Supplementary Figure S5).
Figure 6. Melanocortin 1 receptor (MC1R) deficiency increases kidney infiltration of macrophages in nephrotoxic serum (NTS) nephritis and enhances M1 polarization.
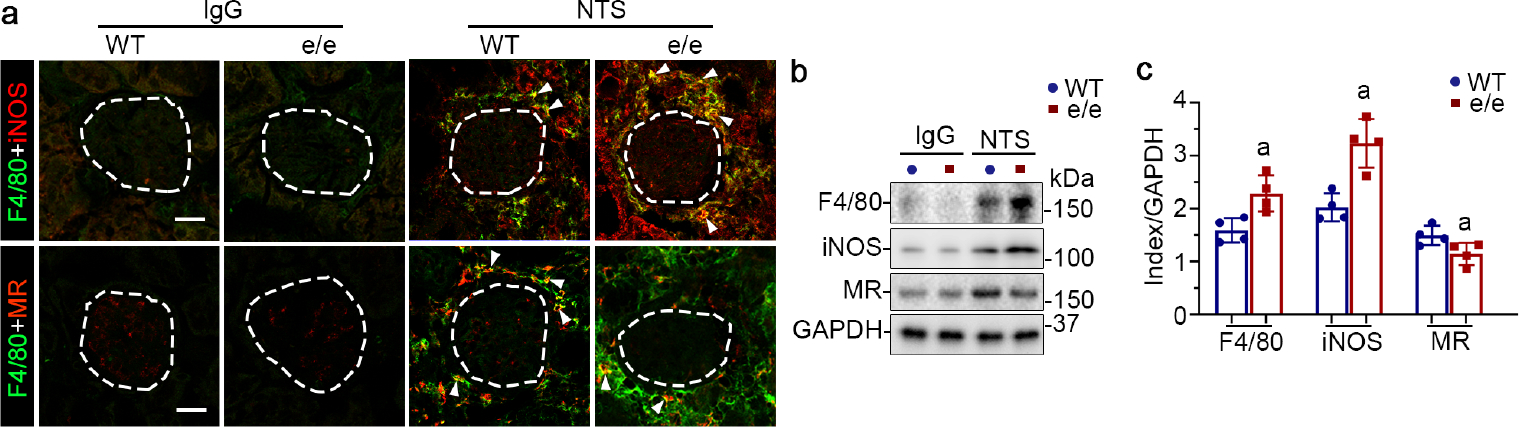
(a) Representative micrographs show dual-color immunofluorescence staining of kidney specimens procured from IgG or NTS-injured and vehicle-treated mice for F4/80 and inducible nitric oxide synthase (iNOS) or mannose receptor (MR), with glomeruli outlined by dashed circles (bar=25 μm). Arrowheads indicate cells positive for dual-color staining. (b) Kidney homogenates were processed for immunoblot analysis for indicated proteins. (c) Estimation of the abundance of indicated proteins in NTS-injured groups by densitometric analyses of immunoblots, expressed as relative levels normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). aP<0.05 versus NTS-injured wild-type (WT) mice for the same protein by unpaired t test (n=4). To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Hematopoietic MC1R signaling protects against NTS nephritis and mediates the beneficial effect of melanocortin therapy
MC1R is expressed in many tissues,33, 34 including hematopoietic cells, such as lymphocytes, dendritic cells, and macrophages.33, 34 To determine if hematopoietic MC1R signaling is involved in NTS nephritis, WT or e/e mice received adoptive transfer of syngeneic BMDCs prepared from WT or e/e mice, followed by NTS injury and treatment with melanocortins or vehicle (Figure 7a). Shown in Figure 7b, BMDCs freshly prepared from donor mice were labelled with PKH26. After NTS injury, PKH26-positive cells were noted in kidney specimens of recipient mice, including renal interstitia and glomeruli, as identified by podocin staining, denoting successful adoptive transfer and tissue engraftment of BMDCs (Figure 7c). The morphologic findings were further verified by PKH26+ donor chimerism in the kidney, the blood, and the spleen (Supplementary Figure S6), as estimated by flow cytometry quantification of the isolated hematopoietic cells positive for CD45 and PKH26. As compared with WT mice that received WT BMDCs, adoptive transfer of e/e BMDC to WT mice worsened proteinuria and kidney dysfunction after NTS injury, shown by the uACR (1.819, 95%CI: 0.06428, 3.574) and BUN levels (11.56, 95%CI: 0.3011, 22.83; Supplementary Figure S7). Conversely, compared with control e/e mice receiving e/e BMDCs, adoptive transfer of WT BMDCs to e/e mice significantly ameliorated albuminuria (Figure 7d; uACR: 7.793, 95%CI: 5.206, 10.38) and kidney dysfunction (Figure 7e; BUN: 18.25, 95%CI: 5.084, 31.41), and attenuated histologic lesions of NTS nephritis (Supplementary Figure S8a), as evaluated by glomerulonephritis scores (0.7333, 95%CI: 0.3571, 1.110), tubulointerstitial nephritis scores (0.750, 95%CI: 0.4091, 1.091), and percentage of crescentic glomeruli (17.22, 95%CI: 11.34, 23.11; Supplementary Figure S8b–d). Consistent with the improvement in proteinuria, podocyte damage in NTS-injured e/e mice was improved by adoptive transfer of WT BMDCs, marked by retention of podocyte-specific markers in glomeruli, including wilms’ tumor-1 and podocin, as determined by fluorescent immunohistochemistry staining (Figure 8a) and immunoblot analysis of isolated glomeruli (Figure 8b–d). In addition, adoptive transfer of WT BMDCs rather than e/e BMDCs reinstated the protective efficacy of RCI, NDP-MSH, or MS05 in NTS-injured e/e mice, marked by an improvement in proteinuria, kidney function and histologic lesions (Figure 7 and Supplementary Figure S8). This effect was accompanied by a restored protection of melanocortins against podocyte injury, as demonstrated by preservation of wilms’ tumor-1 and podocin expression in glomeruli (Figure 8). Moreover, the beneficial effect exerted by adoptive transfer of WT BMDCs was associated with reduction in glomerular deposition of autologous IgG (Figure 9a and b) and C5b-9 (Figure 9a and c).
Figure 7. Adoptive transfer of bone marrow-derived cells (BMDC) prepared from syngeneic wild-type (WT) mice alleviates proteinuria, kidney injury, and dysfunction in e/e (melanocortin 1 receptor -null) mice with nephrotoxic serum (NTS) nephritis, and reinstates the protective efficacy of melanocortin therapy.
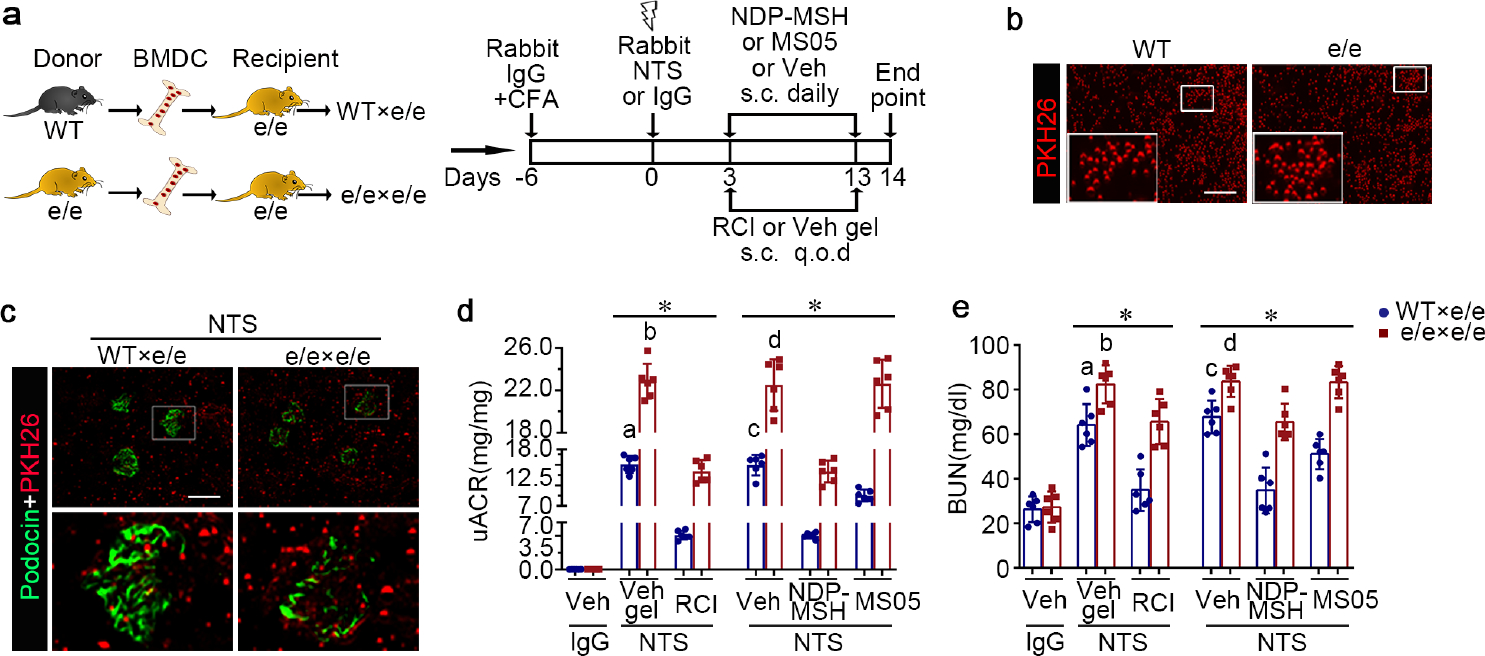
(a) Schematic diagram depicts the animal study design. (b) Representative fluorescent micrographs of red fluorescent dye PKH26-labelled BMDC (red) prior to infusion (bar=100μm). (c) Representative fluorescent micrographs show engraftment of PKH26+ BMDC (red) in renal interstitia and glomeruli, marked by podocin staining (green), in recipient mice 14 days after NTS injury (Scale bar=100μm). (d) Proteinuria was estimated by the urinary albumin-to-creatinine ratios (uACR). (e) Kidney function was assessed by measuring blood urea nitrogen (BUN) levels in sera. *P<0.05 by analysis of variance. aP<0.05 versus NTS-injured mice treated with e/e BMDC and Vehicle (Veh) gel or with WT BMDC and repository corticotropin injection (RCI); bP<0.05 versus NTS-injured mice treated with e/e BMDC and RCI; cP<0.05 versus NTS-injured mice treated with e/e BMDC and Veh or with WT BMDC and pan-melanocortin receptor agonist [Nle, DPhe]-α-MSH (NDP-MSH) or MC1R agonist MS05 (custom-made peptide, GL Biochem); dP<0.05 versus NTS-injured mice treated with e/e BMDC and NDP-MSH; (n=6). CFA, complete Freund’s adjuvant; q.o.d., every other day; s.c., subcutaneous. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Figure 8. Adoptive transfer of bone marrow-derived cells (BMDC) prepared from syngeneic wild-type (WT) mice protects e/e (melanocortin 1 receptor -null) mice against nephrotoxic serum (NTS)-elicited podocyte injury, and restores the podocyte protective efficacy of melanocortin therapy.
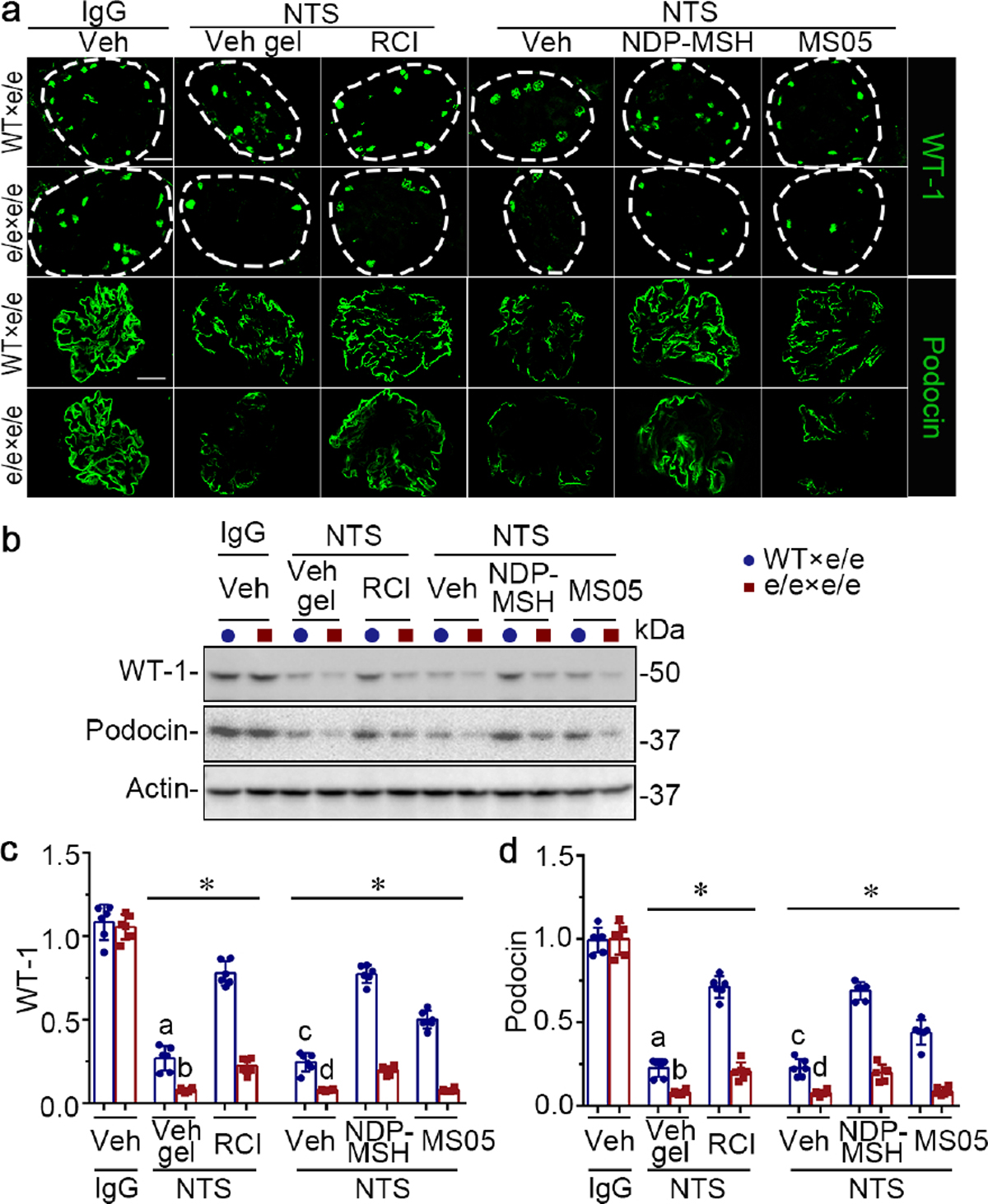
(a) Animals were treated as described in Figure 7. Representative micrographs show immunofluorescence staining of kidney specimens for podocyte-specific markers wild-type (WT) and podocin, with glomeruli outlined by dashed circles (bar =20 μm). (b) Representative blots showing immunoblot analysis of isolated glomeruli for indicated proteins. (c, d) Estimation of the abundance of WT-1 and podocin in glomeruli by densitometric analyses of immunoblots, expressed as relative levels normalized to actin. *P<0.05 by analysis of variance. aP<0.01 versus NTS-injured mice treated with e/e BMDC and Vehicle (Veh) gel or with WT BMDC and repository corticotropin injection (RCI); bP<0.01 versus NTS-injured mice treated with e/e BMDC and RCI; cP<0.05 versus NTS-injured mice treated with e/e BMDC and (Veh) or with WT BMDC and pan-melanocortin receptor agonist [Nle, DPhe]-α-MSH (NDP-MSH) or MC1R agonist MS05 (custom-made peptide, GL Biochem); dP<0.05 versus NTS-injured mice treated with e/e BMDC and NDP-MSH; (n=6). CFA, complete Freund’s adjuvant; q.o.d., every other day; s.c., subcutaneous. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Figure 9. Adoptive transfer of bone marrow-derived cells (BMDC) prepared from syngeneic wild-type (WT) mice diminishes glomerular deposition of mouse IgG and C5b-9 in e/e (melanocortin 1 receptor -null) mice with nephrotoxic serum (NTS) nephritis, and restores the protective effect of melanocortin therapy.
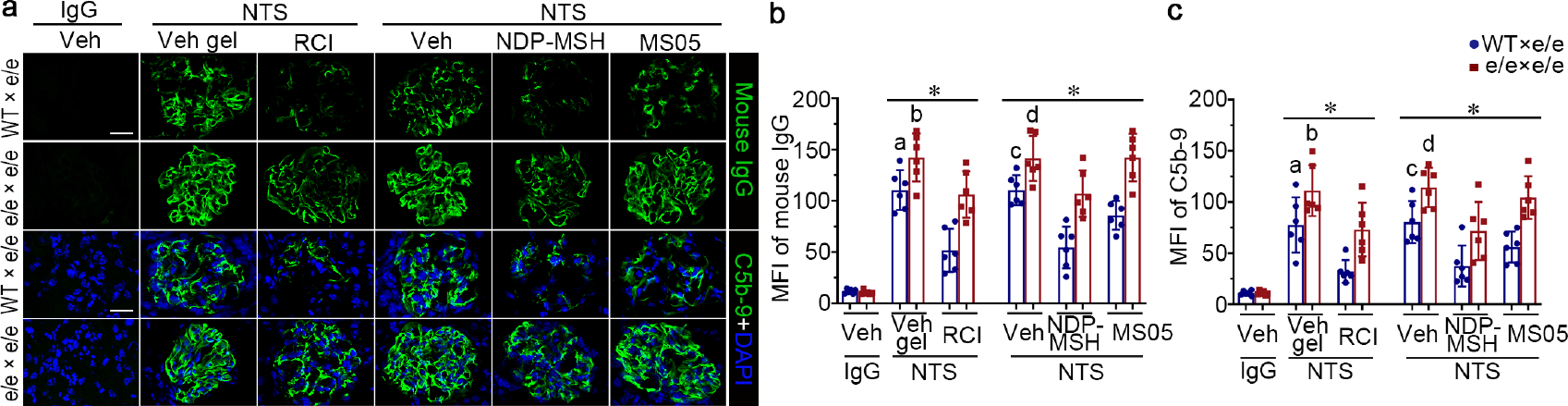
(a) Animals were treated as described in Figure 7. Representative micrographs show immunofluorescence staining of kidney specimens for mouse IgG or C5b-9 (bar =20 μm). (b, c) Arbitrary levels of mean fluorescence intensity (MFI) of (b) mouse IgG staining or (c) C5b-9 staining in glomeruli, as estimated by computerized morphometric analysis. *P<0.05 by analysis of variance. aP<0.05 versus NTS-injured mice treated with e/e BMDC and Vehicle (Veh) gel or with BMDC and repository corticotropin injection (RCI); bP<0.05 versus NTS-injured mice treated with e/e BMDC and RCI; cP<0.05 versus NTS-injured mice treated with e/e BMDC and Vehicle (Veh) or with WT BMDC and pan-melanocortin receptor agonist [Nle, DPhe]-α-MSH (NDP-MSH) or MC1R agonist MS05 (custom-made peptide, GL Biochem); dP<0.05 versus NTS-injured mice treated with e/e BMDC and NDP-MSH; (n=6). CFA, complete Freund’s adjuvant; q.o.d., every other day; s.c., subcutaneous. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
MC1R is expressed by renal leukocytes and modulates adaptive immune response via inhibition of nuclear factor-κB
To characterize the patterns of expression of melanocortin receptor (MCR) in diverse leukocyte subsets in the kidney, a post hoc analysis of single-cell RNA sequencing (scRNAseq) transcriptome of kidney biopsies from lupus patients was performed based on the publicly available data derived from the Accelerating Medicines Partnership (AMP) Phase I Studies of Lupus Nephritis.35 Shown in Supplementary Figure S9a–c, MC1R, but not other MCRs, was expressed in human kidneys with lupus nephritis by diverse immune cells, including T cells, B cells and macrophages. To validate the mRNA expression data, renal leukocytes were isolated from WT mice with NTS nephritis and subjected to flow cytometry analysis for MC1R in combination with a number of leukocyte markers, including CD45, CD3, CD19 and F4/80 (Figure 10a). In agreement with previous reports,36 approximately 57% of CD45+ leukocytes, 46% of CD3+ T lymphocytes, 78% of CD19+ B lymphocytes, and 65% of F4/80+ macrophages in the NTS nephritic kidneys expressed MC1R, suggesting that renal leukocytes are potential target cells for melanocortin hormones via binding to MC1R. The flow cytometry data were further verified by immunoblot analysis of isolated macrophages, T and B lymphocytes for MC1R, which clearly showed MC1R expression in the WT but not the e/e group (Figure 10b).
Figure 10. Melanocortin 1 receptor (MC1R) is expressed by diverse subsets of renal leukocytes and inversely correlates with the nuclear factor (NFκB) pathway.
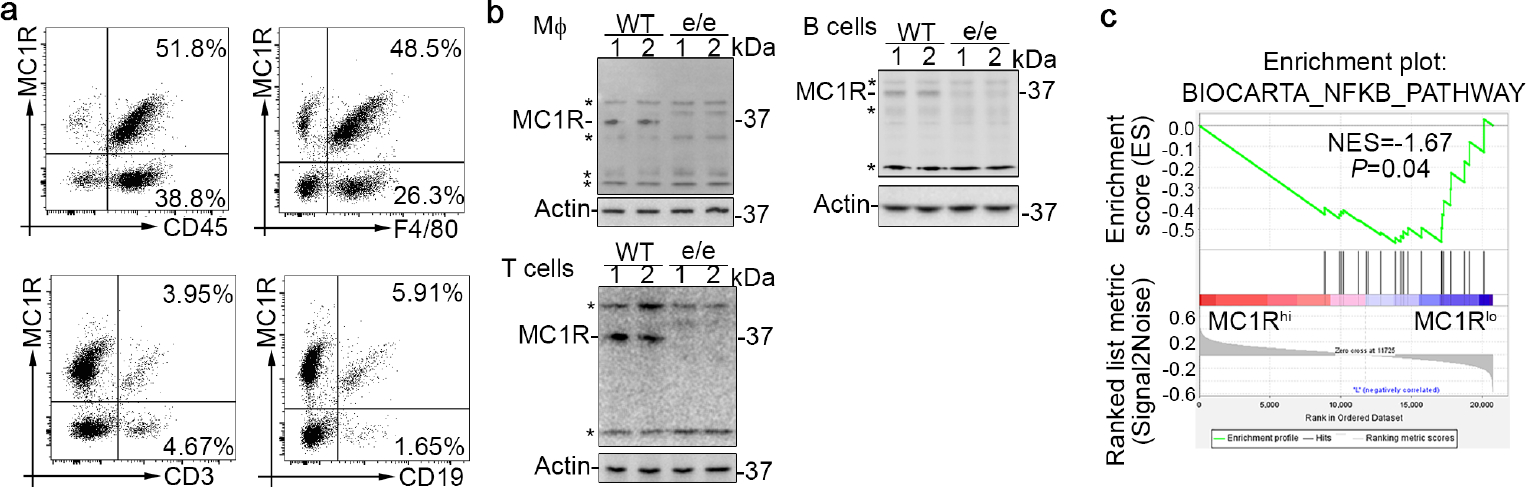
(a) Flow cytometry analysis of renal leukocytes isolated from wild-type (WT) mice with nephrotoxic serum (NTS) nephritis (pool of 3 animals per group) demonstrates MC1R expression in CD45+, F4/80+, CD3+, and CD19+ renal leukocytes. (b) Representative immunoblots show MC1R expression in bone marrow-derived macrophages and splenocyte-derived T and B cells prepared from WT or e/e (melanocortin 1 receptor–null) mice. Asterisks indicate nonspecific bands. (c) Gene set enrichment analysis based on data derived from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database (GEO accession number GSE121239) demonstrated that MC1R expression inversely correlates with the curated gene set “BIOCARTA_NFKB_PATHWAY” in peripheral blood mononuclear cells collected from patients with systemic lupus erythematosus. Normalized enrichment score (NES) and nominal P value are shown.
To further determine the molecular mechanism by which MC1R modulates immune cell activity and immune responses, additional post hoc analyses were performed using the publicly available transcriptome of peripheral blood mononuclear cells (PBMCs) isolated from lupus patients (GEO accession number: GSE121239). A number of gene sets related to immunogenic or tolerogenic regulatory pathways were examined by gene set enrichment analysis (GESA). The curated gene set “BIOCARTA_NFKB_PATHWAY” exhibited significant enrichment in low-expression of MC1R as opposed to high-expression of MC1R in lupus PBMCs (Figure 10c), suggesting that MC1R is negatively associated with NFκB signaling in immune cells.
NFκB has been implicated as a critical modulator of immune responses in diverse immune cells.37 Thus, it is tempting to speculate that MC1R may modulate NFκB signaling and thereby regulate the activity of immune cells in NTS nephritis. To test this hypothesis, splenocytes were prepared from WT and e/e mice 14 days after NTS injury and stimulated in vitro with rabbit IgG in the presence or absence of MS05 (Figure 11a). Enzyme-linked immunosorbent assay (ELISA) analysis of splenocyte supernatants (Figure 11b) revealed that the production of anti-rabbit IgG2c (0.813, 95%CI: −0.001646, 1.628) and IgG3 (0.6773, 95%CI: 0.0731, 1.281) was significantly greater in e/e cells than in WT cells and was suppressed by MS05 in WT cells (Figure 11c), associated with a reduction in NFκB phosphorylation and activation (Figure 11d). In contrast, in e/e splenocytes, these effects of MS05 were only marginally induced but were mimicked by pyrrolidine dithiocarbamate, an NFκB inhibitor (Figure 11c), suggesting that MC1R may regulate in vitro immune responses of splenocytes via NFκB inhibition.
Figure 11. Melanocortin 1 receptor (MC1R) regulates in vitro immune response in cultured splenocytes involving nuclear factor (NFκB) inhibition.
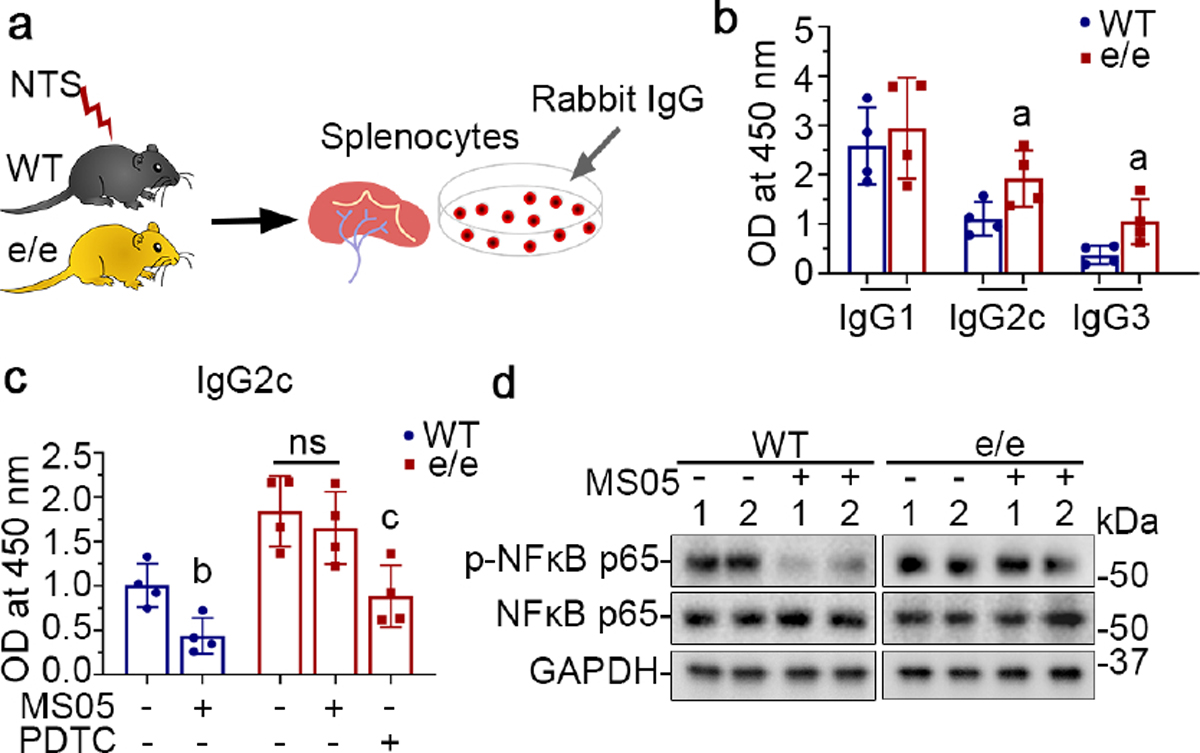
(a) Splenocytes were prepared from WT or e/e mice after NTS injury and were stimulated with rabbit IgG in the presence or absence of MS05 (10−7M) or PDTC (2.5μM). (b) ELISA of rabbit IgG-specific mouse IgG1, IgG2c and IgG3 in the culture supernatants. aP<0.05 versus WT groups for the same IgG subclass by unpaired t test; (n=4). (c) ELISA of rabbit IgG-specific mouse IgG2c in the culture supernatants after indicated treatments. bP<0.05 versus the WT control group; cP<0.05 versus the e/e control group by unpaired t test (n=4); ns, not significant. (d) Representative immunoblots show expression of indicated proteins in splenocytes collected after different treatments.
To determine whether MC1R signaling also regulates macrophage polarization, in vitro studies on macrophages were performed. BMDCs were prepared from WT and e/e mice and incubated with granulocyte-macrophage colony-stimulating factor (GM-CSF) for 7 days to generate bone marrow-derived macrophages (Figure 12a), which were then primed to the M1 phenotype with lipopolysaccharide and interferon-γ for 4 days in the presence or absence of MS05. As shown in Figure 12b, lipopolysaccharide + interferon-γ caused NFκB p65 phosphorylation and activation in macrophages. This effect was abrogated by co-treatment with MS05, concomitant with reduced expression of iNOS and IL-1β but increased expression of MR and arginase-1, consistent with a pro-M2 skewing effect on macrophage polarization. In contrast, in e/e macrophages, the effects of MS05 were barely induced but were mimicked by pyrrolidine dithiocarbamate with a shift from the M1 to M2 phenotype (Figure 12b). Taken together, these findings suggest that MC1R signaling exerts a pro-M2 skewing effect on macrophage polarization via NFκB inhibition.
Figure 12. Melanocortin 1 receptor (MC1R) signaling mitigates proinflammatory M1 activation and facilitates the anti-inflammatory M2 polarization of macrophages via repression of nuclear factor (NFκB) activation.
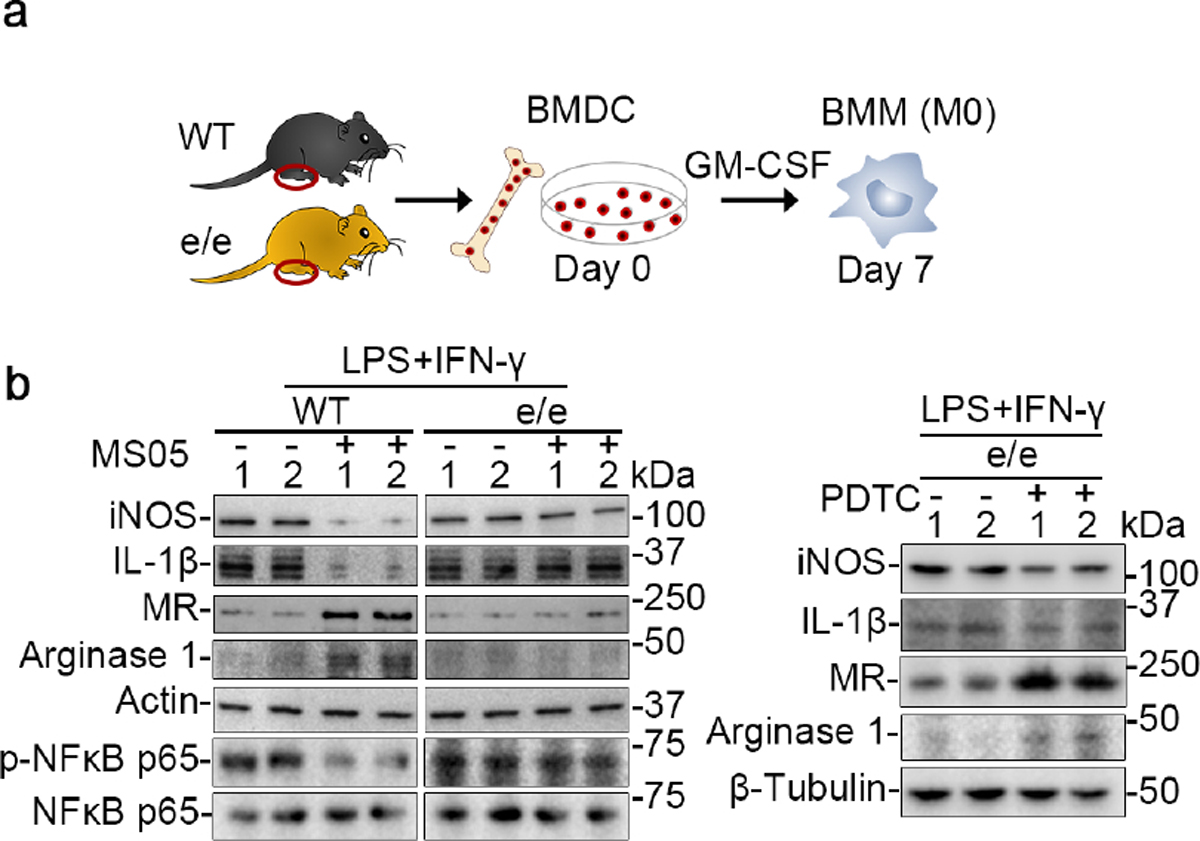
(a) A schematic diagram depicts the preparation of bone marrow-derived macrophages (BMM) from bone marrow-derived cells (BMDC) isolated from wild-type (WT) or e/e (MC1R-null) mice. (b) BMM cells were primed with a mix of lipopolysaccharide (LPS;100ng/mL) and interferon-γ (IFN-γ; 50ng/mL) in the presence or absence of MC1R agonist MS05 (custom-made peptide, GL Biochem; 10−7M) or pyrrolidine dithiocarbamate (PDTC; 2.5μM). Representative immunoblots show expression of diverse proteins, as well as actin or β-tubulin, which served as loading controls. GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; iNOS, inducible nitric oxide synthase; MR, mannose receptor.
Discussion
Clinical and experimental evidence suggests that melanocortin therapy confers a protective effect in glomerular disease.38 Our present study demonstrates that MC1R signaling in hematopoietic cells plays a key role in mediating the beneficial effect of melanocortin therapy in NTS nephritis.
In addition to causing renal cell injury, NTS nephritis is characterized by a robust systemic immune response.39, 40 In our study, melanocortin therapy ameliorated glomerular injury, and this effect was associated with reduced glomerular deposition of autologous IgG and C5b-9. This finding is in agreement with previous reports on other immune-mediated diseases. For instance, in the pristine induced lupus-like murine model, Botte et al41 found that NDP-MSH treatment improved glomerular hypercellularity and renal damage, and this improvement was associated with reduced IgG deposition in glomeruli, diminished plasma levels of IgG1 and IgG2a, and reduced incidence of antinuclear antibody, all consistent with a suppressed humoral immune response. The pathogenesis of the abnormal immune response in diseases like lupus42–44 or NTS nephritis32, 45 is complex, and it depends upon the orchestrated interaction of a number of immune-competent cells, including antigen-presenting cells, and T and B lymphocytes. The exact mechanisms by which the melanocortinergic signaling regulates this process are not fully understood, but our data and previous findings shed light on some potential mechanisms. Immune cells including T and B cells express MCR and may be an important target of melanocortins.36,46 Similarly, we found that MC1R is expressed by a variety of renal leukocyte subsets, suggesting that melanocortins might directly bind to immune cells and modulate their reactivity to autoantigens or xenoantigens as in NTS. In support of this notion, Olsen et al 47 detected a direct effect of melanocortins on human B cells in vitro. They found reduced proliferation and IgG production in peripheral blood B cells that were isolated from healthy volunteers and exposed to Acthar Gel.47 The MCRs that mediate this effect have been uncertain but may involve MC1R or MC3R.48 Our present study demonstrates that MC1R signaling represses autologous antibody generation, although the pathogenic role of the autologous antibodies in NTS nephritis is still under discussion.
Adaptive cellular immune response is also implicated in glomerulonephritis32, and specifically, adaptive Th immunity influences the severity of NTS nephritis.49 Several lines of evidence from our study, including increased renal expression of IFN-γ and augmented levels of antigen-specific IgG2c and IgG3 in e/e mice upon NTS injury, suggest that MC1R deficiency promotes the Th1 immune response. In addition, MC1R deficiency seems to disrupt regulatory T cell homeostasis in NTS nephritis, as shown by reduced numbers of Foxp3+ cells in the diseased kidney. This effect is in agreement with a recent finding that MC1R is essential for the protective effect of NDP-MSH in mice with experimental autoimmune encephalomyelitis - in this context, it suppressed the Th1 response and potentiated Treg function.50
Macrophages also play a role in NTS nephritis51: M1 macrophage cell therapy worsens, whereas M2 macrophage transfusion ameliorates, NTS nephritis.52 Monocytes/macrophages are also known to express MCR and thus may contribute to the beneficial effect of melanocortins53. Our data indicate that macrophage-specific MC1R signaling favors M2 polarization and an anti-inflammatory phenotype. In line with our findings, MC1R-promoted M2 polarization has also been described in infectious inflammation54 and in macrophage foam cell formation in atherosclerotic plaques55. Nevertheless, the functional role of MC1R regulation of macrophage polarization for the protective effect of MC1R signaling in the NTS nephritis is still uncertain and merits further investigation.
Finally, although other signaling mechanisms may also mediate MC1R’s role in NTS nephritis, our findings that MC1R modulates the NFκB pathway, and this is likely involved in MC1R regulation of immune cell activity, are in concert with recent evidence showing MC1R modulates proinflammatory NFκB activation in other cell types and tissues.56, 57 In our study, although MS05 is a selective MC1R agonist, RCI is a steroidogenic pan-MCR agonist that activates all 5 MCRs (MC1–5R), and NDP-MSH is a nonsteroidogenic pan-MCR agonist that activates MC1R and MC3,4,5R. If not only MC1R but also other MCRs influence NTS nephritis, then activation of all MCRs by RCI or NDP-MSH might have greater protective effects than activation of MC1R only by MS05, shown in our study. Moreover, the benefit of MS05 was largely blunted in e/e mice with NTS nephritis, whereas the pan-MCR agonists such as RCI and NDP-MSH, were still effective.
In summary, MC1R deficiency aggravates NTS nephritis in mice and mitigates the beneficial effects of melanocortins. Mechanistically, hematopoietic-specific MC1R signaling seems to protect against NTS nephritis via modulation of the immune response. Our findings may open up new avenues for development of a novel therapeutic approach for immune-mediated glomerular disease, based on targeting of MC1R.
Supplementary Material
Figure S1. The protective efficacy of melanocortin therapy against NTS nephritis and related podocyte injury is blunted in e/e mice with MC1R deficiency.
Figure S2. WT and e/e mice exhibit albuminuria and glomerulopathy to a comparable extent in the early phase of NTS nephritis.
Figure S3. Similar severity of podocyte injury is observed in WT and e/e mice in the early phase of NTS nephritis.
Figure S4. The protective efficacy of melanocortin therapy against glomerular deposition of mouse IgG and C5b-9 in NTS nephritis is diminished in e/e mice with MC1R deficiency.
Figure S5. Melanocortin therapy mitigates macrophage infiltration in NTS nephritis, associated with an M2-skewed macrophage polarization.
Figure S6. PKH26+ donor chimerism is achieved in the blood, the spleen and the kidney of recipient mice with NTS nephritis.
Figure S7. Adoptive transfer of bone marrow-derived cells (BMDC) prepared from syngeneic e/e mice worsens proteinuria and kidney dysfunction in WT mice with NTS nephritis.
Figure S8. Adoptive transfer of bone marrow-derived cells (BMDC) prepared from syngeneic WT mice alleviates kidney injury and dysfunction in e/e mice with NTS nephritis, and reinstates the protective efficacy of melanocortin therapy.
Figure S9. MC1R is expressed by diverse renal leukocyte subsets based on single cell RNA sequencing analysis of kidney biopsies from lupus patients.
Figure S10. Negative controls for immunostainings.
Translational Statement.
The melanocortin neuropeptides, represented by adrenocorticotropic hormone (ACTH), have recently emerged as a novel therapeutic choice for treating refractory glomerular diseases. The molecular mechanism of this action remains uncertain. This study demonstrated, in both in vivo and in vitro models, that hematopoietic-specific melanocortin 1 receptor(MC1R) signaling attenuates nephrotoxic serum nephritis and mediates the beneficial effect of melanocortin therapy. This renoprotective action of MC1R is achieved, at least in part, via a regulatory effect on immune response. These findings may open up new avenues for developing novel therapeutic strategies to treat immune-mediated glomerular disease based on targeting of MC1R.
ACKNOWLEDGEMENTS
The authors are grateful to Dr. Chandra Mohan for providing the nephrotoxic serum. This work was supported in part by a research grant from the Mallinckrodt ARD, LLC. RG was supported in part by the U.S. National Institutes of Health grant DK114006. The funders had no role in the design and conduct of this study, collection and interpretation of the data, or preparation and approval of the manuscript.
Footnotes
DISCLOSURE
Part of this work was presented as an oral presentation at the American Society of Nephrology Kidney Week 2020. R.G. has consulted for Reata, Mallinckrodt and ANI Pharmaceuticals. All the other authors declared no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Meyers CM, Geanacopoulos M, Holzman LB, et al. Glomerular disease workshop. J Am Soc Nephrol 2005; 16: 3472–3476. [DOI] [PubMed] [Google Scholar]
- 2.Riley CM. Nephrotic syndrome. Effect of adenocorticotrophic hormone. Pediatrics 1951; 7: 457–471. [PubMed] [Google Scholar]
- 3.Arneil GC, Wilson HEC. A.C.T.H in Nephrosis. Archives of Disease in Childhood 1953; 28: 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soffer LJ, Levitt MF, Baehr G. Use of cortisone and adrenocorticotropic hormone in acute disseminated lupus erythematosus. AMA archives of internal medicine 1950; 86: 558–573. [DOI] [PubMed] [Google Scholar]
- 5.Gettig J, Cummings JP, Matuszewski K. H.p. Acthar gel and cosyntropin review: clinical and financial implications. P & T : a peer-reviewed journal for formulary management 2009; 34: 250–257. [PMC free article] [PubMed] [Google Scholar]
- 6.Piel CF. Management of nephrosis; the use of long continued hormone therapy. California medicine 1956; 85: 152–156. [PMC free article] [PubMed] [Google Scholar]
- 7.Adams DA, Maxwell MH, Bernstein D. Corticosteroid therapy of glomerulonephritis and the nephrotic syndrome: a review. Journal of chronic diseases 1962; 15: 29–50. [DOI] [PubMed] [Google Scholar]
- 8.Bradham WS, Parker WF, Smythe CM. Adult nephrosis: the role of renal biopsy in predicting response to corticosteroids. Journal of the South Carolina Medical Association 1964; 60: 147–153. [PubMed] [Google Scholar]
- 9.Gong R Leveraging melanocortin pathways to treat glomerular diseases. Advances in chronic kidney disease 2014; 21: 134–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madan A, Mijovic-Das S, Stankovic A, et al. Acthar gel in the treatment of nephrotic syndrome: a multicenter retrospective case series. BMC nephrology 2016; 17: 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lieberman KV, Ettinger L, Picarelli C. Adrenocorticotropic Hormone for Steroid-Resistant and Oral Steroid-Intolerant Children With Minimal Change Nephrotic Syndrome. Annals of the New York Academy of Sciences 2014; 2: 109. [Google Scholar]
- 12.Bomback AS, Canetta PA, Beck LH Jr., et al. Treatment of resistant glomerular diseases with adrenocorticotropic hormone gel: a prospective trial. American journal of nephrology 2012; 36: 58–67. [DOI] [PubMed] [Google Scholar]
- 13.Bomback AS, Tumlin JA, Baranski J, et al. Treatment of nephrotic syndrome with adrenocorticotropic hormone (ACTH) gel. Drug design, development and therapy 2011; 5: 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hogan J, Bomback AS, Mehta K, et al. Treatment of idiopathic FSGS with adrenocorticotropic hormone gel. Clinical journal of the American Society of Nephrology : CJASN 2013; 8: 2072–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hladunewich MA, Cattran D, Beck LH, et al. A pilot study to determine the dose and effectiveness of adrenocorticotrophic hormone (H.P. Acthar® Gel) in nephrotic syndrome due to idiopathic membranous nephropathy. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association 2014; 29: 1570–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ponticelli C, Passerini P, Salvadori M, et al. A randomized pilot trial comparing methylprednisolone plus a cytotoxic agent versus synthetic adrenocorticotropic hormone in idiopathic membranous nephropathy. American journal of kidney diseases : the official journal of the National Kidney Foundation 2006; 47: 233–240. [DOI] [PubMed] [Google Scholar]
- 17.Bagchi S, Behera V, Agarwal SK. ACTH (corticotrophin) therapy in resistant primary membranous nephropathy. Kidney Int 2019; 96: 250–251. [DOI] [PubMed] [Google Scholar]
- 18.Swan AM, Kyaw HH, Aye ZN. Therapeutic response of repository corticotropin injection in treatment of proteinuria due to anca negative pauci-immune crescentic glomerulonephritis. Arch Gen Intern Med 2018; 2: 37–40. [Google Scholar]
- 19.Li X, Golubovsky J, Hui-Yuen J, et al. Adrenocorticotropic hormone gel in the treatment of systemic lupus erythematosus: A retrospective study of patients. F1000Research 2015; 4: 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong R The renaissance of corticotropin therapy in proteinuric nephropathies. Nat Rev Nephrol 2011; 8: 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiao Y, Berg AL, Wang P, et al. MC1R is dispensable for the proteinuria reducing and glomerular protective effect of melanocortin therapy. Sci Rep 2016; 6: 27589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qiao Y, Wang P, Chang M, et al. Melanocortin therapy ameliorates podocytopathy and proteinuria in experimental focal segmental glomerulosclerosis involving a podocyte specific non-MC1R-mediated melanocortinergic signaling. Clin Sci (Lond) 2020; 134: 695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lindskog A, Ebefors K, Johansson ME, et al. Melanocortin 1 receptor agonists reduce proteinuria. Journal of the American Society of Nephrology : JASN 2010; 21: 1290–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elvin J, Buvall L, Lindskog Jonsson A, et al. Melanocortin 1 receptor agonist protects podocytes through catalase and RhoA activation. American journal of physiology Renal physiology 2016; 310: F846–856. [DOI] [PubMed] [Google Scholar]
- 25.Lindskog Jonsson A, Granqvist A, Elvin J, et al. Effects of melanocortin 1 receptor agonists in experimental nephropathies. Plos One 2014; 9: e87816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chadban SJ, Atkins RC. Glomerulonephritis. Lancet (London, England) 2005; 365: 1797–1806. [DOI] [PubMed] [Google Scholar]
- 27.Pippin JW, Brinkkoetter PT, Cormack-Aboud FC, et al. Inducible rodent models of acquired podocyte diseases. American journal of physiology Renal physiology 2009; 296: F213–229. [DOI] [PubMed] [Google Scholar]
- 28.Xie C, Liu K, Fu Y, et al. RANTES deficiency attenuates autoantibody-induced glomerulonephritis. Journal of clinical immunology 2011; 31: 128–135. [DOI] [PubMed] [Google Scholar]
- 29.Murphy J, Summer R, Wilson AA, et al. The prolonged life-span of alveolar macrophages. American journal of respiratory cell and molecular biology 2008; 38: 380–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bergwall L, Wallentin H, Elvin J, et al. Amplification of the Melanocortin-1 Receptor in Nephrotic Syndrome Identifies a Target for Podocyte Cytoskeleton Stabilization. Scientific reports 2018; 8: 15731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Frontiers in immunology 2014; 5: 520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Artinger K, Kirsch AH, Aringer I, et al. Innate and adaptive immunity in experimental glomerulonephritis: a pathfinder tale. Pediatric nephrology (Berlin, Germany) 2017; 32: 943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cone RD. Studies on the physiological functions of the melanocortin system. Endocrine reviews 2006; 27: 736–749. [DOI] [PubMed] [Google Scholar]
- 34.Cone RD, Lu D, Koppula S, et al. The melanocortin receptors: agonists, antagonists, and the hormonal control of pigmentation. Recent progress in hormone research 1996; 51: 287–317; discussion 318. [PubMed] [Google Scholar]
- 35.Arazi A, Rao DA, Berthier CC, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nature Immunology 2019; 20: 902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Catania A The melanocortin system in leukocyte biology. Journal of leukocyte biology 2007; 81: 383–392. [DOI] [PubMed] [Google Scholar]
- 37.Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene 2006; 25: 6758–6780. [DOI] [PubMed] [Google Scholar]
- 38.Chang M, Chen B, Shaffner J, et al. Melanocortin System in Kidney Homeostasis and Disease: Novel Therapeutic Opportunities. Frontiers in physiology 2021; 12: 651236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Togashi K, Yamamoto T, Hara M, et al. Mechanism of inducing autologous antibody formation in nephrotoxic serum nephritis. Acta pathologica japonica 1982; 32: 1059–1065. [DOI] [PubMed] [Google Scholar]
- 40.Unanue ER, Dixon FJ. EXPERIMENTAL GLOMERULONEPHRITIS. VI. THE AUTOLOGOUS PHASE OF NEPHROTOXIC SERUM NEPHRITIS. The Journal of experimental medicine 1965; 121: 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Botte DA, Noronha IL, Malheiros DM, et al. Alpha-melanocyte stimulating hormone ameliorates disease activity in an induced murine lupus-like model. Clinical and experimental immunology 2014; 177: 381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wardowska A The epigenetic face of lupus: Focus on antigen-presenting cells. International immunopharmacology 2020; 81: 106262. [DOI] [PubMed] [Google Scholar]
- 43.Sharabi A, Tsokos GC. T cell metabolism: new insights in systemic lupus erythematosus pathogenesis and therapy. Nature reviews Rheumatology 2020; 16: 100–112. [DOI] [PubMed] [Google Scholar]
- 44.Yap DYH, Chan TM. B Cell Abnormalities in Systemic Lupus Erythematosus and Lupus Nephritis-Role in Pathogenesis and Effect of Immunosuppressive Treatments. International journal of molecular sciences 2019; 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sato T, Kawachi H, Morioka T, et al. Nephrotoxic serum nephritis in nude rats: the roles of host immune reactions in the accelerated type. Clinical and experimental immunology 1993; 91: 131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neumann Andersen G, Nagaeva O, Mandrika I, et al. MC(1) receptors are constitutively expressed on leucocyte subpopulations with antigen presenting and cytotoxic functions. Clin Exp Immunol 2001; 126: 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olsen NJ, Decker DA, Higgins P, et al. Direct effects of HP Acthar Gel on human B lymphocyte activation in vitro. Arthritis Res Ther 2015; 17: 300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cooper A, Robinson SJ, Pickard C, et al. -Melanocyte-Stimulating Hormone Suppresses Antigen-Induced Lymphocyte Proliferation in Humans Independently of Melanocortin 1 Receptor Gene Status. The Journal of Immunology 2005; 175: 4806–4813. [DOI] [PubMed] [Google Scholar]
- 49.Huang XR, Tipping PG, Shuo L, et al. Th1 responsiveness to nephritogenic antigens determines susceptibility to crescentic glomerulonephritis in mice. Kidney international 1997; 51: 94–103. [DOI] [PubMed] [Google Scholar]
- 50.Mykicki N, Herrmann AM, Schwab N, et al. Melanocortin-1 receptor activation is neuroprotective in mouse models of neuroinflammatory disease. Science translational medicine 2016; 8: 362ra146. [DOI] [PubMed] [Google Scholar]
- 51.Chalmers SA, Chitu V, Herlitz LC, et al. Macrophage depletion ameliorates nephritis induced by pathogenic antibodies. Journal of autoimmunity 2015; 57: 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Du Q, Tsuboi N, Shi Y, et al. Transfusion of CD206(+) M2 Macrophages Ameliorates Antibody-Mediated Glomerulonephritis in Mice. The American journal of pathology 2016; 186: 3176–3188. [DOI] [PubMed] [Google Scholar]
- 53.Patel HB, Montero-Melendez T, Greco KV, et al. Melanocortin receptors as novel effectors of macrophage responses in inflammation. Front Immunol 2011; 2: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ji HX, Zou YL, Duan JJ, et al. The synthetic melanocortin (CKPV)2 exerts anti-fungal and anti-inflammatory effects against Candida albicans vaginitis via inducing macrophage M2 polarization. Plos One 2013; 8: e56004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rinne P, Rami M, Nuutinen S, et al. Melanocortin 1 Receptor Signaling Regulates Cholesterol Transport in Macrophages. Circulation 2017; 136: 83–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu X, Fu S, Liu Y, et al. NDP-MSH binding melanocortin-1 receptor ameliorates neuroinflammation and BBB disruption through CREB/Nr4a1/NF-κB pathway after intracerebral hemorrhage in mice. Journal of neuroinflammation 2019; 16: 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moscowitz AE, Asif H, Lindenmaier LB, et al. The Importance of Melanocortin Receptors and Their Agonists in Pulmonary Disease. Frontiers in medicine 2019; 6: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The protective efficacy of melanocortin therapy against NTS nephritis and related podocyte injury is blunted in e/e mice with MC1R deficiency.
Figure S2. WT and e/e mice exhibit albuminuria and glomerulopathy to a comparable extent in the early phase of NTS nephritis.
Figure S3. Similar severity of podocyte injury is observed in WT and e/e mice in the early phase of NTS nephritis.
Figure S4. The protective efficacy of melanocortin therapy against glomerular deposition of mouse IgG and C5b-9 in NTS nephritis is diminished in e/e mice with MC1R deficiency.
Figure S5. Melanocortin therapy mitigates macrophage infiltration in NTS nephritis, associated with an M2-skewed macrophage polarization.
Figure S6. PKH26+ donor chimerism is achieved in the blood, the spleen and the kidney of recipient mice with NTS nephritis.
Figure S7. Adoptive transfer of bone marrow-derived cells (BMDC) prepared from syngeneic e/e mice worsens proteinuria and kidney dysfunction in WT mice with NTS nephritis.
Figure S8. Adoptive transfer of bone marrow-derived cells (BMDC) prepared from syngeneic WT mice alleviates kidney injury and dysfunction in e/e mice with NTS nephritis, and reinstates the protective efficacy of melanocortin therapy.
Figure S9. MC1R is expressed by diverse renal leukocyte subsets based on single cell RNA sequencing analysis of kidney biopsies from lupus patients.
Figure S10. Negative controls for immunostainings.