

Abstract
A positron emission tomography (PET) radioligand for imaging phosphodiesterase 4D (PDE4D) would benefit drug discovery and the investigation of neuropsychiatric disorders. The most promising radioligand to date, namely, [11C]T1650, has shown unstable quantification in humans. Structural elaboration of [11C]T1650 was therefore deemed necessary. High target affinity in the low nM range is usually required for successful PET radioligands. In our PDE4D PET radioligand development, we formulated and optimized an empirical equation (log[IC50 (nM)] = P1 + P2 + P3 + P4) that well described the relationship between binding affinity and empirically derived values (P1–P4) for the individual fragments in four subregions commonly composing each inhibitor (R2 = 0.988, n = 62). This equation was used to predict compounds that would have high inhibitory potency. Fourteen new compounds were obtained with IC50 of 0.3–10 nM. Finally, eight compounds were judged to be worthy of future radiolabeling and evaluation as PDE4D PET radioligands.
Graphical Abstract

INTRODUCTION
Phosphodiesterases (PDE) catalyze the hydrolysis of phosphor-diester bonds. There are 11 families of these enzymes with three of these that are denoted 4, 7, and 8 functioning to hydrolyze the important secondary messenger, cyclic adenosine monophosphate (cAMP). Phosphodiesterase-4 (PDE4) enzymes exist as four subtypes (A, B, C, and D).1,2 Inhibition of PDE4 improves learning and memory in mouse models and shows benefit in major depression and neurodegeneration.3–8 Several non-subtype-selective PDE4 inhibitors (e.g., (R)-rolipram, Figure 1A) are used clinically, but their effective doses are limited by undesirable gastrointestinal side effects, such as nausea, vomiting, and diarrhea.9–11 PDE4 subtypes are highly conserved in their catalytic domains but differ in two different regions: the upstream conserved regions (UCR1 and UCR2) and the control region 3 (CR3) in the C-terminal sequence. Recently, the development of subtype-selective inhibitors has made considerable progress. Novel allosteric inhibitors with PDE4D subtype selectivity have been developed based on targeting the UCR2 region12,13 while PDE4B selective inhibitors have been developed based on targeting the CR3 region.14–16 Studies have shown that PDE4D-selective inhibitors provide not only improved memory and cognition in mouse models but also improved tolerability.12,13 One such PDE4D inhibitor, BPN14770 (Figure 1A), has recently been evaluated for the treatment of Fragile X Syndrome and is in phase III clinical trial.13,17,18 An ability to image and quantify the human brain PDE4D subtype would benefit further drug discovery12,13,16,19–22 and also investigations of neuropsychiatric disorders.
Figure 1.

(A) Chemical structures of (R)-rolipram, BPN14770, and [11C]T1650. (B) Illustration of the interactions of selective inhibitors of PDE4D, exemplified with T1650. (C) Key structural components in PDE4D-selective inhibitors, including one planar scaffold with a H-bond acceptor (P1), and two clamps (P2 and P4) to hold the UCR2 in closed conformation, and a linker (P3). (D) Structural fragments in P1−P4 that were tested in this work. The fragments are presented in ascending order of value, as given below their structures. These values can be added to account for or predict the binding affinity, according to the equation log[IC50 (nM)] = P1 + P2 + P3 + P4.
Positron emission tomography (PET) is a molecular imaging technique that allows quantification of biochemical targets and processes in vivo. PET can be valuable in preclinical and clinical drug development because it can provide critical information about target engagement and occupancy by a drug candidate under specific dose regimens.1,23 The development of radioligands for PET imaging of various PDE enzymes is an active area of research that has provided significant advances for imaging PDEs in families 2, 5, 7, and 4.24,25 At a few research institutes, PDE4 has been imaged in human subjects with PET using the pan-subtype-selective radioligand [11C](R)-rolipram.26–28 A PDE4B-prefering PET radioligand, code-named [18F]PF-06445974, has been developed recently and applied in human study.29–31 However, so far, there is no successful PDE4D-selective radioligand. Recently, we have been seeking to develop PDE4D subtype-selective PET radioligands. Our preceding work explored several chemical series and selected four leads with alkoxypyridinyl cores that were successfully labeled with cyclotron-produced carbon-11 (t1/2 = 20.4 min) and evaluated with PET.22 The best candidate [11C]T1650 ([11C]1; Figure 1A) showed a sizable PDE4D-specific PET signal in monkey brain. However, in subsequent human PET experiments, an accumulation of brain permeable radiometabolites in brain was inferred from the time instability of total volume of distribution (VT) measurements. This accumulation of radiometabolites impedes acquisition of robust output measures of PDE4D density. We concluded that further structural changes to T1650 would be needed to obtain a successful PDE4D radioligand.
Successful PET radioligands must meet very strict requirements.32,33 Foremost, they must have an adequately high affinity for the imaging target, usually represented by a KD or half-maximal inhibitory concentration (IC50) value in the low nM range. As is well known, improving the target binding affinity of lead compounds is a time- and cost-intensive process because it can involve a large number of iterative cycles of design, synthesis, and assay.34,35 Methods for predicting binding affinities are desirable because compounds of predicted inadequate affinity can then be eliminated from consideration and others prioritized for synthesis, thereby increasing development efficiency.
Herein, we further explored structure−activity relationship (SAR) around T1650 in a quest for an inhibitor that could serve for the development of a better-performing PET radioligand. Often in SAR studies, structures are only compared side by side following a single substructure change.12,13,16,20,21,36,37 Even one structural change may have an unpredictable effect. In this systematic study of the SAR emerging around T1650, we discovered an empirical equation that well described the relationship between the nature of structural fragments and PDE4D inhibitory potency (Figure 1C). With this equation, inhibitory potencies for new compounds were predicted well and with high correlation to experimental results (R2 = 0.988). The equation can be rationalized by the aggregation of free energies of binding for four substructure components. Aided by this equation, we successfully obtained more compounds with high inhibitory potencies (IC50 down to 0.3 nM) and some with even superior potency to that of the lead compound T1650 (3.5 nM). Among the potent compounds with IC50 < 10 nM, their selectivity, physiochemical parameters, and amenability for labeling with carbon-11 and fluorine-18 were further evaluated with multiparameter optimization for central nervous system PET radioligands (CNS-PET-MPO).32,33 Finally, eight candidates were judged worthy of future radiolabeling and further evaluation in vivo. Overall, we demonstrated the viability of the empirical equation to predict and prioritize the high-affinity PDE4D inhibitors for further synthesis, thereby greatly saving time and costs. Similarly, the model might be also applicable for the optimization of leads for other PET radioligands.
RESULTS AND DISCUSSION
Structural Design and Synthesis.
An understanding of how PDE4D subtype-selective inhibitors interact with PDE4D enzymes (Figure 1B) is useful for selecting structural changes to be made to a lead inhibitor. The active site of PDE4 is highly conserved among its four subtypes and consists of a hydrophobic pocket, a metal binding pocket, and a solvent-filled side pocket known as the Q, M, and S pockets, respectively.12,38,39 PDE4 isoforms might have two upstream conserved regions (UCR1 and UCR2). UCR1 is responsible for dimerization, whereas UCR2 controls access of cAMP by opening and blocking the active site.12,40 Competitive pan-PDE4 inhibitors interact with the active site by accommodating a planar core with hydrophobic interaction and hydrogen bonding between the H-bond acceptor on the pharmacophore and an enzyme glutamine residue.38
Crystal structures have shown that within the UCR2 region of PDE4D, there is a phenylalanine (Phe271) in place of the tyrosine seen in PDE4A−C.12 This difference has been exploited in the development of inhibitors with PDE4D subtype selectivity.12,13,16,19–22 The pharmacophore in selective PDE4D allosteric inhibitors commonly consists of four parts: a planar scaffold with a H-bond acceptor (P1), two aromatic substituents (P2 and P4), and a linker (P3) (Figure 1C).12 P1 interacts with the hydrophobic Q pocket of the active site. Phe and Ile residues form a clamp to hold P1 while a glutamine residue forms a hydrogen bond with an acceptor in P1. The aromatic P2 and P4 moieties clamp the Phe residue on UCR2 and keep the UCR2 in a closed conformation that traverses the active site and blocks the access of cAMP. The flexible linker (P3) allows the molecule to accommodate itself in the space between the active site and UCR2.
Given these factors, subtle modifications were made to the P1–P4 fragments of T1650 to improve binding affinity while maintaining PDE4D selectivity. Figure 1D lists the fragmental structures of P1–P4 that we tested in this work. Compounds (except 1, 27, 28, and 41) were synthesized from commercially available chemicals by methods13,41 that are depicted in Figures 2 and S1. All of the compounds were characterized with nuclear magnetic resonance (NMR) spectroscopy and high-resolution mass spectroscopy (HRMS) (Figures S7–S423, Supporting Information). They were all confirmed to have over >95% chemical purity with high-performance liquid chromatography (HPLC) before being assayed for PDE4D and PDE4B inhibitory potency.
Figure 2.

Synthetic schemes for the syntheses of PDE4D inhibitors. (A) Inhibitors were synthesized through the Suzuki coupling reactions of alkyl bromide and arylboronic esters/acids. (B) Inhibitors were synthesized through the substitution reactions of alkyl halide. (C) Inhibitors were synthesized through the palladium-mediated coupling of anilines/phenols (ArNH2/ArOH) with aryl chloride. (D) Inhibitor 23 was synthesized as a side product from the reaction of palladium-mediated coupling of α-fluorinated 2-picolinyl triflones with arylboronic acids.
Inhibitory Activity and Assays.
Mutationally activated, dimeric forms of the enzymes PDE4D7-S129D (PDE4D*) and PDE4B1-S133D (PDE4B*) were used for inhibitory activity assays, as reported previously.12,13 In this work, PDE4B was used for selectivity assessment across the PDE4 subtypes because PDE4A−C share the same UCR2. The choice of assay format is critical for measuring inhibitory activity. Here, the real-time, coupled enzyme assay was chosen instead of the method of using a fluorescein-labeled cAMP substrate because the bulky fluorescein label prevents the closure of UCR2 and alters the kinetics of PDE4D inhibitors. The half-maximal inhibitory concentration (IC50) for PDE4D* and PDE4B* of each compound was derived from the plot of inhibition percentage versus inhibitory concentration. Values are listed with structures for compounds 1–47 in the following tables, specifically Table 1 for compounds with P1 and P2 substitutions (1–10), Table 2 for P2 and P3 substitutions (11–23), and Table 3 for P2 and P4 substitutions (24–47). Assays results for compounds 1 (T1650), 27 (T1660), 28 (T1953), and 41 (T2525) are taken from our previous report.22 Selectivity for PDE4D* over PDE4B* was represented by the ratio of PDE4B* IC50 over PDE4D* IC50, or B/D ratio.
Table 1.
Inhibitory Activity of Compounds with P1 and P2 Modificationsa

|
|||||
|---|---|---|---|---|---|
| Comp’d | P1 | P2 | PDE4D* | PDE4B* | Selectivity |
| R1 = | R2 = | IC50(nM) | IC50(nM) | B/D | |
| 1 b | OMe | NO2 | 3.5 | 280 | 80 |
| 2 | OMe | F | 23.8 | 995 | 42 |
| 3 | OMe | Cl | 6.0 | 923 | 154 |
| 4 | OMe | CF3 | 186 | 7850 | 42 |
| 5 | OMe | OCHF2 | 1.4 | 1070 | 793 |
| 6 | OMe | OCF3 | 46 | 7440 | 162 |
| 7 | OMe |

|
13 | 730 | 57 |
| 8 | OCHF2 | F | 7.6 | 1071 | 141 |
| 9 | OCHF2 | OMe | 2.7 | 521 | 193 |
| 10 | F | Cl | 744 | >10000 | >13 |
PDE4D* refers to PDE4D7-S129D, a dimeric isoform of PDE4D that contains a UCR1 mutation (S129D) that mimics protein kinase A (PKA) phosphorylation. PDE4B* refers to PDE4B1-S133D, a dimeric isoform of PDE4B that contains a UCR1 mutation (S133D) that mimics PKA phosphorylation. B/D is the ratio of IC50 for PDE4B* over that for PDE4D*.
The assay result of the compound was adopted from ref 22.
Table 2.
Inhibitory Activity for Compounds with P2 and P3 Modificationsa

|
|||||
|---|---|---|---|---|---|
| Comp’d | P2 | P3 | PDE4D* | PDE4B* | Selectivity |
| R2 = | R3 = | IC50 (nM) | IC50 (nM) | B/D | |
| 11 | F | NH | 529 | 2530 | 5 |
| 12 | Cl | NH | 468 | 3160 | 7 |
| 13 | CF3 | NH | 4310 | >10 000 | >2 |
| 14 | OCHF2 | NH | 188 | 2500 | 13 |
| 15 | F | O | 836 | 4540 | 5 |
| 16 | Cl | O | 306 | 4160 | 14 |
| 17 | CF3 | O | 3540 | 3240 | 0.92 |
| 18 | OCHF2 | O | 1550 | >10 000 | >6 |
| 19 | F | NH | 136 | 1750 | 13 |
| 20 | OMe | NH | 658 | >10 000 | >15 |
| 21 | F | O | 70 | 3110 | 45 |
| 22 | OMe | O | 281 | >10 000 | >36 |
| 23 | OMe | C=O | 873 | 1930 | 2 |
See Table 1 for assay definitions.
Table 3.
Inhibitory Activity of Compounds with P2 and P4 Modificationsa

|
|||||
|---|---|---|---|---|---|
| Comp’d | P2 | P4 | PDE4D* | PDE4B* | Selectivity |
| R2 = | R4 = | IC50 (nM) | IC50 (nM) | B/D | |
| 24 | F |

|
5530 | >10000 | >2 |
| 25 | Cl |

|
1150 | 9170 | 8 |
| 26 | CF3 |

|
>10000 | >10000 | - |
| 27 b | NO2 |

|
2.8 | 1120 | 400 |
| 28 b | Cl |

|
5.1 | 720 | 141 |
| 29 | OCHF2 |

|
11 | 1439 | 137 |
| 30 | OCHF2 |

|
1.06 | 593 | 559 |
| 31 | OCHF2 |

|
785 | >10000 | >13 |
| 32 | OCHF2 |

|
15 | >10000 | >649 |
| 33 | OCHF2 |

|
16 | 1260 | 81 |
| 34 | OCHF2 |

|
21 | >10000 | >472 |
| 35 | OMe |

|
281 | 1060 | 4 |
| 36 | OMe |
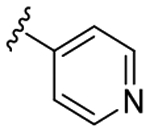
|
3.9 | 621 | 159 |
| 37 | OMe |

|
12.3 | 764 | 62 |
| 38 | OMe |

|
10.2 | 1187 | 116 |
| 39 | OMe |

|
530 | 2270 | 4 |
| 40 | OMe |

|
372 | >10000 | >27 |
| 41 b | Cl |

|
0.5 | 858 | 1672 |
| 42 | OMe |

|
3.5 | 853 | 244 |
| 43 | OMe |

|
2.1 | 513 | 242 |
| 44 | OMe |

|
743 | >10000 | >13 |
| 45 | OMe |

|
42 | >10000 | >238 |
| 46 | OMe |

|
23 | 646 | 28 |
| 47 | OMe |

|
17 | >10000 | >575 |
See Table 1 for assay definitions.
The assay results of the compounds were adopted from ref 22.
Empirical Equation for Describing SAR.
A systematic study of SAR was conducted with 16 compounds, namely, 2–4 and 9 (Table 1), 11–13, 15–17, 20, and 22 (Table 2), and 24–26 and 35 (Table 3). These were arranged in a 4 by 4 matrix of log[PDE4D* IC50 (nM)] values (Figure S2). By comparing the log[PDE4D* IC50 (nM)] between rows and columns, we found that differences in values between paired fragments at the same position are generally consistent with small standard deviations (Figure S2). For example, compounds with N-pyrazolyl groups in P4 (row 2) have a difference of 2.10 ± 0.28 with corresponding compounds having a 3–1H-pyrazolyl group (row 1: 2, 3, 4, and 9). Compounds with −NH− in P3 have a difference of 1.75 ± 0.50 with corresponding compounds having −CH2− (row 3 versus row 1). These observations implied that fragments P1−P4 may be regarded as quite independent in their contributions to enzyme binding and that they work collectively with selectivity (B/D ratio). to influence the binding of inhibitors into the PDE4D enzyme. We considered that such a relationship might be described mathematically with the equation log[IC50 (nM)] = P1 + P2 + P3 + P4, where a value is attributed to each fragment in P1−P4. We tested this possibility with the results for compounds 1–47 in Tables 1–3 (Figures S3 and S4). We solved for the optimized value for each fragment in P1−P4 and listed these values under their structures (Figure S4). With these values, we then calculated the binding affinities of the compounds and plotted them against the experimental values (Figure S5). This plot has a very high linear correlation coefficient (R2 = 0.9885) and a small standard deviation of 0.224 (n = 47). The strong correlation further implied the validity and potential utility of the empirical equation.
Application of the Empirical Equation to DiscoverHigh-Affinity Inhibitors.
Considering the generated empirical equation, small fragment values are clearly desirable for obtaining low PDE4D* IC50 (high inhibitory potency). With this in mind, we sorted the fragments P1−P4 according to increasing value (Figure S4) to allow easy comparison of fragments at each position. Five new compounds 48–52 were then configured from the top-listed fragments and then synthesized and assayed (Table 4). Their experimental log[IC50 (nM)] values differed from predicted results within an acceptable range of ±0.5 (i.e., ×/÷ 3.16 for IC50 (nM)). The lowest IC50 (0.3 nM) value (highest affinity) was obtained for compound 51. This demonstrated the usefulness of the equation for predicting the binding affinities of new compounds.
Table 4.
New Inhibitors Synthesized and Assayed as a Result of Predictiona

| |||||
|---|---|---|---|---|---|
| Comp’d | Predicted Log[PDE4D IC50] | Experimental Log[PDE4D IC50] | PDE4D* | PDE4B* | Selectivity |
| IC50 (nM) | IC50 (nM) | B/D | |||
| 48 | 1.62 | 1.11 | 13 | 2130 | 165 |
| 49 | 0.59 | 0.26 | 1.8 | 665 | 364 |
| 50 | −0.39 | −0.44 | 0.36 | 91 | 254 |
| 51 | −0.90 | −0.52 | 0.30 | 81 | 270 |
| 52 | 0.11 | 0.57 | 3.7 | 198 | 54 |
See Table 1 for assay definitions.
Encouraged by the above results, we further examined the equation with the PDE4D IC50 data on 27 compounds published by Gurney et al in 2019.13 Values for the fragments in P1−P4 were listed under the structures (Figure S6). The correlation coefficient was calculated to be very high (R2 = 0.9866; n = 27). The 4-(3-hydroxypropyl)phenyl and 4-(2-hydroxyethyl)phenyl groups appear as the top two in the list for P4. Therefore, we incorporated these two groups into our subsequent inhibitor design. The resulting four new compounds (53–56) were found to have high binding affinity (PDE4D* IC50 = 4–24 nM) and high selectivity (B/D ratio >420) (Table 5).
Table 5.
New Inhibitors Synthesized and Assayed as a Result of Validation by Published Dataa

|
||||
|---|---|---|---|---|
| Comp’d | P4 | PDE4D* | PDE4B* | Selectivity |
| R4 = | IC50 (nM) | IC50 (nM) | B/D | |
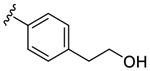
| 53 |
|
4.0 | >10000 | >2519 |
| 54 |

|
16 | >10000 | >621 |
| 55 |

|
6.9 | 4000 | 581 |
| 56 |

|
24 | >10000 | >420 |
See Table 1 for assay definitions.
A series of benzyl compounds had been synthesized during the preparation of phenol precursors for future labeling with [11C]iodomethane. These compounds were also assayed (Table 6). With the results in Tables 1–6, we now had a larger compound set (n = 62) to derive optimized P1−P4 values. These values were listed under their structures in Figure 1D. A plot of predicted values versus experimental values for all compounds listed in Tables 1–6 gave a high correlation coefficient (R2 = 0.9876) and a standard deviation of 0.222 (Table S1 and Figure 3A and 3B), similar to those determined for the compounds in Tables 1–3 alone (1–47; Figure S5). It is noticeable that the absolute values of the fragments changed considerably. However, the relative differences between fragments in P1−P4 were quite well maintained. To address this issue, one can set a standard in each of P1−P4 and compare each fragment to the standard. However, this will complicate the calculation of prediction. Therefore, we kept the values as they were generated. Slight changes were expected in the relative values because, in each calculation, values were optimized to better fit the experimental results of the old compounds and newly added compounds. With more data input, the uncertainty brought by assays would be further reduced.
Table 6.
Inhibitory Activity of Benzyl Compounds (Intermediates for Precursors)a

|
||||
|---|---|---|---|---|
| Comp’d | P4 | PDE4D* | PDE4B* | Selectivity |
| R4 = | IC50 (nM) | IC50 (nM) | B/D | |
| 57 |

|
164 | >10000 | >61 |
| 58 |

|
610 | >7763 | >13 |
| 59 |

|
174 | >10000 | >57 |
| 60 |

|
446 | 274 | 0.61 |
| 61 |

|
171 | 143 | 0.84 |
| 62 |

|
85 | 46 | 0.54 |
See Table 1 for assay definitions.
Figure 3.

(A) Correlation of predicted values and experimental values for all compounds listed in Tables 1–6. (B) Deviation of predicted values from experimental values. (C) Correlation of log[PDE4D binding affinity] with PDE4B binding affinity. (D) Correlation of log[PDE4D binding affinity] with selectivity (B/D ratio).
In Figure 1D, there are 4 fragments in P1, 9 fragments in P2, 4 fragments in P3, and 15 fragments in P4. Theoretically, these fragments can constitute a library of 2160 compounds (4 × 9 × 4 × 15 = 2160). With the equation log[IC50 (nM)] = P1 + P2 + P3 + P4 and 62 assayed compounds, we were able to derive the values for the fragments and predict the binding affinities for 2160 compounds. The empirical model would provide helpful insights into the design and selection of compounds for evaluation, thereby greatly saving the time and cost for arduous syntheses. However, this is not the real case because assays and the model itself would bring uncertainties to the system.
In sum, we formulated a mathematical equation to describe the relationship between the fragments in P1−P4 and the binding affinities of the corresponding compounds. This empirical equation was validated to be effective in predicting the binding affinities of 62 inhibitors and separately 27 already published inhibitors. Compound 51 with the lowest IC50 (0.3 nM) was first predicted and then obtained based on the current knowledge of fragments. The table of fragments in P1−P4 in Figure 1D is expandable with more inputs of assay results and thereby allows more compounds to be assessed before synthesis.
Rationale for the Empirical Equation.
The empirical equation can be understood from the relationship between free energy for ligand binding to a target protein and its equilibrium constant (K), which is expressed to be ΔG = −RT ln K, where R is the gas constant and T is the absolute temperature. In the binding equation of inhibitors and enzymes, K ≈ 1/IC50.34,42 Thus, ΔG ≈ RT ln(IC50) = 2.303RT log(IC50). The empirical equation log[IC50 (nM)] = P1 + P2 + P3 + P4 is therefore easily transformed into the equation ΔGbind(inhibitor) = ΔG(P1) + ΔG(P2) + ΔG(P3) + ΔG(P4). Such an equation implies that the binding free energy is the sum of the discrete binding free energies of the inhibitor fragments. This concept was first introduced and discussed by Page43 who named it the “anchor principle”. This idea has also been utilized by other groups to assist with in silico drug design.34,44,45 Current approaches to compute binding free energies are generally accurate to within 0.5–1.5 kcal/mol of experimental inhibitory activities,44 which is 0.367–1.10 in log(IC50) and ×/÷2.31–12.6 in IC50. The standard deviation of the empirical approach in this work is 0.222 in log(IC50) (×/÷1.67 in IC50), and therefore more accurate in prediction.
With the empirical equation, the solved values can be used to evaluate the contribution of the structural fragments to the inhibitory activity more intuitively, predict the inhibitory activity of new compounds more accurately, and finally lead to the discovery of new high-affinity compounds more efficiently. The reliability of the equation rests on reliable assay results. The structural scope of the model could be increased with the input of more structural fragments. Moreover, with mechanistic understanding, such an empirical equation might be workable for compounds that can be predicted by the aggregate of binding free energies.34,44,45
Selectivity to PDE4D and Other Parameters.
To develop a successful PET radioligand, tackling the problem of binding affinity is only the first step. Its selectivity and other parameters also need to be considered. In Figure 3C and 3D, we plotted compound PDE4B binding affinities (log[PDE4B* IC50 (nM)]) and selectivity (log(B/D ratio)) versus their PDE4D binding affinities (log[PDE4D* IC50 (nM)]). Data with IC50 of >10 000 nM were excluded from the plot. The rest of the data falls into two categories, one has B/D ratio >1 and the other has B/D ratio <1 (17, 60–62). Linear fitting was performed for each category. In Figure 3C, inhibitors without selectivity will show similar binding affinity to PDE4D and PDE4B and result in a slope of 1. The fitting of B/D < 1 has a slope of 1.105 and a correlation coefficient of R2 = 0.9895, suggesting these compounds have a slight preference to inhibit PDE4B. Among compounds with B/ D ratio <1, compound 17 is questionable due to its B/D ratio of 0.92, very close to 1. The other three, 60–62, contain a 1-(3-benzyloxy)-phenyl fragment in P2. This fragment is listed among the last fragments in P2 (Figure 1D) and is deemed to be unsuitable for the design of high-affinity PDE4D ligands. Meanwhile, in Figure 3C, the fitting for data points with B/D ratio >1 has a slope of 0.3532 and a correlation coefficient of R2 = 0.5955, indicating that the preference of these inhibitors to bind to PDE4D increased with high binding affinity. This is more clearly presented in Figure 3D, where the selectivity (log(B/D ratio)) increased with binding affinity to PDE4D* with a higher correlation coefficient of 0.8317 than that with PDE4B* (R2 = 0.1962). This suggests that the increased selectivity can be mainly attributed to the increased binding affinity to PDE4D. Comparing the difference in binding mode to PDE4D* and PDE4B*, the two regions P2 and P4 of the inhibitors play a great role in clamping the phenyl group on the UCR2 of PDE4D* and thus in determining selectivity. Above all, the plots suggest that high affinity is often accompanied with high selectivity to PDE4D in our approach for the discovery of PDE4D inhibitors for PET radioligands development.
One main challenge in developing new PET radioligands for neuroimaging is that they must cross the blood-brain barrier. By statistically analyzing 77 CNS PET radioligands, a CNS PET MPO parameter has been proposed to optimize physiochemical properties in searches for likely successful brain-penetrant radioligands.33,46,47 CNS PET MPO values are calculated by summing transformed functions for CLog P, Clog D7.4, MW, pKa, tPSA, and HBD, across ranges from 0.0 to 6.0. We calculated the six parameters and CNS PET MPO of our compounds accordingly (Table S2) by utilizing Pallas software and plotted their values versus their binding affinities (Figure 4). The more desirable range is painted with an orange background and a less desirable range with a blue background. Most of the compounds fall into the desirable range and there is no significant relationship between the parameters and the binding affinities (Figure 4).
Figure 4.

Relationship of physiochemical properties versus inhibitory potency of PDE4D inhibitors log[PDE4D* IC50]. (A) CLog P, (B) CLog D7.4, (C) MW, (D) tPSA, (E) HBD, (F) pKa. According to the selection criteria of CNS PET radioligand proposed by Zhang et al.,33 the more desirable range is colored with orange background while a less desirable range is colored with blue background. (G) CNS PET MPO. CNS PET MPO is calculated by the summation of the transformed functions of CLog P, Clog D7.4, MW, pKa, tPSA, and HBD.
Potential Candidates for PDE4D Radioligands.
In addition to satisfying criteria for high binding affinity (IC50 < 10 nM) and selectivity (>50-fold), CNS PET MPO scores greater than 3 were considered to optimize physicochemical properties. Also, amenability to labeling with carbon-11 or fluorine-18 at high molar activity had to be considered from a structural perspective. A methoxy group attached to a pyridinyl or phenyl ring is a good structural moiety for labeling at high molar activity through the reaction of phenol precursor with [11C]iodomethane or [11C]methyl triflate.22,48,49 Aryl fluoro groups are feasibly labeled via the reaction of boronic acid, boronic ester, arylstannane, diaryliodonium salt, or aryliodonium ylide precursors with [18F]fluoride ion.48,50,51 Based on these requirements, we selected eight candidates (5, 8, 9, 30, 36, 43, 49, and 52) from the synthesized compounds to be evaluated as PET radioligands (Figure 5). Their potential carbon-11 and fluorine-18 labeling sites are shown in red and blue, respectively. Results from radiolabeling chemistry and the evaluations of prepared radioligands will be published separately.
Figure 5.

Potential candidates for radiolabeling. In addition to criteria of binding affinity (IC50 < 10 nM) and selectivity (>50-fold), CNS PET MPO scores (>3) were used to take physicochemical properties into account.
CONCLUSIONS
In the course of searching for high-affinity PDE4D-selective inhibitors as leads for PET radioligands, we formulated and optimized an empirical equation log[IC50 (nM)] = P1 + P2 + P3 + P4 that well describes the relationship between the binding affinity and structural fragments. The equation was validated with 62 compounds (58 were newly synthesized and assayed) in a systematic SAR study. The inhibitory potencies of compounds were accurately predicted from the equation with a high linear correlation between the experimental and predicted results (R2 = 0.988) and with a standard deviation of 0.222, which is ×/÷1.67 in IC50 and more accurate than might be predicted from in silico predictions. The underlying basis of the equation is that the free energy of the binding of the inhibitor to the enzyme can be calculated from the aggregates of the discrete binding free energies of its fragments. Through this equation, we successfully obtained more than 14 new compounds with high binding affinities (IC50 < 10 nM, with the lowest showing an IC50 of 0.3 nM). Among the high-affinity compounds of IC50 < 10 nM, their selectivity, physiochemical parameters, and amenability for carbon-11 and/or fluorine-18 labeling were further evaluated for CNS PET radioligands. Eight potential candidates were finally found as being worthy of further evaluation in vivo. Above all, we demonstrated the viability of the empirical equation log[IC50 (nM)] = P1 + P2 + P3 + P4 to facilitate the discovery of high-affinity PDE4D inhibitors. The mechanistic explanation suggests similar models could be developed for other drug molecules and would greatly benefit drug and radioligand discovery.
EXPERIMENTAL SECTION
Materials and Methods.
2-Bromo-3-(difluoromethoxy)-6-methylpyridine was synthesized as reported.22 6-Chloro-2-iodopyridin-3-ol was also synthesized as reported.52 Other reagents and solvents were purchased from Sigma-Aldrich, AA Blocks, Astatech, and Enamine. Reagents and solvents were used as received unless otherwise stated. Chromatographic purifications were conducted with silica gel cartridges on a Teledyne Combiflash System. NMR spectra were recorded on a Bruker Avance 400 instrument at room temperature unless otherwise noted. Chemical shifts (δ) were reported in parts per million (ppm) downfield with tetramethylsilane as internal standard. Accurate mass data were obtained using a Waters Xevo G2-XS QTof mass spectrometer. The instrument was operated in positive-ion electrospray ionization (ESI) mode at a resolution of 25 000. The ESI capillary voltage was 2.8 kV, and the desolvation temperature was 280 °C. Accurate masses were determined using sodium trifluoroacetic acid (TFA) as an internal standard. The LC system was a Waters Acquity I-class UPLC. Samples were directly injected into a solvent stream consisting of an 80:20 mixture of MeCN/MeOH with 0.1% TFA and 0.2% formic acid added. cLog D and clog P, pKa values were computed with Pallas (for Windows, software version 3.8.1.2) in default option. tPSA and molecular weight were obtained from ChemDraw version 20.1.
Chemical purities of inhibitors were determined to be over 95% on an analytical reversed-phase high-performance liquid chromatography (HPLC) with the following methods: method A: Luna C18 column (250 × 4.6 mm, 10 μm; Phenomenex; Torrance, CA), isocratic eluent (water−acetonitrile = 50:50 v/v), flow rate = 2 mL/min, absorbance at 254 nm. Method B: Luna C18(2) column (250 × 4.6 mm, 5 μm; Phenomenex; Torrance, CA), isocratic eluent (water−acetonitrile = 50:50 v/v), flow rate = 2 mL/min, absorbance at 254 nm. Method C: Luna C18(2) column (250 × 4.6 mm, 5 μm; Phenomenex; Torrance, CA), isocratic eluent (0.1% TFA in water−acetonitrile = 50:50 v/v), flow rate = 2 mL/min, absorbance at 254 nm.
PDE4 Enzyme Inhibition Assay.
Methods used were as described previously.22
Synthesis of Inhibitors.
6-((1H-Pyrazol-4-yl)methyl)-2-(3-fluorophenyl)-3-methoxypyridine (2).
Step 1: 6-(Bromomethyl)-2-(3fluorophenyl)-3-methoxypyridine (84) (346 mg, 1.17 mmol), 1-(1-ethoxyethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (400 mg, 1.5 mmol), K3PO4 (424 mg, 2 mmol), and Pd(dppf)2Cl2 (20 mg, 0.03 mmol) were added into a 50 mL two-neck round-bottom flask in a glovebox. Then, a degassed solvent mixture of dioxane/water (4:1 v/v, 5 mL) was injected into the flask. The reaction mixture was stirred at 80 °C overnight. After being cooled to room temperature, the reaction mixture was concentrated under reduced pressure. The residue was partitioned between dichloromethane (DCM) and water. The organic layer was separated and dried over Na2SO4. The DCM solution was passed through a pad of celite and concentrated to afford the crude product for direct use in the next step. Step 2: 0.4 mL of 4 M HCl in dioxane/water (v/v = 2:1) was added to the crude product above in MeOH (4 mL). After the mixture was stirred at room temperature overnight, the solvent was removed. Saturated NaHCO3 aqueous solution was added to neutralize the mixture. And the mixture was extracted with DCM thrice. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified on a silica gel column (eluent: 50–70% EtOAc/hexane). The product was obtained as a white solid (130 mg, 39% yield). 1H NMR (400 MHz, CDCl3) δ 10.38 (br, 1H), 7.75 (d, 1H, J = 8.0 Hz), 7.69 (d, 1H, J = 10.8 Hz), 7.51 (s, 2H) 7.42–7.36 (m, 1H), 7.21 (d, 1H, J = 8.4 Hz), 7.09–7.04 (m, 2H), 4.05 (s, 2H), 3.84 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 162.6 (d, J = 242.6 Hz), 152.2, 152.0, 145.4, 139.9 (d, J = 7.9 Hz), 129.4 (d, J = 8.2 Hz), 125.1 (d, J = 2.7 Hz), 122.1, 119.6, 118.9, 116.4 (d, J = 22.6 Hz), 115.0 (d, J = 21.2 Hz), 55.6, 32.4 ppm. 19F NMR (376 MHz, CDCl3) δ −113.9 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H15FN3O+, 284.1199; found 284.1199. Retention time: 4.12 min (HPLC method A).
6-((1H-Pyrazol-4-yl)methyl)-2-(3-chlorophenyl)-3-methoxypyridine (3).
This synthesis was previously reported.41 This compound was prepared from 6-(bromomethyl)-2-(3-chlorophenyl)-3-methoxypyridine (87) (625 mg, 2 mmol) following a procedure similar to that of 2. The product was obtained as a white solid (350 mg, 58% yield). 1H NMR (400 MHz, CDCl3) δ 10.6 (br, 1H), 7.94 (s, 1H), 7.83 (dd, 1H, J1 = 6.0 Hz, J2 = 1.6 Hz), 7.50 (s, 2H), 7.38–7.32 (m, 2H), 7.21 (d, 1H, J = 8.4 Hz), 7.07 (d, 1H, J = 8.4 Hz), 4.05 (s, 2H), 3.84 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.3, 151.9, 145.3, 139.5, 133.8, 129.5, 129.2, 128.2, 127.6, 122.1, 119.5, 118.8, 55.6, 32.3 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H15ClN3O+, 300.0904; found 300.0906. Retention time: 5.67 min (HPLC method A).
6-((1H-Pyrazol-4-yl)methyl)-3-methoxy-2-(3-(trifluoromethyl)-phenyl)pyridine (4).
This compound was prepared from 6-(bromomethyl)-3-methoxy-2-(3-(trifluoromethyl)phenyl)pyridine (85) (692 mg, 2.0 mmol) following a procedure similar to that of 2. The product was obtained as a white solid (254 mg, 38% yield). 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 8.05 (d, 1H, J = 7.6 Hz), 7.52 (d, 1H, J = 7.6 Hz), 7.45–7.39 (m, 3H), 7.12 (d, 1H, J = 8.4 Hz), 6.99 (d, 1H, J = 8.4 Hz), 3.96 (s, 2H), 3.73 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.6, 152.0, 145.1, 138.5, 133.0 (br), 132.8, 130.3 (q, J = 31.9 Hz), 128.5, 126.4 (q, J = 3.9 Hz), 124.8 (q, J = 3.7 Hz), 124.4 (q, J = 270 Hz, including 128.5, 125.7, 123.0, 120.3), 122.4, 119.7, 118.6, 55.7, 32.4 ppm. 19F NMR (376 MHz, CDCl3) δ−63.7 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H15F3N3O+, 334.1167; found 334.1163. Retention time: 7.68 min (HPLC method A).
6-((1H-Pyrazol-4-yl)methyl)-2-(3-(difluoromethoxy)phenyl)-3methoxypyridine (5).
This compound was prepared from 6-(bromomethyl)-2-(3-(difluoromethoxy)phenyl)-3-methoxypyridine (91) (344 mg, 1 mmol) following a procedure similar to that of 2. The product was obtained as a colorless oil (181 mg, 55% yield). 1H NMR (400 MHz, CDCl3) δ 7.81 (d, 1H, J = 8.0 Hz), 7.73 (s, 1H), 7.49 (s, 2H), 7.43 (t, 1H, J = 8.0 Hz), 7.21 (d, 1H, J = 8.4 Hz), 7.13–7.06 (m, 2H), 6.55 (t, 1H, J = 74.0 Hz), 4.04 (s, 2H), 3.83 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.3, 152.0, 151.1 (t, J = 2.5 Hz), 145.4, 139.7, 133.4, 129.3, 126.5, 122.2, 120.5, 119.7, 119.1, 118.7, 116.3 (t, J = 257.3 Hz, including 118.8, 116.3, 113.7), 55.7, 32.3 ppm. 19F NMR (376 MHz, CDCl3) δ −80.3 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H16F2N3O2+, 332.1211; found 332.1213. Retention time: 5.13 min (HPLC method A).
6-((1H-Pyrazol-4-yl)methyl)-3-methoxy-2-(3-(trifluoromethoxy)-phenyl)pyridine (6).
This compound was prepared from 6-(bromomethyl)-3-methoxy-2-(3-(trifluoromethoxy)phenyl)pyridine (92) (362 mg, 1 mmol) following a procedure similar to that of 2. The product was obtained as a colorless oil (202 mg, 58% yield). 1H NMR (400 MHz, CDCl3) δ 10.36 (br, 1H), 7.91 (d, 1H, J = 8.0 Hz), 7.86 (s, 1H), 7.51 (s, 2H), 7.44 (t, 1H, J = 8.0 Hz), 7.22 (d, 1H, J = 8.4 Hz), 7.08 (d, 1H, J = 8.4 Hz), 4.05 (s, 2H), 3.84 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.4, 152.0, 149.0, 145.0, 139.7, 133.5 (br), 129.2, 127.8, 124.4, 122.3, 122.2, 120.6 (q, J = 255.3 Hz, including 124.4, 121.9, 119.3, 116.8), 120.5, 119.6, 118.8, 55.6, 32.4 ppm. 19F NMR (376 MHz, CDCl3) δ −57.6 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H15F3N3O2+, 350.1116; found 350.1117. Retention time: 8.38 min (HPLC method B).
5-(6-((1H-Pyrazol-4-yl)methyl)-3-methoxypyridin-2-yl)benzo[c][1,2,5]oxadiazole (7).
This compound was prepared from 5-(6(bromomethyl)-3-methoxypyridin-2-yl)benzo[c][1,2,5]oxadiazole (90) following a procedure similar to that of 2. The product was obtained as a white solid (203 mg, 66% yield). 1H NMR (400 MHz, CDCl3) δ 8.51 (s, 1H), 8.19 (d, 1H, J = 9.6 Hz), 7.85 (d, 1H, J = 9.2 Hz), 7.53 (s, 2H), 7.30 (d, 1H, J = 8.8 Hz), 7.17 (d, 1H, J = 8.4 Hz), 4.07 (s, 2H), 3.92 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.9, 152.3, 149.7, 148.8, 143.5, 140.4, 134.1, 133.5, 123.2, 119.9, 118.7, 116.5, 115.2, 55.7, 32.3 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H14N5O2+, 308.1147; found 308.1144. Retention time: 4.88 min (HPLC method A).
6-((1H-Pyrazol-4-yl)methyl)-3-(difluoromethoxy)-2-(3fluorophenyl)pyridine (8).
This compound was prepared from 6(bromomethyl)-3-(difluoromethoxy)-2-(3-fluorophenyl)pyridine (88) (332 mg, 1 mmol), 1-(1-ethoxyethyl)-4-(4,4,5,5-tetramethyl1,3,2-dioxaborolan-2-yl)-1H-pyrazole (266 mg, 1 mmol), Na2CO3 (318 mg, 3 mmol), Pd(dppf)Cl2 (71 mg, 0.1 mmol), and a degassed solvent mixture of dioxane/water (2:1 v/v, 7.5 mL) in a 90 °C oil bath following a procedure similar to that of 2. The product was obtained as a colorless oil (52 mg, 16% yield). 1H NMR (400 MHz, CDCl3) δ 9.10 (br, 1H), 7.67 (d, 1H, J = 7.6 Hz), 7.59 (d, 1H, J = 10.0 Hz), 7.52 (s, 2H), 7.49 (s, 1H, J = 8.4 Hz), 7.45–7.38 (m, 1H), 7.14–7.08 (m, 2H), 6.37 (t, 1H, J = 72.8 Hz), 4.10 (s, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ 162.7 (d, J = 243.8 Hz), 158.1, 148.7 (d, J = 2.3 Hz), 143.3 (t, J = 2.7 Hz), 138.6 (d, J = 7.6 Hz), 133.4, 129.8 (d, J =7.9 Hz), 129.4, 125.1 (d, J = 2.9 Hz), 122.4, 117.9, 116.4 (d, J = 22.8 Hz), 115.8 (d, J = 21.1 Hz), 115.7 (t, J = 261.7 Hz, including 118.3, 115.7, 113.1), 32.8 ppm. 19F NMR (376 MHz, CDCl3) δ −81.1, −113.1 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H13F3N3O+, 320.1011; found 320.1016. Retention time: 5.47 min (HPLC method A).
6-((1H-Pyrazol-4-yl)methyl)-3-(difluoromethoxy)-2-(3methoxyphenyl)pyridine (9).
This compound was prepared from 6(bromomethyl)-3-(difluoromethoxy)-2-(3-methoxyphenyl)pyridine (86) (344 mg, 1.0 mmol) following a procedure similar to that of 2. The product was obtained as a pale-yellow oil. (241 mg, 74% yield). 1H NMR (400 MHz, CDCl3) δ 7.43–7.26 (m, 6H), 7.01 (d, 1H, J = 8.4 Hz), 6.89 (d, 1H, J = 8.0 Hz), 6.25 (t, 1H, J = 73.6 Hz), 4.02 (s, 2H), 3.76 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 158.0, 150.0, 143.3, 137.8, 129.5, 129.3, 122.0, 121,8, 118.0, 115.8 (t, J = 261 Hz, including 118.5, 115.8, 113.2), 114.9, 114.7, 55.3, 32.8 ppm. 19F NMR (376 MHz, CDCl3) δ −81.0 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H16F2N3O2+, 332.1211; found 332.1216. Retention time: 4.60 min (HPLC method A).
6-((1H-Pyrazol-4-yl)methyl)-2-(3-chlorophenyl)-3-fluoropyridine (10).
This compound was prepared from 6-(bromomethyl)-2-(3chlorophenyl)-3-fluoropyridine (89) (300 mg, 1 mmol) following a procedure similar to that of 8. The product was obtained as a colorless oil (52 mg, 18% yield). 1H NMR (400 MHz, CDCl3) δ 8.64 (br, 1H), 8.01 (s, 1H), 7.88 (s, 1H), 7.52 (s, 2H), 7.41–7.35 (m, 3H), 7.10 (dd, 1H, J1 = 8.4 Hz, J2 = 3.6 Hz), 4.09 (s, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ 156.6 (d, J = 4.6 Hz), 156.2 (d, J = 257.3 Hz), 143.3 (d, J = 10.5 Hz), 137.2 (d, J = 5.7 Hz), 134.4, 133.5, 129.7, 129.1, 128.8 (d, J = 5.8 Hz), 126.9 (d, J = 6.7 Hz), 124.9 (d, J = 21.0 Hz), 123.0 (d, J = 4.1 Hz), 118.2, 32.7 (d, J = 1.2 Hz) ppm. 19F NMR (376 MHz, CDCl3) δ −127.4 ppm. HRMS ESI [M + H]+ (m/z): calcd for C15H12ClFN3+, 288.0704; found 288.0708. Retention time: 8.75 min (HPLC method A).
6-(3-Fluorophenyl)-5-methoxy-N-(1H-pyrazol-4-yl)pyridin-2amine (11).
This compound was prepared from 6-chloro-2-(3fluorophenyl)-3-methoxypyridine (65) (238 mg, 1 mmol) following a procedure similar to that of 13. The product was obtained as a pale-yellow solid (108 mg, 38% yield). 1H NMR (400 MHz, CDCl3) δ 11.4 (br), 7.76 (d, 1H, J = 7.6 Hz), 7.70 (d, 1H, J = 10.8 Hz), 7.65 (s, 2H), 7.41–7.35 (m, 1H), 7.22 (d, 1H, J = 9.2 Hz), 7.07–7.02 (m, 1H), 6.55 (d, 1H, J = 8.8 Hz), 6.39 (s, 1H), 3.76 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 162.6 (d, J = 242.6 Hz), 151.3, 147.1, 143.5 (d, J = 2.6 Hz), 140.0 (d, J = 7.7 Hz), 129.4 (d, J = 8.1 Hz), 127.7 (br), 124.9 (d, J = 2.8 Hz), 124.3, 123.9, 116.1 (d, J = 22.7 Hz), 114.9 (d, J = 21.1 Hz), 107.2, 56.8 ppm. 19F NMR (376 MHz, CDCl3) δ −113.8 ppm. HRMS ESI [M + H]+ (m/z): calcd for C15H14FN4O+, 285.1152; found 285.1150. Retention time: 4.27 min (HPLC method A).
6-(3-Chlorophenyl)-5-methoxy-N-(1H-pyrazol-4-yl)pyridin-2amine (12).
This compound was prepared from 6-chloro-2-(3chlorophenyl)-3-methoxypyridine (63) (254 mg, 1 mmol) following a procedure similar to that 13. The product was obtained as a yellow oil (78 mg, 26% yield). 1H NMR (400 MHz, CDCl3) δ 10.8 (br, 1H), 7.95 (s, 1H), 7.83 (d, 1H, J = 6.8 Hz), 7.62 (s, 2H), 7.36–7.30 (m, 2H), 7.20 (d, 1H, J = 8.8 Hz), 6.54 (d, 1H, J = 8.8 Hz), 6.48 (s, 1H), 3.74 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 151.4, 147.0, 143.4, 139.6, 133.8, 129.3, 129.3 129.2, 128.1, 127.7 (br), 127.4, 124.3, 123.8, 107.2, 56.8 ppm. HRMS ESI [M + H]+ (m/z): calcd for C15H14ClN4O+, 301.0856; found 301.0855. Retention time: 5.97 min (HPLC method A).
5-Methoxy-N-(1H-pyrazol-4-yl)-6-(3-(trifluoromethyl)phenyl)pyridin-2-amine (13).
Step 1: 6-Chloro-3-methoxy-2-(3(trifluoromethyl)phenyl)pyridine (64) (288 mg, 1 mmol), 1(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-4-amine (167 mg, 1 mmol), tBuOK (135 mg, 1.2 mmol), and Pd(OAc)2/BINAP (40 mg, molar ratio = 1/1.5) were added into a 50 mL two-neck round-bottom flask in a glovebox. Then, degassed anhydrous toluene (5 mL) was injected. The flask was sealed and placed in a 110 °C oil bath. The reaction mixture was stirred overnight. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure. The residue was partitioned between DCM and water. The organic layer was separated and dried over Na2SO4. After passed through a pad of celite, the filtrate was concentrated to afford the crude product for the next step. Step 2: 0.5 mL of 4 M HCl in dioxane/water (v/v = 2:1) was added to the solution of the above crude product in MeOH (5 mL). After the mixture was stirred at room temperature overnight, the volatile solvent was removed. Saturated NaHCO3 aqueous solution was added to neutralize the mixture. Then, the mixture was extracted with DCM thrice. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified with silica gel column (eluent: 50–70% EtOAc/hexane). The product was obtained as a yellow solid (100 mg, 30% yield). 1H NMR (400 MHz, CDCl3) δ 11.4 (br, 1H), 8.27 (s, 1H), 8.16 (d, 1H, J = 7.6 Hz), 7.66 (s, 2H), 7.60 (d, 1H, J = 7.6 Hz), 7.54–7.50 (m, 1H), 7.24 (d, 1H, J = 8.8 Hz), 6.57 (d, 1H, J = 8.8 Hz), 6.36 (s, 1H), 3.76 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 151.4, 147.2, 143.2, 138.5, 132.5, 130.6 (q, J = 31.9 Hz), 128.4, 127.6 (br), 126.1 (q, J = 3.8 Hz), 124.6 (q, J = 3.8 Hz), 124.3 (q, J = 271 Hz, including 128.4, 125.7, 123.0, 120.3), 124.3, 123.8, 107.4, 56.8 ppm. 19F NMR (376 MHz, CDCl3) δ −63.7 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H14F3N4O+, 335.1120; found 335.1122. Retention time: 6.80 min (HPLC method A).
6-(3-(Difluoromethoxy)phenyl)-5-methoxy-N-(1H-pyrazol-4-yl)pyridin-2-amine (14).
This compound was prepared from 6-chloro-2(3-(difluoromethoxy)phenyl)-3-methoxypyridine (69) (286 mg, 1 mmol) following a procedure similar to that of 20. The product was obtained as a pale-yellow glassy solid (71 mg, 21% yield). 1H NMR (400 MHz, CDCl3) δ 7.84 (d, 1H, J = 8.0 Hz), 7.77 (s, 1H), 7.68 (s, 2H), 7.41 (t, 1H, J = 8.0 Hz), 7.24 (d, 1H, J = 9.2 Hz), 7.11 (d, 1H, J = 8.4 Hz), 6.57 (d, 1H, J = 8.8 Hz), 6.55 (t, 1H, J = 74.0 Hz), 6.27 (s, 1H), 3.77 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 151.1 (t, J = 3.4 Hz), 147.2, 143.5, 139.8, 129.3, 127.6, 126.3, 124.3, 123.9, 120.2, 119.1, 116.2 (t, J = 257.3 Hz, including 118.8, 116.2, 113.6), 107.4, 56.8 ppm. 19F NMR (376 MHz, CDCl3) δ−80.3 ppm. HRMS ESI [M + H]+ (m/ z): calcd for C16H15F2N4O2+, 333.1163; found 333.1163. Retention time: 2.32 min (HPLC method C).
6-((1H-Pyrazol-4-yl)oxy)-2-(3-fluorophenyl)-3-methoxypyridine (15).
This compound was prepared from 65 (254 mg, 1 mmol) following a procedure similar to that of 17. The product was obtained as a colorless oil (49.7 mg, 17% yield). 1H NMR (400 MHz, CDCl3) δ 7.80 (d, 1H, J = 8.0 Hz), 7.76–7.36 (m, 3H), 7.40–7.33 (m, 2H), 7.06–7.01 (m, 1H), 6.89 (d, 1H, J = 8.4 Hz), 3.86 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 162.5 (d, J = 242.3 Hz), 155.7, 150.0, 142.0 (d, J = 2.4 Hz), 139.1 (d, J = 8.0 Hz), 138.6, 129.4 (d, J = 8.2 Hz), 125.3 (br), 124.8 (d, J = 2.7 Hz), 124.5, 116.1 (d, J = 23.2 Hz), 115.2 (d, J = 21.1 Hz), 110.3, 56.5 ppm. 19F NMR (376 MHz, CDCl3) δ −113.7 ppm. HRMS ESI [M + H]+ (m/z): calcd for C15H13FN3O2+, 286.0992; found 286.0993. Retention time: 5.38 min (HPLC method A).
6-((1H-Pyrazol-4-yl)oxy)-2-(3-chlorophenyl)-3-methoxypyridine (16).
This compound was prepared from 63 (254 mg, 1 mmol) following a procedure similar to that of 17. The product was obtained as a white solid (136 mg, 45% yield). 1H NMR (400 MHz, CDCl3) δ 11.5 (br, 1H), 8.00 (s, 1H), 7.88–7.86 (m, 1H), 7.72 (s, 2H), 7.34 (d, 1H, J = 8.8 Hz), 7.31–7.29 (m, 2H), 6.86 (d, 1H, J = 8.8 Hz), 3.82 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 155.9, 150.0, 141.9, 138.7, 138.5, 133.8, 129.2, 129.2, 128.3, 127.4, 125.3 (br), 124.4, 110.1, 56.5 ppm. HRMS ESI [M + H]+ (m/z): calcd for C15H13ClN3O2+, 302.0696; found 302.0694. Retention time: 7.30 min (HPLC method A).
6-((1H-Pyrazol-4-yl)oxy)-3-methoxy-2-(3-(trifluoromethyl)phenyl)pyridine (17).
This compound was prepared from 64 (288 mg, 1 mmol) and 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-4-ol (168 mg, 1 mmol) instead of amine following a procedure similar to that of 13. The product was obtained as a white solid (50 mg, 15% yield). 1H NMR (400 MHz, CDCl3) δ 10.8 (br, 1H), 8.31 (s, 1H), 8.18 (d, 1H, J = 7.6 Hz), 7.73 (s, 2H), 7.59 (d, 1H, J = 7.6 Hz), 7.53–7.49 (m, 1H), 7.40 (d, 1H, J = 9.2 Hz), 6.90 (d, 1H, J = 8.8 Hz), 3.87 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 155.9, 150.1, 141.8, 138.6, 137.8, 132.4, 130.3 (q, J = 32.0 Hz), 128.5, 126.1 (q, J = 4.0 Hz), 125.2 (br), 124.9 (q, J = 3.8 Hz), 124.5, 124.3 (q, J = 271 Hz, including 128.4, 125.6, 122.9, 120.2), 110.4, 56.5 ppm. 19F NMR (376 MHz, CDCl3) δ −63.7 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H13F3N3O2+, 336.0960; found 336.0963. Retention time: 9.48 min (HPLC method A).
6-((1H-Pyrazol-4-yl)oxy)-2-(3-(difluoromethoxy)phenyl)-3-methoxypyridine (18).
This compound was prepared from 69 (286 mg, 1 mmol) following a procedure similar to that of 22. The product was obtained as a white solid (53 mg, 16% yield). 1H NMR (400 MHz, CDCl3) δ 7.89 (d, 1H, J = 8.8 Hz), 7.74 (d, 1H, J = 7.6 Hz), 7.67 (s, 1H), 7.49–7.43 (m, 2H), 7.27 (s, 2H), 7.19 (d, 1H, J = 8.4 Hz), 6.58 (t, 1H, J = 74.0 Hz), 3.92 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.2, 151.0, 146.0, 144.5, 142.8, 138.7, 129.7, 126.1, 120.7, 120.4, 119.7, 118.5, 116.0 (t, J = 258.1 Hz, including 118.6, 116.0, 113.4), 114.9, 55.9 ppm. 19F NMR (376 MHz, CDCl3) δ −80.6 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H14F2N3O3+, 334.1003; found 334.1003. Retention time: 5.82 min (HPLC method B).
5-(Difluoromethoxy)-6-(3-fluorophenyl)-N-(1H-pyrazol-4-yl)pyridin-2-amine (19).
This compound was prepared from 6-chloro-3(difluoromethoxy)-2-(3-fluorophenyl)pyridine (68) (274 mg, 1 mmol) following a procedure similar to that of 20. The product was obtained as a pale-yellow solid (156 mg, 49% yield). 1H NMR (400 MHz, CDCl3) δ 7.71 (s, 2H), 7.66 (d, 1H, J = 7.6 Hz), 7.59 (d, 1H, J = 10.0 Hz), 7.44–7.38 (m, 2H), 7.11(td, 1H, J1 = 8.4 Hz, J2 = 2.4 Hz), 6.55 (d, 1H, J = 8.8 Hz), 6.48 (s, 1H), 6.24 (t, 1H, J = 74.0 Hz) ppm. 13C NMR (101 MHz, CDCl3) δ 162.7 (d, J = 243.6 Hz), 154.4, 147.7 (d, J = 2.3 Hz), 138.9 (d, J = 7.9 Hz), 137.4 (t, J = 2.8 Hz), 132.9, 129.7 (d, J = 8.3 Hz), 128.0 (br), 124.9 (d, J = 2.8 Hz), 122.9, 116.1 (d, J = 22.7 Hz), 115.7 (d, J = 21.0 Hz), 116.1 (t, J = 261.0 Hz, including 118.7, 116.1, 113.5), 107.3 ppm. 19F NMR (376 MHz, CDCl3) δ −80.9, −113.1 ppm. HRMS ESI [M + H]+ (m/z): calcd for C15H12F3N4O+, 321.0963; found 321.0960. Retention time: 5.43 min (HPLC method A).
5-(Difluoromethoxy)-6-(3-methoxyphenyl)-N-(1H-pyrazol-4-yl)pyridin-2-amine (20).
This compound was prepared from 6-chloro-3(difluoromethoxy)-2-(3-methoxyphenyl)pyridine (67) (286 mg, 1 mmol), 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-4-amine (168 mg, 1 mmol), Cs2CO3 (430 mg, 1.3 mmol), and Pd(OAc)2/BINAP (40 mg, molar ratio = 1/1.5) in anhydrous dioxane (5 mL) following a procedure similar to that of 13. The product was obtained as a pale-yellow oil (117 mg, 35% yield). 1H NMR (400 MHz, CDCl3) δ 9.97 (br, 1H), 7.63 (s, 2H), 7.44–7.33 (m, 4H), 6.95 (dd, 1H, J1 = 8.0 Hz, J2 = 2.0 Hz), 6.86 (s, 1H), 6.50 (d, 1H, J = 8.8 Hz), 6.19 (t, 1H, J = 74.4 Hz), 3.82 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.6, 154.8, 149.0, 138.1, 137.4 (t, J = 2.9 Hz), 133.1, 129.5, 128.3 (br), 123.1, 121.8, 116.5 (t, J = 260.3 Hz, including 119.1, 116.5, 113.9), 115.0, 114.6, 106.9, 55.5 ppm. 19F NMR (376 MHz, CDCl3) δ −80.9 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H15F2N4O2+, 333.1163; found 333.1159. Retention time: 4.58 min (HPLC method A).
6-((1H-Pyrazol-4-yl)oxy)-3-(difluoromethoxy)-2-(3-fluorophenyl)pyridine (21).
This compound was prepared from 68 (274 mg, 1 mmol) and 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-4-ol (168 mg, 1 mmol) following a procedure similar to that of 22. The product was obtained as a white solid (145 mg, 45% yield). 1H NMR (400 MHz, CDCl3) δ 7.77 (s, 2H), 7.68 (d, 1H, J = 7.6 Hz), 7.64–7.58 (m, 2H), 7.42–7.36 (m, 1H), 7.12–7.07 (m, 1H), 6.93 (d, 1H, J = 8.8 Hz), 6.35 (t, 1H, J = 73.2 Hz) ppm. 13C NMR (101 MHz, CDCl3) δ 162.6 (d, J = 243.7 Hz), 159.0, 146.5, 140.5, 137.9 (d, J = 7.9 Hz), 137.8, 134.1, 129.8 (d, J = 8.1 Hz), 125.4 (br), 124.9 (d, J = 2.9 Hz), 116.2 (d, J = 23.1 Hz), 116.1 (d, J = 21.0 Hz), 115.8 (t, J = 262.2 Hz, including 118.4, 115.8, 113.2), 110.9 ppm. 19F NMR (376 MHz, CDCl3) δ −81.1, −112.9 ppm. HRMS ESI [M + H]+ (m/z): calcd for C15H11F3N3O2+, 322.0803; found 322.0802. Retention time: 6.75 min (HPLC method A).
6-((1H-Pyrazol-4-yl)oxy)-3-(difluoromethoxy)-2-(3methoxyphenyl)pyridine (22).
This compound was prepared from 67 (286 mg, 1 mmol) and 1-(tetrahydro-2H-pyran-2-yl)-1H-pyrazol-4-ol (168 mg, 1 mmol) instead of amine following a procedure similar to that of 20. The product was obtained as a white solid (185 mg, 56% yield). 1H NMR (400 MHz, CDCl3) δ 10.63 (br, 1H), 7.76 (s, 2H), 59(d, 1H, J = 8.8 Hz), 7.49–7.44 (m, 1H), 7.35–7.31 (m, 1H), 6.95–6.88 (m, 2H), 6.30 (t, 1H, J = 73.2 Hz), 3.80 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.6, 159.1, 147.7, 140.7, 138.1, 137.2, 134.3, 129.8, 129.5, 125.6, 121.8, 116.2 (t, J = 261.3 Hz, including 118.8, 116.2, 113.6), 115.4, 114.6, 110.5, 55.4 ppm. 19F NMR (376 MHz, CDCl3) δ −81.1 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H14F2N3O3+, 334.1003; found 334.1003. Retention time: 5.80 min (HPLC method A).
(5-(Difluoromethoxy)-6-(3-methoxyphenyl)pyridin-2-yl)(1H-pyrazol-4-yl)methanone (23).
A mixture of 3-(difluoromethoxy)-2-(3methoxyphenyl)-6-(((trifluoromethyl)sulfonyl)methyl)pyridine (98) (215 mg, 0.5 mmol), 1-(1-ethoxyethyl)-4-(4,4,5,5-tetramethyl-1,3,2dioxaborolan-2-yl)-1H-pyrazole (266 mg, 1 mmol), Pd(OAc)2 (11 mg, 0.05 mmol), DavePhos (60 mg, 0.15 mmol), and K3PO4 (318 mg, 1.5 mmol) in 1,2-dimethoxyethane (DME) (3 mL) was stirred and heated to 90 °C under nitrogen atmosphere.53 After stirring for 24 h, the reaction was quenched with water and extracted with dichloromethane thrice. The combined organic phases were dried over MgSO4 and filtered. The filtrate was evaporated to dryness and was dissolved in MeOH (4 mL). HCl (0.4 mL, M) in dioxane/water (v/v = 2:1) was then added. After the mixture was stirred at room temperature overnight, the solvent was removed. Saturated NaHCO3 aqueous solution was added to neutralize the mixture. And the mixture was extracted with DCM thrice. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified with a silica gel column (eluent: 50–70% EtOAc/hexane). The product was obtained as a white solid (17.5 mg, 10% yield). 1H NMR (400 MHz, CDCl3) δ 11.00 (br, 1H), 8.69 (s, 2H), 8.21 (d, 1H, J = 8.8 Hz), 7.74 (d, 1H, J = 8.4 Hz), 7.50–7.40 (m, 3H), 7.03 (d, 1H, J = 8.0 Hz), 6.53 (t, 1H, J = 72.4 Hz), 3.87 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 184.0, 159.7, 150.9, 149.4, 147.38, 147.35, 139.0 (br), 137.4, 129.5, 128.1, 123.5, 121.8, 121.0, 115.5 (t, J = 262.7 Hz, including 118.1, 115.5, 112.8), 115.1, 115.0, 55.4 ppm. 19F NMR (376 MHz, CDCl3) δ −81.6 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H14F2N3O3+, 346.1003; found 346.1001. Retention time: 5.47 min (HPLC method A).
6-((1H-Pyrazol-1-yl)methyl)-2-(3-fluorophenyl)-3-methoxypyridine (24).
A mixture of 84 (346 mg, 1.17 mmol), pyrazole (140 mg, 2 mmol), and K2CO3 (276 mg, 2 mmol) in MeCN (5 mL) was heated to 80 °C and stirred overnight. The solvent was evaporated to dryness. The residue was taken up with DCM and washed with water thrice. The organic layer was dried over MgSO4 and evaporated. The residue was purified with silica gel column (eluent: 50% EtOAc/hexane). The product was obtained as a colorless oil (226 mg, 68% yield). 1H NMR (400 MHz, CDCl3) δ 7.66 (d, 1H, J = 8.0 Hz), 7.59 (d, 1H, J = 10.0 Hz), 7.48 (m, 2H), 7.32–7.27 (m, 1H), 7.12 (d, 1H, J = 8.4 Hz), 7.00–6.95 (m, 1H), 6.90 (d, 1H, J = 8.4 Hz), 6.22–6.21 (m, 1H), 5.36 (s, 2H), 3.73 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 162.6 (d, J = 242.3 Hz), 153.0, 147.9, 145.6 (d, J = 2.6 Hz), 139.8, 139.5 (d, J = 7.7 Hz), 129.9, 129.4 (d, J = 8.1 Hz), 125.1 (d, J = 2.7 Hz), 121.7, 119.6, 116.4 (d, J = 22.7 Hz), 115.3 (d, J = 21.1 Hz), 106.1, 57.1, 55.7 ppm. 19F NMR (376 MHz, CDCl3) δ −113.8 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H15FN3O+, 284.1199; found 284.1194. Retention time: 7.18 min (HPLC method A).
6-((1H-Pyrazol-1-yl)methyl)-2-(3-chlorophenyl)-3-methoxypyridine (25).
This compound was prepared from 87 (313 mg, 1 mmol) following a procedure similar to that of 24. The product was obtained as a pale-yellow oil (240 mg, 80% yield). 1H NMR (400 MHz, CDCl3) δ 7.93 (s, 1H), 7.84–7.81 (m, 1H), 7.57 (s, 2H), 7.40–7.30 (m, 2H), 7.22 (d, 1H, J = 8.4 Hz), 6.99 (d, 1H, J = 8.4 Hz), 6.33–6.30 (m, 1H), 5.46 (s, 2H), 3.84 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 153.0, 148.0, 145.6, 139.8, 139.1, 133.9, 129.8, 129.5, 129.2, 128.5, 127.6, 121.7, 119.6, 106.1, 57.1, 55.7 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H15ClN3O+, 300.0904; found 300.0903. Retention time: 9.73 min (HPLC method A).
6-((1H-Pyrazol-1-yl)methyl)-3-methoxy-2-(3-(trifluoromethyl)phenyl)pyridine (26).
This compound was prepared from 85 (346 mg, 1 mmol) following a procedure similar to that of 24. The product was obtained as a colorless oil (275 mg, 83% yield). 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1H), 8.05 (d, 1H, J = 7.6 Hz), 7.53 (d, 1H, J = 8.0 Hz), 7.48–7.42 (m, 3H), 7.13 (d, 1H, J = 8.4 Hz), 6.91 (d, 1H, J = 8.8 Hz), 6.22–6.21 (m, 1H), 5.37 (s, 2H), 3.73 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 153.1, 148.1, 145.4, 139.8, 138.0, 132.7, 130.3 (q, J = 32.0 Hz), 129.9, 128.4, 126.4 (q, J = 3.9 Hz), 125.0 (q, J = 3.7 Hz), 124.3 (q, J = 271.2 Hz, including 128.4, 125.7, 123.0, 120.3), 121.9, 119.7, 106.1, 57.0, 55.7 ppm. 19F NMR (376 MHz, CDCl3) δ −63.7 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H15F3N3O+, 334.1167; found 334.1161. Retention time: 13.22 min (HPLC method A).
2-(3-(Difluoromethoxy)phenyl)-3-methoxy-6-((1-methyl-1H-pyrazol-4-yl)methyl)pyridine (29).
This compound was prepared from 5 (66 mg, 0.2 mmol) following a similar procedure to that of 42. The product was obtained as a colorless oil (41 mg, 59% yield). 1H NMR (400 MHz, CDCl3) δ 7.49–7.42 (m, 4H), 7.37 (t, 1H, J = 8.0 Hz), 7.10 (d, 1H, J = 8.4 Hz), 6.97 (dd, 1H, J1 = 8.4 Hz, J2 = 2.4 Hz), 6.33 (t, 1H, J = 73.6 Hz), 4.04 (s, 2H), 3.86 (s, 6H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 158.1, 149.9, 143.3 (t, J = 2.7 Hz), 139.2, 137.8, 129.5, 129.3, 129.1, 121.9, 121.8, 118.7, 115.9 (t, J = 260.8 Hz, including 118.5, 115.9, 113.3), 114.9, 114.7, 55.3, 38.9, 32.9 ppm. 19F NMR (376 MHz, CDCl3) δ −81.03 ppm. HRMS ESI [M + H]+ (m/z): calcd for C18H18F2N3O2+, 346.1367; found 346.1370. Retention time: 6.47 min (HPLC method B).
6-((1H-Imidazol-1-yl)methyl)-2-(3-(difluoromethoxy)phenyl)-3methoxypyridine (30).
This compound was prepared from 91 (172 mg, 0.5 mmol) following a procedure similar to that of 43. The product was obtained as a colorless oil (93 mg, 56% yield). 1H NMR (400 MHz, CDCl3) δ 7.81(d, 1H, J = 7.6 Hz), 7.74 (s, 1H), 7.64 (s, 1H), 7.44 (t, 1H, J = 8.0 Hz), 7.25 (d, 1H, J = 8.0 Hz), 7.16 (d, 1H, J = 8.4 Hz), 7.12 (s, 1H), 7.03 (s, 1H), 6.94 (d, 1H, J = 8.4 Hz), 6.57 (t, 1H, J = 74.0 Hz), 5.24 (s, 2H), 3.87 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 153.2, 151.1, 147.4, 146.1, 139.0, 137.6, 129.9, 129.4, 126.5, 121.1, 120.6, 119.63, 119.60, 119.5, 116.1 (t, J = 257.5 Hz, including 118.7, 116.1, 113.6), 55.7, 52.0 ppm. 19F NMR (376 MHz, CDCl3) δ −80.44 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H16F2N3O2+, 332.1211; found 332.1213. Retention time: 4.55 min (HPLC method B).
2-(3-(Difluoromethoxy)phenyl)-3-methoxy-6-((3-(trifluoromethyl)-1H-1,2,4-triazol-1-yl)methyl)pyridine (31).
This compound was prepared from 91 (172 mg, 0.5 mmol) and 3-(trifluoromethyl)-1H1,2,4-triazole (84 mg, 0.6 mmol) following a procedure similar to that of 43. The product was obtained as a colorless oil (114 mg, 54% yield). 1H NMR (400 MHz, CDCl3) δ 8.38 (s, 1H), 7.76 (d, 1H, J = 7.6 Hz), 7.70 (s, 1H), 7.43 (t, 1H, J = 8.0 Hz), 7.33−7.27 (m, 2H), 7.16 (d, 1H, J = 8.0 Hz), 6.56 (t, 1H, J = 74.0 Hz), 5.50 (s, 2H), 3.89 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 154.2 (q, J = 39.3 Hz, including 154.9, 154.5, 154.1, 153.7), 153.7, 151.0 (t, J = 2.7 Hz), 146.6, 145.1, 144.2, 138.7, 129.4, 126.4, 122.7, 120.6, 119.8, 119.6, 119.1 (q, J = 268.4 Hz, including 123.2, 120.5, 117.8, 115.1), 116.1 (t, J = 258.0 Hz, including 118.7, 116.1, 113.5), 55.8, 55.2 ppm. 19F NMR (376 MHz, CDCl3) δ −63.7, −79.2 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H14F5N4O2+, 401.1037; found 401.1040. Retention time: 14.83 min (HPLC method B).
6-((1H-1,2,4-Triazol-1-yl)methyl)-2-(3-(difluoromethoxy)phenyl)3-methoxypyridine (32).
This compound was prepared from 91 (172 mg, 0.5 mmol) and 1H-1,2,4-triazole (65 mg, 1 mmol) following a procedure similar to that of 43. The product was obtained as a colorless oil (56 mg, 34% yield). 1H NMR (400 MHz, CDCl3) δ 8.29 (s, 1H), 7.98 (s, 1H), 7.78 (d, 1H, J = 8.0 Hz), 7.71 (s, 1H), 7.43 (t, 1H, J = 8.0 Hz), 7.28 (d, 1H, J = 8.4 Hz), 7.19 (d, 1H, J = 8.4 Hz), 7.15 (d, 1H, J = 8.0 Hz), 6.56 (t, 1H, J = 74.0 Hz), 5.47 (s, 2H), 3.87 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 153.4, 152.2, 151.0, 146.2, 145.6, 143.7, 138.9, 129.4, 126.4, 122.3, 120.6, 119.6, 119.5, 116.1 (t, J = 257.5 Hz, including 118.7, 116.1, 113.6), 55.7, 54.6 ppm. 19F NMR (376 MHz, CDCl3) δ −80.46 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H15F2N4O2+, 333.1163; found 333.1165. Retention time: 3.90 min (HPLC method B).
6-((1H-Tetrazol-1-yl)methyl)-2-(3-(difluoromethoxy)phenyl)-3methoxypyridine (33).
This compound was prepared from 91 (172 mg, 0.5 mmol) and 1H-tetrazole (0.45 M in MeCN, 2 mL, 0.9 mmol) following a procedure similar to that of 43. The product was obtained as a colorless oil (90 mg, 54% yield). 1H NMR (400 MHz, CDCl3) δ 8.87 (s, 1H), 7.74 (d, 1H, J = 8.0 Hz), 7.69 (s, 1H), 7.43 (t, 1H, J = 8.0 Hz), 7.31 (s, 2H), 7.17 (d, 1H, J = 8.0 Hz), 6.56 (t, 1H, J = 74.0 Hz), 5.70 (s, 2H), 3.89 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 153.8, 151.0, 146.7, 143.5, 143.0, 138.6, 129.4, 126.4, 122.7, 120.6, 119.8, 119.6, 116.1 (t, J = 258.0 Hz, including 118.6, 116.1, 113.5), 55.8, 52.9 ppm. 19F NMR (376 MHz, CDCl3) δ−80.56 ppm. HRMS ESI [M + H]+ (m/ z): calcd for C15H14F2N5O2+, 334.1116; found 334.1119. Retention time: 5.30 min (HPLC method B).
6-((2H-Tetrazol-2-yl)methyl)-2-(3-(difluoromethoxy)phenyl)-3methoxypyridine (34).
This compound was the byproduct of the synthesis of 33. The product was obtained as a colorless oil (50 mg, 30% yield). 1H NMR (400 MHz, CDCl3) δ 8.56 (s, 1H), 7.80 (d, 1H, J = 8.0 Hz), 7.72 (s, 1H), 7.42 (t, 1H, J = 8.0 Hz), 7.29 (d, 1H, J = 8.4 Hz), 7.17–7.13 (m, 2H), 6.55 (t, 1H, J = 74.0 Hz), 5.98 (s, 2H), 3.88 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 153.6, 153.2, 151.0 (t, J = 2.7 Hz), 146.2, 144.1, 138.8, 129.3, 126.5, 122.2, 120.6, 119.6, 119.5, 116.1 (t, J = 257.6 Hz, including 118.7, 116.1, 113.6), 57.7, 55.7 ppm. 19F NMR (376 MHz, CDCl3) δ−80.44 ppm. HRMS ESI [M + H]+ (m/z): calcd for C15H14F2N5O2+, 334.1116; found 334.1110. HRMS ESI [M − N2]+ (m/z): calcd for C15H14F2N3O2+, 306.1054; found 306.1056. Retention time: 7.70 min (HPLC method B).
6-((1H-Pyrazol-1-yl)methyl)-3-(difluoromethoxy)-2-(3methoxyphenyl)pyridine (35).
This compound was prepared from 86 (344 mg, 1 mmol) following a procedure similar to that of 24. The product was obtained as a yellow oil (274 mg, 83% yield). 1H NMR (400 MHz, CDCl3) δ 7.52 (d, 2H, J = 8.0 Hz), 7.45 (d, 1H, J = 8.4 Hz), 7.38–7.28 (m, 3H), 6.92 (d, 1H, J = 8.4 Hz), 6.46–6.09 (m, 2H), 5.45 (s, 2H), 3.79 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 153.8, 150.2, 144.3, 140.2, 137.3, 130.1, 129.6, 129.4, 121.8, 121.0, 115.7 (t, J = 264 Hz, including 118.3, 115.7, 113.1), 115.2, 114.6, 106.4, 57.1, 55.4 ppm. 19F NMR (376 MHz, CDCl3) δ−81.2 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H16F2N3O2+, 332.1211; found 332.1210. Retention time: 7.33 min (HPLC method A).
3-(Difluoromethoxy)-2-(3-methoxyphenyl)-6-(pyridin-4ylmethyl)pyridine (36).
86 (344 mg, 1 mmol), pyridin-4-ylboronic acid (148 mg, 1.2 mmol), K3PO4 (424 mg, 2 mmol), and Pd(dppf)Cl2 (22 mg, 0.03 mmol) were added into a 50 mL two-neck round-bottom flask in a glovebox. Then, a degassed solvent mixture of dioxane/water (4:1 v/v, 5 mL) was injected. The flask was sealed and placed in an 80 °C oil bath. The reaction mixture was stirred overnight. After cooling to room temperature, the reaction mixture was concentrated under reduced pressure. The residue was partitioned between DCM and water. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified with silica gel column (eluent: 50–70% EtOAc/ hexane). The product was obtained as a light brown oil (26 mg, 7.6% yield). 1H NMR (400 MHz, CDCl3) δ 8.55 (d, 1H, J = 4.4 Hz), 7.51 (d, 1H, J = 8.4 Hz), 7.44 (d, 1H, J = 7.6 Hz), 7.40–7.35 (m, 2H), 7.24 (d, 1H, J = 4.8 Hz), 7.09 (d, 1H, J = 8.4 Hz), 6.98 (dd, 1H, J1 = 8.0 Hz, J2 = 2.4 Hz), 6.36 (t, 1H, J = 73.6 Hz), 4.19 (s, 2H), 3.85 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 155.9, 150.3, 150.0, 148.1, 143.6, 137.6, 137.6, 129.5, 129.3, 124.4, 122.5, 121.8, 115.8 (t, J = 261.3 Hz, including 118.4, 115.8, 113.1), 115.0, 114.7, 55.3, 43.3 ppm. 19F NMR (376 MHz, CDCl3) δ −81.2 ppm. HRMS ESI [M + H]+ (m/z): calcd for C19H17F2N2O2+, 343.1258; found 343.1259. Retention time: 9.68 min (HPLC method A).
3-(Difluoromethoxy)-2-(3-methoxyphenyl)-6-(pyridin-3ylmethyl)pyridine (37).
This compound was prepared from 86 (344 mg, 1 mmol) and pyridin-3-ylboronic acid (148 mg, 1.2 mmol) following a procedure similar to that of 36. The product was obtained as a colorless oil (65 mg, 19% yield). 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 8.50 (d, 1H, J = 4.0 Hz), 7.65 (d, 1H, J = 7.6 Hz), 7.50 (d, 1H, J = 8.4 Hz), 7.45 (d, 1H, J = 7.6 Hz), 7.40–7.35 (m, 2H), 7.26–7.23 (m, 1H), 7.08 (d, 1H, J = 8.4 Hz), 6.98 (dd, 1H, J1 = 8.0 Hz, J2 = 2.4 Hz), 6.34 (t, 1H, J = 73.2 Hz), 4.20 (s, 2H), 3.86 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 156.7, 150.3, 150.2, 148.0, 143.5, 137.6, 136.6, 134.8, 129.6, 129.3, 123.5, 122.3, 121.8, 115.8 (t, J = 261.1 Hz, including 118.4, 115.8, 113.2), 115.0, 114.6, 55.3, 41.2 ppm. 19F NMR (376 MHz, CDCl3) δ −81.1 ppm. HRMS ESI [M + H]+ (m/z): calcd for C19H17F2N2O2+, 343.1258; found 343.1259. Retention time: 9.40 min (HPLC method A).
5-((5-(Difluoromethoxy)-6-(3-methoxyphenyl)pyridin-2-yl)methyl)pyrimidine (38).
This compound was prepared from 86 (344 mg, 1 mmol) and pyrimidin-5-ylboronic acid (149 mg, 1.2 mmol) following a procedure similar to that of 36. The product was obtained as an orange oil (65 mg, 19% yield). 1H NMR (400 MHz, CDCl3) δ 9.11 (s, 1H), 8.76 (s, 2H), 7.54 (d, 1H, J = 8.4 Hz), 7.44–7.34 (m, 2H), 7.15 (d, 1H, J = 8.4 Hz), 6.98 (dd, 1H, J1 = 8.0 Hz, J2 = 2.0 Hz), 6.98 (dd, 1H, J1 = 8.0 Hz, J2 = 2.4 Hz), 6.36 (t, 1H, J = 73.2 Hz), 4.18 (s, 2H), 3.85 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 157.3, 157.2, 155.0, 150.6, 143.7 (t, J = 2.7 Hz), 137.4, 132.6, 129.8, 129.4, 122.2, 121.7, 115.7 (t, J = 261.7 Hz, including 118.3, 115.7, 113.1), 115.2, 114.6, 55.3, 38.5 ppm. 19F NMR (376 MHz, CDCl3) δ −81.2 ppm. HRMS ESI [M + H]+ (m/z): calcd for C18H16F2N3O2+, 344.1211; found 344.1206. Retention time: 5.57 min (HPLC method A).
N-((5-(Difluoromethoxy)-6-(3-methoxyphenyl)pyridin-2-yl)methyl)-1H-pyrazol-4-amine (39).
This compound was prepared from 86 (133 mg, 0.39 mmol) and tert-butyl 4-amino-1H-pyrazole-1carboxylate (92 mg, 0.5 mmol) following a procedure similar to that of 40. The product was obtained as a pale-yellow oil (65 mg, 48% yield). 1H NMR (400 MHz, CDCl3) δ 7.54 (d, 1H, J = 8.4 Hz), 7.45–7.36 (m, 3H), 7.32 (d, 1H, J = 8.4 Hz), 7.17 (s, 2H), 6.98 (dd, 1H, J1 = 8.0 Hz, J2 = 2.0 Hz), 6.36 (t, 1H, J = 73.6 Hz), 5.90 (br, 2H), 4.34 (s, 2H), 3.85 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 155.8, 150.2, 143.9, 137.6, 133.1, 129.4, 122.4 (br), 121.8, 121.3, 115.8 (t, J = 260.3 Hz, including 118.4, 115.8, 113.2), 114.9, 114.8, 55.4, 52.7 ppm. 19F NMR (376 MHz, CDCl3) δ −81.1 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H17F2N4O2+, 347.1320; found 347.1322. Retention time: 3.42 min (HPLC method A).
6-(((1H-Pyrazol-4-yl)oxy)methyl)-3-(difluoromethoxy)-2-(3methoxyphenyl)pyridine (40).
Step 1: A mixture of 86 (133 mg, 0.39 mmol), tert-butyl 4-hydroxy-1H-pyrazole-1-carboxylate (92 mg, 0.5 mmol), and K2CO3 (276 mg, 2 mmol) in MeCN (5 mL) was stirred at room temperature overnight. The mixture was filtered to remove the salts, and solids were washed with DCM. The DCM solution was evaporated to dryness. The residue was purified with a silica gel column (eluent: 50% EtOAc/hexane). Step 2: The above product was dissolved with DCM (2.5 mL) and trifluoroacetic acid (0.5 mL) was added. After the mixture was stirred at room temperature overnight, the solvent was removed. Saturated NaHCO3 aqueous solution was added to neutralize the mixture. And the mixture was extracted with DCM thrice. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified with silica gel column (eluent: 50% EtOAc/ hexane). The product was obtained as a colorless oil (117 mg, 86% yield). 1H NMR (400 MHz, CDCl3) δ 7.62 (d, 1H, J = 8.4 Hz), 7.51 (d, 1H, J = 8.4 Hz), 7.44–7.35 (m, 5H), 6.99 (d, 1H, J = 8.0 Hz), 6.39 (t, 1H, J = 73.6 Hz), 5.90 (br, 1H), 5.14 (s, 2H), 3.86 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 154.0, 150.2, 145.3, 144.2, 137.4, 129.5, 129.4, 121.8, 121.1 (br), 120.8, 115.7 (t, J = 261.6 Hz, including 118.3, 115.7, 113.1), 115.1, 114.7, 73.9, 55.4 ppm. 19F NMR (376 MHz, CDCl3) δ −81.2 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H16F2N3O3+, 348.1160; found 348.1164. Retention time: 5.07 min (HPLC method A).
3-(Difluoromethoxy)-2-(3-methoxyphenyl)-6-((1-methyl-1H-pyrazol-4-yl)methyl)pyridine (42).
A mixture of JMJ-81 (66 mg, 0.2 mmol), iodomethane (50 mg), and K2CO3 (40 mg) in MeCN (3 mL) was stirred at room temperature for 24 h. The solvent was evaporated to dryness. The residue was purified with a silica gel column (eluent: DCM/20% EtOAc). The product was obtained as a colorless oil (32 mg, 46% yield). 1H NMR (400 MHz, CDCl3) δ 7.83 (d, 1H, J = 8.0 Hz), 7.75 (s, 1H), 7.44–7.40 (m, 2H), 7.26 (s, 1H), 7.21 (d, 1H, J = 8.4 Hz), 7.12 (dd, 1H, J1 = 8.0 Hz, J2 = 2.4 Hz), 7.08 (d, 1H, J = 8.4 Hz), 6.56 (t, 1H, J = 74.4 Hz), 3.99 (s, 2H), 3.86 (s, 3H), 3.83 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.4, 152.0, 151.1 (t, J = 2.8 Hz), 145.3, 139.8, 139.2, 129.3, 129.1, 126.5, 122.2, 120.5, 119.6, 119.5, 119.1, 116.3 (t, J = 257.3 Hz, including 118.8, 116.3, 113.7), 55.7, 38.8, 32.5 ppm. 19F NMR (376 MHz, CDCl3) δ−80.25 ppm. HRMS ESI [M + H]+ (m/z): calcd for C18H18F2N3O2+, 346.1367; found 346.1365. Retention time: 6.55 min (HPLC method B).
6-((1H-Imidazol-1-yl)methyl)-3-(difluoromethoxy)-2-(3methoxyphenyl)pyridine (43).
A mixture of 86 (172 mg, 0.5 mmol), 1H-imidazole (68 mg, 1 mmol), and K2CO3 (138 mg, 1 mmol) in MeCN (3 mL) was stirred at room temperature for 2 days. The solvent was evaporated to dryness. The residue was purified with a silica gel column (eluent: 100% EtOAc). The product was obtained as a colorless oil (112 mg, 68% yield). 1H NMR (400 MHz, CDCl3) δ 7.66 (s, 1H), 7.55 (d, 1H, J = 7.6 Hz), 7.45–7.37 (m, 3H), 7.14 (s, 1H), 7.04 (s, 1H), 7.00 (d, 1H, J = 8.0 Hz), 6.91(d, 1H, J = 8.4 Hz), 6.39 (t, 1H, J = 72.8 Hz), 5.30 (s, 2H), 3.87 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 153.0, 150.6, 144.38, 144.35, 137.7, 137.1, 130.1, 129.8, 129.4, 121.7, 120.4, 119.5, 115.6 (t, J = 262 Hz, including 118.2, 115.6, 113.0), 115.3, 114.7, 55.4, 52.0 ppm. 19F NMR (376 MHz, CDCl3) δ −81.4 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H16F2N3O2+, 332.1211; found 332.1211. Retention time: 4.17 min (HPLC method B).
3-(Difluoromethoxy)-2-(3-methoxyphenyl)-6-((3-(trifluoromethyl)-1H-1,2,4-triazol-1-yl)methyl)pyridine (44).
This compound was prepared from 86 (172 mg, 0.5 mmol) and 3-(trifluoromethyl)-1H1,2,4-triazole (84 mg, 0.6 mmol) following a procedure similar to that of 43. The product was obtained as a colorless oil (182 mg, 91% yield). 1H NMR (400 MHz, CDCl3) δ 8.41 (s, 1H), 7.62 (d, 1H, J = 7.6 Hz), 7.39–7.35 (m, 2H), 7.32 (s, 1H), 7.28 (d, 1H, J = 8.4 Hz), 7.02–6.98 (m, 1H), 6.41 (t, 1H, J = 72.8 Hz), 5.55 (s, 2H), 3.85 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 154.5 (q, J = 39.4 Hz, including 155.1, 154.7, 154.3, 153.9), 151.0, 149.7, 145.3, 144.9 (t, J = 2.6 Hz), 136.8, 129.7, 129.4, 121.9, 121.7, 119.1 (q, J = 270 Hz, including 123.1, 120.4, 117.7, 115.1), 115.5 (t, J = 262 Hz, including 118.1, 115.5, 112.9), 115.4, 114.6, 55.3, 55.0 ppm. 19F NMR (376 MHz, CDCl3) δ −60.7, −80.0 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H14F5N4O2+, 401.1037; found 401.1041. Retention time: 13.9 min (HPLC method B).
6-((1H-1,2,4-Triazol-1-yl)methyl)-3-(difluoromethoxy)-2-(3methoxyphenyl)pyridine (45).
This compound was prepared from 86 (172 mg, 0.5 mmol) and 1H-1,2,4-triazole (65 mg, 1 mmol) following a procedure similar to that of 43. The product was obtained as a colorless oil (96 mg, 58% yield). 1H NMR (400 MHz, CDCl3) δ 8.31 (s, 1H), 8.00 (s, 1H), 7.58 (d, 1H, J = 7.6 Hz), 7.42–7.35 (m, 3H), 7.15 (d, 1H, J = 8.4 Hz), 7.00 (d, 1H, J = 8.0 Hz), 6.39 (t, 1H, J = 72.8 Hz), 5.53 (s, 2H), 3.85 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 152.4, 151.3, 150.7, 144.6 (t, J = 2.7 Hz), 144.0, 137.0, 129.6, 129.4, 121.7, 121.5, 115.6 (t, J = 262.3 Hz, including 118.2, 115.6, 113.0), 115.3, 114.6, 55.4, 54.4 ppm. 19F NMR (376 MHz, CDCl3) δ −81.4 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H15F2N4O2+, 333.1163; found 333.1163. Retention time: 3.70 min (HPLC method B).
6-((1H-Tetrazol-1-yl)methyl)-3-(difluoromethoxy)-2-(3methoxyphenyl)pyridine (46).
This compound was prepared from 86 (172 mg, 0.5 mmol) and 1H-tetrazole (0.45 M in MeCN, 2 mL, 0.9 mmol) following a procedure similar to that of 43. The product was obtained as a colorless oil (110 mg, 62% yield). 1H NMR (400 MHz, CDCl3) δ 8.91 (s, 1H), 7.63 (d, 1H, J = 8.4 Hz), 7.39–7.36 (m, 2H), 7.32–7.29 (m, 2H), 7.02–6.98 (m, 1H), 6.42 (t, 1H, J = 72.8 Hz), 5.76 (s, 2H), 3.85 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 151.1, 149.0, 145.0, 143.2, 136.7, 129.7, 129.5, 121.9, 121.6, 115.5 (t, J = 262.9 Hz, including 118.1, 115.5, 112.8), 115.4, 114.6, 55.4, 52.6 ppm. 19F NMR (376 MHz, CDCl3) δ−81.5 ppm. HRMS ESI [M + H]+ (m/z): calcd for C15H14F2N5O2+, 334.1116; found 334.1112. HRMS ESI [M + Na]+ (m/z): calcd for C15H13F2N5O2Na+, 356.0935; found 356.0935. Retention time: 4.98 min (HPLC method B).
6-((2H-Tetrazol-2-yl)methyl)-3-(difluoromethoxy)-2-(3methoxyphenyl)pyridine (47).
This compound was the byproduct of the synthesis of JMJ-174 because 1H- and 2H-tetrazole isomers are tautomers at room temperature. The product was obtained as a colorless oil (56 mg, 34% yield). 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 7.59 (d, 1H, J = 8.4 Hz), 7.43 (d, 1H, J = 8.0 Hz), 7.39–7.34 (m, 2H), 7.09 (d, 1H, J = 8.4 Hz), 7.00–6.97 (m, 1H), 6.40 (t, 1H, J = 73.2 Hz), 6.03 (s, 2H), 3.85 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.5, 153.4, 150.7, 149.7, 144.8 (t, J = 2.7 Hz), 136.9, 129.6, 129.4, 121.8, 121.4, 115.6 (t, J = 262.3 Hz, including 118.2, 115.6, 112.9), 115.4, 114.6, 57.5, 55.3 ppm. 19F NMR (376 MHz, CDCl3) δ −81.40 ppm. HRMS ESI [M + H]+ (m/z): calcd for C15H14F2N5O2+, 334.1116; found 334.1120. Retention time: 7.20 min (HPLC method B).
6-((1H-Pyrazol-4-yl)methyl)-3-methoxy-2-(3-methoxyphenyl)pyridine (48).
6-(Bromomethyl)-3-methoxy-2-(3-methoxyphenyl)pyridine (94) (307 mg, 1 mmol), 1-(1-ethoxyethyl)-4-(4,4,5,5tetramethyl-1,3,2-dioxaborolan-2-yl)-1H-pyrazole (319 mg, 1.2 mmol), Na2CO3 (159 mg, 1.5 mmol), and Pd(PPh3)4 (35 mg, 0.03 mmol) were added into a 50 mL round-bottom flask in a glovebox. Then, a degassed solvent mixture of toluene/EtOH/water (4/3/3 mL) was injected into the sealed flask. The reaction mixture was stirred at 80 °C overnight. The subsequent procedure was similar to that of 2. The product was obtained as a white solid (80 mg, 27% yield). 1H NMR (400 MHz, CDCl3) δ 9.60 (br, 1H), 7.70–7.64 (m, 1H), 7.55–7.43 (m, 5H), 7.34 (t, 1H, J = 8.0 Hz), 7.18 (d, 1H, J = 8.4 Hz), 7.03 (d, 1H, J = 8.4 Hz), 6.92 (dd, 1H, J1= 8.0 Hz, J2 = 2.4 Hz), 4.04 (s, 2H), 3.83 (s, 3H), 3.80 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.2, 152.2, 146.8, 139.1, 133.4 (br), 132.1, 128.7, 122.0, 121.7, 119.5, 118.7, 114.9, 114.0, 55.6, 55.3, 32.3 ppm. HRMS ESI [M + H]+ (m/z): calcd for C17H18N3O2+, 296.1399; found 296.1400. Retention time: 3.42 min (HPLC method B).
2-(3-(Difluoromethoxy)phenyl)-3-methoxy-6-(pyridin-4ylmethyl)pyridine (49).
This compound was prepared from 91 (270 mg, 0.78 mmol) following a procedure similar to that of 58. The product was obtained as a colorless oil (91.2 mg, 34% yield). 1H NMR (400 MHz, CDCl3) δ 8.52 (d, 1H, J = 4.8 Hz), 7.81 (d, 1H, J = 7.6 Hz), 7.74 (s, 1H), 7.42 (t, 1H, J = 8.0 Hz), 7.24–7.22 (m, 3H), 7.14 (d, 1H, J = 8.0 Hz), 7.07 (d, 1H, J = 8.4 Hz), 6.56 (t, 1H, J = 74.4 Hz), 4.15 (s, 2H), 3.86 (s, 3H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.3, 151.1, 150.2, 149.8, 149.0, 145.8, 139.5, 129.3, 126.5, 124.4, 122.8, 120.5, 119.6, 119.3, 116.2 (t, J = 257.4 Hz, including 118.8, 116.2, 113.6), 55.7, 43.0 ppm. 19F NMR (376 MHz, CDCl3) δ −80.3 ppm. HRMS ESI [M + H]+ (m/z): calcd for C19H17F2N2O2+, 343.1258; found 343.1259. Retention time: 8.20 min (HPLC method B).
6-((1H-Pyrazol-4-yl)methyl)-3-(difluoromethoxy)-2-(3nitrophenyl)pyridine (50).
This compound was prepared from 6(bromomethyl)-3-(difluoromethoxy)-2-(3-nitrophenyl)pyridine (96) (359 mg, 1 mmol) and K2CO3 (350 mg, 2.5 mmol) as the base following a procedure similar to that of 48. The product was obtained as a colorless oil (130 mg, 37% yield). 1H NMR (400 MHz, CDCl3) δ 11.4 (br, 1H), 8.77 (s, 1H), 8.25–8.22 (m, 2H), 7.61 (t, 1H, J = 8.0 Hz), 7.55–7.52 (s, 3H), 7.19 (d, 1H, J = 8.4 Hz), 6.47 (t, 1H, J = 72.4 Hz), 4.11 (s, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ 158.4, 148.2, 147.2, 143.5 (t, J = 2.5 Hz), 138.2, 135.4, 133.4 (br), 129.2, 124.5, 123.5, 123.1, 117.6, 115.7 (t, J = 262.5 Hz, including 118.3, 115.7, 113.0), 32.7 ppm. 19F NMR (376 MHz, CDCl3) δ −81.05 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H13F2N4O3+, 347.0956; found 347.0959. Retention time: 4.97 min (HPLC method B).
6-((1H-Imidazol-1-yl)methyl)-3-(difluoromethoxy)-2-(3nitrophenyl)pyridine (51).
This compound was prepared from JMJ187 (180 mg, 0.5 mmol) following a procedure similar to that of 43. The product was obtained as a white solid (107 mg, 62% yield). 1H NMR (400 MHz, CDCl3) δ 8.79 (s, 1H), 8.30 (d, 1H, J = 8.0 Hz), 8.23 (d, 1H, J = 7.6 Hz), 7.68–7.62 (m, 3H), 7.16 (s, 1H), 7.05 (s, 1H), 7.01 (d, 1H, J = 8.4 Hz), 6.51 (t, 1H, J = 72.0 Hz), 5.33 (s, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ 153.6, 148.5, 148.2, 144.6 (t, J = 2.5 Hz), 137.9, 137.7, 135.4, 130.5, 129.8, 129.5, 124.7, 124.1, 121.7, 119.6, 115.5 (t, J = 264 Hz, including 118.2, 115.5, 112.9), 52.0 ppm. 19F NMR (376 MHz, CDCl3) δ −81.5 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H13F2N4O3+, 347.0956; found 347.0957. Retention time: 4.33 min (HPLC method B).
6-((1H-Imidazol-1-yl)methyl)-3-(difluoromethoxy)-2-(3fluorophenyl)pyridine (52).
This compound was prepared from 88 (75 mg, 0.25 mmol) following a procedure similar to that of 43. The product was obtained as a colorless oil (57 mg, 71% yield). 1H NMR (400 MHz, CDCl3) δ 7.66 (d, 1H, J = 8.8 Hz), 7.65 (s, 1H), 7.61–7.56 (m, 2H), 7.47–7.41 (m, 1H), 7.17–7.12 (m, 2H), 6.94 (d, 1H, J = 8.4 Hz), 6.61 (s, 1H), 6.43 (t, 1H, J = 72.4 Hz), 5.30 (s, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ 162.8 (d, J = 244.2 Hz), 153.3, 149.5 (d, J = 2.4 Hz), 144.5 (t, J = 2.5 Hz), 138.1 (d, J = 7.8 Hz), 130.4, 130.1 (d, J = 8.3 Hz), 129.9, 125.2 (d, J = 2.9 Hz), 121.0, 119.6, 116.5 (d, J = 23.1Hz), 116.4 (d, J = 21.0 Hz), 115.6 (t, J = 263.0 Hz, including 118.3, 115.6, 113.0), 52.1 ppm. 19F NMR (376 MHz, CDCl3) δ−81.4, −117.8 ppm. HRMS ESI [M + H]+ (m/z): calcd for C16H13F3N3O+, 320.1011; found 320.1012. Retention time: 4.52 min (HPLC method B).
2-(4-((6-(3-(Difluoromethoxy)phenyl)-5-methoxypyridin-2-yl)methyl)phenyl)ethan-1-ol (53).
This compound was prepared from 91 (184 mg, 0.53 mmol) and (4-(2-hydroxyethyl)phenyl)boronic acid (100 mg, 0.6 mmol) following a procedure similar to that of 58. The product was obtained as a colorless oil (202 mg, 98.9% yield). 1H NMR (400 MHz, CDCl3) δ 7.83 (d, 1H, J = 7.6 Hz), 7.74 (s, 1H), 7.42 (t, 1H, J = 8.0 Hz), 7.26 (d, 2H, J = 7.6 Hz), 7.20–7.10 (m, 4H), 7.03 (d, 1H, J = 8.4 Hz), 6.55 (t, 1H, J = 74.0 Hz), 4.13 (s, 2H), 3.85–3.82 (m, 5H), 2.84 (t, 2H, J = 6.4 Hz), 1.52 (br, 1H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.7, 152.2, 151.3, 145.5, 140.0, 138.4, 136.6, 129.52, 129.47, 129.4, 126.7, 122.9, 120.7, 119.8, 119.3, 116.5 (t, J = 257.1 Hz, including 119.0, 116.5, 113.9), 63.9, 55.8, 43.6, 39.0 ppm. 19F NMR (376 MHz, CDCl3) δ −80.2 ppm. HRMS ESI [M + H]+ (m/z): calcd for C22H22F2NO3+, 386.1568; found 386.1566. Retention time: 11.60 min (HPLC method B).
3-(4-((6-(3-(Difluoromethoxy)phenyl)-5-methoxypyridin-2-yl)methyl)phenyl)propan-1-ol (54).
This compound was prepared from 91 (184 mg, 0.53 mmol) and (4-(3-hydroxypropyl)phenyl)boronic acid (108 mg, 0.6 mmol) following a procedure similar to that of 58. The product was obtained as a colorless oil (166 mg, 78.5% yield). δ 7.83 (d, 1H, J = 8.0 Hz), 7.74 (s, 1H), 7.42 (t, 1H, J = 8.0 Hz), 7.25–7.11 (m, 6H), 7.02 (d, 1H, J = 8.4 Hz), 6.56 (t, 1H, J = 74.4 Hz), 4.13 (s, 2H), 3.82 (s, 3H), 3.66 (t, 2H, J = 6.4 Hz), 2.68 (t, 2H, J = 7.6 Hz), 1.91–1.84 (m, 2H), 1.35 (br, 1H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.9, 152.1, 151.3, 145.5, 140.0, 139.9, 137.8, 129.5, 129.3, 128.8, 126.8, 122.8, 120.7, 119.8, 119.3, 116.5 (t, J = 257.1 Hz, including 119.0, 116.5, 113.9), 62.5, 55.8, 43.6, 34.4, 31.9 ppm. 19F NMR (376 MHz, CDCl3) δ −80.2 ppm. HRMS ESI [M + H]+ (m/z): calcd for C23H24F2NO3+, 400.1724; found 400.1730. Retention time: 15.18 min (HPLC method B).
2-(4-((5-(Difluoromethoxy)-6-(3-methoxyphenyl)pyridin-2-yl)methyl)phenyl)ethan-1-ol (55).
This compound was prepared from 86 (184 mg, 0.53 mmol) and (4-(2-hydroxyethyl)phenyl)boronic acid (100 mg, 0.6 mmol) following a procedure similar to that of 58. The product was obtained as slightly yellow oil (195 mg, 95% yield). 1H NMR (400 MHz, CDCl3) δ 7.46–7.42 (m, 3H), 7.37 (t, 1H, J = 8.0 Hz), 7.26 (d, 2H, J = 8.0 Hz), 7.18 (d, 2H, J = 8.0 Hz), 7.04 (d, 1H, J = 8.4 Hz), 6.97 (dd, 1H, J1 = 8.0 Hz, J2 = 2.4 Hz), 6.31 (t, 1H, J = 73.6 Hz), 4.18 (s, 2H), 3.86–3.81 (m, 5H), 2.84 (t, 2H, J = 6.4 Hz), 1.52 (br, 1H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.7, 158.4, 150.0, 143.50, 138.0, 137.6,136.9, 129.62, 129.59, 129.48, 122.6, 122.0, 116.0 (t, J = 260.8 Hz, including 118.7, 116.0, 113.4), 115.1, 114.9, 63.8, 55.5, 44.0, 39.0 ppm. 19F NMR (376 MHz, CDCl3) δ −81.0 ppm. HRMS ESI [M + H]+ (m/z): calcd for C22H22F2NO3+, 386.1568; found 386.1573. Retention time: 10.43 min (HPLC method B).
3-(4-((5-(Difluoromethoxy)-6-(3-methoxyphenyl)pyridin-2-yl)methyl)phenyl)propan-1-ol (56).
This compound was prepared from 86 (184 mg, 0.53 mmol) and (4-(3-hydroxypropyl)phenyl)boronic acid (108 mg, 0.6 mmol) following a procedure similar to that of 58. The product was obtained as a colorless oil (191 mg, 90% yield). 1H NMR (400 MHz, CDCl3) δ 7.46–7.42 (m, 3H), 7.37 (t, 1H, J = 8.0 Hz), 7.23 (d, 2H, J = 8.0 Hz), 7.15 (d, 2H, J = 8.0 Hz), 7.03 (d, 1H, J = 8.4 Hz), 6.97 (d, 1H, J = 8.0 Hz), 6.31 (t, 1H, J = 73.6 Hz), 4.17 (s, 2H), 3.85 (s, 3H), 3.65 (t, 2H, J = 6.4 Hz), 2.68 (t, 2H, J = 7.6 Hz), 1.90–1.83 (m, 2H), 1.52 (br, 1H) ppm. 13C NMR (101 MHz, CDCl3) δ 159.7, 158.6, 150.0, 143.5, 140.3, 138.0, 136.9, 129.6, 129.5, 129.4, 122.6, 122.1, 116.1 (t, J = 260.7 Hz, including 118.7, 116.1, 113.5), 115.1, 114.9, 62.4, 55.5, 44.0, 34.4, 31.9 ppm. 19F NMR (376 MHz, CDCl3) δ −81.0 ppm. HRMS ESI [M + H]+ (m/z): calcd for C23H24F2NO3+, 400.1724; found 400.1720. Retention time: 13.22 min (HPLC method B).
6-((1H–Pyrazol-4-yl)methyl)-3-benzyloxy)-2-(3(difluoromethoxy)phenyl)pyridine(57).
This compound was prepared from 3-(benzyloxy)-6-(bromomethyl)-2-(3-(difluoromethoxy)phenyl)pyridine (93) (420 mg, 1 mmol) following a procedure similar to that of 2. The product was obtained as a colorless oil (120 mg, 29% yield). 1H NMR (400 MHz, CDCl3) δ 7.85 (d, 1H, J = 8.0 Hz), 7.77 (s, 1H), 7.49 (s, 2H), 7.42–7.27 (m, 6H), 7.25 (d, 1H, J = 8.4 Hz), 7.10 (d, 1H, J = 8.0 Hz), 7.04 (d, 1H, J = 8.8 Hz), 6.42 (t, 1H, J = 74.4 Hz), 5.07 (s, 2H), 4.04 (s, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.8, 151.1 (t, J = 2.3 Hz), 145.8, 139.6, 136.1, 133.5 (br), 129.3, 128.7, 128.2, 127.2, 126.6, 122.2, 121.4, 120.3, 119.3, 118.7, 116.2 (t, J = 256.9 Hz, including 118.8, 116.2, 113.7), 70.8, 32.4 ppm. 19F NMR (376 MHz, CDCl3) δ −80.2 ppm. HRMS ESI [M + H]+ (m/z): calcd for C23H20F2N3O2+, 408.1524; found 408.1520. Retention time: 12.3 min (HPLC method B).
3-(Benzyloxy)-2-(3-(difluoromethoxy)phenyl)-6-(pyridin-4ylmethyl)pyridine (58).
Compound 93 (420 mg, 1 mmol), pyridin-4ylboronic acid (149 mg, 1.2 mmol), Na2CO3 (159 mg, 1.5 mmol), and Pd(PPh3)4 (35 mg, 0.03 mmol) were added into a 50 mL round-bottom flask in a glovebox. Then, a degassed solvent mixture of toluene/EtOH/ water (4/3/3 mL) was injected into the sealed flask. The reaction mixture was stirred at 80 °C overnight. After cooling to room temperature, the reaction mixture was partitioned between DCM and water. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified with silica gel column (eluent: 50–70% EtOAc/hexane). The product was obtained as a light-yellow oil (165 mg, 39% yield). 1H NMR (400 MHz, CDCl3) δ 8.52 (d, 1H, J = 6.0 Hz), 7.86 (d, 1H, J = 7.6 Hz), 7.78 (s, 1H), 7.41 (t, 1H, J = 8.0 Hz), 7.37–7.31 (m, 5H), 7.26–7.21 (m, 2H), 7.12 (dd, 1H, J1 = 8.0 Hz, J2 = 2.0 Hz), 7.03 (d, 1H, J = 8.4 Hz), 6.42 (t, 1H, J = 74.0 Hz), 5.09 (s, 2H), 4.14 (s, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ 151.4, 151.0 (t, J = 2.8 Hz), 150.6, 149.8, 139.4, 136.0, 129.3, 128.7, 128.2, 127.2, 126.5, 124.4, 122.7, 121.3, 120.3, 119.4, 116.2 (t, J = 257.2 Hz, including 118.8, 116.2, 113.6), 70.8, 43.0 ppm. 19F NMR (376 MHz, CDCl3) δ −80.3 ppm. HRMS ESI [M + H]+ (m/z): calcd for C25H21F2N2O2+, 419.1571; found 419.1577. Retention time: 23.67 min (HPLC method B).
6-((1H-Imidazol-1-yl)methyl)-3-(benzyloxy)-2-(3(difluoromethoxy)phenyl)pyridine (59).
This compound was prepared from 93 (210 mg, 0.5 mmol) following a procedure similar to that of 43. The product was obtained as a white solid (134 mg, 66% yield). 1H NMR (400 MHz, CDCl3) δ 7.85 (d, 1H, J = 7.6 Hz), 7.78 (s, 1H), 7.63 (s, 1H), 7.42 (t, 1H, J = 8.4 Hz), 7.40–7.31 (m, 5H), 7.28 (d, 1H, J = 8.0 Hz), 7.14 (dd, 1H, J1 = 8.0 Hz, J2 = 2.0 Hz), 7.11 (s, 1H), 7.02 (s, 1H), 6.89 (d, 1H, J = 8.4 Hz), 6.43 (t, 1H, J = 72.4 Hz), 5.23 (s, 2H), 5.11 (s, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ 152.2, 151.1 (t, J = 2.7 Hz), 147.7, 146.4, 138.9, 137.6, 135.7, 129.9, 129.4, 128.8, 128.4, 127.3, 126.5, 121.3, 121.0, 120.4, 119.7, 119.5, 116.2 (t, J = 257.3 Hz, including 118.7, 116.2, 113.6), 70.9, 52.0 ppm. 19F NMR (376 MHz, CDCl3) δ −80.4 ppm. HRMS ESI [M + H]+ (m/z): calcd for C23H20F2N3O2+, 408.1524; found 408.1525. Retention time: 12.5 min (HPLC method B).
6-((1H-Pyrazol-4-yl)methyl)-2-(3-(benzyloxy)phenyl)-3(difluoromethoxy)pyridine (60).
This compound was prepared from 2(3-(benzyloxy)phenyl)-6-(bromomethyl)-3-(difluoromethoxy)pyridine (95) (420 mg, 1 mmol) as the base following a procedure similar to that of 50. The product was obtained as a colorless oil (234 mg, 57% yield). 1H NMR (400 MHz, CDCl3) δ 10.7 (br, 1H), 7.54–7.42 (m, 7H), 7.38–7.28 (m, 4H), 7.06–7.01 (m, 2H), 6.26 (t, 1H, J = 73.4 Hz), 5.09 (s, 2H), 4.07 (s, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ 158.8, 158.2, 150.0, 143.4 (t, J = 2.7 Hz), 138.0, 137.1, 133.5 (br), 129.6, 129.5, 128.7, 127.7, 122.3, 122.1, 118.0, 116.0, 115.9 (t, J = 261.2 Hz, including 118.5, 115.9, 113.3), 115.8, 70.2, 32.9 ppm. 19F NMR (376 MHz, CDCl3) δ −81.0 ppm. HRMS ESI [M + H]+ (m/z): calcd for C23H20F2N3O2+, 408.1524; found 408.1525. Retention time: 12.43 min (HPLC method B).
2-(3-(Benzyloxy)phenyl)-3-(difluoromethoxy)-6-(pyridin-4ylmethyl)pyridine (61).
This compound was prepared from 95 (420 mg, 1 mmol) following a procedure similar to that of 58. The product was obtained as a yellow oil (140 mg, 33% yield). 1H NMR (400 MHz, CDCl3) δ 8.53 (d, 2H, J = 5.6 Hz), 7.50–7.44 (m, 5H), 7.40–7.30 (m, 4H), 7.22 (d, 2H, J = 5.6 Hz), 7.23–7.03 (m, 2H), 6.30 (t, 1H, J = 73.2 Hz), 5.11 (s, 2H), 4.18 (s, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ 158.7, 155.8, 150.2, 150.0, 148.0, 143.6 (t, J = 2.8 Hz), 137.6, 137.0, 129.5, 129.4, 128.6, 128.0, 127.5, 124.4, 122.5, 122.1, 115.9, 115.7 (t, J = 257.2 Hz, including 118.3, 115.7, 113.1), 115.6, 70.0, 43.3 ppm. 19F NMR (376 MHz, CDCl3) δ −81.1 ppm. HRMS ESI [M + H]+ (m/z): calcd for C25H21F2N2O2+, 419.1571; found 419.1572. Retention time: 23.7 min (HPLC method B).
6-((1H-Imidazol-1-yl)methyl)-2-(3-(benzyloxy)phenyl)-3(difluoromethoxy)pyridine (62).
This compound was prepared from 95 (210 mg, 0.5 mmol) following a procedure similar to that of 43. The product was obtained as a colorless oil (155 mg, 76% yield). 1H NMR (400 MHz, CDCl3) δ 7.64 (s, 1H), 7.53 (d, 1H, J = 8.4 Hz), 7.47–7.31 (m, 8 H), 7.13 (s, 1H), 7.08–7.05 (m, 1H), 7.02 (s, 1H), 6.89 (d, 1H, J = 8.4 Hz), 6.32 (t, 1H, J = 72.8 Hz), 5.27 (s, 2H), 5.13 (s, 2H) ppm. 13C NMR (101 MHz, CDCl3) δ 158.7, 153.0, 150.5, 144.3, 137.7, 137.2, 136.9, 130.1, 129.8, 129.5, 128.6, 128.0, 127.5, 122.0, 120.5, 119.5, 116.2, 115.6, 115.5 (t, J = 262.4 Hz, including 118.2, 115.5, 112.9), 70.1, 52.0 ppm. 19F NMR (376 MHz, CDCl3) δ −81.4 ppm. HRMS ESI [M + H]+ (m/z): calcd for C23H20F2N3O2+, 408.1524; found 408.1519. Retention time: 12.8 min (HPLC method B).
Syntheses of Intermediates.
Synthetic details can be found in the Supporting Information.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the Intramural Research Program of NIH (NIMH) for financial support (ZIA-MH002793), Dr. John Lloyd of the NIDDK Advance Mass Spectrometry Facility for HRMS measurements, Tetra Therapeutics (Drs. Richard A. Nugent, Brian E. Marron, Mark E. Gurney) for PDE4B/D assays, and Dr. Xuesong Li of the NIBIB for the data fitting with MATLAB.
ABBREVIATIONS USED
- B/D
ratio of IC50 for PDE4B* over that for PDE4D*
- cAMP
cyclic adenosine monophosphate
- clog D
calculated distribution coefficient
- clog P
calculated partition coefficient
- CNSPET-MPO
multiparameter optimization for central nervous system PET radioligands
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectroscopy
- NMR
nuclear magnetic resonance
- PDE4
phosphodiesterase-4
- PDE4B*
PDE4B1-S133D, a dimeric isoform of PDE4B that contains a UCR1 mutation (S133D) that mimics PKA phosphorylation
- PDE4D*
PDE4D7-S129D, a dimeric isoform of PDE4D that contains a UCR1 mutation (S129D) that mimics PKA phosphorylation
- PET
positron emission tomography
- SAR
structure−activity relationship
- UCR1 and UCR2
upstream conserved regions
- V T
total volume of distribution
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c01745.
Syntheses of Intermediates, Figures S1−S6, Table S1, and S2; NMR spectra, HRMS spectra, and analytical HPLC chromatograms of the inhibitors and intermediates (Figures S7−S423) (PDF)
PDE4D molecular formula strings with biochemical data (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.2c01745
The authors declare no competing financial interest.
Contributor Information
Meijuan Jiang, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States.
Shuiyu Lu, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States.
Sanjay Telu, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States.
Victor W. Pike, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland 20892-1003, United States
REFERENCES
- (1).Sun J; Xiao Z; Haider A; Gebhard C; Xu H; Luo HB; Zhang HT; Josephson L; Wang L; Liang SH Advances in cyclic nucleotide phosphodiesterase-targeted PET imaging and drug discovery. J. Med. Chem 2021, 64, 7083–7109. [DOI] [PubMed] [Google Scholar]
- (2).Francis SH; Blount MA; Corbin JD Mammalian cyclic nucleotide phosphodiesterases: Molecular mechanisms and physiological functions. Physiol. Rev 2011, 91, 651–690. [DOI] [PubMed] [Google Scholar]
- (3).Wachtel H Potential antidepressant activity of rolipram and other selective cyclic adenosine 3′,5′-monophosphate phosphodiesterase inhibitors. Neuropharmacology 1983, 22, 267–272. [DOI] [PubMed] [Google Scholar]
- (4).Barad M; Bourtchouladze R; Winder DG; Golan H; Kandel E Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc. Natl. Acad. Sci. U.S.A 1998, 95, 15020–15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Vitolo OV; Sant’Angelo A; Costanzo V; Battaglia F; Arancio O; Shelanski M Amyloid beta -peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 13217–13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gong B; Vitolo OV; Trinchese F; Liu S; Shelanski M; Arancio O Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J. Clin. Invest 2004, 114, 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Smith DL; Pozueta J; Gong B; Arancio O; Shelanski M Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc. Natl. Acad. Sci. U.S.A 2009, 106, 16877–16882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Myeku N; Clelland CL; Emrani S; Kukushkin NV; Yu WH; Goldberg AL; Duff KE Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med 2016, 22, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Deeks ED Apremilast: A review in psoriasis and psoriatic arthritis. Drugs 2015, 75, 1393–1403. [DOI] [PubMed] [Google Scholar]
- (10).Spina D PDE4 inhibitors: current status. Br. J. Pharmacol 2008, 155, 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Garnock-Jones KP Roflumilast: A review in COPD. Drugs 2015, 75, 1645–1656. [DOI] [PubMed] [Google Scholar]
- (12).Burgin AB; Magnusson OT; Singh J; Witte P; Staker BL; Bjornsson JM; Thorsteinsdottir M; Hrafnsdottir S; Hagen T; Kiselyov AS; et al. Design of phosphodiesterase 4D (PDE4D) allosteric modulators for enhancing cognition with improved safety. Nat. Biotechnol 2010, 28, 63–70. [DOI] [PubMed] [Google Scholar]
- (13).Gurney ME; Nugent RA; Mo X; Sindac JA; Hagen TJ; Fox D III; O’Donnell JM; Zhang C; Xu Y; Zhang HT; et al. Design and synthesis of selective phosphodiesterase 4D (PDE4D) allosteric inhibitors for the treatment of Fragile X Syndrome and other brain disorders. J. Med. Chem 2019, 62, 4884–4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Naganuma K; Omura A; Maekawara N; Saitoh M; Ohkawa N; Kubota T; Nagumo H; Kodama T; Takemura M; Ohtsuka Y; et al. Discovery of selective PDE4B inhibitors. Bioorg. Med. Chem. Lett 2009, 19, 3174–3176. [DOI] [PubMed] [Google Scholar]
- (15).Fox D 3rd; Burgin AB; Gurney ME Structural basis for the design of selective phosphodiesterase 4B inhibitors. Cell. Signalling 2014, 26, 657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hagen TJ; Mo X; Burgin AB; Fox D; Zhang Z; Gurney ME Discovery of triazines as selective PDE4B versus PDE4D inhibitors. Bioorg. Med. Chem. Lett 2014, 24, 4031–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zhang C; Xu Y; Chowdhary A; Fox D 3rd; Gurney ME; Zhang HT; Auerbach BD; Salvi RJ; Yang M; Li G; O’Donnell JM Memory enhancing effects of BPN14770, an allosteric inhibitor of phosphodiesterase-4D, in wild-type and humanized mice. Neuropsychopharmacology 2018, 43, 2299–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Tetra Discovery Partners. A Study of BPN14770 in Male Adults (Aged 18 to 45) with Fragile X Syndrome, 2022. https://ClinicalTrials.gov/show/NCT05358886.
- (19).Aspiotis R; Deschênes D; Dubé D; Girard Y; Huang Z; Laliberté F; Liu S; Papp R; Nicholson DW; Young RN The discovery and synthesis of highly potent subtype selective phosphodiesterase 4D inhibitors. Bioorg. Med. Chem. Lett 2010, 20, 5502–5505. [DOI] [PubMed] [Google Scholar]
- (20).Brullo C; Massa M; Villa C; Ricciarelli R; Rivera D; Pronzato MA; Fedele E; Barocelli E; Bertoni S; Flammini L; Bruno O Synthesis, biological activities and pharmacokinetic properties of new fluorinated derivatives of selective PDE4D inhibitors. Bioorg. Med. Chem 2015, 23, 3426–3435. [DOI] [PubMed] [Google Scholar]
- (21).Brullo C; Rapetti F; Abbate S; Prosdocimi T; Torretta A; Semrau M; Massa M; Alfei S; Storici P; Parisini E; Bruno O Design, synthesis, biological evaluation and structural characterization of novel GEBR library PDE4D inhibitors. Eur. J. Med. Chem 2021, 223, No. 113638. [DOI] [PubMed] [Google Scholar]
- (22).Wakabayashi Y; Telu S; Dick RM; Fujita M; Ooms M; Morse CL; Liow JS; Hong JS; Gladding RL; Manly LS; et al. Discovery, radiolabeling, and evaluation of subtype-selective inhibitors for positron emission tomography imaging of brain phosphodiesterase-4D. ACS Chem. Neurosci 2020, 11, 1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Hooker JM; Carson RE Human positron emission tomography neuroimaging. Annu. Rev. Biomed. Eng 2019, 21, 551–581. [DOI] [PubMed] [Google Scholar]
- (24).Schröder S; Wenzel B; Deuther-Conrad W; Scheunemann M; Brust P Novel Radioligands for Cyclic Nucleotide Phosphodiesterase Imaging with Positron Emission Tomography: An Update on Developments Since 2012. Molecules 2016, 21, No. 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Schröder S; Scheunemann M; Wenzel B; Brust P Challenges on Cyclic Nucleotide Phosphodiesterases Imaging with Positron Emission Tomography: Novel Radioligands and (Pre-)Clinical Insights since 2016. Int. J. Mol. Sci 2021, 22, No. 3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Fujita M; Hines CS; Zoghbi SS; Mallinger AG; Dickstein LP; Liow JS; Zhang Y; Pike VW; Drevets WC; Innis RB; Zarate CA Downregulation of brain phosphodiesterase type IV measured with 11C-(R)-rolipram positron emission tomography in major depressive disorder. Biol. Psychiatry 2012, 72, 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).DaSilva JN; Lourenco CM; Meyer JH; Hussey D; Potter WZ; Houle S Imaging cAMP-specific phosphodiesterase-4 in human brain with R-[11C]rolipram and positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 1680–1683. [DOI] [PubMed] [Google Scholar]
- (28).Niccolini F; Wilson H; Pagano G; Coello C; Mehta MA; Searle GE; Gunn RN; Rabiner EA; Foltynie T; Politis M Loss of phosphodiesterase 4 in Parkinson disease: Relevance to cognitive deficits. Neurology 2017, 89, 586–593. [DOI] [PubMed] [Google Scholar]
- (29).Zhang L; Chen L; Beck EM; Chappie TA; Coelho RV; Doran SD; Fan KH; Helal CJ; Humphrey JM; Hughes Z; et al. The Discovery of a Novel Phosphodiesterase (PDE) 4B-Preferring Radioligand for Positron Emission Tomography (PET) Imaging. J. Med. Chem 2017, 60, 8538–8551. [DOI] [PubMed] [Google Scholar]
- (30).Wakabayashi Y; Strenkona P; Arakawa R; Yan X; Van Buskirk MG; Jenkins MD; Montero Santamaria JA; Maresca KP; Takano A; Liow JS; et al. First-in-human evaluation of 18F-PF06445974, a PET radioligand that preferentially labels phosphodiesterase-4B. J. Nucl. Med 2022, 63, 1919–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).National Institute of Mental Health; National Institutes of Health Clinical Center. Novel Positron Emission Tomography (PET) Radiotracer to Image Phosphodiesterase-4B (PDE4B), 2017. https://ClinicalTrials.gov/show/NCT03030391.
- (32).Pike VW Considerations in the development of reversibly binding PET radioligands for brain imaging. Curr. Med. Chem 2016, 23, 1818–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhang L; Villalobos A; Beck EM; Bocan T; Chappie TA; Chen L; Grimwood S; Heck SD; Helal CJ; Hou X; et al. Design and selection parameters to accelerate the discovery of novel central nervous system positron emission tomography (PET) ligands and their application in the development of a novel phosphodiesterase 2A PET ligand. J. Med. Chem 2013, 56, 4568–4579. [DOI] [PubMed] [Google Scholar]
- (34).Raman EP; Paul TJ; Hayes RL; Brooks CL Automated, accurate, and scalable relative protein−ligand binding free-energy calculations using lambda dynamics. J. Chem. Theory Comput 2020, 16, 7895–7914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Paul SM; Mytelka DS; Dunwiddie CT; Persinger CC; Munos BH; Lindborg SR; Schacht AL How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discovery 2010, 9, 203–214. [DOI] [PubMed] [Google Scholar]
- (36).Aspiotis R; Deschenes D; Dube D; Girard Y; Huang Z; Laliberte F; Liu S; Papp R; Nicholson DW; Young RN The discovery and synthesis of highly potent subtype selective phosphodiesterase 4D inhibitors. Bioorg. Med. Chem. Lett 2010, 20, 5502–5505. [DOI] [PubMed] [Google Scholar]
- (37).Woodrow MD; Ballantine SP; Barker MD; Clarke BJ; Dawson J; Dean TW; Delves CJ; Evans B; Gough SL; Guntrip SB; et al. Quinolines as a novel structural class of potent and selective PDE4 inhibitors. Optimisation for inhaled administration. Bioorg. Med. Chem. Lett 2009, 19, 5261–5265. [DOI] [PubMed] [Google Scholar]
- (38).Card GL; England BP; Suzuki Y; Fong D; Powell B; Lee B; Luu C; Tabrizizad M; Gillette S; Ibrahim PN; et al. Structural basis for the activity of drugs that inhibit phosphodiesterases. Structure 2004, 12, 2233–2247. [DOI] [PubMed] [Google Scholar]
- (39).D’Ursi P; Guariento S; Trombetti G; Orro A; Cichero E; Milanesi L; Fossa P; Bruno O Further insights in the binding mode of selective inhibitors to human PDE4D enzyme combining docking and molecular dynamics. Mol. Inf 2016, 35, 369–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Cedervall P; Aulabaugh A; Geoghegan KF; McLellan TJ; Pandit J Engineered stabilization and structural analysis of the autoinhibited conformation of PDE4. Proc. Natl. Acad. Sci. U.S.A 2015, 112, E1414–E1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Singh J; Gurney ME; Burgin AB; Sandanayaka V; Kiselyov A; Motta A; Schiltz G; Hategan G; Hagen T Biaryl PDE4 Inhibitors for Treating Inflammatory, Cardiovascular, and CNS Disorders. 2009/0324569A1, 2009.
- (42).Yung-Chi C; Prusoff WH Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (43).Page MI Entropy, binding energy, and enzymic catalysis. Angew. Chem., Int. Ed 1977, 16, 449–459. [Google Scholar]
- (44).Vilseck JZ; Sohail N; Hayes RL; Brooks CL Overcoming challenging substituent oerturbations with multisite λ-dynamics: A case study targeting β-secretase 1. J. Phys. Chem. Lett 2019, 10, 4875–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Vilseck JZ; Armacost KA; Hayes RL; Goh GB; Brooks CL Predicting binding free energies in a large combinatorial chemical space using multisite λ dynamics. J. Phys. Chem. Lett 2018, 9, 3328–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Wager TT; Hou X; Verhoest PR; Villalobos A Moving beyond rules: The development of a central nervous system multiparameter optimization (CNS MPO) approach to enable alignment of druglike properties. ACS Chem. Neurosci 2010, 1, 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wager TT; Chandrasekaran RY; Hou X; Troutman MD; Verhoest PR; Villalobos A; Will Y Defining desirable central nervous system drug space through the alignment of molecular properties, in vitro ADME, and safety attributes. ACS Chem. Neurosci 2010, 1, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Deng X; Rong J; Wang L; Vasdev N; Zhang L; Josephson L; Liang SH Chemistry for positron emission tomography: Recent advances in 11C-, 18F-, 13N-, and 15O-labeling reactions. Angew. Chem., Int. Ed 2019, 58, 2580–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Solbach C; Uebele M; Reischl G; Machulla HJ Efficient radiosynthesis of carbon-11 labelled uncharged Thioflavin T derivatives using [11C]methyl triflate for β-amyloid imaging in Alzheimer’s Disease with PET. Appl. Radiat. Isot 2005, 62, 591–595. [DOI] [PubMed] [Google Scholar]
- (50).Telu S; Siméon FG; Lu S; Pike VW Hypervalent Iodine Compounds as Precursors for Biomedical Radiotracers. In The Chemistry of Hypervalent Halogen Compounds, Patai’s Chemistry of Functional Groups; Olofson B; Marek I; Rappoport Z, Eds.; John Wiley & Sons Ltd.: Chichester, 2019; pp 721–780. [Google Scholar]
- (51).Lu S; Siméon FG; Telu S; Cai L; Pike VW The Chemistry of Labeling Heterocycles with Carbon-11 or Fluorine-18 for Biomedical Imaging. In Advances in Heterocyclic Chemistry; Scriven EFV; Ramsden CA, Eds.; Academic Press: Cambridge, 2020; Vol. 132, pp 241–384. [Google Scholar]
- (52).Němec V; Hylsová M; Maier L; Flegel J; Sievers S; Ziegler S; Schröder M; Berger BT; Chaikuad A; Valčíková B; et al. Furo[3,2-b]pyridine: A privileged scaffold for highly selective kinase inhibitors and effective modulators of the hedgehog pathway. Angew. Chem., Int. Ed 2019, 58, 1062–1066. [DOI] [PubMed] [Google Scholar]
- (53).Nambo M; Yim JC; Freitas LBO; Tahara Y; Ariki ZT; Maekawa Y; Yokogawa D; Crudden CM Modular synthesis of alpha-fluorinated arylmethanes via desulfonylative cross-coupling. Nat. Commun 2019, 10, No. 4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.