

Abstract
Multiple cytosolic innate sensors form large signalosomes after activation, but this assembly needs to be tightly regulated to avoid accumulation of misfolded aggregates. We found that the eIF2α kinase heme-regulated inhibitor (HRI) controls NOD1 signalosome folding and activation through a process requiring eukaryotic initiation factor 2α (eIF2α), the transcription factor ATF4, and the heat shock protein HSPB8.The HRI/eIF2α signaling axis was also essential for signaling downstream of the innate immune mediators NOD2, MAVS, and TRIF but dispensable for pathways dependent on MyD88 or STING. Moreover, filament-forming α-synuclein activated HRI-dependent responses, which suggests that the HRI pathway may restrict toxic oligomer formation. We propose that HRI, eIF2α, and HSPB8 define a novel cytosolic unfolded protein response (cUPR) essential for optimal innate immune signaling by large molecular platforms, functionally homologous to the PERK/eIF2α/HSPA5 axis of the endoplasmic reticulum UPR.
Graphical Abstract
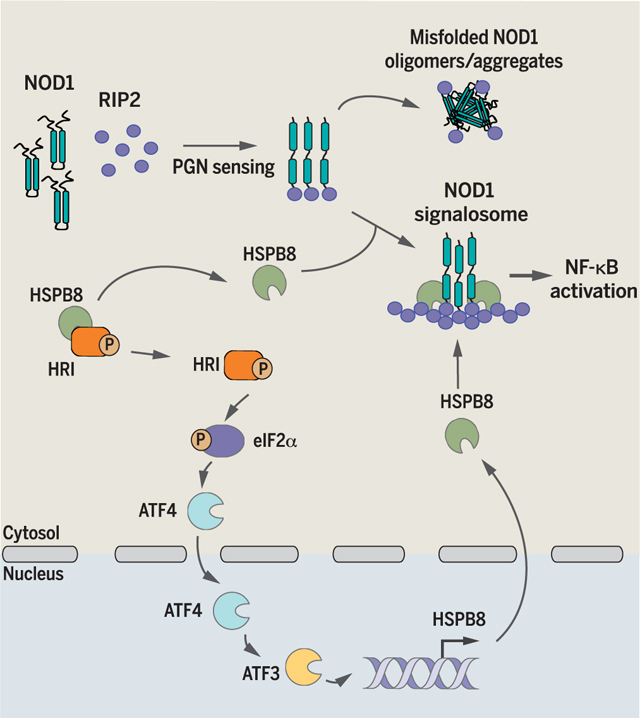
The integrated stress response (ISR) is an evolutionarily conserved signaling cascade through which cells can cope with their environment and adjust their metabolic status. Detection of various cellular stresses by the ISR is performed by four kinases: PKR (protein kinase R), GCN2 (general control non-derepressible 2), PERK (protein kinase RNA-like ER kinase), and HRI (heme-regulated inhibitor). PKR detects viral double-stranded RNA (dsRNA); GCN2 senses uncharged tRNAs that accumulate in cells lacking free amino acids (AA) (1, 2); PERK detects accumulation ofmisfolded proteins in the endoplasmic reticulum (ER), also known as ER stress (3); and HRI responds to a broad range of stresses, including heme deprivation, oxidative stress, and heat shock (4–7), although cytosolic proteotoxicity may represent an overarching trigger for HRI (8). These four kinases phosphorylate eIF2α (eukaryotic initiation factor 2α) on Ser51, which results in a temporary block in translation and transcriptional reprograming to cope with the encountered stress.
The HRI/eIF2α axis is required for inflammatory responses during infection
To characterize the role of the ISR in host responses to bacterial pathogens, we infected mouse embryonic fibroblasts (MEFs), expressing either the wild-type form of eIF2α or a knock-in (KI) mutation encoding the Ser51 → Ala (S51A) substitution, with Salmonella, Shigella, and Listeria, which induce eIF2α phosphorylation (9–11). In line with the established role of eIF2α phosphorylation in stress granule formation, infection with the three pathogens induced stress granule accumulation in wild-type but not KI MEFs (fig. S1, A to C). However, we surprisingly noticed that the secretion of pro-inflammatory cytokines (Fig. 1A and fig. S1, D to H) was blunted in KI MEFs relative to wild-type cells in response to the three pathogens in a manner independent of bacterial replication (fig. S1, I to K). The reduced secretion of Cxcl1 in the KIMEFs correlated with reduced expression of Cxcl1 (Fig. 1B), showing that the ISR is required for transcriptional cytokine responses to infection. Shigella infection induced the expression of ATF3 and ATF4 (fig. S2), and as expected, up-regulation of the ISR-associated gene Atf3 was dependent on eIF2α phosphorylation and ATF4, an essential transcription factor that, upon eIF2α phosphorylation, mediates transcriptional reprogramming during the ISR (12) (fig. S3).
Fig. 1. The HRI/eIF2α axis is required for inflammatory responses during infection.

(A) Cxcl1 secretion from the supernatants of wild-type (WT) and eIF2α S51A knock-in (KI) MEFs after infection with Shigella for the indicated times, as measured by ELISA. (B and C) Expression of Cxcl1 in WT and KI MEFs (B) and WT and Atf4 knockout (Atf4–/–) MEFs (C) left unstimulated (CTR) or after infection with Shigella. (D) HeLa cells infected with Shigella or treated for 1 hour with 300 mM sodium arsenite (Ars) and analyzed by immunoblotting. (E) HeLa cells transduced with lentiviral particles targeting a scrambled sequence (SC), both HRI and GCN2 [double knockdown (DKD)] left untreated (U), infected 1 hour with Shigella (S.f), treated 1 hour with 300 μM Ars or treated with Krebs-Ringer buffer (KRB) for 45 min and analyzed by immunoblotting. (F and G) Expression of ATF3 (F) and IL8 (G) in HeLa cells transduced with lentiviral particles targeting a scrambled sequence (SC), HRI (shHRI), or GCN2 (shGCN2) and infected with Shigella. (H and I) Expression of CXCL1 (H) and IL1α (I) in scrambled, HRI KD, and GCN2 KD HeLa cells infected with Shigella. Data are means ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
In agreement with results obtained in KI cells, Cxcl1 expression after Shigella infection was blunted in Atf4–/– MEFs (Fig. 1C).Down-regulation of Shigella-induced cytokine expression in ISR-deficient cells was not a consequence of exacerbated translation, as reduced translation was observed in Atf4–/– MEFs (fig. S4). Finally, the impact of acute inhibition of the ISR was analyzed by using ISRIB, a specific inhibitor of eIF2α phosphorylation. ISRIB treatment potently inhibited IL8, ATF3, and GADD34 expression in Shigella-infected cells at doses that did not alter translation (fig. S5). Thus, phosphorylation of eIF2α is essential for pro-inflammatory and stress responses during bacterial infection.
We next aimed to identify the eIF2α kinases that mediate the ISR during bacterial infection. Our previous work has shown that invasive bacterial pathogens trigger GCN2 activation (9, 10), but the contribution of the other eIF2α kinases remains undetermined. Upon infection of HeLa cells with Shigella, we observed the accumulation of a slower-migrating form of HRI, indicative of phosphorylation and activation, and this effect increased at later times of infection (Fig. 1D). As a control, treatment with arsenite, a known HRI inducer, resulted in full conversion of HRI to its slower-migrating form (Fig. 1D). HRI activation is also associated with its dissociation from Hsc70 (13); both Shigella infection and arsenite treatment induced HRI/Hsc70 dissociation (fig. S6). We next assessed the relative contributions of HRI and GCN2 in triggering the ISR during infection by knocking down their expression (fig. S7, A and B). Although the formation of stress granules after arsenite treatment or AA starvation depended on HRI and GCN2, respectively (figs. S7C and S8), we observed a partial contribution of each kinase to Shigella-induced stress granule formation and eIF2α phosphorylation (fig. S7, D and E). Transient knockdown of both HRI and GCN2 [double knockdown (DKD)] resulted in near-complete abolishment of stress granule formation and eIF2a phosphorylation after Shigella infection, relative to scrambled cells (Fig. 1E and fig. S7D), thus showing that HRI and GCN2 cooperate to trigger the ISR during Shigella infection.
We next analyzed the relative contributions of HRI and GCN2 to the transcriptional responses induced by Shigella. Whereas ATF3 expression after arsenite treatment or AA starvation was dependent on HRI and GCN2, respectively (fig. S7F), ATF3 induction during Shigella infection was dependent on both HRI and GCN2 (Fig. 1F). Moreover, the oxidative stress response gene HMOX1 was induced in an HRI-dependent manner, whereas the AA metabolism gene ASNS was induced in a GCN2-dependentmanner in Shigella-infected cells (fig. S7, G to J). During infection with Shigella, expression of IL-8, CXCL1, and IL-1α (Fig. 1, G to I) was strongly repressed in HRI knockdown (KD) cells, whereas knockdown of GCN2 had no effect. Down-regulation of Shigella-induced cytokine expression in HRI KD cells was not a consequence of exacerbated translation, as reduced translation was observed relative to scramble control and GCN2 KD cells (fig. S9). Thus, although both HRI and GCN2 are needed for the full ISR during Shigella infection, HRI is the sole eIF2α kinase controlling ISR-dependent pro-inflammatory responses to this pathogen (fig. S10).
HRI is essential for NOD1- and NOD2-driven inflammatory responses
In nonmyeloid cells, the NOD1/2–NF-κB axis is critical for inflammatory signaling in response to Shigella (14–16). After Shigella infection, NF-κB activation was blunted in HRI KD cells (Fig. 2A and fig. S11A). The decreased NF-κB response was not due to a lack of bacterial invasion or replication (fig. S11B) and was independent from translation regulation, because p65 nuclear translocation after Shigella infection occurred prior to translation arrest (fig. S12). In agreement with this finding, the expression of Cxcl1 and Il6 (Fig. 2, B and C) was markedly reduced in Shigella-infected Hri–/– MEFs relative to Hri+/+ cells. Knockdown of HRI in HCT116 cells significantly blunted IL-8 expression in response to the NOD1 and NOD2 ligands (Fig. 2D), which suggests that HRI directly controls NOD1/2-dependent signaling. As a control, we noted that the expression of NOD1 and NOD2 was unaffected by HRI or GCN2 knockdown (fig. S13, A and B). Cxcl1 secretion was also reduced in Hri–/– bone marrow–derived macrophages (BMDMs) stimulated with NOD ligands either directly (Fig. 2E) or in synergy with lipopolysaccharide (LPS) stimulation (fig. S13C). Similarly, Cxcl1 expression was reduced in Hri–/– organoids stimulated with Shigella culture supernatant (Fig. 2F). Intraperitoneal injection of the NOD2 ligand muramyl dipeptide (MDP) triggered a blunted secretion of Cxcl1 in the serum of Hri–/– mice relative to Hri+/+ littermates (Fig. 2G), a reduced infiltration of neutrophils into the peritoneum (fig. S13, D and E), and a smaller release of interleukin (IL)–1β into the peritoneal cavity (fig. S13F). Similar effectswere noted when Hri–/–, Hri+/–, or Hri+/+ mice were injected with theNOD1 agonist FK-156 (fig. S13, G and H). To characterize the role played by HRI in an in vivo model of infection driven partly by NOD1 and NOD2 (17, 18), we infected Hri–/– and Hri+/+ mice with Citrobacter rodentium. Significantly less cecal Cxcl1, Ccl2, and serum Cxcl1 (Fig. 2,Hand I, and fig. S13I), as well as a trend for reduced serum Ccl2 and cecal Il6 (fig. S13, J and K), were observed in C. rodentium–infected Hri–/– mice relative to Hri+/+ littermates, in line with studies demonstrating reduced serum IL-6 in Hri–/– mice during Listeria infection (19). This blunted pro-inflammatory response was not due to differences in colonization, because both groups of mice displayed a similar bacterial burden (fig. S13L). Together, these results demonstrate a key role for HRI in controlling NOD1/2-driven pro-inflammatory responses in cell lines, in primary cells, and in vivo.
Fig. 2. HRI is essential for NOD1- and NOD2-driven inflammatory responses.

(A) Scrambled, HRI KD, and GCN2 KD HeLa cells were infected with Shigella for 30 min; the percentage of cells displaying nuclear NF-κB p65 was quantified by immunofluorescence. (B and C) Expression of Cxcl1 (B) and Il-6 (C) in wildtype (HRI+/+) and HRI knockout (HRI–/–) MEFs infected with Shigella. (D) Expression of IL-8 in scrambled and HCT116 KD cells after 4 hours of stimulation with L18-MDP (10 ng/ml) or C12-iE-DAP (10 μg/ml). (E) Cxcl1 secretion from the supernatants of HRI+/+ and HRI–/– bone marrow–derived macrophages (BMDMs) stimulated for 6 hours with L18-MDP (10 ng/ml) or C12-iE-DAP (10 μg/ml), as measured by ELISA. (F) Expression of Cxcl1 in intestinal organoids derived from HRI+/+ and HRI–/– mice treated for 4 hours with Shigella conditioned medium (CM Shigella). (G) Serum Cxcl1 in HRI+/+, HRI heterozygous (HRI+/–), and HRI–/– mice after intraperitoneal injection with MDP, as measured by ELISA. (H and I) Cxcl1 (H) and CCL2 (I) in the cecum homogenate of HRI+/+ and HRI–/– mice 4 days after infection with Citrobacter rodentium, as measured by ELISA. Graphs represent means ± SD from three to five independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 (two-way ANOVA followed by Tukey multiple-comparisons test).
The HRI signaling axis controls the folding and solubility of NOD1 oligomers
We speculated that HRI might control NOD1/2 signaling at the level of complex formation. Scramble control or shHRI (short hairpin HRI) cells transfected with NOD1-HA (NOD1 fused to hemagglutinin) and either unstimulated or stimulated with C12-iE-DAP were lysed in non-ionic (NP-40) or ionic [radioimmunoprecipitation assay (RIPA)] buffers. Stimulation with C12-iE-DAP resulted in the release of NOD1 into the soluble fraction of NP-40–lysed cells, and analysis by blue native polyacrylamide gel electrophoresis (PAGE) revealed that the released NOD1 was in the form of large oligomers (Fig. 3A, left panels). Interestingly, although NOD1 was expressed normally in shHRI cells, stimulation with C12-iE-DAP failed to release the protein into the soluble fraction, as it remained mainly insoluble (Fig. 3A, left panels). To determine whether NOD1 proteins did not form oligomers at all upon stimulation or whether those oligomers did form but could not be solubilized by NP-40, we performed a similar analysis in cells lysed with RIPA, which strips most non-transmembrane proteins from membranes. Using this buffer, NOD1 complexes could now be solubilized in shHRI cells, and blue native PAGE analysis revealed that they were constitutively found as large complexes (Fig. 3A, right panels). Thus, NOD1 forms large signalling platforms, or signalosomes, that are soluble in both ionic and non-ionic buffers in control cells, but these complexes can only be extracted by an ionic buffer in shHRI cells.
Fig. 3. The HRI signaling axis controls the folding and solubility of NOD1 oligomers.

(A) Scrambled and HRI KD HEK293T cells were stimulated for 30 min with C12-iE-DAP (10 ng/ml) and analyzed by blue native PAGE and SDS-PAGE. (B) HEK293T cells were stimulated with C12-iE-DAP (10 ng/ml) for 30 min, and NOD1-HA immunocomplexes were subjected to SYPRO Orange hydrophobicity measurements. RFU, relative fluorescence units. (C) Scrambled or HRI KD HEK293T cells were stimulated for 30 min with C12-iE-DAP (10 ng/ml) and NOD1-HA immunocomplexes were analyzed as in (B). (D) HEK293T cells with or without ISRIB treatment (1 μg/ml) were stimulated with C12-iE-DAP (10 ng/ml) and NOD1-HA immunocomplexes were analyzed as in (B). Graphs represent means ± SD from three or four independent experiments. *P < 0.05, **P < 0.01; ns, not significant.
To gain better understanding of NOD1 complex formation in HRI-silenced cells, we transfected scramble control and shHRI cells with NOD1-HA and stimulated them with C12-iE-DAP, as above, or left them unstimulated (Fig. 3A). These cells were then lysed in RIPA buffer, and NOD1 complexes were immunoprecipitated and incubated with the dye SYPRO Orange, whose fluorescence increases when the dye binds hydrophobic patches or misfolded proteins. To control for the robustness of the assay, we denatured the NOD1 immunocomplexes and an unrelated purified protein by boiling, resulting in increased fluorescence (Fig. 3B and fig. S14, A and B). Whereas NOD1 oligomers displayed similar fluorescence in scramble versus shHRI cells in control conditions, stimulation with C12-iE-DAP resulted in a significant increase in fluorescence only in shHRI cells (Fig. 3C and fig. S14C), which suggests improper folding of the NOD1 complexes assembled after ligand stimulation in shHRI cells. In agreement with this finding, acute inhibition of eIF2α-dependent signalling with ISRIB also increased the hydrophobicity of NOD1 complexes in cells stimulated with C12-iE-dap (Fig. 3D). Thus, the HRI/eIF2α signaling axis controls the assembly of theNOD1 signalosomes after ligand stimulation.
The HRI-eIF2α-HSPB8 loop is essential for NOD1 and NOD2 signaling
We next aimed to determine how HRI controls NOD signalosome assembly. Heat shock protein (HSP) chaperones regulate the assembly and stability of protein complexes, so we speculated that HRI might regulate NOD signalosome assembly through the expression and action of HSPs. In our earlier microarray analysis of Shigella-induced genes (9), HSPB8 was among the most upregulated genes and was the only HSP regulated by Shigella (fig. S15, A and B), an observation that we confirmed in wild-type MEFs (Fig. 4A). Hspb8 was also up-regulated by the unfolded protein response (UPR)/ER stress inducer thapsigargin (Fig. 4A), whereas the UPR/ER stress chaperone Hspa5 (also called BiP or GRP78) was induced by thapsigargin but not Shigella infection (fig. S16A). Thapsigargin also induced HSPB8 and HSPA5 expression in HCT116 cells (fig. S16, B and C) and primarymurine intestinal organoids (fig. S16, D and E). Thus, both HSPB8 and HSPA5 are induced by UPR/ER stress, but HSPB8 is additionally up-regulated by infection. We also analyzed the kinetics of HSPB8 induction by Shigella and observed that whereas the pro-inflammatory genes IL8 and CXCL1, as well as the stress-induced genes ATF3 and GADD34, were induced in a first wave (as early as 1 hour after infection), HSPB8 was induced in a second wave, at 2 to 4 hours after infection (fig. S16, F to J).
Fig. 4. The HRI-eIF2α-HSPB8 loop is essential for NOD1 and NOD2 signaling.

(A to C) Expression of Hspb8/HSPB8 in WT MEFs (A), Atf4–/– MEFs (B), and scrambled, HRI KD, and GCN2 KD HeLa cells (C) infected with Shigella or treated with 5 μM thapsigargin for 4 hours. (D) Expression of IL8 in scrambled or HSPB8 KD (shHSPB8) HeLa cells infected with Shigella. (E) Expression of IL8 in scrambled or HRI KD HeLa cells infected 4 hours with Shigella with or without transfection of HSPB8-V5, as measured by qPCR. (F) Coimmunoprecipitation experiments with unstimulated (CTR) or Shigella-infected HeLa cell lysates. (G) Coimmunoprecipitation experiments with the lysates of HeLa cells that were transfected with NOD1-HA and HSPB8-V5 and infected with Shigella. (H) Scrambled or HRI KD HEK293T cells transfected with NOD1-HA with or without HSPB8-V5 and analyzed as in Fig. 3A. (I and J) Scrambled or HRI KD HEK293T cells transfected with or without HSPB8-V5 (I) and scrambled or HSPB8 KD HEK293T cells (J) were stimulated for 30 min with C12-iE-DAP (10 ng/ml) and NOD1-HA complexes were subjected to SYPRO Orange hydrophobicity assays. Graphs display means ± SD from three to five independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. Asterisk in (F) denotes a nonspecific band.
To further characterize the pathway that controls HSPB8 expression, we infected Atf4–/– MEFs and CRISPR-engineered ATF3–/– HCT116 cells (fig. S17A) with Shigella or stimulated them with thapsigargin. Shigella-induced up-regulation of HSPB8 was ATF4-dependent (Fig. 4B) as well as ATF3-dependent (fig. S17B), in agreement with kinetics data showing that ATF3 is induced before HSPB8 (fig. S16, F to J), whereas induction by thapsigargin required neither ATF4 nor ATF3 (Fig. 4B and fig. S17B). In contrast, HSPA5 upregulation by thapsigargin stimulation was partially dependent on ATF4 and did not require ATF3 (fig. S17, C and D), in agreement with the known reliance ofHSPA5 expression on the IRE1α and ATF6 branches of the UPR (20, 21). Moreover, Hspb8 up-regulation by Shigella infection was abrogated in eIF2α KI MEFs (fig. S17E), although baseline levels of Hspb8 mRNA and protein (fig. S17F) were higher in KI as compared to wild-type MEFs in uninfected cells, possibly as a result of higher baseline levels of Atf3 (see fig. S3A). Finally, HRI but not GCN2 controlled HSPB8 upregulation after Shigella infection (Fig. 4C); this effect was specific, as HSP90 expression was unaffected by HRI knockdown (fig. S17G).
At baseline, protein levels of HSPB8 were lower in Shigella-infected shHRI cells than in scramble control or shGCN2 cells (fig. S17H). Moreover, by 30 min after Shigella infection, a decline of the protein HSPB8, but not HSP90 or Hsc70, was observed (fig. S17H), which suggests that HSPB8 chaperoning activity was engaged rapidly during infection and may result in its degradation. It is thus possible that the HRI-dependent transcriptional up-regulation of HSPB8 during infection represents a regulatory feedback loop to restore HSPB8 levels. Together, these results identify HSPB8 as a key HSP induced by Shigella infection through the HRI/eIF2α/ATF4/ATF3 signaling axis.
We then asked whether HSPB8 mediated the effects of HRI on NOD-dependent signaling. Short hairpin RNA (shRNA)–mediated silencing of HSPB8 (fig. S17I) strongly inhibited Shigella-induced IL-8 up-regulation (Fig. 4D), whereas overexpression of HSPB8 in HRI KD cells or in HRI CRISPR knockout cells rescued Shigella-induced NF-κB signaling and inflammatory responses (Fig. 4E and figs. S18 and S19). Thus, the HRI signaling axis regulates Shigella-induced pro-inflammatory NF-κB signaling through an HSPB8-dependent process.
We next aimed to uncover the mechanism through which the HRI-HSPB8 pathway regulates Shigella-induced pro-inflammatory NF-κB signaling. Drawing parallels with the UPR pathway in which activation occurs after displacement of chaperones to the proteins requiring refolding (22), we speculated that HRI might interact with HSPB8. Coimmunoprecipitation assays confirmed that HSPB8 interacted with HRI oligomers at the endogenous level, and the proteins rapidly dissociated after infection with Shigella (Fig. 4F). Concomitantly, an interaction between NOD1 immunocomplexes and HSPB8-V5 (HSPB8 fused to a V5 tag)was observed,which peaked at 10 min after infection (Fig. 4G). Thus, displacement of HSPB8 from HRI to forming NOD1 signalosomes may act as a signal for HRI activation. Notably, HSPB8 also interacted with NOD2 complexes and NOD2-HSPB8 interactions increased after MDP stimulation (fig. S20). Furthermore, ectopic expression of HSPB8 was sufficient to rescue the release of NP-40–soluble NOD1 complexes (Fig. 4H) and to reduce the hydrophobicity of NOD1 immunocomplexes in C12-iE-DAP–stimulated HRI KD cells (Fig. 4I). In agreement with these findings, silencing of HSPB8 increased the hydrophobicity Of NOD1 immunocomplexes after stimulation with C12-iE-DAP (Fig. 4J). Thus, the HRIHSPB8 axis regulates NOD1 signaling through the control of signalosome assembly and folding, and HSPB8 displacement from HRI to the NOD1 signalosome likely serves as a signal to trigger an HRI-dependent eIF2α-ATF4-ATF3 pathway inducing transcriptional up-regulation of HSPB8 for homeostatic regulation.
HRI differentially regulates PRM signaling
We next aimed to explore whether the HRI/eIF2α/HSPB8 axis regulates other pattern recognition molecule (PRM) pathways. We first investigated whether Toll-like receptor (TLR) signaling was influenced by HRI. Stimulation of scramble control cells or shHRI HCT116 with the TLR5 ligand flagellin triggered similar expression of IL-8 (fig. S21A). Stimulation of Hri+/+ and Hri–/– BMDMs with the TLR4, TLR5, and TLR2 ligands LPS, flagellin, and Pam3CSK4, respectively, induced similar secretion of Cxcl1 (Fig. 5A). In agreement with this finding, MyD88 signaling was similar in LPS-stimulated Hri+/+ and Hri–/– BMDMs (Fig. 5B and fig. S21, B and C). In contrast, TRIF activation was blunted in LPS-treated Hri–/–BMDMs (Fig. 5C and fig. S21, B and D), which suggests that HRI regulates the TLR4/TRIF but not the TLR4/MyD88 signaling axis. Consistently, intraperitoneal injection of Hri+/+, Hri+/–, or Hri–/– mice with LPS triggered similar levels of serum Cxcl1 (fig. S21E). TLR3-TRIF signaling was also partially impaired in Hri–/– BMDMs (Fig. 5D and fig. S22). Furthermore Hri+/+ and Hri–/– BMDMs had similar expression of Cxcl1 and Ifnb (Fig. 5, E and F) and secreted comparable amounts of interferon (IFN) α/β (fig. S23) after stimulation with inducers of the cGAS-STING pathway. In contrast, Hri–/– BMDMs expressed significantly less Cxcl1 and Ifnb and secreted less type I IFNs in response to RIG-I-MAVS triggers (Fig. 5, E and F, and fig. S23). In agreement, no differences in STING-driven inflammation were observable in Hri+/+ and Hri–/– MEFs or BMDMs after infection with herpes simplex virus (HSV) (fig. S24). In contrast, a strong decrease of these markers, together with a reduced formation of MAVS oligomers, was noted in Hri–/– MEFs and BMDMs infected with the RIG-I-MAVS activator Sendai virus (SeV) (fig. S25). In agreement with a role for HRI in TRIF-dependent and MAVS-dependent responses, but not MyD88-dependent responses, we further noticed that acute inhibition of eIF2α phosphorylation using ISRIB significantly inhibited Il6 and Ifnb induced by polyinosinic-polycytidylic acid [poly(I:C)] addition (TLR3-TRIF trigger) or transfection (MAVS trigger), and that it inhibited Ifnb induced by LPS (TLR4-TRIF trigger) without affecting Il6 (TLR4-MyD88 trigger) (fig. S26). In support of the role of HRI in host defense against RNA viruses, we observed a significant blunting of immune cell recruitment and reduced damage in the lungs of Hri–/– mice at day 6 after infection with influenza A virus (Fig. 5, G to I, and fig. S27). Finally, heptose-1,7-bisphosphate (HBP) sensing by TIFA [tumor necrosis factor receptor–associated factor (TRAF)–interacting protein with forkhead-associated domain] also required HRI, as scramble control cells exhibited a robust induction of IL-8 after stimulation with HBP-containing conditioned media from gmhB– Neisseria meningitidis (14), which was severely reduced in shHRI cells (fig. S28).
Fig. 5. HRI differentially regulates PRM signaling.

(A) Cxcl1 secretion from the supernatant of HRI+/+ or HRI–/– BMDMs stimulated for 4 hours with LPS (1 μg/ml), flagellin (100 ng/ml), or Pam3CSK4 (1 μg/ml), as measured by ELISA. (B and C) Il6 (B) and Ifnβ (C) expression in HRI+/+ or HRI–/– BMDMs treated with LPS (100 ng/ml) for the indicated times, as measured by qPCR. (D) Ifnβ expression in HRI+/+ or HRI–/– BMDMs treated with poly(I:C) (1 μg/ml) for the indicated times, as measured by qPCR. (E and F) Cxcl1 (E) and Ifnβ expression (F) in HRI+/+ and HRI–/– BMDMs after the indicated stimulations, as measured by qPCR. (G to I) Total lung cells (G) and inflammatory monocytes (H) from HRI+/+, HRI+/–, and HRI–/– mice infected with Puerto Rico/8/34 (H1N1) influenza A virus (IAV) at 6 days after infection, as measured by flow cytometry. (I) Representative micrographs (magnification 40×) of H&E-stained lungs from HRI+/+, HRI+/–, and HRI–/– mice infected as in (G) and (H). (J and K) Secretion of IL-1β from the supernatant of HRI+/+ and HRI–/– BMDMs stimulated with LPS (100 ng/ml) for 3 hours alone or subsequently stimulated with 10 μM nigericin or 5mMATP for 45min (J) and BMDMs primed for 4 hours with LPS (100 ng/ml) in Opti-MEM and stimulated with Fugene alone or transfected with poly(dA:dT) for 16 hours (K), as measured by ELISA. (L) HRI+/+ and HRI–/– BMDMs were stimulated for 3 hours with LPS (100 ng/ml) and stimulated with 10 μM nigericin for 45min, then analyzed by blue native PAGE. Bar graphs represent means ± SD from three to five independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 (one-way ANOVA followed by Tukey multiple-comparisons test).
We then tested whether the NLRP3 inflammasome required HRI for signaling; NLRP3 differs from other PRMs tested so far, in that NLRP3 inflammasome assembly triggers rapid cell death and does not induce downstream transcriptional reprogramming. LPS-dependent priming of Hri+/+ and Hri–/– BMDMs, which relies on the TLR4/MyD88 axis, resulted in comparable upregulation of NLRP3 and pro–IL-1β in the lysates of Hri+/+ and Hri–/– BMDMs (fig. S29A), in agreement with the results above. After LPS priming, stimulation with adenosine triphosphate (ATP) or nigericin yielded reduced release of mature IL-1β and cleaved caspase-1 into the supernatant of Hri–/– BMDMs (fig. S29A). In agreement with this result, we observed decreased secretion of IL-1β (Fig. 5J) and IL-18 (fig. S29B) in Hri–/– BMDMs. AIM2 inflammasome activity was also reduced in Hri–/– BMDMs (Fig. 5K and fig. S30). However, cell death induced by LPS transfection, which depends on the caspase-11 inflammasome (23), was unaffected in Hri–/– BMDMs, whereas caspase-1 activation and IL-1β release, which requireNLRP3 (24),were similarly blunted (fig. S31). NLRP3 activation results in the assembly of a well-structured oligomeric inflammasome complex that can be resolved using native gel electrophoresis. Interestingly, stimulation of BMDMs with nigericin resulted in blunted assembly of endogenousNLRP3 inflammasomes in Hri–/– cells (Fig. 5L). Activation of the NLRP3 inflammasome further licenses assembly of ASC (apoptosis-associated speck-like protein containingCARD)prion-like polymers (25), which form specks. The endogenous ASC fibers and specks were reduced in Hri–/– BMDMs stimulated with nigericin (fig. S32).SYPRO Orange hydrophobicity measurements of NLRP3 inflammasome complexes after nigericin stimulation revealed greater hydrophobicity of NLRP3 inflammasomes in Hri–/– BMDMs (fig. S33), suggesting improper folding. However, ISRIB treatment had no impact on NLRP3 activation (fig. S34), which implies that HRI deficiency might induce a cellular state that affects NLRP3 inflammasome assembly but that the eIF2α/HSPB8 arm of the HRI signaling axis is likely not involved in the assembly or stability of the NLRP3 inflammasome. This is not unexpected, given that the NLRP3-ASC complex rapidly leads to pyroptotic cell death and does not induce signaling that modulates transcription. Together, these results reveal thatHRI regulates NLRP3/ASC inflammasome assembly and function, although the underlying mechanism of how this regulation occurs remains to be elucidated.
HRI controls the cellular response to prefibrillar PRM adaptors and α-synuclein
We noticed that the PRM pathways regulated by HRI share the property that their adaptor proteins (RIP2 for NOD1/2, MAVS for RIG-I/MDA-5, TRIF for TLR3/TLR4, ASC forNLRP3/AIM2) can form amyloid-like fibrils in vitro. To begin elucidating the molecular requirements that trigger activation of the HRI/eIF2α/HSPB8 signaling axis and to test directly whether enforced assembly of these adaptors was sufficient to induce HRI-dependent signaling, we transfected scramble CTR versus HRI KD human embryonic kidney (HEK) 293T cells with various PRM adaptors. Overexpression of RIP2, MAVS, and TRIF resulted in an HRI-dependent induction of IL8 and HSPB8 (Fig. 6, A and B), which suggests that the assembly of PRM signalosome adaptors might be an essential step for HRI activation. In contrast, overexpression of MyD88, which assembles “myddosomes” with well-defined stoichiometry that, contrary to “TRIFosomes,” do not form amyloid-like fibrils (26), induced IL8 in an HRI-independent manner and did not up-regulate HSPB8 (Fig. 6, A and B). Overexpression of ASC induced neither IL8 nor HSPB8 (Fig. 6, A and B), in agreement with our observation (see above) that NLRP3 inflammasome assembly is not controlled by the eIF2α/HSPB8 arm of HRI signaling. Moreover, we observed that necroptosis signaling, which involves amyloid-like fibrillar assembly of RIPK1/3 heterodimers (27), was also regulated by HRI (fig. S35). In agreement with this finding, overexpression of α-synuclein, a protein that forms toxic amyloid fibrils that are a pathological hallmark of Parkinson’s disease (28), induced ATF3, CHOP, and HSPB8 (Fig. 6, C to E) expression in an HRI-dependent manner. In the neuroblastoma cell line SH-SY5Y, HRI silencing induced accumulation of dimers of endogenous α-synuclein phosphorylated at Ser129 (Fig. 6F), which is indicative of misfolding (29). Together, these data suggest that HRI-dependent signaling may be essential for controlling the assembly and/or stability of amyloid-like fibrils in the cytosol.
Fig. 6. HRI controls the cellular response to prefibrillar PRM adaptors and α-synuclein.

(A and B) IL8 (A) and HSPB8 (B) expression in HEK293T cells transduced with lentiviral particles targeting a scrambled sequence (SC) and HRI (shHRI) and transfected with the indicated plasmids. (C to E) ATF3 (C), CHOP (D), and HSPB8 (E) expression in SC and shHRI HEK293T cells transfected with the indicated plasmids. (F) SC and shHRI SH-SY5Y cells treated with 5 mM MG132 for 4 hours and analyzed by Western blotting. Graphs represent means ± SD from three to five independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Concluding remarks
Our data reveal that HRI controls the formation and folding of large PRM signalosomes in the cytosol, thereby affecting downstream innate immune signaling. Because HRI differentially regulates PRM signaling, the protein may be sensitive to specific molecular superstructures. This idea is supported by the finding that HRI-regulated PRMs rely on adaptor proteins that, at least in vitro, assemble filamentous structures. Considering that HRI is a stress-sensitive kinase of the ISR pathway, it is tempting to speculate that potentially toxic molecular superstructures, such as self-assembling amyloid-like fibrils or filaments, are direct activators of the HRI signaling axis. If this were the case, HRI could be involved in detecting and restricting the accumulation of cytosolic protein aggregates that are associated with neurodegenerative diseases, as hinted by our preliminary results with α-synuclein. Future biochemical studies should delineate the detailed nature of the (supra-)molecular structures that activate HRI.
The HRI/eIF2α/HSPB8 signaling axis shares a remarkable homology with the PERK-dependent branch of the UPR, which regulates protein folding in the ER. Indeed, the eIF2α kinase PERK senses the accumulation of misfolded proteins in the ER and is activated after dissociation with HSPA5, and earlier studies suggest the presence of a similar mechanism of regulation between the chaperone p58 and PKR (30). This pathway stimulates the up-regulation of stress-associated genes, such as the ER chaperone HSPA5 that encodes BiP/GRP78. We thus propose that HRI and HSPB8 represent functional homologs of PERK and BiP, respectively, and that the HRI/ eIF2α/HSPB8 signaling axis detects certain forms of cytosolic UPR (cUPR).
Materials and methods
Reagents and stimulations
To induce amino acid starvation, cells were rinsed three times with PBS and incubated in Krebs Ringer bicarbonate (KRB) buffer (118.5 mM NaCl, 4.74 mM KCl, 1.18 mM KH2PO4, 23.4 mM NaHCO3, 5 mM glucose, 2.5 mM CaCl2, and 1.18 mM MgSO4, adjusted to pH 7.6 by titration with 1 N NaOH). Unless stated, chemicals and ligands were all obtained from Sigma or Invivogen, respectively. 3′cGAMP or c-di-GMP was delivered into cells by diluting ligands in digitonin permeabilization buffer [5 μg/ml for poly(dA:dT) and poly(I:C), 2 μg/ml for dsRNA] as described (31). For activation of AIM2, poly (dA:dT) was delivered using/FuGene. Supernatant was collected 16 hours after stimulation unless specified.
Cell lines
HeLa, HEK293T, HCT116, MEFs, L929, and SH-SY5Y cells were all cultured in DMEM with 10% FBS and 1% penicillin and streptomycin. Primary MEFs were prepared as described (32). Lentiviral knockdown was performed using the targeting sequences in table S1 as described (33). Bone marrow–derived macrophages (BMDMs) were prepared from the femurs and tibia of mice by obtaining bone marrow and incubating cells in RPMI with 20% conditioned media from L929 cells, 10% FBS, 1% penicillin and streptomycin, and 2 mM l-glutamine for 7 days. EIF2α knockin (KI) MEFs were obtained from R. J. Kaufman. ATF4 knockout MEFs were provided by R. Wek and were grown in DMEM with 10% FBS, 1% penicillin and streptomycin, and supplemented with 55 μM β-mercaptoethanol, nonessential amino acids (NEAA), and essential amino acids. CRISPR/CAS9 knockout cells were generated using the CRISPR-Cas9 system as described (34). THP-1 cells were grown in RPMI with 10% FBS supplemented with penicillin/streptomycin, NEAA, and 2-mercaptoethanol.
Mice
Hri+/– breeder mice were obtained from a previous study (6). Mice were bred and housed under SPF conditions at the Division of Comparative Medicine, University of Toronto. All experiments were performed with 7- to 11-week-old littermate mice from HRI+/– crosses. All mice experiments were approved by the local Animal Ethics Review Committee. Genotyping was performed with the primers shown in table S2.
Bacterial strains and in vitro bacterial infections
Infections with Shigella flexneri, Salmonella Typhimurium, and Listeria monocytogenes were performed as described (9, 10). For Shigella infections where the time is not indicated, infections were performed for 4 hours. For Listeria infections of BMDMs, plates were centrifuged at 500g for 10 min after addition of bacteria, incubated at 37°C/5% CO2 for 30 min, incubated with fresh gentamycin-containing medium (30 mg/ml), then left for an additional 2 hours. To measure bacterial replication, cells were infected as described above and lysed with PBS containing 0.1% Triton; lysate was serial-diluted and plated. Shigella conditioned medium was produced by using stationary phase cultures, pelleting bacteria by centrifugation (10,000g for 5 min) and filtering with a 0.22-μm syringe filter. Neisseria gonorrhoeae conditioned medium was purified as described (35).
Viral infections
Sendai virus and HSV were provided by D. Garcin and D. Boutolleau, respectively. Cells were incubated with Sendai virus (SeV at 40 HA/ml) or HSV (MOI of 5) diluted in serum-free DMEM and 2 hours later replaced with DMEM with 20% calf serum and antibiotics, incubated at 37°C. For influenza experiments, mice were intranasally (in 25 μl PBS) infected with a sublethal dose (50 plaque-forming units) with influenza A/Puerto Rico/8/34 virus provided by J. A. McCullers. All experiments were performed according to the animal research ethics board of McGill University.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Measurements of gene expression (unless otherwise specified) were performed by RT-qPCR. RT-PCR analysis was performed with SYBR green reagents as described (9). Murine and human qPCR primers used are listed in tables S3 and S4, respectively. For influenza infections, RNA was isolated using Qiazol reagent (Qiagen) and AllIn-One RT MasterMix (abm) was used for cDNA synthesis. Table S5 indicates viral primers.
Western blots
Unless otherwise indicated, cells were lysed using RIPA. Lysates were centrifuged at 13,000g to separate out membrane fractions versus insoluble fractions and cytoplasmic proteins versus soluble fractions. The membrane fractions were resuspended in RIPA buffer with Laemmli blue loading buffer and boiled to obtain the insoluble fractions. Samples were run on acrylamide gels and transferred onto PVDF. Membranes were blocked with 5% milk in TBST and incubated with the indicated antibodies: mouse anti-tubulin (#T5168, Sigma, 1/10,000 dilution), rabbit anti-eIF2α (#9722S, NEB, 1/1000), rabbit anti-phospho-eIF2α (#9721, NEB, 1/1000), rabbit anti-HRI (#07–226, Upstate, 1/5000), anti-HRI polyclonal (6) (1/3000) rabbit anti-GCN2 (#ab137543, Abcam, 1/1000), monoclonal anti-HA tag (#G036, Abcam, 1/1000), monoclonal anti-Flag-Tag (#F3165, Sigma, 1/1000), rabbit anti-V5-Tag (#13202, NEB, 1/1000), rabbit anti-HSPB8 (#3059, cell signaling, 1/500), rabbit anti-HSP90 (#ab13492, Abcam, 1/3000), rabbit anti-Hsc70 (#ab51052, Abcam, 1/1000), rabbit anti-ERK (#9102, NEB, 1/4000), mouse anti-phospho-ERK (#9106, NEB, 1/4000), mouse anti-IkBα (#4814, NEB, 1/4000), rabbit anti-IkBα (# 9242, NEB, 1/2000), mouse anti-phospho-IkBα (#9246, NEB, 1/2000),rabbit anti-GAPDH (#G9545,Sigma,1/20,000), mouseanti-caspase-1(p20) (#AG-20B-0042, Adipogen, 1/4000), mouse anti-NLRP3 (#AG-20B-0014, Adipogen, 1/4000), goat anti-mouse IL-1β (#AF-401-NA, R&D Systems, 1/1000), rabbit anti-IRF3 (#4302, NEB, 1/2000), rabbit anti-phospho-IRF3 (Ser396) (#4947, NEB), rabbit anti-NAK/TBK1 (EP611Y) (#ab40676, Abcam, 1/4000), rabbit anti-phospho-TBK1 (Ser172) (EPR2867(2)) (#ab109272, Abcam, 1/2000) or rabbit anti-phospho-TBK1/NAK (Ser172) (#5483, NEB, 1/2000), rabbit anti-MAVS (#4983, NEB, 1/2000) and rabbit anti-MAVS (M-300) (#sc-68881, Santa Cruz, 1/1000), rabbit anti-STING/TMEM173 (#19851–1-AP, Proteintech, 1/2000), rabbit anti-ASC mouse specific (D2W8U) (#67824, NEB, 1/2000), rabbit polyclonal anti-ATF3 (H-90) (#sc22978, Santa Cruz, 1/1000), rabbit anti-ATF4 (D4B8) (#11815, NEB, 1/1000), mouse monoclonal anti-puromycin (4G11) (#MABE342, Millipore, 1/5000), rabbit anti-α-synuclein phosphoS129 (#ab51253, Abcam, 1/3000), mouse anti-α-synuclein (#ab1903, Abcam, 1/3000).
Immunofluorescence
Preparation of samples for immunofluorescence assays was performed as described (11). Antibodies were used as follows: anti-Tia-1, 1:100 (Santa Cruz); anti-p65, 1:100 (Abcam); anti-ASC, 1:400 (NEB).
Surface sensing of translation (SUnSEt) assay
SUnSEt assays were conducted as described (11).
Coimmunoprecipitation and SYPRO Orange experiments
For coimmunoprecipitation experiments, cells were lysed using RIPA buffer on ice and subsequently centrifuged at 13,000g for 3 min. Protein G beads (Pierce) were washed twice with RIPA buffer and centrifuged at 2000g for 10 min before adding lysate and the indicated antibodies. The immunoprecipitation reaction was incubated overnight at 4°C. Next, samples were centrifuged at 10,000g for 5 min, washed with RIPA buffer, resuspended in RIPA buffer with Laemmli blue loading buffer and boiled for 10 min. For hydrophobicity measurements, the immunoprecipitated samples were washed three times with PBS and resuspended in PBS. Samples were then incubated with a final concentration of 5× SYPRO Orange and fluorescence was measured using a RT-qPCR machine (300 nm excitation/470 nm emission) between 20° and 100°C. Hydrophobicity measurements of NOD1 were performed by transfecting HEK293T cells with NOD1-HA plasmid. Each dot on the scatterplots represents a data point from a unique experiment; n = 3 to 5 experiments were performed for each SYPRO Orange assay.
Intraperitoneal injections
Mice were injected intraperitoneally with 50 μg of MDP, 50 μg of FK-156, or 1 μg of LPS. Two hours after injection, mice were killed and whole blood was collected by cardiac puncture. To obtain the peritoneal lavage, 2 ml of PBS was injected into the peritoneal cavity, mixed briefly by massaging and collected. Peritoneal lavage was centrifuged for 10 min at 500g, the supernatant was used for ELISAs and the pelleted cells were used for flow cytometry.
In vivo Citrobacter infections
Citrobacter rodentium strain DBS 100 was cultured in LB medium overnight at 37°C. Bacterial colony-forming units (CFUs) were calculated after inoculation by plating on MacConkey agar. Mice were fasted overnight before being inoculated with 1 × 108 CFU bacteria in 200 μl volume by oral gavage. C. rodentium was obtained from P. Sherman (HSC, Toronto). Mice were killed 4 days after infection and the cecum and blood of mice collected for ELISA experiments.
Flow cytometry
Isolated peritoneal cells were sequentially incubated with the LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (ThermoFisher Scientific), rat anti-mouse CD16/32 monoclonal antibody (clone 43, ThermoFisher), Alexa Fluor 700-conjugated rat anti-mouse Ly-6G (clone RB6–8C5, ThermoFisher), and APC-eFluor 780-conjugated rat anti-mouse CD11b (clone M1/70). For influenza infections, lungs were perfused with 10 ml of PBS, minced and digested in collagenase (150 U/ml) for 1 hour at 37°C and 5% CO2. Single-cell suspensions were obtained by crushing cells through a 70-mm filter and stained with Fixable Viability Dye eFluor506 (eBioscience) for 20 min in PBS at 4°C, washed, then stained with anti-mouse CD16/32 (clone 93, eBioscience) in 0.5% BSA/ PBS to block nonspecific binding for 5 min at 4°C. Finally, cells were stained with BUV395-conjugated rat anti-mouse CD45.2 (clone 104, BDBiosciences),PE-Cy7-conjugated rat anti-mouse CD11b (clone M170, BD Biosciences), Alexa Fluor 700-conjugated rat anti-mouse Ly6G (clone 1A8, BD Biosciences), and PE-conjugated rat antimouse CCR2 (clone 475301, R&D) and FITC-conjugated rat anti-mouse Ly6C (clone AC-21, BD Biosciences) in 0.5% BSA/PBS for 20 min at 4°C. Flow cytometry analysis was performed on an LSRFortessa X-20 Cell Analyzer or LSRFortessa Cell Analyzer (influenza) (BD Biosciences) via FACSDiva acquisition software (BD Biosciences), and analysis performed with FlowJo v10.3 (FlowJo, LLC).
Microarray analysis
Data from a previous report (9) were repurposed.
IFN protein quantification
Quantification of total IFN in the supernatants of BMDMs was performed using B16-Blue IFN α/β reporter cell lines as per the manufacturer’s instructions (Invivogen).
Enzyme-linked immunosorbent assays (ELISAs)
ELISAs were performed according to the manufacturer’s instructions. Cxcl1, IL-1β, IL-6, and murine CCL2 ELISA kits were obtained from R&D Systems. Measurements of IL-1β in response to NLRP3 stimulation and IL-6 measurements in macrophages were performed with kits from Thermofisher Scientific. Murine IFNβ ELISA and IL-18 ELISA kits were obtained from PBL Assay Science and MBL, respectively.
Transfections
For transient expression, transfections were performed with PEI MW25,000. HSPB8-V5 (36) and α-synuclein–GFP plasmids were obtained from Addgene. TIFA-flag was generated by S. D. Gray-Owen. NOD1-HA and NOD2-HA expression vectors were used in a previous study (37).
Luciferase
Luciferase assays were performed as described and adapted to HeLa cells (37).
Organoids
Murine organoid cultures were obtained and grown as described (38).
Histopathology
Lungs were excised and fixed in 10% formalin for 48 hours. Hematoxylin and eosin (H&E) staining was performed by the Histopathology Core of the RI-MUHC of McGill University and scanned up to 40× on the Aperio AT Turbo (Leica).
Blue native PAGE
Cells were lysed using either NP-40 lysis buffer to obtain cytoplasmic proteins and loosely membrane-associated proteins or RIPA to obtain all proteins. After lysis, experiments were performed as described (35).
Semi-denaturing detergent agarose gel electrophoresis (SDD-AGE)
For size estimation of MAVS aggregates, SDD-AGE was performed as described (25).
Cell death assays
Primed BMDMs[Pam3CSK4 (1mg/ml)for 4hours] were transfected with LPS using FuGene. LDH release was measured using LDH Cytotoxicity Detection Kit (Clontech). Percentages were obtained by comparing to Triton-lysed cells (100% cell death) and subtracting untreated controls.
Cell death was also quantified by monitoring propidium iodide (PI) incorporation (5 μg/ml) via fluorescence measurements over 24 hours on a microplate fluorimeter (Fluostar Omega, BMG Labtech).
ASC oligomerization
After stimulation, cells were lysed with cold PBS containing 0.5% Triton X-100 and centrifuged at 6000g for 15 min at 4°C. The pellets were washed twice in PBS and freshly prepared disuccinimidyl suberate (2 mM) was added to the resuspended pellets incubated with mixing for 30 min. The pellets were collected by centrifugation and redissolved in SDS-PAGE sample buffer.
Statistics
For analysis of data in fig. S16, B to E, a Student t test was used. For multiple comparisons, data was analyzed using analysis of variance (ANOVA) with Bonferroni post-test for cellular assays and Tukey post-test for in vivo experiments.
Supplementary Material
ACKNOWLEDGMENTS
Funding:
Supported by Canadian Institutes of Health Research (CIHR) grants PJT-15319 and MOP-12353 (S.E.G.), Agence Nationale de Recherche sur le SIDA (ANRS) and foundation ARC (D.A.), CIHR team grant on chronic inflammation THC-13523 (D.J.P. and S.E.G.), and an Ontario Graduate Scholarship (M.A.N. and J.T.).
Footnotes
Competing interests: R.G.G. and S.D.G. have filed patents concerning the therapeutic potential of heptose 1-phosphate and heptose 1,7-biphosphate.
SUPPLEMENTARY MATERIALS
Data and materials availability:
HRI knockout mice are available from J.J.C. under a material agreement with the Institute of Medical Engineering and Science, Massachusetts Institute of Technology. All data are available in the main text or supplementary materials.
REFERENCES AND NOTES
- 1.Dever TE et al. , Phosphorylation of initiation factor 2a by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68, 585–596 (1992). doi: 10.1016/0092-8674(92)90193-G; [DOI] [PubMed] [Google Scholar]
- 2.Zhang P. et al. , The GCN2 eIF2a kinase is required for adaptation to amino acid deprivation in mice. Mol. Cell. Biol. 22, 6681–6688 (2002). doi: 10.1128/MCB.22.19.6681-6688.2002; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harding HP, Zhang Y, Ron D, Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274 (1999). doi: 10.1038/16729; [DOI] [PubMed] [Google Scholar]
- 4.Ranu RS, Regulation of protein synthesis in rabbit reticulocyte lysates: The hemeregulated protein kinase (HRI) and double stranded RNA induced protein kinase (dRI) phosphorylate the same site(s) on initiation factor eIF-2. Biochem. Biophys. Res. Commun. 91, 1437–1444 (1979). doi: 10.1016/0006-291X(79)91227-0; [DOI] [PubMed] [Google Scholar]
- 5.Yerlikaya A, Kimball SR, Stanley BA, Phosphorylation of eIF2α in response to 26S proteasome inhibition is mediated by the haem-regulated inhibitor (HRI) kinase. Biochem. J. 412, 579–588 (2008). doi: 10.1042/BJ20080324; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.A. P. Han et al. , Heme-regulated eIF2α kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 20, 6909–6918 (2001). doi: 10.1093/emboj/20.23.6909; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McEwen E. et al. , Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 280, 16925–16933 (2005). doi: 10.1074/jbc.M412882200; [DOI] [PubMed] [Google Scholar]
- 8.Ron D, Translational control in the endoplasmic reticulum stress response. J. Clin. Invest. 110, 1383–1388 (2002). doi: 10.1172/JCI0216784; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tattoli I. et al. , Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11, 563–575 (2012). doi: 10.1016/j.chom.2012.04.012; [DOI] [PubMed] [Google Scholar]
- 10.Tattoli I. et al. , Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. 32, 3066–3078 (2013). doi: 10.1038/emboj.2013.234; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsalikis J. et al. , Intracellular Bacterial Pathogens Trigger the Formation of U Small Nuclear RNA Bodies (U Bodies) through Metabolic Stress Induction. J. Biol. Chem. 290, 20904–20918 (2015). doi: 10.1074/jbc.M115.659466; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harding HP et al. , An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619–633 (2003). doi: 10.1016/S1097-2765(03)00105-9; [DOI] [PubMed] [Google Scholar]
- 13.Thulasiraman V. et al. , Evidence that Hsc70 negatively modulates the activation of the heme-regulated eIF-2α kinase in rabbit reticulocyte lysate. Eur. J. Biochem. 255, 552–562 (1998). doi: 10.1046/j.1432-1327.1998.2550552.x; [DOI] [PubMed] [Google Scholar]
- 14.Gaudet RG et al. , Innate Recognition of Intracellular Bacterial Growth Is Driven by the TIFA-Dependent Cytosolic Surveillance Pathway. Cell Rep. 19, 1418–1430 (2017). doi: 10.1016/j.celrep.2017.04.063; [DOI] [PubMed] [Google Scholar]
- 15.Girardin SE et al. , Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300, 1584–1587 (2003). doi: 10.1126/science.1084677; [DOI] [PubMed] [Google Scholar]
- 16.Girardin SE et al. , CARD4/Nod1 mediates NF-κB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2, 736–742 (2001). doi: 10.1093/embo-reports/kve155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geddes K. et al., Identification of an innate T helper type 17 response to intestinal bacterial pathogens. Nat. Med. 17, 837–844 (2011). doi: 10.1038/nm.2391; [DOI] [PubMed] [Google Scholar]
- 18.Kim YG et al. , The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 34, 769–780 (2011). doi: 10.1016/j.immuni.2011.04.013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bahnan W. et al. , The eIF2α Kinase Heme-Regulated Inhibitor Protects the Host from Infection by Regulating Intracellular Pathogen Trafficking. Infect. Immun. 86, e00707–17 (2018). doi: 10.1128/IAI.00707-17; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P, Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb. Perspect. Biol. 5, a013169 (2013). doi: 10.1101/cshperspect.a013169; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Celli J, Tsolis RM, Bacteria, the endoplasmic reticulum andthe unfolded protein response: Friends or foes? Nat. Rev. Microbiol. 13, 71–82 (2015). doi: 10.1038/nrmicro3393; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walter P, Ron D, The unfolded protein response: From stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011). doi: 10.1126/science.1209038; [DOI] [PubMed] [Google Scholar]
- 23.Crowley SM, Vallance BA, Knodler LA, Noncanonical inflammasomes: Antimicrobial defense that does not play by the rules. Cell. Microbiol. 19, e12730 (2017). doi: 10.1111/cmi.12730; [DOI] [PubMed] [Google Scholar]
- 24.Kayagaki N et al., Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011). doi: 10.1038/nature10558; pmid: 22002608 [DOI] [PubMed] [Google Scholar]
- 25.Cai X. et al. , Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156, 1207–1222 (2014). doi: 10.1016/j.cell.2014.01.063; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bryant CE, Symmons M, Gay NJ, Toll-like receptor signalling through macromolecular protein complexes. Mol. Immunol. 63, 162–165 (2015). doi: 10.1016/j.molimm.2014.06.033; [DOI] [PubMed] [Google Scholar]
- 27.Li J. et al. , The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 (2012). doi: 10.1016/j.cell.2012.06.019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dikiy I, Eliezer D, Folding and misfolding of α-synuclein on membranes. Biochim. Biophys. Acta 1818, 1013–1018 (2012). doi: 10.1016/j.bbamem.2011.09.008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oueslati A, Implication of a-Synuclein Phosphorylation at S129 in Synucleinopathies: What Have We Learned in the Last Decade? J. Parkinsons Dis. 6, 39–51 (2016). doi: 10.3233/JPD-160779; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polyak SJ, Tang N, Wambach M, Barber GN, Katze MG,The P58 cellular inhibitor complexes with the interferon-induced, double-stranded RNA-dependent protein kinase, PKR, to regulate its autophosphorylation and activity. J. Biol. Chem. 271, 1702–1707 (1996). doi: 10.1074/jbc.271.3.1702; [DOI] [PubMed] [Google Scholar]
- 31.Woodward JJ, Iavarone AT, Portnoy DA, c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705 (2010). doi: 10.1126/science.1189801; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Travassos LH et al. , Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62 (2010). doi: 10.1038/ni.1823; [DOI] [PubMed] [Google Scholar]
- 33.Sorbara MT et al. , The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity 39, 858–873 (2013). doi: 10.1016/j.immuni.2013.10.013; [DOI] [PubMed] [Google Scholar]
- 34.Sorbara MT et al. , Complement C3 Drives Autophagy-Dependent Restriction of Cyto-invasive Bacteria. Cell Host Microbe 23, 644–652.e5 (2018). doi: 10.1016/j.chom.2018.04.008; [DOI] [PubMed] [Google Scholar]
- 35.Gaudet RG et al. , Cytosolic detection of the bacterial metabolite HBP activates TIFA-dependent innate immunity. Science 348, 1251–1255 (2015). doi: 10.1126/science.aaa4921; [DOI] [PubMed] [Google Scholar]
- 36.Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH,HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2a phosphorylation. J. Biol. Chem. 284, 5523–5532 (2009). doi: 10.1074/jbc.M807440200; [DOI] [PubMed] [Google Scholar]
- 37.Lee J. et al. , pH-dependent internalization of muramyl peptides from early endosomes enables Nod1 and Nod2 signaling. J. Biol. Chem. 284, 23818–23829 (2009). doi: 10.1074/jbc.M109.033670; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tattoli I. et al. , NLRX1 Acts as an Epithelial-Intrinsic Tumor Suppressor through the Modulation of TNF-Mediated Proliferation. Cell Rep. 14, 2576–2586 (2016). doi: 10.1016/j.celrep.2016.02.065; [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
HRI knockout mice are available from J.J.C. under a material agreement with the Institute of Medical Engineering and Science, Massachusetts Institute of Technology. All data are available in the main text or supplementary materials.