

Abstract
Mutations of the voltage-gated sodium channel SCN1A gene (MIM#182389) are among the most clinically relevant epilepsy-related genetic mutations and present variable phenotypes, from the milder genetic epilepsy with febrile seizures plus to Dravet syndrome, a severe developmental and epileptic encephalopathy. Qualitative neuroimaging studies have identified malformations of cortical development in some patients and mild atrophic changes, partially confirmed by quantitative studies. Precise correlations between MRI findings and clinical variables have not been addressed. We used morphometric methods and network-based models to detect abnormal brain structural patterns in 34 patients with SCN1A-related epilepsy, including 22 with Dravet syndrome. By measuring the morphometric characteristics of the cortical mantle and volume of subcortical structures, we found bilateral atrophic changes in the hippocampus, amygdala, and the temporo-limbic cortex (P-value < 0.05). By correlating atrophic patterns with brain connectivity profiles, we found the region of the hippocampal formation as the epicenter of the structural changes. We also observed that Dravet syndrome was associated with more severe atrophy patterns with respect to the genetic epilepsy with febrile seizures plus phenotype (r = −0.0613, P-value = 0.03), thus suggesting that both the underlying mutation and seizure severity contribute to determine atrophic changes.
Keywords: SCN1A mutation, hippocampus, epilepsy, Dravet syndrome, limbic formation
Introduction
Mutations of the voltage-gated sodium channel SCN1A gene (MIM#182389) are among the most clinically relevant epilepsy-related genetic mutations and are associated with variable phenotypes. Milder forms, i.e. genetic epilepsy with febrile seizures plus (GEFS+), are characterized by recurrent febrile seizures that occur beyond age 6 years and variable afebrile seizure types that usually go into remission within adolescence (Camfield and Camfield 2015). The more severe phenotype, Dravet syndrome, is characterized by recurrent, often prolonged, febrile and afebrile seizures starting in the first year of life, followed by multiple seizures types resistant to various anti-seizure medications, associated with cognitive, behavioral, and motor impairment (Dravet 2011). The observed trajectories of cognitive decline and long-term outcome suggest that Dravet syndrome might be considered as a paradigm of epileptic encephalopathy in which the epileptic activity itself contributes to impaired brain function (Cetica et al. 2017; Mei et al. 2019).
Animal models suggest that the loss of function and hypoexcitability of at least some subtypes of GABAergic interneurons are the basic pathological mechanism whereby SCN1A mutations activate epileptogenesis (Mantegazza and Broccoli 2019; Guerrini et al. 2023) and highlight the important role played by SCN1A in pyramidal neurons, particularly in the cortico-hippocampal circuit, during the chronic phase of the disease (Mattis et al. 2022).
Brain MRI findings in SCN1A-related epilepsies include scattered reports of brain atrophy and hippocampal sclerosis (Striano et al. 2007; Ruffolo et al. 2020) and co-occurring malformations of cortical development (Barba et al. 2014). However, these observations remain rare and insufficient for addressing genotype–phenotype correlations (Guerrini et al. 2011). Quantitative neuroimaging studies showed large-scale developmental brain changes in patients with epilepsy and SCN1A mutations (Pérez et al. 2014; Lee et al. 2017), but did not investigate possible correlations with clinical features.
More advanced neuroimaging approaches with the potential of detecting alterations in the network organization of the brain (Sisodiya et al. 2020) have not yet been applied to Dravet syndrome. Although initial quantitative studies suggest that atrophic changes occur in Dravet syndrome (Lee et al. 2017), a more precise network-based characterization might uncover patterns of atrophy that relate to the connectome architecture of SCN1A-mutated epilepsy. Characterization of morphologic abnormalities and involved networks in an etiologically homogeneous epilepsy subgroup may in fact be less challenging compared with larger heterogeneous epilepsy populations (Alhusaini et al. 2016; Bathelt et al. 2016). For instance, genotype–phenotype correlations have emerged at a structural level in PCDH19-gene-related encephalopathy that well correlate with gene expression patterns and clinical manifestations (Lenge et al. 2020). In patients with SCN1A mutations and Dravet syndrome, quantitative neuroimaging studies are of interest since the etiologic role of the underlying genetic defect, which underlies phenotypic heterogeneity, and of the superimposed epilepsy, remain unclear (Guerrini et al. 2011; Nabbout et al. 2013), as is the anatomic counterpart of progressive cognitive deterioration. Our hypothesis is that the underlying neurophysiological dysfunction caused by the SCN1A mutation might confer to the brain a unique profile of vulnerability whose consequences are not easily disclosed by classical neuropathology (Catarino et al. 2011; Guerrini et al. 2011). We used morphometric methods and network-based models to detect abnormal structural patterns in brain structures and networks in patients with SCN1A-related epilepsy and comparatively analyzed patients’ subgroups stratified by different clinical severity.
Materials and methods
Participants
This study includes 34 patients and 34 controls retrospectively evaluated at 3 tertiary pediatric neurology centers (Meyer Children’s Hospital, 18 patients and 34 controls; Verona University Hospital, 11 patients; Besta Neurological Institute, 5 patients). All patients (mean age 9.4 years, median 7.1, range 1.0–28.8) carried a pathogenic SCN1A (NM_001165963.1) gene mutation identified by Sanger sequencing or next-generation sequencing. To explore a possible correlation between the MRI findings we analyzed and the predicted probability (in %) to develop Dravet syndrome, we used the SCN1A Epilepsy Prediction Model (https://scn1a-prediction-model.broadinstitute.org/; Brunklaus et al. 2022). Eight patients had been diagnosed with GEFS+, and 22 with Dravet syndrome. Controls were matched with patients for sex and age (±1 year, mean age 9.97 years, median 7.9, range 1.0–29.0) and were otherwise healthy individuals who had received an MRI for uncomplicated headache. Clinical and genetic data of the 34 patients are summarized in Supplementary Table 1, and pathogenic variants for each patient are reported in Supplementary Fig. 1.
MRI data acquisition
Participants underwent MRI acquisitions using nonstandardized imaging protocols on MR systems (3 T Achieva, Philips Healthcare, The Netherlands at Meyer Hospital; 3 T Magnetom Skyra, Siemens Healthineers, Germany at Verona University Hospital; 1.5 T Avanto, Siemens Healthineers, Germany at Besta Hospital). For the MRI assessment of brain structure, all scans included at least a 3D T1-weigthed fast-spoiled gradient echo, 3D T2-weighted Fluid Attenuation Inversion Recovery (3D T2-FLAIR), T2-weighted Fast Spin-Echo, T2*-weighted Gradient-eco, and 2D T1-weigthed acquisitions. The study was approved by the Pediatric Ethics Committee of the Tuscany Region, Italy, in the context of the DESIRE project (FP7 EU call, grant agreement 602531) and its extension by the DECODE-EE (Tuscany Region Call for Health 2018) and the Human Brain Optical Mapping (Fondazione Cassa di Risparmio di Firenze 2021) projects, and the experiments were undertaken with the understanding and written consent of each subject or their guardian.
MRI data analysis
Visual analysis
Brain MRI images were reviewed searching for structural abnormalities at cortical and subcortical levels. Artifacts at risk of hampering computational analyses were reported.
Cortical and subcortical brain analysis
The morphometric analysis of cortical and subcortical gray-matter (GM) structures was conducted by processing T1-3D MR images of patients and controls using the FreeSurfer pipeline (version 7.2.0, https://surfer.nmr.mgh.harvard.edu). The processing pipeline included the following steps: segmentation of subcortical structures (Fischl et al. 2002), cortical segmentation and surface reconstruction of boundaries between white and GM (white surface) and between GM and cerebrospinal fluid (CSF, pial surface; Fischl 2012), and parcellation of the cortical surfaces in different gyral and sulcal regions (Fischl et al. 2004). 3D high-resolution reconstructions of white and pial surfaces were obtained for each hemisphere. Subcortical segmentations and cortical surface reconstructions were visually inspected and manually corrected, where needed. We measured the volume of subcortical structures and the intracranial, subcortical, and cortical volumes (CV) of the whole brain.
We assessed the morphometric properties of the cortex at region of interest (ROI) level. To investigate the atrophy process, cortical thickness (CT), surface area (SA), and CV were measured for each vertex of the cortical surfaces. CT is assessed by measuring the distance between white and pial surfaces (Fischl and Dale 2000) and reflects neural migration/postmigrational organization (Rakic 1988) and GM pruning (Huttenlocher 1990). SA is calculated as the sum of the area of the triangles surrounding a vertex and has been demonstrated to be a highly heritable trait (Panizzon et al. 2009) and associated with cognitive abilities (Vuoksimaa et al. 2015). CV is the multiplication of CT and SA. We calculated the average values of morphometric features (CT, SA, and CV) for the ROIs of the parcellated cortex (Desikan et al. 2006).
For 3 patients who had follow-up MR scans at different ages, we conducted a longitudinal comparative analysis of percentage volumetric changes.
Analysis of subfields in subcortical structures
The FreeSurfer processing pipeline included advanced modules for the segmentations of subcortical structures, in particular the hippocampal subfields (Iglesias et al. 2015), thalamic nuclei (Iglesias et al. 2018), and amygdala nuclei (Saygin et al. 2017). Such algorithms are optimized for the subfield segmentations by adopting a computational atlas with a novel atlas building algorithm based on Bayesian inference and working on T1-weighted images. To obtain more reliable segmentations, we included in the processing pipeline additional 3D T2-FLAIR MRIs. We visually inspected and manually corrected subfield segmentations (Sämann et al. 2020), when necessary.
For each subject, we calculated the average volume of the segmented substructures of the hippocampus, amygdala, and thalamic complex.
Epicenter mapping
As a second-step analysis, we adopted the processing pipeline proposed by Larivière et al. (2020) to identify the structural and functional connectivity profiles significantly correlated with the disease-specific abnormality map and provide an epicenter mapping. We used both the CT and SA values to identify cortical and subcortical regions with significant atrophy in SCN1A mutation carriers and find epicenters. The epicenters are regions strongly connected to high-atrophy regions and weakly connected to low-atrophy regions. The processing pipeline to identify epicenters included the following steps (https://enigma-toolbox.readthedocs.io): loading z-value atrophy CT and SA maps, calculated as indicated in the previous sections; loading high-resolution structural (derived from diffusion-weighted tractography) and functional (derived from resting-state functional MRI) connectivity matrices from a cohort of healthy adults of the Human Connectome Project, parcellated according Desikan et al. (2006); extraction of seed-based structural and functional connectivity matrices, using as seed each cortical and subcortical region; assessment of the spatial correlation between the structural/functional connectivity matrices and z-value atrophy maps (epicenters are represented by regions with strong negative correlation); and assessment of the statistical significance of each spatial correlation between seed-based cortico-cortical and subcortico-cortical connectivity and atrophic measurements using spin permutation tests (number of rotation 1,000, P-value < 0.1).
Statistical analysis
We carried out statistical analyses on morphometric features using FreeSurfer (Fischl 2012) and FSL (Jenkinson et al. 2012) tools and a script developed in MATLAB R2020a (Statistics and Machine Learning Toolbox Version 11.6, MathWorks Inc., Natick, MA).
Age-related statistical analysis
We performed an ROI-based statistical analysis to identify age-related patterns in morphometric features (CT, SA, and CV). To evaluate the effect of the diagnostic group (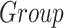 ) and age (
) and age (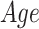 ) on morphometric features (
) on morphometric features (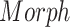 ), we performed a group analysis between SCN1A mutation carriers and controls by a multivariable linear regression model, using the center of acquisition (
), we performed a group analysis between SCN1A mutation carriers and controls by a multivariable linear regression model, using the center of acquisition (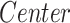 ) and the age (
) and the age (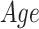 ) as covariates and age-by-group variable as interaction term (
) as covariates and age-by-group variable as interaction term (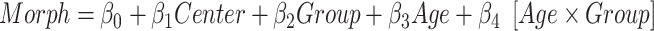 ). We adopted the center (
). We adopted the center (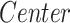 ) as confounding variable to model the variability introduced by the different MRI scanners. By the same model, we also assessed the main effect on morphometric values of the age at MRI and investigated the possible effects of age-by-group interactions (
) as confounding variable to model the variability introduced by the different MRI scanners. By the same model, we also assessed the main effect on morphometric values of the age at MRI and investigated the possible effects of age-by-group interactions (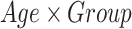 ). In subcortical structures, to evaluate the effect of the diagnostic group and age on volumetric measurements (
). In subcortical structures, to evaluate the effect of the diagnostic group and age on volumetric measurements (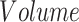 ), we similarly performed a group analysis adding the estimated intracranial volume (
), we similarly performed a group analysis adding the estimated intracranial volume (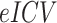 ) as a covariate (
) as a covariate (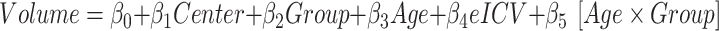 ).
).
Since control subjects were recruited in a single center (Meyer Hospital), in order to control for possible unbalance, we provided additional group analysis considering only the 18 patients of the Meyer cohort and 18 age- and sex-matched controls acquired at the same center.
Relationship with clinical assessment
For each patient, we investigated the effects of average morphometric values on clinical variables (age at seizure onset, SCN1A Epilepsy Prediction Model score, and the diagnosis [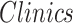 ]) by the following linear regression model:
]) by the following linear regression model: 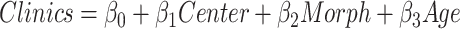
Results
There were no significant age differences between individuals with SCN1A and controls (t = −0.2, P = 0.84).
MRI visual analysis
After visual inspection of diagnostic MRI sequences, we excluded from the morphometric analysis 2 patients (nos 32 and 34) who could not be evaluated because of movement artifacts. In 7 patients, visual analysis uncovered atrophic patterns, with mild to severe atrophy in cerebral or cerebellar areas, with no recurrent area-specific distribution (Supplementary Table 1).
Morphometric analysis of cortical structures
In the morphometric analysis (Table 1 and Fig. 1), we found the cortex to be thinner in the parahippocampal gyri with respect to controls (t = −2.84, P = 0.006, CT on the left side; t = −3.85, P < 0.001, CT on the right side). We also observed a reduced SA extending beyond the same region to include the entorhinal cortex bilaterally (t = −3.22, P = 0.002, on the left side; t = −2.14, P = 0.037, on the right side), the fusiform (t = −1.99, P = 0.050, on the left side; t = −2.55, P = 0.013, on the right side), and the right inferior temporal (t = −2.31, P = 0.025) gyri, with a corresponding reduction of CV. Such volumetric reduction did not contribute to a significant reduction at whole-brain level (Table 2) but was observed in the subcortical structures, bilaterally involving the hippocampus (t = −2.67, P = 0.010, on the left side; t = −2.61, P = 0.011, on the right side) and amygdala (t = −2.98, P = 0.004, on the left side; t = −2.12, P = 0.038, on the right side). Atrophic patterns were confirmed by the higher volumetric values of the lateral ventricles (t = +1.87, P = 0.066, on the left side; t = +1.11, P = 0.271, on the right side) and CSF (t = +3.39, P = 0.001).
Table 1.
Effect of the group on the cortical thickness, surface area, and cortical volume of temporal structures of the cortex, located in the left (L) and right (R) hemispheres. Data were analyzed by a multivariate linear regression model (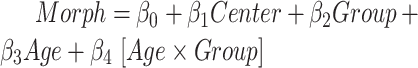 ]) to explore the effect on morphometric values because of the center of acquisition (
]) to explore the effect on morphometric values because of the center of acquisition (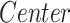 ), group (
), group (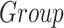 ), and age (Age).
), and age (Age).
| Morphometric variables | |||||||
|---|---|---|---|---|---|---|---|
| Cortical thickness (mm) | Surface area (mm2) | Cortical volume (mm3) | |||||
| Region | Side |
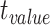
|
P value |
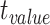
|
P value |
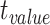
|
P value |
| Entorhinal | L | 0.93 | 0.358 | −3.22 | 0.002 | −2.31 | 0.025 |
| R | −1.08 | 0.283 | −2.14 | 0.037 | −2.03 | 0.047 | |
| Fusiform | L | −1.38 | 0.174 | −1.96 | 0.055 | −2.53 | 0.014 |
| R | −0.11 | 0.913 | −2.54 | 0.014 | −2.12 | 0.038 | |
| Parahippocampal | L | −2.84 | 0.006 | −1.88 | 0.066 | −3.32 | 0.002 |
| R | −3.85 | <0.001 | −2.10 | 0.041 | −3.42 | 0.001 | |
| Superior temporal | L | −1.39 | 0.170 | −0.68 | 0.499 | −1.36 | 0.180 |
| R | −0.70 | 0.488 | −0.62 | 0.540 | −0.84 | 0.403 | |
| Middle temporal | L | −0.24 | 0.814 | −1.22 | 0.226 | −0.91 | 0.366 |
| R | −1.47 | 0.146 | −1.19 | 0.241 | −1.61 | 0.114 | |
| Inferior temporal | L | −0.09 | 0.931 | −1.33 | 0.189 | −0.80 | 0.427 |
| R | 1.13 | 0.262 | −2.31 | 0.025 | −1.65 | 0.105 | |
| Temporal pole | L | −0.46 | 0.650 | −1.95 | 0.056 | −1.43 | 0.160 |
| R | −0.72 | 0.473 | −1.94 | 0.058 | −2.09 | 0.041 | |
| Frontal pole | L | 1.88 | 0.066 | −2.18 | 0.033 | 0.244 | 0.191 |
| R | −0.88 | 0.380 | 0.15 | 0.884 | −1.08 | 0.285 | |
| Medial orbitofrontal | L | 2.44 | 0.018 | −2.18 | 0.033 | 1.18 | 0.244 |
| R | −2.55 | 0.014 | 2.52 | 0.015 | −1.24 | 0.221 | |
| Caudal middle frontal | L | 1.59 | 0.117 | −1.14 | 0.259 | −0.54 | 0.590 |
| R | 1.42 | 0.160 | −0.20 | 0.841 | 0.21 | 0.830 | |
| Lateral orbitofrontal | L | −0.41 | 0.681 | −0.58 | 0.560 | −0.13 | 0.892 |
| R | 1.81 | 0.075 | −0.35 | 0.728 | 0.96 | 0.341 | |
| Rostral middle frontal | L | 1.14 | 0.258 | −1.16 | 0.250 | −0.59 | 0.552 |
| R | 1.09 | 0.277 | −0.39 | 0.699 | −0.06 | 0.946 | |
| Pars opercularis | L | 0.49 | 0.622 | −0.87 | 0.386 | −0.18 | 0.852 |
| R | 0.59 | 0.554 | −0.75 | 0.451 | −0.51 | 0.611 | |
| Pars orbitalis | L | −0.06 | 0.948 | −1.46 | 0.149 | −1.21 | 0.231 |
| R | 0.17 | 0.865 | −0.79 | 0.433 | −0.43 | 0.667 | |
| Pars triangularis | L | 0.42 | 0.675 | −1.64 | 0.106 | −1.33 | 0.186 |
| R | 0.58 | 0.558 | −0.84 | 0.403 | −0.56 | 0.577 | |
| Superior frontal | L | 0.59 | 0.557 | −1.03 | 0.303 | −0.77 | 0.438 |
| R | 0.27 | 0.787 | −1.28 | 0.205 | −1.27 | 0.208 | |
| Precentral | L | −1.09 | 0.278 | −0.60 | 0.548 | −1.07 | 0.285 |
| R | 0.04 | 0.966 | −1.23 | 0.225 | −1.11 | 0.270 | |
| Postcentral | L | −0.41 | 0.681 | −0.91 | 0.364 | −0.80 | 0.425 |
| R | 0.49 | 0.624 | −0.66 | 0.512 | −0.23 | 0.816 | |
| Paracentral | L | −0.72 | 0.473 | −1.34 | 0.182 | −1.94 | 0.056 |
| R | −0.23 | 0.819 | −0.91 | 0.365 | −1.40 | 0.167 | |
| Supramarginal | L | 0.15 | 0.874 | 0.04 | 0.961 | 0.11 | 0.910 |
| R | −0.45 | 0.649 | −0.84 | 0.406 | −1.86 | 0.068 | |
| Superior parietal | L | 1.06 | 0.292 | −1.17 | 0.246 | −0.30 | 0.761 |
| R | 0.56 | 0.576 | −1.05 | 0.296 | −0.60 | 0.546 | |
| Cuneus | L | −0.06 | 0.947 | −1.97 | 0.053 | −1.60 | 0.114 |
| R | −0.12 | 0.905 | −1.48 | 0.143 | −0.98 | 0.327 | |
| Precuneus | L | −0.30 | 0.759 | −1.38 | 0.171 | −1.28 | 0.203 |
| R | 0.17 | 0.860 | −1.55 | 0.125 | −1.17 | 0.247 | |
| Lateral occipital | L | 1.80 | 0.076 | −2.03 | 0.046 | −0.78 | 0.433 |
| R | −0.849 | 0.400 | −2.21 | 0.031 | −2.11 | 0.038 | |
| Insula | L | −0.66 | 0.509 | −2.63 | 0.011 | −2.54 | 0.014 |
| R | −1.11 | 0.271 | −2.26 | 0.028 | −2.72 | 0.009 | |
| Whole cortex | L | 0.23 | 0.822 | −1.50 | 0.139 | −0.46 | 0.643 |
| R | −0.09 | 0.931 | −1.41 | 0.165 | −0.67 | 0.503 | |
Bold indicates significantly altered measures (P < 0.05).
Fig. 1.

Statistical whole-brain group analysis of SCN1A-patients compared with matched controls. The results of the statistical analysis on CT a) and SA b) are represented by z-value maps superimposed on the pial surfaces of left and right hemispheres. We observed statistically significant lower values of CT and SA in left and right temporo-limbic cortices, and higher but not significant values of CT in orbitofrontal, superior frontal, and occipital cortices (for further details, please see the results of the ROI analysis reported in Table 1).
Table 2.
Effect of the group (Group) on the volume (mm3) calculated in the subcortical regions, located in the left (L) and right (R) hemispheres. Data were analyzed by a multivariate linear regression model (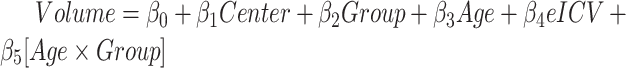 ]) to explore the effect on the volume because of the center of acquisition (
]) to explore the effect on the volume because of the center of acquisition (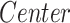 ), group (
), group (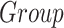 ), and age (Age) and estimated intracranial volume (
), and age (Age) and estimated intracranial volume (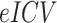 ).
).
| Controls | SCN1A | ||||||
|---|---|---|---|---|---|---|---|
| Region | Side |
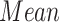
|
STD |
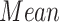
|
STD |
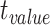
|
P value |
| Thalamus | L | 6,924.13 | 1,082.3 | 6,495.01 | 1,040.3 | −0.17928 | 0.85844 |
| R | 6,681.64 | 1,034.0 | 6,299.39 | 984.62 | 0.07828 | 0.93792 | |
| Caudate | L | 3,437.92 | 600.5 | 3,293.65 | 576.86 | −0.42025 | 0.67610 |
| R | 3,670.4 | 675.2 | 3,412.49 | 560.89 | −0.70391 | 0.48475 | |
| Putamen | L | 5,030.57 | 711.1 | 4,441.63 | 920.41 | −1.84098 | 0.07156 |
| R | 5,030.96 | 900.6 | 4,508.99 | 1,006.96 | −1.02209 | 0.31166 | |
| Pallidum | L | 1,748.24 | 333.0 | 1,614.8 | 325.48 | −0.39306 | 0.69595 |
| R | 1,735.89 | 313.7 | 1,546.12 | 321.45 | −1.25073 | 0.21686 | |
| Lateral Ventricle | L | 5,866.04 | 2,443.5 | 6,740.77 | 5,028.7 | 1.87409 | 0.06677 |
| R | 5,742.72 | 3,273.5 | 5,308.56 | 4,099.19 | 1.11265 | 0.27118 | |
| Hippocampus | L | 3,821.97 | 610.7 | 3,355.8 | 605.71 | −2.67232 | 0.01014 |
| R | 3,893.04 | 655.2 | 3,433.38 | 581.21 | −2.61288 | 0.01183 | |
| Amygdala | L | 1,623.68 | 301.0 | 1,318.18 | 311.52 | −2.97858 | 0.00446 |
| R | 1,721.31 | 334.2 | 1,485.01 | 305.46 | −2.12237 | 0.03878 | |
| Cerebrospinal fluid | 860.69 | 187.2 | 982.09 | 318.51 | 3.39211 | 0.00136 | |
| Cerebellum WM | L | 11,984.9 | 3,436.4 | 11,729.9 | 4,231.88 | −0.80358 | 0.42545 |
| R | 11,660.59 | 3,166.4 | 10,733.0 | 2,754.9 | −1.16642 | 0.24898 | |
| Cerebellum GM | L | 52,681.66 | 7,322.3 | 51,699.4 | 7,489.02 | −0.25929 | 0.79648 |
| R | 5,2312.13 | 7,504.3 | 51,299.0 | 6,197.94 | −0.16717 | 0.86791 | |
| Brain stem | 17,274.5 | 3,821.4 | 15,403.9 | 3,451.02 | −1.85682 | 0.06923 | |
| Brain volume | L | 259,389.85 | 35,933.16 | 248,384.3 | 26,291.7 | −0.4655 | 0.64360 |
| R | 260,128.49 | 36,132.94 | 247,862.8 | 26,020.7 | −0.67424 | 0.50327 | |
| Estimated total intracranial volume | 1,401,639.91 | 191,759.33 | 1,246,690.76 | 210,111.75 | −0.72676 | 0.4707 | |
Bold indicates significantly altered measures (P < 0.05).
In the cortical morphometric analysis conducted on the Meyer cohort (Supplementary Table 2), we found similar atrophic patterns in the parahippocampal gyri (t = −1.69, P = 0.099 on the left side, t = −3.59, P = 0.001 on the right side) and in surrounding temporal cortex. An atrophic trend was also detected in the subcortical structures (Supplementary Table 3) of the hippocampus (t = −2.13, P = 0.040, on the left; t = −1.74, P = 0.091, on the right) and amygdala (t = −1.77, P = 0.085, on the left; t = −0.64, P = 0.525, on the right).
Longitudinal analysis on single individuals
Of the 3 patients whose follow-up MRI scans were available for analysis (Supplementary Fig. 2), 2 (patients 1 and 17) were children with GEFS+ assessed between age 6 and 8 years, and 1 (patient 16) was an infant with Dravet syndrome whose MRIs were acquired at 1.1 and 2.5 years.
Patient 1. The first MRI of Patient 1 (GEFS+) was performed at age 6.9 years and the second scan at 7.4 years. Longitudinal volumetric analysis revealed an atrophic pattern, with reduced total brain volume (rate of percent change −2.7%), mainly because of volume reduction of gray (−2.9%; basal ganglia −3.7%, cortical −2.1%), and white matter (−3.9%) bilaterally. Atrophy also involved the hippocampal formation (hippocampus: −2.4% on the left, −1.2% of the right; amygdala: −1.6% on the left; −1.1% on the right) and was confirmed by the enlargement of the lateral ventricles (+54.3% on the left, +56.0% on the right) and increased CSF (+31.3%).
Patient 17
MRI studies were performed at 6.8 and 8.8 years of age. This patient (GEFS+) presented a similar, though less pronounced, trend of atrophy with respect to patient 1. Total brain volume was reduced at follow-up (−1.15%), with the atrophic changes involving the cortex (−2.9%) and the basal ganglia (−1.3%) of both hemispheres. An atrophic pattern was also suggested by the enlargement of the lateral ventricles (+7.9% on the left and +6.3% on the right) with CSF increase (+8.2%). The hippocampal formation followed a positive growth pattern (hippocampus: +2.6% left, +3.2% right; amygdala: +6.0% left, +6.0% right).
Patient 16
In this patient with Dravet syndrome, scanned at 1.1 and 2.5 years, we observed a mild increase in total brain volume (+1.9%), because of increased GM volume (+4.0%), particularly in the cortical GM (+4.1%), but reduction of WM volume (−4.9%). Hippocampus and amygdala were also volumetrically increased at follow-up (hippocampus: +11.5% left, +14.2% right; amygdala: +19.1% left, +19.8% right). In spite of the volumetric increase of these structures during follow-up, an overall atrophic pattern was still present with enlarged lateral ventricles (+109.7% left and +91.4% right) and increased CSF space (+18.3%). The atrophic pattern was also noted in the volumetric assessment of the cortex that revealed a reduction (−11.1%) when compared with the fitting curve (Supplementary Fig. 3g).
Volumetric analysis of subcortical structures
The group analysis of subcortical measurements revealed volumetric reductions bilaterally involving several hippocampal subfields in the SCN1A group (Supplementary Table 4), in particular the section no. 3 of the Cornus Ammonis, in the head (t = −2.37, P = 0.021, on the left side; t = −2.47, P = 0.017, on the right side) and body (t = −3.44, P = 0.001, on the left side; t = −2.74, P = 0.008, on the right side). The amygdalar nuclei were reduced in volume (Supplementary Table 5), although not to a degree reaching statistical significance, except in the right central nucleus (t = −2.23, P = 0.030). The thalamic nuclei were not significantly altered (Supplementary Table 6).
Epicenter analysis
Based on atrophic regions identified by morphometry, we performed a mapping of the epicenters and observed a statistically significant structural correlation (Supplementary Table 7 and Fig. 2a) of CT reduction in the left entorhinal cortex (r = −0.336, P < 0.001) and right fusiform gyrus (r = −0.265, P < 0.043). Structural atrophic patterns of SA (Supplementary Table 7 and Fig. 3a) involved a wide area that included bilaterally the entorhinal (r = −0.321, P = 0.034 on the left; r = −0.298, P = 0.005 on the right), fusiform (r = −0.335, P = 0.028 on the left; r = −0.227, P = 0.001 on the right), and parahippocampal gyri (r = −0.275, P = 0.050 on the left; r = −0.182, P = 0.038 on the right), whereas functional SA epicenters (Supplementary Table 8) included only the insula bilaterally (r = −0.31, P = 0.065 on the left; r = −0.30, P = 0.078 on the right). At the subcortical level, the structural epicenters (Supplementary Tables 9 and Fig. 2c) were located in the right hippocampus for CT (r = −0.26, P = 0.043), and involved bilaterally the hippocampus (r = −0.39, P = 0.017 on the left; r = −0.28, P = 0.009 on the right) and amygdala (r = −0.46, P < 0.001 on the left; r = −0.30, P = 0.004 on the right) for SA (Fig. 2d). Functional SA epicenters (Supplementary Tables 9 and 10 and Fig. 3d) were located in the hippocampus bilaterally (for CT: r = −0.19, P = 0.074 on left, r = −0.22, P = 0.069 on right).
Fig. 2.
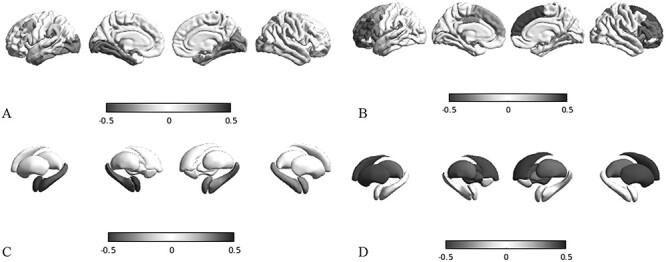
Epicenter mapping of CT. Spatial correlations between CT patterns and seed-based cortico-cortical a, b) and subcortico-cortical c, d), structural a, c, blue-black colormap), and functional b, d, red-black colormap) connectivity profiles. In this example, we use as seeds the left entorhinal gyrus a, b) and the left hippocampus c, d).
Fig. 3.
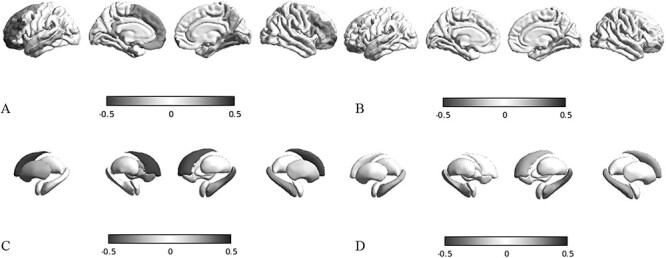
Epicenter mapping of SA. Spatial correlations between SA patterns and seed-based cortico-cortical a, b) and subcortico-cortical c, d), structural a, c, blue-black colormap), and functional b, d, red-black colormap) connectivity profiles. In this example, we use as seeds the left entorhinal gyrus a, b) and the left hippocampus c, d).
Age-related patterns
A significant effect of age on CT was observed in many different regions of the subcortical structures (Supplementary Fig. 3) and cortex (Supplementary Fig. 4). The variation of volume was strongly related to age in the group of patients with SCN1A mutations. In particular, volumes of the subcortical structures increased with age (Supplementary Table 11), whereas the brain volume globally decreased (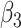 = −1,780.55, P = 0.027 on left;
= −1,780.55, P = 0.027 on left; 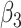 = −1,809.80, P = 0.025 on right), possibly in relation to the volumetric reduction observed at the cortical level. Indeed, CT was reduced with increasing age in the entire temporal lobe (Supplementary Table 12), in particular in the right entorhinal cortex (β3 = −0.73, P < 0.001), left fusiform (β3 = −0.02, P < 0.001), and right parahippocampal gyri (β3 = −0.54, P = 0.006). Mean thickness of the entire left hemisphere was reduced (β3 = −0.03, P < 0.001).
= −1,809.80, P = 0.025 on right), possibly in relation to the volumetric reduction observed at the cortical level. Indeed, CT was reduced with increasing age in the entire temporal lobe (Supplementary Table 12), in particular in the right entorhinal cortex (β3 = −0.73, P < 0.001), left fusiform (β3 = −0.02, P < 0.001), and right parahippocampal gyri (β3 = −0.54, P = 0.006). Mean thickness of the entire left hemisphere was reduced (β3 = −0.03, P < 0.001).
Relationship between morphometric and clinical assessments
By applying the multivariate statistical model to clinical variables, we found a trend toward more pronounced atrophy in patients with more severe clinical conditions (i.e. lower age at seizure onset and higher pSCN1A Epilepsy Prediction Model, as indicated in Table 3). In patients with Dravet syndrome (Table 3), we found more severe cortical thinning in the right parahippocampal region (β3 = −0.061, P = 0.030). We also observed a significantly negative correlation between age at seizure onset and volume of specific subcortical structures (Table 4, putamen and pallidum, bilaterally; left amygdala, right thalamus, and left accumbens).
Table 3.
Effect on clinical variables of the cortical thickness (CT) of temporal structures in SCN1A-related epilepsy patients. Data were analyzed by a multivariate linear regression model (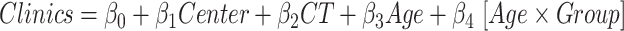 ) to explore the effect on the clinical variables of patients because of the center of acquisition (
) to explore the effect on the clinical variables of patients because of the center of acquisition (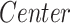 ), group (
), group (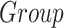 ), and age (Age).
), and age (Age).
| Age at seizure onset | pSCN1A | Clinical diagnosis (DS vs GEFS+) | |||||
|---|---|---|---|---|---|---|---|
| Region | Side |
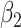
|
P value |
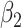
|
P value |
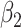
|
P value |
| Entorhinal | L | −0.292 | 0.1563 | 0.0109 | 0.174 | −0.0307 | 0.405 |
| R | −0.360 | 0.0943 | 0.0141 | 0.098 | −0.0189 | 0.614 | |
| Fusiform | L | −0.324 | 0.1929 | 0.0090 | 0.309 | −0.0785 | 0.088 |
| R | −0.462 | 0.0574 | 0.0147 | 0.120 | −0.0329 | 0.507 | |
| Parahippocampal | L | −0.264 | 0.1914 | 0.0130 | 0.114 | −0.0356 | 0.226 |
| R | −0.293 | 0.1566 | 0.0136 | 0.096 | −0.0613 | 0.030 | |
| Superior temporal | L | −0.400 | 0.0552 | 0.0172 | 0.039 | −0.0218 | 0.545 |
| R | −0.409 | 0.0543 | 0.0197 | 0.024 | −0.0169 | 0.650 | |
| Middle temporal | L | −0.418 | 0.0570 | 0.0157 | 0.067 | −0.0367 | 0.380 |
| R | −0.392 | 0.0577 | 0.0170 | 0.045 | −0.0251 | 0.515 | |
| Inferior temporal | L | −0.429 | 0.0409 | 0.0173 | 0.042 | −0.0305 | 0.416 |
| R | −0.413 | 0.0536 | 0.0160 | 0.064 | −0.0340 | 0.393 | |
| Temporal pole | L | −0.411 | 0.0485 | 0.0161 | 0.062 | −0.0192 | 0.614 |
| R | −0.419 | 0.0390 | 0.0171 | 0.030 | −0.0199 | 0.611 | |
| Frontal pole | L | −0.440 | 0.08459 | 0.0167 | 0.107 | −0.0464 | 0.409 |
| R | −0.632 | 0.0112 | 0.0200 | 0.056 | −0.0571 | 0.340 | |
| Insula | L | −0.364 | 0.11663 | 0.0141 | 0.130 | −0.0368 | 0.355 |
| R | −0.362 | 0.0775 | 0.0147 | 0.064 | −0.0230 | 0.519 | |
| Mean thickness | L | −0.432 | 0.06522 | 0.0143 | 0.109 | −0.0473 | 0.332 |
| R | −0.477 | 0.0353 | 0.0160 | 0.075 | −0.0335 | 0.494 | |
Bold indicates significantly altered measures (P < 0.05).
Table 4.
Effect on clinical variables of the volume of subcortical structures in SCN1A-related epilepsy patients. Data were analyzed by a multivariate linear regression model (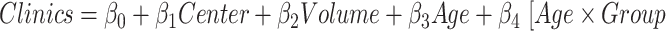 ]) to explore the effect on the clinical variables of SCN1A patients because of the center of acquisition (
]) to explore the effect on the clinical variables of SCN1A patients because of the center of acquisition (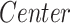 ), group (
), group (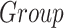 ), and age (Age).
), and age (Age).
| Age at seizure onset | pSCN1A | Clinical Diagnosis (DS vs GEFS+) | |||||
|---|---|---|---|---|---|---|---|
| Region | Side |
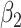
|
P value |
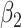
|
P value |
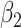
|
P value |
| Thalamus | L | 0.0031 | 0.0583 | −5.240E−05 | 0.3882 | −2.168E−04 | 0.1030 |
| R | 0.0034 | 0.0497 | −6.775E−05 | 0.3072 | −2.216E−04 | 0.1266 | |
| Caudate | L | 0.0019 | 0.5455 | −3.367E−05 | 0.7585 | −2.818E−04 | 0.2646 |
| R | 0.0029 | 0.3500 | −7.583E−05 | 0.4886 | −3.840E−04 | 0.1265 | |
| Putamen | L | 0.0038 | 0.0334 | −9.970E−05 | 0.1286 | −2.811E−04 | 0.0843 |
| R | 0.0039 | 0.0146 | −9.844E−05 | 0.1023 | −2.853E−04 | 0.0584 | |
| Pallidum | L | 0.0144 | 0.0155 | −2.609E−04 | 0.2681 | −8.989E−04 | 0.0652 |
| R | 0.0149 | 0.0134 | −3.170E−04 | 0.1910 | −8.589E−04 | 0.0903 | |
| Hippocampus | L | 0.0028 | 0.3450 | −3.029E−05 | 0.7882 | −4.012E−04 | 0.2694 |
| R | 0.0042 | 0.1822 | −7.724E−05 | 0.5266 | −3.371E−04 | 0.2411 | |
| Amygdala | L | 0.0105 | 0.0472 | −2.885E−04 | 0.1154 | −5.874E−04 | 0.2421 |
| R | 0.0048 | 0.3938 | −1.179E−04 | 0.5668 | −3.447E−04 | 0.4569 | |
| Accumbens | L | 0.0253 | 0.0096 | −5.987E−04 | 0.0853 | −2.146E−03 | 0.0072 |
| R | 0.0174 | 0.1222 | −3.392E−04 | 0.4157 | −1.392E−03 | 0.1293 | |
Bold indicates significantly altered measures (P < 0.05).
Discussion
We examined quantitative neuroimaging features of cortical and subcortical structures in patients with SCN1A-related epilepsy and in controls and found patients to exhibit a bilateral reduction of CT and SA in the mesio-temporal regions, i.e. parahippocampal and entorhinal cortex. We also observed bilateral volumetric reductions in subcortical regions associated with the hippocampus and amygdala. As observed in other epilepsy-related conditions, the abnormal pattern of CT detected in these regions might result from severe and chronic seizure activity (Whelan et al. 2018). However, a reduced SA might also reflect a defective brain development (Llinares-Benadero and Borrell 2019; Richards et al. 2021), as demonstrated by diagnostic MRI in other sodium channelopathies (Baasch et al. 2014; Syrbe et al. 2016; Zaman et al. 2020).
Previous studies highlighted large-scale structural alterations of the brain in children with SCN1A mutations (Lee et al. 2017), especially in Dravet syndrome (Pérez et al. 2014). The atrophic patterns we identified bear similarities in topography with respect to those earlier studies but were more pronounced in structures of the hippocampal formation. Our network-based analysis investigating which cortical and subcortical structures could be considered as an anatomic epicenter for SCN1A-related epilepsy identified CT and SA abnormalities in the mesial temporal lobe. To study how epileptic seizures might model the atrophic patterns in cortical and subcortical brain structures in common epilepsies, Larivière et al. (2020) proposed a network-based modeling of the atrophic patterns and found that in temporal lobe epilepsy (TLE) ipsilateral temporo-limbic cortices and several ipsilateral subcortical regions could be identified as disease epicenters. The investigational hypothesis was that GM atrophy patterns in TLE and idiopathic generalized epilepsy are related to the brain connectome architecture, which concentrates the effects of TLE in the limbic regions. In TLE, the most severe cortical thinning was associated with longer epilepsy duration (Whelan et al. 2018). A study investigating longitudinal MRI scans performed at least 6 months apart in individuals with focal epilepsy showed that the annualized rate of atrophy within brain regions structurally connected to the ipsilateral hippocampus exceeded the rate expected for healthy aging (Galovic et al. 2019). Our study suggests that earlier age at seizure onset might have an effect toward a more severe volumetric reduction at cortical level. Lower hippocampal volumes have also been reported in nonaffected siblings of patients with TLE, suggesting a genetic origin of hippocampal vulnerability (Long et al. 2020).
We conducted a genotype–phenotype analysis to establish whether the observed atrophic patterns are more likely the consequence of specific mutations or epilepsy severity, e.g. seizure burden. Brain atrophy might also correlate with the use of some antiseizure medications (Tondelli et al. 2020), but information on specific medication use at the time of MRI could not be systematically retrieved. We found that atrophic changes were more severe in patients with Dravet syndrome with respect to GEFS+, suggesting that seizure frequency may at least partly contribute to volume loss. However, we found no evidence of association between mutation type/location and severity of atrophy.
Cortico-hippocampal circuits are an elective site of pathology in SCN1A-mutated mice (Mattis et al. 2022) where circuit dysfunction may be because of excessive excitation rather than impaired inhibition. Studies in induced pluripotent stem (iPS) cells generated from patients with Dravet syndrome showed that SCN1A mutations specifically reduced the excitability of inhibitory neurons in the hippocampus, whereas the derived excitatory neurons remained functionally normal (Stein et al. 2019). In our study, longitudinal assessment of 3 patients confirmed that the SCN1A mutations they carried influenced the regular growth patterns of the brain. In 2 children with GEFS+, we observed specific patterns of atrophy, whereas an infant with Dravet syndrome exhibited signs of impaired developmental processes at whole-brain level. Brain development is critical for later cognitive outcomes and vulnerable to early neurodevelopmental disorders, in particular during the first 2 postnatal years as highlighted by longitudinal studies (Wang et al. 2019). Our cross-sectional analysis confirmed a tendency for an atrophic pattern in the hippocampal formation, which was more pronounced in patients with more severe clinical conditions. In particular, patients with Dravet syndrome exhibited more severe atrophic changes, corroborating the hypothesis that a structural abnormality underlies the clinical disorder (Guerrini et al. 2011). Extrapyramidal signs and gait abnormalities that are observed in adults with Dravet syndrome (Selvarajah et al. 2022) are in line with the progressive atrophic changes that emerged from our analysis.
This study has some limitations. Patients were retrospectively recruited in a relatively small cohort with a wide age range. However, age effects were considered and evaluated in the multivariate statistical models. MR images had not been obtained using a standardized protocol, and the recruitment of controls only at one center may have introduced unbalances in the analysis. However, we adopted a validated pipeline for the morphometric approaches that attenuated discrepancies between MRI protocols adopted in different centers. Finally, the structural covariance of atrophic patterns was calculated by adopting a novel method based on standardized connectomics data sets of adults. More advanced neuroimaging studies are warranted by adopting dedicated protocols for the assessment of the connectivity profiles at the single-subject level.
Supplementary Material
Acknowledgments
Meyer Children’s Hospital IRCCS, IRCCS Istituto Neurologico Besta, and Neuropsichiatria Infantile Verona are Full Members of the ERN EpiCare.
Meyer Children’s Hospital IRCCS, Fondazione IRCCS Neurologico Carlo Besta, and University of Verona are members of the European Reference Network for rare and complex epilepsies EpiCARE.
Contributor Information
Matteo Lenge, Neuroscience Department, Meyer Children’s Hospital IRCCS, 50139, Florence, Italy.
Simona Balestrini, Neuroscience Department, Meyer Children’s Hospital IRCCS, 50139, Florence, Italy.
Davide Mei, Neuroscience Department, Meyer Children’s Hospital IRCCS, 50139, Florence, Italy.
Letizia Macconi, Neuroradiology Unit, Meyer Children’s Hospital IRCCS, 50139, Florence, Italy.
Maria Eugenia Caligiuri, Neuroscience Research Center, Department of Medical and Surgical Sciences, Magna Grecia University, 88100, Catanzaro, Italy.
Valeria Cuccarini, Neuroradiology Unit, Fondazione IRCCS Neurologico Carlo Besta, 20100, Milan, Italy.
Domenico Aquino, Neuroradiology Unit, Fondazione IRCCS Neurologico Carlo Besta, 20100, Milan, Italy.
Federica Mazzi, Neuroradiology Unit, Fondazione IRCCS Neurologico Carlo Besta, 20100, Milan, Italy.
Ludovico d’Incerti, Neuroradiology Unit, Meyer Children’s Hospital IRCCS, 50139, Florence, Italy.
Francesca Darra, Child Neuropsychiatry Unit, Department of Engineering for Innovation Medicine University of Verona, 37100, Verona, Italy.
Bernardo Dalla Bernardina, Child Neuropsychiatry Unit, Department of Engineering for Innovation Medicine University of Verona, 37100, Verona, Italy; Pediatric Epilepsy Research Center (CREP), Azienda Ospedaliera Universitaria Integrata, 37100, Verona, Italy.
Renzo Guerrini, Neuroscience Department, Meyer Children’s Hospital IRCCS, 50139, Florence, Italy.
Author contributions
Matteo Lenge (Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Validation, Visualization, Writing—original draft), Simona Balestrini (Writing—review & editing), Davide Mei (Data curation, Visualization), Letizia Macconi (Data curation, Methodology), Maria E. Caligiuri (Data curation, Methodology), Valeria Cuccarini (Writing—review & editing), Domenico Aquino (Data curation), Federica Mazzi (Data curation), Ludovico d’Incerti (Data curation, Writing—review & editing), Francesca Darra (Writing—review & editing), Bernardo D. Bernardina (Writing—review & editing), and Renzo Guerrini (Funding acquisition, Supervision, Writing—review & editing)
Funding
The study was supported by the DESIRE project (FP7 EU call, grant agreement 602531) and its extension by the DECODE-EE (Tuscany Region Call for Health 2018) and the Human Brain Optical Mapping (Fondazione Cassa di Risparmio di Firenze 2021) projects. This project was also supported by the European Union - Next Generation EU - NRRP M6C2 - Investment 2.1 Enhancement and strengthening of biomedical research in the NHS; and by a grant from the Italian Ministry of Health and Tuscany Region (grant code RF-2021-12372804, 2023-2026) Use of Transcranial Magnetic Stimulation (TMS) as a surrogate of Pathophysiology in genetic epilepsies - TMSpath. It has also been supported by Dravet Italia Onlus.
Conflict of interest statement: None declared.
Data availability
Data are available from the corresponding author upon reasonable request.
References
- Alhusaini S, Whelan CD, Sisodiya SM, Thompson PM. Quantitative magnetic resonance imaging traits as endophenotypes for genetic mapping in epilepsy. NeuroImage Clin. 2016:12:526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baasch AL, Hüning I, Gilissen C, Klepper J, Veltman JA, Gillessen-Kaesbach G, Hoischen A, Lohmann K. Exome sequencing identifies a de novo SCN2A mutation in a patient with intractable seizures, severe intellectual disability, optic atrophy, muscular hypotonia, and brain abnormalities. Epilepsia. 2014:55(4):25–29. [DOI] [PubMed] [Google Scholar]
- Barba C, Parrini E, Coras R, Galuppi A, Craiu D, Kluger G, Parmeggiani A, Pieper T, Schmitt-Mechelke T, Striano P, et al. Co-occurring malformations of cortical development and SCN1A gene mutations. Epilepsia. 2014:55(7):1009–1019. [DOI] [PubMed] [Google Scholar]
- Bathelt J, Astle D, Barnes J, Raymond FL, Baker K. Structural brain abnormalities in a single gene disorder associated with epilepsy, language impairment and intellectual disability. NeuroImage Clin. 2016:12:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunklaus A, Pérez-Palma E, Ghanty I, Xinge J, Brilstra E, Ceulemans B, Chemaly N, de Lange I, Depienne C, Guerrini R, et al. Development and validation of a prediction model for early diagnosis of SCN1A-related epilepsies. Neurology. 2022:98(11):E1163–E1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camfield P, Camfield C. Febrile seizures and genetic epilepsy with febrile seizures plus (GEFS+). Epileptic Disord. 2015:17(2):124–133. [DOI] [PubMed] [Google Scholar]
- Catarino CB, Liu JYW, Liagkouras I, Gibbons VS, Labrum RW, Ellis R, Woodward C, Davis MB, Smith SJ, Cross JH, et al. Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology. Brain. 2011:134(10):2982–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetica V, Chiari S, Mei D, Parrini E, Grisotto L, Marini C, Pucatti D, Ferrari A, Sicca F, Specchio N, et al. Clinical and genetic factors predicting Dravet syndrome in infants with SCN1A mutations. Neurology. 2017:88(11):1037–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage. 2006:31(3):968–980. [DOI] [PubMed] [Google Scholar]
- Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011:52(SUPPL. 2):3–9. [DOI] [PubMed] [Google Scholar]
- Fischl B. FreeSurfer. NeuroImage. 2012:62(2):774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A. 2000:97(20):11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, Van Der Kouwe A, Killiany R, Kennedy D, Klaveness S, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002:33(3):341–355. [DOI] [PubMed] [Google Scholar]
- Fischl B, van der Kouwe A, Destrieux C, Halgren E, Ségonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, et al. Automatically parcellating the human cerebral cortex. Cereb Cortex. 2004:14(1):11–22. [DOI] [PubMed] [Google Scholar]
- Galovic M, Van Dooren VQH, Postma TS, Vos SB, Caciagli L, Borzì G, Cueva Rosillo J, Vuong KA, De Tisi J, Nachev P, et al. Progressive cortical thinning in patients with focal epilepsy. JAMA Neurol. 2019:76(10):1230–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini R, Striano P, Catarino C, Sisodiya SM. Neuroimaging and neuropathology of Dravet syndrome. Epilepsia. 2011:52(SUPPL. 2):30–34. [DOI] [PubMed] [Google Scholar]
- Guerrini R, Conti V, Mantegazza M, Balestrini S, Galanopoulou AS, Benfenati F. Developmental and epileptic encephalopathies: from genetic heterogeneity to phenotypic continuum. Physiol Rev. 2023:103(1):433–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenlocher PR. Morphometric study of human cerebral cortex development. Neuropsychologia. 1990:28(6):517–527. [DOI] [PubMed] [Google Scholar]
- Iglesias JE, Augustinack JC, Nguyen K, Player CM, Player A, Wright M, Roy N, Frosch MP, McKee AC, Wald LL, et al. A computational atlas of the hippocampal formation using ex vivo, ultra-high resolution MRI: application to adaptive segmentation of in vivo MRI. NeuroImage. 2015:115:117–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias JE, Insausti R, Lerma-Usabiaga G, Bocchetta M, Van Leemput K, Greve DN, van der Kouwe A, Fischl B, Caballero-Gaudes C, Paz-Alonso PM. A probabilistic atlas of the human thalamic nuclei combining ex vivo MRI and histology. NeuroImage. 2018:183:314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson M, Beckmann CF, Behrens TEJJ, Woolrich MW, Smith SM. Fsl. NeuroImage. 2012:62(2):782–790. [DOI] [PubMed] [Google Scholar]
- Larivière S, Rodríguez-Cruces R, Royer J, Caligiuri ME, Gambardella A, Concha L, Keller SS, Cendes F, Yasuda C, Bonilha L, et al. Network-based atrophy modeling in the common epilepsies: a worldwide ENIGMA study. Sci Adv. 2020:6(47):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y-JJ, Yum M-SS, Kim M-JJ, Shim W-HH, Yoon HM, Yoo IH, Lee J, Lim BC, Kim KJ, Ko T-SS. Large-scale structural alteration of brain in epileptic children with SCN1A mutation. NeuroImage Clin. 2017:15:594–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenge M, Marini C, Canale E, Napolitano A, de Masi S, Trivisano M, Mei D, Longo D, Espagnet MCR, Lucenteforte E, et al. Quantitative MRI-based analysis identifies developmental limbic abnormalities in PCDH19 encephalopathy. Cereb Cortex. 2020:30(11):6039–6050. [DOI] [PubMed] [Google Scholar]
- Llinares-Benadero C, Borrell V. Deconstructing cortical folding: genetic, cellular and mechanical determinants. Nat Rev Neurosci. 2019:20(3):161–176. [DOI] [PubMed] [Google Scholar]
- Long L, Galovic M, Chen Y, Postma T, Vos SB, Xiao F, Wu W, Song Y, Huang S, Koepp M, et al. Shared hippocampal abnormalities in sporadic temporal lobe epilepsy patients and their siblings. Epilepsia. 2020:61(4):735–746. [DOI] [PubMed] [Google Scholar]
- Mantegazza M, Broccoli V. SCN1A/NaV1.1 channelopathies: mechanisms in expression systems, animal models, and human iPSC models. Epilepsia. 2019:60(S3):S25–S38. [DOI] [PubMed] [Google Scholar]
- Mattis J, Somarowthu A, Goff KM, Jiang E, Yom J, Sotuyo N, McGarry LM, Feng H, Kaneko K, Goldberg EM. Corticohippocampal circuit dysfunction in a mouse model of Dravet syndrome. eLife. 2022:11:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei D, Cetica V, Marini C, Guerrini R. Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies. Epilepsia. 2019:60(S3):S2–S7. [DOI] [PubMed] [Google Scholar]
- Nabbout R, Chemaly N, Chipaux M, Barcia G, Bouis C, Dubouch C, Leunen D, Jambaqué I, Dulac O, Dellatolas G, et al. Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J Rare Dis. 2013:8(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panizzon MS, Fennema-Notestine C, Eyler LT, Jernigan TL, Prom-Wormley E, Neale M, Jacobson K, Lyons MJ, Grant MD, Franz CE, et al. Distinct genetic influences on cortical surface area and cortical thickness. Cereb Cortex. 2009:19(11):2728–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez A, García-Pentón L, Canales-Rodríguez EJ, Lerma-Usabiaga G, Iturria-Medina Y, Román FJ, Davidson D, Alemán-Gómez Y, Acha J, Carreiras M. Brain morphometry of Dravet syndrome. Epilepsy Res. 2014:108(8):1326–1334. [DOI] [PubMed] [Google Scholar]
- Rakic P. Specification of cerebral cortical areas. Science. 1988:241(80):170–176. [DOI] [PubMed] [Google Scholar]
- Richards K, Jancovski N, Hanssen E, Connelly A, Petrou S. Atypical myelinogenesis and reduced axon caliber in the Scn1a variant model of Dravet syndrome: an electron microscopy pilot study of the developing and mature mouse corpus callosum. Brain Res. 2021:1751:147157. [DOI] [PubMed] [Google Scholar]
- Ruffolo G, Martinello K, Labate A, Cifelli P, Fucile S, Di Gennaro G, Quattrone A, Esposito V, Limatola C, Giangaspero F, et al. Modulation of GABAergic dysfunction due to SCN1A mutation linked to hippocampal sclerosis. Ann Clin Transl Neurol. 2020:7(9):1726–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sämann PG, Iglesias JE, Gutman B, Grotegerd D, Leenings R, Flint C, Dannlowski U, Clarke-Rubright EK, Morey RA, van Erp TGM, et al. FreeSurfer-based segmentation of hippocampal subfields: a review of methods and applications, with a novel quality control procedure for ENIGMA studies and other collaborative efforts. Hum Brain Mapp. 2020:43(1):207–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saygin ZM, Kliemann D, Iglesias JE, van der Kouwe AJW, Boyd E, Reuter M, Stevens A, Van Leemput K, McKee A, Frosch MP, et al. High-resolution magnetic resonance imaging reveals nuclei of the human amygdala: manual segmentation to automatic atlas. NeuroImage. 2017:155:370–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvarajah A, Gorodetsky C, Marques P, Zulfiqar Ali Q, Berg AT, Fasano A, Andrade DM. Progressive worsening of gait and motor abnormalities in older adults with Dravet syndrome. Neurology. 2022:98(22):E2204–E2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodiya SM, Whelan CD, Hatton SN, Huynh K, Altmann A, Ryten M, Vezzani A, Caligiuri ME, Labate A, Gambardella A, et al. The ENIGMA-epilepsy working group: mapping disease from large data sets. Hum Brain Mapp. 2020:43(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein RE, Kaplan JS, Li J, Catterall WA. Hippocampal deletion of NaV1.1 channels in mice causes thermal seizures and cognitive deficit characteristic of Dravet syndrome. Proc Natl Acad Sci U S A. 2019:116(33):16571–16576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striano P, Mancardi MM, Biancheri R, Madia F, Gennaro E, Paravidino R, Beccaria F, Capovilla G, Bernardina BD, Darra F, et al. Brain MRI findings in severe myoclonic epilepsy in infancy and genotype-phenotype correlations. Epilepsia. 2007:48(6):1092–1096. [DOI] [PubMed] [Google Scholar]
- Syrbe S, Zhorov BS, Bertsche A, Bernhard MK, Hornemann F, Mütze U, Hoffmann J, Hörtnagel K, Kiess W, Hirsch FW, et al. Phenotypic variability from benign infantile epilepsy to Ohtahara syndrome associated with a novel mutation in SCN2A. Mol Syndromol. 2016:7(4):182–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tondelli M, Vaudano AE, Sisodiya SM, Meletti S. Valproate use is associated with posterior cortical thinning and ventricular enlargement in epilepsy patients. Front Neurol. 2020:11(622):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuoksimaa E, Panizzon MS, Chen C-H, Fiecas M, Eyler LT, Fennema-Notestine C, Hagler DJ, Fischl B, Franz CE, Jak A, et al. The genetic association between neocortical volume and general cognitive ability is driven by global surface area rather than thickness. Cereb Cortex. 2015:25(8):2127–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Lian C, Wu Z, Zhang H, Li T, Meng Y, Wang L, Lin W, Shen D, Li G. Developmental topography of cortical thickness during infancy. Proc Natl Acad Sci U S A. 2019:116(32):15855–15860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan CD, Altmann A, Botía JA, Jahanshad N, Hibar DP, Absil J, Alhusaini S, Alvim MKM, Auvinen P, Bartolini E, et al. Structural brain abnormalities in the common epilepsies assessed in a worldwide ENIGMA study. Brain. 2018:141(2):391–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman T, Helbig KL, Clatot J, Thompson CH, Kang SK, Stouffs K, Jansen AE, Verstraete L, Jacquinet A, Parrini E, et al. SCN3A-related neurodevelopmental disorder: a Spectrum of epilepsy and brain malformation. Ann Neurol. 2020:88(2):348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding author upon reasonable request.