

Summary
Spinal and bulbar muscular atrophy (SBMA) is a neuromuscular disease with substantial mitochondrial and metabolic dysfunctions. SBMA is caused by polyglutamine (polyQ) expansion in the androgen receptor (AR). Activating or increasing the NAD+-dependent deacetylase, SIRT3, reduced oxidative stress and death of cells modeling SBMA. However, increasing diminished SIRT3 in AR100Q mice failed to reduce acetylation of the SIRT3 target/antioxidant, SOD2, and had no effect on increased total acetylated peptides in quadriceps. Yet, overexpressing SIRT3 resulted in a trend of motor recovery, and corrected TCA cycle activity by decreasing acetylation of SIRT3 target proteins. We sought to boost blunted SIRT3 activity by replenishing diminished NAD+ with PARP inhibition. Although NAD+ was not affected, overexpressing SIRT3 with PARP inhibition fully restored hexokinase activity, correcting the glycolytic pathway in AR100Q quadriceps, and rescued motor endurance of SBMA mice. These data demonstrate that targeting metabolic anomalies can restore motor function downstream of polyQ-expanded AR.
Subject areas: Biological sciences, Biochemistry, Molecular biology, Cell biology
Graphical abstract
Highlights
-
•
Peptide acetylation is elevated and SIRT3 protein and activity are diminished in SBMA
-
•
Increasing SIRT3 corrects TCA cycle activity in SBMA mouse quadriceps
-
•
PARP inhibition does not rescue reduced NAD+ in SBMA mouse skeletal muscle
-
•
Increased SIRT3 + PARP inhibition rescues hexokinase activity and exercise endurance
Biological sciences; Biochemistry; Molecular biology; Cell biology
Introduction
Spinal and bulbar muscular atrophy (SBMA) is an adult-onset progressive neuromuscular disease that is caused by a CAG repeat expansion in the androgen receptor (AR) gene, encoding a polyglutamine (polyQ) tract.1,2 In response to androgens, the polyQ-expanded AR misfolds and aggregates within the nucleus, inciting toxicity, primarily in lower motor neurons and skeletal muscle.3,4,5,6 This manifests in men as weakness and atrophy of mainly proximal/distal limbs, and bulbar musculature, resulting in difficulty with walking, speaking, and swallowing.1,7,8
The polyQ-expanded AR causes SBMA pathogenesis by disrupting multiple cellular processes, including mitochondrial function, DNA repair, metabolism, and energy production.3,9,10 Previous interventions that have improved either mitochondrial health11,12 or modulated metabolic processes3,9,13,14 have rescued motor dysfunction in mouse models of SBMA. Importantly, there is a substantial reduction in NAD+ in quadriceps of male SBMA knock-in (KI) mice immediately following exercise.3 NAD+ is a critical component of metabolism, and it is necessary for the activity of specific enzymes involved in many cellular functions, such as sirtuins and poly-ADP ribose polymerases (PARPs).
Sirtuins (SIRT) are a family of NAD+-dependent enzymes with relevant functions in aging and age-related diseases (reviewed in15), including SBMA.16 SIRT3 is the main deacetylase within the mitochondria and serves to protect against oxidative stress by deacetylating and activating SOD2, the main mitochondrial redox regulator.17 SIRT3 is also involved in modulating metabolism to support increased energy demands during various stress events, as it deacetylates (regulates) a number of enzymes in the tricarboxylic acid (TCA) cycle, fatty acid β-oxidation, and components of the electron transport chain (reviewed in18,19). SIRT3 has garnered much attention as a target for treating many conditions ranging from neurodegeneration to metabolic disorders, including Huntington’s disease20,21,22 (reviewed in23), which also involves polyQ expansion.
PARPs are NAD+-dependent enzymes involved in DNA repair and, in particular, PARP-1 can utilize the majority of cellular NAD+ when active.24 Furthermore, a high rate of NAD+ consumption promotes increased NAD+ synthesis and diminishes ATP reserves.25,26 A recent study found that the NAD Salvage I/II and NAD biosynthesis II pathways are upregulated in SBMA patient iPSC-derived motor neurons27, further indicating that NAD+ metabolism is dysregulated in SBMA. The study also revealed numerous metabolic anomalies in these motor neurons, including upregulation of genes involved in the TCA cycle.27 Although PARPs are best known for their role in DNA repair and transcriptional modulation, they are also involved in metabolic regulation by influencing mitochondrial function and oxidative metabolism (reviewed in28); more specifically, PARP-1 inhibits glycolysis, causing ATP loss.26
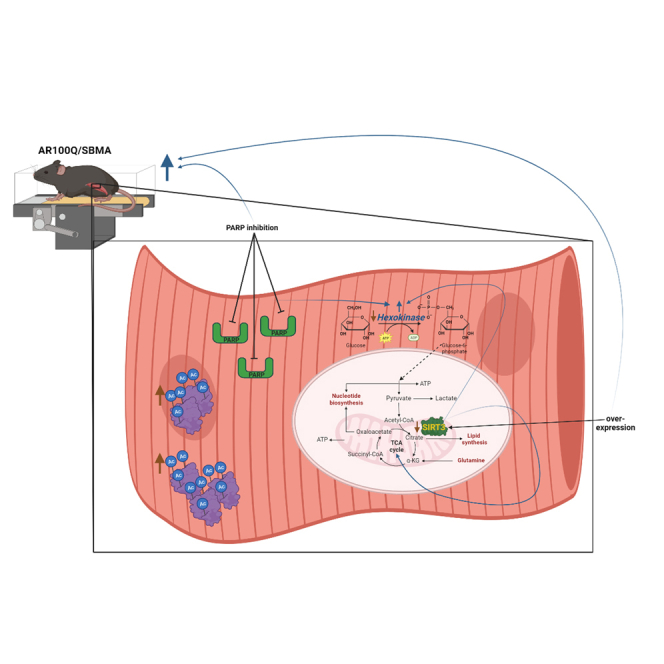
Herein, we found that SIRT3 protein levels are reduced in skeletal muscle of SBMA mice, and a combinatorial approach of increasing SIRT3 protein and inhibiting PARPs rescues exercise endurance of these mice. Acetylome analysis of AR100Q mouse quadriceps revealed increased TCA cycle activity that was corrected with SIRT3 overexpression, along with modulation of other metabolic pathways essential for energy production. The addition of the PARP inhibitor, olaparib, to increased SIRT3, fully restored the activity of hexokinase, the first enzyme in the glycolytic pathway, further imparting correction of energy producing metabolism. This treatment did not impact monomeric or aggregated polyQ-expanded AR protein levels. Thus, these studies highlight the critical impact of metabolic aberrations in the pathogenesis of SBMA and support the idea of a combined approach to correct cellular dysfunctions downstream of toxic disease-initiating proteins.
Results
SOD2 and SIRT3 proteins are reduced in the skeletal muscle of AR100Q mice
Previous studies have identified several dysfunctions of mitochondria, including increased ROS in models of SBMA.3,9,27,29,30 One of the main antioxidants, SOD2, is diminished at both the transcript and protein levels in a number of SBMA models,29,31,32 suggesting that its function may be compromised in SBMA. The transgenic mouse model of SBMA used in these studies expresses human AR with a polyQ tract of 100 glutamine residues (AR100Q), and reflects pathologies observed in human patients, with androgen-dependent and progressive muscle atrophy and motor dysfunction.30 We have also included non-glutamine expanded AR24Q transgenic mice in these studies to control for AR overexpression and, to note, these AR24Q males exhibit no motor deficits due to overexpression of AR.30 At late-stage disease (11 weeks) SOD2 was substantially decreased in the skeletal muscle of AR100Q mice compared to wild type (WT) siblings (Figure 1A, top panel). Since SOD2 acetylation renders the protein inactive,33 we evaluated the percent of SOD2 acetylated at lysine 68, which was unchanged compared to controls (WT and AR24Q) (Figure 1A, bottom panel).
Figure 1.

SIRT3 and SOD2 proteins are reduced in AR100Q mice, and activating and increasing SIRT3 reduces ROS and cell death in vitro
(A) Western analysis of quadriceps from 11 week-old WT, AR24Q, and AR100Q male mice (n = 3). Top, total SOD2 with tubulin loading control, and densitometry analysis of total SOD2 compared to tubulin. Bottom, a second SDS-PAGE gel probed for acetylated (acetyl-K68) SOD2 with tubulin loading control, and densitometry analysis of acetyl-K68/total SOD2 (from A top) after each were normalized to tubulin loading control.
(B) Western analysis of quadriceps from 11 week-old WT, AR24Q, and AR100Q male mice (n = 3). Endogenous SIRT3 and tubulin loading control with densitometry analysis of SIRT3 normalized to tubulin loading control.
(C) ROS levels in AR112Q expressing PC12 cells (n = 12) treated with 10 nM DHT plus either DMSO (vehicle control), 1 μM viniferin, 1 μM honokiol, or 100 μM melatonin for 48 h (representative image from 3 experiments).
(D) Cell death of AR100Q expressing C2C12 myotubes (n = 3) treated with either DMSO (vehicle control), 1 μM viniferin, 1 μM honokiol, or 100 μM melatonin for 10 days (representative image from 3 experiments).
(E) Left, immunoblot of MYC-tagged SIRT3, and tubulin in AR112Q SIRT3-MYC-overexpressing PC12 cells (n = 3). Right, ROS levels in AR112Q SIRT3-MYC-overexpressing PC12 cells (n = 12) after 48 h of DHT treatment (representative image from 3 experiments).
(F) Left, immunoblot of MYC-tagged SIRT3, and tubulin loading control from AR100Q SIRT3-MYC-overexpressing C2C12 myoblasts (n = 3). Right, percent cell death of AR100Q SIRT3-MYC-overexpressing C2C12 myoblasts (n = 3) (LDH assay) (representative image from 3 experiments). ∗∗ = Nested two-way t-test, p ≤ 0.01.
(A–F) All statistical analysis show mean ± SD, and used a two-way ANOVA and Tukey’s post-hoc (∗ = p ≤ 0.05, ∗∗ = p ≤ 0.01, ∗∗∗ = p ≤ 0.001) unless otherwise noted. WT = wild type, AR24Q = non-polyQ-expanded AR, AR100Q = polyQ-expanded AR, EV = empty vector, SIRT3 = SIRT3-myc-his overexpression, SIRT3-H248Y = SIRT3-myc-his overexpression (deacetylase inactive).
The mitochondrial NAD-dependent deacetylase, SIRT3, deacetylates SOD2 at lysine 68 and 122,33 leading to its activation.33 Others have reported the transcript for SIRT3 is diminished in the tibialis anterior of AR100Q mice.32 We found that SIRT3 is significantly reduced compared to WT mice (Figure 1B). These observations reveal diminished levels of two key proteins for muscle metabolism and homeostasis, SOD2 and SIRT3.
SIRT3 activation or overexpression reduces ROS and death of cells modeling SBMA
Enhancing SIRT3 is protective in many disorders with mitochondrial dysfunction,20,21,34,35 and since SIRT3 is reduced in SBMA, we assessed the effects of activating SIRT3 with viniferin,20 honokiol,34 and melatonin35,36 on ROS and cell death in two cell models of SBMA. We used Tet-inducible PC12 cells harboring polyQ-expanded AR (AR112Q), which form nuclear aggregates of polyQ AR37 and die in response to dihydrotestosterone (DHT) treatment.16 AR112Q expressing PC12 cells were found to have increased ROS compared to non-polyQ-expanded AR-expressing (AR10Q) controls (Figure S1A), which was significantly reduced by all 3 SIRT3 activating compounds (Figure 1C). We also used C2C12 myotubes which stably express AR100Q and atrophy in response to DHT treatment.38 AR100Q expressing C2C12 myotubes have decreased viability compared to myotubes expressing a normal glutamine tract (AR24Q) (Figure S1B), and all of the SIRT3 activators reduced death of AR100Q myotubes (Figure 1D).
Next, we sought to determine whether increasing the SIRT3 protein would also be of benefit in cell models of SBMA. We generated Tet-inducible AR112Q PC12 clonal cell lines that stably and constitutively overexpress human SIRT3-myc-his (WT), human SIRT3-H248Y-myc-his (deacetylase inactive),39 or contain an empty vector (EV) to serve as the control (Figure 1E). AR112Q-SIRT3-WT and AR112Q-SIRT3-H248Y clonal PC12 cell lines were analyzed by immunoblot for myc expression (SIRT3), and two pairs of cell lines with matched myc levels (SIRT3-WT and SIRT3-H248Y) were chosen for further experimentation (Figures 1E and S1C). ROS levels were reduced in AR112Q-expressing PC12 cell lines with over-expressed SIRT3-WT, and increased in AR112Q-expressing PC12 cells with over-expressed deacetylase deficient SIRT3-H248Y (Figure 1E), indicating that the deacetylase activity of SIRT3 is necessary for the protective function of SIRT3 in these cells. Additionally, we generated AR100Q C2C12 clonal cell lines that stably and constitutively overexpress human SIRT3-myc-his (WT) or EV control (Figures 1F and S1D). We evaluated C2C12 myoblast viability since the high passage number resulting from the generation of clonal lines prevented these cells from differentiating into myotubes. SIRT3 overexpression substantially and significantly reduced death of AR100Q C2C12 myoblasts (Figure 1F). Altogether, these results indicate that SIRT3 activation or overexpression reduces polyQ-expanded AR-induced ROS and toxicity in vitro.
SIRT3 overexpression increases the mass of AR100Q mouse quadriceps
Since SIRT3 activation and overexpression suppressed oxidative stress and toxicity in cell models of SBMA, we sought to determine whether overexpression of SIRT3 protein could rescue muscle pathology and motor deficits, or increase the lifespan of male mice modeling SBMA. We thus crossed transgenic mice over-expressing FLAG-tagged mouse M1 isoform of SIRT3 with AR100Q mice and evaluated the wet weights of the quadriceps and gastrocnemius. Both the quadriceps and gastrocnemius have substantial pathology in SBMA, which is prominent in this AR100Q model.30 SIRT3 overexpression in AR100Q mice significantly increased quadriceps muscle mass compared to AR100Q at age 8 weeks (Figure 2A). There was also a trend of an increase in the percent of total mass of the gastrocnemius in SIRT3-overexpressing AR100Q mice compared to AR100Q (Figure S2A).
Figure 2.

Overexpression of flag-tagged SIRT3 (mouse isoform 1) in AR100Q mice increases quadriceps muscle mass but does not rescue motor function. Exogenous SIRT3 and total SOD2 proteins are reduced in the AR100Q quadriceps
(A) Muscle mass (left) and percent of total body mass (right) of quadriceps from WT, SIRT3, AR100Q, and AR100Q-SIRT3 male mice (n = 3) at age 8 weeks.
(B) Behavioral cohort composed of 10 X WT, 10 X SIRT3 only, 10 X AR100Q, and 8 X AR100Q-SIRT3 male mice underwent analysis as indicated. Top, accelerating rotarod analysis was performed weekly, starting at age 6 weeks. Bottom, accelerating treadmill analysis at ages 8, 11, and 12 weeks. ∗ = difference between WT vs. AR100Q and AR100Q-SIRT3.
(C) Left, immunoblots of endogenous SIRT3 and flag-tagged SIRT3, and tubulin loading control in quadriceps muscle of WT, SIRT3, AR100Q, and AR100Q-SIRT3 male mice (n = 3) at age 11 weeks. Note that endogenous SIRT3 cannot be seen in WT or AR100Q because of early exposure time. Right, densitometry analysis of flag-tagged SIRT3 normalized to tubulin loading control. ∗ = Two-way t-test, p ≤ 0.05.
(D) Left, immunoblots of total and acetylated (acetyl-K68) SOD2 and tubulin loading controls (2 separate gels) in quadriceps muscle of WT, SIRT3, AR100Q, and AR100Q-SIRT3 male mice (n = 3) at age 11 weeks. Right, densitometry analysis of total SOD2/tubulin and acetyl-K68/total SOD2 after each were normalized to tubulin loading control.
(E) Acetylome analysis of SOD2 acetyl-K68 (left) and acetyl-K122 (right) intensity of quadriceps from WT, SIRT3, AR24Q, AR100Q, and AR100Q-SIRT3 male mice (n = 3) at age 11 weeks. (n = 2 for SIRT3 and AR100Q-SIRT3 due to outliers below the −3 correlation Z score cut off).
(A–E) All statistical analysis show mean ± SD, and used a two-way ANOVA and Tukey’s post-hoc (∗ = p ≤ 0.05, ∗∗ = p ≤ 0.01, ∗∗∗ = p ≤ 0.001) unless otherwise noted. WT, wild type; SIRT3, SIRT3-flag overexpression; AR24Q, non-polyQ-expanded; AR100Q, polyQ-expanded AR; AR100Q-SIRT3, polyQ-expanded AR with SIRT3-flag overexpression.
SIRT3 overexpression promotes a trend in motor function recovery in AR100Q mice
The increased skeletal muscle mass suggests that SIRT3 overexpression is providing some protection to skeletal muscle, therefore, we evaluated whether this would translate into enhanced motor function. A cohort of mice was established and evaluated for total body weight, grip strength, and their ability to remain on an accelerating rotarod and treadmill. WT and SIRT3 male mice exhibited similar motor function, with no difference in weight, percent weight change, or lifespan (Figures S2B–S2D). SIRT3 overexpression did not modify total body weight loss over time, grip strength deficits, or lifespan of AR100Q male mice (Figures S2B–S2D). However, the AR100Q-SIRT3 mice did have a trend of increased motor function on a rotarod and motor endurance on the treadmill, when compared to the AR100Q mice (Figure 2B). These results suggest that SIRT3 overexpression is not sufficient to ameliorate polyQ-expanded pathology in vivo, at least in this aggressive mouse model of SBMA. However, the increased quadriceps mass and the trend of increased rotarod and treadmill performance may be indicative of some protection afforded by increasing the SIRT3 protein in SBMA.
Exogenously overexpressed SIRT3 protein is decreased in AR100Q mouse quadriceps
We confirmed that in the quadriceps of SIRT3 and AR100Q-SIRT3 mice the exogenous SIRT3-flag (mouse isoform-1) is detected by anti-SIRT3, and anti-flag antibodies (Figure 2C). Unexpectedly, we observed there to be a substantial reduction (11-fold) of the exogenous SIRT3-flag in the AR100Q-SIRT3 mice compared to the SIRT3-flag only mice (Figure 2C). A reduction of endogenous SIRT3 in the AR100Q muscle can be explained by the reduced SIRT3 transcript identified in tibialis of similarly aged AR100Q mice.32 However, the 1.5-fold reduction of SIRT3 transcript cannot fully explain the 17-fold reduction of endogenous SIRT3 protein in AR100Q quadriceps (Figure 1B). Since the reduction of the exogenous SIRT3-flag protein is not due to reduced transcription, as it is not transcriptionally regulated by the mouse’s endogenous SIRT3 promoter, this further supports the idea that the half-life of the SIRT3 protein is diminished in the face of the polyQ-expanded AR protein. The enhanced SIRT3 protein turnover in AR100Q skeletal muscle may be due to exacerbated oxidative and metabolic stress, as has been observed in a number of studies evaluating cellular stress and the modulation of SIRT3 (reviewed in40). The reduced exogenous SIRT3-flag could be one mechanism by which this method of overexpression of SIRT3 is not sufficient to ameliorate motor dysfunction of AR100Q mice, as there may still not be enough SIRT3 protein to circumvent the toxicity of the AR100Q protein.
Total and acetylated SOD2 are not modified by overexpression of SIRT3 in AR100Q quadriceps
To evaluate the functional activity of SIRT3, we analyzed the acetylation status of SOD2.33 Again, total SOD2 was decreased in AR100Q mice, and this was unchanged in the presence of exogenously overexpressed SIRT3 (Figure 2D, top panel). Acetylation of SOD2 at lysine 68 also remained unchanged with overexpressed SIRT3 (Figure 2D, bottom panel). We used acetylome analysis as a broader approach to determine the acetylation status of peptides/proteins within quadriceps of a parallel cohort of 11-week-old mice. Again, there was no change in acetylated SOD2-K68 with SIRT3 overexpression (Figure 2E, left panel). However, we did note that there was an increase in acetylated (inactivated) SOD2-K122 in AR100Q quadriceps compared to WT (Figure 2E, right panel), which may infer reduced SIRT3 activity in the face of stress induced by AR100Q. Since acetylome analysis does not take into account total protein levels (and since we observed dramatically reduced total SOD2 in AR100Q skeletal muscle at this age), this increase in acetylation of SOD2-K122 is likely underestimated. Moreover, it indicates that SOD2 function is reduced, suggesting blunted SIRT3 activity.
NAD+ levels are substantially decreased with or without SIRT3 overexpression in AR100Q skeletal muscle
Further investigation into why exogenous SIRT3 failed to rescue motor function in AR100Q mice led us to consider SIRT3’s metabolic co-factor, NAD+, which drives its deacetylase activity. We know that the deacetylase function of SIRT3 is essential for its protective effects in cell models of SBMA (Figure 1E), and that NAD+ levels in quadriceps are greatly diminished after exercise in the knock-in (KI) mouse model of SBMA.3
At age 11 weeks NAD+ levels in both quadriceps and gastrocnemius muscles of AR100Q mice were substantially and significantly reduced compared to WT and AR24Q mice (Figures 3A and 3B). In the quadriceps, this deficiency was solely dependent upon the polyQ-expansion (Figure 3A). Evaluation of NAD+ levels in quadriceps and gastrocnemius from 11-week-old mice in the SIRT3 cohort replicated the dramatic reduction in NAD+ in AR100Q compared to WT controls previously observed (Figure 3A), and revealed that overexpressing SIRT3 contributes to further reduction of this metabolite (Figure 3B). This may be explained by enhanced consumption of NAD+ by the exogenously overexpressed SIRT3.
Figure 3.

Targeting diminished NAD+ with PARP inhibition in cells and skeletal muscle harboring polyQ-expanded AR
(A) Levels of NAD+ in quadriceps (left) and gastrocnemius (right) from WT, AR24Q, and AR100Q male mice (n = 3) at age 11 weeks.
(B) Levels of NAD+ in quadriceps (left) and gastrocnemius (right) from WT, SIRT3, AR100Q, and AR100Q-SIRT3 male mice (n = 3) at age 11 weeks.
(C) Levels of NAD+ in AR10Q-, and AR112Q-expressing PC12 cells (n = 3) treated with EtOH DHT, or DHT + olaparib (representative image from 3 experiments).
(D) ROS levels of PC12 polyQ-expanded AR112Q expressing cells (n = 12) treated with either DMSO (vehicle control), or 1 μM olaparib for 48 h (representative image from 3 experiments). ∗∗∗ = Nested two-way t-test, p ≤ 0.001.
(E) Cell death of AR100Q expressing C2C12 myotubes (n = 3) treated with either DMSO (vehicle control), or 1 μM olaparib for 10 days (representative image from 3 experiments). ∗ = Nested two-way t-test, p ≤ 0.05.
(F) Muscle mass (left) and percent of total mass (right) of gastrocnemius from WT and AR100Q male mice (n = 3) at age 8 weeks fed no-drug compounded or olaparib-compounded food.
(G) Levels of NAD+ in quadriceps (left) and gastrocnemius (right) from WT and AR100Q male mice (n = 4) at age 8 weeks fed no-drug compounded food or olaparib-compounded food.
(A–G) All statistical analysis show mean ± SD, and used a two-way ANOVA and Tukey’s post-hoc (∗ = p ≤ 0.05, ∗∗ = p ≤ 0.01, ∗∗∗ = p ≤ 0.001) unless otherwise noted. EtOH, ethanol; WT, wild type; SIRT3, SIRT3-flag overexpression; AR100Q, polyQ-expanded AR; AR100Q-SIRT3, polyQ-expanded AR with SIRT3-flag overexpression; AR10Q, non-polyQ-expanded AR; AR112Q, polyQ-expanded AR; olaparib, PARP inhibitor (1150 g of olaparib in 1 kg of feed).
Inhibition of PARPs restores NAD+, reduces ROS, and decreases death of cells modeling SBMA
Since NAD+ is diminished in SBMA, and NAD+ is critical to SIRT3 activity and overall metabolism, we focused on restoring NAD+ in SBMA. We chose to inhibit the activity of PARPs, with the potent and FDA-approved PARP-1 and PARP-2 inhibitor, olaparib, since PARP-1 is a major consumer of cellular NAD+ when active.25,26 AR112Q-expressing PC12 cells had reduced NAD+ in response to DHT treatment, and olaparib restored NAD+ levels (Figure 3C) and reduced ROS (Figure 3D). Furthermore, olaparib also reduced the death of AR100Q C2C12 myotubes (Figure 3E). Altogether, these results suggest that PARP inhibition may abrogate ROS, and prevent cell death of polyQ-expanded AR-expressing cells, while restoring NAD+.
Inhibition of PARPs increases gastrocnemius mass, but fails to rescue motor function or increase NAD+ in skeletal muscle of AR100Q mice
We evaluated whether PARP inhibition with olaparib could rescue muscle pathology and motor deficits of male mice modeling SBMA. A small cohort of AR100Q and WT male mice were fed olaparib-compounded food, and compared to mice fed control food. The percent total mass of gastrocnemius muscle from olaparib-treated AR100Q mice was increased (Figure 3F). Using a larger cohort, we found that olaparib treatment had no effect on total body weight, motor function, or lifespan of WT mice (Figures S3A–S3D). Unfortunately, olaparib also had no effect on body weight, grip strength, lifespan, or quadriceps mass of AR100Q mice (Figures S3A, S3B, S3D, and S3E). However, the olaparib-treated AR100Q mice did have a trend of increased motor function on the rotarod and motor endurance on the treadmill, when compared to the AR100Q non-treated mice (Figure S3C). These results suggest that olaparib does not substantially modulate polyQ-expanded AR-induced motor deficits in vivo as a single agent therapeutic at this dose. However, the increased gastrocnemius mass and the trend of increased rotarod and treadmill performance suggest that olaparib may be imparting some protection in mice modeling SBMA.
Olaparib treatment did not increase NAD+ in WT or AR100Q mice (Figure 3G). To ensure that the lack of an effect on NAD+ was not merely due to inefficient olaparib dosing and PARP inhibition, we obtained PARP-1 knockout (KO) mice and evaluated the effect of either a single allele loss of PARP-1, or a full KO of PARP-1 on the NAD+ levels of AR100Q quadriceps and gastrocnemius. Neither condition modulated AR100Q NAD+ in either muscle type (Figures S3F and S3G). Thus, PARPs, and specifically PARP-1, have no effect on NAD+ levels in the skeletal muscle of AR100Q male mice.
Increasing SIRT3 protein, in combination with PARP inhibition further reduces ROS in vitro and rescues motor endurance of AR100Q mice
Although olaparib treatment of AR100Q male mice did not result in recovery of NAD+ in skeletal muscle, the fact that both olaparib and SIRT3 overexpression each increased muscle mass and induced trends of increased motor function (Figures 2A, 2B, 3F, and S3C) encouraged us to investigate the effect of combining these treatments. Olaparib treatment of AR112Q-SIRT3-expressing PC12 cells further reduced ROS than either treatment alone (Figure 4A, left panel). Moreover, in the presence of tert-butyl hydroperoxide (TBHP), an organic compound that induces oxidative stress, only the AR112Q-SIRT3 cells treated with olaparib had a reduction in ROS (Figure 4A, right panel). These results suggested that a combination of SIRT3 activation and PARP inhibition might have a more substantial effect in reducing pathology in SBMA.
Figure 4.

Overexpressing SIRT3 plus PARP inhibition with olaparib further reduces ROS in vitro and rescues treadmill running without affecting AR100Q protein levels
(A) ROS in AR112Q SIRT3-MYC-overexpressing PC12 cells (n = 12) treated with either DMSO (vehicle control), or 1 μM olaparib for 48 h with (right) or without (left) ROS induction with 250 μM TBHP (representative image from 3 experiments).
(B) Muscle mass and percent of total mass of quadriceps (left) and gastrocnemius (right) from WT, AR100Q, and AR100Q-SIRT3 male mice (n = 3) at age 8 weeks fed no-drug compounded or olaparib-compounded feed.
(C) Behavioral cohort composed of 8 X WT fed no-drug compounded food, 11 X AR100Q fed no-drug compounded food, and 11 X AR100Q-SIRT3 male mice fed olaparib-compounded food. Left, accelerating rotarod analysis starting at age 6 weeks; ∗ = difference between WT vs. AR100Q and AR100Q-SIRT3. Right, accelerating treadmill analysis at 11 weeks of age.
(D) Left, immunoblot of total PAR following a PAR affinity resin assay from 8-week-old mouse quadriceps. WT lanes are oversaturated. Right-top, analysis of total protein lysates utilized in the PAR assay showing AR100Q monomer, and SDS-insoluble HMW aggregated AR100Q and tubulin loading control. Right-bottom, densitometry analysis of AR monomer and SDS-insoluble HMW aggregated AR normalized to tubulin loading control.
(A–D) All statistical analysis show mean ± SD, and used a two-way ANOVA and Tukey’s post-hoc (∗ = p ≤ 0.05, ∗∗ = p ≤ 0.01, ∗∗∗ = p ≤ 0.001). TBHP, tert-butyl hydroperoxide; WT, wild type; AR100Q, polyQ-expanded AR; AR100Q-SIRT3, polyQ-expanded AR with SIRT3-flag overexpression; HMW, high molecular weight; olaparib, PARP inhibitor (1150 g of olaparib in 1 kg of feed); PAR, poly-ADP-ribose.
Thus, we established a cohort of WT and AR100Q mice fed control food, and AR100Q-SIRT3 mice fed olaparib-compounded food to evaluate whether this combinatorial approach would ameliorate SBMA pathogenies. Treating AR100Q-SIRT3 mice with olaparib resulted in a trend of increased muscle weights (Figure 4B); however, this combinatorial treatment had no effect on total body weight, grip strength, or lifespan of AR100Q mice (Figures S4A–S4C). But, in keeping with what we observed in the SIRT3 (Figure 2B) and olaparib only cohorts (Figure S3C), AR100Q-SIRT3 mice treated with olaparib also had a trend toward enhanced rotarod function (Figure 4C, left panel). Additionally, evaluation of the quadriceps revealed that overexpression of SIRT3 with olaparib did not affect total or acetyl-SOD2 protein when compared to WT and AR100Q mice with no drug (Figure S4E). Nevertheless, AR100Q-SIRT3 mice treated with olaparib had a substantial and significant recovery of motor endurance, as measured via accelerating treadmill running (Figure 4C, right panel). With this encouraging result, we went on to evaluate potential mechanisms by which increasing SIRT3 and inhibiting PARPs may improve aerobic exercise in SBMA.
Olaparib eliminates PAR in AR100Q-SIRT3 quadriceps and neither SIRT3 overexpression nor PARP inhibition have any effect on monomeric or SDS-insoluble aggregated species in AR100Q mice quadriceps
We previously observed no impact on NAD+ levels in the skeletal muscle of AR100Q mice following PARP inhibition with olaparib (Figure 3G) and the same results were observed when AR100Q mice overexpressing SIRT3 were treated with olaparib (Figure S4D). Additionally, the lack of change of NAD+ levels in the AR100Q skeletal muscle with genetic KO of one or both alleles of PARP-1 (Figures S3F and S3G) supports the notion that the unchanged NAD+ levels in AR100Q mice are not due to an inefficient dosage of olaparib. However, we had yet to ensure that the dose of olaparib utilized in these studies was effectively penetrating skeletal muscle and inhibiting PARPs.
The activity of PARPs can be measured by evaluating poly(ADP-ribose (PAR)), which is one of the chemical moieties that PARPs covalently attach to their target proteins, furthermore, PARP-1, itself, is one of its main targets in a process called autoPARylation.24 Thus, we set up an additional cohort of WT and AR100Q mice with the no drug-compounded food and AR100Q-SIRT3 mice with the olaparib-compounded food to evaluate PAR. We precipitated PAR with WWE domain (PAR) affinity resin from the quadriceps of these mice and performed immunoblot analysis for PAR (Figure 4D, left panel). Of note, we detected a PAR positive band around 116 kD, which is the molecular weight of PARP-1, and may correspond with autoparylated PARP-1. This band was most prominent in the WT lanes, while there was substantially less band intensity in the AR100Q no drug lanes, and there were no detectable bands in the AR100Q-SIRT3 olaparib lanes, indicating that olaparib did penetrate the muscle and effectively inhibited PARPs.
Further analysis of the total lysates from the mice utilized in the PAR assay revealed a lack of change in the AR100Q monomer and AR100Q SDS-insoluble aggregates (Figure 4D, right panel). This indicates that neither SIRT3 overexpression nor olaparib reduces AR protein; therefore, this combinatorial treatment enhances motor function by abrogating toxicity downstream of polyQ-expanded AR.
Acetylome analysis reveals enhanced overall peptide acetylation in AR100Q quadriceps, and protein acetylation is modulated by overexpressed SIRT3
As briefly mentioned in Figure 2E, we aged a cohort of WT, AR24Q, SIRT3 only, AR100Q, and AR100Q-SIRT3 mice to 11 weeks (late stage disease for AR100Q), and performed global acetylome analysis of their quadriceps via mass spectroscopy. Initial analysis revealed a substantial and significant increase in the total intensity of acetylated peptides in the AR100Q samples compared to WT, SIRT3, and AR24Q (Figure 5A). Although there was a slight decrease of intensity in the AR100Q-SIRT3 samples, it was not significant.
Figure 5.

Acetylome analysis reveals substantially increased acetylation of peptides within AR100Q mouse quadriceps, with highly dysregulated metabolic pathways that are partially corrected by overexpression of SIRT3
WT, SIRT3-M1-flag (SIRT3), AR24Q, AR100Q, AR100Q-SIRT3 male mice (n = 3) were aged to 11 weeks. Quadriceps from these mice were digested with trypsin and acetylated peptides were enriched using Cell Signaling PTMScan Acetyl-Lysine Motif antibody. Enriched peptides were analyzed by LC-MS/MS on a Q Ex active HF mass spectrometer. For analysis n = 2 for SIRT3 and AR100Q-SIRT3 due to outliers below the −3 correlation Z score cut off.
(A) The levels of total acetylated sites in peptides are indicated by the intensity values. Statistical analysis shows mean ± SD; ∗∗∗ = two-way ANOVA and Tukey’s post-hoc, p ≤ 0.001.
(B) Differential expression of acetylated proteins between groups. Limma was used to find protein acetylation significantly different between groups. Direction of fold changes are listed in the top row. The number of proteins within the false discovery rate of <5% (estimated % of genes that satisfy f-value threshold that may be false positives) are listed in the bottom row.
(C) Heatmap of the top 30 proteins significantly changed in AR100Q and AR100Q-SIRT3 quadriceps compared to WT, SIRT3Q, and AR24Q controls. Left, upregulated acetylation of proteins. Right, downregulated acetylation of proteins. Red = increased acetylation. Blue = decreased acetylation.
(D) Ingenuity Pathway Analysis comparing AR100Q vs. AR24Q (top), and AR100Q vs. SIRT3/AR100Q (bottom). Red = activated pathway. Blue = inhibited pathway. Red font indicates gene names of proteins that have been identified as targets of SIRT3’s deacetylase activity (cited in results section). WT, wild type; S, SIRT3-flag overexpression; 24, AR24Q (non-polyQ-expanded AR); 100, AR100Q (polyQ-expanded AR); C, CROSS or AR100Q-SIRT3 (polyQ-expanded AR with SIRT3-flag overexpression).
We next evaluated differences in protein acetylation. When comparing AR100Q to AR24Q control quadriceps, we identified that AR100Q quadriceps had a 151-fold change in proteins with enhanced acetylation and an 86-fold change in proteins with decreased acetylation (Figure 5B). When comparing the AR100Q-SIRT3 to the AR100Q quadriceps, AR100Q-SIRT3 had a 26-fold change in proteins with enhanced acetylation and a 67-fold change in proteins with decreased acetylation (Figure 5B). Thus, the polyQ-expansion in AR dramatically alters the acetylome of quadriceps, and increasing SIRT3 further alters acetylation, and notably reduces the acetylation of a large number of proteins.
Overexpression of SIRT3 in AR100Q quadriceps corrects aberrant TCA cycle activity
Within the list of the top 30 hyperacetylated proteins in AR100Q quadriceps resides a protein that is essential for mitochondrial beta-oxidation of long chain fatty acids, which generates acetyl-CoA, a major precursor of ketogenesis and the TCA cycle. Hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha (HADHA) is the alpha subunit of the mitochondrial trifunctional protein responsible for catalyzing three of the last four steps of the mitochondrial beta-oxidation pathway. HADHA is highly acetylated (37 sites) and highly regulated, with 16 sites deacetylated by SIRT3.41 Overexpression of SIRT3 in AR100Q quadriceps substantially reduced acetylation of this protein (Figure 5C), leading us to inquire about the function of metabolic pathways in these muscles.
We employed Ingenuity Pathway Analysis to evaluate the acetylome data from the WT, SIRT3, AR24Q, AR100Q, and AR100Q-SIRT3 mouse quadriceps. We initially evaluated the AR100Q compared to the WTs, and from the list of altered pathways we determined which proteins remained changed when AR100Q were compared to the AR24Q mice, to account for effects due to overexpressed human AR. In the skeletal muscle of SBMA KI mice, fatty acid metabolism is increased,9 and our acetylome data also showed increased activity of the “Fatty Acid Beta Oxidation I″ pathway in the AR100Q quadriceps, albeit, without significance (Figure 5D). Overexpression of SIRT3 significantly reduced the activity of the “Fatty Acid Beta Oxidation I″ pathway, as well as the “Oxidative Phosphorylation” pathway (Figure 5D). As others have reported,3,9,27,32 we also observed inhibition of the “Glycolysis 1” pathway via the acetylome analysis. However, overexpression of SIRT3 did not have any effect on glycolysis (Figure 5D). As previously observed in the KI mouse model of SBMA,9 the “TCA Cycle II (Eukaryotic)” was also elevated in AR100Q quadriceps, and this could indicate reduced cellular NADH and ATP.42 The activity of the TCA cycle was reduced with SIRT3 overexpression (Figure 5D). Of note, there are four proteins in the TCA Cycle II pathway that were hyperacetylated and are targets of SIRT3’s deacetylase activity3,43,44,45; furthermore, the acetylation of these proteins was substantially reduced in the AR100Q-SIRT3 quadriceps (Figure 5D). Therefore, although SIRT3 failed to reduce acetylation of SOD2 (Figure 2E), and had no impact on the overall acetylation of peptides (Figure 5A) in the AR100Q quadriceps; SIRT3 significantly modified the function of multiple metabolic pathways in AR100Q quadriceps through its direct effect to deacetylate several of its target proteins (Figure 5D).
PARP inhibition coupled with overexpression of SIRT3 rescues hexokinase activity in AR100Q quadriceps
Glycolysis is substantially reduced in many models of SBMA.3,9,27 Our acetylome analysis of AR100Q quadriceps also highlights this deficit (Figure 5D), but SIRT3 overexpression did not improve glycolysis in the AR100Q quadriceps via this pathway analysis. Due to the significant rescue of exercise endurance of AR100Q-SIRT3 mice treated with the PARP inhibitor, olaparib (Figure 4C), we sought to determine whether this dual treatment could further correct energy producing pathways in AR100Q mice. PARP-1 has been identified to negatively regulate glycolysis by inhibiting hexokinase-1, independent of its function to reduce cellular NAD+.26 This was intriguing to us since inhibiting PARPs did not restore NAD+ (Figure 3G), and hexokinase-II is reduced at both the transcript and protein level in multiple mouse models of SBMA (including the AR100Q model).3,32 We performed an assay that revealed a significant decrease in hexokinase activity in AR100Q mice compared to WT, and that increasing SIRT3 and plus olaparib fully restored hexokinase activity in the quadriceps of AR100Q mice (Figure 6). Hexokinase activity was only partially increased in the AR100Q quadriceps with either of these treatments alone (Figures S5A and S5B). Since hexokinase initiates the first step in glycolysis, this dual treatment rescue of hexokinase activity may be restoring glycolytic flux in AR100Q muscle, to in turn improve exercise capacity.
Figure 6.
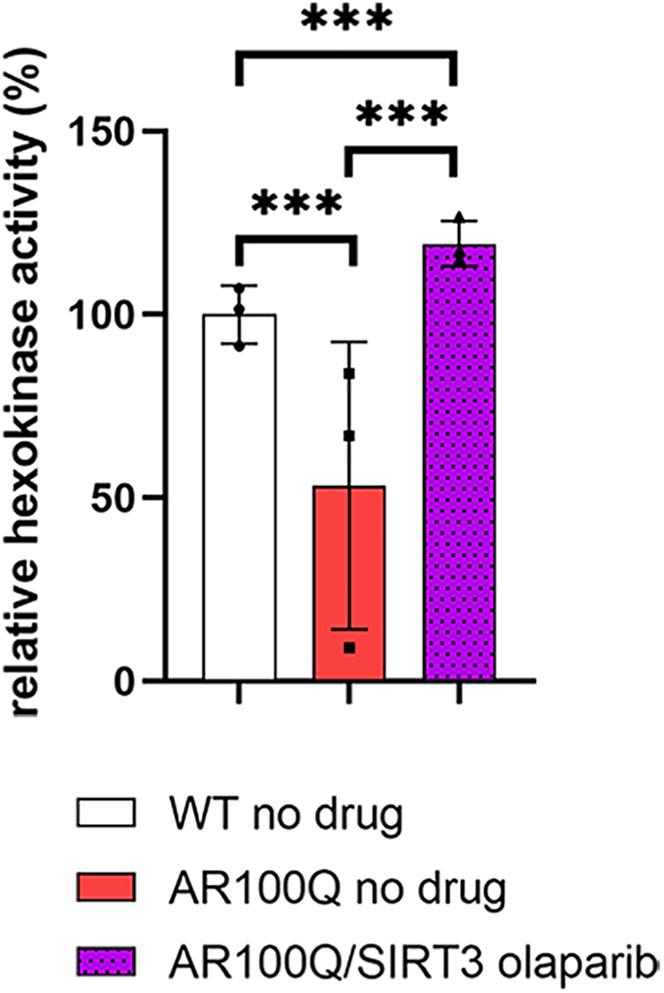
Overexpressing SIRT3 plus PARP inhibition with olaparib fully restores hexokinase activity in quadriceps of AR100Q mice
Levels of Hexokinase in quadriceps from WT, AR100Q, and AR100Q-SIRT3 male mice (n = 3) at age 11 weeks fed no-drug compounded or olaparib-compounded food. All statistical analysis show mean ± SD, and used a two-way ANOVA and Tukey’s post-hoc (∗∗∗ = p ≤ 0.001). WT, wild type; SIRT3, SIRT3-flag overexpression; AR100Q, polyQ-expanded AR; AR100Q-SIRT3, polyQ-expanded AR with SIRT3-flag overexpression; olaparib, PARP inhibitor (1150 g of olaparib in 1 kg of feed).
Discussion
Our studies identified that correcting diminished SIRT3 protein, while also inhibiting PARPs, restores dysregulated metabolism and supports aerobic exercise of mice modeling SBMA. Previous studies have identified the critical impact that polyQ-expanded AR has on modulating metabolic pathways within skeletal muscles.3,9,27,30 In addition to their motor dysfunctions, SBMA patients may also suffer from a general metabolic disorder. Blood panels from SBMA patients reveal elevated fasting glucose, total cholesterol, low-density lipoproteins, and triglycerides46; while MRI analysis further reveals non-alcoholic fatty liver disease47 (reviewed in48). Patient muscle biopsies contain reduced transcripts of proteins involved in carbohydrate metabolism,9,49 and substantial mitochondrial and metabolic dysregulation has also been documented in skeletal muscle and motor neurons of multiple SBMA models, including the mice used in these studies (AR100Q).3,9,27,30
A recent study identified that iPSC-derived motor neurons from an SBMA patient have reduced intracellular acetyl-CoA and repression of metabolic transcripts coupled with reduced promoter site histone H3K27 acetylation.27 Additionally, RNA-seq pathway analysis revealed diminished glycolysis,27 which may point to a reduced production of acetyl CoA. This is supported by the diminished pyruvate, the precursor to acetyl CoA, along with the decrease in transcripts of proteins necessary for glycolysis during exercise in the quadriceps of SBMA KI mice.3 Our acetylome analysis reiterates this notion by also identifying diminished glycolysis within AR100Q quadriceps, even without the stress of exercise. Furthermore, the increase in peptide acetylation in the AR100Q quadriceps may be another contributing factor to diminished cellular acetyl-CoA in SBMA.
A study of SBMA KI mice also identified a substantial reduction in NAD+ in quadriceps after exercise,3 which we also observed in the AR100Q quadriceps without exercise, suggesting that there is depletion of cellular NAD+ pools in muscles harboring polyQ-expanded AR both during exercise and at rest. Furthermore, reduced NAD+ levels can also impair glycolysis, since NAD+ is a key cofactor of this pathway.50 With the diminished NAD+ in SBMA skeletal muscle, we postulated that NAD+-dependent sirtuins, and specifically SIRT3, might have blunted activity in SBMA. Diminished sirtuin activity may also be responsible for the overabundance of acetylated peptides we observed in the AR100Q quadriceps.
Further evaluation of the acetylome data from AR100Q quadriceps also supports the idea of reduced SIRT3 function, since multiple SIRT3 target proteins are hyperacetylated. Compellingly, overexpression of SIRT3 managed to reverse the hyperacetylation of these SIRT3 targets; leading to the modulation of several metabolic pathways that may be responsible for rescuing aerobic exercise capacity of the olaparib fed AR100Q-SIRT3 mice.
PARP inhibition with olaparib, and genetic reduction/KO of the PARP-1 gene all failed to restore NAD+ in AR100Q skeletal muscle, discounting PARP-1 overutilization of NAD+ as one of the main mechanisms of NAD+ loss in SBMA. This indicates that the protection afforded by olaparib in vivo cannot be due to improving NAD+ availability. We postulate that increasing NAD+ has potential to further correct metabolism and motor function in SBMA, although further studies are necessary to validate this hypothesis. But, indeed, in a number of studies, restoring diminished NAD+ improves metabolism and motor function with aging and in various diseases51; for instance in the case of mice modeling Duchene’s muscular dystrophy (DMD). In mice modeling DMD, NAD+ is reduced and NAD+ repletion improves muscle function.52 In this DMD model, the reduced NAD+ is coupled with enhanced PARP activity, which is counter to our findings of PARP activity in the AR100Q SBMA mice. We found a dramatic reduction of PAR in AR100Q quadriceps compared to WT mice, which suggests that PARP-1 activity is reduced in SBMA, and likely explains how PARP inhibition with olaparib did not lead to a recovery of NAD+ in AR100Q skeletal muscle. However, even in the face of reduced PARP-1 activity in AR100Q muscle, olaparib inhibition of PARPs coupled with SIRT3 overexpression rescued the capacity of AR100Q mice to perform aerobic exercise. This suggests that reducing PARP activity with olaparib may be either further correcting metabolic anomalies, or inhibiting the PARylation of proteins involved in SBMA pathogenies. The full recovery of hexokinase activity by olaparib in the SIRT3 overexpressing mice supports this first suggestion, since PARP-1 can inhibit hexokinase and thus glycolysis.26 Recovery of hexokinase activity could restore glycolytic flux, increasing pyruvate production, and thus enhancing energy production through the TCA cycle.
Additionally, PARP-1 and PARP-2 (both inhibited by olaparib) are recruited to sites of AR occupancy in chromatin and regulate AR-dependent transcriptional activity,53,54 therefore, reducing PARP activity may also be modulating the transcriptional output of polyQ-expanded AR in a manner that enhances skeletal muscle function. Additional studies into the function of PARPs in SBMA pathogenesis are warranted. Moreover, since olaparib had no effect on NAD+ in AR100Q mice, identification of an intervention that can increase NAD+ may have potential to further correct metabolic dysregulation and promote a more substantial rescue of motor function in SBMA. Finally, the observation that SIRT3 overexpression with PARP inhibition does not affect the quantity of either monomeric or SDS-insoluble aggregated AR100Q protein underscores the impact of the function of metabolic correction downstream of disease initiation.
This study identified SIRT3 and PARPs as targets for therapeutic development in SBMA due to their capacity to modulate and correct metabolism while enhancing aerobic exercise. We predict that this therapeutic strategy of increasing SIRT3 activity while impeding PARP activity also has the potential to improve metabolic functions critical to other neurodegenerative or muscle disorders. Lastly, since therapies in development are targeting disease-initiating proteins, we predict that correcting metabolic dysfunction by way of modulating SIRT3 and PARPs in the setting of reducing the disease-initiating protein would result in a more robust amelioration of disease.
Limitations of the study
We identified SIRT3 and PARPs as critical mediators of SBMA pathogenesis using the established PC12 and C2C12 cell models of SBMA and transgenic AR100Q mouse model of SBMA. Ubiquitous overexpression of SIRT3 protein and peripheral PARP inhibition together rescued motor endurance of AR100Q male mice. However, the relevance of this therapeutic approach in modulating disease in SBMA human patients is not known. These studies did not identify why olaparib treatment failed to increase NAD+ in skeletal muscle of AR100Q mice. Moreover, we have yet to determine whether increasing NAD+ coupled with increased SIRT3, or increasing NAD+ alone, can improve SIRT3 function and ameliorate SBMA pathogenesis. Understanding the mechanisms for NAD+ loss and restoring this metabolite in SBMA requires further investigation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse α-tubulin IgG1 | Sigma-Aldrich | Cat# T6199; RRID: AB_477583 |
| mouse-myc IgG1 | Thermo Fisher Scientific | Cat# R950-25; RRID: AB_2556560 |
| mouse SOD2-A2 IgG2b | Santa Cruz | Cat# sc-133134; RRID: AB_2191814 |
| rabbit acetyl K68 SOD2 IgG | Abcam | Cat# ab137037; RRID: AB_2784527 |
| mouse anti-Poly-ADP-Ribose IgG3 | Trevigen | Cat# 4335-MC-100; RRID: AB_2572318 |
| rabbit SIRT3 | Cell Signaling | Cat# 5490; RRID: AB_10828246 |
| rabbit mAb androgen receptor | Cell Signaling | Cat# 5153; RRID: AB_10691711 |
| mouse ANTI-FLAG® M2 | Sigma | Cat# F1804; RRID: AB_262044 |
| Goat anti-rabbit IgG-HRP | Thermo Fisher Scientific | Cat# 65-6120; RRID: AB_2533967 |
| Goat anti-mouse IgG1-HRP | Thermo Fisher Scientific | Cat# A10551; RRID: AB_2534048 |
| Goat anti-mouse IgG2B-HRP | Novus | Cat# NB7521; RRID: AB_10124376 |
| Goat anti-mouse IgG3-HRP | Thermo Fisher Scientific | Cat# M32607; RRID: AB_2536651 |
| Chemicals, peptides, and recombinant proteins | ||
| honokiol | Tocris | 4590 |
| viniferin | Carbosynth | FV30429 |
| melatonin | Tocris | 3550 |
| olaparib | Selleck Chemicals | AZD2281 |
| tert-butyl hydroperoxide solution | Sigma | 75-91-2 |
| DHT (5A-ANDROSTAN-17B-OL-3-ONE) | Sigma | A8380-1G |
| EtOH (200 proof) | Electron Microscopy Sciences | 15055 |
| DMSO | Fisher BioReagents™ | BP231100 |
| protease inhibitor tablets | Thermo Scientific | A32963 |
| SuperSignal TM West Pico Plus Chemiluminescent Substrate | Thermo Scientific | 34577 |
| Critical commercial assays | ||
| DCFDA/H2DCFDA kit | Abcam | ab113851 |
| LDH assay kit | Cayman Chemical | 601170 |
| NAD+ assay kit | BioAssay Systems | E2ND-100 |
| Negative Control Magnetic Resin | Tulip Biolabs | 2427 |
| PAR WWE Domain Magnetic Affinity Resin assay | Tulip Biolabs | 2438 |
| Hexokinase assay kit | Abcam | ab136957 |
| DC protein assay kit | Biorad | 5000112 |
| Experimental models: Cell lines | ||
| Tet-inducible AR112Q and AR24Q PC12 cell lines | Walcott and Merry37 | N/A |
| AR100Q and AR24Q C2C12 cell lines | Milioto et al.38 | N/A |
| Experimental models: Organisms/strains | ||
| pCAGGS-AR100Q and pCAGGS-AR24Q transgenic mice | Chivet et al.30 | N/A |
| CAG-SIRT3M1 transgenic mice | N/A | N/A |
| PARP-1 KO mice (129S-Parp1tm1Zqw/J) | Wang et al.55 | JAX: 002779 |
| Recombinant DNA | ||
| pCDNA4-Myc-HisB-Sirt3 | Ahn et al.39 | Addgene: 24918 |
| pCDNA4-Myc-HisA-H248Y-Sirt3 | Ahn et al.39 | Addgene: 24917 |
| Software and algorithms | ||
| Graph Pad Software Prism 8 | ||
| Limma package | ||
| Ingenuity Pathway Analysis software | ||
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Heather L. Montie (heathermon@pcom.edu).
Materials availability
Tet-inducible AR112Q created to overexpress Human SIRT3 WT or human SIRT3-H248Y (deacetylase inactive), AR100Q C2C12 cells created to overexpress Human SIRT3 WT, and empty plasmid control cell lines generated for these studies can be obtained from the lead contact upon request.
Experimental model and study participant details
Creation of AR112Q PC12 cells stably overexpressing human SIRT3 and maintaining PC12 cells
PC12 cells that express human AR112Q and AR10Q following doxycycline-treatment (Tet-inducible) were generously provided by Diane Merry, PhD (Thomas Jefferson University).37 AR10Q and AR112Q PC12 cells were maintained in DMEM with 4.5 g/L glucose, L-glutamine & sodium pyruvate (Corning) supplemented with 10% heat-inactivated horse serum (HS), 5% heat-inactivated fetal bovine serum (FBS), 2% glutamine, 1% penicillin/streptomycin (Sigma-Aldrich), 100 μg/mL G418 (Sigma-Aldrich), and 200 μg/mL hygromycin (Gemini Bio).
Human SIRT3 WT (Addgene), human SIRT3-H248Y (deacetylase inactive) (Addgene),39 or an empty vector (pcDNA4/myc-His B backbone) control constructs were stably transfected into AR112Q PC12 cells with Lipofectamine 2000 (Invitrogen). Clonal lines were selected with 200 μg/μL zeocin (Invitrogen) containing media and single colonies were collected. Clonal SIRT3-WT and SIRT3-H248Y lines were paired based on matched myc (SIRT3 tag) protein expression.
PC12 cells were plated in charcoal-stripped (CS) serum containing media 24 h (hrs) before experiments began and continued in CS serum containing media throughout each experiment. AR10Q, AR112Q, AR112Q-SIRT3, and AR112Q-SIRT3 (H245Y) expressing PC12 cells were treated with doxycycline (DOX) (Fisher Bioreagents) to induce equivalent AR expression, plus either EtOH or 10 nM DHT (Sigma-Aldrich). Cells were treated with honokiol (Tocris), viniferin (Carbosynth), melatonin (Tocris), olaparib (Selleck Chemicals), or a DMSO control for 48 or 96 h before undergoing specific assays. Cells were re-treated every 48 h.
Creation of AR100Q C2C12 cells stably overexpressing SIRT3 and maintaining C2C12 cells
C2C12 cells constitutively expressing human AR100Q and AR24Q were kindly provided by were kindly provided by Maria Pennuto, PhD (University of Padova).38 C2C12 cells (myoblasts) were cultured in DMEM supplemented with 10% FBS, 1% glutamine, 1% penicillin/streptomycin and kept below 70% confluence at all times unless differentiating. To differentiate C2C12 myoblasts into myotubes, cells were plated into polyD-lysine coated dishes at high density in regular 10% FBS containing media for the first 24 h. After 24 h the media was changed to low serum (2% CS-HS), and 75% of the media was changed every 48–96 h. On the day 3, DHT and drugs were added to the media. All cells were maintained at 37°C in a 5% CO2 incubator.
Human SIRT3-WT in the pcDNA-myc-his B plasmid backbone (Addgene)39 or an empty vector (EV) control were stably transfected into AR100Q C2C12 cells with Lipofectamine 2000 (Invitrogen). Clonal lines were selected with 200 μg/μL zeocin (Invitrogen) containing media and single colonies were collected. Cells were maintained thereafter in media with 50 μg/μL zeocin (Invitrogen).
PC12 cells were derived from a male rat. C2C12 cells were derived from female mouse. Neither of these cell lines or the newly created PC12 and C2C12 cell lines have been authenticated for these studies.
Mouse models utilized in these studies
All mice were housed in a conventional/non-barrier animal facility in individually ventilated cages, with up to 5 adults per cage. Temperature was maintained between 68°C and 72°C and humidity ranged from 30 to 70%. Mice had access to water and food ad libitum. Mice were fed Purina 5015 mouse diet, except for trials including olaparib-compounded feed, wherein which controls were fed Dyets # 110700 (AIN-93G) Purified Rodent Diet and olaparib treated mice received olaparib 1150 mg of olaparib (Selleck Chemicals, AZD2281) compounded per kilogram of mouse feed (Dyets # 110700 (AIN-93G)) (as outlined in more detail in “Method Details”). Mice that were breeding were supplemented with Love Mash Rodent Reproductive Diet - Gamma Irradiated (Bio-Serv).
Mice expressing heterozygous human normal Q-length AR, AR24Q (AR24Q+/−) and polyQ-expanded AR, AR100Q (AR100Q+/−), under the control of the cytomegalovirus immediate-early enhancer and the chicken beta-actin (pCAGGS) promoter were kindly provided by Maria Pennuto, PhD (University of Padova).30 These transgenic lines (males and females) were maintained and the colony expanded by breeding with C57BL/6J (Jackson Laboratory) WT mice.
Mice over-expressing FLAG-tagged mouse M1 isoform of SIRT3, driven by the CAG promoter, were kindly provided by Qian Tong, PhD (Baylor College of Medicine) and maintained on the C57BL/6NJ background (Jackson Laboratory). Both males and females were utilized for breeding purposes. For the creation of AR100Q-SIRT3 overexpressing mice, AR100Q mice were bred with SIRT3 overexpressing mice. This cross generated mice on a 50:50 C57BL/6J and C57BL/6NJ background with the following possible genotypes: SIRT3 +/−, AR100 +/−, SIRT3+/AR100+, WT −/−.
PARP-1 KO mice (129S-Parp1tm1Zqw/J, on a SvImJ background, strain #00277955) were obtained from Jackson Laboratory and maintained on the 129S1/SvImJ background. Both males and females were utilized for breeding purposes. Breeding PARP-1 KO mice with AR100Q mice resulted in AR100Q-PARP-1 het mice (50% C57BL/6J/50% 129S1/SvImJ), and WT-PARP-1 het controls which were used to maintain the same background. AR100Q mice were bred with SvImJ WT mice to produce AR100Q and WT with PARP-intact (50% C57BL/6J/50% 129S1/SvImJ) controls. AR100Q-PARP-1 het mice were bred with PARP-1 KO mice to produce AR100Q-PARP-1 KO mice and WT-PARP-1 KO (25% C57BL/6J/75% 129S1/SvImJ). AR100Q (50% C57BL/6J/50% 129S1/SvImJ) were bred with 129S1/SvImJ WT mice to produce AR100Q and WT PARP-1 intact (25% C57BL/6J/75% 129S1/SvImJ) controls. Mice were weaned 3 weeks after birth and ears were clipped for identification and genotyping.
For experiments only male mice were utilized due to the need for high levels of male hormones (androgens) to produce SBMA phenotype. The male mice used in tissue experiments were sacrificed at 8 or 11 weeks of age. The male mice used in motor function, survival assays underwent analysis beginning at 5 weeks of age until death. This is noted in the survival charts; in general AR100Q males become moribund and need to be sacrificed between 12 and 15 weeks of age. After all AR100Q males in an experiment had succumbed to disease, the non-AR100Q male mice in the study were also all sacrificed at that time. All procedures involving mice were approved by the Institutional Animal Care and Use Committee of Philadelphia College of Osteopathic Medicine, in accordance with the NIH Guidelines for the Care and Use of Experimental Animals.
Method details
Cell harvesting for protein analysis
For western analysis PC12 and C2C12 cells were harvested in modified RIPA (50 mM Tris pH 8.0, 0.15 M NaCl, 1% NP40, 0.5% DOC, 0.5% SDS) plus 50 μg/mL PMSF, and one protease inhibitor tablet (Thermo Scientific) per 10 mL of lysis buffer. Before harvesting, cells were washed three times with 1X PBS and then scrapped into ice-cold lysis buffer. Lysates were immediately frozen in a 70% ethanol and dry ice bath before being stored at −80°C.
Lactate dehydrogenase (LDH assay)
C2C12 cells were plated on polyD-lysine coated plates/wells and cultured in differentiation media (method described above). After the 5th (AR100Q-SIRT3 overexpressing or AR100Q-EV lines only) or 10th day of treatment the LDH levels were quantified with an LDH assay kit (Cayman Chemical, 601170) using the manufacturer’s protocol. Samples of media were taken from the same well at the end of the treatment period (for experimental sample reading) and after inducing cell death by adding 10% Triton X-100 to media (for 100% cell death control reading). At the end of the assay, samples were evaluated in a colorimetric plate reader (iMarkTM microplate reader, Bio-Rad) at 450 nm. Data generated for LDH concentrations for the experimental samples were compared to the LDH values from the “100% cell death control” to determine the percent cell death.
ROS assay
PC12 cells were plated into the wells of a 96-well plate and a ROS assay (DCFDA/H2DCFDA kit (Abcam, ab113851) was performed with 12 biological replicates for each cell type/condition. After 48 h the media was removed and the wells were washed with buffer provided with the kit and then incubated with DCFDA solution for 45 min (min.) at 37°C in the dark. After staining, the buffer was added and the plate was moved into a fluorescence plate reader (Fluoroskan Ascent CF, Labsystems) set at Ex/Em = 485/535 nm. For experiments where ROS were induced, tert-butyl hydroperoxide (250 μM) was added to each well, and a reading was taken after 15 min.
NAD+ assay
PC12 cells were plated per 6-well plate and treated for 96 h, and then pelleted in cold PBS. NAD+ extraction buffer was added to cell pellets or pulverized frozen mouse muscle which was homogenized with a handheld homogenizer (PRO200 Bio-Gen Series). Samples were then handled per the manufacturer’s protocol (BioAssay Systems, E2ND-100). Samples were evaluated in a colorimetric plate reader (iMarkTM microplate reader, Bio-Rad) at 570 nm for a time “zero” and after 15 min incubation at room temperature (RT) for a time “15 min” time point. The NAD+ concentration was calculated by using the following formula: [NAD+] = (OD at 15 min. – OD at zero)/(slope of standard curve).
Mouse PCR identification
Mouse genotypes were determined by PCR of DNA extracted from mouse ear clips. A REDExtract-N-Amp Tissue PCR kit (Sigma-Aldrich, 647-014-00-9) was used to extract mouse-ear DNA following the manufacturer’s instructions. PCR was performed using the following primers: human AR transgene (forward 5′-CTTCTGGCGTGTGACCGGCG, reverse 5′-TGAGCTTGGCTGAATCTTCC), SIRT3-flag (forward 5′-GCAACGTGCTGGTTATTGTG, reverse 5′-GTGGAGGAGCCTCAGGAAAGT), WT PARP-1 reverse (5′-CCAGCGCAGCTCAGAGAA GCCA), PARP-1 common forward (5′-CATGTTCGATGGGAAAGTCCC), PARP-1 KO reverse (5′-AGGTGAGATGACAGGAGATC), and BCl2 control (forward 5′-ATGGCGCAAGCCGGGAGAACA, reverse 5′-CCGGTTCAGGTACTCAGTCAT).
Olaparib-compounded mouse feed
Eleven hundred and fifty milligrams of olaparib (Selleck Chemicals, AZD2281) per kilogram of mouse feed was compound by Dyets Inc. All mice on drug studies were given Dyets feed without drug from 4 to 5 weeks of age, and mice were divided into no-drug or olaparib-containing feed at 5 weeks of age, through the end of life. The concentration of olaparib to compound into the feed was determined by using 2.14 g of chow per day (average amount food AR100Q male mice consume between 6 and 12 weeks of age, from previous studies) and the conversion value (human to mouse) as outlined “HED (mg/kg) = Animal dose (mg/kg) × (Animal Km/Human Km)”56 of the highest human daily dose of olaparib approved by the FDA for human use (600 mg). The no-drug and olaparib-compounded feed were stored at −20°C, and freshly thawed feed was provided to mice every 7 days. Weekly feed intake/drug exposure was recorded for each cage of mice.
Mouse tissue
Male mice of various genotypes were randomized into experimental groups. Only male mice were utilized for experiments due to the critical dependence upon SBMA pathogenesis on male hormones. Mice were aged to 8 or 11 weeks and then sacrificed and tissue was dissected. Quadriceps and gastrocnemius muscles were weighed (wet weight) and then flash-frozen in liquid (LN2), and then stored long term at −80°C.
Mouse muscle protein lysate preparation
Previously dissected and flash-frozen in LN2 and stored long term at −80°C mouse muscle was pulverized with a frozen metal mortar and pestle on dry ice. The following lysis buffers were utilized for mouse muscle: modified RIPA (as outlined in cell lysate section), PAR assay RIPA buffer, or acetylome lysis buffer, plus 1 mM PMSF, 5 mM sodium butyrate (deacetylase inhibitor), 10 μM tannic acid (PARG inhibitor), and one protease inhibitor tablet (Thermo Scientific, A32963) per 10 mL of lysis buffer. Lysis buffer was added to the pulverized muscle (10–20 μL per mg tissue) in a pre-chilled flat bottom 2 mL microcentrifuge tube and homogenized (PRO200 Bio-Gen Series) until the muscle was homogeneous in liquid (∼15 s (sec.) at second to max setting). After homogenization, samples were sonicated for 5 min and then centrifuged at 4,000 g for 15 min at RT. The supernatant was collected and a DC protein assay kit (Bio-Rad) was used to determine protein concentration.
SDS-PAGE electrophoresis and transfer
After protein quantification, protein lysates were diluted in laemmli buffer, boiled at 95°C for 5 min and run on SDS-PAGE (7.5% or 12% TGX gels; 4–20% tris-acetate gels (Bio-Rad)). Samples were run at 125 V in a Mini-PROTEAN or Midi-Criterion apparatus (Bio-Rad) and transferred to a PVDF membrane using a Mini- or Midi-Trans-Blot Cell (Bio-Rad) at filled with cold transfer buffer (47.8 mM Tris, glycine 386.31 mM, and 20% methanol = Mini gels; 25 mM Tris, 192 mM glycine, and 20% methanol = Midi gels) at 100 V for 1 h.
Western analysis and antibodies
PVDF membranes were blocked in 5% non-fat dry milk in TTBS for 30–60 min. Primary antibodies were diluted in 5% non-fat dry milk in TTBS and the PVDF membrane was incubated at 4°C for 24–48 h. Secondary-HRP antibodies were also diluted in 5% non-fat dry milk in TTBS and the membrane was incubated for 1 h (hr) at RT. The membrane was submersed in SuperSignal TM West Pico Plus Chemiluminescent Substrate (Thermo Fisher Scientific, 34577) following the manufacturer’s protocol and a ChemidocTM MP imaging system (Bio-Rad) was used to develop the membranes.
Primary antibodies used include: mouse α-tubulin IgG1 (Sigma-Aldrich, T6199), mouse-myc IgG1 (Thermo Fisher, R950-25), mouse SOD2-A2 IgG2b (Santa Cruz, sc-133134), rabbit acetyl K68 SOD2 IgG (Abcam, ab137037), mouse anti-Poly-ADP-Ribose IgG3 (Trevigen, 4335-MC-100), rabbit SIRT3 (Cell Signaling, D22A3), rabbit mAb androgen receptor (Cell Signaling, D6F11), mouse ANTI-FLAG M2 (Sigma, F1804).
Secondary antibodies used include: Goat anti-rabbit IgG-HRP (Invitrogen, 65–6120), Goat anti-mouse IgG1-HRP (Invitrogen, A10551), Goat anti-mouse IgG2B-HRP (Novus, P33), Goat anti-mouse IgG3-HRP (Invitrogen, M32607)
PAR assay
Previously dissected and flash-frozen in LN2 stored long term at −80°C mouse quadriceps were pulverized with a frozen metal motar and pestle on dry ice. Pulverized quadricep samples were lysed in RIPA buffer (50 mM Tris, pH 7.5, 0.4M NaCl, 1 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM DTT), plus 50 μg/mL PMSF, 10 μM tannic acid and one protease inhibitor tablet (Thermo Scientific, A32963) per 10 mL of lysis buffer and prepared as outlined above. Lysates were pre-cleared on a rotator overnight at 4°C with Negative Control Magnetic Resin (Tulip Biolabs, #2427). The next day, the beads were removed and PAR WWE Domain Magnetic Affinity Resin assay (Tulip Biolabs, #2438) beads were added to the samples. Samples were incubated overnight at 4°C on a rotator. The lysates were removed, the resin was washed with the lysis buffer 4 times and once with PBS, laemmli buffer was added, boiled at 95°C for 1 min, and samples were removed from the resin.
Hexokinase assay
Hexokinase assay buffer from (Abcam, ab136957) was added to pulverized frozen mouse quadriceps which were homogenized with a handheld homogenizer (PRO200 Bio-Gen Series). Samples were then handled per the manufacturer’s protocol. Samples were evaluated in a colorimetric plate reader (iMarkTM microplate reader, Bio-Rad) at 450 nm for a time “zero” and after 35 min incubation at room temperature (RT) for a time “35 min” time point. The hexokinase absorbance was calculated by using the following formula: [hexokinase absorbance] = (OD at 35 min. – OD at zero)/(slope of standard curve). Specific hexokinase activity was obtained by the following formula: [hexokinase activity] = (hexokinase absorbance)/((reaction time) (sample volume)) × dilution factor. The wild-type control samples were made to be 100% and the other samples were graphed as a percent of the wild-type control.
Mouse behavioral assays
Male mice of various genotypes were randomized into experimental groups. Only male mice were utilized for experiments due to the critical dependence upon SBMA pathogenesis on male hormones. Staff and students performing behavioral studies with mice were blinded to the genotype and drug treatment.
A grip strength apparatus (Ugo Basile) was utilized to measure the force exerted by the forepaws, or all four paws of mice once per week. Individual mice grasped a metal bar (forepaws) or wire mesh (all four paws) and were gently pulled by the base of their tail to measure their maximum force exerted to maintain their grip. The grip strength assay was conducted 6 times per mouse, for both forepaws only and all four paws. Grubbs outlier test (GraphPad Prism) was run to remove outliers with an alpha value ≤0.05.
Mice were run on an accelerating rotarod apparatus (Ugo Basile) (4–40 rpm over 5 min) for a total of 10 min (or less if they fell before the end of 10 min). The rotarod apparatus was placed inside a laminar flow hood. The amount of time each mouse maintained themselves on the accelerating rotarod was measured until mice dropped from the rotarod or if a mouse spun two consecutive rotations without moving. The mice were trained similarly over two consecutive days the week of or the week before the first week of testing. Each week, each mouse was run on the accelerating rotarod four times (with at least 15 min break between runs), and their average time spent on the rotarod was used for analysis.
A Five Lane Touchscreen Treadmill for Mice (Panlab Harvard Apparatus) was utilized to analyze endurance. Treadmill training was performed for 2 days before the first week of testing (10 weeks of age). The training protocol on day 1 consisted of 5 min at 8 m/min, while day 2 consisted of 5 min at 8 m/min followed by 5 min at 10 m/min (as in3). For the experimental trial (11 and/or 12 weeks of age) had individual mice running on a flat treadmill with a graded acceleration protocol consisting of 10 m (m)/min. for the first 40 min, with the speed increasing thereafter by increments of 1 m/min every 10 min for 30 min, and then finally increasing speed by increments of 1 m/min every 5 min until mice were exhausted (as in3). The exhaust was determined by 5 consecutive sec. or 20 overall sec. of failure to exercise (by staying on shock rods at the start of the treadmill). All testing events had a gradient warm-up period of 1 m/min to the starting speed for 2 min.
Acetylome analysis
A cohort of WT, SIRT3-M1-flag (SIRT3), AR24Q, AR100Q, AR100Q-SIRT3 (n = 3 each group) were aged to 11 weeks. Mice were sacrificed and both quadriceps from each mouse were flash frozen in LN2. Quadriceps were homogenized in acetylome lysis buffer containing 100 mM Tris, 9 M urea, 20 mM glycine, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 5 mM sodium butyrate, and one protease inhibitor tablet (Thermo Scientific, A32963) per 10 mL of lysis buffer. Samples were sonicated for 5 min, centrifuged at 4,000 g for 15 min at RT, and the protein concentration of the supernatant was measured via a D/C assay. Ten milligrams of total protein from each sample was frozen on dry ice and stored at −80°C.
Frozen lysates were transferred to the Proteomics & Metabolomics Facility where they were reduced with dithiothreitol, alkylated with iodoacetamide, and digested in-solution with trypsin (final 2M urea, 1:50 enzyme:protein). Starting materials and digests were run on SDS-gels to confirm trypsin digestion. Only peptides were observed after post-digest (PD). Digested peptides were cleaned up using Waters SepPak C18 columns. Acetylated peptides were then enriched using Cell Signaling PTMScan Acetyl-Lysine Motif antibody. Enriched peptides were analyzed by LC-MS/MS on a Q Exactive HF mass spectrometer using an extended 4 h. LC gradient. MS data were searched with full tryptic specificity against the UniProt mouse proteome database (10/02/2020) using MaxQuant 1.6.17.0. The “Match between runs” feature was used to help transfer identifications across experiments to minimize missing values. Peptides and sites identified only by matching (no MS/MS information) are indicated in the “Identification Type” columns. Variable modifications searched include: Acetylation (+42.01056) on K, Oxidation (+15.99491) on M, Deamidation (+0.98402) on N, and Carbamylation (+43.00581) on K and N-terminus. Fixed modifications searched include Carbamidomethylation (+57.02146) on C. Modified peptides were required to have a minimum score of 40 and delta score of 6. The false discovery rates for protein, peptide and sites identifications were set at 1%. The levels of acetylated sites are indicated by the Intensity values. Intensity values are summed up extracted ion current of all isotopic clusters associated with the identified sequence, i.e., the acetylated site intensity was consolidated from different peptide versions containing the same acetylated site. Acetylated sites not detected in at least two of the triplicates (in any group) were removed. Fold changes between groups were calculated using non-normalized Intensity, and after normalizing to the median Intensity of each sample. To calculate the fold change between groups, acetylated site Intensity values were processed by: 1) Log2 transformation of the Intensity values and replacing missing values (site not identified) with the minimum value of the dataset.
Acetylome data produced by the Proteomics & Metabolomics Facility (The Wistar Institute) were transferred to the Bioinformatics facility (The Wistar Institute) for further analysis. The following comparisons were made between groups: WT vs. SIRT3 only, WT vs. AR24Q, WT vs. AR100Q, AR100Q vs. AR24Q, and AR100Q-SIRT3 vs. AR100Q. Intensity levels from “Acetyl (K) Sites_Expanded” sheet was used for analysis. Analysis: 1) Overview (clustering, multidimensional plotting), 2) Protein numbers, 3) Overlaps, 4) Enrichment analysis. Ingenuity Pathway Analysis software was used for enrichment analyses using all detected proteins in the experiment as a background.
Quantification and statistical analysis
Statistics
Data were analyzed using paired two-tailed t-test, one-way ANOVA, two-way ANOVA, or a nested two-way ANOVA with Tukey’s multiple comparison test in Prism 8 (GraphPad). Specified data was analyzed with Grubb’s outlier test and excluded from the study. Statistical significance was considered p ≤ 0.05, and differentiation was made between p ≤ 0.05, p ≤ 0.01, and p ≤ 0.001. At a specified time point, statistical analysis comparisons included multiple groups (ex. WT versus multiple AR100Q conditions).
For the acetylome analysis, the false discovery rates for protein, peptide and sites identifications were set at 1%. For comparison between two sample groups, Log2 ratio, Fold change and Student’s t-test p value were calculated using the Log2 Intensity (or Log2 Normalized Intensity). The more stringent q-value (t-test p value adjusted to account for multiple testing using Benjamini-Hochberg FDR) was also calculated. The |FC|>2, q < 0.05 column shows acetylated site with significant change (absolute fold change >2, minimum adjusted p value of 0.05). Zero values were floored to the 1% min detected intensity level. Quantile normalization was performed. Quality control was performed and 2 samples were deemed technical outliers and removed from analysis. Limma package was used to estimate significance between pairs of groups. Ingenuity Pathway Analysis software was used for enrichment analyses using all detected proteins in the experiment as a background.
Acknowledgments
We thank Christine Hammond, MS, for expertise and training for various laboratory equipment and techniques utilized in these studies. We thank Hsin Yao Tang, PhD, and the Proteomics & the Metabolomics Facility (The Wistar Institute, Philadelphia, PA) for performing the acetylome analysis, and Andrew Kossenkov, PhD, and the Bioinformatics facility (The Wistar Institute, Philadelphia, PA) for bioinformatics analysis of the acetylome data. We thank Robert Barsotti, PhD, Jocelyn Lippman-Bell, PhD, Grace Farber, PhD, and Bela Peethambaran, PhD, for their review and comments of the manuscript draft. The Philadelphia College of Osteopathic Medicine (PCOM) provided monetary support for experiments through the Division of Research (DOR) and the Center for Chronic Disorders of Aging (CCDA) (to H.L.M.). The Guinta Family Research Scholarship supported fellowship and research supplies for D.R.G. This work was also supported by the USDA (CRIS 3092-5-001-059 to Q.T.).
Author contributions
Conceptualization, D.R.G., J.R.M., E.M.H., D.S., C.L., and H.L.M.; Methodology, D.R.G., J.R.M., E.M.H., D.S., C.L., K.H.H., and H.L.M.; Investigation, D.R.G., J.R.M., E.M.H., D.S., R.S.P., C.L., K.H., P.M., D.M., M.V.L., E.N., K.H.H., T.F., and H.L.M.; Formal Analysis, D.R.G., J.R.M., E.M.H., D.S., C.L., and H.L.M.; Writing – Original Draft. D.R.G. and H.L.M.; Writing – Review and Editing D.R.G., D.S., C.L., K.H., M.V.L., E.M.H., E.N., D.E.M., Q.T., M.P., and H.L.M.; Funding Acquisition, H.L.M. and D.R.G.; Resources D.E.M., Q.T., M.P.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: July 22, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107375.
Supplemental information
This excel sheet contains the raw data from the acetylome analysis of 11 week old mouse quadriceps. The first sheet (Summary) is a summary of the sample names and how they were prepared for acetylation-specific LC-MS/MS and how the data from the LC-MS/MS was analyzed. The second sheet (Acetyl (K) sites) contains a list of identified Acetyl (K) sites with the levels of acetylated sites indicated by intensity values for each sample. The third sheet (Acetyl (K)Sites_Expanded) has Intensity ___1, ___2, and ___3 columns transposed into separate rows. This is the main data sheet. Acetylated sites not detected in at least two of the triplicates (in any group) were removed. Fold changes between groups were calculated using non-normalized intensity values (columns AS to CF), and after normalizing to the median Intensity of each sample (columns CG to DT). The |FC|>2, q<0.05 column shows acetylated site with significant change (absolute fold change >2, minimum adjusted p-value of 0.05). The fourth sheet (Mod. Peptide) shows the peptides identified. The fifth sheet (Protein group) shows the protein identifications.
Data and code availability
-
•
Experimental data reported in this manuscript will be shared by the lead contact upon request. The raw acetyl (K)Sites for the acetylome analysis are available as a supplemental excel file (Data S1: Acetylome raw data (Acetyl (K) Sites), related to Figure 5).
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Kennedy W.R., Alter M., Sung J.H. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968;18:671–680. doi: 10.1212/wnl.18.7.671. [DOI] [PubMed] [Google Scholar]
- 2.La Spada A.R., Wilson E.M., Lubahn D.B., Harding A.E., Fischbeck K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 3.Giorgetti E., Yu Z., Chua J.P., Shimamura R., Zhao L., Zhu F., Venneti S., Pennuto M., Guan Y., Hung G., Lieberman A.P. Rescue of Metabolic Alterations in AR113Q Skeletal Muscle by Peripheral Androgen Receptor Gene Silencing. Cell Rep. 2016;17:125–136. doi: 10.1016/j.celrep.2016.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adachi H., Katsuno M., Minamiyama M., Waza M., Sang C., Nakagomi Y., Kobayashi Y., Tanaka F., Doyu M., Inukai A., et al. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain. 2005;128:659–670. doi: 10.1093/brain/awh381. [DOI] [PubMed] [Google Scholar]
- 5.Sorarù G., D'Ascenzo C., Polo A., Palmieri A., Baggio L., Vergani L., Gellera C., Moretto G., Pegoraro E., Angelini C. Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J. Neurol. Sci. 2008;264:100–105. doi: 10.1016/j.jns.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Johansen J.A., Yu Z., Mo K., Monks D.A., Lieberman A.P., Breedlove S.M., Jordan C.L. Recovery of function in a myogenic mouse model of spinal bulbar muscular atrophy. Neurobiol. Dis. 2009;34:113–120. doi: 10.1016/j.nbd.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhodes L.E., Freeman B.K., Auh S., Kokkinis A.D., La Pean A., Chen C., Lehky T.J., Shrader J.A., Levy E.W., Harris-Love M., et al. Clinical features of spinal and bulbar muscular atrophy. Brain. 2009;132:3242–3251. doi: 10.1093/brain/awp258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atsuta N., Watanabe H., Ito M., Banno H., Suzuki K., Katsuno M., Tanaka F., Tamakoshi A., Sobue G. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain. 2006;129:1446–1455. doi: 10.1093/brain/awl096. [DOI] [PubMed] [Google Scholar]
- 9.Rocchi A., Milioto C., Parodi S., Armirotti A., Borgia D., Pellegrini M., Urciuolo A., Molon S., Morbidoni V., Marabita M., et al. Glycolytic-to-oxidative fiber-type switch and mTOR signaling activation are early-onset features of SBMA muscle modified by high-fat diet. Acta Neuropathol. 2016;132:127–144. doi: 10.1007/s00401-016-1550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malik B., Devine H., Patani R., La Spada A.R., Hanna M.G., Greensmith L. Gene expression analysis reveals early dysregulation of disease pathways and links Chmp7 to pathogenesis of spinal and bulbar muscular atrophy. Sci. Rep. 2019;9:3539. doi: 10.1038/s41598-019-40118-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iida M., Katsuno M., Nakatsuji H., Adachi H., Kondo N., Miyazaki Y., Tohnai G., Ikenaka K., Watanabe H., Yamamoto M., et al. Pioglitazone suppresses neuronal and muscular degeneration caused by polyglutamine-expanded androgen receptors. Hum. Mol. Genet. 2015;24:314–329. doi: 10.1093/hmg/ddu445. [DOI] [PubMed] [Google Scholar]
- 12.Bott L.C., Badders N.M., Chen K.L., Harmison G.G., Bautista E., Shih C.C.Y., Katsuno M., Sobue G., Taylor J.P., Dantuma N.P., et al. A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2016;25:1979–1989. doi: 10.1093/hmg/ddw073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palazzolo I., Stack C., Kong L., Musaro A., Adachi H., Katsuno M., Sobue G., Taylor J.P., Sumner C.J., Fischbeck K.H., Pennuto M. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron. 2009;63:316–328. doi: 10.1016/j.neuron.2009.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rinaldi C., Bott L.C., Chen K.L., Harmison G.G., Katsuno M., Sobue G., Pennuto M., Fischbeck K.H. Insulinlike growth factor (IGF)-1 administration ameliorates disease manifestations in a mouse model of spinal and bulbar muscular atrophy. Mol. Med. 2012;18:1261–1268. doi: 10.2119/molmed.2012.00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jęśko H., Wencel P., Strosznajder R.P., Strosznajder J.B. Sirtuins and Their Roles in Brain Aging and Neurodegenerative Disorders. Neurochem. Res. 2017;42:876–890. doi: 10.1007/s11064-016-2110-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montie H.L., Pestell R.G., Merry D.E. SIRT1 modulates aggregation and toxicity through deacetylation of the androgen receptor in cell models of SBMA. J. Neurosci. 2011;31:17425–17436. doi: 10.1523/JNEUROSCI.3958-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou X., Chen M., Zeng X., Yang J., Deng H., Yi L., Mi M.T. Resveratrol regulates mitochondrial reactive oxygen species homeostasis through Sirt3 signaling pathway in human vascular endothelial cells. Cell Death Dis. 2014;5:e1576. doi: 10.1038/cddis.2014.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kincaid B., Bossy-Wetzel E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013;5:48. doi: 10.3389/fnagi.2013.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finley L.W.S., Haigis M.C. Metabolic regulation by SIRT3: implications for tumorigenesis. Trends Mol. Med. 2012;18:516–523. doi: 10.1016/j.molmed.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu J., Jin J., Cichewicz R.H., Hageman S.A., Ellis T.K., Xiang L., Peng Q., Jiang M., Arbez N., Hotaling K., et al. trans-(-)-epsilon-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington Disease. J. Biol. Chem. 2012;287:24460–24472. doi: 10.1074/jbc.M112.382226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naia L., Carmo C., Campesan S., Fão L., Cotton V.E., Valero J., Lopes C., Rosenstock T.R., Giorgini F., Rego A.C. Mitochondrial SIRT3 confers neuroprotection in Huntington's disease by regulation of oxidative challenges and mitochondrial dynamics. Free Radic. Biol. Med. 2021;163:163–179. doi: 10.1016/j.freeradbiomed.2020.11.031. [DOI] [PubMed] [Google Scholar]
- 22.Tyagi A., Nguyen C.U., Chong T., Michel C.R., Fritz K.S., Reisdorph N., Knaub L., Reusch J.E.B., Pugazhenthi S. SIRT3 deficiency-induced mitochondrial dysfunction and inflammasome formation in the brain. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-35890-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McDonnell E., Peterson B.S., Bomze H.M., Hirschey M.D. SIRT3 regulates progression and development of diseases of aging. Trends Endocrinol. Metabol. 2015;26:486–492. doi: 10.1016/j.tem.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnakumar R., Kraus W.L. The PARP side of the nucleus: molecular actions, physiological outcomes, and clinical targets. Mol. Cell. 2010;39:8–24. doi: 10.1016/j.molcel.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andrabi S.A., Umanah G.K.E., Chang C., Stevens D.A., Karuppagounder S.S., Gagné J.P., Poirier G.G., Dawson V.L., Dawson T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. USA. 2014;111:10209–10214. doi: 10.1073/pnas.1405158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fouquerel E., Goellner E.M., Yu Z., Gagné J.P., Barbi de Moura M., Feinstein T., Wheeler D., Redpath P., Li J., Romero G., et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014;8:1819–1831. doi: 10.1016/j.celrep.2014.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pourshafie N., Masati E., Bunker E., Nickolls A.R., Thepmankorn P., Johnson K., Feng X., Ekins T., Grunseich C., Fischbeck K.H. Linking epigenetic dysregulation, mitochondrial impairment, and metabolic dysfunction in SBMA motor neurons. JCI Insight. 2020;5 doi: 10.1172/jci.insight.136539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai P., Cantó C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metabol. 2012;16:290–295. doi: 10.1016/j.cmet.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 29.Ranganathan S., Harmison G.G., Meyertholen K., Pennuto M., Burnett B.G., Fischbeck K.H. Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2009;18:27–42. doi: 10.1093/hmg/ddn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chivet M., Marchioretti C., Pirazzini M., Piol D., Scaramuzzino C., Polanco M.J., Romanello V., Zuccaro E., Parodi S., D'Antonio M., et al. Polyglutamine-Expanded Androgen Receptor Alteration of Skeletal Muscle Homeostasis and Myonuclear Aggregation Are Affected by Sex, Age and Muscle Metabolism. Cells. 2020;9 doi: 10.3390/cells9020325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beitel L.K., Alvarado C., Mokhtar S., Paliouras M., Trifiro M. Mechanisms mediating spinal and bulbar muscular atrophy: investigations into polyglutamine-expanded androgen receptor function and dysfunction. Front. Neurol. 2013;4:53. doi: 10.3389/fneur.2013.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim W.F., Forouhan M., Roberts T.C., Dabney J., Ellerington R., Speciale A.A., Manzano R., Lieto M., Sangha G., Banerjee S., et al. Gene therapy with AR isoform 2 rescues spinal and bulbar muscular atrophy phenotype by modulating AR transcriptional activity. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abi6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y., Zhang J., Lin Y., Lei Q., Guan K.L., Zhao S., Xiong Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011;12:534–541. doi: 10.1038/embor.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J.X., Shen S.N., Tong Q., Wang Y.T., Lin L.G. Honokiol protects hepatocytes from oxidative injury through mitochondrial deacetylase SIRT3. Eur. J. Pharmacol. 2018;834:176–187. doi: 10.1016/j.ejphar.2018.07.036. [DOI] [PubMed] [Google Scholar]
- 35.Reiter R.J., Tan D.X., Rosales-Corral S., Galano A., Jou M.J., Acuna-Castroviejo D. Melatonin Mitigates Mitochondrial Meltdown: Interactions with SIRT3. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19082439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song C., Peng W., Yin S., Zhao J., Fu B., Zhang J., Mao T., Wu H., Zhang Y. Melatonin improves age-induced fertility decline and attenuates ovarian mitochondrial oxidative stress in mice. Sci. Rep. 2016;6 doi: 10.1038/srep35165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walcott J.L., Merry D.E. Ligand promotes intranuclear inclusions in a novel cell model of spinal and bulbar muscular atrophy. J. Biol. Chem. 2002;277:50855–50859. doi: 10.1074/jbc.M209466200. [DOI] [PubMed] [Google Scholar]
- 38.Milioto C., Malena A., Maino E., Polanco M.J., Marchioretti C., Borgia D., Pereira M.G., Blaauw B., Lieberman A.P., Venturini R., et al. Beta-agonist stimulation ameliorates the phenotype of spinal and bulbar muscular atrophy mice and patient-derived myotubes. Sci. Rep. 2017;7 doi: 10.1038/srep41046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahn B.H., Kim H.S., Song S., Lee I.H., Liu J., Vassilopoulos A., Deng C.X., Finkel T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA. 2008;105:14447–14452. doi: 10.1073/pnas.0803790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcus J.M., Andrabi S.A. SIRT3 Regulation Under Cellular Stress: Making Sense of the Ups and Downs. Front. Neurosci. 2018;12:799. doi: 10.3389/fnins.2018.00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rardin M.J., Newman J.C., Held J.M., Cusack M.P., Sorensen D.J., Li B., Schilling B., Mooney S.D., Kahn C.R., Verdin E., Gibson B.W. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc. Natl. Acad. Sci. USA. 2013;110:6601–6606. doi: 10.1073/pnas.1302961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martínez-Reyes I., Chandel N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020;11:102. doi: 10.1038/s41467-019-13668-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sawant Dessai A., Dominguez M.P., Chen U.I., Hasper J., Prechtl C., Yu C., Katsuta E., Dai T., Zhu B., Jung S.Y., et al. Transcriptional Repression of SIRT3 Potentiates Mitochondrial Aconitase Activation to Drive Aggressive Prostate Cancer to the Bone. Cancer Res. 2021;81:50–63. doi: 10.1158/0008-5472.CAN-20-1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cui X.X., Li X., Dong S.Y., Guo Y.J., Liu T., Wu Y.C. SIRT3 deacetylated and increased citrate synthase activity in PD model. Biochem. Biophys. Res. Commun. 2017;484:767–773. doi: 10.1016/j.bbrc.2017.01.163. [DOI] [PubMed] [Google Scholar]
- 45.Finley L.W.S., Haas W., Desquiret-Dumas V., Wallace D.C., Procaccio V., Gygi S.P., Haigis M.C. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakatsuji H., Araki A., Hashizume A., Hijikata Y., Yamada S., Inagaki T., Suzuki K., Banno H., Suga N., Okada Y., et al. Correlation of insulin resistance and motor function in spinal and bulbar muscular atrophy. J. Neurol. 2017;264:839–847. doi: 10.1007/s00415-017-8405-3. [DOI] [PubMed] [Google Scholar]
- 47.Guber R.D., Takyar V., Kokkinis A., Fox D.A., Alao H., Kats I., Bakar D., Remaley A.T., Hewitt S.M., Kleiner D.E., et al. Nonalcoholic fatty liver disease in spinal and bulbar muscular atrophy. Neurology. 2017;89:2481–2490. doi: 10.1212/WNL.0000000000004748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Francini-Pesenti F., Vitturi N., Tresso S., Sorarù G. Metabolic alterations in spinal and bulbar muscular atrophy. Rev. Neurol. (Paris) 2020;176:780–787. doi: 10.1016/j.neurol.2020.03.020. [DOI] [PubMed] [Google Scholar]
- 49.Polanco M.J., Parodi S., Piol D., Stack C., Chivet M., Contestabile A., Miranda H.C., Lievens P.M.J., Espinoza S., Jochum T., et al. Adenylyl cyclase activating polypeptide reduces phosphorylation and toxicity of the polyglutamine-expanded androgen receptor in spinobulbar muscular atrophy. Sci. Transl. Med. 2016;8:370ra181. doi: 10.1126/scitranslmed.aaf9526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Houtkooper R.H., Cantó C., Wanders R.J., Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Covarrubias A.J., Perrone R., Grozio A., Verdin E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021;22:119–141. doi: 10.1038/s41580-020-00313-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ryu D., Zhang H., Ropelle E.R., Sorrentino V., Mázala D.A.G., Mouchiroud L., Marshall P.L., Campbell M.D., Ali A.S., Knowels G.M., et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci. Transl. Med. 2016;8:361ra139. doi: 10.1126/scitranslmed.aaf5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schiewer M.J., Goodwin J.F., Han S., Brenner J.C., Augello M.A., Dean J.L., Liu F., Planck J.L., Ravindranathan P., Chinnaiyan A.M., et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012;2:1134–1149. doi: 10.1158/2159-8290.cd-12-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gui B., Gui F., Takai T., Feng C., Bai X., Fazli L., Dong X., Liu S., Zhang X., Zhang W., et al. Selective targeting of PARP-2 inhibits androgen receptor signaling and prostate cancer growth through disruption of FOXA1 function. Proc. Natl. Acad. Sci. USA. 2019;116:14573–14582. doi: 10.1073/pnas.1908547116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Z.Q., Auer B., Stingl L., Berghammer H., Haidacher D., Schweiger M., Wagner E.F. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9:509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- 56.Nair A.B., Jacob S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016;7:27–31. doi: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This excel sheet contains the raw data from the acetylome analysis of 11 week old mouse quadriceps. The first sheet (Summary) is a summary of the sample names and how they were prepared for acetylation-specific LC-MS/MS and how the data from the LC-MS/MS was analyzed. The second sheet (Acetyl (K) sites) contains a list of identified Acetyl (K) sites with the levels of acetylated sites indicated by intensity values for each sample. The third sheet (Acetyl (K)Sites_Expanded) has Intensity ___1, ___2, and ___3 columns transposed into separate rows. This is the main data sheet. Acetylated sites not detected in at least two of the triplicates (in any group) were removed. Fold changes between groups were calculated using non-normalized intensity values (columns AS to CF), and after normalizing to the median Intensity of each sample (columns CG to DT). The |FC|>2, q<0.05 column shows acetylated site with significant change (absolute fold change >2, minimum adjusted p-value of 0.05). The fourth sheet (Mod. Peptide) shows the peptides identified. The fifth sheet (Protein group) shows the protein identifications.
Data Availability Statement
-
•
Experimental data reported in this manuscript will be shared by the lead contact upon request. The raw acetyl (K)Sites for the acetylome analysis are available as a supplemental excel file (Data S1: Acetylome raw data (Acetyl (K) Sites), related to Figure 5).
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.