

Summary
Genome-wide association studies along with expression quantitative trait locus (eQTL) mapping have identified hundreds of single-nucleotide polymorphisms (SNPs) and their target genes in prostate cancer (PCa), yet functional characterization of these risk loci remains challenging. To screen for potential regulatory SNPs, we designed a CRISPRi library containing 9,133 guide RNAs (gRNAs) to cover 2,166 candidate SNP loci implicated in PCa and identified 117 SNPs that could regulate 90 genes for PCa cell growth advantage. Among these, rs60464856 was covered by multiple gRNAs significantly depleted in screening (FDR < 0.05). Pooled SNP association analysis in the PRACTICAL and FinnGen cohorts showed significantly higher PCa risk for the rs60464856 G allele (p value = 1.2 × 10−16 and 3.2 × 10−7, respectively). Subsequent eQTL analysis revealed that the G allele is associated with increased RUVBL1 expression in multiple datasets. Further CRISPRi and xCas9 base editing confirmed that the rs60464856 G allele leads to elevated RUVBL1 expression. Furthermore, SILAC-based proteomic analysis demonstrated allelic binding of cohesin subunits at the rs60464856 region, where the HiC dataset showed consistent chromatin interactions in prostate cell lines. RUVBL1 depletion inhibited PCa cell proliferation and tumor growth in a xenograft mouse model. Gene-set enrichment analysis suggested an association of RUVBL1 expression with cell-cycle-related pathways. Increased expression of RUVBL1 and activation of cell-cycle pathways were correlated with poor PCa survival in TCGA datasets. Our CRISPRi screening prioritized about one hundred regulatory SNPs essential for prostate cell proliferation. In combination with proteomics and functional studies, we characterized the mechanistic role of rs60464856 and RUVBL1 in PCa progression.
Keywords: post-GWAS function analysis, eQTL, prostate cancer risk, cohesin, CTCF, RUVBL1, CRISPRi screening, SILAC proteomics, noncoding variation, base editing
Graphical abstract
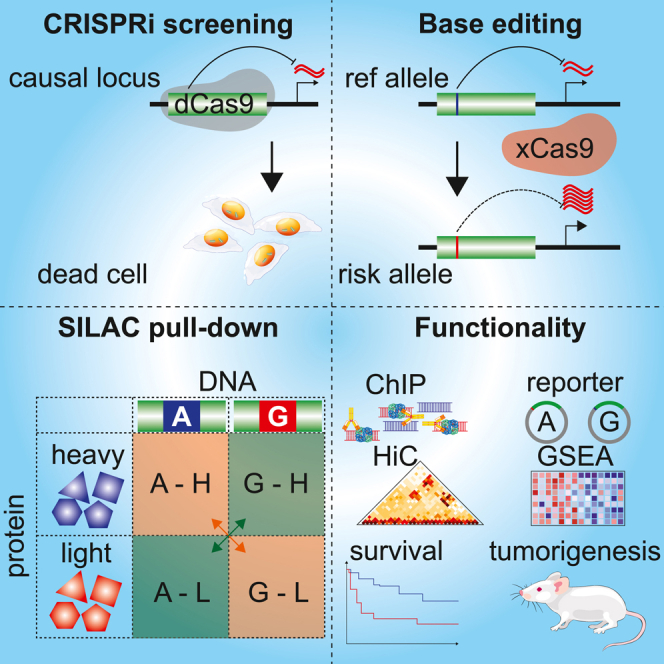
A major goal of post-GWAS studies is to functionally characterize causal SNPs that confer increased risk to disease phenotypes. Here, we applied CRISPRi and proteomics screening to identify regulatory SNPs at prostate-cancer risk loci and functionally characterized the impact of the rs60464856-RUVBL1 locus via in vitro and in vivo approaches.
Introduction
Among all cancer types, prostate cancer (PCa) accounted for 26% of 970,250 new cancer cases and caused 11% of 319,420 cancer-related deaths in US males in 2021.1 As a cancer type with strong genetic predispositions, PCa has been extensively investigated in genome-wide association studies (GWASs)2 in which researchers aim to determine susceptible variants associated with increased disease risk and aggressiveness.2,3 It has been reported that the contribution of GWAS-identified loci to PCa risk is nearly 20%.4 Although GWASs have been highly productive, only a few risk loci have been functionally characterized.5,6,7,8 Thus far, because these risk single-nucleotide polymorphisms (SNPs) are found in non-coding portions of the genome, it is believed that many of them (or their closely linked SNPs) alter the activities of regulatory elements and quantitatively change gene expression rather than directly mutating protein sequences.9,10,11,12 With tremendous large-scale GWAS findings, especially those of high reproducibility, it is believed that some non-coding variants play a subtle but profound role in PCa initiation, progression, and metastasis by modulating the expression of discrete susceptibility genes.
To dissect functional variants and their target genes in PCa, researchers apply bioinformatic and benchtop approaches to prioritize and validate the causal variants.13,14,15 Curated databases such as ChIP-atlas,16 ENCODE,17 JASPAR,18 and GTEx19 provide abundant resources for estimating the genetic contribution of GWAS variants to PCa and other cancer susceptibilities. However, large-scale genetic assays are often needed for interrogation of endogenous variant loci and direct characterization of consequent phenotype changes to determine the biological effect.20,21,22 One such assay is lentiviral-based Cas9-mediated screening, which has emerged as a powerful tool for evaluating the biological significance of genes of interest on a large scale.23,24 Compared with canonical wild-type Cas9-based screening, dead Cas9 (dCas9) forms steric hindrance according to the gRNA sequence and induces transcription repression if fused to repressor peptides KRAB (Krüppel-associated box). dCas9-KRAB can specifically decrease target gene expression without strand cleavage and is used for screening regulatory elements in mammalian cells.25,26
Most SNPs are believed to function as regulatory switches in the human genome. With the eliminated nuclease activity,26 we hypothesized that dCas9-based CRISPRi could be used to interfere with regulatory sequences at SNP loci and faithfully mimick the transcription alteration caused by single-base differences. To test this hypothesis, we first established multiple stable dCas9-KRAB prostate cell lines and designed an unbiased, highly reproducible gRNA library that targeted candidate SNPs at PCa risk loci. We then performed negative selection for potential SNPs conferring a growth advantage. Finally, we provided a detailed analysis of an SNP-gene pair for its functional role by using prostate cell lines and a mouse model and performed a successful proteomics identification of transcriptional regulators that mediate the variant's regulatory change. (Figure 1A) Our results support the use of CRISPRi-based approaches at disease risk loci for regulatory SNP screening.
Figure 1.

CRISPRi screening in prostate cell lines decodes regulatory SNPs from eQTL analyses
(A) Study design of the current project, including eQTL target selection, CRISPRi screening, and functional validation.
(B) Overall SNP candidates in BPH1 (red), DU145 (green), and PC3 (blue) cells over 3-week screening.
(C–E) Comparison of the gRNA fold change between the selected and unselected SNPs in BPH1 (C), DU145 (D), and PC3 (E) cells. The gRNA fold change was calculated from the average of the two biological replicates. The bar represents the standard error of the fold changes within each category.
(F) Enrichment of the selected compared to unselected SNPs in transcription start sites (TSSs).
(G) Enrichment of the selected compared to unselected SNPs in allele-specific binding (ASB) annotation according to the ANANASTRA database.
(H–J) Demonstration of gRNA representation for the selected SNPs in BPH1 (H), DU145 (I), and PC3 (J) cells. gRNAs targeting the main validation SNP rs60464856 are highlighted in red.
Methods
eQTL-based SNP selection at prostate-cancer risk loci
The rationale for the regulatory screening is explained in Figures S1A and S1B. In brief, to select candidate SNPs, we first retrieved cis-eQTL data from our benign prostate tissues27 and identified all SNPs with gene-wise FDR ≤ 1 × 10−3. We then applied ENCODE annotations (including histone modification, common transcription factor binding, and DNase hypersensitivity in prostate cell lines) to filter for candidate functional SNPs.
gRNA selection and library pool amplification for candidate SNPs
To design gRNAs for the candidate SNPs, we retrieved DNA sequences surrounding each SNP (±23bp). We used the CRISPOR program (http://crispor.tefor.net/) for in-silico selection. We assembled the gRNA oligo pool into the lentiGuide-Puro (RRID: Addgene_52963) backbone and transformed the pooled oligos into highly sensitive Endura ElectroCompetent Cells (Lucigen) to generate plasmid libraries.
dCas9/KRAB stable cell lines and gRNA library processing
To establish stable cell lines, we packaged lenti-dCas9-KRAB-blast (RRID: Addgene_89567) into lentiviral particles with HEK293FT cells and used 10-fold concentrated virus particles to transduce several human cells, including RWPE1, BPH1, 22Rv1, PC3, DU145 and HEK293FT cell lines. After blasticidin selection, stable dCas9 expression was verified with immunoblots. The gRNA virus library (packaged from lentiGuide-Puro) was titrated in three dCas9 stable prostate cell lines, including BPH1 (originated from benign prostatic hyperplasia), DU145 (originated from prostatic brain metastasis) and PC3 (originated from prostatic bone metastasis) for the screening. We excluded RWPE1 because the serum-free culture condition conflicts with the virus supernatant, and we excluded 22Rv1 because the small cell size influenced the accuracy of cell counting during screening. To achieve low multiplicity of infection (MOI), we optimized the cell number and virus amount over 72-h puromycin selection, such that the non-infected group would be eliminated by 95%–99%, whereas the library group retained 30%–40% viability. The cell viability was measured with the CellTiter-Glo Luminescent Cell Viability Assay (Promega G7570). After confirming low MOI integration, we removed puromycin from the medium and continued the cell culture for 21 days. We isolated genomic DNA at baseline D1 (day 1) and endpoint D21 (day 21).
gRNA readout sequencing
We used the Illumina HiSeq platform to sequence the gRNA readout amplicons. We aimed for at least a 500-fold library size depth for each replicate to ensure quantification accuracy.
Data QC and analysis
To quantify the representation of each gRNA, we used a Python script “count_spacer.py” developed by Feng Zhang’s lab to scan the FASTQ file for perfectly matched hits and generate raw read counts for each experiment. We then used principal-component analysis (PCA) to evaluate the similarity of the plasmid library, baseline, and screening endpoint gRNA representation. To determine SNP alleles conferring growth advantage in these cell lines, we used RIGER (RNAi gene enrichment ranking) extension (https://software.broadinstitute.org/GENE-E/extensions.html) to calculate a rank list for SNPs or alleles with the most depleted gRNA representation.28 The RIGER program ranks gRNAs according to their depletion effects and then identifies the SNP targeted by the shRNAs.
Plasmid construction and siRNA design
To enable fluorescence-based cell sorting, we assembled copGFP ORF into base editor plasmid29 xCas9(3.7)-ABE(7.10) (RRID: Addgene_108382) with NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs). A 223-bp flanking sequencing surrounding rs60464856 was amplified from RWPE1 cells (rs60464856 heterozygote) and further subcloned into the pGL3-basic vector between NheI and XhoI sites with NEBuilder HiFi DNA Assembly Master Mix. Small hairpin RNA (shRNA) targeting RUVBL1, primers, and gRNA sequences are listed in Table S1.
Reagents and cell culture
Antibody against Cas9 protein (844302) was purchased from BioLegend. Antibody against RUVBL1 (10210-2-AP) was purchased from Proteintech. Antibody against β-actin (4970) was purchased from Cell Signaling Technology. SMC3 (ab9263), H3K4me1 (ab8895), and IgG isotype control antibodies (ab171870 for rabbit, ab37355 for mouse) were purchased from Abcam. SMC1A (61067), CTCF (61311), and H3K27ac (39034) antibodies were purchased from Active Motif. CTCFL (MABE1125) and H3K4me3 (c15410003) antibodies were purchased from Sigma-Aldrich and Diagenode, respectively. DU145 (CVCL_0105), PC3 (RRID: CVCL_0035), 22Rv1 (RRID: CVCL_1045), and RWPE1 (RRID: CVCL_3791) cells were obtained from the ATCC. BPH1 (RRID: CVCL_1091) cells were purchased from Sigma-Aldrich. HEK293FT cells (RRID: CVCL_6911) were purchased from Thermo Fisher Scientific. Cell lines were disposed of and replaced with low passage aliquots after being subcultured 15 times. Unless specified otherwise, all cell culture reagents were obtained from ThermoFisher Scientific. BPH1, DU145, PC3, and 22Rv1 cells were grown in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS). HEK293FT cells were grown in DMEM medium supplemented with 10% FBS and 500 μg/mL geneticin selective antibiotics. RWPE1 cells were grown in Keratinocyte Serum-Free Medium. All cell lines were examined for mycoplasma contamination with Venor GeM Mycoplasma Detection Kit (Sigma-Aldrich).
CRISPR base editing
To change the rs60464856 A allele to G allele, we created a GFP-labeled xCas9(3.7)-ABE(7.10) plasmid based on the backbone from David Liu’s lab.29 A gRNA template was synthesized in a gblock fragment with an hU6 promoter and a scaffold (https://benchling.com/protocols/10T3UWFo/detailed-gblocks-based-crispr-protocol). Because rs60464856 is located in the base editing window, the adenine base editor can be directed to the SNP site and catalyze A into the G allele. We co-transfected 2.5 μg xCas9(3.7)-ABE7.10 plasmid and 1.2 μg gRNA gblock into cells 80% confluent in each well of the six-well plate. After 48 h, GFP-positive cells were sorted by flow cytometry and collected for allele dosage quantification. The GFP-positive cells were seeded into single clones once editing efficiencies were above 5%. After expanding the single clones for ten days, we used amplification-refractory mutation system (ARMS) PCR for genotyping from the direct lysate. We also used Sanger sequencing to verify the germline change of each single clone.
Real-time PCR and immunoblot analysis
Total RNA was extracted from cells via the Direct-zol RNA Miniprep Kit (Zymo Research). One microgram of total RNA was reverse transcribed by iScript cDNA Synthesis Kit (BioRad). Quantification reactions were performed with PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) on the CFX96 Touch real-time PCR system (BioRad). The primers are listed in Table S1. Total protein was extracted and electrophoresed as described previously;30 minor modification was performed with Mini-PROTEAN Precast Gels (BioRad). SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific) produced luminescent signals on the LICOR imaging system. Captured images were aligned in Photoshop and assembled in Illustrator.
Luciferase reporter assay
The cells were seeded into a 24-well plate. After 12 to 16 h, Lipofectamine 3000 was used for transfecting 500 ng of pGL3 reporter plasmids to each well. The media were replaced after transfection for 24 h. After 48 h of transfection, the cells were lysed for the luciferase assay according to the Dual-Luciferase Reporter Assay (E1960, Promega) protocol. The luminescence signals were measured with the GlowMax plate reader. After normalization to Renilla luciferase readout, relative firefly luciferase activities driven by corresponding promoters were represented by fold changes in lumiescence.
HiC data analysis
We downloaded the processed data for DU145, PC3, VCaP, and LNCaP cells from the GSE17209931 supplementary file collection and used the ICED (iterative correction and eigenvector decomposition) normalized data matrix for visualization and analysis. We used a 20 kb bin for DU145 and PC3 cells and a 40 kb bin for VCaP and LNCaP cells; these sizes are the smallest bin size in the processed data. We used PERL modules from the existing pipeline cworld-dekker (https://github.com/dekkerlab/cworld-dekker) to generate the heatmap and to sub-slice the ICED data matrix. For demonstration convenience, we highlighted the method to calculate the left-to-right ratio (L/R) in Figures S6A–S6E. After visualizing the heatmap, we decided to focus on two hotspots surrounding the rs60464856 locus (chr3: 127840001–127860000 [hg19]) in DU145 and PC3 cells, including three 20-kb bins on the left (chr3: 127780001–127800000 [hg19] to chr3: 127820001–127840000 [hg19]) and three 20-kb bins on the right (chr3: 12786000–127880000 [hg19] to chr3: 127900001–127920000 [hg19]), both interacting with a distant ten 20-kb-bin region near the 3-prime end of RPN1 (chr3: 128120001–128140000 [hg19] to chr3: 128300001–128320000 [hg19]). We aggregated the ICED count in the hotspot and divided the left to the right to obtain the L/R ratio in each cell line. For VCaP and LNCaP, we applied the same strategy to calculate the L/R ratio, except for the larger bin size included for each interaction spot. Because the bin count for each hotspot was arbitrarily decided, we also calculated the L/R ratio for different options in Figure S6E. The results showed that the current selection represents an average estimation among the multiple options.
Allele-specific proteomics screening with stable isotope labeling by amino acid in cell culture (SILAC)
The BPH1, DU145, and PC3 cells were grown in SILAC RPMI 1640 medium (ThermoFisher 88365) for five passages before cells were harvested for nuclear protein extraction (Active Motif 40010). After confirming that heavy amino acid labeling efficiency reached 99.9%, we applied the nuclear extracts to the desalting spin column (ThermoFisher 89882) to remove excessive ions. The DNA baits harboring rs60464856 A and G alleles were produced according to methods in a previous publication.32 For each binding reaction, 2 μg of purified DNA baits were conjugated to 25 μL Streptavidin Dynabeads (ThermoFisher 65001). The clean conjugated beads were incubated with 12.5 μL precleared nuclear protein at 4° overnight. The incubated beads were washed five times and combined for two parallel quantitative-mass-spectrometry runs to provide the allelic protein binding ratio. Qualified proteins with allelic binding were defined as (1) concordant allele ratio changes: log2(A/G) × log2(G/A) < 0 and (2) drastic allele ratio differences: |log2(A/G) − log2(G/A)| ≥ 2. For proteins with allelic hits, we further narrowed them down to those with known DNA binding functions according to UniProt databases (https://www.uniprot.org/).33
Chromatin immunoprecipitation qPCR assays
Because the rs60464856 locus was demonstrated to reside in an insulation region in an analysis of previously published Hi-C data, we adapted our chromatin immunoprecipitation (ChIP) qPCR protocols for chromosome conformation capturing.34 Compared with conventional ChIP assay protocol, our protocol applied dual crosslinking to maximally preserve chromatin contacts.35 Each ChIPed DNA sample was tested in four qPCR reactions, including (1) rs60464856 locus enrichment primer pair amplifying a 223-bp fragment centering on rs60464856; (2) rs60464856 locus control primer pair amplifying a 183-bp fragment 2.5 kb upstream to the SNP; (3) rs60464856 A allele-specific primer pair; and (4) rs60464856 G allele-specific primer pair. To quantify the histone modification profiling in the subclones, we performed ChIP reactions with H3K4me1, H3K4me3, and H3K27ac antibodies and quantified the rs60464856 locus enrichment over the upstream control primer. We further normalized the fold change against the input DNA to obtain an enrichment score for each modification.
RNA-seq and gene set enrichment analysis (GSEA)
Total RNA sample was extracted with the Zymo Direct-zol microprep kit, with the on-column DNase digestion step included. The mRNA was purified from total RNA via poly-T oligo-attached magnetic beads. After fragmentation, the first-strand cDNA was synthesized with random hexamer primers followed by the second-strand cDNA synthesis. The library was ready after end repair, A-tailing, adapter ligation, size selection, amplification, and purification. A total of 6G genomic data were targeted for each sample, which guaranteed roughly 20 million 150 bp paired-end RNA reads. The FASTQ file was trimmed with cutadapt and quantified with the RSEM package36 for gene expression. To determine which pathway was associated with RUVBL1 expression, we used interactive GSEA software37 to find which gene set showed statistically significant enrichment. For the TCGA prostate cancer tissue, we used the FPKM data from the NCI GDC data portal for the GSEA analysis.
In vivo xenograft mouse model
Animal experiments were performed according to the protocol approved by the Institutional Animal Care and Use Committee of Fudan University. Nude mice (6-week-old males) were purchased from GemPharmatech (Jiangsu, China) and maintained in a pathogen-free environment. PC3 was grown in RPMI1640 containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified CO2 incubator. Before injection into the mice, the cells were harvested by trypsinization and washed two times with PBS. PC3 cells were then resuspended in 100 μL serum-free medium, mixed with 50% Matrigel (BD Biosciences), and injected (5 × 106/site) subcutaneously into the hind flank of each mouse. Tumor volume was measured with a digital caliper once a week and calculated according to the formula: V = (L × W2)/2 (L = length; W = width; all parameters are in millimeters). After 4 weeks, the mice were sacrificed, and tumors were taken for weight measurement. Mouse tumor models and protocols were approved by the Animal Experiment Review Board (20210302-071) of the School of Basic Medical Sciences, Fudan University, Shanghai, China.
Results
Candidate SNP selection, gRNA searching, and screening library production
We first screened the ENCODE database and identified 2664 SNPs (TARGET) with strong epigenomic signals (Figure S1C). We then applied a Bayesian framework by using summary statistics to calculate a posterior inclusion probability (PIP) to predict SNP functionality. We included five SNPs with the highest PIP score for each eQTL-associated gene as another PIP SNP category (n = 194). Finally, we included 641 control SNPs (CTL) that showed strong eQTL signals but without epigenomic features. After removing duplicated SNPs, we selected 3,408 SNPs for gRNA design. We scanned these SNPs with the CRISPOR program and eventually designed 9,133 gRNAs, including 100 control sequences that did not target any genome loci (NCG) (Figure S1D). The exact sequence design can be found in Table S2.
We used immunoblots to confirm stable expression of dCas9 after one month of transduction (Figure S2A). We ensured low MOI infection by measuring gRNA lentiviral library function titer in 72 h (Figure S2B) and visualized a 261-bp gRNA amplicon for each sample on agarose gel for quantification by high-throughput sequencing (Figure S2C). The sequencing summary for each sample is shown in Figure S2D. We also visualized the normalized count in each replicate endpoint and observed high correlations in all three cell lines (Figures S2E–S2G).
CRISPRi screening identified top candidates of regulatory SNPs
We first performed PCA analysis and found tight clustering between baseline and plasmid libraries (Figure S3A), suggesting faithful representations of original gRNA libraries in transfected cell lines. This analysis also found highly diverse but cell-line-dependent distribution in the end libraries, indicating gRNA profile changes by the selection process. We then normalized the read count by using the total noncount with perfect sequence match in each sample and calculated the fold change by dividing the read count in the end library by the read count in the baseline library. The subsequent RIGER analysis showed 779 gRNAs targeting 117 SNPs with permutation test FDR ≤0.1 in both replicates (Figure 1B). Further analysis did not find significant correlations between the fold change and gRNA specificity score (Figures S3B–S3D), suggesting minimal off-target effects of these selected gRNAs. When comparing end and baseline libraries, we found significant gRNA depletion in BPH1, DU145, and PC3 screening experiments (Figures 1C–1E). When comparing relative gRNA changes between different categories of candidate SNPs, we found significantly higher growth depletion only in BPH1 cells with gRNA targeting the PIP and TARGET SNPs (Figures S3E–S3G). We also found that a significantly higher proportion of the SNPs selected as screening hits resided in transcription start sites of human genes (Figure 1F) and tended to be allelically bound to transcriptional-factor binding according to ANANASTRA annotation (Figure 1G).38 We plotted the gRNA representation before and after the growth selection and highlighted representative SNPs in each cell line (Figures 1H–1J). The raw and normalized gRNA count and the eQTL mapped with the candidate SNP are listed in Table S2. The RIGER analysis output is listed in Table S3.
rs60464856 displays a regulatory role in RUVBL1 expression underpinning susceptibility for prostate cancer
Among the 117 SNPs showing significant growth inhibition over a 3-week screening, the SNP rs60464856 was consistently selected in all tested prostate cell lines. The SNP sequence was targeted by tengRNAs for the A and six gRNAs for the G allele. We observed significant A allele depletion (fold change ≤0.75) in ten gRNAs in BPH1, five gRNAs in DU145, and ten gRNAs in PC3 cells (Figure 2A). rs60464856 is located in a previously identified risk locus, and the G allele was associated with a 10% increased risk of prostate cancer in 107,247 cases and 127,006 controls (p value = 1.2 × 10−16) (Figure 2B).39 Consistently, a phenome-wide association analysis (PheWAS) in the FinnGen cohort (n = 342,499) with 2,202 endpoints revealed the strongest association of rs60464856 with malignant neoplasm of prostate (11,590 cases and 110,189 controls; p value = 3.2 × 10−7) (Figure S4A).40 We further showed that among the RUVBL1 eQTL loci, rs60464856 resided in a linkage disequilibrium block with the second-best significance (Figure 2C). Additionally, the rs60464856 G allele was significantly associated with elevated RUVBL1 expression in GTEx (https://gtexportal.org/home/),19 Mayo,27,41,42 and TCGA prostate eQTL43 datasets (Figure 2D). Interestingly, we found the most abundant isoform of RUVBL1 was associated with the rs60464856 G allele in the TCGA PCa splicing QTL44,45 and Mayo cohorts27,41,42 (Figures S4B–S4D). We also used three gRNAs targeting the rs60464856 locus to transiently transfect the dCas9 stable cells and observed a consistent knockdown effect on RUVBL1 expressions in all three prostate cell lines (Figure 2E). To evaluate the nuances of endogenous allele transition contributing to downstream gene expression, we applied nickase Cas9 (xCas9) base editing technology, which features high conversion efficiency and a minimal indel rate, to precisely substitute the rs60464856 allele in prostate cells (Figure 2F). Finally, we generated multiple isogenic subclones with accurate base conversion and confirmed significant increases of endogenous RUVBL1 expression by the G allele in BPH1 and DU145, but not in PC3 cells (Figure 2G).
Figure 2.

The rs60464856-RUVBL1 locus shows a regulatory role underpinning susceptibility to prostate cancer
(A) Screening fold change of rs60464856 gRNA in BPH1, DU145, and PC3 cells. The Gray bar indicates the rs60464856 locus in the germline, and colored strips above or below the bar indicate gRNA binding Crick or Watson strands of the germline DNA. Numbers in the center show the average fold change for two screening replicates.
(B) Locus plots for rs60464856 locus PCa GWAS significance in the PRACTICAL cohort.
(C) Locus plots for RUVBL1 eQTL significance in the Mayo prostate dataset.
(D) eQTL associations between RUVBL1 expression and rs60464856 genotypes in GTEx, Mayo, and TCGA prostate cohorts.
(E) RUVBL1 expression in dCas9-KRAB stable prostate cells transfected with non-targeting control guide (NCG) and rs60464856 targeting gRNAs. The individual points indicate different gRNA clones. The inhibition fold changes were calculated from three biological replicates. The bar represents standard error.
(F) Schematic figure showing xCas9-based A-to-G base editing in prostate cell lines.
(G) RUVBL1 expression in rs60464856 base-edited clones in BPH1, DU145, and PC3 cells. The bar represents the mean and standard error.
rs60464856 binds cohesin subunits allelically in a manner mediated by chromatin interactions
To evaluate potential allelic protein binding at rs60464856, we searched for ChIP-seq data collections, such as Cistrome Data Browser (http://cistrome.org/db/#/)46 and ChIP-Atlas (https://chip-atlas.org/),16 for potential transcription factor bindings. However, we did not find highly convincing evidence showing allelic TF binding on this locus. Since multiple datasets report histone modification signals on the rs60464856 locus, we then performed ChIP-qPCR to evaluate the locus-specific enrichment for active histone modification markers (H3K4me1, H3K4me3, and H3K27ac) in the base-edited subclones. We found distinct histone modification status in PC3 clones, especially for H3K27ac on the rs60464856 locus (Figure 3A). Consistently, we found the enrichment of H3K4me1, H3K4me3, and H3K27ac modifications on rs60464856 in multiple cell lines (Figure 3B), suggesting a robust regulatory potential of this locus in the prostate. Intriguingly, in an rs60464856 heterozygous cell line, RWPE1, we identified higher H3K4me3 and H3K27ac coverage for G than A allele, suggesting stronger transcription activity driven by the risk allele. In prostate cell lines and TCGA prostate cancer tissue47 (Figure S5), we also identified consistent open chromatin signals near the rs60464856 locus, further supporting its regulatory potential on RUVBL1. With a recent HiC dataset,31 we also visualized the long-range interactions surrounding the rs60464856 locus. By summing the normalized count from the interaction hot spot on both sides of the rs60464856 locus, we calculated the left-to-right ratio (L/R) in each cell line (Figures S6A–S6E). This analysis showed that PC3 cells had only 60% interaction compared to DU145 cells (Figure 3C). In contrast, the interactions L/R ratios were roughly equal in VCaP and LNCaP cells (Figure 3D). To confirm the transcription activity of the rs60464856 locus, we tested its flanking sequence (chr3: 128,123,257-128,123,479, negative strand) using a reporter assay and found higher promoter activity of the G allele than the A allele in these cell lines (Figure 3E).
Figure 3.

Involvement of the rs60464856 locus with the RUVBL1 enhancer, long-range interaction, and allelic binding to cohesin subunits
(A) H3K4me1, H3K4me3, and H3K27ac modifications on rs60464856 locus in BPH1, DU145, and PC3 clones.
(B) ChIP-seq signals of H3K4me1, H3K4me3, and H3K27ac modifications on the rs60464856 locus in prostate cells.
(C and D) HiC interaction heatmap in PC3 (C top), DU145 (C bottom), VCaP (D top), and LNCaP (D bottom) cells. Each L and R indicate interaction ICED normalized read count on the left and right sides of the rs60464856 bin. The red and green labeled boxes in the contact matrix represent the interaction hotspot on the left and right side of the rs60464856 locus, respectively. The red and green arrows in the gene-model illustration highlight the interactions between the left and right region, respectively, of rs60464856 and a distant region downstream of RPN1. The dotted red arrow demonstrates weakened left interactions in PC3 compared with DU145 cells.
(E) Luciferase reporter assay comparing rs60464856 allele transcription activities in BPH1, DU145, and PC3 cells. The bar represents the standard error across multiple technical replicates, and the p values represent simple t test results.
(F) SILAC-based DNA pull-down proteomics pipeline for identifying allelic protein binding on the rs60464856 locus.
(G) Allelic proteomics with BPH1 SILAC extracts.
(H–K) ChIP-qPCR measuring rs60464856 locus enrichment and allele-specific binding to SMC1A, SMC3, CTCF, and CTCFL proteins in BPH1 (H and I) and PC3 (J and K) base-edited populations. Each dot represents experimental data from an independent immunoprecipitation biological replicate. The bar represents the standard error, and the p value significance (∗∗p value <0.001; ∗p value <0.05; N.S. = not significant) represents the Mann Whitney test between the ChIPed sample and the input.
We then applied SILAC-based proteomics to detect possible transcription factors or DNA-binding proteins. This assay took advantage of isotype-labeled nuclear extract in DNA pull-down reactions, thus converting the protein binding difference into the ratio of different molecular weights (Figure 3F). This proteomics analysis identified increased cohesin subunits bound to the rs60464856 A rather than the G allele within the BPH1 nuclear extract (Figure 3G). To further elucidate whether cohesin could bind endogenous rs60464856 locus allelically, we used ChIP assays specific to the rs60464856 locus from BPH1 and PC3 base-edited populations. In BPH1 cells, we found significant locus enrichment (Figure 3H) for SMC1A, SMC3, and CTCF, and the A allele preference for SMC3 and CTCF (Figure 3I). In PC3 cells, we only found minor locus enrichment for SMC3 and CTCF (Figure 3J), and no allele preference was observed for all tested antibodies (Figure 3K). We also explored the unique allelic binding role of SMC3, using existing ChIP-seq datasets48 (GSE49402 and GSE36578) that quantified SMC1A and SMC3 binding in the human genome (Figure S7A). We performed STREME motif scan with the private peak region of SMC1A and SMC3, and found that only SMC3 private peaks included an outstanding significant motif in both cell lines (Figure S7B). Through STREME -TOMTOM comparison, we also found that this motif was highly similar to the CTCF binding site (MA0139.1) (Figure S7C). More importantly, the rs60464856 A allele is located in the CTCF zinc finger seven interaction domain and is consistently preferred by multiple versions of the CTCF motif.49 This result suggests a potential mechanism about how the rs60464856 protective allele mediates cohesin-CTCF complex formation and supports our observation in the HiC datasets (Figures 3C and 3D).
RUVBL1 knockdown inhibits prostate-cell proliferation by downregulating cell-cycle-related pathways
To evaluate the oncogenic role of RUVBL1, we examined the perturbation effect of RUVBL1 by CRISPR or RNAi screening in the DepMap portal (https://depmap.org/portal/). We found that RUVBL1 was a common essential gene in most human cell lines and had a stronger dependency score than the median of all essential genes for the prostate cell lines used in our screening, including BPH1, DU145, and PC3 cells (Figure 4A). To further characterize the function of RUVBL1, we generated stable cell lines infected with small hairpin RNA (shRNA) lentiviral particles and verified that RUVBL1 had been successfully knocked down at the mRNA and protein expression (Figures S8 and 4B). We further monitored the growth of the stable cell lines with daily Incucyte scans and found that RUVBL1 knockdown by both shRNAs significantly suppressed proliferation in BPH1 (Figure 4C), DU145 (Figure 4D), and PC3 (Figure 4E) cell lines. We also observed drastic reductions in colony formation for the RUVBL1 knockdown group in BPH1, DU145, and PC3 cells (Figure 4F). To characterize the transcriptome alteration caused by RUVBL1 downregulation, we quantified RNA profiling of BPH1 and PC3 cells and calculated the GSVA score for the HALLMARK gene set collection. Interestingly, multiple cell-cycle-related pathways, including MYC targets, E2F targets, and G2M checkpoint genes, showed significant enrichment with RUVBL1 expression (Figures 4G–4H). We further visualized the changes in gene expression in these significantly enriched pathways and found consistent trends with RUVBL1 expression (Figure 4I). To further explore the RUVBL1-related transcriptomic alterations induced by the rs60464856 risk allele, we performed mRNA-seq in ten BPH-1 clones, including five with AAA and five with AGG genotypes. The RNA-seq result shows 1.35-fold higher RUVBL1 expression in AGG clones than in AAA clones; PPDE (posterior probabilities of being differentially expressed) is equal to 0.99999995 (Figure S9A). We also performed GSEA analysis with the normalized RNA-seq data in the HALLMARK gene set collection and found pathway changes similar to those in RUVBL1 knockdown experiments and TCGA prostate-cancer profiling (Figure S9B). To characterize the tumorigenic effect of RUVBL1 in vivo, we performed xenograft mouse experiments with the PC3 stable cell lines. We found that the RUVBL1 knockdown significantly inhibited tumor growth (Figure 4J) and reduced endpoint tumor weight in the mice model (Figure 4K).
Figure 4.

Cell-cycle arrest mediated by RUVBL1 knockdown significantly inhibits prostate cell proliferation
(A) Dependency score of RUVBL1 for CRISPR and RNAi screening from CCLE human cell line collections. A score of 0 indicates that a gene is not essential, whereas a score of −1 is comparable to the median of all pan-essential genes.
(B) Measuring RUVBL1 protein expression after shRNA knockdown with lentiviral infection (low MOI). The numbers below each lane indicate RUVBL1 protein intensity normalized to that of the ACTB internal control.
(C–E) Cell proliferation curve comparing cell-growth differences after RUVBL1 knockdown in BPH1 (C), DU145 (D), and PC3 (E) cell lines. Each bar represents experimental data from six independent technical replicates. The experiment has been repeated twice starting from lentiviral infection.
(F) Colony-formation assay comparing clonogenic potential alterations after RUVBL1 knockdown in BPH1, DU145, and PC3 cell lines. Each well represents experimental data from four technical replicates. The experiment has been repeated twice starting from lentiviral infection.
(G) GSVA on RNA-seq data in BPH1 and PC3 cells after RUVBL1 knockdown on HALLMARK collection pathways.
(H) GSEA summary for the most significant RUVBL1-driven gene sets in BPH1 and PC3 cells.
(I) Representative heatmap showing significantly enriched HALLMARK pathways, including MYC target, E2F target, and G2M checkpoint. The RNA-seq was performed in two technical replicates within each group.
(J and K) Tumor volume changes (J) and tumor weight (K) comparison in xenograft experiments with the PC3 cell line. Each xenograft subgroup included six biological replicates.
RUVBL1 expression increases aggressiveness and predicts a worse prognosis in prostate cancer
To characterize the malignant potential associated with RUVBL1 in clinical samples, we retrieved three indices of genome instabilities, including the altered fraction of the genome, mutation count, and aneuploidy score for 488 TCGA prostate cancer. We found that RUVBL1 expression was positively associated with these indices (Figures 5A–5C). Additionally, prostate cancer with higher RUVBL1 expression tended to have a more advanced T stage and Gleason score (Figure 5D). We also calculated the GSVA score for the HALLMARK gene set collection with the TCGA prostate-cancer RNA profiling and found that the cell-cycle-related pathways showed up consistently in the eight most significantly enriched gene sets (Figure 5E). We further demonstrated the enrichment score (Figures 5F–5G) and FDR q value for these significantly enriched pathways and found consistent positive enrichment with RUVBL1 expression. We performed a Kaplan-Meier analysis to see whether RUVBL1 could serve as a prognostic marker and found significantly worse progression-free (Figure 5H) survival in individuals with higher RUVBL1 expression. To determine whether the RUVBL1-enriched genes could serve a prognostic role, we also used k-means clustering methods with the leading-edge gene to separate the prostate-cancer patients into groups with different risks (Figure 5I). The Kaplan-Meier analysis showed that individuals with increased risk tended to have significantly worse progression-free survival (Figures 5J–5L). Additionally, RUVBL1 expression was consistently upregulated in PCa primary and metastasis tumor tissue (Figures S10A–S10C).50,51,52 We also demonstrated a positive association between RUVBL1 expression and elevated prediagnostic PSA level,52 higher Gleason score,53 and worse biochemical recurrence-free survival54 in existing cohorts (Figures S10D–S10F).
Figure 5.

Association between RUVBL1 expression and aggressiveness in prostate cancer
(A–C) Associations in TCGA prostate cancer cohort between RUVBL1 expression and genome instability metrics, including the altered fraction of genome (A), the mutation count (B), and the aneuploidy score (C).
(D) RUVBL1 expression in TCGA prostate tissue with different T stages and Gleason scores. The p value tested the linear trends between the RUVBL1 expression with the incremental stages or scores. The dashed lines indicate the quartiles and the medians of each group.
(E) GSVA on RNA-seq data in TCGA prostate cancer tissue by ranking RUVBL1 expression on HALLMARK collection pathways.
(F) GSEA enrichment plots showing significantly enriched HALLMARK pathways in TCGA prostate cohort.
(G) Normalized enrichment score and FDR q value for significantly enriched HALLMARK pathways in the TCGA prostate cohort.
(H) Kaplan-Meier analysis of progression-free survival in TCGA prostate cancer patients stratified by RUVBL1 gene expression.
(I) Heatmap with RUVBL1 leading-edge genes; the side bar shows k-means-stratified groups with different risks related to significantly enriched HALLMARK pathways, including MYC target V1, E2F targets, and G2M checkpoint, in the TCGA prostate cohort.
(J–L) Kaplan-Meier analysis on progression-free survival in TCGA prostate cancer patients stratified by the GSEA leading-edge gene enriched with RUVBL1 in each pathway, including MYC targets V1 (J), E2F targets (K), and G2M checkpoints (L).
Discussion
Over the past decade, GWASs and eQTL analyses have been highly productive in finding PCa risk loci and susceptibilty genes.55,56,57 Despite contributing to a better understanding of the biological significance of risk predisposition, these analyses did not directly demonstrate the regulatory role of individual loci and the functional consequence of each causal gene. To better delineate the functionality of these genetic findings, a large-scale functional evaluation of target risk loci is highly warranted. Because of the non-coding nature of most risk loci, these variants are believed to play a regulatory role. Therefore, we applied the dCas9-based CRISPRi assay to target SNP sequences at risk loci and aimed to mimick the regulatory alteration caused by single base substitutions. This genome-wide screening at PCa risk loci revealed 117 SNPs showing a regulatory role in cell proliferation. Interestingly, these proliferation-related SNPs are enriched in gene transcription start sites, suggesting that the majority of the phenotypic changes are related to transcription alterations caused by dCas9 interference.
This study characterized the regulatory role of a risk SNP, rs60464856. We observed consistent growth inhibition by multiple gRNAs targeting this locus. More importantly, with multiple gRNA targeting both alleles at the same genomic locations, we observed interesting phenotypical changes related to seed region mismatches in multiple prostate cell lines. As demonstrated by previous studies,58,59 any mismatches in the 7-bp seed region of the gRNA could cause a rapid rejection of these targets by the dCas9 protein. These facts might explain the A-allele-specific depletion effect of gRNA on positions −5 to −7, as shown in our data (see Figure 2A). When the mismatch is located outside the seed region, the gRNA with SNP at the −8 position showed depletion effects for both alleles. To further validate the regulatory role of rs60464856, we created multiple subclones carrying converted G alleles. As expected, the G-allele-carrying subclones showed elevated RUVBL1 expression in BPH1 and DU145 cells. However, this elevated expression was not observed in PC3 subclones, possibly as a result of the distinct status of histone modification and chromatin interaction in PC3 cells.
This study also showed allele-specific binding of SMC3, which is different from SMC1A, at the rs60464856 locus. As the major subunits of human cohesin, both SMC1A and SMC3 mediate multiple biological processes, including DNA looping, chromosome condensation, and chromosome segregation, by forming heterodimers.60,61 Intriguingly, there have been several unique observations about the distinct phenotypic changes brought about by SMC1A or SMC3 knockdown. Magdalena et al.62 identified that SMC3 knockdown rendered SMC1A unstable and led to less cytoplasmic accumulation, whereas SMC1A knockdown did not influence SMC3 stability or cytoplasmic accumulation. A recent study63 reported that SMC1A and SMC3 ATPase active sites had differential effects on cohesin ATPase function and that SMC3 has a unique function in DNA tethering. Our database mining showed significant signal overlap between SMC1A and SMC3 binding sites in the genome, but only private peaks from SMC3 enriched CTCF motifs in the two cell lines studied. As an insulator that can block enhancers to regulate target genes, CTCF was first discovered as a transcriptional repressor and believed to execute a hub role in controlling gene expression.49,64 In our findings, CTCF might also play a crucial role in governing RUVBL1 gene regulation, potentially through insulating the rs60464856 loci allelically.
RUVBL1, also known as RuvB-like AAA ATPase 1 or TIP49, possesses an ATP-dependent DNA helicase activity and has been reported to regulate a wide range of cellular processes,65 including chromatin decondensation,66,67 misfolded protein aggregation,68 and transcription regulation.69 In addition to the previously reported mTORC1 pathway,70 our enrichment analysis demonstrated that RUVBL1 expression was consistently correlated with cell-cycle regulation and MYC signaling activities in both cell lines and tumor tissues. This result demonstrates a potential use of RUVBL1-selective inhibitors in treating prostate cancer.71,72 The result also suggests using the rs60464856 genotype to stratify a target population for future clinical trials.
One potential limitation of this study is inconsistent results in some tests among different cell lines. Although these inconsistencies might attribute to genetic heterogeneity, we also want to highlight that some hits found exclusively in DU145 cells are reported to be functional in prostate cells—for instance, the established functional variants residing in the binding sites of the transcription factors TMPRSS2-ERG and HNF1B.73 Additionally, to increase our gRNA library coverage, we can use novel CRISPR systems with expanded PAM site compatibility. With stringent library preparation and screening processes, the biological implications for the functional variants discovered exclusively in only a singular cell line are still worth investigating, which might uncover unique SNP and gene functions specific to the subline of interest. In summary, we applied CRISPRi screening technology to screen for survival-essential SNPs at the genome scale. We identified more than a hundred functional SNPs that regulate cell proliferation. We further characterized the rs60464856 risk variant for its regulatory role in the prostate context and target gene RUVBL1 for its functional role in prostate-cancer cell proliferation and disease progression. This result will enrich our knowledge of PCa predisposition and provide insight into the cancer risk classification and potential therapeutic targets for personalized treatment.
Acknowledgments
We thank Flow Cytometry; Molecular Genomic; Proteomics and Metabolomics; and Analytic Microscopy core facilities at the Moffitt Cancer Center, an NCI-designated Comprehensive Cancer Center (P30CA076292). The authors also acknowledge the participants and investigators of the FinnGen study (www.finngen.fi). This work was supported by the National Institutes of Health (R01CA250018 and R01CA212097 to L. Wang, R01CA263494 to L. Wu) and National Natural Science Foundation of China (82073082) as well as the Jane and Aatos Erkko Foundation, the Finnish Cancer Foundation, and the Sigrid Juséliuksen Säätiö to G.-H.W. This study was also supported by the Medical Science Data Center of Fudan University. We thank Dr. Stephen N. Thibodeau at Mayo Clinic for providing the eQTL candidate list for designing this screening. We also thank the PRACTICAL consortium for providing GWAS resources supporting this project. The full funding information and the author affiliation for the consortium are listed in the supplemental information.
Author contributions
Y.T. and L.W. contributed to study design and performed CRISPRi and proteomics screening, statistical analysis and functional validation. D.D., Z.W. and G.-H.W. performed functional analysis and mouse work. L.Wu. and J.P. contributed to collecting GWAS summary statistics, G.-H.W. and L.W. supervised the study and contributed to study design and data interpretation. Y.T., D.D., G.-H.W., and L.W. co-wrote the manuscript. All authors read, commented on, and approved the final version.
Declaration of interests
The authors declare no competing interests.
Published: August 3, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.07.003.
Contributor Information
Gong-Hong Wei, Email: gonghong_wei@fudan.edu.cn.
Liang Wang, Email: liang.wang@moffitt.org.
Web resources
ChIP-Atlas, https://chip-atlas.org/
count_spacer.py, https://github.com/fengzhanglab/Screening_Protocols_manuscript
cworld-dekker, https://github.com/dekkerlab/cworld-dekker
Depmap, https://depmap.org/portal/
FinnGen PheWAS, https://r8.finngen.fi/
NCI GDC Data Portal, https://portal.gdc.cancer.gov/
Sequence Read Archive (SRA), https://www.ncbi.nlm.nih.gov/sra/
TCGA chromatin accessibility landscape, https://gdc.cancer.gov/about-data/publications/ATACseq-AWG
TCGA Pan-Cancer Splicing Quantitative Trait Loci, http://www.cancersplicingqtl-hust.com/#/
TCGA PancanQTL, http://bioinfo.life.hust.edu.cn/PancanQTL/
TCGA SpliceSeq, https://bioinformatics.mdanderson.org/TCGASpliceSeq/
Uniprot, https://www.uniprot.org/
Supplemental information
Data and code availability
The accession number for the results reported in this paper is GEO: GSE224654, which includes CRISPRi screening gRNA readout (GEO: GSE224653) and RNA-seq expression for the RUVBL1 knockdown experiment (GEO: GSE224646). The gRNA sequence design, raw and normalized count, and the eQTL mapped with the candidate SNP are listed in Table S2. The RIGER analysis output is listed in Table S3. The publicly available datasets used are listed in Table S4. The SILAC proteomics sequencing result is listed in Table S5. Detailed information about the rs60464856 base editing can be accessed in the github repository (https://github.com/Yijun-Tian/Base_Editing-rs60464856).
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer Statistics, 2021. CA. Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Schaid D.J., Sinnwell J.P., Batzler A., McDonnell S.K. Polygenic risk for prostate cancer: Decreasing relative risk with age but little impact on absolute risk. Am. J. Hum. Genet. 2022;109:900–908. doi: 10.1016/j.ajhg.2022.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E., et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47:D1005–D1012. doi: 10.1093/nar/gky1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stadler Z.K., Thom P., Robson M.E., Weitzel J.N., Kauff N.D., Hurley K.E., Devlin V., Gold B., Klein R.J., Offit K. Genome-wide association studies of cancer. J. Clin. Oncol. 2010;28:4255–4267. doi: 10.1200/JCO.2009.25.7816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farashi S., Kryza T., Clements J., Batra J. Post-GWAS in prostate cancer: from genetic association to biological contribution. Nat. Rev. Cancer. 2019;19:46–59. doi: 10.1038/s41568-018-0087-3. [DOI] [PubMed] [Google Scholar]
- 6.Gallagher M.D., Chen-Plotkin A.S. The Post-GWAS Era: From Association to Function. Am. J. Hum. Genet. 2018;102:717–730. doi: 10.1016/j.ajhg.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edwards S.L., Beesley J., French J.D., Dunning A.M. Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 2013;93:779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tian P., Zhong M., Wei G.H. Mechanistic insights into genetic susceptibility to prostate cancer. Cancer Lett. 2021;522:155–163. doi: 10.1016/j.canlet.2021.09.025. [DOI] [PubMed] [Google Scholar]
- 9.Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M., et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trynka G., Sandor C., Han B., Xu H., Stranger B.E., Liu X.S., Raychaudhuri S. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 2013;45:124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shetty A., Seo J.H., Bell C.A., O'Connor E.P., Pomerantz M.M., Freedman M.L., Gusev A. Allele-specific epigenetic activity in prostate cancer and normal prostate tissue implicates prostate cancer risk mechanisms. Am. J. Hum. Genet. 2021;108:2071–2085. doi: 10.1016/j.ajhg.2021.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu Q., Ge D., Heinzen E.L., Dickson S.P., Urban T.J., Zhu M., Maia J.M., He M., Zhao Q., Shianna K.V., Goldstein D.B. Prioritizing genetic variants for causality on the basis of preferential linkage disequilibrium. Am. J. Hum. Genet. 2012;91:422–434. doi: 10.1016/j.ajhg.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fritsche L.G., Patil S., Beesley L.J., VandeHaar P., Salvatore M., Ma Y., Peng R.B., Taliun D., Zhou X., Mukherjee B. Cancer PRSweb: An Online Repository with Polygenic Risk Scores for Major Cancer Traits and Their Evaluation in Two Independent Biobanks. Am. J. Hum. Genet. 2020;107:815–836. doi: 10.1016/j.ajhg.2020.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kichaev G., Bhatia G., Loh P.R., Gazal S., Burch K., Freund M.K., Schoech A., Pasaniuc B., Price A.L. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am. J. Hum. Genet. 2019;104:65–75. doi: 10.1016/j.ajhg.2018.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oki S., Ohta T., Shioi G., Hatanaka H., Ogasawara O., Okuda Y., Kawaji H., Nakaki R., Sese J., Meno C. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018;19 doi: 10.15252/embr.201846255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis C.A., Hitz B.C., Sloan C.A., Chan E.T., Davidson J.M., Gabdank I., Hilton J.A., Jain K., Baymuradov U.K., Narayanan A.K., et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46:D794–D801. doi: 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranašić D., et al. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020;48:D87–D92. doi: 10.1093/nar/gkz1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.GTEx Consortium The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gurumurthy C.B., Grati M., Ohtsuka M., Schilit S.L.P., Quadros R.M., Liu X.Z. CRISPR: a versatile tool for both forward and reverse genetics research. Hum. Genet. 2016;135:971–976. doi: 10.1007/s00439-016-1704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang L., Chan A.K.N., Miyashita K., Delaney C.D., Wang X., Li H., Pokharel S.P., Li S., Li M., Xu X., et al. High-resolution characterization of gene function using single-cell CRISPR tiling screen. Nat. Commun. 2021;12:4063. doi: 10.1038/s41467-021-24324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan F., Li J., Milosevic J., Petroni R., Liu S., Shi Z., Yuan S., Reynaga J.M., Qi Y., Rico J., et al. KAT6A and ENL Form an Epigenetic Transcriptional Control Module to Drive Critical Leukemogenic Gene-Expression Programs. Cancer Discov. 2022;12:792–811. doi: 10.1158/2159-8290.CD-20-1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gasperini M., Findlay G.M., McKenna A., Milbank J.H., Lee C., Zhang M.D., Cusanovich D.A., Shendure J. CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions. Am. J. Hum. Genet. 2017;101:192–205. doi: 10.1016/j.ajhg.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wojtal D., Kemaladewi D.U., Malam Z., Abdullah S., Wong T.W.Y., Hyatt E., Baghestani Z., Pereira S., Stavropoulos J., Mouly V., et al. Spell Checking Nature: Versatility of CRISPR/Cas9 for Developing Treatments for Inherited Disorders. Am. J. Hum. Genet. 2016;98:90–101. doi: 10.1016/j.ajhg.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H., Guimaraes C., Panning B., Ploegh H.L., Bassik M.C., et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A., et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thibodeau S.N., French A.J., McDonnell S.K., Cheville J., Middha S., Tillmans L., Riska S., Baheti S., Larson M.C., Fogarty Z., et al. Identification of candidate genes for prostate cancer-risk SNPs utilizing a normal prostate tissue eQTL data set. Nat. Commun. 2015;6:8653. doi: 10.1038/ncomms9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo B., Cheung H.W., Subramanian A., Sharifnia T., Okamoto M., Yang X., Hinkle G., Boehm J.S., Beroukhim R., Weir B.A., et al. Highly parallel identification of essential genes in cancer cells. Proc. Natl. Acad. Sci. USA. 2008;105:20380–20385. doi: 10.1073/pnas.0810485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z., Liu D.R. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian Y., Liu Q., Yu S., Chu Q., Chen Y., Wu K., Wang L. NRF2-Driven KEAP1 Transcription in Human Lung Cancer. Mol. Cancer Res. 2020;18:1465–1476. doi: 10.1158/1541-7786.MCR-20-0108. [DOI] [PubMed] [Google Scholar]
- 31.San Martin R., Das P., Dos Reis Marques R., Xu Y., Roberts J.M., Sanders J.T., Golloshi R., McCord R.P. Chromosome compartmentalization alterations in prostate cancer cell lines model disease progression. J. Cell Biol. 2022;221 doi: 10.1083/jcb.202104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butter F., Davison L., Viturawong T., Scheibe M., Vermeulen M., Todd J.A., Mann M. Proteome-wide analysis of disease-associated SNPs that show allele-specific transcription factor binding. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coudert E., Gehant S., de Castro E., Pozzato M., Baratin D., Neto T., Sigrist C.J.A., Redaschi N., Bridge A., UniProt Consortium Annotation of biologically relevant ligands in UniProtKB using ChEBI. Bioinformatics. 2023;39 doi: 10.1093/bioinformatics/btac793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lafontaine D.L., Yang L., Dekker J., Gibcus J.H. Hi-C 3.0: Improved Protocol for Genome-Wide Chromosome Conformation Capture. Curr. Protoc. 2021;1:e198. doi: 10.1002/cpz1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akgol Oksuz B., Yang L., Abraham S., Venev S.V., Krietenstein N., Parsi K.M., Ozadam H., Oomen M.E., Nand A., Mao H., et al. Systematic evaluation of chromosome conformation capture assays. Nat. Methods. 2021;18:1046–1055. doi: 10.1038/s41592-021-01248-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boytsov A., Abramov S., Aiusheeva A.Z., Kasianova A.M., Baulin E., Kuznetsov I.A., Aulchenko Y.S., Kolmykov S., Yevshin I., Kolpakov F., et al. ANANASTRA: annotation and enrichment analysis of allele-specific transcription factor binding at SNPs. Nucleic Acids Res. 2022;50:W51–W56. doi: 10.1093/nar/gkac262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schumacher F.R., Al Olama A.A., Berndt S.I., Benlloch S., Ahmed M., Saunders E.J., Dadaev T., Leongamornlert D., Anokian E., Cieza-Borrella C., et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018;50:928–936. doi: 10.1038/s41588-018-0142-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurki M.I., Karjalainen J., Palta P., Sipilä T.P., Kristiansson K., Donner K.M., Reeve M.P., Laivuori H., Aavikko M., Kaunisto M.A., et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature. 2023;613:508–518. doi: 10.1038/s41586-022-05473-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tian Y., Soupir A., Liu Q., Wu L., Huang C.C., Park J.Y., Wang L. Novel role of prostate cancer risk variant rs7247241 on PPP1R14A isoform transition through allelic TF binding and CpG methylation. Hum. Mol. Genet. 2022;31:1610–1621. doi: 10.1093/hmg/ddab347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larson N.B., McDonnell S., French A.J., Fogarty Z., Cheville J., Middha S., Riska S., Baheti S., Nair A.A., Wang L., et al. Comprehensively evaluating cis-regulatory variation in the human prostate transcriptome by using gene-level allele-specific expression. Am. J. Hum. Genet. 2015;96:869–882. doi: 10.1016/j.ajhg.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gong J., Mei S., Liu C., Xiang Y., Ye Y., Zhang Z., Feng J., Liu R., Diao L., Guo A.Y., et al. PancanQTL: systematic identification of cis-eQTLs and trans-eQTLs in 33 cancer types. Nucleic Acids Res. 2018;46:D971–D976. doi: 10.1093/nar/gkx861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tian J., Wang Z., Mei S., Yang N., Yang Y., Ke J., Zhu Y., Gong Y., Zou D., Peng X., et al. CancerSplicingQTL: a database for genome-wide identification of splicing QTLs in human cancer. Nucleic Acids Res. 2019;47:D909–D916. doi: 10.1093/nar/gky954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryan M., Wong W.C., Brown R., Akbani R., Su X., Broom B., Melott J., Weinstein J. TCGASpliceSeq a compendium of alternative mRNA splicing in cancer. Nucleic Acids Res. 2016;44:D1018–D1022. doi: 10.1093/nar/gkv1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng R., Wan C., Mei S., Qin Q., Wu Q., Sun H., Chen C.H., Brown M., Zhang X., Meyer C.A., Liu X.S. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019;47:D729–D735. doi: 10.1093/nar/gky1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corces M.R., Granja J.M., Shams S., Louie B.H., Seoane J.A., Zhou W., Silva T.C., Groeneveld C., Wong C.K., Cho S.W., et al. The chromatin accessibility landscape of primary human cancers. Science. 2018;362 doi: 10.1126/science.aav1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan J., Enge M., Whitington T., Dave K., Liu J., Sur I., Schmierer B., Jolma A., Kivioja T., Taipale M., Taipale J. Transcription factor binding in human cells occurs in dense clusters formed around cohesin anchor sites. Cell. 2013;154:801–813. doi: 10.1016/j.cell.2013.07.034. [DOI] [PubMed] [Google Scholar]
- 49.Yin M., Wang J., Wang M., Li X., Zhang M., Wu Q., Wang Y. Molecular mechanism of directional CTCF recognition of a diverse range of genomic sites. Cell Res. 2017;27:1365–1377. doi: 10.1038/cr.2017.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu Y.P., Landsittel D., Jing L., Nelson J., Ren B., Liu L., McDonald C., Thomas R., Dhir R., Finkelstein S., et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J. Clin. Oncol. 2004;22:2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 51.Varambally S., Yu J., Laxman B., Rhodes D.R., Mehra R., Tomlins S.A., Shah R.B., Chandran U., Monzon F.A., Becich M.J., et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8:393–406. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 52.Taylor B.S., Schultz N., Hieronymus H., Gopalan A., Xiao Y., Carver B.S., Arora V.K., Kaushik P., Cerami E., Reva B., et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Setlur S.R., Mertz K.D., Hoshida Y., Demichelis F., Lupien M., Perner S., Sboner A., Pawitan Y., Andrén O., Johnson L.A., et al. Estrogen-dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J. Natl. Cancer Inst. 2008;100:815–825. doi: 10.1093/jnci/djn150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ross-Adams H., Lamb A.D., Dunning M.J., Halim S., Lindberg J., Massie C.M., Egevad L.A., Russell R., Ramos-Montoya A., Vowler S.L., et al. Integration of copy number and transcriptomics provides risk stratification in prostate cancer: A discovery and validation cohort study. EBioMedicine. 2015;2:1133–1144. doi: 10.1016/j.ebiom.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eeles R.A., Durocher F., Edwards S., Teare D., Badzioch M., Hamoudi R., Gill S., Biggs P., Dearnaley D., Ardern-Jones A., et al. Linkage analysis of chromosome 1q markers in 136 prostate cancer families. The Cancer Research Campaign/British Prostate Group U.K. Familial Prostate Cancer Study Collaborators. Am. J. Hum. Genet. 1998;62:653–658. doi: 10.1086/301745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu J. Combined analysis of hereditary prostate cancer linkage to 1q24-25: results from 772 hereditary prostate cancer families from the International Consortium for Prostate Cancer Genetics. Am. J. Hum. Genet. 2000;66:945–957. doi: 10.1086/302807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schaid D.J., McDonnell S.K., Blute M.L., Thibodeau S.N. Evidence for autosomal dominant inheritance of prostate cancer. Am. J. Hum. Genet. 1998;62:1425–1438. doi: 10.1086/301862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boyle E.A., Andreasson J.O.L., Chircus L.M., Sternberg S.H., Wu M.J., Guegler C.K., Doudna J.A., Greenleaf W.J. High-throughput biochemical profiling reveals sequence determinants of dCas9 off-target binding and unbinding. Proc. Natl. Acad. Sci. USA. 2017;114:5461–5466. doi: 10.1073/pnas.1700557114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zheng T., Hou Y., Zhang P., Zhang Z., Xu Y., Zhang L., Niu L., Yang Y., Liang D., Yi F., et al. Profiling single-guide RNA specificity reveals a mismatch sensitive core sequence. Sci. Rep. 2017;7 doi: 10.1038/srep40638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun M., Nishino T., Marko J.F. The SMC1-SMC3 cohesin heterodimer structures DNA through supercoiling-dependent loop formation. Nucleic Acids Res. 2013;41:6149–6160. doi: 10.1093/nar/gkt303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Deardorff M.A., Kaur M., Yaeger D., Rampuria A., Korolev S., Pie J., Gil-Rodríguez C., Arnedo M., Loeys B., Kline A.D., et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 2007;80:485–494. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laugsch M., Seebach J., Schnittler H., Jessberger R. Imbalance of SMC1 and SMC3 cohesins causes specific and distinct effects. PLoS One. 2013;8 doi: 10.1371/journal.pone.0065149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Çamdere G., Guacci V., Stricklin J., Koshland D. The ATPases of cohesin interface with regulators to modulate cohesin-mediated DNA tethering. Elife. 2015;4 doi: 10.7554/eLife.11315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Filippova G.N., Fagerlie S., Klenova E.M., Myers C., Dehner Y., Goodwin G., Neiman P.E., Collins S.J., Lobanenkov V.V. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol. Cell Biol. 1996;16:2802–2813. doi: 10.1128/MCB.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dauden M.I., López-Perrote A., Llorca O. RUVBL1-RUVBL2 AAA-ATPase: a versatile scaffold for multiple complexes and functions. Curr. Opin. Struct. Biol. 2021;67:78–85. doi: 10.1016/j.sbi.2020.08.010. [DOI] [PubMed] [Google Scholar]
- 66.Gentili C., Castor D., Kaden S., Lauterbach D., Gysi M., Steigemann P., Gerlich D.W., Jiricny J., Ferrari S. Chromosome Missegregation Associated with RUVBL1 Deficiency. PLoS One. 2015;10 doi: 10.1371/journal.pone.0133576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Magalska A., Schellhaus A.K., Moreno-Andrés D., Zanini F., Schooley A., Sachdev R., Schwarz H., Madlung J., Antonin W. RuvB-like ATPases function in chromatin decondensation at the end of mitosis. Dev. Cell. 2014;31:305–318. doi: 10.1016/j.devcel.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 68.Zaarur N., Xu X., Lestienne P., Meriin A.B., McComb M., Costello C.E., Newnam G.P., Ganti R., Romanova N.V., Shanmugasundaram M., et al. RuvbL1 and RuvbL2 enhance aggresome formation and disaggregate amyloid fibrils. EMBO J. 2015;34:2363–2382. doi: 10.15252/embj.201591245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang H., Li B., Zuo L., Wang B., Yan Y., Tian K., Zhou R., Wang C., Chen X., Jiang Y., et al. The transcriptional coactivator RUVBL2 regulates Pol II clustering with diverse transcription factors. Nat. Commun. 2022;13:5703. doi: 10.1038/s41467-022-33433-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shin S.H., Lee J.S., Zhang J.M., Choi S., Boskovic Z.V., Zhao R., Song M., Wang R., Tian J., Lee M.H., et al. Synthetic lethality by targeting the RUVBL1/2-TTT complex in mTORC1-hyperactive cancer cells. Sci. Adv. 2020;6 doi: 10.1126/sciadv.aay9131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Assimon V.A., Tang Y., Vargas J.D., Lee G.J., Wu Z.Y., Lou K., Yao B., Menon M.K., Pios A., Perez K.C., et al. CB-6644 Is a Selective Inhibitor of the RUVBL1/2 Complex with Anticancer Activity. ACS Chem. Biol. 2019;14:236–244. doi: 10.1021/acschembio.8b00904. [DOI] [PubMed] [Google Scholar]
- 72.Zhang G., Wang F., Li S., Cheng K.W., Zhu Y., Huo R., Abdukirim E., Kang G., Chou T.F. Discovery of small-molecule inhibitors of RUVBL1/2 ATPase. Bioorg. Med. Chem. 2022;62 doi: 10.1016/j.bmc.2022.116726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Giannareas N., Zhang Q., Yang X., Na R., Tian Y., Yang Y., Ruan X., Huang D., Yang X., Wang C., et al. Extensive germline-somatic interplay contributes to prostate cancer progression through HNF1B co-option of TMPRSS2-ERG. Nat. Commun. 2022;13:7320. doi: 10.1038/s41467-022-34994-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the results reported in this paper is GEO: GSE224654, which includes CRISPRi screening gRNA readout (GEO: GSE224653) and RNA-seq expression for the RUVBL1 knockdown experiment (GEO: GSE224646). The gRNA sequence design, raw and normalized count, and the eQTL mapped with the candidate SNP are listed in Table S2. The RIGER analysis output is listed in Table S3. The publicly available datasets used are listed in Table S4. The SILAC proteomics sequencing result is listed in Table S5. Detailed information about the rs60464856 base editing can be accessed in the github repository (https://github.com/Yijun-Tian/Base_Editing-rs60464856).