

Summary
By converting physical forces into electrical signals or triggering intracellular cascades, stretch-activated ion channels allow the cell to respond to osmotic and mechanical stress. Knowledge of the pathophysiological mechanisms underlying associations of stretch-activated ion channels with human disease is limited. Here, we describe 17 unrelated individuals with severe early-onset developmental and epileptic encephalopathy (DEE), intellectual disability, and severe motor and cortical visual impairment associated with progressive neurodegenerative brain changes carrying ten distinct heterozygous variants of TMEM63B, encoding for a highly conserved stretch-activated ion channel. The variants occurred de novo in 16/17 individuals for whom parental DNA was available and either missense, including the recurrent p.Val44Met in 7/17 individuals, or in-frame, all affecting conserved residues located in transmembrane regions of the protein. In 12 individuals, hematological abnormalities co-occurred, such as macrocytosis and hemolysis, requiring blood transfusions in some. We modeled six variants (p.Val44Met, p.Arg433His, p.Thr481Asn, p.Gly580Ser, p.Arg660Thr, and p.Phe697Leu), each affecting a distinct transmembrane domain of the channel, in transfected Neuro2a cells and demonstrated inward leak cation currents across the mutated channel even in isotonic conditions, while the response to hypo-osmotic challenge was impaired, as were the Ca2+ transients generated under hypo-osmotic stimulation. Ectopic expression of the p.Val44Met and p.Gly580Cys variants in Drosophila resulted in early death. TMEM63B-associated DEE represents a recognizable clinicopathological entity in which altered cation conductivity results in a severe neurological phenotype with progressive brain damage and early-onset epilepsy associated with hematological abnormalities in most individuals.
Keywords: epilepsy, infantile spasms, abnormal myelination, white matter abnormality, ion channels, leak cation currents, epileptic encephalopathy, hemolytic anemia, osmotic stress
Graphical abstract
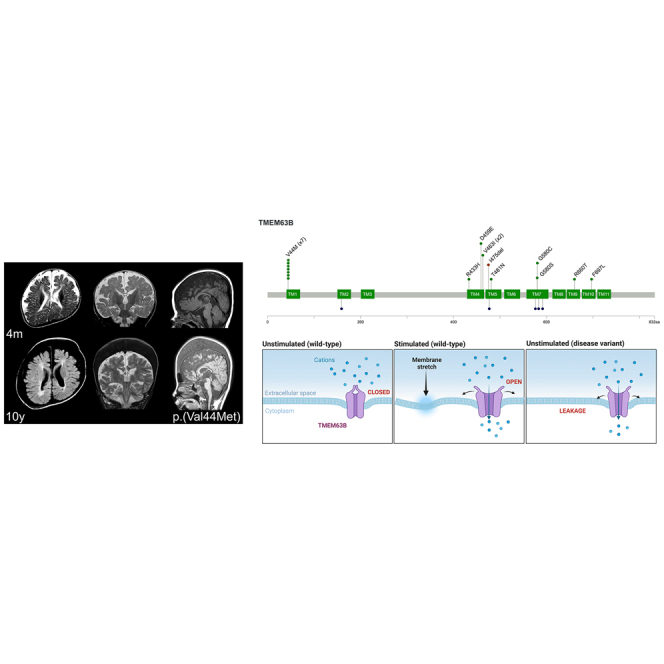
Combining in silico evaluation, in vitro electrophysiology and Ca2+ imaging, and in vivo modeling in Drosophila, Vetro et al. demonstrate that gain-of-function variants of the stretch-activated ion channel TMEM63B cause a severe neurodevelopmental disorder with early-onset epilepsy and progressive brain damage associated with hematological abnormalities in most individuals.
Introduction
TMEM63B, with its two paralogs TMEM63A and C, belongs to a gene family initially identified as the closest homologs of the OSCA proteins, which represent the largest family of stretch-activated ion channels conserved across eukaryotes.1,2 In plants, members of the OSCA family sense osmotic stress-induced mechanical forces across the plasma membrane and activate a signaling pathway responsible for regulating water transpiration and root growth.1,3 In mammals, members of the TMEM63 family mediate cation currents in response to osmotic and mechanical stimuli affecting membrane tension.1,4 This process is crucial for cell volume regulation and viability, as changes in osmolarity may cause water influx or efflux through the plasma membrane, resulting in cell swelling or shrinkage.5
Other members of the TMEM63 family have been associated with monogenic disorders, namely transient infantile hypomyelinating leukodystrophy-19 (HLD19 [MIM: 618688]), with developmental delay of variable severity caused by heterozygous TMEM63A variants,6,7,8 and autosomal recessive spastic paraplegia-87 (SPC-87 [MIM: 619966]), caused by biallelic truncating variants of TMEM63C.9 The TMEM63B gene still lacks a clear association with human diseases and is not yet listed either in the OMIM’s Morbid Map of the Human Genome or in the ClinGen Gene-Disease Validity database (https://www.clinicalgenome.org/). The gene is ubiquitously expressed in humans, and, according to the GTEx portal (https://www.gtexportal.org/home/), multiple TMEM63B mRNA isoforms occur with different tissue specificity. Multiple Tmem63b mRNA isoforms have also been identified in mice, with post-transcriptional modifications contributing to mRNA diversity.10 A brain-specific Tmem63b isoform, exhibiting alternative splicing of exon 4 and glutamine to arginine change (Q/R) at exon 20, has been shown to regulate Ca2+ permeability and osmosensitivity of the channel.10
We identified ten distinct heterozygous de novo variants of TMEM63B in 17 unrelated individuals with early-onset developmental and epileptic encephalopathy (DEE), all associated with white matter disease, corpus callosum abnormalities, and variable cortical, cerebellar, and hematological abnormalities.
After determining the most represented brain TMEM63B isoform in humans, we tested in vitro the effect of selected variants on the protein localization and function by immunocytochemistry, whole-cell patch clamp, and calcium imaging in transfected Neuro2a cells. We also modeled in vivo in Drosophila the effects of the ectopic expression of two variants. Our findings indicate that heterozygous variants of TMEM63B result in a clinically recognizable DEE syndrome whose pathophysiology lies in altered functional properties of the channel.
Material and methods
Research cohort
Our initial discovery cohort consisted of 600 consecutive individuals referred to the Neuroscience Department of the Meyer Children’s Hospital to investigate the genetic causes of DEEs. Within this cohort, we identified by whole-exome sequencing (WES)11 de novo heterozygous variants of TMEM63B in three individuals (IDs 1–3) with a homogeneous clinical and neuroimaging phenotype and promoted an international collaboration through GeneMatcher12 identifying 14 additional individuals carrying de novo TMEM63B variants (IDs 4–17). We reviewed medical records, electroencephalograms (EEGs), and brain magnetic resonance imaging (MRI) scans. We classified seizure types following the International League Against Epilepsy (ILAE) criteria13 whenever applicable and used more descriptive terms when seizure phenomenology could not fit classification terminology. We obtained written informed consent for the study from all participants or their legal guardians, according to local requirements. The study was approved by the Pediatric Ethics Committees of the Tuscany Region, Italy, in the context of the DESIRE FP7 EU project and its extension by the DECODE-EE project.
Detailed methods for MRI investigations, genetic and structural protein analyses, functional characterization of TMEM63B variants, and Drosophila modeling are reported in the supplemental information.
Results
Clinical findings
Clinical, EEG, and MRI findings of the 17 individuals are summarized in Tables 1 and S2. They were all unrelated subjects who exhibited a markedly overlapping DEE phenotype with early-onset drug-resistant epilepsy (17/17, 100%), severe developmental delay (17/17, 100%), early generalized hypotonia evolving to spastic quadriparesis (13/17, 76%), nystagmus and central visual impairment (11/17, 65%). Epilepsy onset ranged from birth to 3 years but occurred within the first year in 14/17 (82%) and in the first month of life in 6/17 (35%). A common pattern of the epilepsy phenotype was early onset of focal seizures (11/17, 65%), often manifested as apnoeic episodes in newborns, followed over months by epileptic spasms (5/17, 29%) or by different types of focal and generalized onset seizures (7/17, 41%). Infantile epileptic spasms, which were the initial manifestation of epilepsy in three additional individuals (3/17, 18%), had therefore been present in 9/17 individuals (53%). In two remaining individuals (2/17, 12%), onset was at 2 and 3 years with focal seizures with impaired awareness. Epilepsy was severe at onset in all individuals, with episodes of status epilepticus in three (3/17, 18%). At last follow-up, five individuals (5/17, 29%) no longer had severe epilepsy, including three who had experienced prolonged seizure freedom on medication (3/17, 18%).
Table 1.
Clinical features of the 17 individuals with TMEM63B variants
| Individuals’ ID/gender | TMEM63B varianta(cDNA and protein) | Age at last follow-up/death | Age at seizure onset/type | Seizure types/severity during follow-up | Treatment ever tried (+ efficacy, +/− transient efficacy, − worsening or not tolerated) | EEG | Brain magnetic resonance imaging | Clinical neurological phenotype | Hematological findings |
|---|---|---|---|---|---|---|---|---|---|
| 1/M | c.130G>A (p.Val44Met) |
8 years/deceased at 9 years (pneumonia) | 6 months/infantile spasms | spasms, myoclonic, focal with impaired awareness/daily | CZP, ESM, PB, VGB | 6 months–8 years: slow background, bilateral independent discharges; epileptic spasms and myoclonic szs | 5 months, 4 years: thin CC, colpocephaly, abnormal myelination, dysmorphic lateral ventricles, enlarged extracerebral spaces, progressive mild cerebellar atrophy, and watershed areas WM abnormality | threatened preterm labor at 35 weeks, global profound DD, generalized hypotonia, plagiocephaly, nystagmus, dysphagia (PEG 17 months), dyskinesias | mild anemia |
| 2/M | c.1298G>A (p.Arg433His) |
10 years | birth/focal | bilateral independent focal motor with impaired awareness, focal to bilateral tonic-clonic/weekly | CBZ, CLB, CZP, LEV, LCM, MDZ, PB, PHT, STP, TPM | 7−10 years: slow background, multifocal discharges; focal szs recorded | 6 years, 10 years: thin CC, multifocal WM abnormalities, ventricular asymmetry, progressive cerebellar atrophy | global profound DD, ataxic gait, lower limb hypertonia, nystagmus | mild abnormalities of RBCt, MCV, MCH |
| 3/M | c.1442C>A (p.Thr481Asn) |
15 years | 2 months/focal motor with asymmetric posturing | focal with impaired awareness and asymmetric posturing, focal to bilateral tonic-clonic/weekly | CBZ, CLB, PB, PER, STP, VGB, VNS | 15 years: slow background, focal epileptiform discharges | 4 years: thin CC, posterior predominant multifocal WM abnormalities, dysmorphic asymmetric lateral ventricles, enlarged cortical sulci | global profound DD, spastic asymmetric quadriparesis, severe cortical visual impairment | none |
| 4/Mb | c.130G>A (p.Val44Met) |
12 years/deceased at 12.5 years (pneumonia) | 4 months/focal | focal with impaired awareness; 9 months on: spasms and focal to bilateral tonic-clonic/daily; single episode of SE (14 months) | CBZ, CLB, KD, LTG, LEV, PRED, RFM, VGB, VPA, VNS | 8 months–10 years: slow background, bilateral independent or multifocal discharges; epileptic spasms recorded | 5 months, 7 years: thin CC, multifocal WM abnormalities, dysmorphic enlarged lateral ventricles, mild cortical and cerebellar atrophy, progressive trabecular bone thickening | global profound DD, wheelchair bound, cortical visual impairment, by 10 years knee fixed flexion contractures, dysphagia (PEG 5 years) | severe macrocytic anemia transfusion dependent |
| 5/M | c.130G>A (p.Val44Met) |
8 years | day 2/apnoeic | 4 months: epileptic spasms; 7 years: focal hyperkinetic motor with impaired awareness/sz-free | CBZ(+), CLB, LEV, PB(+), TPM(+) | 7 years: slow background, bilateral independent or multifocal discharges; focal szs recorded | 1 week, 7 months, 2 years: thin CC, abnormal myelination, dysmorphic asymmetric lateral ventricles, enlarged extracerebral spaces, progressive posteriorly predominant WM abnormality, and cerebellar atrophy | global profound DD, severe cortical visual impairment, nystagmus, spastic quadriparesis, dysphagia (PEG 2 years) | jaundice at birth |
| 6/Fb | c.130G>A (p.Val44Met) |
15 months/deceased at 23 months (pneumonia in progressive respiratory failure) | day 10/apnoeic | focal onset with impaired awareness, focal to bilateral tonic-clonic, epileptic spasms/daily | Alimemazine, CBZ, CLB, PB(+/−), paraldehyde, PRED, TPM, VGB, VPA, VNS(+), biotin, pyridoxine | 2–23 months: slow background, multifocal or generalized discharges with burst suppression; focal szs recorded | 2 months: thin CC, enlarged extracerebral spaces, diffusely abnormal myelination | global profound DD, quadriparesis with generalized hypotonia, cortical visual impairment, nystagmus, dysphagia (PEG 16 months) | macrocytic anemia, transfusion dependent |
| 7/Ma | c.130G>A (p.Val44Met) |
20 months | birth/apnoeic | 2 weeks: stiffening episodes; 9 months: epileptic spasms/sz-free | LEV, steroids, VGB | birth–20 months: normal background, then hypsarrhythmia, slow background, focal discharges | 1 week, 5 months, 11 months: thin CC, widespread WM abnormalities, enlarged dysmorphic lateral ventricles, mild progressive cerebellar atrophy | global profound DD, asymmetric quadriparesis, generalized hypotonia, cortical visual impairment, nystagmus, dysphagia (no PEG yet) | scleralicterus |
| 8/F | c.130G>A (p.Val44Met) |
17 years | 4 months/infantile spasms | 1 year: generalized tonic, focal motor with impaired awareness/daily | ACTH, CBZ, CLB, CZP, ESM, GBP, ivIg, LCM, LEV, PB, PER, PIR, PLP, PRM, STP, TPM, VPA, ZNS | 4 months–12 years: hypsarrhythmia, then slow background, multifocal epileptiform discharges | 4 months, 2 years, 10 years: thin CC, abnormal myelination, dysmorphic asymmetric lateral ventricles, enlarged extracerebral spaces, progressive widespread WM abnormality, ventricular dilatation, cerebellar atrophy, and trabecular bone thickening | global profound DD, quadriparesis, dysphagia (PEG 12 years) | severe anemia requiring occasional transfusions |
| 9/M | c.130G>A (p.Val44Met) |
15 years | 2 years 6 months/febrile seizures | 3 years: epileptic spasms in small clusters/weekly | CZP, VPA | slow background, no epileptiform discharges in 4 EEG recordings | 4 months: thin CC with absent splenium and delayed myelination; 6 years 7 months: increased signal of WM in watershed areas, generalized decrease of white matter volume | global profound DD, wheelchair bound, quadriparesis, pseudobulbar signs, jerky involuntary movements, stereotypical movements and behavior, dysphagia (PEG 3 years), cortical visual impairment, nystagmus | mild anemia |
| 10/F | c.1377C>G (p.Asp459Glu) |
3 years | 2 weeks/focal | bilateral independent focal, epileptic spasms/daily | CBD, KD, LEV, VGB, VNS, ZNS | 2–9 months: slow background, hypsarrhythmia, multifocal discharges; focal szs and epileptic spasms recorded | 2 months: thin CC, enlarged extracerebral spaces, abnormal myelination more pronounced posteriorly, Rathke cleft cyst | global profound DD, quadriparesis, generalized hypotonia, cortical blindness with roving eye movements, dysphagia | mild macrocytic anemia |
| 11/M | c.1387G>A (p.Val463Ile) |
7 years | 2 years/focal | occasional focal with impaired awareness/during fever | VPA | 4–6 years: normal background, bilateral independent discharges | 6 years: multifocal, posteriorly predominant WM abnormality | global severe DD, motor impairment, generalized hypotonia | none |
| 12/M | c.1387G>A (p.Val463Ile) |
23 months | first few weeks/myoclonic jerks | 14 months; intractable focal seizures | LEV, CLB, ESM, CLZ, TPM(+/−), VPA(−) | 14 months: normal background, generalized and focal discharges | 14 months: mild prominence of ventricular system and extra-axial CSF spaces | global DD, dysphagia | none |
| 13/F | c.1424_1426del (p.Ile475del) |
16 years | 6 months/infantile spasms | tonic/daily; GTCs/monthly | ACTH, CBD, CLB, CZP, LEV, LTG, OXC, TPM(−), VGB(−), VPA | 4 months–16 years: hypsarrhythmia, then slow background, bilateral independent or multifocal discharges; epileptic spasms and tonic szs recorded | 7 months, 2 years, 4 years, 8 years: thin CC, multifocal WM abnormalities, asymmetric dysmorphic lateral ventricles, progressive trabecular bone thickening | global profound DD, generalized hypotonia, microcephaly, visual impairment, nystagmus, squint, spastic quadriparesis, dysphagia (PEG 12 years) | severe hemolytic anemia, transfusion dependent |
| 14/M | c.1738G>A (p.Gly580Ser) |
5 years | 10 months/focal | GTCs/occasional during fever | VPA | 2 years: slow background, focal discharges | 1 year 7 months: thin CC, widespread WM abnormality, especially periventricular, asymmetric dysmorphic lateral ventricles, mild cerebellar atrophy | global profound DD, spastic quadriparesis, axial hypotonia, upper limb dystonia, visual impairment, dysphagia (PEG 3 years) | none |
| 15/F | c.1738G>T (p.Gly580Cys) |
30 years | 3 years/focal | focal with impaired awareness, GTCs/yearly | CBZ, CZP, PHT(+), VPA, ZNS | 15 years: slow background, focal discharges; focal szs recorded | 15 years, 21 years, 24 years, 27 years, 28 years: thin CC, widespread WM abnormality, especially periventricular/posterior, progressive cerebellar atrophy, and trabecular bone thickening | global moderate DD, cerebellar ataxia, tremor, dysarthria, limited mobility, bipolar disorder | mild hyperbilirubinemia |
| 16/F | c.1979G>C (p.Arg660Thr) |
25 years | 11 months/focal | focal with posturing and impaired awareness, recurrent SE/yearly | CBZ(−), LMT(−), OXC, PB(−), TPM, VNS(+) | 17 months–22 years: background mildly abnormal, diffuse beta activity, multifocal discharges; focal szs recorded | 1 year, 2 years, 10 years: thin CC, multifocal WM abnormality | global moderate DD, spastic quadriparesis, ASD | macrocytic anemia |
| 17/M | c.2089T>C (p.Phe697Leu) |
15 years | 11 months/focal | focal with eye deviation; jerking, GTCs, recurrent SE/sz-free at last FU | CLB, LTG(−), LEV(−), OXC, PHT, VPA | 5–17 months: normal background; focal discharges | 1 year, 12 years: thin CC, abnormal myelination, colpocephaly, dysmorphic lateral ventricles, increased WM signal with posterior/periventricular predominance | global, severe DD, toe walking, ASD | none |
Abbreviations and symbols: ASD, autism spectrum disorder; CBD, cannabidiol; CBZ, carbamazepine; CC, corpus callosum; CLB, clobazam; CZP, clonazepam; DD, developmental delay; ESM, ethosuximide; F, female; GBP, gabapentin; GTCS, generalized tonic-clonic seizures; ivIg, intravenous immunoglobulin; KD, ketogenic diet; LCM, lacosamide; LEV, levetiracetam; LTG, lamotrigine; M, male; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; MDZ, midazolam; NA, not available; OXC, oxcapazepine; PB, phenobarbital; PEG, percutaneous endoscopic gastrostomy; PER, perampanel; PHT, phenytoin; PIR, piracetam; PLP, pyridoxal 5′-phosphate; PRED, prednisolone; PRM, primidone; RBCs, red blood cells; RFM, rufinamide; SE, status epilepticus; STP, stiripentol; sz, seizure; TGP, targeted gene panel; TPM, topiramate; VGB, vigabatrin; VNS, vagus nerve stimulation; VPA, valproic acid; WM, white matter; ZNS, zonisamide.
Variants’ nomenclature is based on the GenBank: NM_018426.3 reference sequence.
IDs 4, 6, and 7 are mentioned in Cacheiro et al.14 (Table S8: DDD subject 1, 100KGP subject 2, and100KGP subject 1); treatment ever tried: +, efficacy; +/−, transient efficacy; −, worsening or not tolerated.
EEGs showed slow background activity with bilateral independent or multifocal epileptiform discharges in most individuals.
All individuals (17/17, 100%) had global developmental delay, with moderate-to-profound intellectual disability and severe motor impairment; only two had developed communicative language skills (2/17, 12%). Severe dysphagia was present in 11 individuals (11/17, 65%), requiring a percutaneous endoscopic gastrostomy (PEG) insertion in eight (8/17, 47%). In 12 individuals (12/17, 71%), clinical history revealed hematological abnormalities resulting in abnormal hemoglobin levels, often with macrocytosis and signs of hemolysis (hyperbilirubinemia, jaundice, hepatosplenomegaly) with a fluctuating course. In four of the 12 individuals (4/12, 33%) with hematological abnormalities, three of whom were carrying the recurrent c.130G>A (p.Val44Met) variant, anemia was severe and required blood transfusions (IDs 4, 6, 8, and 13). In ID 4, peripheral blood film showed abnormally shaped red blood cells (RBCs) with elliptocytes and increased stomatocytes. Bone-marrow biopsy was performed in two individuals (IDs 4 and 8) with signs of hemolysis in both and evidence for hematopoiesis and myelodysplastic syndrome with aplastic anemia in one (ID 8, Figure S1). Auxological parameters at birth were normal in most individuals (Table S2). Three individuals (3/17, 18%) had died prematurely due to pneumonia (IDs 1, 4, and 6; Table 1).
Prompted by the evidence for progressive hearing loss in Tmem63b-deleted mice,4 we investigated whether any signs of hearing impairment were present in our cohort. Eleven out of 17 (65%) individuals received formal hearing tests (Table S2) with abnormal results in two (IDs 15 and 16; 2/11, 18%). In a further individual (ID 10), hearing impairment was suspected on clinical ground but not formally tested.
MRI findings
All individuals had at least one brain MRI scan at 1.5 or 3T between ages 1 week and 28 years, and 11 individuals were studied with two or more scans taken 6 months to 13 years apart (Table 1 and Figure 1). MRI revealed an association of multiregional or widespread white matter signal abnormalities, dysmorphic lateral ventricles, thinning of corpus callosum (17/17, 100%), cerebellar atrophy (8/17, 47%), and atrophy of the cerebral cortex (8/17, 47%).
Figure 1.

Brain MRI in 16 individuals with TMEM63B pathogenic variants
Individuals’ ID is shown in the upper left corner. For each individual, the corresponding TMEM63B variant is reported in the lower right corner. For IDs 1, 4, 5, 7, 8, 15, and 17, two sets of comparative images are presented, taken respectively from the initial and follow-up investigation. For IDs 2, 3, 6, 10, 11–14, and 16, only one set of images is presented. For ID 2 we included additional images, illustrating suspected areas of focal cortical dysplasia (circles). Structural abnormalities include a combination of white matter (WM) abnormalities, dysmorphic lateral ventricles, thinning of corpus callosum (CC), and cortical and cerebellar atrophy that are variably distributed. In ID 1, MRI at age 5 months shows increased extracerebral spaces with enlarged cortical sulci, thin CC with colpocephaly, and high signal intensity of the hemispheric WM, consistent with abnormal myelination. At age 4, enlarged extracerebral spaces and thinning of the CC are less prominent but there is a definite high signal abnormality of the WM, with dysmorphic lateral ventricles, and mild atrophy of the cerebellar cortex. In ID 4, after a first MRI at age 5 months only showing reduced volume of the frontal lobes, a follow-up scan at 7 years revealed a progressive change in shape of the lateral ventricles and CC, revealing WM suffering with mild atrophy of the cerebellar cortex. In IDs 5 and 8, changes that occurred from age 7 months to age 2 years (ID 5) and from age 4 months to 10 years (ID 8) are comparable with those observed in ID 1. The abnormal ventricular shape causes ventricular asymmetry in these individuals. Cerebellar atrophy is indicated by arrows. In ID 7, a second MRI at age 11 months confirms the severe WM abnormality with dilated ventricles and thin CC and reveals mild progressive cerebellar atrophy (arrow). In ID 15, imaged twice 7 years apart, MRI shows as WM changes and cerebellar atrophy continued to progress after age 21 (arrow). In ID 17, only minor changes occurred between age 1 year and 12 years. At 1 year, there were areas of abnormal myelination, especially on the right hemisphere, with thin CC and colpocephaly. At age 12, the CC remained thin but because of maturation processes was thicker than before and ventricular dilatation less prominent, with mild peritrigonal high signal intensity. In ID 2, brain MRI taken at 10 years shows bilateral patchy WM abnormalities highlighted by a circle in the left frontal and right temporal regions. This finding, in association with focal seizures, had raised the suspicion of areas of focal cortical dysplasia. Additional findings include ventricular asymmetry, thin CC, and progressive cerebellar atrophy. In ID 3, imaged at 4 years, there are multifocal high signal WM changes with dysmorphic and asymmetric lateral ventricles, thin CC, and enlarged cortical sulci. IDs 6 and 10, both imaged at age 2 months, exhibited increased extracerebral spaces with enlarged sulci, thin CC, and reduced signal intensity of the WM, consistent with abnormal myelination (ID 10, circles). In ID 11, at age 6 years, there were multifocal areas of WM abnormalities without atrophic changes. In ID 12, at age 14 months, there is already an obvious combination of dilated extracerebral spaces and ventricles with thin CC and high signal abnormality in the posterior WM. IDs 13, 14, and 16 imaged between 1 year 7 months and 10 years show similar findings, slightly varying in severity and including a thin CC, areas of abnormal signal intensity of the WM with posterior predominance in all, dilated asymmetric ventricles (IDs 13 and 14), and mild cerebellar atrophy (ID 14). In IDs 4, 8, 13, 15, and 16, thickening of the trabecular (spongy) bone of the skull (indicated by the asterisks) is suggestive of a blood disorder.
All the 11 individuals for whom repeated MRIs were available for comparison (IDs 1, 2, 4, 5, 7–9, 13, and 15–17) exhibited clear morphological or signal signs resulting from the combination of maturational and disease-related progressive changes. For example, when cerebellar atrophy was present, there had always been evidence of atrophy progression from initial to follow-up imaging (IDs 1, 2, 4, 5, 7, 8, and 15). White matter signal abnormalities (always present) appeared in early scans as severely delayed myelination that was better appreciated with T2-weighted imaging and later evident as diffuse or more circumscribed areas of high-signal intensity changes in fluid attenuated inversion recovery (FLAIR) (see IDs 1, 5, 7, and 8 in Figure 1). In the only individual who had received control MRI scans at different adult ages (ID 15), cerebellar and white matter changes continued to worsen between age 21 and 28. Overall, white matter abnormalities, ventricular shape, and thinning of the corpus callosum exhibited similar longitudinal changes. Signs of progression of cortical atrophy were minor, if any, and proportional to overall volume shrinking. MRI also showed progressive thickening of the trabecular (spongy) bone of the skull in five individuals (5/17, 29%; IDs 4, 8, 13, and 15 in Figure 1; ID 9, not shown), four of whom had anemia (IDs 4, 8, 9, and 13), with a myelodysplastic disorder in three (IDs 4, 8, and 13), while one had experienced episodes of hyperbilirubinemia (ID 15).
Genetic findings
In these 17 individuals, we identified ten distinct TMEM63B variants (Table 1 and Figure 2) including nine missense substitutions and one in-frame deletion. None of them were present in publicly available allele frequency databases, such as gnomAD and TOPMed, or in our internal dataset, and all were predicted to be damaging by multiple prediction tools (Table S3). Available gene constraint scores from gnomAD indicate that TMEM63B is globally highly intolerant to both loss-of-function (LoF; pLI score = 1.00; observed/expected [o/e] LoF = 0.07) and missense (Z score = 4.22; o/e missense = 0.475) variants. In addition, all variants were in regions of the protein predicted to be intolerant to missense variations according to region-level metrics (Figures 2B and S2) and involved amino acids highly conserved among the vertebrate orthologs of the protein (Figure 2C, Table S3), as well as the paralogs TMEM63A and TMEM63C (Figure S3).
Figure 2.

Genetic results
(A) The lollipop diagram shows the distribution of the TMEM63B variants observed in our cohort on the linear protein map and relative to the TMEM63B exons (top, based on NM_018426.3 reference sequence). The variants are represented as green (missense substitutions) or brown (in-frame deletion) dots and all map in the transmembrane helices TM1, TM4–7, and TM9–10 (green boxes). The p.Val44Met (V44M) and the p.Val463I (V463I) are recurrent in seven and two individuals, respectively, as illustrated by the number of green dots. At the bottom of the diagram, the dark blue dots represent the residues affected by missense variants in TMEM63A (five variants in six unrelated individuals: p.Gly168Glu [G168E], p.Ile462Asn [I462N], p.Gly553Val [G553V], p.Tyr559His [Y559H], p.Gly567Ser [G567S]).6,7,8
(B) Tolerance landscape plot of the TMEM63B protein provided by the MetaDome web server (https://stuart.radboudumc.nl/metadome/). The tool identifies regions of low tolerance to missense variations based on the local non-synonymous over synonymous variants ratio from gnomAD.15 All variants in our cohort are contained in intolerant/highly intolerant regions (in red) of the landscape.
(C) Multiple sequence alignment shows the protein sequence of the human TMEM63B protein (NP_060896.1) and of its orthologs in five different vertebrate species (Pan troglodytes, Sus scrofa, Mus musculus, Gallus gallus, Danio rerio) with the mutated residues in bold. The details of the TMEM63B variants in the cohort are displayed above the alignments. The asterisk below the sequence indicates positions that have a single, fully conserved residue between all the input sequences, the colon indicates conservation between groups of strongly similar properties, and the period indicates conservation between groups of weakly similar properties.
We noticed a complementary linear distribution on the protein sequence of the TMEM63B variants in our cohort versus the gene variants in the reference population from gnomAD. The latter were enriched in exons 3–14 and 22–24 and substantially depleted in exons 15–21, where our variants clustered (Figures 2B and S2). This observation, supported by the local constraint metrics provided by multiple tools, suggests an increased selective pressure in the region of the gene corresponding to the seventh transmembrane (TM) domain region RSN1_7TM (PF02714). This domain is conserved among osmosensitive calcium-permeable cation channels.16
The p.Val44Met variant was recurrent in seven unrelated individuals (IDs 1, 4–9), the c.1387G>A (p.Val463Ile) in two (IDs 11 and 12), and two different changes affected the same residues in two individuals (c.1738G>A [p.Gly580Ser] in ID 14 and c.1738G>T [p.Gly580Cys] in ID 15). Eight of the affected residues are fully conserved from human to zebrafish (Val44, Arg433, Asp459, Val463, Thr481, Gly580, Arg660, and Phe697), and one is fully conserved up to chicken (Ile475). In addition, six out of nine affected residues are fully conserved among all the three paralogs’ sequences (Val44, Asp459, Val463, Gly580, Arg660, and Phe697), and three (Arg433, Thr481, and Ile475) are fully conserved in two among three paralogs and they maintain similar properties in the third paralog (Figure S3).
The TMEM63B variant occurred de novo in the 16 individuals for whom parental DNA was tested. Individual 10 was born through egg donation; therefore, maternal DNA was not available for analysis. The variant was absent from the DNA of the father and of the healthy dizygotic twin.
Based on both in silico analyses using sequence data and the functional studies described below, we interpreted all variants as detrimental for the function of the protein.
In 13/17 (77%) individuals, we did not identify additional clinically relevant variants (Table S2). In ID 12, a de novo TSC1 variant was associated with macrocephaly with no other signs of tuberous sclerosis complex (TSC1 [MIM: 191100]). In three more individuals (IDs 9, 16, and 17), additional variants were identified, which were either classified as variants of uncertain significance (VOUSs) or reported as unsolicited findings not related to the phenotypic spectrum presented here (Table S2).
Structural considerations
Because crystallographic data are not yet available for TMEM63B, we can only access information about the protein topology based on its partial homology with the plants’ OSCAs for a subset of whom crystallographic data are available.2,17,18 As the percentage of identity in protein sequence between mammals TMEM63A–C proteins and OSCAs is around 20%,1 mapping our variants on these structures would be inaccurate. Aware of the limitations of prediction tools in regions of low homology, we mapped our variants on the structure exhibiting more homogeneous resolution across the protein, including both the TM helices and the intracellular domains (Figures 3 and S4). In our model, 81% of residues had >90% confidence, including all those affected by variants in our cohort, all affecting a predicted TM helix (Figures 2A, 3A, and 3B). Five variants fell in TM4 (c.1298G>A [p.Arg433His], c.1377C>G [p.Asp459Glu], and c.1387G>A [p.Val463Ile]) or TM5 (c.1424_1426del [p.Ile475del] and c.1442C>A [p.The481Asn]) (Figures 3A and 3B, blue helices), whose tilting and rearrangement upon osmotic stimulus plays a role in channel opening in Oryza sativa OSCA1.2 protein.18 The remaining variants mapped in TM1 (p.Val44Met), TM7 (p.Gly580Ser and p.Gly580Cys), TM8 (c.1979G>C [p.Arg660Thr]), and TM9 (c.2089T>C [p.Phe697Leu]). All but the recurrent p.Val44Met variant are in the Pfam-classified domain RSN1_7TM,16 and six of them affect the predicted pore-forming TM4–TM8 helices.2
Figure 3.

Structural consideration of TMEM63B pathogenic variants
(A and B) View of the predicted tridimensional protein structure of TMEM63B from the membrane plane (A) and the extracellular side (B). All the variants in our cohort map into a transmembrane (TM) helix: p.Val44Met (V44M) in TM1 (dark green helix), p.Arg433His (R433H), p.Asp459Glu (D459E), p.Val463Ile (V463I), p.Ile475del (I475del), and p.Thr481Asn (T481N) in TM4 and TM5 (blue helices), p.Gly580Ser (G580S) in TM7 (orange helix), p.Arg660Thr (R660T) in TM8 (pink helix), and p.Phe697Leu (F697L) in TM9 (light green helix). Dotted line in (A) indicates the plasma membrane, OUT the extracellular side and IN the intracellular (cytoplasmic) side. Details of selected variants are provided in the inlets.
(C) Predicted structural change induced by the D459E substitution. The OD2 atom of Asp459 is predicted to form a buried salt bridge with NZ atom of Lys460 (K460). The substitution of an aspartic acid with a glutamic acid at position 459 increases the distance between the NZ atom of Lys460 and the closer oxygen atom (OE2) available to make a salt bridge, breaking this bond.
(D) Predicted structural change induced by the G580S substitution. The substitution of a glycine (green) with a bulkier amino acid (serine, orange) changes the RSA of the amino acid at position 580 (5.9%–3.8%). In addition, OG atom of Ser580 might form a salt bridge with NE1 atom of Trp485 (W485) and help in stabilizing the structure of the pore.
(E) Predicted structural change induced by the G580C substitution. The substitution of a glycine (green) with a bulkier amino acid (cysteine, orange) changes the RSA of the amino acid at position 580 (5.9%–3.7%). Although the substitution introduces an amino acid with a free SH group that can make disulphide bonds with other amino acids with free SH groups (depicted as yellow spheres), the distance between C580 and the closer amino acid with a free SH group (C486, 10.519 Å) is too big to allow the making of such type of bond.
(F) Predicted structural change induced by the R660T substitution. The substitution of a buried charged residue (arginine) with an uncharged residue (threonine) at position 660 disrupts a salt bridge formed by NH2 atom of Arg660 and Asp137 (D137).
The p.Val44Met (IDs 1 and 4–9) lies in a region with limited sequence homology between OSCAs and TMEM63A–C and for which available OSCA structures have low resolution.2,17 The valine to methionine substitution involves two amino acids with similar properties (Grantham distance 21; scale 0–215) and mass. The TM1 helix is located on the external surface of the channel and is not directly involved in the predicted pore, but it seems to be rather involved, together with TM7, in the sensitivity to membrane stretching (Figure 2).2,17
In a FoldX-based model, Met44 is predicted to make van der Waals contacts with Gln477, Phe478, and Thr481, all mapping on the TM4 helix (Figure S5). Because the free energy change associated with p.Val44Met is negative (−1.46, calculated by FoldX), we hypothesize that this substitution may stabilize the protein structure.
We estimated evolutionary conservation and role of the mutated residues by ConSurf, which confirmed that all changes affected highly conserved residues with predicted functional and/or structural role (Figure S6; Table S4).
We also evaluated the structural impact of the nine distinct missense variants in our cohort by using the Missense3D tool19 and found that eight of nine may affect the protein structure, although limitedly, via changes in cavities volume (7/8), residues accessibility (3/8), and breakage of non-covalent bonds (2/8) (Table S4). The p.Asp459Glu substitution (ID 10) disrupts a salt bridge formed between OD2 atom of Asp459 and NZ atom of Lys460 (distances: 4.168 Å between OD2 atom of Asp450 and NZ atom of Lys460, 5.503 Å between OE2 atom of Glu450 and NZ atom of Lys460) (Figure 3C). The p.Gly580Ser substitution (ID 14) replaces a buried glycine, with a residue solvent accessibility (RSA) of 5.9%, with a buried serine with an RSA of 3.8%. In addition, OG atom of Ser580 might form a salt bridge with NE1 atom of Trp485 (W485), possibly stabilizing the structure of the pore (Figure 3D). Substitution of the Gly580 amino acid with a cysteine (p.Gly580Cys, ID 15) introduces a bulkier residue at position 580 and changes its RSA from 5.9% to 3.7%. Although the free thiol (SH) residue of Cys580 can make disulphide bonds with other amino acids with free SH residues, the distance between Cys580 and Cys486, which is the closer amino acid with a free SH residue (10.519 Å; Figure 3E), does not allow the making of disulphide bonds. The p.Arg660Thr variant (ID 16) replaces a buried charged arginine (RSA 5.6%) with an uncharged threonine. This substitution also disrupts a salt bridge between NH2 group of Arg660 and OD1 atom of Asp137 (distance: 4.787 Å) (Figure 3F). For p.Val44Met, p.Arg433His (ID 2), p.Val463Ile (IDs 11 and 12), and p.Phe697Leu (ID 17), Missense3D suggests only mild alterations of the cavity volume (<70 Å3) without significant structural changes, which might still influence the overall stability of the protein (Table S4). It has indeed been demonstrated that cavities in membrane proteins play a pivotal role in balancing stability and flexibility, impacting protein function.20 For the p.The481Asn (ID 3), which according to ConSurf affects a predicted structural residue, Missense3D did not indicate clear structural damage but could calculate cavity volume in the mutant structure. As for the in-frame deletion variant p.Ile475del (ID 13), Phyre2 modeling did not predict gross alterations of the secondary structure.
Post-transcriptional editing in TMEM63B mRNA from human cerebral cortex and selection of variants for in vitro functional studies
In the mouse brain, the Tmem63b isoform lacking exon 4 is the most expressed during all developmental stages, and the Q/R editing at exon 20 is absent in the early brain but increases with age, reaching ∼80% in Tmem63b mRNA in the adult cortex.10
To characterize TMEM63B expression profile in humans, we reverse-transcribed RNA from an adult human cerebral cortex sample into cDNA and amplified by PCR the region around exon 4 (Figure S7A). We found that exon 4 was missing in 80% of TMEM63B mRNA (Figures S7B and S7C). We further characterized the Q/R editing at exon 20 in long and short TMEM63B isoforms by PCR and Sanger sequencing and found that the editing occurred only in the short isoform (Figures S7D and S7E). Both the exon 4 splicing and Q/R editing findings are in line with previous observations in mice,10 except for lower Q/R editing occurrence in the adult human cortex (∼40%) (Figure S7E).
Collectively, these data prompted us to study the effects of selected TMEM63B variants in vitro by modeling them in the short non-edited isoform, which is likely the most abundant in the developing cortex.
For functional studies in Neuro2A cells, we selected six missense variants (p.Val44Met, p.Arg433His, c.1442C>A [p.Thr481Asn], p.Gly580Ser, c.1979G>C [p.Arg660Thr], and c.2089T>C [p.Phe697Leu]), each affecting a distinct TM domain of the channel (Figure 2A). We also included in the experiments the truncating variant c.973C>T (p.Arg325∗) as a negative control. We became aware of such variant through our matchmaking initiative, as it was found at the homozygous state in an individual with an unrelated phenotype and was heterozygous in both parents who were healthy (unpublished data). The p.Arg325∗ is located in exon 12 of 24 and predicted to result in nonsense-mediated decay (NMD) of the aberrant mRNA.
p.Val44Met, p.Arg433His, p.Thr481Asn, p.Gly580Ser, p.Arg660Thr, and p.Phe697Leu do not affect TMEM63B localization at the plasma membrane
The TMEM63B channel normally localizes at the plasma membrane level.21,22 To address the impact of p.Val44Met, p.Arg433His, p.Thr481Asn, p.Gly580Ser, p.Arg660Thr, and p.Phe697Leu on TMEM63B localization, we performed immunocytochemistry in Neuro2A cells transfected with GCaMP6f plasmids overexpressing either wild-type (WT) or mutant hemagglutinin (HA)-tagged TMEM63B and GCaMP6f empty vector as control.
In WT- and mutant-expressing Neuro2A cells, TMEM63B was correctly localized at the membrane level (Figure 4). As expected, we did not observe TMEM63B expression in control GCAMP6f and in p.Arg325∗-expressing cells. These findings suggest that the six missense variants we studied exert their effect by altering the channel function without impacting localization on the plasma membrane.
Figure 4.

Immunocytochemistry to assess TMEM63B localization at the plasma membrane
Confocal microscopy photographs of Neuro2A cells transfected with GCaMP6f empty vector, TMEM63B WT, or mutant plasmids and analyzed 48 h post-transfection. Transfected cells express the GCaMP6f protein, which fluoresces in the green channel. Cells were stained with primary anti-HA tag antibody, secondary Alexa Fluor 555 antibody, and DAPI. Scale bar = 10 μm.
p.Val44Met, p.Arg433His, p.Thr481Asn, p.Gly580Ser, p.Arg660Thr, and p.Phe697Leu affect TMEM63B conductance
In physiological conditions, TMEM63B operates as a non-selective cationic channel activated by mechanical and osmotic stimuli.1,4 To address the impact of the selected p.Val44Met, p.Arg433His, p.Thr481Asn, p.Gly580Ser, p.Arg660Thr, and p.Phe697Leu variants on channel properties, we applied a set of electrophysiological protocols previously implemented to characterize WT TMEM63B in vitro.4 In transfected Neuro2A cells, we initially recorded whole-cell currents elicited by a −80 to +80 mV voltage ramp protocol in isotonic conditions (300 mOsm/L extracellular solution) and then imposed a hypo-osmotic stimulus by switching to a 170 mOsm/L solution.
In cells expressing each of the selected TMEM63B variants, a −80 mV step imposed under isotonic conditions elicited a stable inward current that was absent in TMEM63B WT, control GCAMP6f, and p.Arg325∗-expressing cells (Figures 5A and 5B). Outward currents elicited at +80 mV were also enhanced in cells expressing p.Val44Met, p.Arg433His, p.Thr481Asn, p.Gly580Ser, p.Arg660Thr, and p.Phe697Leu compared with controls (Figure 5C). These six variants also caused a significant depolarization of the reversal potential, as shown by the current-voltage (I/V) relationship (Figures 5A and 5D).
Figure 5.

Electrophysiological recordings on transfected Neuro2A cells
(A) Left: representative raw traces of TMEM63B-mediated currents registered under isotonic condition in Neuro2A cells transfected with GCaMP6f, TMEM63B WT, or mutant plasmids. Cells were held at 0 mV and recorded with a ramp protocol from −80 mV to +80 mV, 100 ms duration, 0.1 Hz. Right: current-voltage (I/V) relationship showing variant-induced change in reversal potential.
(B and C) Quantification of whole-cell current density at −80mV (B) and +80mV (C) (GCaMP6f = 16 cells, TMEM63B WT = 18 cells, TMEM63B R325∗, V44M, R433H, T481N, G580S, R660T, and F697L = 17 cells; ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05, ns = not significant, Kruskal-Wallis and Dunn’s multiple comparisons tests).
(D) Quantification of reversal potential (GCaMP6f = 16 cells, TMEM63B WT = 18 cells, TMEM63B R325∗, V44M, R433H, T481N, G580S, R660T, and F697L = 17 cells; ∗∗∗∗p < 0.0001, one-way ANOVA and Tukey’s multiple comparison test). Data are expressed as mean ± SEM.
Under hypo-osmotic conditions, a −80 mV step elicited an inward current in cells expressing either the WT or mutant TMEM63B, while no current was generated in control cells (Figure S8). In most cases, cells underwent massive swelling and eventually burst, as previously reported.4 WT- and mutant-expressing cells that sustained the hypo-osmotic shock and reached the plateau currents exhibited similar current amplitudes (Figures S8A and S8B). The overall increase in currents observed at −80 mV, expressed as the delta (Δ) between currents measured under hypo-osmotic and isotonic conditions, was larger in WT- than in mutant TMEM63B-expressing cells (Figure S8C), as might be expected by an inward leak current under isotonic conditions in the mutant.
Overall, these results show that p.Val44Met, p.Arg433His, p.Thr481Asn, p.Gly580Ser, p.Arg660Thr, and p.Phe697Leu alter channel conductance, resulting in partial channel activation in physiological conditions, without altering the maximal channel conductance under hypo-osmotic stimulation.
p.Val44Met, p.Arg433His, and p.Thr481Asn affect calcium permeability
Previous experiments conducted in Neuro2A cells demonstrated that hypo-osmotic challenges trigger cationic currents across the TMEM63B channel with hyposmolarity-induced Ca2+ influx.4 To determine whether the TMEM63B variants affect the hyposmolarity-induced Ca2+ response, we co-expressed in Neuro2A cells the Ca2+ sensor GCaMP6f and either WT TMEM63B or one of the three mutants p.Val44Met, p.Arg433His, and p.Thr481Asn. We selected these variants based on their recurrence in multiple individuals with overlapping clinical features (p.Val44Met) and location in critical TM helices TM4 (p.Arg433His) and TM5 (p.Thr481Asn), whose rearrangement is implicated in channel opening.18
We performed calcium imaging under a confocal microscope in a recording chamber equipped with a perfusion system to deliver the test hypo-osmotic solutions to the cells. As previously reported,4 a hypo-osmotic stress provided by decreasing the osmolarity of the perfusion solution from 300 mOsm/L to 170 mOsm/L triggered a Ca2+ response (Figure 6A) in about 35% of cells expressing WT TMEM63B, while only a small fraction of control cells transfected with GCaMP6f responded (Figure 6B). Neuro2A cells expressing p.Val44Met showed reduced responsivity with respect to the WT-expressing cells. We also tested the response to weaker osmotic stresses that are more likely to be representative of physiological changes by shifting solution osmolarity from 300 mOsm/L down to 255 mOsm/L. The percentage of responsive cells decreased, and all the three mutants expressing cells showed a reduced responsivity score, which was significant for p.Arg433His (Figure 6B). The time course of representative responses plotted in Figures 6A and 6C shows a heterogeneous behavior of cells, ranging from transient Ca2+ pulses to sustained increments.
Figure 6.

Mutations of TMEM63B impair the Ca2+ response to hypotonic stress
(A) Representative time-lapse sequence of Neuro2A cells co-transfected with WT and p.Val44Met TMEM63B after exposure to hypo-osmotic solution (170 mOsm/L). For each genotype, we show two cells characterized by transient or steady responses. The green dots on the traces indicate the timing of the images. Calibration bar is 10 μm in all images.
(B) Fraction of Neuro2A cells transfected with WT, p.Val44Met, p.Arg433His, or p.Thr481Asn TMEM63B presenting a Ca2+ response within 10 min from exposure to hypo-osmotic solutions. The number of analyzed cells is on top of each column, with the number of replicates in parentheses.
(C) Representative traces showing the change of fluorescence for several cells transfected with WT, p.Val44Met, p.Arg433His, or p.Thr481Asn TMEM63B.
(D) Cumulative results indicating the peak amplitude of the Ca2+ response to hypo-osmotic stimulus (255 mOsm/L). Numbers indicate the responding cells in each group. Total number of analyzed cells and replicates as indicated in (B).
(E) Integral of the Ca2+ change for the indicated experimental groups. In this analysis, we included only the cells that returned to baseline within 150 s from the transient onset. All mutants showed a drastically reduced response. Abbreviations and symbols: ∗∗∗, p < 0.001; ∗∗, p < 0.01; ∗, p < 0.05 (chi-squared test); GCaM, control cells transfected with GCaMP6f empty vector. Data are expressed as box plots ranging from 25th percentile to 75th percentile, while the whiskers indicate the range of the outliers with a coefficient of 1.5.
Given the role of Ca2+ in orchestrating the cell response to osmotic stress,4 we next analyzed the amplitude and dynamics of the observed Ca2+ changes. Cells expressing the three variants showed smaller Ca2+ transient compared with those expressing WT TMEM63B. This is true when considering the maximal amplitude of the response (Figure 6D) and when considering the integral of the Ca2+ change computed for cells that returned to baseline within 150 s from the response onset (Figure 6E).
p.Val44Met and p.Gly580Cys cause lethal toxicity in Drosophila
About two-thirds of the vital genes in the Drosophila genome are involved in eye development, including genes required for general cellular processes. For this reason, the fly eye provides an excellent experimental system to study the role of target genes in cellular function and development and in neurodevelopment/degeneration.23 To evaluate the potential impact of selected variants on the eye morphology, we generated transgenic flies expressing the human TMEM63B gene. We designed transgenes for the WT TMEM63B, the recurrent p.Val44Met, and the p.Gly580Cys variants by using the GMR-Gal4 ectopic expression system, which is mostly expressed in the retina.24 A schematic representation of the transgene construct is shown in Figure S9A. The Gal4-UAS system25 induces TMEM63B expression by the binding of the yeast transcription factor Gal4 to the upstream activating sequence (UAS). The expression level of the Gal4/UAS system increases in a temperature-dependent manner24 and can therefore be modulated by changing the fly-rearing temperatures. In WT TMEM63B-expressing flies, which were viable and reached the adult stage, we did not observe eye abnormalities (Figures S9B and S9C). However, when expressing the p.Val44Met and p.Gly580Cys TMEM63B transgenes, we did not obtain any adult fly as both variants caused early lethality. We could not obtain any adult fly even after reducing the expression level of the two transgenes to the minimum by lowering the rearing temperature down to 18°C. In adult flies expressing WT TMEM63B by c739-Gal4, we performed immunohistochemistry and observed dotted signals on the cell membrane (Figure S9D), thus demonstrating correct expression of the transgene in a neuronal cell type, Kenyon cells. We observed the same lethal phenotype by expressing the TMEM63B transgenes under either the GMR-Gal4 or the c739-Gal4 drivers, demonstrating that both variants caused lethal toxicity in Drosophila. Because the lack of either Kenyon cells or photoreceptors is not sufficient in itself to cause death in flies, we expect this phenotype to be caused by the expression of the p.Val44Met and p.Gly580Cys TMEM63B transgenes in other cell types indispensable during developmental stages.
Discussion
This series of 17 individuals with pathogenic heterozygous de novo variants of TMEM63B defines the phenotypic features of an autosomal dominant early-onset DEE syndrome. All individuals exhibited global developmental delay, moderate-to-profound intellectual disability, severe motor impairment, and severe epilepsy with onset from birth to 3 years. Three individuals had died prematurely, and those who had reached adolescent or adult age remained completely dependent. Most had central visual impairment and swallowing dysfunction requiring PEG insertion. Epilepsy was a prominent feature and was quite severe at onset, featuring recurrent episodes of status in some individuals, although a variable degree of seizure control was reported, with three individuals achieving seizure freedom on treatment. We noticed a common trajectory of the epilepsy phenotype, especially with the recurrent p.Val44Met variant, characterized by neonatal onset of apnoeic/focal seizures, evolving to epileptic spasms around the age of 4 to 6 months, and then continuing with focal, generalized seizures, or both. EEGs were consistent with an epileptic encephalopathy pattern at onset, including multifocal interictal epileptiform abnormalities and multiple seizure types in most. MRIs showed a consistent pattern of widespread or multiregional white matter abnormalities, with posterior periventricular predominance, dysmorphic lateral ventricles, thin corpus callosum, variably accompanied by global cerebellar atrophy (eight individuals), and areas of cortical atrophy (eight individuals). Overall, initial MRI findings in most individuals could be misdiagnosed as consequence of perinatal hypoxic-ischemic cerebral injury (see for example IDs 1, 3, 5, 7, 8, and 12–16). However, longitudinal neuroimaging with relatively long time intervals (up to 13 years) often revealed signs of progression, especially affecting the white matter and cerebellum. Over a clinical monitoring period up to ages 20 months to 30 years, three individuals died at ages 23 months, 9, and 12 years due to pneumonia. In the remaining 14 individuals, clinical findings were consistent with a DEE akin to cerebral palsy in most, with limited signs of progression in ten (IDs 1, 2, 5, 7–9, 13, and 15–17) and of a milder encephalopathy in four (IDs 3, 10, 11, and 14).
Overall, clinical and imaging findings are consistent with a progressive neurodegenerative clinical course and indicate, in most individuals, prenatal central nervous system impairment leading to onset of symptoms early after birth or within the first year of life. Hematological abnormalities might have contributed to brain damage. In 12 individuals, we found macrocytosis or signs of chronic hemolysis, often from birth, with a fluctuating course (Tables 1 and S2). Although such abnormalities were mostly not clinically significant or only caused jaundice in the neonatal period, in four individuals, the anemia was so severe it required periodic transfusions. In four of the individuals with anemia, MRI showed progressive thickening of the trabecular (spongy) bone of the skull (IDs 4, 8, and 13 in Figure 1 and individual 9, not shown), a finding often associated with severe hematological disorders.26
TMEM63B is evolutionarily conserved and highly intolerant to both LoF and missense variants in the general population. The International Mouse Phenotyping Consortium (IMPC, https://www.mousephenotype.org/) lists Tmem63b among the essential genes in database because its homozygous knockout causes pre-weaning lethality in the C57BL/6N strain.27 Heterozygous Tmem63b mutant mice from the IMPC exhibit a neurodevelopmental phenotype and increased circulating bilirubin level, both consistent with observations in our individuals. No further significant hematological or sensorial abnormalities were observed in this mouse model, but susceptibility to epilepsy (e.g., by electroconvulsive threshold testing phenotypic assays) was not assessed. In a more vital C57BL/6N-FVB/N mixed breed, surviving Tmem63b knockout mice developed progressive hearing loss.4 Hearing impairment was described in 2/11 (18%) individuals who could be formally tested and in one more individual who was not tested. Because individuals with TMEM63B-associated DEE might be at increased risk of hearing impairment and this clinical problem is easily overlooked in the severely disabled, objective testing should be performed in this disorder.
Mendelian disease genes associated with autosomal dominant disorders are overrepresented among genes whose loss causes early development lethality (DL) in mice.14 TMEM63B was prioritized among the potential candidates for human developmental disorders based on the overlap between genes that cause DL in mouse and those carrying de novo variants in large-scale human rare disease sequencing datasets.14 Of five individuals with de novo TMEM63B variants included in the Deciphering Developmental Disorders (DDD28) and 100KGP29 studies, with minimal phenotypic information, we could retrieve detailed clinical data in three, which we added to this series (IDs 4, 6, and 7; Table 1).
All ten variants were distributed in a TM domain conserved among osmosensitive calcium-permeable cation channels and affected residues under selective pressure in the general population, consistently with their predicted pathogenicity. None were a clear LoF/haploinsufficient variant (e.g., causing frameshift or premature stop codon). Heterozygous deletions including TMEM63B are rarely reported in the Database of Genomic Variants, where at least two individuals carry truncating deletions removing most of the gene (http://dgv.tcag.ca/; supporting variants nssv1153700 and nssv538990). Through our matchmaking initiative, we became aware of an individual with an unrelated phenotype and a homozygous p.Arg325∗ truncating variant of TMEM63B present in both healthy heterozygous parents (unpublished data). As we demonstrated in transfected Neuro2A, this variant abolished the expression of the mutated TMEM63B, in line with the predicted NMD.
These observations argue against haploinsufficiency being the obvious pathogenic mechanism in our cohort, as also supported by structural modeling and functional data. Structural modeling suggested mild changes in eight of ten of the variants without a disruptive effect on the protein structure. All ten variants affected TM helices, including TM4–TM8, constituting the channel pore, or TM1 and TM7, involved in sensing membrane tension,2 and were predicted to cause minimal structural changes, such as disruption or creation of single salt bridges and changes in cavity volume. The predicted mutational consequences might therefore imply altered channel function, e.g., by stabilizing the pore opening or affecting ion permeability and selectivity. In line with this hypothesis, and as expected for variants sparing protein folding, none of the six variants we tested in vitro impaired protein localization in the plasma membrane.
The p.Val44Met was recurrent in seven individuals, and the remaining variants clustered across a ∼270-residue TM region. Recurring mutations, as well as mutations with spatial clustering patterns, may exert their pathogenic effect through disease mechanisms other than LoF, such as gain of function (GoF) with enhanced activity or dominant-negative effects.30,31
We cannot exclude that variants in domains other than the TM helices or differently affecting the TMEM63B function, including LoF, might cause not-yet-described heterogeneous disorders.
In two independent Drosophila models, loss of Tmem63 resulted in viable flies not exhibiting gross defects in coordination but lacking the ability to discriminate food texture or humidity.32,33 In one of these models, the ectopic expression of the human TMEM63B gene in knockout flies rescued the defective phenotype in moisture attraction, demonstrating functional conservation of the two orthologs.33 We thus decided to model the p.Val44Met and p.Gly580Cys variants in Drosophila. However, both p.Val44Met and p.Gly580Cys TMEM63B transgenes caused lethal toxicity in our models. In contrast, the phenotype of the WT TMEM63B-expressing flies was indistinguishable from the control animals. These observations suggest that the variants we tested cause a toxic GoF of TMEM63B rather than LoF.
Heterozygous variants of TMEM63A have been associated with developmental delay and hypomyelination.6,7,8 Disease-associated missense variants are enriched at amino acid sites that are conserved across paralogs,34 and differences in the clinical consequences of variants in paralogs may be due to different expression patterns and novel functions emerged with adaptive evolution.35 When aligning protein sequence of and comparing variants positions in TMEM63B and TMEM63A (Figures 1A and S2), we found that the glycine 580 mutated to serine in ID 14 and to cysteine in ID 15 corresponds to the glycine 567 recurrently mutated to serine in TMEM63A in two unrelated individuals with transient hypomyelination.6 The p.Gly580Ser affects a buried glycine in TM7. In the Arabidopsis AtOSCA1.1 channel, the corresponding glycine 528 is located in a bending of the TM6 helix.2 Targeted mutagenesis of glycine 528 to alanine or proline reduces the pressure necessary to elicit a current, suggesting that channel activation might involve straightening of M6 around glycine 528 to relieve the blockage of the ion channel pore.2 According to in silico prediction and structural modeling, we might expect that both variants of glycine 580 stabilize the structure of the pore.
Multiple expression datasets indicate a complementary expression pattern between TMEM63A and TMEM63B, with the first being mainly expressed in oligodendrocytes and the second strongly expressed in neurons and to a lesser extent in astrocytes and oligodendrocyte precursor cells (OPCs) (Figure S10; https://singlecell.broadinstitute.org; https://www.proteinatlas.org/). The six individuals reported to date with TMEM63A variants had follow-ups of variable duration and exhibited phenotypes that were either similar to our individuals’7,8 or milder in those whose white matter changes improved with age,6 a phenomenon we did not observe.
For in vitro functional studies, we focused on the TMEM63B mRNA isoform most represented in the human cerebral cortex, the main generator of epileptogenic activity, and, in line with previous approaches,4 we used Neuro2A cells to model six selected variants (p.Val44Met, p.Arg433His, p.Thr481Asn, p.Gly580Ser, p.Arg660Thr, and p.Phe697Leu). In physiologic isotonic conditions, the −80 mV step revealed inward leak currents in cells expressing mutants but not WT TMEM63B or control cells, indicating that the variants lead to a gain in conductance even in the absence of the hypo-osmotic stimulus gating the channel.
Under a hypo-osmotic challenge, we measured similar values of inward current amplitude in both mutant- and WT TMEM63B-expressing Neuro2A cells, suggesting that the variants do not alter the maximal channel conductance. However, the overall increase between currents measured in hypo-osmotic and isotonic conditions was larger in WT- than in mutant TMEM63B-expressing cells. Similarly, when we examined cells expressing the p.Val44Met, p.Arg433His, and p.Thr481Asn variants by calcium imaging, the Ca2+ transients generated under hypo-osmotic stimulation were smaller across the mutant channels compared with the WT. One possible explanation for these observations is that those three variants result in a selective reduction of the relative permeability for Ca2+ in favor of Na+.
Inward cationic leak currents in neural cells expressing TMEM63B mutants may lead to altered neuronal excitability and/or impaired Ca2+ homeostasis. In C57BL/6N-FVB/N Tmem63b knockout mice, progressive hearing loss due to necroptosis of the outer hair cells (OHCs),4 which face severe shape- and volume-changing conditions, was suggested to reflect an abnormal response to osmotic and mechanical stimuli, ultimately leading to cell death.4 Brain shrinking with white matter changes, the main imaging finding in our series, might indicate neuronal loss, defective myelination, and possibly defective oligodendrocyte development. Clinical and imaging findings suggest that cell damage is already present at birth but further progresses postnatally. Already during prenatal life, several factors may determine osmotic challenges to the brain, including hydration changes, electrolyte imbalance, and mechanical stress. In a brain where neural cells are exceedingly vulnerable to even trivial volume changes and electrolytic imbalances, recurrent seizures may, in a vicious circle, make TMEM63B-defective neurons particularly susceptible to osmotic imbalance. During seizure activity, the extracellular environment surrounding axons and oligodendrocytes undergoes changes in volume and osmolarity,36 and given the very small volume of the extracellular space surrounding myelinated axons, oligodendrocytes might be subjected to continuous changes in local osmolarity. Unfortunately, our understanding of the homeostasis of the extracellular space is still in its infancy.37 In line with the above considerations, the epilepsy phenotype observed in our cohort is considerably more severe compared with other neurological conditions featuring structural damage of similar magnitude (i.e., cerebral palsy due to vascular injury).38
We hypothesize that the severe epileptic encephalopathy present in our individuals is caused by multiple converging factors including alteration of electrogenesis due to diffusely impaired ion currents and susceptibility to osmotic imbalance, resulting in structural brain damage, which can in turn be epileptogenic.
In 12 individuals, we also observed hematological abnormalities of variable severity, with signs of chronic hemolysis and myelodysplasia. In inherited hemolytic anemias, abnormally shaped RBCs may reflect disorders of cation permeability in the membrane, resulting in cellular over- or dehydration. Dehydrated hereditary stomatocytosis (DHSt), a rare congenital hemolytic anemia, is caused by dominant GoF mutations of PIEZO1, encoding for a stretch-activated ion channel.39,40 In some individuals with DHSt, hematologic abnormalities are subtle,39,40 as also seen in most of our individuals in whom they were revealed. In DHSt, PIEZO1 mutations keep the channel in an open conformation with prolonged activity, resulting in Ca2+ influx and consequent K+ efflux though Ca-sensitive Gardos channels, decreasing intracellular osmolarity and causing dehydration of RBCs.40,41 Although the role of TMEM63B in RBCs remains to be elucidated, it is known to be expressed in the RBC membrane (http://rbcc.hegelab.org/), such as PIEZO1, and might act similarly.
Erythrocytes are highly deformable cells. The intense mechanical solicitations they face when circulating through the brain microvasculature require highly effective mechanosensory feedback mechanisms. Under normal healthy conditions, deformability of their membrane allows RBCs to flow through vessels of diameter less than the cell diameter (7–8 μm), ensuring robust tissue perfusion and oxygen delivery.42 This RBC feature is particularly critical in the brain microvascular network, where the diameter of cortical capillaries is around 4–5 μm.43 RBCs also play an active role in stabilizing neurovascular flow dynamics, both by their physical properties44 and by targeting the vascular endothelium with vasoactive molecules.45 Chronic anemic changes and the physical vulnerability of TMEM63B-defective RBCs might contribute to white matter abnormalities, which often predominate in watershed vascular areas in the individuals reported here (Figure 1). Similar cerebrovascular complications are known to occur in individuals with hereditary hemolytic anemia.46
In conclusion, TMEM63B DEE represents a recognizable clinicopathological entity deriving from the dysfunctional behavior of a highly conserved stretch-activated ion channel. TMEM63B variants cause a gain in conductance with impaired Ca2+ homeostasis and are associated with neurodegenerative changes that start during prenatal brain development and slowly progress during postnatal development and adulthood, affecting the cerebral white and gray matter and cerebellum. The clinical counterpart of such widespread anatomic abnormalities includes severe early-onset epilepsy, associated with moderate-to-profound intellectual disability and severe motor and cortical visual impairment. Although a clear genotype-phenotype correlation is not yet possible, the phenotype associated with the recurrent p.Val44Met variant is uniformly severe. Concomitant hematological changes make this a complex syndrome also featuring potentially life-threatening acute hemolytic episodes occurring without obvious triggers.
Consortia
The TMEM63B collaborators are Francesca Pochiero, Francesco Mari, Venkateswaran Ramesh, Valeria Capra, Margherita Mancardi, Boris Keren, Cyiril Mignot, Matteo Lulli, Kendall Parks, Helen Griffin, Melanie Brugger, Telethon Undiagnosed Diseases Program (TUDP) consortium, Vincenzo Nigro, Yuko Hirata, Reiko Koichihara, Borut Peterlin, Yuko Hirata, Ryuto Maki, and Yohei Nitta.
The members of the Genomics England Research Consortium are J.C. Ambrose, P. Arumugam, R. Bevers, M. Bleda, F. Boardman-Pretty, C.R. Boustred, H. Brittain, M.A. Brown, M.J. Caulfield, G.C. Chan, A. Giess, J. Griffin, A. Hamblin, S. Henderson, T.J.P. Hubbard, R. Jackson, L.J. Jones, D. Kasperaviciute, M. Kayikci, A. Kousathanas, L. Lahnstein, A. Lakey, S.E A. Leigh, I.U.S. Leong, F.J. Lopez, F. Maleady-Crowe, M. McEntagart, F. Minneci, J. Mitchell, L. Moutsianas, M. Mueller, N. Murugaesu, A.C. Need, P. O’Donovan, C.A. Odhams, C. Patch, D. Perez-Gil, M. B. Pereira, J. Pullinger, T. Rahim, A. Rendon, T. Rogers, K. Savage, K. Sawant, R.H. Scott, A. Siddiq, A. Sieghart, S.C. Smith, A. Sosinsky, A. Stuckey, M. Tanguy, A.L. Taylor Tavares, E.R.A. Thomas, S.R. Thompson, A. Tucci, M.J. Welland, E. Williams, K. Witkowska, S.M. Wood, and M. Zarowiecki.
Acknowledgments
We are grateful to all the research subjects and family members for their participation in this study. This work was generated within the European Reference Networks EpiCARE and ITHACA. This work was supported by grants to R.G. from the Tuscany Region Call for Health 2018 (grant DECODE-EE) and Fondazione Cassa di Risparmio di Firenze (Human Brain Optical Mapping Project). Support to E.A. in sequencing and analysis was provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) and by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung, and Blood Institute grant UM1 HG008900 and in part by National Human Genome Research Institute grant R01 HG009141. A.T. was supported by Fondazione Telethon, Telethon Undiagnosed Diseases Program (grant GSP15001). N.M. received support from the Japan Agency for Medical Research and Development (AMED) under grant numbers JP22ek0109486, JP22ek0109549, and JP22ek0109493 and the Takeda Science Foundation. T.R. was supported by the Australian NHMRC Centre for Research Excellence in Neurocognition (1117394).
Author contributions
R.G. designed the research study, acquired and analyzed clinical and genetic data, and drafted the manuscript; A.V. participated in designing the research study, analyzed genetic data, and drafted the manuscript; S.B. acquired and analyzed clinical data and participated in drafting the manuscript; C.P. analyzed functional data, performed statistical analysis, and participated in drafting the manuscript; G.M.R. participated in designing functional studies in transfected cells, analyzed calcium imaging data, and performed statistical analysis; V.C. participated in designing functional studies and in genetic and protein structural data analysis; A.M., G.M., R.P., and S.G. performed experimental analyses in transfected cells; K.H. and H.O. participated in protein structural data analysis; A.D., C.v.S., E.V., G.B., J.J.v.d.S., M.K., M.P.M., M. Sakamoto, N.M., R.B., R.R., S.H., T.B., and T.R. acquired clinical and genetic data; E.A., E.K.B., J.T., K.W., M. Scala, R.K., S.M., and S.V. acquired clinical data; A.T., D.M., D.S., E.H.S., K.M., K.v.G., M.J.V.H., P.C., and T.C. acquired genetic data; A.S. and T.S. produced and analyzed Drosophila models.
Declaration of interests
The authors have declared that no conflict of interest exists.
Published: July 7, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.06.008.
Contributor Information
Renzo Guerrini, Email: r.guerrini@meyer.it.
TMEM63B collaborators:
Francesca Pochiero, Francesco Mari, Venkateswaran Ramesh, Valeria Capra, Margherita Mancardi, Boris Keren, Cyiril Mignot, Matteo Lulli, Kendall Parks, Helen Griffin, Melanie Brugger, Vincenzo Nigro, Yuko Hirata, Reiko Koichihara, Borut Peterlin, Yuko Hirata, Ryuto Maki, and Yohei Nitta
The Genomics England Research Consortium:
John C. Ambrose, Prabhu Arumugam, Roel Bevers, Marta Bleda, Freya Boardman-Pretty, Christopher R. Boustred, Helen Brittain, Matthew A. Brown, Mark J. Caulfield, Georgia C. Chan, Adam Giess, John N. Griffin, Angela Hamblin, Shirley Henderson, Tim J.P. Hubbard, Rob Jackson, Louise J. Jones, Dalia Kasperaviciute, Melis Kayikci, Athanasios Kousathanas, Lea Lahnstein, Anna Lakey, Sarah E.A. Leigh, Ivonne U.S. Leong, Javier F. Lopez, Fiona Maleady-Crowe, Meriel McEntagart, Federico Minneci, Jonathan Mitchell, Loukas Moutsianas, Michael Mueller, Nirupa Murugaesu, Anna C. Need, Peter O’Donovan, Chris A. Odhams, Christine Patch, Daniel Perez-Gil, Marina B. Pereira, John Pullinger, Tahrima Rahim, Augusto Rendon, Tim Rogers, Kevin Savage, Kushmita Sawant, Richard H. Scott, Afshan Siddiq, Alexander Sieghart, Samuel C. Smith, Alona Sosinsky, Alexander Stuckey, Mélanie Tanguy, Ana Lisa Taylor Tavares, Ellen R.A. Thomas, Simon R. Thompson, Arianna Tucci, Matthew J. Welland, Eleanor Williams, Katarzyna Witkowska, Suzanne M. Wood, and Magdalena Zarowiecki
Supplemental information
Data and code availability
The data supporting the findings of this study are available within the article and/or its supplemental information. The exome datasets supporting this study have not been deposited in a public repository due to privacy and ethical/legal issues. TMEM63B genetic variants identified in our study have been submitted to DECIPHER (https://www.deciphergenomics.org; accession IDs 512417 and 512438–512452). Any additional raw data are available on request from the corresponding author.
References
- 1.Murthy S.E., Dubin A.E., Whitwam T., Jojoa-Cruz S., Cahalan S.M., Mousavi S.A.R., Ward A.B., Patapoutian A. OSCA/TMEM63 are an evolutionarily conserved family of mechanically activated ion channels. Elife. 2018;7 doi: 10.7554/eLife.41844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang M., Wang D., Kang Y., Wu J.-X., Yao F., Pan C., Yan Z., Song C., Chen L. Structure of the mechanosensitive OSCA channels. Nat. Struct. Mol. Biol. 2018;25:850–858. doi: 10.1038/s41594-018-0117-6. [DOI] [PubMed] [Google Scholar]
- 3.Yuan F., Yang H., Xue Y., Kong D., Ye R., Li C., Zhang J., Theprungsirikul L., Shrift T., Krichilsky B., et al. OSCA1 mediates osmotic-stress-evoked Ca2+ increases vital for osmosensing in Arabidopsis. Nature. 2014;514:367–371. doi: 10.1038/nature13593. [DOI] [PubMed] [Google Scholar]
- 4.Du H., Ye C., Wu D., Zang Y.Y., Zhang L., Chen C., He X.Y., Yang J.J., Hu P., Xu Z., et al. The Cation Channel TMEM63B Is an Osmosensor Required for Hearing. Cell Rep. 2020;31:107596. doi: 10.1016/j.celrep.2020.107596. [DOI] [PubMed] [Google Scholar]
- 5.Lang F., Busch G.L., Ritter M., Völkl H., Waldegger S., Gulbins E., Häussinger D. Functional significance of cell volume regulatory mechanisms. Physiol. Rev. 1998;78:247–306. doi: 10.1152/physrev.1998.78.1.247. [DOI] [PubMed] [Google Scholar]
- 6.Yan H., Helman G., Murthy S.E., Ji H., Crawford J., Kubisiak T., Bent S.J., Xiao J., Taft R.J., Coombs A., et al. Heterozygous Variants in the Mechanosensitive Ion Channel TMEM63A Result in Transient Hypomyelination during Infancy. Am. J. Hum. Genet. 2019;105:996–1004. doi: 10.1016/j.ajhg.2019.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukumura S., Hiraide T., Yamamoto A., Tsuchida K., Aoto K., Nakashima M., Saitsu H. A novel de novo TMEM63A variant in a patient with severe hypomyelination and global developmental delay. Brain Dev. 2022;44:178–183. doi: 10.1016/j.braindev.2021.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Tonduti D., Mura E., Masnada S., Bertini E., Aiello C., Zini D., Parmeggiani L., Cantalupo G., Talenti G., Veggiotti P., et al. Spinal cord involvement and paroxysmal events in “Infantile Onset Transient Hypomyelination” due to TMEM63A mutation. J. Hum. Genet. 2021;66:1035–1037. doi: 10.1038/s10038-021-00921-1. [DOI] [PubMed] [Google Scholar]
- 9.Tábara L.C., Al-Salmi F., Maroofian R., Al-Futaisi A.M., Al-Murshedi F., Kennedy J., Day J.O., Courtin T., Al-Khayat A., Galedari H., et al. TMEM63C mutations cause mitochondrial morphology defects and underlie hereditary spastic paraplegia. Brain. 2022;145:3095–3107. doi: 10.1093/brain/awac123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu D., Zang Y.Y., Shi Y.Y., Ye C., Cai W.M., Tang X.H., Zhao L., Liu Y., Gan Z., Chen G.Q., et al. Distant coupling between RNA editing and alternative splicing of the osmosensitive cation channel tmem63b. J. Biol. Chem. 2020;295:18199–18212. doi: 10.1074/jbc.RA120.016049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vetro A., Nielsen H.N., Holm R., Hevner R.F., Parrini E., Powis Z., Møller R.S., Bellan C., Simonati A., Lesca G., et al. ATP1A2-and ATP1A3-associated early profound epileptic encephalopathy and polymicrogyria. Brain. 2021;144:1435–1450. doi: 10.1093/brain/awab052. [DOI] [PubMed] [Google Scholar]
- 12.Sobreira N., Schiettecatte F., Valle D., Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015;36:928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheffer I.E., Berkovic S., Capovilla G., Connolly M.B., French J., Guilhoto L., Hirsch E., Jain S., Mathern G.W., Moshé S.L., et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017;58:512–521. doi: 10.1111/epi.13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cacheiro P., Muñoz-Fuentes V., Murray S.A., Dickinson M.E., Bucan M., Nutter L.M.J., Peterson K.A., Haselimashhadi H., Flenniken A.M., Morgan H., et al. Human and mouse essentiality screens as a resource for disease gene discovery. Nat. Commun. 2020;11:655. doi: 10.1038/s41467-020-14284-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wiel L., Baakman C., Gilissen D., Veltman J.A., Vriend G., Gilissen C. MetaDome: Pathogenicity analysis of genetic variants through aggregation of homologous human protein domains. Hum. Mutat. 2019;40:1030–1038. doi: 10.1002/humu.23798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou C., Tian W., Kleist T., He K., Garcia V., Bai F., Hao Y., Luan S., Li L. DUF221 proteins are a family of osmosensitive calcium-permeable cation channels conserved across eukaryotes. Cell Res. 2014;24:632–635. doi: 10.1038/cr.2014.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maity K., Heumann J.M., McGrath A.P., Kopcho N.J., Hsu P.K., Lee C.W., Mapes J.H., Garza D., Krishnan S., Morgan G.P., et al. Cryo-EM structure of OSCA1.2 from Oryza sativa elucidates the mechanical basis of potential membrane hyperosmolality gating. Proc. Natl. Acad. Sci. USA. 2019;116:14309–14318. doi: 10.1073/pnas.1900774116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X., Wang J., Sun L. Structure of the hyperosmolality-gated calcium-permeable channel OSCA1.2. Nat. Commun. 2018;9:5060. doi: 10.1038/s41467-018-07564-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ittisoponpisan S., Islam S.A., Khanna T., Alhuzimi E., David A., Sternberg M.J.E. Can Predicted Protein 3D Structures Provide Reliable Insights into whether Missense Variants Are Disease Associated? J. Mol. Biol. 2019;431:2197–2212. doi: 10.1016/j.jmb.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo R., Cang Z., Yao J., Kim M., Deans E., Wei G., Kang S.G., Hong H. Structural cavities are critical to balancing stability and activity of a membrane-integral enzyme. Proc. Natl. Acad. Sci. USA. 2020;117:22146–22156. doi: 10.1073/pnas.1917770117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marques M.C., Albuquerque I.S., Vaz S.H., Bernardes G.J.L. Overexpression of Osmosensitive Ca2+-Permeable Channel TMEM63B Promotes Migration in HEK293T Cells. Biochemistry. 2019;58:2861–2866. doi: 10.1021/acs.biochem.9b00224. [DOI] [PubMed] [Google Scholar]
- 22.Zhao X., Yan X., Liu Y., Zhang P., Ni X. Co-expression of mouse TMEM63A, TMEM63B and TMEM63C confers hyperosmolarity activated ion currents in HEK293 cells. Cell Biochem. Funct. 2016;34:238–241. doi: 10.1002/cbf.3185. [DOI] [PubMed] [Google Scholar]
- 23.Iyer J., Wang Q., Le T., Pizzo L., Grönke S., Ambegaokar S.S., Imai Y., Srivastava A., Troisí B.L., Mardon G., et al. Quantitative Assessment of Eye Phenotypes for Functional Genetic Studies Using Drosophila melanogaster. G3. 2016;6:1427–1437. doi: 10.1534/g3.116.027060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kramer J.M., Staveley B.E. GAL4 causes developmental defects and apoptosis when expressed in the developing eye of Drosophila melanogaster. Genet. Mol. Res. 2003;2:43–47. [PubMed] [Google Scholar]
- 25.Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 26.Andreu-Arasa V.C., Chapman M.N., Kuno H., Fujita A., Sakai O. Craniofacial manifestations of systemic disorders: CT and MR imaging findings and imaging approach. Radiographics. 2018;38:890–911. doi: 10.1148/rg.2018170145. [DOI] [PubMed] [Google Scholar]
- 27.Dickinson M.E., Flenniken A.M., Ji X., Teboul L., Wong M.D., White J.K., Meehan T.F., Weninger W.J., Westerberg H., Adissu H., et al. High-throughput discovery of novel developmental phenotypes. Nature. 2016;537:508–514. doi: 10.1038/nature19356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Firth H.V., Richards S.M., Bevan A.P., Clayton S., Corpas M., Rajan D., Van Vooren S., Moreau Y., Pettett R.M., Carter N.P. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 2009;84:524–533. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turnbull C., Scott R.H., Thomas E., Jones L., Murugaesu N., Pretty F.B., Halai D., Baple E., Craig C., Hamblin A., et al. The 100 000 Genomes Project: Bringing whole genome sequencing to the NHS. BMJ. 2018;361:k1687. doi: 10.1136/bmj.k1687. [DOI] [PubMed] [Google Scholar]
- 30.Lelieveld S.H., Wiel L., Venselaar H., Pfundt R., Vriend G., Veltman J.A., Brunner H.G., Vissers L.E.L.M., Gilissen C. Spatial Clustering of de Novo Missense Mutations Identifies Candidate Neurodevelopmental Disorder-Associated Genes. Am. J. Hum. Genet. 2017;101:478–484. doi: 10.1016/j.ajhg.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkie A.O. The molecular basis of genetic dominance. J. Med. Genet. 1994;31:89–98. doi: 10.1136/jmg.31.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Q., Montell C. Mechanism for food texture preference based on grittiness. Curr. Biol. 2021;31:1850–1861.e6. doi: 10.1016/j.cub.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li S., Li B., Gao L., Wang J., Yan Z. Humidity response in Drosophila olfactory sensory neurons requires the mechanosensitive channel TMEM63. Nat. Commun. 2022;13:3814. doi: 10.1038/s41467-022-31253-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lal D., May P., Perez-Palma E., Samocha K.E., Kosmicki J.A., Robinson E.B., Møller R.S., Krause R., Nürnberg P., Weckhuysen S., et al. Gene family information facilitates variant interpretation and identification of disease-associated genes in neurodevelopmental disorders. Genome Med. 2020;12:28. doi: 10.1186/s13073-020-00725-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guschanski K., Warnefors M., Kaessmann H. The evolution of duplicate gene expression in mammalian organs. Genome Res. 2017;27:1461–1474. doi: 10.1101/gr.215566.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lux H.D., Heinemann U., Dietzel I. Ionic changes and alterations in the size of the extracellular space during epileptic activity. Adv. Neurol. 1986;44:619–639. [PubMed] [Google Scholar]
- 37.Sætra M.J., Einevoll G.T., Halnes G. An electrodiffusive neuron-extracellular-glia model for exploring the genesis of slow potentials in the brain. PLoS Comput. Biol. 2021;17:e1008143. doi: 10.1371/journal.pcbi.1008143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cooper M.S., Mackay M.T., Dagia C., Fahey M.C., Howell K.B., Reddihough D., Reid S., Harvey A.S. Epilepsy syndromes in cerebral palsy: varied, evolving and mostly self-limited. Brain. 2023;146:587–599. doi: 10.1093/brain/awac274. [DOI] [PubMed] [Google Scholar]
- 39.Andolfo I., Alper S.L., De Franceschi L., Auriemma C., Russo R., De Falco L., Vallefuoco F., Esposito M.R., Vandorpe D.H., Shmukler B.E., et al. Multiple clinical forms of dehydrated hereditary stomatocytosis arise from mutations in PIEZO1. Blood. 2013;121:3925–3935. doi: 10.1182/blood-2013-02-482489. [DOI] [PubMed] [Google Scholar]
- 40.Albuisson J., Murthy S.E., Bandell M., Coste B., Louis-Dit-Picard H., Mathur J., Fénéant-Thibault M., Tertian G., De Jaureguiberry J.P., Syfuss P.Y., et al. Dehydrated hereditary stomatocytosis linked to gain-of-function mutations in mechanically activated PIEZO1 ion channels. Nat. Commun. 2013;4:1884. doi: 10.1038/ncomms2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caulier A., Garçon L. PIEZO1, sensing the touch during erythropoiesis. Curr. Opin. Hematol. 2022;29:112–118. doi: 10.1097/MOH.0000000000000706. [DOI] [PubMed] [Google Scholar]
- 42.Ebrahimi S., Bagchi P. A computational study of red blood cell deformability effect on hemodynamic alteration in capillary vessel networks. Sci. Rep. 2022;12:4304. doi: 10.1038/s41598-022-08357-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmid F., Barrett M.J.P., Jenny P., Weber B. Vascular density and distribution in neocortex. Neuroimage. 2019;197:792–805. doi: 10.1016/j.neuroimage.2017.06.046. [DOI] [PubMed] [Google Scholar]
- 44.Schmid F., Barrett M.J.P., Obrist D., Weber B., Jenny P. Red blood cells stabilize flow in brain microvascular networks. PLoS Comput. Biol. 2019;15:e1007231. doi: 10.1371/journal.pcbi.1007231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richardson K.J., Kuck L., Simmonds M.J. Beyond oxygen transport: active role of erythrocytes in the regulation of blood flow. Am. J. Physiol. Heart Circ. Physiol. 2020;319:H866–H872. doi: 10.1152/AJPHEART.00441.2020. [DOI] [PubMed] [Google Scholar]
- 46.Patel R., Sabat S., Kanekar S. Imaging Manifestations of Neurologic Complications in Anemia. Hematol. Oncol. Clin. North Am. 30. 2016;30:733–756. doi: 10.1016/j.hoc.2016.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available within the article and/or its supplemental information. The exome datasets supporting this study have not been deposited in a public repository due to privacy and ethical/legal issues. TMEM63B genetic variants identified in our study have been submitted to DECIPHER (https://www.deciphergenomics.org; accession IDs 512417 and 512438–512452). Any additional raw data are available on request from the corresponding author.