

Summary
Heterogeneous nuclear ribonucleoprotein C (HNRNPC) is an essential, ubiquitously abundant protein involved in mRNA processing. Genetic variants in other members of the HNRNP family have been associated with neurodevelopmental disorders. Here, we describe 13 individuals with global developmental delay, intellectual disability, behavioral abnormalities, and subtle facial dysmorphology with heterozygous HNRNPC germline variants. Five of them bear an identical in-frame deletion of nine amino acids in the extreme C terminus. To study the effect of this recurrent variant as well as HNRNPC haploinsufficiency, we used induced pluripotent stem cells (iPSCs) and fibroblasts obtained from affected individuals. While protein localization and oligomerization were unaffected by the recurrent C-terminal deletion variant, total HNRNPC levels were decreased. Previously, reduced HNRNPC levels have been associated with changes in alternative splicing. Therefore, we performed a meta-analysis on published RNA-seq datasets of three different cell lines to identify a ubiquitous HNRNPC-dependent signature of alternative spliced exons. The identified signature was not only confirmed in fibroblasts obtained from an affected individual but also showed a significant enrichment for genes associated with intellectual disability. Hence, we assessed the effect of decreased and increased levels of HNRNPC on neuronal arborization and neuronal migration and found that either condition affects neuronal function. Taken together, our data indicate that HNRNPC haploinsufficiency affects alternative splicing of multiple intellectual disability-associated genes and that the developing brain is sensitive to aberrant levels of HNRNPC. Hence, our data strongly support the inclusion of HNRNPC to the family of HNRNP-related neurodevelopmental disorders.
Keywords: heterogeneous ribonucleoprotein C, HNRNPC, HNRNP, neurodevelopmental disorder, NDD, intellectual disability, RNA processing, alternative splicing, induced pluripotent stem cells, iPSCs
Graphical abstract
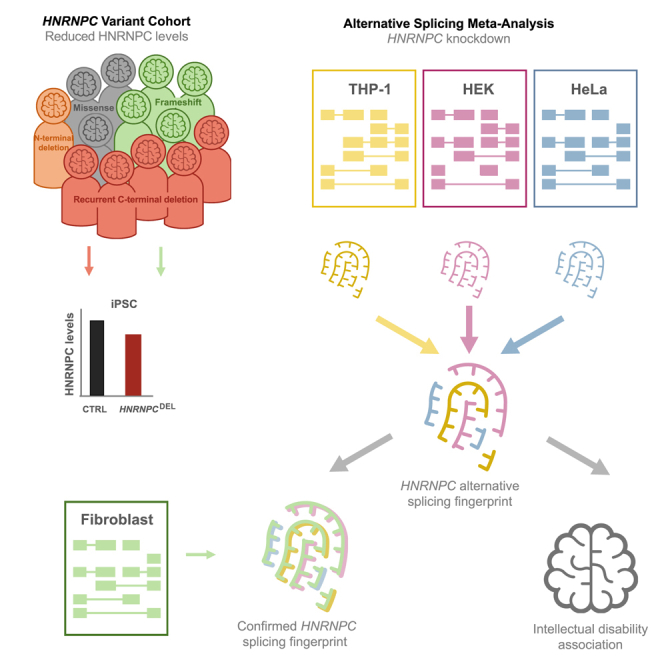
We identified genetic variants of HNRNPC in 13 individuals with intellectual disability and global developmental delay. Through a meta-analysis of multiple cell types, we found that loss of HNRNPC affects alternative splicing, in particular of intellectual disability-associated genes. In vivo assays confirmed that neurodevelopment was affected by aberrant HNRNPC levels.
Introduction
Heterogeneous nuclear ribonucleoprotein C (HNRNPC [MIM: 164020]) encodes a member of the ubiquitous HNRNP family, consisting of 33 distinct RNA-binding proteins.1 These proteins play diverse roles in various aspects of mRNA processing, making them key regulators of gene expression.2,3 Six members of the HNRNP family have previously been associated with neurodevelopmental disorders (NDDs) (HNRNPH1 [MIM: 601035],4 HNRNPH2 [MIM: 300610],5 HNRNPK [MIM: 600712],6 HNRNPR [MIM: 607201],7 HNRNPU [MIM: 602869],8,9 SYNCRIP/HNRNPQ [MIM: 616686]10). Through an extensive meta-analysis, an additional seven members of the HNRNP family have recently been identified as candidate genes for NDDs.1 All 13 HNRNP members are associated with neurobehavioral phenotypes, encompassing intellectual disability, developmental delay, behavioral issues, hypotonia, seizures, and structural brain abnormalities. In most cases, variants in HNRNP with high pLI scores, indicating low tolerability of loss-of-function variants, were correlated with an NDD phenotype. This suggests a particularly crucial role for these HNRNP family proteins in neurodevelopment.1 Notably, two individuals with heterozygous de novo HNRNPC variants (Ind9 and Ind13 in our study) were previously identified in a meta-analysis of genetic variants in developmental disorders11 and HNRNP-associated developmental disorders.1 However, HNRNPC did not meet the criteria to be included as an NDD-associated HNRNP gene at the time of publication, likely due to the minimum requirement of three probands per candidate gene in this study.1
The HNRNP family members all contain one or more RNA-binding domains, such as an RNA-recognition motif (RRM) or a basic-leucine zipper (bZIP) motif,12,13 and all are involved in various aspects of RNA processing.2 Several studies have shown that HNRNPC specifically plays an important role in RNA splicing by facilitating alternative exon usage.14,15,16 Furthermore, HNRNPC functions as a molecular ruler, aiding in the export of mRNA transcripts longer than 700 nucleotides from the nucleus to the cytoplasm. Consequently, the loss of HNRNPC leads to the accumulation of mRNA in the nucleus through the U snRNA pathway.17 HNRNPC has also been implicated in IRES-related translation, where its binding to poly U stretches facilitates the assembly of the translational machinery. Notably, the translation of target proteins such as MYC (alias c-myc),18 NR1H2 (alias Unr),19 PDGFB (alias c-sis),20 and XIAP21 correlates with HNRNPC levels. Lastly, the regulatory post-transcriptional process known as N6-methyladenine (m6A) modification has been associated with HNRNPC function,22,23 reporting HNRNPC as a “reader” of m6A modification.
In this study, we describe a cohort of 13 individuals with heterozygous germline variants in HNRNPC, including a recurrent de novo in-frame deletion in five individuals (GenBank: NC_000014.9:g.21211238_21211264del, equivalent to c.850_876del [p.Arg284_Asp292del] for HNRNPC-iso1 and c.889_915del [p.Arg297_Asp305del] for HNRNPC-iso2), further referred to as HNRNPCDEL. This report delineates the molecular and phenotypic spectrum of a HNRNPC-related neurodevelopmental disorder, characterized by global developmental delay, intellectual disability, behavioral abnormalities, and subtle facial dysmorphic features in most individuals. The molecular function of HNRNPC was assessed in vitro, utilizing induced pluripotent stem cells (iPSCs) and fibroblasts derived from affected individuals. The study focused on investigating RNA processing-related functions such as alternative splicing. In addition, the effect of altered HNRNPC levels on (murine) neuronal function is assessed in vivo and in vitro. Taken together, our data provide evidence that HNRNPC variants underlie the neurodevelopmental phenotype in these individuals, supporting the inclusion of HNRNPC in the family of HNRNP-related neurodevelopmental disorders.
Material and methods
Ethics approval
The generation of iPSCs of Ind1 and control has been approved by the Erasmus MC ethics commision (METC, NL60886.078.17). Ind8 was identified through participation in the Undiagnosed Diseases Network Study, which was approved by the National Institutes of Health Intramural Institutional Review Board. The remaining individuals with HNRNPC variants from this cohort were identified in a diagnostic setting, except for Ind9 and Ind13 who had been previously reported in literature. Therefore, no additional ethics approval was required for this retrospective study.1,11
Consent
Consent was obtained regarding clinical information and details of the HNRNPC variant for all individuals from this retrospective cohort study. In addition, for Ind1 from this study additional consent was obtained regarding the construction of iPSC lines. Consent to participate and to publish pictures/clinical details was obtained for all included individuals by their treating clinician.
Identification of Ind1 with HNRNPC variant via whole-exome sequencing
Whole-exome sequencing (WES) was performed with Agilent Sureselect Capture (Clinical Research Exome V2) and run on HiSeq (101 bp paired-end, Illumina). Data were demultiplexed by the Illumina Software CASAVA.24 Reads were mapped to hg19 using the program BWA.25 Variants were detected with the Genome Analysis Toolkit (GATK).26 Variants were filtered with the Cartagenia software package (Agilent technologies) on quality (read depth ≥10), minor allele frequency (≥0.1% in 200 alleles in dbSNP, ESP6500, the 1000 Genome project, GoNLor the ExAC database), and location (within an exon or first/last 10 bp of introns). Variants were further selected based on three inheritance models (de novo autosomal dominant, autosomal recessive, and X-linked recessive), and classified with Alamut Visual.
Cloning and lentiviral generation
shRNA constructs for knockdown
The shRNA expression plasmids targeting either human HNRNPC or mouse Hnrnpc are cloned into the pLKO.1 backbone (Addgene, 8453). Ready-made plasmids were purchased from the shRNA Mission Library (Sigma) via the Erasmus MC Biomics facility. Targeting sequences of the shRNA can be found in Table S1.
Cloning HNRNPC-iso1, HNRNPC-iso2, and deletion constructs
As part of our routine pipeline for rapid screening of variants of unknown significance in candidate ID genes (PRiSM, www.functionalgenomics.nl/), we generated expression constructs for HNRNPC-iso1 and HNRNPC-iso2.
HNRNPC-iso1 was amplified from human cDNA using Phusion polymerase (New England Biolabs) with primer P3608 introducing an AscI site and Kozak sequence at the 5′ end of the gene and primer P3600 introducing a NotI and a PacI site at the 3′ end of the gene. The recurrent variant (c.889_915del [p.Arg297_Asp305del]) was cloned using primers P3608 and P3601 introducing the 27 bp (9 aa) deletion at the 3′ end of HNRNPC-iso1 as well as a NotI and a PacI site. Both PCR fragments were cloned into a TOPO backbone and their sequence verified (Macrogen). Using AscI and PacI restriction sites, HNRNPC-iso1 and HNRNPC-iso1DEL were cloned into a dual expression vector27 which expresses the gene of interest under the CAG promoter and tdTomato under the PGK promoter. This vector lacking the gene of interest was used as a negative control (empty vector).
HNRNPC-iso2 was generated by amplification of HNRNPC-iso1 or HNRNPC-iso1DEL using Phusion polymerase with two sets of primers, to enable insertion of the C2 domain. The 5′ end was amplified using primer P1971 which hybridizes with the CAG promoter of the HNRNPC-iso1 construct and primer P5549 introducing the C2 domain and a silent mutation resulting in a BamHI site. The 3′ end of HNRNPC-iso1 or HNRNPC-iso1DEL was amplified using primer P5550 introducing a silent mutation resulting in a BamHI site and primer p5551 introducing a NotI and PacI site. PCR-fragments were digested with AscI-BamHI and BamHI-NotI respectively and cloned into the expression vector using AscI and NotI restriction sites. All purified plasmids (Midi plasmid kit, QIAGEN) were verified by sequencing the gene of interest (Macrogen).
To obtain constructs without tdTomato, the HNRNPC variants were cloned into a backbone with a CAG promoter but without the PGK promoter and tdTomato using AscI and NotI restriction sites. A list of all primers can be found in Table S2 and a list of plasmids in Table S3.
Tagged HNRNPC constructs
N-terminal tags were cloned into the HNRNPC-iso1 and HNRNPC-iso1DEL constructs using the EcoRI and AscI sites preceding the HNRNPC start codon sequence. For the FLAG tag at the 5′ end of HNRNPC, a start codon sequence followed by a 3xFLAG-tag sequence and a Gly-Ala-Pro sequence were introduced. This was achieved using dimerized primers P5288 and P5289, which had EcoRI and AscI sticky ends at the 5′ and 3′ end of the dimer, respectively. For the HA-tag at the 5′ end of HNRNPC, a start codon followed by the 3xHA-tag sequence with a Ser-Gly-Ala-Pro linker sequence were generated using two dimerized primer sets, P5436 and P5437, which had a 5′ EcoRI overhang and a 3′ BsiWI restriction site (BsiWI digested after dimerization), and P5438 and P5439, which had a 5′ BsiWI overhang and 3′ AscI overhang. For the V5-tag at the 5′ end of HNRNPC, a start codon followed by a V5-tag sequence and Ser-Gly-Ala-Pro sequence were accomplished using dimerized primers P5440 and P5441, with EcoRI and AscI overhangs at the 5′ and 3′ ends of the dimer, respectively.
The eGFP fragment was amplified from a plasmid containing the eGFP sequence using primer P5442, which introduced a EcoRI site at the 3′ end, and primer P5443, which removed the original stop codon sequence and introduced a Ser-Gly-Ala-Pro linker sequence followed by an AscI restriction site at the 5′ end. To generate the HNRNPC-iso2 and HNRNPC-iso2DEL constructs, HNRNPC-iso1 was replaced by HNRNPC-iso2 or HNRNPC-iso2DEL using the AscI and NotI restriction sites. All purified plasmids (Midi plasmid kit, QIAGEN) were verified by sequencing of the gene of interest (Macrogen). A list of all primers can be found in Table S2, and a list of plasmids is provided in Table S3.
Lentivirus generation
Plasmids pMD2.G (Addgene, 12259) and psPAX2 (Addgene, 12260) (both gifted by Didier Trono) were used to manufacture the shRNA containing lentiviruses. Lentiviruses were produced in HEK293-T cells as previously described by Addgene (https://www.addgene.org/protocols/lentivirus-production/). In brief, HEK293-T cells were co-transfected with envelope plasmid (Addgene #12259), lentiviral packaging plasmid (Addgene #12260), and shRNA constructs. 72 h post-transfection the virus was harvested from the cell culture medium, spun down in filter tubes (4,000 × g, 20 min; Millipore, #UFC910024), snap frozen, and stored at −80°C.
PRiSM screen
Mice
Female FvB/NHsD (Envigo) mice were crossed either with FvB/NHsD males to perform primary neuronal culture experiments or with C57BL6/J males (Charles River) for in utero electroporation experiments. All animals were group housed in IVC cages (Sealsafe 1145T, Tecniplast) and fed ad libitum with food pellets (801727CRM(P) from Special Dietary Service) with ad libitum water supply. Cages contained bedding material (Lignocel BK 8/15 from Rettenmayer) and were kept on a 12/12 h light/dark cycle at 21°C (±1°C) with humidity between 40% and 70%. All animal experiments were approved by the Local Animal Experimentation Ethical Committee, in accordance with Institutional Animal Care and Use Committee guidelines.
In utero electroporation
IUE (in utero electroporation) was performed as previously described.28 In brief, pyramidal layer 2/3 progenitor cells from mouse embryos were electroporated at gestational age E14.5. The construct of interest was co-electroporated with a tdTomato or eGFP reporter plasmid to fluorescently label targeted cells. The DNA constructs were diluted in fast green (0.05%) to a final concentration of 2 μg/μL and were subsequently injected into the lateral ventricle with a glass pipette. Tweezer-type electrodes conducted a 50 ms pulse/150 ms interpulse electrical square pulses of 45 V, generated by a pulse generator (ECM 830, BTX Harvard Apparatus). The positive pole targeted the developing somatosensory cortex (SScx). Female and male pups were used for histological processing.
Primary hippocampal cultures
Primary hippocampal cultures were prepared from FvB/NHsD wild-type mice as previously described.29 In brief, murine hippocampi were isolated from E16.5 embryos and incubated in pre-warmed trypsin/EDTA solution (Invitrogen) for tissue dissociation at 37°C for 20 min. Next, cells were resuspended in Neurobasal medium (Gibco, #21103-049) supplemented with 2% B27 (Gibco, #17504044), 1% penicillin/streptomycin (Gibco), and 1% GlutaMAX (Invitrogen). Finally, dissociated cells were plated on poly-d-lysine (25 mg/mL, Sigma)-coated 15 mm glass coverslips at a density of 1 × 106 cells per coverslip. Primary cultures were cultured at 37°C in 5% CO2.
Immunohistochemistry
Mice (P1 pups) were euthanized with an overdose of pentobarbital and perfused transcardially with 4% paraformaldehyde (PFA). Brain tissue was dehydrated in 10% sucrose overnight and embedded in 12% gelatin and 30% sucrose (P1 pups) or 10% sucrose (P7 pups) in 0.1 M Phosphate buffer (PB). Coronal sections of 40 μm were cut with a freezing microtome (SM2000R; Leica Microsystems). DNA was stained by 4′,6-diamidino-2-phenylindole solution (DAPI, 1:10,000, Invitrogen) for 10 min. Tissue slices were mounted on 24 × 40 mm coverslips with Mowiol (Sigma-Aldrich). Images were taken on a LSM700 Zeiss Confocal Laser Scanning Microscope and analyzed using Fiji.
Transfection of primary hippocampal neurons
Primary murine neurons were transfected with a total of 1.8 μg DNA per 12-well with Lipofectamine 2000 (Invitrogen, #11668-019) according to manufacturer’s instructions.
Neuronal morphology
For morphological analysis, primary murine neurons were transfected at 1 or 7 days in vitro (DIV1, DIV7) and fixed in 4% PFA/sucrose (10 min at room temperature [RT]) 3- or 5-day post-transfection. Confocal images (LSM700×Zeiss Confocal Laser Scanning, 20× objective, 0.5 zoom, 1,024 × 1,024 pixels) of transfected neurons were exported for further analysis in SynD, a published MATLAB script.30 Total neurite length and arborization were measured and a Sholl analysis was performed.
Cell culture: iPSC
iPSC generation
Peripheral mononuclear blood cells (PMBCs) were extracted and enriched from EDTA blood of Ind1 who bears the HNRNPCDEL variant (GenBank: NC_000014.9:g.21211238_21211264del), as well as an age- and gender-matched control subject. Consecutively, the PBMCs were enriched for erythroid progenitors and reprogrammed toward human induced pluripotent stem cells (iPSCs) by the Erasmus MC IPS facility. In brief, the Yamanaka transcription factors MYC (alias c-myc), KLF4, POU5F1 (alias OCT4), and SOX231 were transduced via the CytoTune-iPS 2.0 Sendai Reprogramming Kit according to manufacturer’s instructions (Invitrogen A16517). Subsequently, single colonies were selected, expanded, and cultured in complete StemFlex medium (Thermo Fisher, A3349401) according to manufacturer’s instructions. iPSC quality and pluripotency were assessed by karyotyping, qPCR of pluripotency markers, immunocytochemistry, and differentiation toward meso-, endo-, and ectoderm lineages with STEMdiff trilineage differentiation kit (STEMCELL Technologies, 05230). Confluent cultures were dissociated in 0.5 mM EDTA and passaged to Geltrex (Thermo Fisher, A1413201)-coated culture plates.
Transfection
iPSCs were transfected in a suspension of DNA (1.6 μg DNA/1 million cells), Lipofectamine 2000 (2.5 μL; Invitrogen, #11668-019) in Opti-MEM (Thermo Fisher, #31985062), as described previously.32
Lentiviral transduction
iPSCs were transduced with shRNA constructs for 80 h to achieve sufficient HNRNPC knockdown. Approximately 2.5 μL/mL viral constructs were complemented with 10 μg/mL Polybrene (Millipore, # TR-1003-G) for efficient transduction. Medium was refreshed 6 h post-transduction and every other day thereafter.
Cell culture: HEK293-T, U-2 OS
General culture
The HEK293-T (human embryonic kidney) and U-2 OS (osteoblastoma) cells were cultured at 37°C under 5% CO2 in DMEM (Gibco, #11965084) with GlutaMAX (Invitrogen, #31331093), supplemented with 10% fetal bovine serum (Capricorn scientific, #CP18-2112) and 1% penicillin/streptomycin (Sigma, #P0781). Medium was refreshed every 2–3 days and cells were passaged at ±90% confluence using Trypsin/EDTA (3 min). HEK293-T cells were purchased from ATCC, U-2 OS cells were kindly gifted by Mario van der Stelt (Molecular Physiology, Leiden University, original source ATCC). Cells were regularly tested for mycoplasma contamination. Cultures were discarded after 2–3 months of use.
Transfection
One day prior to transfection, cells were seeded at appropriate density (±60% confluence). A 3:1 (m/m) mixture of polyethyleneimine (PEI; Polyscience Inc., #24765-1) and plasmid DNA (1–1.5 μg/well in 12-well, 3 μg/well in 6-well, 10 μg/plate in 10 cm plates) was prepared in serum free DMEM (Gibco, #11965084) and incubated for 15 min at room temperature. Transfection was performed by dropwise addition of the PEI/DNA mixture to the cells. Culture medium was refreshed 6 h post-transfection and cells were fixed or harvested at 24–48 h post-transfection (as indicated in figure legends).
Immunocytochemistry
Cells were fixed on the glass coverslips with either PFA/sucrose (4% PFA; 0.4% NaOH; 1.6% NaH2PO4 Monobasic; 4% sucrose) or 4% PFA. For mRNA localization studies, cells were subsequently permeabilized with ice-cold methanol (10 min) and rehydrated in 70% ethanol (minimum 10 min). To detect poly(A)+ RNA, the cells were incubated with 1 ng/μL 5′-Cy3-poly(dT)30 probe (IDT, Integrated DNA Technologies) in hybridization buffer (25% formamide, 2× saline sodium citrate [SSC] buffer [0.3 M sodium chloride; 30 mM trisodium citrate; pH 7]; 1 mg/mL yeast tRNA, 10% dextran sulfate, in DEPC H2O) (1 h, 37°C).
Primary antibody staining was performed O/N at 4°C or 2 h at room temperature (RT). The following primary antibodies were used: rabbit-anti-HNRNPC antibody (1:1,000, Proteintech, #11760-1-AP), rabbit-anti-HNRNPC antibody (for endogenous murine HNRNPC, 1:500, Thermo scientific, #PA5-22280), guinea pig-anti-MAP2 antibody (1:750, OSynaptic Systems, #188004), rat-anti-Tubulin (1:200, Thermo scientific, #MA1-80017), and mouse-anti-KI-67 antibody (1:500, Millipore, #MAB4190). Fluorescent secondary antibodies were used for detection (1:200, 1 h at RT): donkey-anti-mouse-Alexa488 (Jackson ImmunoResearch, #715545150), donkey-anti-rabbit-Cy3 (Jackson ImmunoResearch, #711-165-152, 1:200), donkey-anti-rabbit-488 (Jackson ImmunoResearch, #711-545-152, 1:200), donkey-anti-rabbit-Alexa647 (Jackson ImmunoResearch, #711-605-152, 1:200), donkey-anti-guinea pig-647 (Jackson ImmunoResearch, #706-605-148, 1:200), donkey-anti-rat-647 (Jackson ImmunoResearch, #712-605-153, 1:200). DNA was stained by 4′,6-diamidino-2-phenylindole solution (DAPI, 1:10,000, 10 min) (Invitrogen). Coverslips were mounted on glass slides with Mowiol (Sigma-Aldrich). Images were taken on a LSM700 Zeiss Confocal Laser Scanning Microscope and analyzed using Fiji.
Western blot
For protein analysis, cells were detached according to culture protocol and pelleted (1,000 × g, 3 min). Cell pellets were lysed by sonication (30 s, probe sonicator) or in lysis buffer (50 mM Tris-HCl [pH 7.6], 100 mM NaCl, 50 mM NaF, 1% Triton X-100). Protein concentration was determined with a BCA assay (Thermo Fisher Scientific, 23225). Samples were denatured in 0.1 M DDT (Sigma, #D9779-5G) and 1× XT Sample buffer (Bio-Rad, #1610791) (5 min, 95°C). Proteins (20 μg/sample) were resolved by SDS-PAGE on precast 4%–12% Criterion XT Bis-Tris (Bio-Rad) or 4%–16% Tris-Glycin (Bio-Rad) gels along with PageRuler Plus Protein Marker (Thermo Scientific, 26620). Proteins were transferred to 0.2 μm nitrocellulose membranes by Trans-Blot Turbo Transfer system (BioRad). Membranes were blocked in 5% milk in TBS-T (50 mM Tris, 150 mM NaCl, 0.05% Tween 20) (30 min at RT) and incubated with the following antibodies: rabbit-anti-HNRNPC (1:1,000 in 2% milk TBS-T, O/N, 4°C; Proteintech, #11760-1-AP) and mouse-anti-actin (1:20,000 in 2% milk TBS-T, O/N, 4°C; Chemicon, MAB1501R), secondary goat-anti-rabbit (1:15,000 in 2% milk TBS-T, 1 h at RT; LI-COR Biosciences, IRDye 800CW,926-32211) and secondary goat-anti-mouse (1:15,000 in 2% Milk TBS-T, 1 h at RT; LI-COR Biosciences, IRDye 800CW, 926-32210).The membranes were scanned on the Odyssey CLx (LI-COR Biosciences) and quantified using the Image studio light (LI-COR Biosciences) software.
Co-immunoprecipitation
Transfection
Transfections were performed according to standard protocol (see section transfection). Sequences encoding the bait protein (FLAG-HNRNPC-iso1 or FLAG-HNRNPC-iso1DEL) and the prey protein (HA-HNRNPC-iso1 or HA-HNRNPC-iso1DEL or HA-HNRNPC-iso2 or HA-HNRNPC-iso2DEL) were co-transfected (5 μg each per 10 cm plate). Cells were harvested 24 h post-transfection by scraping in PBS, pelleted (5 min, 1,000 × g; 90% of sample for co-IP, 10% for regular western blot) and stored at −80°C.
Co-IP and sample preparation
Cell pellets were lysed in 500 μL cold co-IP buffer (20 mM Tris-HCl [pH 8.0], 0.5% NP-40, 150 mM NaCl, 1x PhosStop [Roche], 1x Complete protease inhibitor [Roche]) by sonication (2 cycles, 3 s at 5-micron amplitude). Protein concentration was determined by BCA assay (Thermo Fisher Scientific, 23225) and samples were diluted to 1 mg/mL in co-IP buffer.
Co-immunoprecipitation procedure was adapted from manufacturer’s protocol. In brief, anti-FLAG M2 Magnetic Beads (25 μL of 50% slurry per sample; Sigma-Aldrich) were diluted in wash buffer (75 μL per sample; 20 mM Tris-HCl [pH 8.0], 0.5% NP-40, 150 mM NaCl) in 1.5 mL Eppendorf tubes in a magnetic rack. Beads were washed twice in 250 μL of wash buffer.
Cell lysate (500 μL at 1 mg/mL protein) was added to the magnetic beads and 20 μL of sample was taken apart (INPUT sample). Lysates were incubated with beads with end-over-end rotation (O/N, 4°C). Subsequently, 20 μL supernatant was collected (UNBOUND sample) and the beads were washed three times with 500 μL of wash buffer. Protein was eluted from beads by boiling in 25 μL of 2x XT sample buffer (Bio-Rad, #1610791) (10 min, 95°C) (BOUND sample) and supplemented with DTT (100 mM). INPUT and UNBOUND samples were supplemented with XT sample buffer (1× final concentration) and DTT (100 mM). Proteins were denatured (5 min, 95°C) and resolved by SDS-PAGE (15 μg protein for INPUT and UNBOUND = 3% of total input; all for BOUND = 100% of total input).
SDS-PAGE and western blot
Samples were resolved on a 4%–20% Criterion TGX gel by SDS-PAGE (15 min at 100 V, 70 min at 150 V in XT-MOPS) along with PageRuler Plus Protein Marker (Thermo Scientific). Proteins were transferred to 0.2 μm nitrocellulose membranes by Trans-Blot Turbo Transfer system (Bio-Rad). Membranes were blocked with 5% milk in TBS-T (50 mM Tris, 150 mM NaCl, 0.05% Tween 20) (45 min at RT). Membranes were subsequently incubated with HRP-coupled antibody rat-anti-HA-HRP (1:1,000 in 2% milk TBS-T, 1 h at RT, Roche, AB_390917) for the co-IP or primary antibodies mouse-anti-FLAG (1:1,000 in 2% milk TBS-T; Sigma-Aldrich, F1804), mouse-anti-actin (1:20,000 in 2% milk TBS-T, 1 h at RT; Chemicon, MAB1501R), rabbit-anti-HA (1:1,000 in 2% milk TBS-T, 1 h at RT; CST3742; Cell Signaling Technologies). Membranes were subsequently rinsed with TBS-T and incubated with fluorescent secondary antibodies goat-anti-mouse (LI-COR Biosciences, IRDye 800CW, 926-32210) and goat-anti-rabbit (LI-COR Biosciences, IRDye 800CW,926-32211) (both 1:15,000 in 2% milk TBS-T, 45 min at RT). After rinsing the membrane with TBS-T and TBS, chemiluminescence was detected with ECL reagent (Thermo-Fisher Scientific) or by fluorescence scanning on the Odyssey CLx.
RNA sequencing of iPSCs
RNA isolation
RNA was extracted from iPSCs of one 70% confluent well of a 6-well plate (approximately 1 × 106 cells), pr cell line. The cells were washed with PBS twice and collected in Trizol (Invitrogen, #15596-026). Total RNA was isolated using the PureLink RNA Mini Kit (ThermoScientific, #12183018A), according to manufacturer’s instructions. Library preparation and sequencing were completed at Biomics at the Erasmus MC.
RNA-seq library preparation
RNA-seq libraries were prepared according to the Illumina TruSeq stranded mRNA protocol (www.illumina.com). In brief, 200 ng of total RNA was purified using poly-T oligo-attached magnetic beads to end up with poly(A)-containing mRNA. The poly(A)-tailed mRNA was fragmented, and cDNA was synthesized using SuperScript II and random primers in the presence of Actinomycin D. cDNA fragments were end repaired, purified with AMPure XP beads, and A-tailed using Klenow exo-enzyme in the presence of dATP. Paired end adapters with dual index (Illumina) were ligated to the A-tailed cDNA fragments and purified using AMPure XP beads. The resulting adapter-modified cDNA fragments were enriched by PCR using Phusion polymerase as follow: 30 s at 98°C, followed by 15 cycles of 10 s at 98°C, 30 s at 60°C, 30 s at 72°C, followed by 5 min at 72°C. PCR products were purified using AMPure XP beads and eluted in 30 μL of resuspension buffer. One microliter was loaded on an Agilent Technologies 2100 Bioanalyzer using a DNA 1000 assay to determine the library concentration and for quality check.
RNA sequencing
Sequencing has been performed on Illumina HiSeq2500 sequencer, in Rapid run mode, for paired-end reads 50 bp in length, at least 40M clusters per sample.
For RNA-seq analysis, the raw sequencing data (.fastq) of a published HeLa dataset,16 HEK dataset,22 and THP-1 dataset,15 as well as the iPSCs dataset generated by the Biomics facility at the Erasmus MC and the dataset of fibroblasts were imported into the Galaxy platform.33 CutAdapt (Galaxy v.1.16.5) was used to trim reads of low sequencing quality (threshold of 20), filtering out reads with a read length <50 nucleotides. After read quality was ensured (FastQC; Galaxy v.0.72+galaxy1; http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), reads were mapped to the human reference genome GRCh38 utilizing default settings of the STAR algorithm (Galaxy v.2.7.2b).34 Transcript assembly was guided by the reference annotation file Gencode version V36.35 To assess counts per gene, we analyzed the mapped datasets with the feature Counts tool (Galaxy v.1.6.4+galaxy1).36 For the principal component analysis (PCA), normalized gene expression counts were analyzed with the DESeq2 tool (Galaxy v.2.11.40.6+galaxy1).37 Differentially expressed (DE) genes were defined by an adjusted p value < 0.05 and a fold change abs (log2 (FC)) >0.5. Ultimately, the alternative exons reported in the discovery dataset16,14 were confirmed in the HEK and THP-1 dataset and accessed in the iPSCs and Fibroblast dataset by manual calculation of the spanning reads from.bam files. Moreover, we performed an unbiased splicing analysis using the Modeling Alternative Junction Inclusion Quantification (MAJIQ) tool.38
Shared DE genes and AS were visualized in a Venn diagram. A pathway analysis was performed via QIAGEN IPA analysis.39 Functional annotation of AS events was performed with DAVID.40,41 Heatmaps were blotted with the heatmap2 tool (Galaxy v.3.1.3+galaxy0).
Statistical analysis
Statistical analyses are performed using GraphPad Prism following the statistical methods as mentioned in the results section and figure legends.
Results
Identification of 13 individuals with heterozygous variants in HNRNPC
Description of primary case subject
A 12-year-old boy (individual 1, Ind1) was referred to our outpatient clinic for genetic evaluation because of unexplained developmental delay and mild intellectual disability. He is the second child of healthy, non-consanguineous parents of European descent. Pregnancy and birth were uneventful. His developmental milestones were all slightly delayed. He walked his first steps at 18 months of age and spoke his first words at 2 years of age and his first sentences at 2.5 years of age. Fine motor skills were below average. He was clumsy from time to time but could participate in sports. He had articulation problems for which he received speech therapy. His total intelligence quotient (IQ) was determined at 54 (verbal IQ 65 and performance IQ 55). He is a friendly and sociable child, showing happy behavior. Concentration was poor for which methylphenidate was prescribed. Falling asleep was difficult and he often woke up very early. Physical examination demonstrated some subtle facial dysmorphisms: brachycephaly, high frontal hairline, slight hypotelorism, flaring of the eyebrows (lateral side), thin upper lip, and slightly smooth philtrum (Figure 1B). There was a remarkable stiffness of the joints, and a lordosis was observed. A skin examination demonstrated two café-au-lait spots, multiple small nevi, and cutis marmorata (chest). Growth parameters were all normal (Tables 1 and S4).
Figure 1.

Identified HNRNPC variants mapped to HNRNPC functional domains and dysmorphic facial features of individuals bearing HNRNPC variants
(A) Schematic representation of HNRNPC-iso1 and HNRNPC-iso2 and their functional domains: RRM (RNA-recognition motif), C2 (isoform C2-specific domain), bZLM (basic region zipper-like motif), CLZ (leucine-zipper like oligomerization domain), and CTD (C-terminal domain). Variant annotation is based on HNRNPC-iso1. The recurrent (red), frameshift (yellow), N-terminal deletion (blue), and missense (black) variants are indicated at the affected amino acid location. The nucleotide sequence of the recurrent variant (HNRNPCDEL) is indicated with the repeat sequence highlighted in red.
(B) Photos of seven individuals with HNRNPC variants, illustrating shared dysmorphic features including thin upper lip, smooth philtrum, and mildly deep-set eyes (in some). The facial appearance of Ind10 (132 kb deletion including three coding exons at the N terminus of HNRNPC) did not clearly overlap with the other individuals from this cohort.
Table 1.
Summary of clinical features of individuals with HNRNPC variants
| Recurrent-variant c.889-915del (n = 5) | Frameshift variants (n = 4) | N-terminal deletion (n = 1) | Missense variants (n = 3) | Total | |
|---|---|---|---|---|---|
| Development | |||||
| Intellectual disability | 2/2 (100%) | 3/3 (100%) | 1/1 (100%) | 3/3 (100%) | 9/9 (100%) |
| Speech delay/problems | 5/5 (100%) | 3/3 (100%) | 1/1 (100%) | 3/3 (100%) | 12/12 (100%) |
| Gross motor delay | 5/5 (100%) | 3/3 (100%) | 0/1 (0%) | 3/3 (100%) | 11/12 (92%) |
| Fine motor delay | 5/5 (100%) | 3/3 (100%) | 1/1 (100%) | 3/3 (100%) | 12/12 (100%) |
| Growth/feeding | |||||
| Short stature | 0/5 (0%) | 2/3 (67%) | 0/1 (0%) | 0/3 (0%) | 3/12 (25%) |
| Low weight | 0/5 (0%) | 1/3 (33%) | 0/1 (0%) | 0/3 (0%) | 1/12 (8%) |
| Microcephaly | 3/5 (60%) | 1/4 (25%) | 0/1 (0%) | 0/3 (0%) | 4/13 (31%) |
| Feeding/GI problems | 4/5 (80%) | 3/3 (100%) | 1/1 (100%) | 1/3 (33%) | 9/12 (75%) |
| Neurological | |||||
| Seizures | 0/5 (0%) | 2/4 (50%) | 0/1 (0%) | 1/3 (50%) | 3/13 (23%) |
| Hypotonia | 3/5 (60%) | 3/3 (100%) | 0/1 (0%) | 1/3 (50%) | 7/12 (58%) |
| Movement disorder | 2/5 (40%) | 1/4 (25%) | 0/1 (0%) | 0/2 (0%) | 3/12 (25%) |
| Brain abnormalities | 3/4 (75%) | 3/3 (100%) | 1/1 (100%) | 3/3 (100%) | 10/11 (91%) |
| Behavior | |||||
| Happy demeanor | 3/5 (60%) | 2/3 (67%) | 0/1 (0%) | 2/3 (67%) | 7/12 (58%) |
| Sleeping problems | 4/5 (80%) | 3/3 (100%) | 1/1 (100%) | 1/3 (33%) | 9/12 (75%) |
| Poor concentration | 4/5 (80%) | 1/3 (33%) | 1/1 (100%) | 1/3 (33%) | 7/12 (58%) |
| Dysmorphic features | |||||
| Deep-set eyes | 3/5 (60%) | 2/3 (67%) | 0/1 (0%) | 2/3 (100%) | 7/12 (58%) |
| Thin upper lip | 4/5 (80%) | 2/3 (67%) | 1/1 (100%) | 3/3 (100%) | 10/12 (83%) |
| Smooth philtrum | 3/5 (60%) | 2/3 (67%) | 0/1 (0%) | 1/3 (33%) | 6/12 (50%) |
| Other | |||||
| Recurrent ear infections | 1/5 (20%) | 1/3 (33%) | 0/1 (0%) | 1/3 (33%) | 3/12 (25%) |
Identification of a de novo heterozygous HNRNPC variant
Trio whole-exome sequencing (WES) identified a de novo heterozygous variant in HNRNPC (GenBank: NC_000014.9:g.21211238_21211264del). HNRNPC is located on chromosome 14 (14q11.2) and encodes two major isoforms: HNRNPC-iso2 (GenBank: NM_031314.3, 306 aa) and the smaller but more abundant HNRNPC-iso1 (GenBank: NM_004500.4, 293 aa), which lacks the C2 domain. The recurrent HNRNPC variant in HNRNPC-iso1 (GenBank: NM_004500.4:c.850_876del, GenBank: NP_004491.2:p.Arg284_Asp292del) and HNRNPC-iso2 (GenBank: NM_031314.3:c.889_915del, GenBank: NP_112604.2:p.Arg297_Asp305del) are further referred to as HNRNPCDEL.
The HNRNPCDEL variant has been reported in ClinVar (accession: RCV001249428.1). The individual reported on ClinVar was included in our cohort as Ind2. The same variant was also reported in the gnomAD population database.42 To the best of our knowledge, the individual in our cohort does not correspond to the individual in gnomAD. Possibly, the individual in gnomAD was mosaic or only mildly affected, resulting in incorrect assignment to the non-neuro classification in the database.
Majority of LoF variants reported on gnomAD do not affect canonical HNRNPC transcript
The pLI scores indicate the probability of a gene being intolerant to loss-of-function (LoF) variants, with scores of 0.9–1.0 indicating extreme intolerance toward LoF.42 The pLI score of 0.98 for HNRNPC thus indicated significant constraint against LoF for this gene.
In total, 18 HNRNPC variants were listed as potential loss of function in gnomAD (15 on gnomAD v.2.1.1 and 10 on gnomAD v.3.1.2, including 7 overlapping variants). However, as is detailed in Table S5, only a minority of these variants were predicted to result in LoF in the two isoforms that are primarily expressed in the human brain: HNRNPC-iso1 (ENST00000553300) and HNRNPC-iso2 (ENST00000554455). Specifically, three of them were confidently predicted to result in loss of function in the HNRNPC-iso2 isoform but not in the HNRNPC-iso1 isoform.
Other individuals bearing HNRNPC variants
We identified 10 other individuals with heterozygous variants in HNRNPC through Genematcher.43 Their variant was identified either via WES trio (Ind1–Ind5, Ind11, and Ind12), WES (Ind6, Ind7, Ind9, and Ind13), or WGS (Ind8, Ind10, and Ind13), as indicated in Table S4. Of note, two individuals (Ind9 and Ind13) were previously identified in the Deciphering Developmental Disorders cohort (n = 31,058 parent-offspring trios of individuals with developmental disorders)11 and described in recent literature1 (Table S4). All variants were annotated to the MANE transcript HNRNPC-iso1 (GenBank: NM_004500.4). In addition, genomic, protein, and HNRNPC-iso2 annotations are stated in Table 2.
Table 2.
Variant annotations for HNRNPC following HGVS standards
| Individual |
HNRNPC |
HNRNPC-iso1 |
HNRNPC-iso2 |
||
|---|---|---|---|---|---|
| NC_000014.9 | NM_004500.4 | NP_004491.2 | NM_031314.3 | NP_112604.2 | |
| C-term deletion | |||||
| 1–5 | g.21211238_21211264del | c.850_876del | p.Arg284_Asp292del | c.889_915del | p.Arg297_Asp305del |
| Frameshift | |||||
| 6 | g.21234140_21234141del | c.54_55del | p.Phe19Hisfs∗13 | c.54_55del | p.Phe19Hisfs∗13 |
| 7 | g.21211284dup | c.825dup | p.Glu276Argfs∗3 | c.864dup | p.Glu289Argfs∗3 |
| 8 | g.21211454del | c.754del | p.Asp252Thrfs∗18 | c.793del | p.Asp265Thrfs∗18 |
| 9 | g.21211475_21211481del | c.724_730del | p.Gly242Glnfs∗26 | c.763_769del | p.Gly255Glnfs∗26 |
| N-term deletion | |||||
| 10 | g.21220392_21352183del | c.−82945_366-7272del | – | c.-82945_405-7272del | – |
| Missense | |||||
| 11 | g.21231018C>T | c.296G>A | p.Arg99Gln | c.296G>A | p.Arg99Gln |
| 12 | g.21230992C>T | c.317+5G>A | – | c.322G>A | p.Val108Ile |
| 13 | g.21234004G>A | c.190C>T | p.Arg64Trp | c.190C>T | p.Arg64Trp |
Interestingly, four individuals (Ind2–Ind5) harbored the exact same de novo HNRNPC variant as Ind1 (GenBank: NM_004500.4:c.850_876del). The high frequency of this recurrent variant could indicate a dominant-acting effect on protein function or might be the result of a recurrent mutational event arising from a modest repeat sequence in the 3′ end of HNRPNC (Figure 1A).
Four individuals of our cohort had frameshift variants: Ind6 (GenBank: NM_004500.4:c.54_55del), Ind7 (GenBank: NM_004500.4:c.825dup), Ind8 (GenBank: NM_004500.4:c.754del), and Ind9 (GenBank: NM_004500.4:c.724_730del).27 These four frameshift variants were predicted to result in a premature termination codon resulting in a truncation of HNRNPC, as indicated in Table S4. Since these variants were in regions with limited susceptibility to nonsense-mediated decay (NMD) (within the 5′ most 150 nucleotides or within the last exon or the last 50 nucleotides of the penultimate exon), we predicted that none of these frameshift variants were affected by NMD.44 All variants were assessed according to their likelihood of pathogenicity using ACMG variant classification45,46 or MetaDome predictor.47 Where applicable, potential NMD escape was assessed via NMDEscPredictor (Table S4).48
One individual (Ind10) was diagnosed with a large (±132 kb) deletion (GenBank: NM_004500.4:c.−82945_366_7272del) spanning the first three coding exons of the HNRNPC as well as RPGR ineracting protein 1 (RPGRIP1 [MIM: 605446]) and the 3′ UTR of SPT16 homolog, facilitates chromatin remodeling subunit (SUPT16H [MIM: 605012]).
The remaining three individuals had heterozygous HNRNPC missense variants: Ind11 (GenBank: NM_004500.4:c.296G>A, GenBank: NP_004491.2:p.Arg99Gln), Ind12 (GenBank: NM_004500.4:c.317+5G>A), and Ind13 (GenBank: NM_004500.4:c.190C>T, GenBank: NP_004491.2:p.Arg64Trp). Notably, the HNRNPC variant of Ind12 was predicted to abolish a splice donor site in HNRNPC-iso1, but encodes a missense variant in the less abundant HNRNPC-iso2 isoform (GenBank: NM_031314.3:c.322G>A, GenBank: NP_112604.2:p.Val108Ile).49,50
Phenotypic features of individuals with HNRNPC variants
All 13 individuals in our HNRNPC cohort were assessed by their local clinical geneticists and presented with overlapping clinical phenotypes (Tables 1 and S4), including a global developmental delay in all individuals, as well as a mild to severe intellectual disability (observed in those old enough to be assessed). Of note, limited clinical data were available for Ind9, which might lead to an underrepresentation in all clinical aspects. This individual was previously reported in a large developmental disorder cohort.11
Delays in fine and gross motor skills were observed in most individuals of our cohort (12/12 and 11/12, respectively). Speech and language development were delayed in all individuals. Most of them were able to speak and communicate, but some spoke very few words or were non-verbal.
Articulation problems and dysarthria were described as well. Hypotonia was reported in 7/12 individuals with HNRNPC variants, epilepsy was present in 3/13 individuals, and movement disorders (gait ataxia, tremors, tics) were reported in 3/12 individuals. Microcephaly was observed in 4/12 individuals, with 3 of them bearing the HNRNPC recurrent variant (Tables 1 and S4).
Several individuals displayed subtle overlapping dysmorphic features, including deep-set eyes (7/12), thin upper lip (10/12), and a smooth philtrum (6/12), but a clearly recognizable facial gestalt was not apparent in this small cohort (Figure 1B). A few individuals in this cohort had more significant facial dysmorphisms and congenital malformations (Tables 1 and S4). Notably, individual 8 had a more dysmorphic phenotype than the other individuals (no photo available).
Behavioral abnormalities were reported for most individuals and included poor concentration/ADHD (7/12) and anxiety problems in one individual (Ind5). Notably, 7/12 individuals were reported to have a (very) happy demeanor. Sleeping problems, including sleep apnea, were observed in most individuals (9/12), as were feeding problems (9/12). Growth problems did not seem to be a core component of the phenotype of this HNRNPC cohort. Growth parameters were calculated as standard deviation (SD) according to each clinician’s growth reference charts (for Ind8 the US reference was used51). However, two out of three individuals in the frameshift variant subgroup had a short stature. Two individuals had exotropia/esotropia (Ind7, Ind8) and another individual had bilateral colobomatous microphthalmia (Ind11), but most affected individuals did not exhibit ophthalmologic concerns. Hearing loss was reported in two individuals (Ind8, Ind11).
Functional characterization of iPSCs and fibroblasts of affected individuals
HNRNPCDEL abundance is reduced in HNRNPCDEL iPSCs with retained isomerization capacity
We set out to analyze the molecular consequences of the recurrent HNRNPCDEL variant by assessing the effect of this variant on previously described functions of HNRNPC.13,14,16,33 To that end, we generated iPSCs from peripheral blood mononuclear cells (PBMCs) obtained from individual 1 (HNRNPCDEL iPSC) (EMCi225: hpscreg.eu/cell-line/EMCi225-A, hpscreg.eu/cell-line/EMCi225-B, hpscreg.eu/cell-line/EMCi225-C, hpscreg.eu/cell-line/EMCi225-D) and compared those to a sex- and age-matched control iPSC line (EMCi169: hpscreg.eu/cell-line/EMCi169-A, hpscreg.eu/cell-line/EMCi169-B, hpscreg.eu/cell-line/EMCi169-C). As indicated on the registry, all iPSC lines were characterized according to a state-of-the-art protocol and expressed common pluripotency markers. No apparent growth or morphological differences were observed between HNRNPCDEL and control iPSCs.
Western blot analysis of these iPSCs indicated the abundance of a faster migrating HNRNPC band in HNRNPCDEL cells (Figure 2A), likely representing the truncated HNRNPCDEL. This was confirmed by the absence of the faster migrating band in control samples and its correspondence in size with recombinantly expressed HNRNPC-iso1DEL in HEK293T cells (Figure 2A). Notably, the truncated protein (HNRNPC-iso1DEL) was less abundant than the full-length HNRNPC isoform, resulting in significantly reduced levels of total HNRNPC to 45.15% (Figure 2B; t(9) = 3.571, p = 0.006). Reduced abundance (16% reduction) was observed on normalized counts of total HNRNPC mRNA level via RNA sequencing (RNA-seq), but this difference was not statistically significant (Table S6, DESeq2 analysis, log 2(FC) = −0.23, adjusted p value = 0.81).
Figure 2.

The HNRNPCDEL variant results in reduced levels of HNRNPC but does not affect HNRNPC or mRNA localization
(A) Western blot of recombinant HNRNPC-iso1 and the recurrent HNRNPC-iso1DEL variant in HEK293-T cells as well as endogenous HNRNPC levels in iPSCs from Ind1 (HNRNPCDEL) or a control subject. ACTIN served as a housekeeping protein for normalization of the HNRNPC levels.
(B) Quantification of total HNRNPC levels as determined by Western blot, normalized to ACTIN for protein loading. Data are calculated relative to control iPSCs levels mean ± SEM, t test: ∗∗∗p < 0.001.
(C) Representative z stack maximum projections of HNRNPCDEL and control iPSCs and control iPSCs transduced with HNRNPC-targeting shRNAs. Cells were stained for endogenous HNRNPC (red), mRNA (oligoDT-Cy3, grayscale, false-colored), and DNA (DAPI, blue). Arrowheads indicate cells with HNRNPC knockdown, based on HNRNPC staining. Scale bars represent 50 μm.
(D and E) Quantification of HNRNPC knockdown in control iPSCs from maximum projections of Z-stacks in (C) show a significant HNRNPC knockdown by HNRNPC-targeting shRNAs (D) and slightly reduced oligoDT signal in HNRNPC knockdown cells (E) (mean ± SEM, one-way ANOVA: ns, not significant, ∗p < 0.05, ∗∗∗∗p < 0.0001).
(F) Recombinant over-expression of HNRNPC-iso1, HNRNPC-iso2, HNRNPC-iso1DEL, or HNRNPC-iso2DEL in control iPSCs, stained for HNRNPC (red, false colored from Alexa 647 signal) and mRNA (OligoDT-Cy3, grayscale, false colored) reveals altered mRNA localization upon elevated HNRNPC levels. Scale bars represent 50 μm.
(G) Quantification of oligoDT signal in iPSCs overexpressing HNRNPC (from F) shows significantly increased oligoDT signal in targeted cells (mean ± SEM, t test, ∗∗∗∗p < 0.0001). All experiments were performed on at least 2 independent cell lines (N = 2) and at least 2 independent transfections or transductions (n = 2).
We next investigated whether the 9 amino acid deletion affected the nuclear targeting of HNRNPC. Identical localization of HNRNPC was observed in HNRNPCDEL and control iPSCs (Figures S1A and S1B), showing distinct nuclear localization with exclusion from the nucleoli as shown by KI-67 counterstaining52 during interphase. During mitosis, HNRNPC localizes in the cytoplasm with exclusion from the chromatids, as shown by DAPI chromatid staining. A similar HNRNPC localization pattern was observed for recombinant GFP-tagged HNRNPC-iso1 and HNRNPC-iso2 and the HNRNPC-iso1DEL and HNRNPC-iso2DEL variants in U-2 OS and HEK293T cells (Figures S2A and S2B).
HNRNPC has been shown to function as a heterotetramer with a (C1)3(C2)1 stoichiometry.53 Its C-terminal domain (CTD), located within the in frame-deletion of amino acids 284–292 and 297–305 for HRNRNPC-iso1 and HNRNPC-iso2, respectively (Figure 1A), is thought to affect tetramer stability, as HNRNPC variants lacking parts of the CTD region (amino acid 241–290 of HNRNPC-iso1) show impaired tetramerization.12,13 We therefore assessed whether the association of the HNRNPCDEL variant was affected using a co-immunoprecipitation approach on recombinant tagged HNRNPC and HNRNPCDEL in U-2 OS cells. Interaction of either HNRNPC isoform was maintained for both HNRNPCDEL isoforms (Figure S3), indicating that HNRNPCDEL has not lost its ability to form HNRNPC oligomers.
Excessive HNRNPC abundance traps poly(A)-RNA in the nucleus
Previously, loss of functional HNRNPC was associated with mRNA accumulation in the nucleus.17 We therefore compared mRNA localization between control and HNRNPCDEL iPSCs by fluorescence in situ hybridization (FISH) with an oligoDT-Cy3 probe. Surprisingly, mRNA localization was not altered in the HNRNPCDEL iPSCs (Figure 2C). Moreover, in contrast to literature, shRNA-mediated knockdown of HNRNPC (Table S1) did not alter mRNA localization in iPSCs (Figure 2C, arrowheads), despite a significant reduction in HNRNPC levels in iPSCs by lentiviral transduction with shRNA (73.33%, F(3,58) = 138.8; p < 0.0001) (Figure 2D). Quantification of the oligoDT signal as z-scores per analyzed image did not reveal differences between shRNA-targeted (t(28) = 0.2928, p = 0.7718) or non-targeted cells (t(28) = 0.2317, p = 0.8184) in iPSCs derived from affected individuals or control subjects (Figure 2E). Similarly, mRNA localization was not affected by shRNA-mediated knockdown of HNRNPC in U-2 OS cells (Figure S4), based on oligoDT staining. This lack of mRNA accumulation observed in iPSCs derived from affected individuals strongly suggests that the HNRNPCDEL variant does not encode a hyperactive gain-of-function protein.
In contrast, recombinant expression of either of the two HNRNPC isoforms or the recurrent HNRNPCDEL variants induced a significant mRNA accumulation in the nucleus in iPSCs (t(98) = 18.51, p < 0.0001) (Figures 2F and 2G). Of note, the effect of HNRNPC-iso1, HNRNPC-iso2, HNRNPC-iso1DEL, or HNRNPC-iso2DEL was not significantly different between the overexpressed isoforms or variants (F(3,46) = 0.1147; p = 0.9511). To exclude bleed-through effects of nuclear HNRNPC signal to the Cy3 channel (OligoDT), we confirmed our findings with Alexa 647 and Alexa 488 secondary antibodies against the primary anti-HNRNPC, for which similar effects were observed (Figure S1C). Since overexpression of the HNRNPCDEL variant was indistinguishable from overexpression of wild-type HNRNPC, these results indicate that the HNRNPCDEL variant is unlikely to result in a loss-of-function protein for this biological process.
Taken together, these iPSC-based experiments did not reveal a specific effect of the recurrent HNRNPCDEL variant on localization or oligomerization. Furthermore, we demonstrated that mRNA localization was highly sensitive to increased levels of HNRNPC, independently of the isoform or variant. These data thus suggest that the HNRNPCDEL variant does not act as a dominant-negative or gain-of-function protein, but rather that the HNRNPCDEL-associated pathogenicity results from reduced HNRNPC levels.
Meta-analysis of HNRNPC knockdown RNA-seq datasets reveals an HNRNPC-dependent signature of alternative exon and ALU inclusion and exclusion
To investigate whether HNRNPC haploinsufficiency could be responsible for HNRNPC pathogenicity, we investigated the HNRNPC function as a regulator of alternative exon and ALU inclusion or exclusion.14,15,16 To this end, we compared three published datasets of knockdown of HNRNPC in HeLa cells,14 HEK cells,22 and THP-1 cells15 (Figure S5A). All three published datasets show substantial knockdown of HNRNPC: 96% in HeLa (DESeq2 analysis, log 2(FC) = −4.62, adjusted p value = 9,32E−58), 98% in HEK (DESeq2 analysis, log 2(FC) = −5.56, adjusted p value = 0), and 53% in THP-1 (DESeq2 analysis, log 2(FC) = −1.08, adjusted p value = 1.28E−65) (Figure 3A). A principal component (PCA) analysis of these datasets shows that control samples cluster distinctly from HNRNPC knockdown, but in some cases with large variance between samples. This suggests that the changes in differential gene expression are in fact small (Figure S5B). Differential gene expression (DESeq2, adjusted p value < 0.05, Table S6) revealed an overlap of 682 differentially expressed genes between the three knockdown datasets (Figure S5C). Although an ingenuity pathway analysis (IPA) revealed an overlap in a number of canonical pathways for the HEK and HeLa dataset, only a very limited overlap was identified among all three knockdown datasets (Figure S5D). Thus, despite a distinct clustering in the PCA blot of control cells and cells with reduced levels of HNRNPC, no evident HNRNPC-dependent signature of differentially expressed genes could be identified.
Figure 3.

Meta-analysis of RNA-seq data examining the effect of HNRNPC loss on alternative exon usage or ALU splicing
(A) HNRNPC RNA expression in investigated datasets show significantly reduced levels in HEK,22 HeLa,14 and THP-115 cells upon knockdown. Normalized to control (%), mean ± SEM.
(B) Visual assessment of abundance of alternative exons or ALU sequences identified by Zarnack et al.14 revealed an overlap of 62 target exons/ALUs between the three cancer datasets. Unbiased alternative splicing analysis via MAJIQ of AS targets with a probability of change ≥0.95 in at least two datasets was analyzed with MAJIQ (C–E, G–H).
(C) AS targets with a probability of change ≥0.95 in all three datasets shows an overlap of 555 targets.
(D) These targets consist of cassette exons (42.16%), multi exon spanning (28.83%), alternative introns (8.83%), alternative first (4.86%)/last (4.50%) exons, tandem cassettes (3.96%), and others (6.85%).
(E) Independent clustering on PSI score shows separation of control and HNRNPC-KD samples independent of cell type.
(F) Visual assessment of abundance of alternative exons or ALU sequences identified by Zarnack et al.14 revealed an overlap of 28 target exons/ALUs between the three cancer datasets and fibroblasts.
(G) Probability of change of AS targets plotted for all 5 datasets on 2,070 targets identified in (D, targets overlapping in at least 2 datasets).
(H) PSI scores of AS targets with a probability of change ≥0.95 in at least two cancer datasets and >0.5 in fibroblasts. Fibroblasts of Ind8 cluster with HNRNPC-KD samples.
Using an elegant individual-nucleotide resolution UV cross-linking and immunoprecipitation (iCLIP) method, König and colleagues16 identified multiple alternative exons or ALU sequences as HNRNPC splicing targets, including targets in CD55 molecule (Cromer blood group) (CD55 [MIM: 25240]), helicase lymphoid species (HELLS [MIM: 603946]), RIC1 homolog, RAB6A GEF complex partner 1 (RIC1 [MIM: 610354]), methyl CpG-binding domain protein 3 (MBD3 [MIM: 602573]), mitochondrial tRNA translation optimization 1 (MTO1 [MIM: 614667]), peroxisome biogenesis factor 14 (PEX14 [MIM: 601792]), WRN RecQ like helicase (WRN [MIM: 604611]), and zinc-finger protein X-linked (ZFX [MIM: 314980]). These targets were experimentally confirmed by RNA-seq and RT-PCR in a follow-up study using HeLa cells with a HNRNPC knockdown.14 Using their published RNA-seq dataset as well as the two aforementioned published RNA-seq datasets of HNRNPC knockdown in HEK cells22 and THP-115 cells, we performed a meta-analysis to investigate whether there was a shared signature of alternative splicing across multiple studies and cell lines. To that end, we calculated the AS target abundance (Fisher’s exact test, Table S7) for the previously reported list of 63 alternative exon or ALU sequences (Supplemental Table S2 in Zarnack et al.14 which we manually curated to 73 targets, as per visual inspection of the mapped reads where multiple alternative exons were detected, Table S7) and found a significant overlap of 85% (62 of 73 targets) between the published datasets (Figure 3B).
In addition, we performed an unbiased approach to investigate alternative splicing in the aforementioned datasets with the MAJIQ tool.38 The results of the analysis were collected in a junction file (Table S8) which lists the probability of change and the percent selected index, Ψ∈[0,1] (PSI) score per AS target and dataset. This PSI score captures the marginal fraction of isoforms that utilize the investigated splicing junction. In addition, all AS targets were summarized in Table S9, generated by MAJIQ.
Even though Zarnack and colleagues mentioned approximately 1,141 deregulated AS targets in their DEXSeq analysis, they provide details on only 63 targets,14 which we investigated in detail here. The overlapping genes between this dataset of 63 targets and our MAJIQ analysis (threshold: probability of change ≥ 0.95) of the HeLa dataset were 33% (21 out of 63), while the overlap with the AS target genes described in the summary file was 57% (36 out of 63) (Figure S6A). This comparison indicates that the MAJIQ tool reproduces a substantial number of AS targets reported previously. The extent of overlap between the identified targets for AS between the two methods depends on the strictness of filters. For the subsequent analysis, we continued using the MAJIQ, since it identifies the AS type and can include multiple datasets.
Therefore, we next examined the common splicing targets between all published datasets (probability of change > 0.95 in at least two out of three datasets, HEK,22 HeLA,14 and THP-115) and found 555 AS targets overlapping in all three datasets and 2,070 AS targets deregulated in at least two datasets (Figure 3C). Of these 555 AS targets, the majority (71%) represents either Cassette exons (42%) or multi exon spanning (29%) splicing events (Figure 3D). The PSI score per AS event of the 2,070 targets significantly (>0.95) affected in at least two datasets were plotted in a heatmap (Figure 3E). An automatically generated summary file of the MAJIQ analysis reported 1,189 modules (1,106 genes) affected by AS as a result of loss of HRNNPC.
Of note, despite using non-neuronal cells, an analysis using the DAVID tool40,41 on the set of 1,106 genes (out of which 1,059 were recognized by DAVID) revealed a significant association (p = 0.036) of intellectual disability within the list of AS-targeted genes, primarily attributed to the loss of HNRNPC. The 60 AS-targeted genes associated with intellectual disability were reported in Table S7.
Fibroblasts derived from affected individuals show the presence of the HNRNPC-dependent fingerprint of alternative spliced exons
Having identified a signature of alternative splice events as a functional readout of HNRNPC activity, we investigated whether this signature could also be observed in the HNRNPCDEL iPSC line derived from Ind1. Unfortunately, in iPSCs most of the identified alternative exons or ALU sequences were either not expressed or unchanged (Figures 3F and 3G, Table S8), indicating that iPSCs likely have a very different exon or ALU sequence abundance profile.
Since alternative exon inclusion and exclusion analysis relies on relative values of exon abundance compared to other exons within a given transcript rather than absolute expression of the transcript itself, it can be used as a functional readout of HNRNPC function in just a single RNA-seq sample. We therefore included RNA-seq data from fibroblasts from Ind8 (GenBank: NM_004500.4:c.754del, GenBank: NP_04491.2; p.Asp252Thrfs∗18) and a sex-matched control subject.
Based on our manual analysis of the targets reported by Zarnack et al.,14 we calculated (Fisher’s exact test) the relative abundance of alternative exons or ALU sequences for the 73 aforementioned selected targets in the fibroblast dataset (Figure 3F, Table S7). Of these, 12 targets could not be assessed in the fibroblasts since the gene, exon, or ALU was not expressed, hence reducing the total to 61 targets. Of all identified alternative exons or ALU sequences, 28 out of 61 (46%) showed the same alternative splicing pattern as the HNRNPC signature identified in the meta-analysis (Figure 3F). For the remaining alternative exons or ALU sequences, 29 (48%) showed no change and only 3 (5%) showed an opposite effect (Figure 3F, Table S7).
The relative abundance of alternative exons or ALU sequences was visualized for the previously reported most extensively characterized differentially spliced exons in CD55, HELLS, RIC1, MBD3, MTO1, PEX14, WRN, and ZFX;16,14 Figure S6B). Subsequently, we used the 2,070 identified AS targets in the unbiased MAJIQ meta-analysis (targets in at least two datasets) and plotted their probability of AS scores in the fibroblast and iPSC datasets via unsupervised clustering (Figure 3G). Interestingly, we could confirm that most targets (2,038; 99%) were not changed in the iPSCs dataset while 331 (16%) targets showed a probability of change of 0.5 or higher in fibroblasts. Of note, we set the threshold for probability of change at 0.5, since the expected protein abundance in these cells is above 50% (due to NMD escape). Hence, to detect a change in AS we lowered this threshold. Lastly, with unsupervised clustering, we plotted the PSI scores of these 331 targets in all but the iPSC datasets and show that fibroblasts from the affected individual (Ind8) cluster together with the KD samples while all control samples cluster distinctly (Figure 3H).
Some examples of alternatively spliced genes that show a strong AS signal in fibroblasts—CD55, GINS Complex Subunit 3 (GINS3 [MIM: 610610]), DNA polymerase delta 3, accessory subunit (POLD3 [MIM: 611415]), RNA Binding Motif 26 (RBM26 [MIM: 620081]), SGT1 homolog, MIS12 kinetochore complex assembly cochaperone (SUGT1 [MIM: 604098])—were visualized with voila38 in Figure S6C.
Taken together, this unbiased approach confirms the overlap of alternative splicing in fibroblasts from Ind8 and the identified HNRNPC-dependent signature of alternatively spliced exons in our meta-analysis of HNNRPC-knockdown cell lines and provides further evidence that HNRNPC haploinsufficiency leads to aberrant splicing, in particular affecting genes involved in intellectual disability.
Aberrant HNRNPC levels affect neuronal morphology, migration, and cell survival
HNRNPC is crucial for neuronal morphology and cell survival
Considering that the recurrent HNRNPCDEL variant leads to reduced protein levels, along with our observation of HNRNPC-dependent change of exon inclusion in the fibroblasts from Ind8, and the fact that 50% of these affected genes are expressed in the brain, it is plausible that HNRNPC haploinsufficiency is underlying the neurodevelopmental phenotype in these individuals. In line with this hypothesis, the pLI score of 0.9842 for HNRNPC indicates low tolerance for loss-of function mutations. The pLI score is a measure of tolerance for loss of function calculated based on the number of protein-truncating variants in a database of 141,456 individuals of diverse ancestries42 and provides an important indicator of potential pathogenicity. Notably, homozygous loss of HNRNPC is non-viable in mice.54 We therefore sought to assess the effects of reduced HNRNPC expression and recombinant expression of the HNRNPCDEL variant on neuronal function.
To this end, we selected two mouse-specific shRNAs that specifically target HNRNPC for degradation. The ability of the selected shRNAs to reduce murine HNRNPC levels was tested in primary neuronal cultures, derived from the embryonic (E16.5) mouse brains. HNRNPC levels were quantified by immunocytochemistry analysis of targeted (tdTomato+) cells (maximum projections of z stack images). Cells were transfected with the shRNA constructs 20 h after setup (days in vitro 1 [DIV1]) and assessed for HNRNPC levels in targeted cells 144 h after knockdown (DIV7), as the reported half-life of HNRNPC is 47 h in cultured primary neurons55 (Figures 4A and S7A). HNRNPC levels were significantly reduced by shRNA_1 (to 55%) and shRNA_2 (to 67%) and by using a combination of both shRNAs (to 52%) as compared to a scrambled control shRNA (F(4,168) = 70.68, p < 0.0001) (Figure 4C).
Figure 4.

Changes in HRNPC level affect neuronal morphology
(A) Representative maximum projections of z stack confocal images of murine neurons targeted with shRNA constructs (tdTomato+, red), stained for HNRNPC (green, white arrows indicate targeted neurons). Scale bars: 50 μm.
(B) Representative maximum projections of z stack confocal images of murine neurons targeted with HNRNPC-iso1, HNRNPC-iso1DEL (tdTomato+), stained for HNRNPC (green) and MAP2 (gray). Scale bars represents 50 μm.
(C) Quantification of HNRNPC knockdown efficiency (% of CTRL shRNA) in primary murine neurons targeted with HNRNPC shRNA or a scramble control (CTRL) shRNA 7 days after transfection (one-way ANOVA).
(D and E) Total neurite length (μm) (D) and neurite arborization measured by Sholl analysis (E) of primary murine neurons targeted with shRNAs (1, 2, 1 + 2) for HNRNPC knockdown and scramble control (CTRL).
(F–H) Soma size (μm) (F), total neurite length (μm) (G), and neurite arborization measured by Sholl analysis (H) were significantly reduced in HNRNPC-overexpressing neurons (one-way ANOVA, mixed-effects analysis).
(C–H) All measurements were performed on at least 2 individual plugs (n = 2), 2 individual transfections per construct (n = 2), and 10 images per condition (n = 10). Error bars indicate mean ± SEM. ns, not significant, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
The soma size of the neurons was unaffected by the knockdown of HNRNPC (CTRL shRNA_1 [F(3,116) = 2; p = 0.3837], shRNA_2 [F(3,116) = 2; p = 0.4124], and the combination 1 + 2 [F(3,116) = 2; p = 0.7681]) (Figure S7B). However, total neurite length was substantially reduced in cells with HNRNPC knockdown via shRNA_2 (F(3,116) = 3.88; p = 0.0062) and a combination of 1 + 2 (F(3,116) = 3.88; p = 0.0223) (Figure 4D). For shRNA_1 the total neurite length shows a strong trend toward reduction (F(3,116) = 3.88; p = 0.0719). Moreover, neuronal arborization was significantly altered when HNRNPC levels were decreased by shRNA_1 (F(1,58) = 5.371; p = 0.024), shRNA_2 (F(1,58) = 12.85; p = 0.0007), and a combination of both (F(1,58) = 8.846; p = 0.0043) (Figure 4E).
Since we observed a strong effect of HNRNPC and HNRNPCDEL overexpression on nuclear mRNA accumulation in our cellular studies (Figures 2F and S2C), we set out to further investigate the functional impact of elevated HNRNPC-iso1 and HNRNPC-iso1DEL levels in mice by in vitro neuronal morphological assessment. Although the recombinant expression results in non-physiological levels of HNRNPC, the assay enables assessment of potential gain of function for HNRNPC-iso1DEL in developing neurons. Similar to our observations in iPSCs, recombinant HNRNPC-iso1 and HNRNPC-iso1DEL in primary murine neurons showed a predominantly nuclear localization (Figure 4B). Morphological characterization of neurons expressing either HNRNPC-iso1 or HNRNPC-iso1DEL (DIV10 at 72 h post-transfection) revealed substantially reduced soma area (F(2,57) = 18.81; p < 0.0001) (Figure 4F), total neurite length (F(2,57) = 13.94; p (HNRNPC) < 0.0001 and p (HNRNPCDEL) = 0.001) (Figure 4G), and dendrite arborization (F(1,38) = 59.21; p < 0.0001 for HNRNPC-iso1 and F(1,38) = 47.16; p < 0.0001 for HNRNPC-iso1DEL) (Figure 4H). Importantly, no differences were observed between overexpression of HNRNPC-iso1 and HNRNPC-iso1DEL in the morphological characterization. Strikingly, 5 days post-transfection (DIV7), the dendrites of cells overexpressing HNRNPC-iso1 or HNRNPC-iso1DEL deteriorated dramatically and could no longer be detected, while cells transfected with the control construct (empty vector, tdTomato) developed normally (Figure S7D).
Taken together, these results strongly support the notion that HNRNPC levels need to be tightly controlled for normal neuronal function.
Altered levels of HNRNPC in vivo affect neuronal migration in the IUE assay
Given that neuronal migration of the developing cortex is very sensitive to improper neuronal functioning, we made use of in utero electroporation of E14.5 mouse embryos to express HNRNPC shRNAs as well as HNRNPC-iso1 and HNRNPC-iso1DEL in immature neurons of the subventricular zone. These cells migrate within the cortical plate and are destined to ultimately form layer 2/3 of the cortex. Neuronal migration of targeted cells in the somatosensory cortex (SScx) (identified by tdTomato abundance) was assessed at postnatal day 1 (P1), a timepoint at which almost all targeted cells should have migrated out toward the cortical plate56,57 (Figure 5A). The migration pattern was analyzed as the cumulative distribution of targeted cells over the entire cortex, which is spatially divided into 10 bins of equal size between the pia (bin 1) and the ventricle (bin 10). The cumulative distribution patterns of cells targeted with HNRNPC-degrading shRNA were significantly different compared to those targeted with a scramble control shRNA (D(2) = 0.3349, p < 0.0001) (Figures 5B and 5C). Reduced HNRNPC levels thus affect the ability of targeted neurons to compete with non-targeted neurons to migrate to the outer cortical layers (layer 2/3).
Figure 5.

Altered HNRNPC expression delays neuronal migration of targeted cells in the IUE assay
(A) Schematic representation of the in utero electroporation (IUE) procedure. Cells that will form the somatosensory cortex (SScx) are targeted with the expression constructs at embryonic day 14.5 (E14.5) via in utero electroporation (IUE). These cells migrate from the intermediate zone (IZ) toward more superficial layers of the cortex such as the cortical plate (CP) and marginal zone (MZ).
(B and D) Representative confocal images of the somatosensory cortex (SSCx) of histological slices at P1 of cells targeted with CTRL or HNRNPC targeting shRNAs (B) or with HNRNPC-iso1, HNRNPC-iso1DEL, or tdTomato (D). Targeted cells (tdTomato+) are shown in red, cortical layers are indicated with dotted lines based on nuclear staining (DAPI, blue). IZ, intermediate zone; scale bars: 150 μm.
(C and E) Localization of tdTomato+ cells across the SSCx layers calculated as the percentage of cells per bin and displayed in cumulative neuronal migrations blots. Two-sample Komolov-Smirnov p values are indicated.
(B–E) IUE: All measurements were performed on at least 3 individual animals (n = 3) and 3 images per animal (n = 3). Error bars indicate mean ± SEM. ∗∗∗∗p < 0.0001.
At postnatal day 1 (P1), neuronal migration in the developing somatosensory cortex (SSCx) was significantly reduced (D(2) = 0.5701, p < 0.0001) in cells targeted for overexpression of HNRNPC-iso1 (Figures 5D and 5E). In line with our finding that the HNRNPC-iso1DEL variant does not appear to affect HNRNPC function, we found the same phenotype upon overexpression of the HNRNPC-iso1DEL variant (D(2) = 0.5611, p < 0.0001) (Figures 5D and 5E). In contrast, most cells targeted with the control protein (tdTomato+) migrated out to the cortical layers 2/3 (L2/3) (Figure 5D). Interestingly, this delay is not detectable at postnatal day 7 (P7) (Figure S7C).
Combined, these data illustrate that both enhanced and decreased levels of HNRNPC affect neuronal function, resulting in delayed neuronal migration of targeted cells during early murine cortical development. Moreover, these experiments provide further support that the HNRNPCDEL variant behaves like the wild-type protein and suggest that the pathogenicity of this variant arises from decreased abundance.
Discussion
A wide variety of heterozygous variants in HNRNPC result in a NDD phenotype
Here, we present a cohort of 13 individuals with heterozygous HNRNPC variants with shared features of neurodevelopmental delay and minor facial dysmorphic features, partially overlapping with those of other previously published HNRNP syndromes.1 Behavioral problems including poor concentration and attention span were also noted. Interestingly, 54% of our cohort had a very happy demeanor. Sleeping problems were reported in most of individuals (83%). We divided the cohort into four separate groups: (1) recurrent variant c.889_915del; (2) frameshift variants predicted to escape NMD; (3) large N-terminal deletion variant; and (4) missense variants. No clear genotype-phenotype correlations were observed between these sub-cohorts (Tables 1 and S4). However, seizures occurred more often in the group with frameshift and missense variants, and microcephaly was present in the group with the HNRNPC-recurrent variant (3/5) and for one of the frameshift variants.
Nine different HNRNPC variants affecting various HNRNPC domains were identified, including frameshift (c.54_55delAT, c.793delG, c.864dupA, c.763_769del), missense (c.190C>T, c.296G>A, c.322G>A), and in-frame deletion (c.889_915del) variants and a large deletion affecting HNRNPC as well as two other genes (GenBank: NC_000014.9:21220392_21352183del; SUPT16H, RPGRIP1). Generally, frameshift variants cause amino acid changes followed by a premature termination codon (PTC), which typically results in NMD of the RNA.58 However, NMD efficiency is decreased in the 5′-most 150 nucleotides of the coding region as well as in the last exon.44 This escape from NMD is supported by RNA-sequencing data of Ind8 (Figure S8), but is not experimentally assessed for Ind6, Ind7, and Ind9.
We expect the large deletion (Ind10) to result in haploinsufficiency for HNRNPC. The identified missense variants may affect HNRNPC folding and/or function due to their location in or proximity to the RNA recognition motif (RRM) and the C2 domain (Ind12 and Ind13, p.Val108Ile and p.Arg64Trp, respectively). Mutations that affect the RRM and C2 domains have been shown to affect RNA-binding specificity.59 We anticipate that the Ind13 variant p.Arg64Trp is likely to affect HNRNPC folding (Figures 6A and 6B) as the positively charged arginine is exchanged for a bulky, hydrophobic tryptophan, thus likely disrupting the predicted alpha helix in this highly structured RRM region (entry: O77768; alphafold.ebi.ac.uk). No accurate structure predictions were available for the Ind11 and Ind12 variants (p.Arg99Gln and p.Val108Ile, respectively) (Figures 6A, 6C, and 6D). For the latter, we expect limited disruption of the protein structure, due to high molecular similarity between the valine and isoleucine residues. Of note, the Ind12 variant is predicted to weaken the splice donor site of HNRNPC-iso1, which suggests that the expression of HNRNPC-iso2 is relatively enhanced, thus disrupting the (C1)3(C2)1 stoichiometry.
Figure 6.

HNRNPC-iso2 3D structure prediction in AlphaFold
(A) HNRNPC-iso2 3D structure predictions were obtained from alphafold.ebi.ac.uk(entry: O77768); reported HNRNPC variants are indicated with gray arrows.
(B–D) Detailed illustrations of the missense variants: c.190C>T (p.Arg64Trp) in the RRM (B), c.296G>A (p.Arg99Gln) near the RRM and C2 domain (C), and c.332G>A (p.Val108Ile) in the C2 domain (D).
HNRNPCDEL iPSCs did not reveal functional effects of the recurrent HNRNPCDEL variant
The recurrent HNRNPCDEL variant, as seen in Ind1, was detected in five unrelated individuals. Hence, we initially favored a model in which the deletion of the C-terminal nine amino acids affected the activity of HNRNPC. The deletion of these nine amino acids is the result of an in-frame microdeletion, c.889_915del (based on the longer C2 isoform), near the C-terminal domain (CTD; aa 240–290). The CTD is thought to affect HNRNPC tetramer stability, and HNRNPC variants lacking parts of the CTD region have been shown deficient in tetramer formation.12,13 However, we did not observe a change in association of HNRNPC isoforms with the deletion variant in a co-immunoprecipitation setup.
iPSCs derived from Ind1 as well as control subjects enabled us to further study the effect of HNRNPCDEL in an endogenous setting. Although we were able to verify the abundance of the HNRNPCDEL variant in cells from an affected individual, we did not find alterations in subcellular localization or other reported cellular functions of HNRNPC, including mRNA transport and alternative splicing.
Downregulation of HNRNPC has been shown to cause nuclear accumulation of mRNA in HeLa cells via impairment of an HNRNPC-dependent nuclear mRNA export mechanism.17 Although we observed a significant effect on nuclear accumulation of mRNA upon HNRNPC overexpression, efficient HNRNPC knockdown (approximately 70% in iPSCs) did not affect mRNA localization. This finding indicates that these cells were not suitable for utilizing mRNA localization as a readout to detect impaired HNRNPC function. HeLa cells may potentially demonstrate increased levels of HNRNPC, as has been noted in several cancer types,15,60,61 and any functional role described might be cell type dependent or cancer specific.62 However, we were not able to replicate the reported change in mRNA localization upon HNRNPC knockdown in U-2 OS (osteosarcoma) cells, either. One could speculate that excess levels of HNRNPC may retain an extensive amount of RNA by binding and stabilizing it in the nucleus. This is in line with our overexpression studies in which we observed a nuclear mRNA staining pattern upon overexpression of all assessed HNRNPC isoforms and variants in iPSCs (Figure 2F).
Previous studies have shown that HNRNPC is involved in cell-cycle-dependent translation via internal ribosome entry sites (IRES) during the G2/M phase, when cap-dependent translation is partly inhibited.18,19 This cell-cycle-specific function of HNRNPC in IRES translation may cause altered protein abundance, specifically of proteins dependent on IRES translation during neurodevelopment.63 In line with these findings, we clearly observed altered subcellular localization of HNRNPC in mitotic cells.
The absence of a phenotype in iPSCs derived from an affected individual (Ind1) as well as control iPSCs in which we induced a strong HNRNPC knockdown might be due to the use of undifferentiated iPSCs. Recently, RNA structural changes have been reported in neurogenesis, which correlates with the openness of RNA structure for HNRNPC binding.64 Hence, the exten of neuronal differentiation may affect the target binding capacity of HNRNPC in either neural progenitor cells (NPCs) or neurons and might thus be affected differently than in iPSCs. Therefore, functional assessment of HNRNPC and HNRNPCDEL in NPCs or iPSC-derived neurons may lead to the discovery of disease-relevant phenotypes and provide a potential pathogenic molecular mechanism. Of note, samples suitable for iPSC generation were available only for individual 1, although similar results are expected for iPSCs from the other individuals with the same recurrent variant.
Loss of HNRNPC affects differential splicing
Analysis of differentially expressed genes showed an effect of loss of HNRNPC but did not reveal any remarkable dysregulated pathway (Figure S5D). In mammalian cells, the levels of RNA-binding proteins (RBPs) such as HNRNPC is regulated by other RBPs. Recently, a reciprocal expression regulation of HNRNPC and CELF2 has been reported in T cells.65 In the meta-analysis, CELF2 expression remained unaffected across all analyzed datasets (DESeq2 result, Table S6), leading us to assume that the role of HNRNPC in gene expression is cell type specific.
Previous RNA-seq analysis has identified RNA-binding sites of HNRNPC and suggests a regulatory role for HNRNPC in alternative exon or ALU sequence inclusion and exclusion.16,14 We combined this dataset with datasets of two additional cancer cell lines (HEK22 and THP-115) in a meta-analysis and identified a strong effect of loss of HNRNPC function on alternative splicing, regardless of the cell line. Interestingly, this meta-analysis showed that both a full knockdown in HEK and HeLa cells as well as a 50% knockdown in THP-1 cells show the same pattern. This suggests the effect of homo- and heterozygous loss of HNRNPC to be highly similar and supports HNRNPC haploinsufficiency as a potential cause of pathogenicity in our cohort.
Interestingly, the DAVID analysis of genes affected by alternative splicing (AS) due to the loss of HNRNPC in the MAJIQ meta-analysis revealed an enrichment for genes associated with intellectual disability (ID). This indicates the vast potential and value of in silico analysis of published datasets and their relevance for hypothesis testing.
RNA-seq data of fibroblasts obtained from Ind8 (bearing the p.Asp265Thrfs∗18 variant) and a sex-matched control subject enabled us to establish the presence of the fingerprint of HNRNPC-dependent alternative splicing as identified in our meta-analysis. Via the manual analysis, we report about 52% of the alternative exon or ALU sequences identified in the HNRNPC knockdown meta-analysis to be similarly altered in the fibroblasts from affected individual Ind8, which strongly supports the notion that HNRNPC haploinsufficiency underlies the observed pathogenicity. This result is further confirmed with the unbiased MAJIQ analysis. In contrast, the iPSCs from Ind1, which showed a reduction to 45% of the total HNRNPC levels, did not show alterations in any of the alternative exons identified upon HNRNPC downregulation as reported in literature16,14 and our meta-analysis. This discrepancy is potentially explained by the difference in RNA structure and openness in pluri- and/or multipotent cells as compared to differentiated cells.64
Dysregulation of HNRNPC levels affect neuronal function
Based on the neurodevelopmental delay observed in our cohort and the conserved dysregulation of splicing upon loss of HNRNPC, as suggested by the meta-analysis, we assessed the functional role of HNRNPC in our murine in vivo and in vitro screen for gene variants associated with neurodevelopmental disorders (PRiSM, www.functionalgenomics.nl). Our in vivo approach revealed aberrant neuronal migration in the somatosensory cortex (SSCx) in the mouse brain upon shRNA-mediated knockdown of HNRNPC. Also, neuronal morphology was altered upon reduced HNRNPC abundance. The observed reduced neuronal migration of shRNA-targeted cells in our IUE assay indicates an important role for HNRNPC in (early) neurodevelopment and strongly supports the notion that pathogenic variants leading to HNRNPC haploinsufficiency cause NDD. Impaired cell migration has also been reported with knockdown of HNRNPC in neural crest cells.6 However, the migration deficits observed in the IUE assay are not necessarily predictive for the presence of cortical migration deficits in affected individuals, as the knockdown of HNRNPC likely results in HNRNPC levels that are lower as compared to haploinsufficiency. In addition, the targeted cells have to compete with the surrounding non-targeted cells, which may amplify the migration deficit. Yet, it is notable that structural brain changes were observed in several MRIs of Ind2, Ind8, and Ind12.7 Possibly, such migrations deficits arise from loss of heterozygosity in a subset of cells. Notably, overexpression of HNRNPC-iso1 in the developing mouse brain had similar effects on neuronal migration and additionally resulted in aberrant neuronal morphology and neuronal toxicity, suggesting correct HNRNPC dosage is crucial for brain development.
The similarity between neuronal migration deficits upon overexpression of HNRNPC-iso1 versus HNRNPC-iso1DEL suggests that the HNRNPCDEL variant does not abolish HNRNPC function. This is in line with our finding that overexpression of either HNRNPC-iso1 or HNRNPC-iso1DEL in iPSCs causes nuclear mRNA accumulation. Hence, we believe that the pathogenic effect of the HNRNPCDEL variant is caused by its reduction in HNRNPC levels. We expect the relatively high prevalence of the c.889_915del variant to be a result of a nucleic acid sequence repeat (c.906 _915 is homologous to c.879_888; GenBank: NM_031314.3), which are typically more prone to mutation due to general instability of repetitive DNA sequences.66
In conclusion, although the exact molecular mechanism underlying the pathogenic effect of the HNRNPC missense variants described remains to be identified, our study indicates that the developing brain is sensitive to aberrant levels of HNRNPC and that haploinsufficiency of HNRNPC results in aberrant splicing, in particular enriched for ID genes, ultimately leading to a neurodevelopmental disorder. This demonstrates that the HNRNPC should be added to the list of HNRNP-related neurodevelopmental disorders.
Acknowledgments
First and foremost, we are grateful for all individuals and parents for their cooperation and permission to publish their clinical and genetic information in this manuscript.
The inclusion of individual 11 was made possible through access to the data and findings generated by the 100,000 Genomes Project. The 100,000 Genomes Project is managed by Genomics England Limited (a wholly owned company of the Department of Health and Social Care). The 100,000 Genomes Project is funded by the National Institute for Health Research and NHS England. The Wellcome Trust, Cancer Research UK, and the Medical Research Council have also funded research infrastructure. The 100,000 Genomes Project uses data provided by individuals and collected by the National Health Service as part of their care and support.
Further, we would like to thank the IPS core facility at the Erasmus MC for generating the IPS lines, registering them to hPSCreg, and providing help and support throughout the project. Also, we are appreciative for the RNA sequencing of the IPS lines by the Biomics facility at the Erasmus MC. Furthermore, we want to acknowledge the Erasmus MC Bioinformatics And Computational Network (BACON) for their instant help and support. Lastly, we would like to thank all funding bodies who made this work possible. A detailed description can be found in the supplemental acknowledgments.
Author contributions
A.B., A.C.M.v.E., E.N., and Y.E. conceptualized, designed, and drafted the work. A.B. was responsible for the identification of individual 1, which initiated the study. A.B. was involved in gathering clinical data, as provided by A.E.B., C.L.M., D.H.G., D.A.S., D.Z., D.W., E.W., F.V., I.K., I.T., J.C.-S., J.B., J.M.E., J.P., L.C.B., M. Balasubramanian, M. Bertrand, M.A.G., M.J., M.M., M.S., N.H.R., P.J.B., S.B., S.M.L., S.E.R., S.S., and T.B.H. A.B. and M.W. were involved in the interpretation of the clinical data. The conception of the molecular and cellular studies was the responsibility of E.N., A.C.M.v.E., and Y.E. M.E. was responsible for cloning the HNRNPC-iso1 cDNAs and generating expression vectors for HNRNPC-iso1 and HNRNPC-iso1DEL (pYE1573, pYE1574). Other (tagged) HNRNPC constructs were generated by A.C.M.v.E.. E.D.R.O.H.-M. contributed to the analysis and interpretation of the data. I.W. performed and analyzed in vitro and in vivo mouse experiments. E.N., J.P., J.A., L.C.B., and R.M.H. were involved in the gathering, analysis, and interpretation of RNA-seq data. A.B., A.C.M.v.E., E.N., and Y.E. were involved in drafting, writing, and editing the manuscript with significant contributions from L.C.B., M.W., M.A.G., and R.M.H. Scientific guidance on the molecular studies was provided by A.C.M.v.E. and Y.E. All authors revised the manuscript critically for intellectual content, approved the final manuscript for publication, and agreed to be accountable for their published work.
Declaration of interests
The authors declare no competing interests.
Published: August 3, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.07.005.
Contributor Information
Arjan Bouman, Email: a.bouman@erasmusmc.nl.
Ype Elgersma, Email: y.elgersma@erasmusmc.nl.
Genomics England Research Consortium:
J.C. Ambrose, P. Arumugam, R. Bevers, M. Bleda, F. Boardman-Pretty, C.R. Boustred, H. Brittain, M.A. Brown, M.J. Caulfield, G.C. Chan, A. Giess, J.N. Griffin, A. Hamblin, S. Henderson, T.J.P. Hubbard, R. Jackson, L.J. Jones, D. Kasperaviciute, M. Kayikci, A. Kousathanas, L. Lahnstein, A. Lakey, S.E.A. Leigh, I.U.S. Leong, F.J. Lopez, F. Maleady-Crowe, M. McEntagart, F. Minneci, J. Mitchell, L. Moutsianas, M. Mueller, N. Murugaesu, A.C. Need, P. O‘Donovan, C.A. Odhams, C. Patch, D. Perez-Gil, M.B. Pereira, J. Pullinger, T. Rahim, A. Rendon, T. Rogers, K. Savage, K. Sawant, R.H. Scott, A. Siddiq, A. Sieghart, S.C. Smith, A. Sosinsky, A. Stuckey, M. Tanguy, A.L. Taylor Tavares, E.R.A. Thomas, S.R. Thompson, A. Tucci, M.J. Welland, E. Williams, K. Witkowska, S.M. Wood, and M. Zarowiecki
Undiagnosed Diseases Network:
Maria T. Acosta, David R. Adams, Raquel L. Alvarez, Justin Alvey, Aimee Allworth, Ashley Andrews, Euan A. Ashley, Ben Afzali, Carlos A. Bacino, Guney Bademci, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak-Toydemir, Anita Beck, Alan H. Beggs, Edward Behrens, Gill Bejerano, Hugo J. Bellen, Jimmy Bennet, Jonathan A. Bernstein, Gerard T. Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Devon Bonner, Lorenzo Botto, Lauren C. Briere, Gabrielle Brown, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Peter Byers, William E. Byrd, John Carey, Olveen Carrasquillo, Thomas Cassini, Ta Chen Chang, Sirisak Chanprasert, Hsiao-Tuan Chao, Ivan Chinn, Gary D. Clark, Terra R. Coakley, Laurel A. Cobban, Joy D. Cogan, Matthew Coggins, F. Sessions Cole, Heather A. Colley, Heidi Cope, Rosario Corona, William J. Craigen, Andrew B. Crouse, Michael Cunningham, Precilla D'Souza, Hongzheng Dai, Surendra Dasari, Joie Davis, Jyoti G. Dayal, Margaret Delgado, Esteban C. Dell'Angelica, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Argenia L. Doss, Emilie D. Douine, Dawn Earl, David J. Eckstein, Lisa T. Emrick, Christine M. Eng, Marni Falk, Elizabeth L. Fieg, Paul G. Fisher, Brent L. Fogel, Irman Forghani, Jiayu Fu, William A. Gahl, Ian Glass, Page C. Goddard, Rena A. Godfrey, Alana Grajewski, Meghan C. Halley, Rizwan Hamid, Neal Hanchard, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M. Hisama, Ingrid A. Holm, Jason Hom, Martha Horike-Pyne, Alden Huang, Yan Huang, Sarah Hutchison, Wendy Introne, Rosario Isasi, Kosuke Izumi, Gail P. Jarvik, Jeffrey Jarvik, Suman Jayadev, Orpa Jean-Marie, Vaidehi Jobanputra, Emerald Kaitryn, Shamika Ketkar, Dana Kiley, Gonench Kilich, Shilpa N. Kobren, Isaac S. Kohane, Jennefer N. Kohler, Susan Korrick, Deborah Krakow, Donna M. Krasnewich, Elijah Kravets, Seema R. Lalani, Byron Lam, Christina Lam, Brendan C. Lanpher, Ian R. Lanza, Kimberly LeBlanc, Brendan H. Lee, Roy Levitt, Richard A. Lewis, Pengfei Liu, Xue Zhong Liu, Nicola Longo, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, AudreyStephannie Maghiro, Rachel Mahoney, May Christine Malicdan, Laura A. Mamounas, Teri A. Manolio, Rong Mao, Ronit Marom, Gabor Marth, Beth A. Martin, Martin G. Martin, Julian A. Martínez-Agosto, Shruti Marwaha, Jacob McCauley, Allyn McConkie-Rosell, Alexa T. McCray, Elisabeth McGee, Matthew Might, Danny Miller, Ghayda Mirzaa, Eva Morava, Paolo Moretti, Marie Morimoto, John J. Mulvihill, Mariko Nakano-Okuno, Stanley F. Nelson, Shirley Nieves-Rodriguez, Donna Novacic, Devin Oglesbee, James P. Orengo, Laura Pace, Stephen Pak, J. Carl Pallais, Jeanette C. Papp, Neil H. Parker, Leoyklang Petcharet, John A. Phillips, III, Jennifer E. Posey, Lorraine Potocki, Barbara N. Swerdzewski, Aaron Quinlan, Deepak A. Rao, Anna Raper, Wendy Raskind, Genecee Renteria, Chloe Reuter, Lynette Rives, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Elizabeth Rosenthal, Francis Rossignol, Maura Ruzhnikov, Marla Sabaii, Ralph Sacco, Jacinda B. Sampson, Mario Saporta, Judy Schaechter, Timothy Schedl, Kelly Schoch, Daryl A. Scott, Elaine Seto, Prashant Sharma, Vandana Shashi, Emily Shelkowitz, Sam Sheppeard, Jimann Shin, Edwin Silverman, Janet Sinsheimer, Kathy Sisco, Edward Smith, Kevin Smith, Lilianna Solnica-Krezel, Ben Solomon, Rebecca Spillmann, Andrew Stergachis, Joan Stoler, Kathleen Sullivan, Jennifer Sullivan, Shirley Sutton, David A. Sweetser, Virginia Sybert, Holly K. Tabor, Queenie K.-G. Tan, Amelia L. Tan, Arjun Tarakad, Mustafa Tekin, Fred Telischi, Willa Thorson, Cynthia Tifft, Camilo Toro, Alyssa A. Tran, Rachel A. Ungar, Tiina K. Urv, Adeline Vanderver, Matt Velinder, Dave Viskochil, Tiphanie P. Vogel, Colleen E. Wahl, Melissa Walker, Nicole M. Walley, Jennifer Wambach, Jijun Wan, Lee-kai Wang, Michael F. Wangler, Patricia A. Ward, Daniel Wegner, Monika Weisz, Mark Wener, Tara Wenger, Monte Westerfield, Matthew T. Wheeler, Jordan Whitlock, Lynne A. Wolfe, Shinya Yamamoto, Zhe Zhang, and Stephan Zuchner
Web resources
Alamut™ Visual Plus v.1.6.1, https://www.sophiagenetics.com/platform/alamut-visual-plus/
CASAVA software, https://github.com/zhanglabtools/CASAVA
gnomAD browser, https://gnomad.broadinstitute.org/
Image studio light software, https://www.licor.com/bio/support/answer-portal/software/image-studio.html
IPA analysis software, https://digitalinsights.qiagen.com/IPA
Supplemental information
Data and code availability
All relevant data have been incorporated in the manuscript and its supplemental information. The ethics committee of the Erasmus MC does not allow sharing of individual or control genotype information in the public domain; however, the data are available upon reasonable request. RNA-seq data of HNRNPC knockdown in HeLa cells,14 HEK,22 and THP-115 was accessed via E-MTAB-1147, GSE56010, and GSE176012, respectively. All iPSC lines used in this study have been registered to hPSCreg.eu. Lines derived from Ind1 are desposited as https://hpscreg.eu/cell-line/EMCi225-A, https://hpscreg.eu/cell-line/EMCi225-B, https://hpscreg.eu/cell-line/EMCi225-C, and https://hpscreg.eu/cell-line/EMCi225-D. The sex- and age-matched control lines are registered as https://hpscreg.eu/cell-line/EMCi169-A, https://hpscreg.eu/cell-line/EMCi169-B, and https://hpscreg.eu/cell-line/EMCi169-C.
References
- 1.Gillentine M.A., Wang T., Hoekzema K., Rosenfeld J., Liu P., Guo H., Kim C.N., De Vries B.B.A., Vissers L.E.L.M., Nordenskjold M., et al. Rare deleterious mutations of HNRNP genes result in shared neurodevelopmental disorders. Genome Med. 2021;13:63. doi: 10.1186/s13073-021-00870-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geuens T., Bouhy D., Timmerman V. The hnRNP family: insights into their role in health and disease. Hum. Genet. 2016;135:851–867. doi: 10.1007/s00439-016-1683-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Low Y.H., Asi Y., Foti S.C., Lashley T. Heterogeneous Nuclear Ribonucleoproteins: Implications in Neurological Diseases. Mol. Neurobiol. 2020;58:631–646. doi: 10.1007/S12035-020-02137-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reichert S.C., Li R., A Turner S., van Jaarsveld R.H., Massink M.P.G., van den Boogaard M.J.H., del Toro M., Rodríguez-Palmero A., Fourcade S., Schlüter A., et al. HNRNPH1-related syndromic intellectual disability: Seven additional cases suggestive of a distinct syndromic neurodevelopmental syndrome. Clin. Genet. 2020;98:91–98. doi: 10.1111/cge.13765. [DOI] [PubMed] [Google Scholar]
- 5.Bain J.M., Cho M.T., Telegrafi A., Wilson A., Brooks S., Botti C., Gowans G., Autullo L.A., Krishnamurthy V., Willing M.C., et al. Variants in HNRNPH2 on the X Chromosome Are Associated with a Neurodevelopmental Disorder in Females. Am. J. Hum. Genet. 2016;99:728–734. doi: 10.1016/j.ajhg.2016.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lange L., Pagnamenta A.T., Lise S., Clasper S., Stewart H., Akha E.S., Quaghebeur G., Knight S.J.L., Keays D.A., Taylor J.C., Kini U. A de novo frameshift in HNRNPK causing a Kabuki-like syndrome with nodular heterotopia. Clin. Genet. 2016;90:258–262. doi: 10.1111/CGE.12773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duijkers F.A., McDonald A., Janssens G.E., Lezzerini M., Jongejan A., van Koningsbruggen S., Leeuwenburgh-Pronk W.G., Wlodarski M.W., Moutton S., Tran-Mau-Them F., et al. HNRNPR Variants that Impair Homeobox Gene Expression Drive Developmental Disorders in Humans. Am. J. Hum. Genet. 2019;104:1040–1059. doi: 10.1016/J.AJHG.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yates T.M., Vasudevan P.C., Chandler K.E., Donnelly D.E., Stark Z., Sadedin S., Willoughby J., Broad Center for Mendelian Genomics. DDD study. Balasubramanian M. De novo mutations in HNRNPU result in a neurodevelopmental syndrome. Am. J. Med. Genet. 2017;173:3003–3012. doi: 10.1002/AJMG.A.38492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bramswig N.C., Lüdecke H.J., Hamdan F.F., Altmüller J., Beleggia F., Elcioglu N.H., Freyer C., Gerkes E.H., Demirkol Y.K., Knupp K.G., et al. Heterozygous HNRNPU variants cause early onset epilepsy and severe intellectual disability. Hum. Genet. 2017;136:821–834. doi: 10.1007/S00439-017-1795-6/TABLES/2. [DOI] [PubMed] [Google Scholar]
- 10.Semino F., Schröter J., Willemsen M.H., Bast T., Biskup S., Beck-Woedl S., Brennenstuhl H., Schaaf C.P., Kölker S., Hoffmann G.F., et al. Further evidence for de novo variants in SYNCRIP as the cause of a neurodevelopmental disorder. Hum. Mutat. 2021;42:1094–1100. doi: 10.1002/HUMU.24245. [DOI] [PubMed] [Google Scholar]
- 11.Kaplanis J., Samocha K.E., Wiel L., Zhang Z., Arvai K.J., Eberhardt R.Y., Gallone G., Lelieveld S.H., Martin H.C., McRae J.F., et al. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature. 2020;586:757–762. doi: 10.1038/S41586-020-2832-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shahied L., Braswell E.H., LeStourgeon W.M., Krezel A.M. An antiparallel four-helix bundle orients the high-affinity RNA binding sites in hnRNP C: A mechanism for RNA chaperonin activity. J. Mol. Biol. 2001;305:817–828. doi: 10.1006/jmbi.2000.4331. [DOI] [PubMed] [Google Scholar]
- 13.McAfee J.G., Shahied-Milam L., Soltaninassab S.R., Lestourgeon W.M. A major determinant of hnRNP C protein binding to RNA is a novel bZIP- like RNA binding domain. RNA. 1996;2:1139–1152. [PMC free article] [PubMed] [Google Scholar]
- 14.Zarnack K., König J., Tajnik M., Martincorena I., Eustermann S., Stévant I., Reyes A., Anders S., Luscombe N.M., Ule J. Direct Competition between hnRNP C and U2AF65 Protects the Transcriptome from the Exonization of Alu Elements. Cell. 2013;152:453–466. doi: 10.1016/J.CELL.2012.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herzner A.M., Khan Z., van Nostrand E.L., Chan S., Cuellar T., Chen R., Pechuan-Jorge X., Komuves L., Solon M., Modrusan Z., et al. ADAR and hnRNPC deficiency synergize in activating endogenous dsRNA-induced type I IFN responses. J. Exp. Med. 2021;218 doi: 10.1084/JEM.20201833/212507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.König J., Zarnack K., Rot G., Curk T., Kayikci M., Zupan B., Turner D.J., Luscombe N.M., Ule J. ICLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCloskey A., Taniguchi I., Shinmyozu K., Ohno M. hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science. 2012;335:1643–1646. doi: 10.1126/SCIENCE.1218469. [DOI] [PubMed] [Google Scholar]
- 18.Kim J.H., Paek K.Y., Choi K., Kim T.-D., Hahm B., Kim K.-T., Jang S.K. Heterogeneous Nuclear Ribonucleoprotein C Modulates Translation of c- myc mRNA in a Cell Cycle Phase-Dependent Manner. Mol. Cell Biol. 2003;23:708–720. doi: 10.1128/MCB.23.2.708-720.2003/ASSET/85F59AE9-DBC7-496B-98AC-08ADF6C4BA98/ASSETS/GRAPHIC/MB02310435DE.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schepens B., Tinton S.A., Bruynooghe Y., Parthoens E., Haegman M., Beyaert R., Cornelis S. A role for hnRNP C1/C2 and Unr in internal initiation of translation during mitosis. EMBO J. 2007;26:158–169. doi: 10.1038/SJ.EMBOJ.7601468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sella O., Gerlitz G., Le S.-Y., Elroy-Stein O. Differentiation-Induced Internal Translation of c- sis mRNA: Analysis of the cis Elements and Their Differentiation-Linked Binding to the hnRNP C Protein. Mol. Cell Biol. 1999;19:5429–5440. doi: 10.1128/MCB.19.8.5429/FORMAT/EPUB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holcík M., Gordon B.W., Korneluk R.G. The Internal Ribosome Entry Site-Mediated Translation of Antiapoptotic Protein XIAP Is Modulated by the Heterogeneous Nuclear Ribonucleoproteins C1 and C2. Mol. Cell Biol. 2003;23:280–288. doi: 10.1128/MCB.23.1.280-288.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu N., Dai Q., Zheng G., He C., Parisien M., Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564. doi: 10.1038/NATURE14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. N6-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/J.CELL.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao Z., Huang Y., Duan R., Jin P., Qin Z.S., Zhang S. Disease category-specific annotation of variants using an ensemble learning framework. Brief. Bioinform. 2022;23:bbab438. doi: 10.1093/bib/bbab438. [DOI] [PubMed] [Google Scholar]
- 25.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/GR.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reijnders M.R.F., Kousi M., van Woerden G.M., Klein M., Bralten J., Mancini G.M.S., van Essen T., Proietti-Onori M., Smeets E.E.J., van Gastel M., et al. Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat. Commun. 2017;8:1052. doi: 10.1038/S41467-017-00933-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saito T., Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 2001;240:237–246. doi: 10.1006/DBIO.2001.0439. [DOI] [PubMed] [Google Scholar]
- 29.Banker G., Goslin K. 1991. Culturing nerve cells; p. 453. [DOI] [Google Scholar]
- 30.Schmitz S.K., Hjorth J.J.J., Joemai R.M.S., Wijntjes R., Eijgenraam S., de Bruijn P., Georgiou C., de Jong A.P.H., van Ooyen A., Verhage M., et al. Automated analysis of neuronal morphology, synapse number and synaptic recruitment. J. Neurosci. Methods. 2011;195:185–193. doi: 10.1016/j.jneumeth.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 31.Takahashi K., Yamanaka S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 32.Ma Y., Jin J., Dong C., Cheng E.C., Lin H., Huang Y., Qiu C. High-efficiency siRNA-based gene knockdown in human embryonic stem cells. RNA. 2010;16:2564–2569. doi: 10.1261/rna.2350710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Afgan E., Baker D., Batut B., Van Den Beek M., Bouvier D., Cech M., Chilton J., Clements D., Coraor N., Grüning B.A., et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46:W537–W544. doi: 10.1093/NAR/GKY379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J., et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47:D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/BIOINFORMATICS/BTT656. [DOI] [PubMed] [Google Scholar]
- 37.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaquero-Garcia J., Barrera A., Gazzara M.R., González-Vallinas J., Lahens N.F., Hogenesch J.B., Lynch K.W., Barash Y. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife. 2016;5:e11752. doi: 10.7554/ELIFE.11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krämer A., Green J., Pollard J., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–530. doi: 10.1093/BIOINFORMATICS/BTT703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sherman B.T., Hao M., Qiu J., Jiao X., Baseler M.W., Lane H.C., Imamichi T., Chang W. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update) Nucleic Acids Res. 2022;50:W216–W221. doi: 10.1093/NAR/GKAC194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang D.W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/NPROT.2008.211. [DOI] [PubMed] [Google Scholar]
- 42.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sobreira N., Schiettecatte F., Valle D., Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015;36:928–930. doi: 10.1002/HUMU.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Supek F., Lehner B., Lindeboom R.G.H. To NMD or Not To NMD: Nonsense-Mediated mRNA Decay in Cancer and Other Genetic Diseases. Trends Genet. 2021;37:657–668. doi: 10.1016/J.TIG.2020.11.002. [DOI] [PubMed] [Google Scholar]
- 45.Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Riggs E.R., Andersen E.F., Cherry A.M., Kantarci S., Kearney H., Patel A., Raca G., Ritter D.I., South S.T., Thorland E.C., et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen) Genet. Med. 2020;22:245–257. doi: 10.1038/s41436-019-0686-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiel L., Baakman C., Gilissen D., Veltman J.A., Vriend G., Gilissen C. MetaDome: Pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum. Mutat. 2019;40:1030–1038. doi: 10.1002/HUMU.23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coban-Akdemir Z., White J.J., Song X., Jhangiani S.N., Fatih J.M., Gambin T., Bayram Y., Chinn I.K., Karaca E., Punetha J., et al. Identifying Genes Whose Mutant Transcripts Cause Dominant Disease Traits by Potential Gain-of-Function Alleles. Am. J. Hum. Genet. 2018;103:171–187. doi: 10.1016/j.ajhg.2018.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hebsgaard S.M., Korning P.G., Tolstrup N., Engelbrecht J., Rouzé P., Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24:3439–3452. doi: 10.1093/NAR/24.17.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yeo G., Burge C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]
- 51.Rollins J.D., Collins J.S., Holden K.R. United States Head Circumference Growth Reference Charts: Birth to 21 Years. J. Pediatr. 2010;156:907–913.e2. doi: 10.1016/j.jpeds.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 52.Scholzen T., Gerdes J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 53.Barnett S.F., Friedman D.L., LeStourgeon W.M. The C proteins of HeLa 40S nuclear ribonucleoprotein particles exist as anisotropic tetramers of (C1)3 C2. Mol. Cell Biol. 1989;9:492–498. doi: 10.1128/mcb.9.2.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williamson D.J., Banik-Maiti S., DeGregori J., Ruley H.E. hnRNP C Is Required for Postimplantation Mouse Development but Is Dispensable for Cell Viability. Mol. Cell Biol. 2000;20:4094–4105. doi: 10.1128/MCB.20.11.4094-4105.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dörrbaum A.R., Kochen L., Langer J.D., Schuman E.M. Local and global influences on protein turnover in neurons and glia. Elife. 2018;7 doi: 10.7554/ELIFE.34202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Molyneaux B.J., Arlotta P., Menezes J.R.L., Macklis J.D. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 2007;8:427–437. doi: 10.1038/NRN2151. [DOI] [PubMed] [Google Scholar]
- 57.Dehay C., Kennedy H. Cell-cycle control and cortical development. Nat. Rev. Neurosci. 2007;8:438–450. doi: 10.1038/NRN2097. [DOI] [PubMed] [Google Scholar]
- 58.Nickless A., Bailis J.M., You Z. Control of gene expression through the nonsense-mediated RNA decay pathway. Cell Biosci. 2017;7:26. doi: 10.1186/S13578-017-0153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koloteva-Levine N., Amichay M., Elroy-Stein O. Interaction of hnRNP-C1/C2 proteins with RNA: Analysis using the yeast three-hybrid system. FEBS Lett. 2002;523:73–78. doi: 10.1016/S0014-5793(02)02938-1. [DOI] [PubMed] [Google Scholar]
- 60.Wu Y., Zhao W., Liu Y., Tan X., Li X., Zou Q., Xiao Z., Xu H., Wang Y., Yang X. Function of HNRNPC in breast cancer cells by controlling the dsRNA-induced interferon response. EMBO J. 2018;37 doi: 10.15252/EMBJ.201899017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fischl H., Neve J., Wang Z., Patel R., Louey A., Tian B., Furger A. hnRNPC regulates cancer-specific alternative cleavage and polyadenylation profiles. Nucleic Acids Res. 2019;47:7580–7591. doi: 10.1093/NAR/GKZ461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Han N., Li W., Zhang M. The function of the RNA-binding protein hnRNP in cancer metastasis. J. Cancer Res. Ther. 2013;9:S129–S134. doi: 10.4103/0973-1482.122506. [DOI] [PubMed] [Google Scholar]
- 63.Norris K., Hopes T., Aspden J.L. Ribosome heterogeneity and specialization in development. Wiley Interdiscip Rev RNA. 2021;12:e1644. doi: 10.1002/WRNA.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang J., Zhang T., Yu Z., Tan W.T., Wen M., Shen Y., Lambert F.R.P., Huber R.G., Wan Y. Genome-wide RNA structure changes during human neurogenesis modulate gene regulatory networks. Mol. Cell. 2021;81:4942–4953.e8. doi: 10.1016/J.MOLCEL.2021.09.027. [DOI] [PubMed] [Google Scholar]
- 65.Mallory M.J., McClory S.P., Chatrikhi R., Gazzara M.R., Ontiveros R.J., Lynch K.W. Reciprocal regulation of hnRNP C and CELF2 through translation and transcription tunes splicing activity in T cells. Nucleic Acids Res. 2020;48:5710–5719. doi: 10.1093/NAR/GKAA295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bzymek M., Lovett S.T. Instability of repetitive DNA sequences: The role of replication in multiple mechanisms. Proc. Natl. Acad. Sci. USA. 2001;98:8319–8325. doi: 10.1073/PNAS.111008398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data have been incorporated in the manuscript and its supplemental information. The ethics committee of the Erasmus MC does not allow sharing of individual or control genotype information in the public domain; however, the data are available upon reasonable request. RNA-seq data of HNRNPC knockdown in HeLa cells,14 HEK,22 and THP-115 was accessed via E-MTAB-1147, GSE56010, and GSE176012, respectively. All iPSC lines used in this study have been registered to hPSCreg.eu. Lines derived from Ind1 are desposited as https://hpscreg.eu/cell-line/EMCi225-A, https://hpscreg.eu/cell-line/EMCi225-B, https://hpscreg.eu/cell-line/EMCi225-C, and https://hpscreg.eu/cell-line/EMCi225-D. The sex- and age-matched control lines are registered as https://hpscreg.eu/cell-line/EMCi169-A, https://hpscreg.eu/cell-line/EMCi169-B, and https://hpscreg.eu/cell-line/EMCi169-C.