

ABSTRACT
The prevalence of drug-resistant Mycobacterium tuberculosis infections has prompted extensive efforts to exploit new drug targets in this globally important pathogen. ClpC1, the unfoldase component of the essential ClpC1P1P2 protease, has emerged as one particularly promising antibacterial target. However, efforts to identify and characterize compounds that impinge on ClpC1 activity are constrained by our limited knowledge of Clp protease function and regulation. To expand our understanding of ClpC1 physiology, we employed a coimmunoprecipitation and mass spectrometry workflow to identify proteins that interact with ClpC1 in Mycolicibacterium smegmatis, a surrogate for M. tuberculosis. We identify a diverse panel of interaction partners, many of which coimmunoprecipitate with both the regulatory N-terminal domain and the ATPase core of ClpC1. Notably, our interactome analysis establishes MSMEI_3879, a truncated gene product unique to M. smegmatis, as a novel proteolytic substrate. Degradation of MSMEI_3879 by ClpC1P1P2 in vitro requires exposure of its N-terminal sequence, reinforcing the idea that ClpC1 selectively recognizes disordered motifs on substrates. Fluorescent substrates incorporating MSMEI_3879 may be useful in screening for novel ClpC1-targeting antibiotics to help address the challenge of M. tuberculosis drug resistance.
IMPORTANCE Drug-resistant tuberculosis infections are a major challenge to global public health. Much effort has been invested in identifying new drug targets in the causative pathogen, Mycobacterium tuberculosis. One such target is the ClpC1 unfoldase. Compounds have been identified that kill M. tuberculosis by disrupting ClpC1 activity, yet the physiological function of ClpC1 in cells has remained poorly defined. Here, we identify interaction partners of ClpC1 in a model mycobacterium. By building a broader understanding of the role of this prospective drug target, we can more effectively develop compounds that inhibit its essential cellular activities.
KEYWORDS: Clp protease, Mycobacterium, interactome, proteolysis, proteomics
INTRODUCTION
Tuberculosis is responsible for greater global mortality than any other bacterial pathogen, causing an estimated 1.5 million deaths in 2020 alone (1). While rates of infection and mortality have declined over the past decade, the prevalence of multidrug-resistant tuberculosis infections remains a persistent challenge to global public health. There is consequently an urgent need to develop new drugs and exploit new drug targets in the causative pathogen, Mycobacterium tuberculosis. The essential mycobacterial Clp proteases have emerged as one promising class of targets (2–7).
Clp proteases are well-studied proteolytic machines that unravel and hydrolyze native protein substrates (8, 9). These large oligomeric complexes consist of a hexameric Clp unfoldase that recognizes substrates, unfolds them using energy from ATP hydrolysis, and spools them into an associated peptidase barrel for degradation (10). The mycobacterial Clp peptidase is composed of two paralogous heptamers, ClpP1 and ClpP2, that assemble into a catalytically active hetero-oligomeric ClpP1P2 tetradecamer (11–16). Two alternative unfoldases, ClpC1 and ClpX, dock on the ClpP2 face of the peptidase to form the functional Clp protease (13, 14, 17, 18). Substrates are recognized by directly interacting with the unfoldase (19–21), with the aid of proteolytic adaptors (22, 23), or via posttranslational phosphorylation (24–26).
All components of M. tuberculosis Clp proteases are strictly essential for viability (19, 27–31), making these enzymes attractive targets for novel drug development. Multiple classes of antimicrobials inhibit or dysregulate Clp protease activity by targeting the peptidase (2, 5, 13, 14, 29, 32–34). However, several compound classes have been shown to cross-target human mitochondrial CLPP (35–37), which complicates efforts to develop ClpP-targeting pharmacophores into viable antibacterial therapeutics. By contrast, ClpC1 has no direct homologs in animals, and thus provides an avenue for inhibiting Clp protease activity in the pathogen with lower risk of off-target effects in humans. Cyclic peptides have been identified that kill M. tuberculosis by targeting ClpC1, including cyclomarin A, metamarin, lassomycin, ecumicin (ECU) and rufomycin (RUF), all of which bind to the N-terminal domain (NTD) (3, 4, 38–43).
Bacterial Clp proteases participate in various physiological processes, including protein quality control, response to stress, and regulation of virulence (19, 44–53). Although Clp proteases are known to be essential in mycobacteria, the breadth of their physiological roles is poorly understood. One path to elucidating the functions of Clp proteases is to identify their proteolytic substrates and interaction partners. Prior studies have used targeted capture approaches to identify Clp protease substrates in several bacteria, including Escherichia coli, Bacillus subtilis, Staphylococcus aureus, and Caulobacter crescentus (48, 52, 54–56).
In this study, we use coimmunoprecipitation and mass spectrometry to identify cellular proteins that interact with the ClpC1 unfoldase in Mycolicibacterium smegmatis, a nonpathogenic relative of M. tuberculosis. Importantly, we identify and characterize a novel substrate of the ClpC1P1P2 protease in M. smegmatis.
RESULTS
Identification of interaction partners of full-length ClpC1.
We sought to design a set of ClpC1 constructs that maximized our likelihood of identifying diverse interaction partners. Some interaction partners likely bind to wild-type ClpC1 (ClpC1WT) irrespective of its nucleotide-bound state: for example, to the flexible NTD. However, substrate binding to wild-type AAA+ unfoldases is often transient, due to unfolding/degradation, and ATP dependent (8, 9, 57–59). To increase our ability to capture ATP-dependent interactions and prevent degradation of bona fide substrates, we mutated conserved Glu residues within the D1 and D2 Walker B motifs to Gln (E288Q and E626Q, respectively), yielding ClpC1EQ (Fig. 1A and B) (60). Analogous mutations in related AAA+ enzymes stabilize ATP binding but impair nucleotide hydrolysis and substrate unfolding (56, 61, 62). To facilitate coimmunoprecipitation with interaction partners, all constructs incorporated a C-terminal 3×FLAG tag (DYKDHDG-DYKDHDI-DYKDDDDK). We confirmed that addition of the C-terminal tag did not impair the ATPase activity of ClpC1 or protease activity of ClpC1P1P2 in vitro (see Fig. S1A and B in the supplemental material). We additionally confirmed that that FLAG-tagged ClpC1 can substitute for endogenous ClpC1 in M. smegmatis, albeit with a growth defect (Fig. S1C to H), indicating that this construct retains at least partial ability to support proteolysis in cooperation with ClpP1P2.
FIG 1.
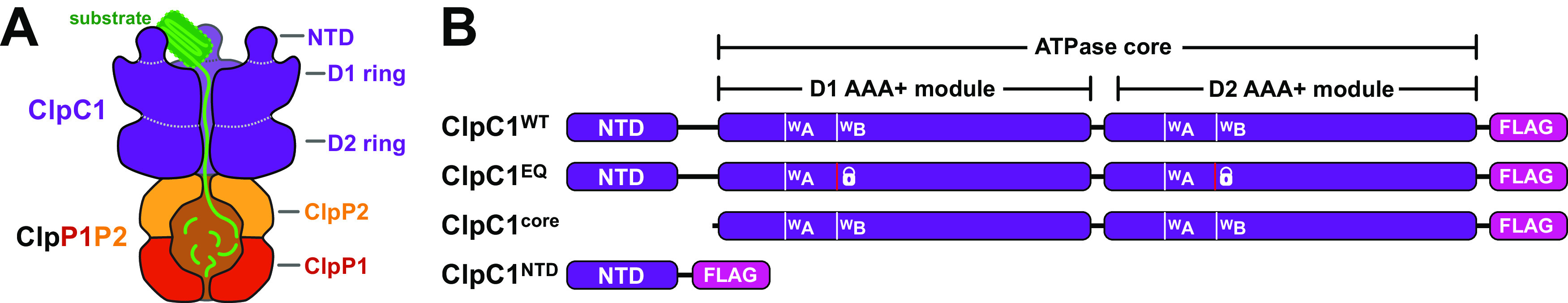
A ClpC1 Walker B double mutant substrate trap was generated in Mycolicibacterium smegmatis (strain ATCC 700084/MC2155). (A) General architecture of the mycobacterial Clp protease. The ATP-dependent unfoldase ClpC1 is responsible for recognizing (via the NTD) and unfolding protein substrates in an ATP-dependent manner (via AAA+ domains D1 and D2), and translocating them into the peptidase barrel composed of catalytic heptamers ClpP1 and ClpP2, wherein degradation occurs. ClpC1EQ associates stably with protein substrates but is unable to unfold or translocate them. (B) Linear sequence models showing the ClpC1 constructs used in this study to identify cellular proteins interacting with specific ClpC1 components such as the NTD and AAA+ core.
ClpC1 constructs, or an empty vector control, were expressed in M. smegmatis MC2155. ClpC1 and interaction partners were coimmunoprecipitated from lysates in the presence of 10 mM ATP using anti-FLAG beads and separated by SDS-PAGE (Fig. 2). Samples were prepared by in-gel trypsin digestion and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using higher-energy collisional dissociation (HCD) fragmentation. Proteomic analysis was performed in the Proteome Discoverer software suite against the M. smegmatis MC2155 proteome using Sequest HT. Proteins observed in at least two of three biological replicates were considered for further analysis. Five proteins were observed in the empty vector control (Fig. 3A), presumably due to nonspecific interactions with beads. Occurrences of these proteins were ignored in all other samples. In total, 163 and 93 proteins were identified in ClpC1WT and ClpC1EQ data sets, respectively (Fig. 3A; see Tables S1 and S2 in the supplemental material).
FIG 2.

Workflow for identification of ClpC1 interaction partners. Mycolicibacterium smegmatis (ATCC 700084/MC2155) cells were grown up to and A600 of ≈0.6 to 1.0, and ClpC1 expression was induced following addition of 28 mM ε-caprolactam. Identification of the interactome was based on gel electrophoresis, trypsin digestion, and LC-MS/MS run using higher-energy collisional dissociation (HCD) fragmentation. All ClpC1 test construct and control experiments were subjected to the same workflow and were carried out in biological replicates.
FIG 3.

Comparative analysis of the ClpC1EQ and ClpC1WT interactome. (A) Bar graph showing the number of proteins identified in the ClpC1EQ and ClpC1WT data sets; (B) Venn diagram showing the number of Mycolicibacterium smegmatis cellular proteins that interacted with ClpC1EQ and ClpC1WT constructs, respectively. The numbers in panels A and B exclude ClpC1 itself. (C) Volcano plot showing enrichment of proteins captured in the M. smegmatis ClpC1 mutant trap versus wild type by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The y axis shows the log2 ratio of normalized scores in the ClpC1EQ data set to those in the ClpC1WT data set, while the x axis shows log2 average number of PSMs for the proteins. Red circles represent proteins enriched in ClpC1EQ with a P value of <0.05 by Student’s t test.
We expected substrates to bind more stably to ClpC1EQ than to ClpC1WT because the wild-type enzyme can presumably unfold substrates in the pulldown conditions. Within the ClpC1EQ data set, 28 proteins matched these criteria and were absent from the ClpC1WT data (Fig. 3B and Table 1). These include cell wall synthesis protein Wag31, succinate dehydrogenase iron-sulfur protein SdhB, aconitate hydratase A (AcnA), and ATP synthase epsilon chain AtpC. There were 65 proteins observed in both the ClpC1WT and ClpC1EQ data sets (Fig. 3A and B). A semiquantitative comparison indicates that 20 of these were enriched in ClpC1EQ over ClpC1WT by at least a ratio of 2:1 in average normalized score (Fig. 3C; Table S2). However, only three proteins were enriched significantly (with a P value of <0.05): the DNA repair and stress response protein RecA, electron transport chain component ubiquinol-cytochrome c reductase cytochrome b subunit (MSMEG_4263), and a MarR family protein transcriptional regulator (MSMEG_2538) (Fig. 3B). The co-occurrence of these in ClpC1WT and ClpC1EQ data sets suggests that these interactions occur stably regardless of ATPase and unfolding activity, as would be expected for adaptors, regulators, or other non-substrate interactors.
TABLE 1.
Proteins that coimmunoprecipitate with ClpC1EQ but not ClpC1WT
| UniProt accession no. | Gene name(s) | Protein description | Normalized avg scorea | Avg no. of PSMs |
|---|---|---|---|---|
| A0R461 | MSMEG_5715, MSMEI_5564 | Bac_luciferase domain-containing protein | 0.390 | 10.667 |
| A0QWT6 | MSMEG_3058, MSMEI_2982 | Lipoprotein | 0.144 | 3.000 |
| A0QSP8 | rplM, MSMEG_1556, MSMEI_1519 | 50S ribosomal protein L13 | 0.085 | 6.667 |
| A0QSD2 | rplD, MSMEG_1437, MSMEI_1401 | 50S ribosomal protein L4 | 0.048 | 5.000 |
| A0QRD2 | MSMEG_1073, MSMEI_1041 | Oxidoreductase, short-chain dehydrogenase/reductase family protein | 0.091 | 9.333 |
| A0R1Z9 | atpC, MSMEG_4935, MSMEI_4808 | ATP synthase epsilon chain (ATP synthase F1 sector epsilon subunit) | 0.060 | 3.000 |
| A0QVU2 | MSMEG_2695, MSMEI_2629 | 35-kDa protein | 0.083 | 2.500 |
| A0QWK5 | ppiB, MSMEG_2974, MSMEI_2900 | Peptidyl-prolyl cis-trans isomerase (PPIase) | 0.085 | 2.500 |
| A0QZ33 | MSMEG_3880, MSMEI_3790 | Nitrilase/cyanide hydratase and apolipoprotein N-acyltransferase | 0.130 | 17.500 |
| A0R1H2 | MSMEG_4752 | Uncharacterized protein | 0.062 | 3.333 |
| A0R5K8 | MSMEG_6227, MSMEI_6066 | Transcriptional regulator, PadR family protein | 0.057 | 8.000 |
| A0R006 | wag31, ag84, MSMEG_4217, MSMEI_4119 | Cell wall synthesis protein Wag31 (antigen 84) | 0.075 | 5.667 |
| A0R2B0 | MSMEG_5048, MSMEI_4921 | Uncharacterized protein | 0.035 | 5.500 |
| A0QPX3 | MSMEG_0550, MSMEI_0535 | Sulfonate binding protein | 0.085 | 3.500 |
| A0QSG6 | rpsE, MSMEG_1472, MSMEI_1436 | 30S ribosomal protein S5 | 0.075 | 12.333 |
| A0R1B5 | MSMEG_4692, MSMEI_4575 | Uncharacterized protein MSMEG_4692/MSMEI_4575 | 0.082 | 4.500 |
| A0QTM5 | pcaD, MSMEG_1897, MSMEI_1857 | 3-Oxoadipate enol-lactonase (EC 3.1.1.24) | 0.102 | 4.500 |
| A0QU11 | MSMEG_2037, MSMEI_1991 | Bac_luciferase domain-containing protein | 0.030 | 3.000 |
| A0QS62 | rplJ, MSMEG_1364, MSMEI_1325 | 50S ribosomal protein L10 | 0.078 | 8.500 |
| A0QX20 | acnA, can, MSMEG_3143, MSMEI_3062 | Aconitate hydratase A (ACN) (aconitase) (EC 4.2.1.3) | 0.055 | 14.500 |
| A0QR90 | MSMEG_1029, MSMEG_2309 | Probable transcriptional regulatory protein | 0.095 | 7.500 |
| A0QT07 | sdhB, MSMEG_1669 | Succinate dehydrogenase, iron-sulfur protein (EC 1.3.99.1) | 0.059 | 6.500 |
| A0QS46 | rplA, MSMEG_1347, MSMEI_1309 | 50S ribosomal protein L1 | 0.053 | 12.000 |
| A0QZJ0 | MSMEG_4042, MSMEI_3947 | Transcriptional regulator, GntR family protein | 0.050 | 6.000 |
| A0QS97 | rpsG, MSMEG_1399, MSMEI_1361 | 30S ribosomal protein S7 | 0.082 | 14.500 |
| Q3L885 | pks, MSMEG_0408, MSMEI_0398 | Polyketide synthase (type I modular polyketide synthase) | 0.023 | 43.000 |
| A0QSG5 | rplR, MSMEG_1471, MSMEI_1435 | 50S ribosomal protein L18 | 0.032 | 6.500 |
| A0R616 | MSMEG_6391, MSMEI_6223 | Propionyl-CoA carboxylase beta chain (EC 6.4.1.3) | 0.039 | 9.500 |
The normalized score is the percentage of the total ion score of a single run attributable to a given protein. Normalized scores were averaged across replicates.
We additionally cross-referenced the 191 proteins observed across ClpC1WT and ClpC1EQ data sets with M. tuberculosis Clp protease interaction partners identified elsewhere by bacterial two-hybrid screening (23) or by LC-MS/MS upon knockdown of ClpP1 and ClpP2 (21). Thirty M. smegmatis proteins in our full-length ClpC1 interactome have M. tuberculosis homologs identified in these prior studies (see Table 3 below).
TABLE 3.
ClpC1 interaction partners for which M. tuberculosis homologs were identified in prior ClpC1 interaction studies
| UniProt accession no. | Gene name(s) | Protein description | Identified in prior studies | Mean normalized score | Mean no. of PSMs | Data set(s) observed in |
|---|---|---|---|---|---|---|
| A0QQD0 | dnaJ, dnaJ1, MSMEG_0711, MSMEI_0694 | Chaperone protein DnaJ1 | ClpC1/P2 KDa | 0.676 | 38.500 | WT, EQ, NTD, CORE |
| A0R203 | atpFH, atpF, atpH, MSMEG_4939, MSMEI_4812 | ATP synthase subunit b-delta | ClpC1/P2 KDa | 0.568 | 51.667 | WT, EQ, NTD, CORE |
| I7G417 | MSMEI_3879 | 5-Oxoprolinase | ClpC1/P2 KDa | 0.480 | 20.500 | WT, EQ, NTD, CORE |
| A0R3L1 | MSMEG_5512 | Magnesium chelatase | ClpC1/P2 KD,a BACTHb | 0.410 | 28.333 | WT, EQ, NTD, CORE |
| A0QQA8 | MSMEG_0688, MSMEI_0671 | Aspartate aminotransferase | ClpC1/P2 KDa | 0.292 | 30.500 | WT, NTD, CORE |
| A0R079 | glnA, glnA1, MSMEG_4290, MSMEI_4189 | Glutamine synthetase (GS) | ClpC1/P2 KDa | 0.162 | 14.000 | WT, NTD |
| A0R617 | pks13, MSMEG_6392, MSMEI_6224 | Polyketide synthase | ClpC1/P2 KDa | 0.160 | 35.333 | WT, NTD |
| A0R200 | atpD, MSMEG_4936, MSMEI_4809 | ATP synthase subunit beta | ClpC1/P2 KDa | 0.158 | 11.667 | WT, EQ, NTD, CORE |
| A0R0T1 | MSMEG_4497, MSMEI_4386 | PhoH family protein | BACTHb | 0.129 | 9.500 | WT, EQ, NTD, CORE |
| A0QV32 | ffh, MSMEG_2430, MSMEI_2369 | Signal recognition particle protein (fifty-four homolog) | ClpC1/P2 KDa | 0.126 | 39.500 | WT, NTD, CORE |
| A0R202 | atpA, MSMEG_4938, MSMEI_4811 | ATP synthase subunit alpha | ClpC1/P2 KDa | 0.124 | 14.667 | WT, EQ, NTD |
| A0QQV4 | gabD2, MSMEG_0889, MSMEI_0868 | Aldehyde dehydrogenase | ClpC1/P2 KDa | 0.123 | 15.417 | WT, EQ, NTD, CORE |
| A0QQQ1 | sodC, MSMEG_0835, MSMEI_0816 | Superoxide dismutase | ClpC1/P2 KDa | 0.100 | 6.500 | WT, NTD, CORE |
| A0R0T8 | dnaJ2, MSMEG_4504, MSMEI_4392 | Chaperone protein DnaJ2 | ClpC1/P2 KDa | 0.094 | 21.000 | WT, CORE |
| A0QYD6 | ndh, MSMEG_3621, MSMEI_3536 | NADH dehydrogenase (EC 1.6.99.3) | ClpC1/P2 KDa | 0.093 | 27.333 | WT |
| A0QR33 | hemL, MSMEG_0969, MSMEI_0943 | Glutamate-1-semialdehyde 2,1-aminomutase (GSA) | ClpC1/P2 KDa | 0.086 | 18.500 | WT |
| A0QVU2 | MSMEG_2695, MSMEI_2629 | 35-kDa protein | ClpC1/P2 KDa | 0.083 | 2.500 | EQ, NTD |
| A0QSY1 | MSMEG_1642, MSMEI_1603 | ABC transporter, ATP-binding protein | ClpC1/P2 KD,a BACTHb | 0.083 | 30.500 | WT |
| A0QX24 | moxR, MSMEG_3147 | ATPase, MoxR family protein | ClpC1/P2 KDa | 0.073 | 5.500 | WT, EQ, NTD |
| A0QZ40 | tatA, MSMEG_3887, MSMEI_3797 | Sec-independent protein translocase protein TatA | ClpC1/P2 KDa | 0.060 | 4.500 | WT, EQ, NTD |
| A0R1Z9 | atpC, MSMEG_4935, MSMEI_4808 | ATP synthase epsilon chain | ClpC1/P2 KDa | 0.060 | 3.000 | EQ |
| A0QT07 | sdhB, MSMEG_1669, MSMEI_1629 | Succinate dehydrogenase, iron-sulfur protein | ClpC1/P2 KDa | 0.059 | 6.500 | EQ |
| A0QNN8 | pntA, MSMEG_0110, MSMEI_0106 | NAD(P) transhydrogenase, alpha subunit (EC 1.6.1.1) | BACTHb | 0.059 | 18.000 | WT |
| P71533 | secA1, MSMEG_1881, MSMEI_1840 | Protein translocase subunit SecA 1 | ClpC1/P2 KDa | 0.056 | 28.833 | WT, EQ, NTD, CORE |
| A0QZJ0 | MSMEG_4042, MSMEI_3947 | Transcriptional regulator, GntR family protein | BACTHb | 0.050 | 6.000 | EQ, CORE |
| A0R616 | MSMEG_6391, MSMEI_6223 | Propionyl-CoA carboxylase beta chain | ClpC1/P2 KDa | 0.039 | 9.500 | EQ |
| Q3L885 | pks, MSMEG_0408, MSMEI_0398 | Polyketide synthase (type I modular polyketide synthase) | ClpC1/P2 KDa | 0.023 | 43.000 | EQ |
| A0QQY3 | MSMEG_0918, MSMEI_0896 | Transcriptional regulator, XRE family protein | BACTHb | 0.019 | 4.667 | WT |
| A0QSL0 | MSMEG_1516, MSMEI_1480 | Thioredoxin reductase | ClpC1/P2 KDa | 0.012 | 9.500 | WT, NTD, CORE |
| A0QR26 | MSMEG_0962, MSMEI_0936 | TetR family protein transcriptional regulator | ClpC1/P2 KD,a BACTHb | 0.004 | 42.000 | WT |
Subcomponents of ClpC1 interact with specific cellular proteins.
The ClpC1 NTD is a discrete folded module thought to interact with substrates and adaptors, such as the N-end rule adaptor ClpS (22), but is dispensable for hexamer formation, ATP hydrolysis, and unfolding activities. We hypothesized that the NTD and the D1/D2 ATPase core of ClpC1 can bind to different interaction partners. To discriminate between interactions with the NTD and ClpC1 core, we created two truncated ClpC1 constructs: one consisting of only the NTD (ClpC1NTD) and another lacking the NTD but possessing the full D1 and D2 core (ClpC1CORE) (Fig. 1B). We repeated our pulldown and proteomics workflow with these constructs.
We observed 243 proteins and 116 proteins that coimmunoprecipitate with ClpC1NTD and ClpC1CORE, respectively (Tables S1 and S2). Comparing data sets, we found 42 proteins that interact with all core-bearing ClpC1 constructs (ClpC1WT, ClpC1EQ, and ClpC1CORE) (Fig. 4A). Notably, 72 out of 116 proteins (62%) that interact with the ClpC1CORE were found in at least one other core-bearing data set, which provides additional confidence that these are bona fide interaction partners (Fig. 4A; Table S2). Of these, only 15 occurred exclusively in core-containing data sets, while the remaining 57 were also found in the ClpC1NTD data set. Appearance in both ClpC1NTD and ClpC1CORE data sets may indicate that these proteins bind nonspecifically. Alternatively, they may genuinely interact with both the NTD and core, for which there is precedence in other organisms. For example, the Bacillus subtilis proteolytic adaptor MecA contacts both the ClpC NTD and M domain (63).
FIG 4.
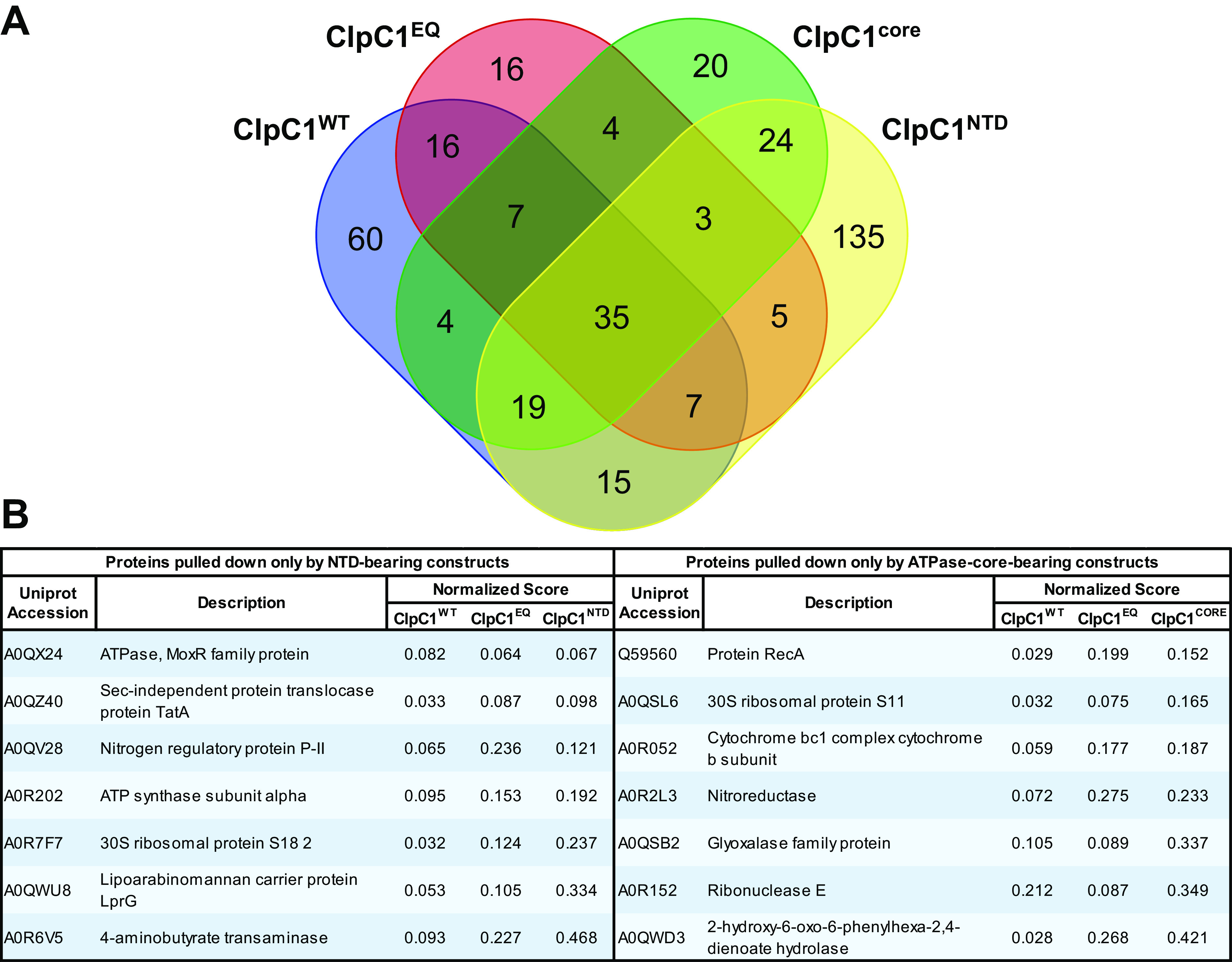
Comparative analysis of the ClpC1 interactome based on the different unfoldase components. (A) A Venn diagram shows protein targets observed to interact with one or more ClpC1 constructs. (B) The tabulated proteins were found to coimmunoprecipitate with all constructs bearing the amino-terminal domain (NTD) or the AAA+ ATPase core. Only 7 proteins were observed to interact with NTD alone. Similarly, as shown on the right only 7 proteins were observed interacting with the core-containing constructs and not in the NTD data sets.
Forty-two proteins were found to interact with all NTD-bearing constructs (ClpC1WT, ClpC1EQ, and ClpC1NTD), although only 7 of these were exclusive to NTD-bearing constructs and absent from ClpC1CORE. Interestingly, only 84 proteins (35%) identified in the ClpC1NTD data set were observed in at least one of the other NTD-bearing ClpC1WT or ClpC1EQ data sets, suggesting differences in how proteins interact with an isolated monomeric NTD and an NTD in the context of a ClpC1 hexamer.
Our analysis identified few proteins that consistently and uniquely interact with NTD-containing or core-containing ClpC1 constructs. Only 7 proteins coimmunoprecipitated with all of the NTD-bearing constructs and were absent from the ClpC1CORE data set. Seven separate proteins interacted with all of the core-bearing constructs, but not ClpC1NTD (Fig. 4B). Surprisingly, 35 proteins were found in all four data sets, suggesting either that some ClpC1-interacting proteins bind to both NTD and core regions or that truncated constructs are able to coassemble with endogenous ClpC1. Overall, our data suggest that many physiological ClpC1 interaction partners exist and that these may bind to multiple regions on the unfoldase.
Mycobacterial ClpC1 has a diverse and far-reaching interactome.
To assess whether specific classes of proteins are preferentially targeted by M. smegmatis ClpC1, we performed Gene Ontology (GO) annotation analysis on the interactome using the Blast2GO software suite (64). GO terms were applied to M. smegmatis (strain ATCC 700084/MC2155) proteins based on the closest homologs present in the Swiss-Prot/UniProt database. We found that ClpC1 potentially regulates proteins with a diverse range of cellular functions (Fig. 5 and Table 2). Of particular interest were protein annotation groups that were highly represented in the full-length ClpC1 interactome. The most represented group of proteins were those associated with nucleic acid binding and metabolism, including transcriptional regulators (Hup, Rho, RpoC, Rne, RpoA, Pnp, IleS, RpoB, TopA, DnaX, SigA and transcriptional regulators belonging to the MarR, PadR, GntR, XRE, Crp/Fnr, TetR families, among other proteins). Additionally, proteins involved in redox reactions were identified, such as GabD2, IspG, Pks, DapB, among others. Another well-represented group were proteins involved in cellular transport (including SecA1, SecF, Ffh, LprG, TatA). Some of these proteins are transmembrane, but have cytoplasmic regions through which proteolytic regulation might occur (65, 66). In addition to nucleic acid metabolism, observed proteins were involved in other forms of metabolism, such as carbohydrate metabolism (GlmS, Eno, SucB, GlpK, SdhA, SdhB, AcnA, Mqo, Kgd), electron transport chain (Ndh, AtpA, AtpC, AtpD, AtpFH), amino acid metabolism (HemL, SerA, GabT, HisG, CarB, GlnA, MSMEG_3973, MSMEG_0688, MSMEI_3879), and lipid metabolism (fatty acid synthase MSMEG_4757, AcpM, acyl coenzyme A [acyl-CoA] synthases MSMEG_0599 and MSMEG_3767). Taken together, these observations suggest that ClpC1 plays a central role in the framework of mycobacterial cellular physiology by potentially regulating metabolic processes and/or ensuring quality control of metabolic enzymes (31).
FIG 5.

Full-length ClpC1 interacts with diverse groups of proteins in Mycolicibacterium smegmatis. Functional categorization of cellular proteins interacting with ClpC1WT and/or ClpC1EQ is shown. Also shown is the distribution of these proteins by Gene Ontology (GO) annotation based mostly on biological process. Functional classification was performed using Blast2GO and was based on annotation of the closest homologs.
TABLE 2.
Proteins that interact with full-length ClpC1 (ClpC1WT or ClpC1EQ)
| UniProt accession no. | Gene name(s) | Protein description | Normalized score (mean ± SD) |
|
|---|---|---|---|---|
| ClpC1WT | ClpC1EQ | |||
| Amino acid metabolism | ||||
| A0QR33 | hemL, MSMEG_0969, MSMEI_0943 | Glutamate-1-semialdehyde 2,1-aminomutase (GSA) | 0.09 ± 0.02 | |
| A0QUY2 | serA, MSMEG_2378, MSMEI_2318 | d-3-Phosphoglycerate dehydrogenase (EC 1.1.1.95) | 0.48 ± 0.64 | |
| A0R6V5 | gabT, MSMEG_6685, MSMEI_6505 | 4-Aminobutyrate transaminase (EC 2.6.1.19) | 0.09 ± 0.1 | 0.23 ± 0.23 |
| A0QZX1 | hisG, MSMEG_4180, MSMEI_4082 | ATP phosphoribosyltransferase (ATP-PRT) (ATP-PRTase) | 0.04 ± 0.03 | |
| A0QWS5 | carB, MSMEG_3047, MSMEI_2973 | Carbamoyl-phosphate synthase large chain (EC 6.3.5.5) | 0.03 ± 0.02 | |
| A0R079 | glnA, glnA1, MSMEG_4290, MSMEI_4189 | Glutamine synthetase (GS) (EC 6.3.1.2) | 0.16 ± 0.21 | |
| A0QZC4 | MSMEG_3973, MSMEI_3878 | N-Methylhydantoinase | 0.39 ± 0.19 | 0.7 ± 1.05 |
| A0QQA8 | MSMEG_0688, MSMEI_0671 | Aspartate aminotransferase | 0.29 ± 0.31 | |
| I7G417 | MSMEI_3879 | 5-Oxoprolinase (ATP-hydrolyzing) (EC 3.5.2.9) | 0.32 ± 0.27 | 0.64 ± 0.53 |
| Carbohydrate metabolism | ||||
| O68956 | glmS, MSMEG_1568, MSMEI_1531 | Glutamine-fructose-6-phosphate aminotransferase | 0.05 ± 0.04 | |
| A0R3B8 | eno, MSMEG_5415, MSMEI_5267 | Enolase (EC 4.2.1.11) (2-phospho-d-glycerate hydrolyase) | 0.13 ± 0.06 | |
| A0R364 | MSMEG_5358, MSMEI_5212 | Acetamidase/formamidase family protein | 0.17 ± 0.16 | 0.06 ± 0.03 |
| A0R266 | MSMEG_5002, MSMEI_4876 | AAA_27 domain-containing protein | 0.06 ± 0.01 | |
| A0R072 | sucB, MSMEG_4283, MSMEI_4182 | Dihydrolipoamide acetyltransferase component of PDH complex | 0.1 ± 0.14 | |
| A0R729 | glpK, MSMEG_6759, MSMEI_6577 | Glycerol kinase (EC 2.7.1.30) | 0.17 ± 0.25 | 0.22 ± 0.13 |
| A0QT08 | sdhA, MSMEG_1670, MSMEI_1630 | Succinate dehydrogenase flavoprotein subunit (EC 1.3.5.1) | 0.12 ± 0.06 | 0.06 ± 0.05 |
| A0QT07 | sdhB, MSMEG_1669, MSMEI_1629 | Succinate dehydrogenase, iron-sulfur protein (EC 1.3.99.1) | 0.06 ± 0.08 | |
| A0QX20 | acnA, can, MSMEG_3143, MSMEI_3062 | Aconitate hydratase A (ACN) (aconitase) (EC 4.2.1.3) | 0.06 ± 0.08 | |
| A0QVL2 | mqo, MSMEG_2613, MSMEI_2551 | Probable malate:quinone oxidoreductase | 0.36 ± 0.16 | |
| A0R2B1 | kgd, sucA, MSMEG_5049, MSMEI_4922 | Multifunctional 2-oxoglutarate metabolism enzyme | 0.12 ± 0.12 | |
| Cell envelope-associated proteins | ||||
| A0QTT7 | MSMEG_1959, MSMEI_1915 | UPF0182 protein MSMEG_1959/MSMEI_1915 | 0.14 ± 0.1 | |
| A0QP93 | MSMEG_0317 | Uncharacterized protein | 0.19 ± 0.21 | |
| A0QYF6 | MSMEG_3641, MSMEI_3556 | VWFA domain-containing protein | 0.04 ± 0.02 | |
| A0QWT6 | MSMEG_3058, MSMEI_2982 | Lipoprotein | 0.14 ± 0.06 | |
| A0R1H2 | MSMEG_4752 | Uncharacterized protein | 0.06 ± 0.06 | |
| A0R006 | wag31, ag84, MSMEG_4217, MSMEI_4119 | Cell wall synthesis protein Wag31 (antigen 84) | 0.07 ± 0.04 | |
| A0R5I3 | MSMEG_6201 | Transglycosylase | 0.04 ± 0.01 | |
| Electron transport chain | ||||
| A0QYD6 | ndh, MSMEG_3621, MSMEI_3536 | NADH dehydrogenase (EC 1.6.99.3) | 0.09 ± 0.12 | |
| A0R052 | MSMEG_4263, MSMEI_4163 | Ubiquinol-cytochrome c reductase cytochrome b subunit | 0.06 ± 0.03 | 0.18 ± 0.02 |
| A0R057 | ctaC, MSMEG_4268, MSMEI_4167 | Cytochrome c oxidase subunit 2 (EC 1.9.3.1) | 0.07 ± 0.01 | 0.21 ± 0.2 |
| A0R203 | atpFH, atpF, atpH, MSMEG_4939, MSMEI_4812 | ATP synthase subunit b-delta | 0.59 ± 0.42 | 0.55 ± 0.31 |
| A0R200 | atpD, MSMEG_4936, MSMEI_4809 | ATP synthase subunit beta | 0.19 ± 0.07 | 0.12 ± 0.04 |
| A0R202 | atpA, MSMEG_4938, MSMEI_4811 | ATP synthase subunit alpha | 0.1 ± 0.04 | 0.15 ± 0.06 |
| A0R051 | qcrA, MSMEG_4262, MSMEI_4162 | Ubiquinol-cytochrome c reductase iron-sulfur subunit | 0.14 ± 0.07 | |
| A0QQB0 | MSMEG_0690, MSMEI_0673 | Iron-sulfur cluster-binding protein | 0.09 ± 0.06 | |
| A0R1Z9 | atpC, MSMEG_4935, MSMEI_4808 | ATP synthase epsilon chain | 0.06 ± 0.04 | |
| Lipid metabolism | ||||
| A0QPE8 | fadA2, MSMEG_0373, MSMEI_0366 | 3-Ketoacyl-CoA thiolase (EC 2.3.1.16) | 0.06 ± 0.04 | |
| A0R1H7 | MSMEG_4757, MSMEI_4637 | Fatty acid synthase | 0.4 ± 0.39 | |
| A0R5R5 | MSMEG_6284, MSMEI_6119 | Cyclopropane-fatty-acyl-phospholipid synthase | 0.05 ± 0.03 | 0.07 ± 0.02 |
| A0R2E8 | fadD6, MSMEG_5086, MSMEI_4960 | Very-long-chain acyl-CoA synthetase (EC 6.2.1.−) (EC 6.2.1.3) | 0.09 ± 0.08 | |
| A0R0B3 | acpM, MSMEG_4326, MSMEI_4226 | Meromycolate extension acyl carrier protein (ACP) | 0.07 ± 0.08 | 0.11 ± 0.02 |
| A0R042 | MSMEG_4254, MSMEI_4153 | AMP-binding enzyme | 0.03 ± 0.03 | |
| A0QYS3 | MSMEG_3767, MSMEI_3678 | Acyl-CoA synthase | 0.07 ± 0.09 | |
| A0R616 | accD4, MSMEG_6391, MSMEI_6223 | Propionyl-CoA carboxylase beta chain (EC 6.4.1.3) | 0.04 ± 0.05 | |
| A0QQ22 | fadD2, MSMEG_0599, MSMEI_0583 | Acyl-CoA synthase | 0.01 ± 0.02 | |
| A0R0B4 | kasA, MSMEG_4327, MSMEI_4227 | 3-Oxoacyl-[acyl-carrier-protein] synthase 1 (EC 2.3.1.41) | 0.06 ± 0.06 | |
| Noncanonical pathways | ||||
| A0R5C0 | MSMEG_6137, MSMEI_5978 | Nonribosomal peptide synthetase | 0.01 ± 0 | |
| A0QPH5 | MSMEG_0400 | Peptide synthetase | 0.11 ± 0.11 | |
| A0QY29 | eis, MSMEG_3513, MSMEI_3433 | Uncharacterized N-acetyltransferase MSMEG_3513 (EC 2.3.1.−) | 0.05 ± 0.04 | |
| A0QV52 | MSMEG_2450, MSMEI_2388 | Adenosylmethionine-8-amino-7-oxononanoate transaminase | 0.03 ± 0.02 | |
| A0QWD3 | MSMEG_2900, MSMEI_2827 | 2-Hydroxy-6-oxo-6-phenylhexa-2,4-dienoate hydrolase (EC 3.7.1.−) | 0.03 ± 0.02 | 0.27 ± 0.24 |
| A0QZ01 | lipT, MSMEG_3848, MSMEI_3758 | Carboxylic ester hydrolase (EC 3.1.1.−) | 0.02 ± 0.02 | |
| A0QZ33 | MSMEG_3880, MSMEI_3790 | Nitrilase/cyanide hydratase | 0.13 ± 0.07 | |
| A0QTM5 | pcaD, MSMEG_1897, MSMEI_1857 | 3-Oxoadipate enol-lactonase (EC 3.1.1.24) | 0.1 ± 0.05 | |
| A0R617 | pks13, MSMEG_6392, MSMEI_6224 | Polyketide synthase | 0.16 ± 0.14 | |
| A0QTE1 | accA3, MSMEG_1807, MSMEI_1762 | Acetyl/propionyl-coenzyme A carboxylase alpha chain | 0.13 ± 0.17 | |
| A0QV28 | glnB, MSMEG_2426, MSMEI_2365 | Nitrogen regulatory protein P-II | 0.06 ± 0.08 | 0.24 ± 0.16 |
| Nucleic acid binding and metabolism, including transcriptional regulators | ||||
| A0QTQ8 | rhlE, MSMEG_1930, MSMEI_1889 | DEAD/DEAH box helicase | 0.19 ± 0.09 | 0.16 ± 0.07 |
| A0R7J3 | parB, MSMEG_6938, MSMEI_6746 | ParB-like partition protein | 0.1 ± 0.09 | |
| A0QT91 | lhr, MSMEG_1757, MSMEI_1715 | DEAD/DEAH box helicase | 0.07 ± 0.02 | |
| Q9ZHC5 | hup, hlp, MSMEG_2389, MSMEI_2329 | DNA-binding protein HU homolog (histone-like protein) (Hlp) | 0.14 ± 0.16 | 0.18 ± 0 |
| A0R656 | MSMEG_6431, MSMEI_6263 | HTH cro/C1-type domain-containing protein | 0.01 ± 0.01 | |
| A0QYG0 | MSMEG_3645, MSMEI_3559 | BFN domain-containing protein | 0.06 ± 0.0009 | |
| A0R218 | rho, MSMEG_4954, MSMEI_4827 | Transcription termination factor Rho (EC 3.6.4.−) | 0.6 ± 0.5 | 0.54 ± 0.02 |
| A0QS66 | rpoC, MSMEG_1368, MSMEI_1329 | DNA-directed RNA polymerase subunit beta′ | 0.45 ± 0.57 | 0.36 ± 0.29 |
| A0R152 | rne, MSMEG_4626, MSMEI_4509 | Ribonuclease E (RNase E) (EC 3.1.26.12) | 0.21 ± 0.18 | 0.09 ± 0.06 |
| A0QSL8 | rpoA, MSMEG_1524, MSMEI_1488 | DNA-directed RNA polymerase subunit alpha | 0.05 ± 0.02 | |
| A0QVQ5 | pnp, gpsI, MSMEG_2656, MSMEI_2593 | Polyribonucleotide nucleotidyltransferase | 0.03 ± 0.05 | |
| A0QX46 | ileS, MSMEG_3169, MSMEI_3087 | Isoleucine-tRNA ligase (EC 6.1.1.5) (isoleucyl-tRNA synthetase) | 0.02 ± 0.02 | |
| P60281 | rpoB, MSMEG_1367, MSMEI_1328 | DNA-directed RNA polymerase subunit beta | 0.24 ± 0.29 | |
| A0R5D9 | topA, MSMEG_6157, MSMEI_5999 | DNA topoisomerase 1 (EC 5.6.2.1) (DNA topoisomerase I) | 0.15 ± 0.19 | |
| A0QW71 | MSMEG_2839, MSMEI_2765 | Transcriptional accessory protein | 0.05 ± 0.04 | |
| A0R5R6 | dnaX, MSMEG_6285, MSMEI_6120 | DNA polymerase III subunit gamma/tau (EC 2.7.7.7) | 0.05 ± 0.02 | |
| A0R3L1 | MSMEG_5512 | Magnesium chelatase | 0.32 ± 0.13 | 0.5 ± 0.6 |
| A0QVD7 | MSMEG_2538, MSMEI_2478 | MarR-family protein transcriptional regulator | 0.03 ± 0.01 | 0.08 ± 0.01 |
| A0R5K8 | MSMEG_6227, MSMEI_6066 | Transcriptional regulator, PadR family protein | 0.06 ± 0.05 | |
| A0QR90 | MSMEG_1029, MSMEG_2309 | Probable transcriptional regulatory protein | 0.09 ± 0.13 | |
| A0QZJ0 | MSMEG_4042, MSMEI_3947 | Transcriptional regulator, GntR family protein | 0.05 ± 0.07 | |
| A0QW02 | sigA, rpoD, MSMEG_2758, MSMEI_2690 | RNA polymerase sigma factor SigA | 0.11 ± 0.05 | |
| A0R5H1 | MSMEG_6189, MSMEI_6029 | Transcriptional regulator, Crp/Fnr family protein | 0.08 ± 0.03 | 0.27 ± 0.17 |
| A0QQY3 | MSMEG_0918, MSMEI_0896 | Transcriptional regulator, XRE family protein | 0.02 ± 0.01 | |
| A0QR26 | MSMEG_0962, MSMEI_0936 | TetR family protein transcriptional regulator | 0.01 ± 0.01 | |
| Phosphorylation/phosphate signaling | ||||
| A0R5N8 | ask, MSMEG_6257, MSMEI_6095 | Aspartokinase (EC 2.7.2.4) | 0.2 ± 0.23 | 0.09 ± 0.04 |
| A0QU86 | MSMEG_2116, MSMEI_2068 | PTS system, glucose-specific IIBC component | 0.03 ± 0.04 | |
| A0QNG5 | ppp, MSMEG_0033, MSMEI_0035 | Protein phosphatase 2C | 0.02 ± 0 | |
| A0QNG2 | pknA, MSMEG_0030, MSMEI_0032 | Serine/threonine protein kinase PknA (EC 2.7.1.−) | 0.01 ± 0.01 | |
| A0QV12 | MSMEG_2410, MSMEI_2349 | Putative serine-threonine protein kinase | 0.05 ± 0.01 | 0.16 ± 0.12 |
| A0R0T1 | MSMEG_4497, MSMEI_4386 | PhoH family protein | 0.1 ± 0.12 | 0.16 ± 0.14 |
| A0QX24 | moxR, MSMEG_3147 | ATPase, MoxR family protein | 0.08 ± 0.06 | 0.06 ± 0.09 |
| Protein quality control | ||||
| A0QQU5 | groEL2, groL2, MSMEG_0880, MSMEI_0859 | 60-kDa chaperonin 1 (GroEL protein 1) (protein Cpn60 1) | 1.68 ± 1.5 | 2.41 ± 0.76 |
| A0QSS4 | groEL1, groL1, MSMEG_1583, MSMEI_1545 | 60-kDa chaperonin 2 (GroEL protein 2) (protein Cpn60 2) | 1.04 ± 0.77 | 0.99 ± 0.13 |
| A0QW35 | MSMEG_2792, MSMEI_2723 | Clp amino-terminal domain protein | 0.09 ± 0.05 | 0.29 ± 0.1 |
| A0R085 | MSMEG_4296, MSMEI_4195 | Protease | 0.05 ± 0 | |
| A0QSH0 | sppA, MSMEG_1476, MSMEI_1440 | Signal peptide peptidase SppA, 67K type (EC 3.4.−.−) | 0.06 ± 0.04 | 0.14 ± 0.05 |
| A0QQM2 | MSMEG_0806, MSMEI_0787 | Hydrolase | 0.05 ± 0.04 | |
| A0R196 | clpX, MSMEG_4671, MSMEI_4553 | ATP-dependent Clp protease ATP-binding subunit ClpX | 0.03 ± 0.01 | |
| A0QTT5 | MSMEG_1957, MSMEI_1913 | Uncharacterized protein | 0.09 ± 0.11 | 0.09 ± 0.01 |
| A0R6J9 | est, MSMEG_6575, MSMEI_6397 | Beta-lactamase | 0.04 ± 0.03 | |
| A0QQD0 | dnaJ, dnaJ1, MSMEG_0711, MSMEI_0694 | Chaperone protein DnaJ1 | 0.53 ± 0.45 | 0.83 ± 0.94 |
| A0R0T8 | dnaJ, dnaJ2, MSMEG_4504, MSMEI_4392 | Chaperone protein DnaJ2 | 0.09 ± 0.01 | |
| A0QQC8 | dnaK, MSMEG_0709, MSMEI_0692 | Chaperone protein DnaK (HSP70) | 0.25 ± 0.3 | |
| A0R199 | tig, MSMEG_4674, MSMEI_4557 | Trigger factor (TF) (EC 5.2.1.8) (PPIase) | 0.16 ± 0.21 | |
| A0QWK5 | ppiB, MSMEG_2974, MSMEI_2900 | Peptidyl-prolyl cis-trans isomerase (PPIase) (EC 5.2.1.8) | 0.09 ± 0 | |
| Redox processes | ||||
| A0QPE7 | fabG4, MSMEG_0372, MSMEI_0365 | Oxidoreductase, short-chain dehydrogenase/reductase family protein | 0.35 ± 0.25 | 0.33 ± 0.04 |
| A0QR89 | MSMEG_1028, MSMEG_2308 | Geranylgeranyl reductase | 0.25 ± 0.25 | 0.25 ± 0.22 |
| A0R696 | MSMEG_6471, MSMEI_6299 | Glycine/d-amino acid oxidase | 0.05 ± 0.01 | |
| A0QR91 | MSMEG_1030, MSMEG_2310 | Monooxygenase | 0.12 ± 0.09 | |
| A0R4Q0 | gabD2, MSMEG_5912, MSMEI_5752 | Putative succinate-semialdehyde dehydrogenase | 0.01 ± 0 | |
| A0R730 | glpD2, MSMEG_6761, MSMEI_6578 | Glycerol-3-phosphate dehydrogenase (EC 1.1.5.3) | 0.09 ± 0.11 | |
| A0R226 | MSMEG_4962, MSMEI_4836 | RemO protein | 0.02 ± 0.02 | |
| A0QSL0 | MSMEG_1516, MSMEI_1480 | Thioredoxin reductase | 0.01 ± 0 | |
| A0QVH9 | ispG, MSMEG_2580, MSMEI_2518 | 4-Hydroxy-3-methylbut-2-en-1-yl diphosphate synthase (flavodoxin) | 0.01 ± 0.01 | |
| A0QSB2 | MSMEG_1417, MSMEI_1382 | Glyoxalase family protein | 0.11 ± 0.12 | 0.09 ± 0.01 |
| A0QQV4 | gabD2, MSMEG_0889, MSMEI_0868 | Aldehyde dehydrogenase | 0.14 ± 0 | 0.11 ± 0.03 |
| A0R2L3 | MSMEG_5155, MSMEI_5022 | Nitroreductase | 0.07 ± 0.04 | 0.28 ± 0.07 |
| A0QYH8 | MSMEG_3663, MSMEI_3576 | Oxidoreductase | 0.06 ± 0.01 | |
| A0QSB9 | mftD, lldD1, MSMEG_1424, MSMEI_1389 | FMN-dependent dehydrogenase | 0.02 ± 0.03 | |
| A0R461 | MSMEG_5715, MSMEI_5564 | Bac_luciferase domain-containing protein | 0.39 ± 0.31 | |
| A0QRD2 | MSMEG_1073, MSMEI_1041 | Oxidoreductase, short-chain dehydrogenase/reductase family protein | 0.09 ± 0.05 | |
| A0QU11 | MSMEG_2037, MSMEI_1991 | Bac_luciferase domain-containing protein | 0.03 ± 0.04 | |
| Q3L885 | pks, MSMEG_0408, MSMEI_0398 | Polyketide synthase (type I modular polyketide synthase) | 0.02 ± 0.03 | |
| A0QXA1 | gltB, MSMEG_3225, MSMEI_3143 | Ferredoxin-dependent glutamate synthase 1 (EC 1.4.7.1) | 0.03 ± 0.01 | |
| A0QXI7 | dapB, MSMEG_3317, MSMEI_3231 | Dihydrodipicolinate reductase, N-terminal domain protein | 0.06 ± 0.06 | |
| A0R221 | MSMEG_4957, MSMEI_4830 | Homoserine dehydrogenase (EC 1.1.1.3) | 0.24 ± 0.18 | 0.2 ± 0.09 |
| A0R0W1 | MSMEG_4527, MSMEI_4414 | Ferredoxin sulfite reductase (EC 1.8.7.1) | 0.03 ± 0.02 | |
| Stress response | ||||
| A0QQQ1 | sodC, MSMEG_0835, MSMEI_0816 | Superoxide dismutase [Cu-Zn] (EC 1.15.1.1) | 0.1 ± 0.02 | |
| A0QS28 | recD, MSMEG_1325, MSMEI_1288 | RecBCD enzyme subunit RecD (EC 3.1.11.5) | 0.02 ± 0.01 | |
| A0QUM3 | cstA, MSMEG_2259, MSMEI_2203 | Carbon starvation protein A | 0.02 ± 0.02 | |
| Q59560 | recA, MSMEG_2723, MSMEI_2656 | Protein RecA (recombinase A) | 0.03 ± 0.04 | 0.2 ± 0.03 |
| Translation | ||||
| A0QS98 | tuf, MSMEG_1401, MSMEI_1363 | Elongation factor Tu (EF-Tu) | 0.24 ± 0.18 | 0.33 ± 0.28 |
| A0QVM7 | infB, MSMEG_2628, MSMEI_2565 | Translation initiation factor IF-2 | 0.46 ± 0.46 | |
| A0QYY6 | rpsA, MSMEG_3833, MSMEI_3743, LJ00_19040 | 30S ribosomal protein S1 | 0.16 ± 0.2 | |
| A0R7F7 | rpsR2, rpsR1, MSMEG_6895, MSMEI_6711 | 30S ribosomal protein S18 2 | 0.03 ± 0.01 | 0.12 ± 0.11 |
| A0QSE0 | rpsQ, MSMEG_1445, MSMEI_1409 | 30S ribosomal protein S17 | 0.02 ± 0.01 | |
| A0QSD7 | rpsC, MSMEG_1442, MSMEI_1406 | 30S ribosomal protein S3 | 0.23 ± 0.19 | 0.36 ± 0.32 |
| A0QV03 | rpmB, rpmB-3, MSMEG_2400, MSMEI_2340 | 50S ribosomal protein L28 | 0.02 ± 0.01 | |
| A0QSL6 | rpsK, MSMEG_1522, MSMEI_1486 | 30S ribosomal protein S11 | 0.03 ± 0.03 | 0.07 ± 0.07 |
| A0QSD1 | rplC, MSMEG_1436, MSMEI_1400 | 50S ribosomal protein L3 | 0.1 ± 0.14 | 0.25 ± 0.09 |
| A0R3I9 | rpmF, MSMEG_5489, MSMEI_5337 | 50S ribosomal protein L32 | 0.03 ± 0.03 | 0.15 ± 0.02 |
| A0QSL5 | rpsM, MSMEG_1521, MSMEI_1485 | 30S ribosomal protein S13 | 0.04 ± 0.04 | |
| A0R150 | rpmA, MSMEG_4624, MSMEI_4507 | 50S ribosomal protein L27 | 0.02 ± 0.02 | |
| A0QVB9 | tsf, MSMEG_2520, MSMEI_2461 | Elongation factor Ts (EF-Ts) | 0.03 ± 0.04 | |
| A0QSD0 | rpsJ, MSMEG_1435, MSMEI_1399 | 30S ribosomal protein S10 | 0.11 ± 0.02 | 0.17 ± 0.24 |
| A0QSD6 | rplV, MSMEG_1441, MSMEI_1405 | 50S ribosomal protein L22 | 0.03 ± 0.01 | |
| A0QSG0 | rplX, MSMEG_1466, MSMEI_1430 | 50S ribosomal protein L24 | 0.02 ± 0.02 | |
| A0QYU7 | rpmI, MSMEG_3792, MSMEI_3704 | 50S ribosomal protein L35 | 0.01 ± 0.01 | |
| A0QSP8 | rplM, MSMEG_1556, MSMEI_1519 | 50S ribosomal protein L13 | 0.08 ± 0.07 | |
| A0QSD2 | rplD, MSMEG_1437, MSMEI_1401 | 50S ribosomal protein L4 | 0.05 ± 0.07 | |
| A0QSG6 | rpsE, MSMEG_1472, MSMEI_1436 | 30S ribosomal protein S5 | 0.07 ± 0.03 | |
| A0QS62 | rplJ, MSMEG_1364, MSMEI_1325 | 50S ribosomal protein L10 | 0.08 ± 0.03 | |
| A0QS46 | rplA, MSMEG_1347, MSMEI_1309 | 50S ribosomal protein L1 | 0.05 ± 0.07 | |
| A0QS97 | rpsG, MSMEG_1399, MSMEI_1361 | 30S ribosomal protein S7 | 0.08 ± 0.03 | |
| A0QSG5 | rplR, MSMEG_1471, MSMEI_1435 | 50S ribosomal protein L18 | 0.03 ± 0.04 | |
| Transport | ||||
| A0QQ65 | MSMEG_0643, MSMEI_0627 | Extracellular solute-binding protein, family protein 5, putative | 0.56 ± 0.25 | 0.63 ± 0.32 |
| A0QXC0 | MSMEG_3247, MSMEI_3164 | Branched-chain amino acid ABC transporter substrate-binding protein | 0.57 ± 0.34 | 0.41 ± 0.18 |
| A0QWL3 | MSMEG_2982, MSMEI_2907 | Putative periplasmic binding protein | 0.46 ± 0.3 | 0.44 ± 0.04 |
| P71533 | secA1, MSMEG_1881, MSMEI_1840 | Protein translocase subunit SecA 1 | 0.07 ± 0.06 | 0.04 ± 0.05 |
| A0QXF3 | MSMEG_3280, MSMEI_3196 | Polyamine-binding lipoprotein | 0.06 ± 0.03 | |
| A0QRB1 | MSMEG_1052, MSMEG_2332 | Amino acid carrier protein | 0.13 ± 0.13 | 0.1 ± 0.07 |
| A0QYK4 | MSMEG_3689, MSMEI_3602 | Sodium:solute symporter | 0.05 ± 0.03 | 0.1 ± 0.03 |
| A0R261 | MSMEG_4999, MSMEI_4871 | Bacterial extracellular solute-binding protein, family protein 5 | 0.05 ± 0.03 | |
| A0QWJ3 | secF, MSMEG_2962, MSMEI_2888 | Protein-export membrane protein SecF | 0.02 ± 0 | |
| A0R5T7 | MSMEG_6307, MSMEI_6142 | Glutamine-binding periplasmic protein | 0.05 ± 0.06 | |
| A0R2C0 | sugC, MSMEG_5058, MSMEI_4931 | ABC transporter, ATP-binding protein SugC | 0.02 ± 0.01 | |
| A0QT21 | MSMEG_1683, MSMEI_1642 | Cytosine/purine/uracil/thiamine/allantoin permease family protein | 0.02 ± 0.02 | |
| A0QVX3 | MSMEG_2727, MSMEI_2660 | Glutamate-binding protein | 0.22 ± 0.03 | 0.39 ± 0.03 |
| A0QXB0 | MSMEG_3235, MSMEI_3153 | ABC-type amino acid transport system, secreted component | 0.23 ± 0.09 | 0.24 ± 0.2 |
| A0R0W7 | MSMEG_4533, MSMEI_4420 | Sulfate-binding protein | 0.16 ± 0.01 | 0.34 ± 0.23 |
| A0QV32 | ffh, MSMEG_2430, MSMEI_2369 | Signal recognition particle protein (fifty-four homolog) | 0.13 ± 0.05 | |
| A0QWU8 | lprG, MSMEG_3070, MSMEI_2993 | Lipoarabinomannan carrier protein LprG | 0.05 ± 0.02 | 0.1 ± 0.02 |
| A0QSV2 | MSMEG_1612, MSMEI_1573 | Extracellular solute-binding protein, family protein 3 | 0.09 ± 0.05 | 0.11 ± 0.02 |
| A0QZ40 | tatA, MSMEG_3887, MSMEI_3797 | Sec-independent protein translocase protein TatA | 0.03 ± 0 | 0.09 ± 0.01 |
| A0QPX3 | MSMEG_0550, MSMEI_0535 | Sulfonate binding protein | 0.09 ± 0.02 | |
| A0QNN8 | pntA, MSMEG_0110, MSMEI_0106 | NAD(P) transhydrogenase, alpha subunit (EC 1.6.1.1) | 0.06 ± 0.01 | |
| A0QSY1 | MSMEG_1642, MSMEI_1603 | ABC transporter, ATP-binding protein | 0.08 ± 0.09 | |
| Uncharacterized proteins | ||||
| A0R3L0 | MSMEG_5511, MSMEI_5359 | von Willebrand factor, type A | 0.05 ± 0 | 0.08 ± 0.02 |
| A0QPE3 | MSMEG_0368, MSMEI_0361 | Uncharacterized protein | 0.03 ± 0.01 | |
| A0R576 | lsr2, MSMEG_6092, MSMEI_5934 | Lsr2 protein | 0.05 ± 0.03 | 0.15 ± 0.21 |
| A0QNZ9 | MSMEG_0222, MSMEI_0215 | DUF2786 domain-containing protein | 0.04 ± 0.03 | |
| I7FPJ2 | MSMEI_4181 | Uncharacterized protein | 0.01 ± 0.01 | |
| A0R6E9 | MSMEG_6524, MSMEI_6350 | ABC polyamine/opine/phosphonate transporter | 0.07 ± 0.09 | |
| Q3I5Q7 | MSMEG_0919, MSMEI_0897 | HBHA-like protein (heparin-binding hemagglutinin) | 0.14 ± 0.13 | 0.25 ± 0.21 |
| A0QTG7 | MSMEG_1835, MSMEI_1793 | TobH protein | 0.22 ± 0.07 | 0.31 ± 0.15 |
| A0QYR3 | MSMEG_3754, MSMEI_3665 | TPR-repeat-containing protein | 0.13 ± 0.02 | 0.07 ± 0.04 |
| A0QZY3 | MSMEG_4192, MSMEI_4094 | Uncharacterized protein | 0.07 ± 0.05 | 0.05 ± 0.08 |
| A0R562 | MSMEG_6078, MSMEI_5918 | LpqE protein | 0.04 ± 0.03 | |
| A0R6C7 | MSMEG_6502, MSMEI_6330 | Uncharacterized protein | 0.04 ± 0 | |
| A0R2T1 | MSMEG_5223 | Uncharacterized protein | 0.04 ± 0.01 | |
| A0QVU2 | MSMEG_2695, MSMEI_2629 | 35-kDa protein | 0.08 ± 0 | |
| A0R2B0 | MSMEG_5048, MSMEI_4921 | Uncharacterized protein | 0.03 ± 0.05 | |
| A0R1B5 | MSMEG_4692, MSMEI_4575 | Uncharacterized protein MSMEG_4692/MSMEI_4575 | 0.08 ± 0.02 | |
Proteins involved in protein quality control, such as protein folding and unfolding, proteome turnover, and protein homeostasis in general, were also important constituents of the interactome (Fig. 5 and Table 2). These included chaperone proteins GroL1, GroL2, DnaJ1, DnaJ2, DnaK, and Tig and Clp proteins ClpX and ClpC2 (MSMEG_2792). ClpC1 is thus named because of the existence of the orthologous ClpC2 which possesses homology to the ClpC1 N-terminal domain, but lacks AAA+ ATPase modules (22, 67). As ClpC1 is itself involved in protein homeostasis and protein fate in general, it is not surprising to see its interaction with these proteins. Also, as noted above, components of the ribosome and other proteins involved in translation were abundant as well, making up ~13% of the total data set (Fig. 5). It is possible that nascent ClpC1 stays in contact with the ribosome during folding, either directly or indirectly through other elements of folding machinery. Alternatively, ribosomal proteins are commonly observed as contaminants in cell-based mass spectrometry experiments (68).
It is also important to point out that for the proteins specifically interacting with either ClpC1 NTD or core alone (Fig. 4), there was some functional diversity. For the NTD-interacting proteins, there were proteins involved in transport (TatA and LprG), phosphorylation/dephosphorylation (MoxR), the electron transport chain (AtpA), amino acid metabolism (GabT), noncanonical pathways (MSMEG_2426), and translation (RpsR2). The ClpC1 core-interacting proteins had roles in redox processes (MSMEG_5155 and MSMEG_1417), stress response (RecA), translation (RpsK), the electron transport chain (MSMEG_4263), nucleic acid binding and metabolism (Rne), and noncanonical pathways (MSMEG _2900).
Taken together, we observe a functionally diverse set of cellular proteins that interact with M. smegmatis ClpC1, revealing a broad diversity of putative substrates and interaction partners of the ClpC1P1P2 protease. This buttresses just how far-reaching its regulatory effects on cellular proteins could be and may help account for the essentiality of these proteases in mycobacteria (19, 27–31).
Physiochemical analysis of termini of interacting proteins.
Some Clp protease substrates are recognized by short terminal degron sequences of various lengths and compositions. M. smegmatis ClpC1 recognizes model substrates bearing a C-terminal SsrA sequence (ADSNQRDYALAA) (13). M. tuberculosis ClpC1 recognizes Hsp20 via the C-terminal sequence TQAQRIAITK (21) and PanD via the C-terminal sequence NAGELLDPRLGVG (20). In addition, substrates displaying some hydrophobic N-terminal residues are delivered to ClpC1 by the adaptor ClpS as part of the N-end rule proteolytic pathway (23, 69).
Because the sequence determinants required for recognition by ClpC1 are not well defined, we compared terminal sequences of proteins pulled down by full-length ClpC1 (ClpC1WT or ClpC1EQ) to the terminal sequences found across the entire M. smegmatis proteome (Fig. 6). The average charge and hydrophobicity of the first and last 10 amino acids (aa) of protein sequences varied widely. We saw no statistically significant differences in these parameters for residues at the N terminus. However, C-terminal regions of interactome proteins were, on average, less positively charged (closer to neutral) and more hydrophilic than equivalent regions from the full proteome. These differences were statistically significant, although the magnitude of the shift was much smaller than the standard deviation (SD) in either data set. The apparent preference for hydrophilic C-terminal character is surprising, as ATP-dependent proteases are often thought to recognize exposed hydrophobic regions as markers for protein misfolding (8). This analysis suggests either a slight preference for uncharged polar termini or that polar termini are more exposed to solvent and more available for ClpC1 binding. Additionally, the discrepancy between expected and observed physicochemical parameters might be explained by a preponderance of ClpC1-interacting proteins that are not proteolytic substrates, and thus may not be subject to the same recognition trends as the substrates.
FIG 6.

Physicochemical analysis of terminal sequences of full-length ClpC1-interacting proteins. (A) Violin plots illustrate the average charges of the first (N-terminal) and last (C-terminal) amino acids in the ClpC1 interactome and in the entire M. smegmatis proteome. Central lines indicate median values, with error bars indicating 1 SD. (B) Mean hydrophobicity of terminal residues in the ClpC1 interactome and control data sets is shown. For both metrics, the values plotted are the average residue value per terminus. Significance was assessed by two-tailed Welch’s t test. *, P < 0.05; **, P < 0.01; ns, not significant.
ClpC1P1P2 recognizes MSMEI_3879 as a proteolytic substrate.
We selected several hits from our data set that (i) were expected to be soluble cytosolic proteins, (ii) had M. tuberculosis homologs identified in prior screens for Clp protease interaction partners (Table 3) (21, 23), and (iii) could be readily expressed and purified from E. coli. These included chaperones DnaJ1 and DnaJ2, transcriptional regulators GntR and XRE, magnesium chelatase, and the apparent pseudogene product MSMEI_3879. Five of these (DnaJ1, GntR, XRE, Mg chelatase, MSMEI_3879) produced clear binding curves to ClpC1 by microscale thermophoresis, with Kapp values in the low micromolar range (Fig. S2), which validates the ability of our overall method to identify bona fide interaction partners. We tested whether these hits were recognized as substrates by ClpC1P1P2 in vitro and found that only MSMEI_3879 was degraded, with ~75% of the protein hydrolyzed within 40 min (Fig. 7A and B). These in vitro results provide preliminary evidence of the substrate status of MSMEI_3879.
FIG 7.
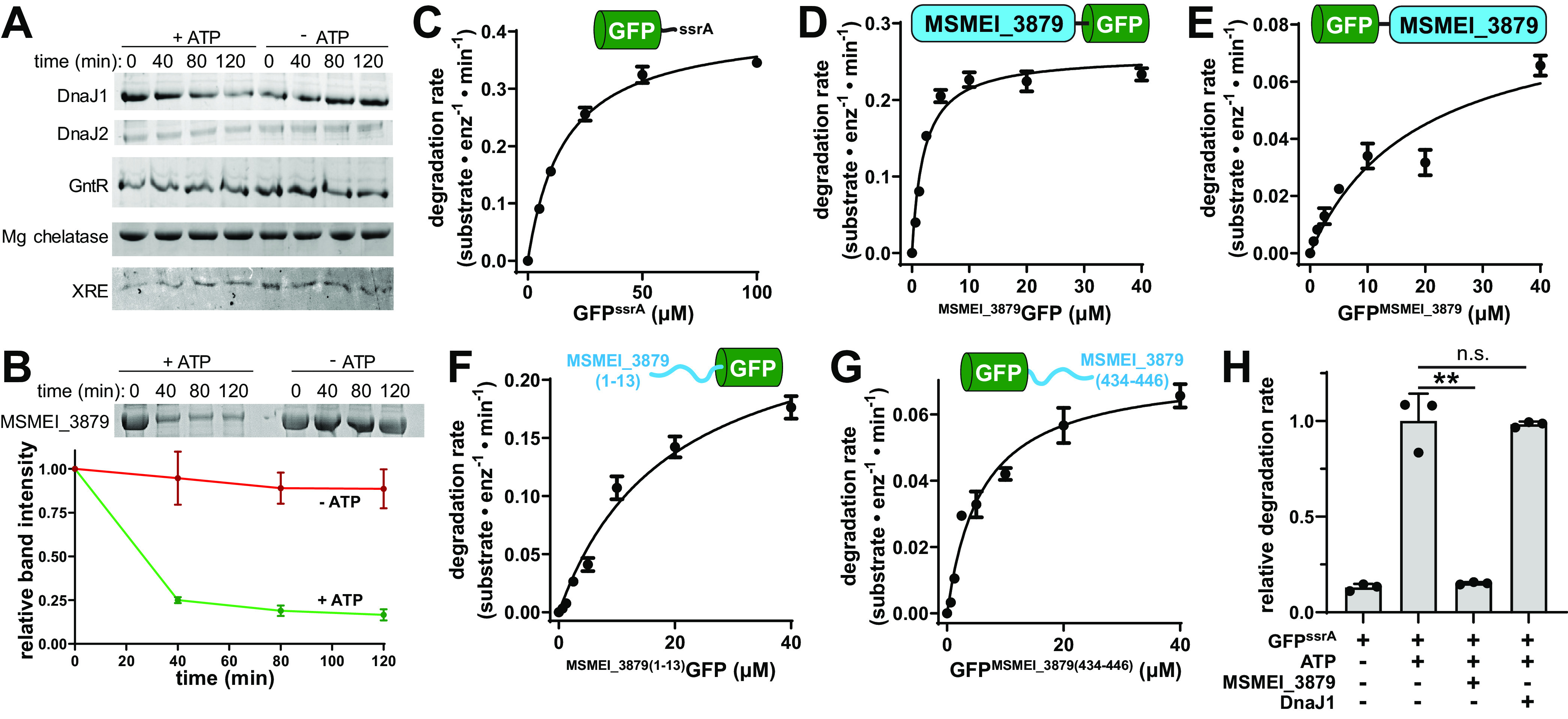
MSMEI_3879 is a substrate of M. smegmatis ClpC1. (A) In vitro degradation assays of DnaJ1, DnaJ2, GntR, magnesium chelatase, and XRE by 1 μM ClpC1 and 1 μM ClpP1P2, monitored by SDS-PAGE. (B) ATP-dependent degradation of 10 μM MSMEI_3879 by ClpC1P1P2 was observed in vitro by SDS-PAGE, with gel densitometry of three replicate assays shown. (C) Michaelis-Menten analysis of GFPssrA proteolysis by 1 μM ClpC1P1P2, as a function of substrate concentration, reveals a kcat of 0.41 ± 0.02 substrate · min−1 · enzyme−1 and a Km of 16.04 ± 1.28 μM. (D) MSMEI_3879GFP was degraded by 1 μM ClpC1P1P2 with a kcat of 0.26 ± 0.01 substrate · min−1 · enzyme−1 and Km of 2.06 ± 0.43 μM. (E) GFPMSMEI_3879 was degraded with a kcat of 0.09 ± 0.02 substrate · min−1 · enzyme−1 and a Km of 18.8 ± 9.6 μM. (F) GFP preceded by the first 13 residues of MSMEI_3879 was degraded with kcat of 0.268 ± 0.033 substrate · min−1 · enzyme−1 and Km of 19.1 ± 4.9 μM. (G) GFP appended with the last 13 residues of MSMEI_3879 was degraded with a kcat of 0.0737 ± 0.0053 substrate · min−1 · enzyme−1 and Km of 5.97 ± 1.3 μM. (H) Degradation of 10 μM GFPssrA by 1 μM ClpC1P1P2 was monitored in the presence and absence of ATP, 10 μM MSMEI_3879, or 10 μM DnaJ1. MSMEI_3879 reduces GFPssrA degradation to the level observed in the absence of ATP. DnaJ1 does not significantly alter the rate of GFPssrA proteolysis. Values are averages of three replicates (n = 3) ± 1 SD. P values were calculated by unpaired two-tailed Student's t test. **, P < 0.01; n.s., not significant.
The MSMEI_3879 locus encodes a 446-aa product with homology to ATP-hydrolyzing hydantoinase/oxoprolinase enzymes in the hydantoinase A family, which catalyze ring-opening reactions on lactam substrates (70–72). Phylogenetic analysis (Fig. S3) reveals a cluster of orthologs with >90% sequence identity to MSMEI_3879 in several related species, including Mycolicibacterium fortuitum, Mycolicibacterium peregrinum, and Mycolicibacterium brisbanense. More distantly related homologs occur across Mycobacteriaceae (73). The closest homolog in M. tuberculosis is OplA, which appears to belong to a separate subclass of enzymes, sharing only 34% sequence identity with MSMEI_3879 and incorporating a C-terminal fusion with a hydantoinase B enzyme.
Notably, Mycolicibacterium orthologs of MSMEI_3879 are generally longer, at about 690 aa. Comparison of the M. smegmatis genome to those of other Mycolicibacterium species reveals a frameshift caused by a single nucleotide insertion in codon 233 (Fig. S4). The MSMEI_3879 open reading frame arises from an alternative start site at position 242 (with respect to the typical Mycolicibacterium start codon), and the resulting polypeptide corresponds to only the latter two-thirds of orthologous hydantoinase/oxoprolinase enzymes. (There is no MSMEG annotation that directly corresponds to MSMEI_3879. The closest analog, MSMEG_3974, describes the entire frameshifted locus.) Comparison of AlphaFold2 (74) predictions of MSMEI_3879 and M. fortuitum AcxA, which share 93% sequence identity and possesses an intact N terminus, suggests that truncation removes a set of surface helices in MSMEI_3879 (Fig. S5). Loss of these helices is predicted to expose hydrophobic residues on two small structural elements that project outward from the body of the protein. In spite of this, MSMEI_3879 expressed well in E. coli, was straightforward to purify and remained stable throughout purification without precipitation or aggregation.
Many Clp protease substrates are recognized by short terminal degron sequences (13, 20, 21, 23). To test whether ClpC1 recognizes MSMEI_3879 by a particular terminus, we engineered green fluorescent protein (GFP) constructs with MSMEI_3879 fused to either the N or C terminus and assayed proteolysis by ClpC1P1P2. Michaelis-Menten analysis of the resulting degradation rates showed that the construct with MSMEI_3879 at the N terminus (MSMEI_3879GFP) is degraded with a kcat of ≈0.26 substrate · min−1 · enzyme−1 and a Km of ≈2 μM (Fig. 7C). For comparison, under the same assay conditions, GFP with an M. smegmatis ssrA tag (GFPssrA) was degraded faster (kcat ≈ 0.4 substrate · min−1 · enzyme−1) but with a much higher Km of ≈16 μM (Fig. 7D), similar to the Km reported for degradation of GFPssrA by M. tuberculosis ClpC1 (23). A construct carrying MSMEI_3879 at the C terminus (GFPMSMEI_3879) was degraded at a substantially lower rate (kcat ≈ 0.09 substrate · min−1 · enzyme−1) and higher Km (~19 μM) than MSMEI_3879GFP (Fig. 7E). The 9-fold lower Km of MSMEI_3879GFP suggests that the N terminus of MSMEI_3879 contributes to efficient degradation by ClpC1, but is not the sole determinant of recognition. Neither GFP fusion was degraded by ClpXP1P2 (Fig. S1B), demonstrating that MSMEI_3879 recognition is specific to ClpC1. We also assessed whether sequences at the beginning or end of MSMEI_3879 function as simple degrons by adding its first 13 residues to the beginning of GFP or its final 13 residues to the end of GFP. Neither construct was degraded with as low a Km or high a kcat as MSMEI_3879GFP. While we cannot rule out that longer sequences would be recognized more robustly, our results suggest that efficient recognition involves multivalent contacts on the folded MSMEI_3879 module, likely through the hydrophobic regions exposed by the truncation (Fig. S5). We tested this by assessing proteolysis of “full-length” MSMEI_3879, incorporating a restored N terminus created by removing the nucleotide insertion that causes a frameshift. As expected, this construct was not degraded by ClpC1P1P2 in vitro (Fig. S6), confirming that the truncation leads to recognition by ClpC1.
Given the lower Km for degradation of MSMEI_3879 compared to GFPssrA, we tested whether untagged MSMEI_3879 can compete with GFPssrA degradation and found that equimolar MSMEI_3879 effectively blocks GFPssrA proteolysis (Fig. 7F). Inhibition was a specific feature of MSMEI_3879, as DnaJ1, a non-substrate protein from our interactome data set (Fig. 7A), had no effect on degradation (Fig. 7F). Taken together, these data indicate MSMEI_3879 is a novel and robustly degraded ClpC1P1P2 substrate and that its N-terminal sequence contributes to its recognition.
Chemoinhibition of ClpC1 blocks MSMEI_3879 degradation.
We sought to assess whether MSMEI_3879 constructs would have utility as reporters for small molecule disruption of ClpC1. We used two well-characterized mycobacterial ClpC1 dysregulators, ecumicin (ECU) and rufomycin (RUF), which bind the ClpC1 NTD and dysregulate the ability of ClpC1 to degrade protein substrates (75–77). We assayed proteolysis of MSMEI_3879GFP and GFPssrA by ClpC1P1P2 in vitro in the presence and absence of 10 μM of each compound. Compared to the untreated control, both ECU and RUF reduced the rate of MSMEI_3879GFP degradation by about 60% (Fig. 8A). Inhibition of GFPssrA degradation was also observed, but with different magnitude for each compound: ECU inhibited by ~80%, while RUF inhibited by ~45% (Fig. 8B). Taken together, these results demonstrate that known ClpC1 dysregulators inhibit proteolysis of MSMEI_3879 by ClpC1P1P2.
FIG 8.

Effect of ClpC1-targeting antibiotics on proteolysis by ClpC1P1P2. Inclusion of 10 μM ECU or RUF inhibits degradation of 10 μM (A) MSMEI_3879GFP and (B) GFPssrA by 1 μM ClpC1P1P2 relative to a nontreatment control. Values are averages from three replicates (n = 3) ± 1 SD. P values were calculated by unpaired two-tailed Student’s t test. * and ** represent P values of <0.05 and 0.01, respectively.
DISCUSSION
It is now well established that Clp protease components, including ClpC1, are essential for mycobacterial viability (19, 27–31), and these enzymes have consequently emerged as promising antibiotic targets. Here, we sought to better understand the physiological roles of ClpC1P1P2 by identifying partners that interact with full-length and truncated ClpC1 constructs. In total, we found 370 unique cellular proteins that interact with one or more ClpC1 construct. The diversity of the identified ClpC1 interactome is consistent with a multifaceted role for this enzyme in mycobacterial physiology. Moreover, our data align with recent explorations of the M. tuberculosis ClpC1 interactome/degradome (21, 23), as 30 proteins found here possess orthologs identified in those screens.
Several factors likely limit our ability to capture the full breadth of cellular interaction partners. Indeed, some known partners were absent from our data set, including substrates WhiB1 and CarD (30) and the proteolytic adaptor ClpS (23). Our approach was generally biased toward abundant and stably bound partners. Proteolytic substrates—which are actively degraded by the protease and interact with ClpC1 only while ATP is abundant—may be more challenging to detect due to dissociation during capture and wash steps. Clp proteases themselves are known to assemble dynamically and disassemble when substrate or ATP is exhausted (13), which likely occurs during sample preparation. Finally, overexpression presumably creates a stoichiometric excess of ClpC1FLAG over ClpP1P2, which may bias capture toward certain classes of partners.
While ClpC1 likely participates in multiple proteolytic programs in mycobacteria, we note that not all interacting proteins identified here are substrates. While five of six candidates tested directly bind ClpC1 in vitro (see Fig. S2 in the supplemental material), only one, MSMEI_3879, proved to be a bona fide substrate (Fig. 7). Non-substrate interaction partners may play a role in regulating ClpC1 activity, may selectively interact with protease-deficient higher-order ClpC1 oligomers (25, 78, 79), or may simply bind nonspecifically under our capture conditions. It is also notable that capture experiments utilizing ClpC1EQ, a construct carrying Walker B mutations expected to stabilize interactions with proteolytic substrates (56, 60–62), yielded fewer identified interaction partners (93 proteins) than experiments with ClpC1WT (163 proteins) (Fig. 3, Table 1, and Table 2; see Table S2 in the supplemental material). This suggests that many of these interaction partners are not engaged as the substrates and instead adopt different modes of interaction that are dependent on nucleotide binding, ATP hydrolysis, and perhaps oligomeric state. A diverse set of dynamic interaction partners would plausibly allow mycobacterial cells to nimbly modulate ClpC1 activity across different growth conditions by altering the oligomeric state of ClpC1, changing substrate preferences, or tuning the stability of the entire Clp protease.
ClpC1 is composed of a globular N-terminal domain that binds to substrates and adaptors and an ATPase core consisting of two AAA+ modules that carry out chemomechanical substrate unfolding and translocation (8, 10, 80–82). Surprisingly, our studies identified few interaction partners that bind exclusively to either the NTD or the ATPase core. This suggests that many ClpC1-interacting proteins make multivalent binding to both components. Alternatively, this could result from truncated constructs (ClpC1NTD and ClpC1CORE) assembling with endogenous ClpC1 in the cell. Regardless of the mechanism involved, this illustrates that ClpC1 acts through the cooperation of its constituent parts.
Importantly, our interactome analysis led us to identify MSMEI_3879 as a bona fide ClpC1P1P2 substrate (Fig. 7). This protein joins a short list of verified mycobacterial Clp protease substrates (20, 21, 23, 24). MSMEI_3879 is degraded relatively efficiently: a GFP construct carrying an N-terminal MSMEI_3879 fusion was degraded with a Km ~7-fold lower than that of the model substrate GFPssrA (13, 17). As expected for this lower Km, degradation of GFPssrA was effectively blocked by an equimolar amount of MSMEI_3879 (Fig. 7F). Our experiments indicate that recognition of MSMEI_3879 by ClpC1 likely involves elements exposed as a result of its N-terminal truncation, due to a unique frameshift mutation (Fig. S4 and S5). Indeed, ClpC1 has been proposed to selectively recognize some substrates via hydrophobic regions and disordered termini (21). It is unclear whether MSMEI_3879 carries out any significant enzymatic function, or whether its proteolysis by ClpC1 plays any role in M. smegmatis physiology. Regardless, MSMEI_3879-based substrates may serve as useful tools for probing ClpC1P1P2 activity and dysregulation. As a proof of concept, we show that known ClpC1-targeting antimicrobials inhibit MSMEI_3879GFP proteolysis in vitro (Fig. 8). Similar substrates could serve as the basis for cell-based screens to identify novel ClpC1-targeting antibiotics.
MATERIALS AND METHODS
Plasmid and strain construction.
Full-length ClpC1 (ClpC1WT), the ClpC1 NTD alone (aa 1 to 147 [ClpC1NTD]), and ClpC1 lacking the NTD (aa 158 to 848 [ClpC1CORE]) were amplified from Mycolicibacterium smegmatis (strain ATCC 700084/MC2155) genomic DNA (ATCC) and cloned into a modified episomal pNIT expression vector (83), followed by a C-terminal 3×FLAG tag. Mutations to Walker B motifs in the D1 (E288Q) and D2 (E626Q) rings of ClpC1WT were introduced by sequential polymerase chain reactions, yielding an ATPase inactive “trap” variant (ClpC1EQ) (56). Plasmid sequences were verified by Sanger sequencing (Genewiz).
In vivo substrate trapping.
Plasmids encoding M. smegmatis ClpC1 constructs were electroporated into M. smegmatis (ATCC 700084/MC2155) at 5 kV in a MicroPulser (Bio-Rad). Liquid starter cultures were made in Middlebrook broth base (HiMedia) supplemented with 0.2% (vol/vol) glycerol (Fisher Scientific), 0.2% (wt/vol) glucose (TCI), and 0.05% (vol/vol) Tween 80 and then (with orbital shaking) grown for 60 h at 37°C. At a starting A600 of 0.05, starter cultures were subcultured into 200 mL of fresh medium. The cultures were then grown at 37°C until the mid-log phase (A600 ≈ 0.6 to 1.0), at which point expression was induced by 28 mM ε-caprolactam (Sigma-Aldrich) for 20 h. All constructs continued to grow similarly over the course of expression. For cell harvesting, centrifugation was carried out at 9,000 × g for 20 min at 4°C and pellets were resuspended in 5 mL of lysis buffer (25 mM HEPES, 10 mM magnesium chloride, 200 mM potassium chloride, and 0.1 mM EDTA, supplemented with 10 mM ATP [pH 7.5]). Cell lysis was done in a microfluidizer (Microfluidics), and subsequent clarification of lysates was performed at 16,000 × g for 30 min at 4°C. Cell supernatants were stored at −80°C. A Bradford assay (Bio-Rad) was performed to estimate total protein content.
Coimmunoprecipitation.
Forty microliters of EZView Red anti-FLAG M2-affinity gel beads (Sigma-Aldrich) was equilibrated and washed twice in 0.5 mL lysis buffer by centrifugation at 8,200 × g for 30 s. To pull down the expressed 3×FLAG-tagged M. smegmatis ClpC1 constructs and interacting proteins, the equilibrated beads were incubated with 1 mL of lysate for 1 h at 4°C with gentle agitation. After incubation, the bead-lysate slurry was spun at 8,200 × g for 30 s and the bead pellet was subsequently washed with lysis buffer (containing 10 mM ATP). To elute, the washed beads were then mixed with 20 μL of 2× Laemmli sample buffer (10% glycerol, 4% SDS, 167 mM Tris-HCl, 0.02% bromophenol blue) in the absence of reducing agent and boiled for 5 min. Samples were then vortexed briefly and centrifuged, and the supernatant containing the eluate was stored at −80°C prior to further analysis. As a negative control, lysates of cells containing empty pNIT vector were processed by the same workflow.
Mass spectrometry sample preparation.
Proteins in the eluate were analyzed on a 6 to 15% SDS-PAGE gradient gel. Each lane was diced, and gel pieces were put into 1.5-mL microcentrifuge tubes. The samples were prepared by adapting standard protocols for in-gel mass spectrometry sample preparation (84). Incubation with 10 mM dithiothreitol (DTT) was carried out for 45 min at 55°C in order to reduce disulfide bonds. Afterwards, carbamidomethylation of cysteines was with 55 μM iodoacetamide for 30 min at room temperature (in the dark). Gel pieces were washed with gel wash buffer (25 mM ammonium bicarbonate and 50% acetonitrile) and dehydrated with 100% acetonitrile prior to drying in a SpeedVac vacuum centrifuge (Thermo). Trypsin digestion was carried out in 25 mM ammonium bicarbonate containing 10 μg mL−1 MS-grade trypsin protease (Pierce), and the mixture was incubated overnight at 37°C. Peptides were subsequently extracted by a two-step process, which was repeated twice: incubation of gel pieces with 5% formic acid (Sigma-Aldrich), followed by 100% acetonitrile. To remove residual salts, the peptide samples were desalted using a HyperSep C18 column (Thermo Fisher Scientific) according to a previously described procedure (84, 85). C18 column-eluted samples were dried by vacuum centrifugation and frozen prior to mass spectrometry.
LC-MS/MS.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis was performed using a Q Exactive Orbitrap interfaced with Ultimate 3000 Nano-LC system (Thermo Fisher Scientific). Trypsin-digested samples were loaded on an Acclaim PepMap rapid-separation liquid chromatography (RSLC) column (75-μm by 15-cm nanoViper) using an autosampler. Analysis of samples was done using a 150-min gradient running from 2% to 95% buffer B (0.1% formic acid in acetonitrile) in buffer A (0.1% formic acid in water) at a flow rate of 0.3 μL min−1. MS data acquisition was done using a data-dependent top10 method, with the most abundant precursor ions from the survey scan chosen for higher-energy collisional dissociation (HCD) fragmentation using stepped normalized collision energies of 28, 30, and 35 eV. Survey scans were acquired at a resolution of 70,000 at m/z 200 on the Q Exactive. LS-MS/MS data were collected in independent biological triplicates.
MS data analysis.
For proteomic analysis, extraction of raw data was performed in the Proteome Discoverer software suite (version 1.4; Thermo Fisher Scientific). The raw data were searched against Mycolicibacterium smegmatis (strain ATCC 700084/MC2155) UniProt Reference Proteome (Proteome ID UP000000757) using Sequest HT (University of Washington and Thermo Fisher Scientific). Iodoacetamide-mediated cysteine carbamidomethylation was set as a static modification. Precursor mass tolerance was set at 10 ppm, while allowing for fragment ion mass deviation of 0.6 Da for the HCD data, and full trypsinization with a maximum of two missed cleavages. Peptide-spectrum match (PSM) validation was done using Percolator, with false-discovery rates (FDRs) of 1% and 5% for stringent and relaxed validation, respectively. Gene Ontology (GO) annotation analysis on the data sets was performed using the Blast2GO software suite (64). Some GO annotation terms were binned into a finite number of final terms.
Expression and purification of recombinant proteins.
N-terminally H7-SUMO-tagged ClpC1 (with or without a C-terminal FLAG tag), N-terminally H7-SUMO-tagged ClpX, and C-terminal H6-tagged ClpP1 and ClpP2 were cloning into pET22b-derived vectors using Gibson Assembly (86). N-terminal H7-SUMO-tagged putative substrates and interaction partners were obtained as synthetic gene constructs (Twist Bioscience) and cloned into pET29b(+). All constructs were expressed in E. coli ER2566 (NEB). Cultures were grown in 1.5×YT at 37°C to exponential phase (A600 of 0.8 to 1.0), and overexpression was induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG), followed by incubation at 30°C for 4 h. Cells were harvested at 4,000 × g for 30 min, and pellets were resuspended in 25 mL His tag lysis buffer (25 mM HEPES [pH 7.5], 300 mM NaCl, 10 mM imidazole [pH 7.5], 10% glycerol), supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF) and 100 μL of EDTA-free protease inhibitor cocktail (Thermo Fisher). After sonication and clarification at 15,000 × g for 30 min, lysates were loaded onto a Ni-nitrilotriacetic acid (NTA) column (MCLAB), washed with 25 mM imidazole, and eluted in 300 mM imidazole. The eluate was spin concentrated (10,000-molecular-weight-cutoff [MWCO]; Amicon, MilliporeSigma) at 4,000 × g. Protein samples were further purified by anion-exchange chromatography (Source 15Q 10/100; Cytiva). H7-SUMO tags were removed by incubation with Saccharomyces cerevisiae SUMO protease Ulp1 (87) or left intact for microscale thermophoresis studies. Constructs were additionally purified by gel filtration (HiLoad 16/600 Superdex 200; Cytiva) into protein degradation buffer (25 mM HEPES, 200 mM potassium chloride, 10 mM magnesium chloride, 0.1 mM EDTA [pH 7.5]).
In vitro assays.
In vitro degradation assays containing 1 μM ClpC1 (hexamer), 1 μM ClpP1 (tetradecamer), 1 μM ClpP2 (tetradecamer), and 10 μM substrate were performed in ClpC1 protein degradation buffer. All degradation assays were carried out in the presence of 50 μM activator peptide Z-Leu-Leu-Nva-CHO (benzyloxycarbonyl-l-leucyl-l-leucyl-l-norvalinal) (12, 13), with a total of 15 mM ATP, along with an ATP regeneration system consisting of 187.5 U mL−1 pyruvate kinase and 50 mM phosphoenolpyruvate (Sigma). For gel degradation assays, 14-μL aliquots were taken at each time point, mixed with 7 μL 2× Laemmli sample buffer (containing 10% β-mercaptoethanol), and analyzed by SDS-PAGE. Gels were stained by 0.1% Coomassie brilliant blue and quantified by ImageJ (88). Plate reader assays were carried out on a Tecan Spark instrument. Degradation of GFP-substrate fusions was monitored by loss of 511-nm emission following excitation at 450 nm. ATPase assays utilized 1 μM ClpC1, 10 mM ATP, and an NADH-coupled ATP regeneration system (89). Consumption of ATP was followed by monitoring the decrease in NADH absorbance at 340 nm. Microscale thermophoresis was performed in a Monolith NT.115 (NanoTemper) using 0.1 μM H7-SUMOClpC1FLAG (hexamer) and 0.1 μM His Lite OG488-Tris-NTA-Ni dye (AAT Bioquest). Data were fit to a Hill-form binding equation in Prism (GraphPad).
CRISPRi assays.
Integrative plasmid PLJR962 (Addgene) (90), which encodes dCas9 machinery driven by an anhydrotetracycline (aTc)-responsive promoter for CRISPR interference (CRISPRi) in M. smegmatis, was modified by Gibson Assembly to contain either a nontargeting (GAGACGATTAATGCGTCTCG) or clpC1-targeting (ATGAGCGCGTCGTCGTCGCCGAA) small guide RNA (sgRNA). Versions of these plasmids were constructed by Gibson Assembly to contain a secondary clpC1 (or clpC1FLAG, clpC1EQ,FLAG) locus downstream of the CRISPRi machinery. The plasmid-borne copy of clpC1 was modified to incorporate strategic codon substitutions to the sgRNA-targeted region that prevent sgRNA binding and thus escape transcriptional knockdown. Plasmids were transformed into M. smegmatis, and growth was monitored on Middlebrook agar plates at 37°C at various levels of aTc induction.
Data availability.
The mass spectrometry data from this work have been submitted to the ProteomeXchange Consortium via the PRIDE partner repository (91) and assigned the identifier PXD030385.
ACKNOWLEDGMENTS
We gratefully thank R. Neunuebel and V. Parashar for the use of instrumentation. We thank Papa-Nii Asare-Okai for technical support. We also thank S. Cho and S. Franzblau of the Institute for Tuberculosis Research at the University of Illinois at Chicago for their gift of ecumicin and rufomycin.
P.C.B. was supported by T32GM133395. K.R.S. was supported by NIH NIGMS award P20GM104316. The UD Department of Chemistry and Biochemistry Mass Spectrometry core was additionally supported by NIH NIGMS award P30GM110758-02. This content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
E.C.O. and K.R.S. designed research. E.C.O., H.R.A, P.C.B., and P.B. performed research. E.C.O. and K.R.S. analyzed data. E.C.O. and K.R.S. wrote the paper.
Footnotes
Supplemental material is available online only.
Contributor Information
Karl R. Schmitz, Email: schmitzk@udel.edu.
Emily Weinert, The Pennsylvania State University.
REFERENCES
- 1.WHO. 2021. Global tuberculosis report 2021. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Compton CL, Schmitz KR, Sauer RT, Sello JK. 2013. Antibacterial activity of and resistance to small molecule inhibitors of the ClpP peptidase. ACS Chem Biol 8:2669–2677. doi: 10.1021/cb400577b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gavrish E, Sit CS, Cao S, Kandror O, Spoering A, Peoples A, Ling L, Fetterman A, Hughes D, Bissell A, Torrey H, Akopian T, Mueller A, Epstein S, Goldberg A, Clardy J, Lewis K. 2014. Lassomycin, a ribosomally synthesized cyclic peptide, kills Mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem Biol 21:509–518. doi: 10.1016/j.chembiol.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gao W, Kim JY, Anderson JR, Akopian T, Hong S, Jin YY, Kandror O, Kim JW, Lee IA, Lee SY, McAlpine JB, Mulugeta S, Sunoqrot S, Wang Y, Yang SH, Yoon TM, Goldberg AL, Pauli GF, Suh JW, Franzblau SG, Cho S. 2015. The cyclic peptide ecumicin targeting ClpC1 is active against Mycobacterium tuberculosis in vivo. Antimicrob Agents Chemother 59:880–889. doi: 10.1128/AAC.04054-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moreira W, Ngan GJ, Low JL, Poulsen A, Chia BC, Ang MJ, Yap A, Fulwood J, Lakshmanan U, Lim J, Khoo AY, Flotow H, Hill J, Raju RM, Rubin EJ, Dick T. 2015. Target mechanism-based whole-cell screening identifies bortezomib as an inhibitor of caseinolytic protease in mycobacteria. mBio 6:e00253-15. doi: 10.1128/mBio.00253-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Famulla K, Sass P, Malik I, Akopian T, Kandror O, Alber M, Hinzen B, Ruebsamen-Schaeff H, Kalscheuer R, Goldberg AL, Brotz-Oesterhelt H. 2016. Acyldepsipeptide antibiotics kill mycobacteria by preventing the physiological functions of the ClpP1P2 protease. Mol Microbiol 101:194–209. doi: 10.1111/mmi.13362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moreno-Cinos C, Goossens K, Salado IG, Van Der Veken P, De Winter H, Augustyns K. 2019. ClpP protease, a promising antimicrobial target. Int J Mol Sci 20:2232. doi: 10.3390/ijms20092232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sauer RT, Baker TA. 2011. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 9.Baker TA, Sauer RT. 2012. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta 1823:15–28. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beardslee PC, Dhamdhere G, Jiang J, Ogbonna EC, Presloid CJ, Prorok M, Bheemreddy P, Sullivan CD, Vorn JC, Schmitz KR. 2021. Clp proteases, p 292–306. In Jez J (ed), Encyclopedia of biological chemistry, 3rd ed. Elsevier, Oxford, United Kingdom. [Google Scholar]
- 11.Wang J, Hartling JA, Flanagan JM. 1997. The structure of ClpP at 2.3 Å resolution suggests a model for ATP-dependent proteolysis. Cell 91:447–456. doi: 10.1016/s0092-8674(00)80431-6. [DOI] [PubMed] [Google Scholar]
- 12.Akopian T, Kandror O, Raju RM, Unnikrishnan M, Rubin EJ, Goldberg AL. 2012. The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J 31:1529–1541. doi: 10.1038/emboj.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmitz KR, Sauer RT. 2014. Substrate delivery by the AAA+ ClpX and ClpC1 unfoldases activates the mycobacterial ClpP1P2 peptidase. Mol Microbiol 93:617–628. doi: 10.1111/mmi.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmitz KR, Carney DW, Sello JK, Sauer RT. 2014. Crystal structure of Mycobacterium tuberculosis ClpP1P2 suggests a model for peptidase activation by AAA+ partner binding and substrate delivery. Proc Natl Acad Sci USA 111:E4587–E4595. doi: 10.1073/pnas.1417120111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li M, Kandror O, Akopian T, Dharkar P, Wlodawer A, Maurizi MR, Goldberg AL. 2016. Structure and functional properties of the active form of the proteolytic complex, ClpP1P2, from Mycobacterium tuberculosis. J Biol Chem 291:7465–7476. doi: 10.1074/jbc.M115.700344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vahidi S, Ripstein ZA, Juravsky JB, Rennella E, Goldberg AL, Mittermaier AK, Rubinstein JL, Kay LE. 2020. An allosteric switch regulates Mycobacterium tuberculosis ClpP1P2 protease function as established by cryo-EM and methyl-TROSY NMR. Proc Natl Acad Sci USA 117:5895–5906. doi: 10.1073/pnas.1921630117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leodolter J, Warweg J, Weber-Ban E. 2015. The Mycobacterium tuberculosis ClpP1P2 protease interacts asymmetrically with its ATPase partners ClpX and ClpC1. PLoS One 10:e0125345. doi: 10.1371/journal.pone.0125345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alhuwaider AAH, Dougan DA. 2017. AAA+ machines of protein destruction in mycobacteria. Front Mol Biosci 4:49. doi: 10.3389/fmolb.2017.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raju RM, Unnikrishnan M, Rubin DH, Krishnamoorthy V, Kandror O, Akopian TN, Goldberg AL, Rubin EJ. 2012. Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog 8:e1002511. doi: 10.1371/journal.ppat.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gopal P, Sarathy JP, Yee M, Ragunathan P, Shin J, Bhushan S, Zhu J, Akopian T, Kandror O, Lim TK, Gengenbacher M, Lin Q, Rubin EJ, Grüber G, Dick T. 2020. Pyrazinamide triggers degradation of its target aspartate decarboxylase. Nat Commun 11:1661. doi: 10.1038/s41467-020-15516-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lunge A, Gupta R, Choudhary E, Agarwal N. 2020. The unfoldase ClpC1 of Mycobacterium tuberculosis regulates the expression of a distinct subset of proteins having intrinsically disordered termini. J Biol Chem 295:9455–9473. doi: 10.1074/jbc.RA120.013456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marsee JD, Ridings A, Yu T, Miller JM. 2018. Mycobacterium tuberculosis ClpC1 N-terminal domain is dispensable for adaptor protein-dependent allosteric regulation. Int J Mol Sci 19:3651. doi: 10.3390/ijms19113651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziemski M, Leodolter J, Taylor G, Kerschenmeyer A, Weber-Ban E. 2021. Genome-wide interaction screen for Mycobacterium tuberculosis ClpCP protease reveals toxin-antitoxin systems as a major substrate class. FEBS J 288:99–114. doi: 10.1111/febs.15335. [DOI] [PubMed] [Google Scholar]
- 24.Barik S, Sureka K, Mukherjee P, Basu J, Kundu M. 2010. RseA, the SigE specific anti-sigma factor of Mycobacterium tuberculosis, is inactivated by phosphorylation-dependent ClpC1P2 proteolysis. Mol Microbiol 75:592–606. doi: 10.1111/j.1365-2958.2009.07008.x. [DOI] [PubMed] [Google Scholar]
- 25.Weinhäupl K, Brennich M, Kazmaier U, Lelievre J, Ballell L, Goldberg A, Schanda P, Fraga H. 2018. The antibiotic cyclomarin blocks arginine-phosphate-induced millisecond dynamics in the N-terminal domain of ClpC1 from Mycobacterium tuberculosis. J Biol Chem 293:8379–8393. doi: 10.1074/jbc.RA118.002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ogbonna EC, Anderson HR, Schmitz KR. 2022. Identification of arginine phosphorylation in Mycolicibacterium smegmatis. Microbiol Spectr 10:e02042-22. doi: 10.1128/spectrum.02042-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 28.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ollinger J, O'Malley T, Kesicki EA, Odingo J, Parish T. 2012. Validation of the essential ClpP protease in Mycobacterium tuberculosis as a novel drug target. J Bacteriol 194:663–668. doi: 10.1128/JB.06142-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raju RM, Jedrychowski MP, Wei JR, Pinkham JT, Park AS, O'Brien K, Rehren G, Schnappinger D, Gygi SP, Rubin EJ. 2014. Post-translational regulation via Clp protease is critical for survival of Mycobacterium tuberculosis. PLoS Pathog 10:e1003994. doi: 10.1371/journal.ppat.1003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, Boutte CC, Rubin EJ, Schnappinger D, Ehrt S, Fortune SM, Sassetti CM, Ioerger TR. 2017. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio 8:e02133-16. doi: 10.1128/mBio.02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brotz-Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, Schroeder W, Hinzen B, Raddatz S, Paulsen H, Henninger K, Bandow JE, Sahl HG, Labischinski H. 2005. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med 11:1082–1087. doi: 10.1038/nm1306. [DOI] [PubMed] [Google Scholar]
- 33.Schmitz KR, Handy EL, Compton CL, Gupta S, Bishai WR, Sauer RT, Sello JK. 2023. Acyldepsipeptide antibiotics and a bioactive fragment thereof differentially perturb Mycobacterium tuberculosis ClpXP1P2 activity in vitro. ACS Chem Biol 18:724–733. doi: 10.1021/acschembio.9b00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Compton CL, Carney DW, Groomes PV, Sello JK. 2015. Fragment-based strategy for investigating and suppressing the efflux of bioactive small molecules. ACS Infect Dis 1:53–58. doi: 10.1021/id500009f. [DOI] [PubMed] [Google Scholar]
- 35.Wong KS, Mabanglo MF, Seraphim TV, Mollica A, Mao YQ, Rizzolo K, Leung E, Moutaoufik MT, Hoell L, Phanse S, Goodreid J, Barbosa LRS, Ramos CHI, Babu M, Mennella V, Batey RA, Schimmer AD, Houry WA. 2018. Acyldepsipeptide analogs dysregulate human mitochondrial ClpP protease activity and cause apoptotic cell death. Cell Chem Biol 25:1017–1030.e9. doi: 10.1016/j.chembiol.2018.05.014. [DOI] [PubMed] [Google Scholar]
- 36.Kang SG, Dimitrova MN, Ortega J, Ginsburg A, Maurizi MR. 2005. Human mitochondrial ClpP is a stable heptamer that assembles into a tetradecamer in the presence of ClpX. J Biol Chem 280:35424–35432. doi: 10.1074/jbc.M507240200. [DOI] [PubMed] [Google Scholar]
- 37.Kang SG, Ortega J, Singh SK, Wang N, Huang NN, Steven AC, Maurizi MR. 2002. Functional proteolytic complexes of the human mitochondrial ATP-dependent protease, hClpXP. J Biol Chem 277:21095–21102. doi: 10.1074/jbc.M201642200. [DOI] [PubMed] [Google Scholar]
- 38.Kirstein J, Moliere N, Dougan DA, Turgay K. 2009. Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat Rev Microbiol 7:589–599. doi: 10.1038/nrmicro2185. [DOI] [PubMed] [Google Scholar]
- 39.Schmitt EK, Riwanto M, Sambandamurthy V, Roggo S, Miault C, Zwingelstein C, Krastel P, Noble C, Beer D, Rao SP, Au M, Niyomrattanakit P, Lim V, Zheng J, Jeffery D, Pethe K, Camacho LR. 2011. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew Chem Int Ed Engl 50:5889–5891. doi: 10.1002/anie.201101740. [DOI] [PubMed] [Google Scholar]
- 40.Vasudevan D, Rao SP, Noble CG. 2013. Structural basis of mycobacterial inhibition by cyclomarin A. J Biol Chem 288:30883–30891. doi: 10.1074/jbc.M113.493767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bürstner N, Roggo S, Ostermann N, Blank J, Delmas C, Freuler F, Gerhartz B, Hinniger A, Hoepfner D, Liechty B, Mihalic M, Murphy J, Pistorius D, Rottmann M, Thomas JR, Schirle M, Schmitt EK. 2015. Gift from nature: cyclomarin A kills mycobacteria and malaria parasites by distinct modes of action. Chembiochem 16:2433–2436. doi: 10.1002/cbic.201500472. [DOI] [PubMed] [Google Scholar]
- 42.Jung IP, Ha NR, Kim AR, Kim SH, Yoon MY. 2017. Mutation analysis of the interactions between Mycobacterium tuberculosis caseinolytic protease C1 (ClpC1) and ecumicin. Int J Biol Macromol 101:348–357. doi: 10.1016/j.ijbiomac.2017.03.126. [DOI] [PubMed] [Google Scholar]
- 43.Li L, MacIntyre LW, Ali T, Russo R, Koirala B, Hernandez Y, Brady SF. 2021. Biosynthetic interrogation of soil metagenomes reveals metamarin, an uncommon cyclomarin congener with activity against Mycobacterium tuberculosis. J Nat Prod 84:1056–1066. doi: 10.1021/acs.jnatprod.0c01104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaillot O, Pellegrini E, Bregenholt S, Nair S, Berche P. 2000. The ClpP serine protease is essential for the intracellular parasitism and virulence of Listeria monocytogenes. Mol Microbiol 35:1286–1294. doi: 10.1046/j.1365-2958.2000.01773.x. [DOI] [PubMed] [Google Scholar]
- 45.Frees D, Qazi SN, Hill PJ, Ingmer H. 2003. Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence. Mol Microbiol 48:1565–1578. doi: 10.1046/j.1365-2958.2003.03524.x. [DOI] [PubMed] [Google Scholar]
- 46.Kwon HY, Ogunniyi AD, Choi MH, Pyo SN, Rhee DK, Paton JC. 2004. The ClpP protease of Streptococcus pneumoniae modulates virulence gene expression and protects against fatal pneumococcal challenge. Infect Immun 72:5646–5653. doi: 10.1128/IAI.72.10.5646-5653.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ES, Siddiqui SM, Wah DA, Baker TA. 2004. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 119:9–18. doi: 10.1016/j.cell.2004.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neher SB, Villen J, Oakes EC, Bakalarski CE, Sauer RT, Gygi SP, Baker TA. 2006. Proteomic profiling of ClpXP substrates after DNA damage reveals extensive instability within SOS regulon. Mol Cell 22:193–204. doi: 10.1016/j.molcel.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 49.Frees D, Sorensen K, Ingmer H. 2005. Global virulence regulation in Staphylococcus aureus: pinpointing the roles of ClpP and ClpX in the sar/agr regulatory network. Infect Immun 73:8100–8108. doi: 10.1128/IAI.73.12.8100-8108.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Estorninho M, Smith H, Thole J, Harders-Westerveen J, Kierzek A, Butler RE, Neyrolles O, Stewart GR. 2010. ClgR regulation of chaperone and protease systems is essential for Mycobacterium tuberculosis parasitism of the macrophage. Microbiology (Reading) 156:3445–3455. doi: 10.1099/mic.0.042275-0. [DOI] [PubMed] [Google Scholar]
- 51.Fernandez L, Breidenstein EB, Song D, Hancock RE. 2012. Role of intracellular proteases in the antibiotic resistance, motility, and biofilm formation of Pseudomonas aeruginosa. Antimicrob Agents Chemother 56:1128–1132. doi: 10.1128/AAC.05336-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trentini DB, Suskiewicz MJ, Heuck A, Kurzbauer R, Deszcz L, Mechtler K, Clausen T. 2016. Arginine phosphorylation marks proteins for degradation by a Clp protease. Nature 539:48–53. doi: 10.1038/nature20122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhandari V, Wong KS, Zhou JL, Mabanglo MF, Batey RA, Houry WA. 2018. The role of ClpP protease in bacterial pathogenesis and human diseases. ACS Chem Biol 13:1413–1425. doi: 10.1021/acschembio.8b00124. [DOI] [PubMed] [Google Scholar]
- 54.Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. 2003. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell 11:671–683. doi: 10.1016/s1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 55.Feng J, Michalik S, Varming AN, Andersen JH, Albrecht D, Jelsbak L, Krieger S, Ohlsen K, Hecker M, Gerth U, Ingmer H, Frees D. 2013. Trapping and proteomic identification of cellular substrates of the ClpP protease in Staphylococcus aureus. J Proteome Res 12:547–558. doi: 10.1021/pr300394r. [DOI] [PubMed] [Google Scholar]
- 56.Graham JW, Lei MG, Lee CY. 2013. Trapping and identification of cellular substrates of the Staphylococcus aureus ClpC chaperone. J Bacteriol 195:4506–4516. doi: 10.1128/JB.00758-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamada-Inagawa T, Okuno T, Karata K, Yamanaka K, Ogura T. 2003. Conserved pore residues in the AAA protease FtsH are important for proteolysis and its coupling to ATP hydrolysis. J Biol Chem 278:50182–50187. doi: 10.1074/jbc.M308327200. [DOI] [PubMed] [Google Scholar]
- 58.Barkow SR, Levchenko I, Baker TA, Sauer RT. 2009. Polypeptide translocation by the AAA+ ClpXP protease machine. Chem Biol 16:605–612. doi: 10.1016/j.chembiol.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang F, Wu Z, Zhang P, Tian G, Finley D, Shi Y. 2009. Mechanism of substrate unfolding and translocation by the regulatory particle of the proteasome from Methanocaldococcus jannaschii. Mol Cell 34:485–496. doi: 10.1016/j.molcel.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walker JE, Saraste M, Runswick MJ, Gay NJ. 1982. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J 1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weibezahn J, Schlieker C, Bukau B, Mogk A. 2003. Characterization of a trap mutant of the AAA+ chaperone ClpB. J Biol Chem 278:32608–32617. doi: 10.1074/jbc.M303653200. [DOI] [PubMed] [Google Scholar]
- 62.Montandon C, Friso G, Liao JR, Choi J, van Wijk KJ. 2019. In vivo trapping of proteins interacting with the chloroplast CLPC1 chaperone: potential substrates and adaptors. J Proteome Res 18:2585–2600. doi: 10.1021/acs.jproteome.9b00112. [DOI] [PubMed] [Google Scholar]
- 63.Wang F, Mei Z, Qi Y, Yan C, Hu Q, Wang J, Shi Y. 2011. Structure and mechanism of the hexameric MecA-ClpC molecular machine. Nature 471:331–335. doi: 10.1038/nature09780. [DOI] [PubMed] [Google Scholar]
- 64.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 65.Prabudiansyah I, Kusters I, Caforio A, Driessen AJ. 2015. Characterization of the annular lipid shell of the Sec translocon. Biochim Biophys Acta 1848:2050–2056. doi: 10.1016/j.bbamem.2015.06.024. [DOI] [PubMed] [Google Scholar]
- 66.Miller BK, Zulauf KE, Braunstein M. 2017. The Sec pathways and exportomes of Mycobacterium tuberculosis. Microbiol Spectr doi: 10.1128/microbiolspec.TBTB2-0013-2016. [DOI] [PubMed] [Google Scholar]
- 67.Ribeiro-Guimarães ML, Pessolani MC. 2007. Comparative genomics of mycobacterial proteases. Microb Pathog 43:173–178. doi: 10.1016/j.micpath.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 68.Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY, Halim VA, Bagshaw RD, Hubner NC, Al-Hakim A, Bouchard A, Faubert D, Fermin D, Dunham WH, Goudreault M, Lin ZY, Badillo BG, Pawson T, Durocher D, Coulombe B, Aebersold R, Superti-Furga G, Colinge J, Heck AJ, Choi H, Gstaiger M, Mohammed S, Cristea IM, Bennett KL, Washburn MP, Raught B, Ewing RM, Gingras AC, Nesvizhskii AI. 2013. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods 10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tobias JW, Shrader TE, Rocap G, Varshavsky A. 1991. The N-end rule in bacteria. Science 254:1374–1377. doi: 10.1126/science.1962196. [DOI] [PubMed] [Google Scholar]
- 70.Marjanovic A, Rozeboom HJ, de Vries MS, Mayer C, Otzen M, Wijma HJ, Janssen DB. 2021. Catalytic and structural properties of ATP-dependent caprolactamase from Pseudomonas jessenii. Proteins 89:1079–1098. doi: 10.1002/prot.26082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Van der Werf P, Orlowski M, Meister A. 1971. Enzymatic conversion of 5-oxo-l-proline (l-pyrrolidone carboxylate) to l-glutamate coupled with cleavage of adenosine triphosphate to adenosine diphosphate, a reaction in the -glutamyl cycle. Proc Natl Acad Sci USA 68:2982–2985. doi: 10.1073/pnas.68.12.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim JM, Shimizu S, Yamada H. 1987. Amidohydrolysis of N-methylhydantoin coupled with ATP hydrolysis. Biochem Biophys Res Commun 142:1006–1012. doi: 10.1016/0006-291x(87)91514-2. [DOI] [PubMed] [Google Scholar]
- 73.Gupta RS, Lo B, Son J. 2018. Phylogenomics and comparative genomic studies robustly support division of the genus Mycobacterium into an emended genus Mycobacterium and four novel genera. Front Microbiol 9:67. doi: 10.3389/fmicb.2018.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Choules MP, Wolf NM, Lee H, Anderson JR, Grzelak EM, Wang Y, Ma R, Gao W, McAlpine JB, Jin YY, Cheng J, Lee H, Suh JW, Duc NM, Paik S, Choe JH, Jo EK, Chang CL, Lee JS, Jaki BU, Pauli GF, Franzblau SG, Cho S. 2019. Rufomycin targets ClpC1 proteolysis in Mycobacterium tuberculosis and M. abscessus. Antimicrob Agents Chemother 63:e02204-18. doi: 10.1128/AAC.02204-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gao X, Yeom J, Groisman EA. 2019. The expanded specificity and physiological role of a widespread N-degron recognin. Proc Natl Acad Sci USA 116:18629–18637. doi: 10.1073/pnas.1821060116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wolf NM, Lee H, Choules MP, Pauli GF, Phansalkar R, Anderson JR, Gao W, Ren J, Santarsiero BD, Lee H, Cheng J, Jin YY, Ho NA, Duc NM, Suh JW, Abad-Zapatero C, Cho S. 2019. High-resolution structure of ClpC1-rufomycin and ligand binding studies provide a framework to design and optimize anti-tuberculosis leads. ACS Infect Dis 5:829–840. doi: 10.1021/acsinfecdis.8b00276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taylor G, Frommherz Y, Katikaridis P, Layer D, Sinning I, Carroni M, Weber-Ban E, Mogk A. 2022. Antibacterial peptide CyclomarinA creates toxicity by deregulating the Mycobacterium tuberculosis ClpC1/ClpP1P2 protease. J Biol Chem 298:102202. doi: 10.1016/j.jbc.2022.102202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maurer M, Linder D, Franke KB, Jäger J, Taylor G, Gloge F, Gremer S, Le Breton L, Mayer MP, Weber-Ban E, Carroni M, Bukau B, Mogk A. 2019. Toxic activation of an AAA+ protease by the antibacterial drug Cyclomarin A. Cell Chem Biol 26:1169–1179.e4. doi: 10.1016/j.chembiol.2019.05.008. [DOI] [PubMed] [Google Scholar]
- 80.Schirmer EC, Glover JR, Singer MA, Lindquist S. 1996. HSP100/Clp proteins: a common mechanism explains diverse functions. Trends Biochem Sci 21:289–296. doi: 10.1016/S0968-0004(96)10038-4. [DOI] [PubMed] [Google Scholar]
- 81.Kar NP, Sikriwal D, Rath P, Choudhary RK, Batra JK. 2008. Mycobacterium tuberculosis ClpC1: characterization and role of the N-terminal domain in its function. FEBS J 275:6149–6158. doi: 10.1111/j.1742-4658.2008.06738.x. [DOI] [PubMed] [Google Scholar]
- 82.Snider J, Thibault G, Houry WA. 2008. The AAA+ superfamily of functionally diverse proteins. Genome Biol 9:216. doi: 10.1186/gb-2008-9-4-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pandey AK, Raman S, Proff R, Joshi S, Kang CM, Rubin EJ, Husson RN, Sassetti CM. 2009. Nitrile-inducible gene expression in mycobacteria. Tuberculosis (Edinb) 89:12–16. doi: 10.1016/j.tube.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gundry RL, White MY, Murray CI, Kane LA, Fu Q, Stanley BA, Van Eyk JE. 2009. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr Protoc Mol Biol Chapter 10:Unit10.25. doi: 10.1002/0471142727.mb1025s88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wiśniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat Methods 6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 86.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 87.Malakhov MP, Mattern MR, Malakhova OA, Drinker M, Weeks SD, Butt TR. 2004. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. J Struct Funct Genomics 5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- 88.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nørby J. 1988. Coupled assay of Na+,K+-ATPase activity. Methods Enzymol 156:116–119. doi: 10.1016/0076-6879(88)56014-7. [DOI] [PubMed] [Google Scholar]
- 90.Rock JM, Hopkins FF, Chavez A, Diallo M, Chase MR, Gerrick ER, Pritchard JR, Church GM, Rubin EJ, Sassetti CM, Schnappinger D, Fortune SM. 2017. Programmable transcriptional repression in mycobacteria using an orthogonal CRISPR interference platform. Nat Microbiol 2:16274. doi: 10.1038/nmicrobiol.2016.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Pérez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yilmaz S, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A, Vizcaíno JA. 2019. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download spectrum.04548-22-s0001.xlsx, XLSX file, 0.09 MB (95.5KB, xlsx)
Supplemental material. Download spectrum.04548-22-s0002.xlsx, XLSX file, 0.3 MB (262.9KB, xlsx)
Supplemental material. Download spectrum.04548-22-s0003.pdf, PDF file, 14.9 MB (14.9MB, pdf)
Data Availability Statement
The mass spectrometry data from this work have been submitted to the ProteomeXchange Consortium via the PRIDE partner repository (91) and assigned the identifier PXD030385.