

Significance
Understanding genetic heterogeneity and the dynamics of AMR associated with clinically important bacterial pathogens is crucial, as they vary geographically and with time. Here, combining whole-genome sequencing and proteome analysis, we decoded ARGs’ heterogeneity and characterized their potency and functionality in E. coli, K. pneumoniae, A. baumannii, P. aeruginosa, and S. enterica. The findings provide insights into the pathogen-specific signatures of ARGs, as well as genome dynamics at a highly resolved taxonomic level. The current research will lay a foundation to support the development of diagnostic and therapeutic procedures, as well as the formulation of an acceptable antibiotic usage policy.
Keywords: antimicrobials, antimicrobial resistance, Gram-negative pathogens, genomics, proteomics
Abstract
Microbes evolve rapidly by modifying their genomes through mutations or through the horizontal acquisition of mobile genetic elements (MGEs) linked with fitness traits such as antimicrobial resistance (AMR), virulence, and metabolic functions. We conducted a multicentric study in India and collected different clinical samples for decoding the genome sequences of bacterial pathogens associated with sepsis, urinary tract infections, and respiratory infections to understand the functional potency associated with AMR and its dynamics. Genomic analysis identified several acquired AMR genes (ARGs) that have a pathogen-specific signature. We observed that blaCTX-M-15, blaCMY-42, blaNDM-5, and aadA(2) were prevalent in Escherichia coli, and blaTEM-1B, blaOXA-232, blaNDM-1, rmtB, and rmtC were dominant in Klebsiella pneumoniae. In contrast, Pseudomonas aeruginosa and Acinetobacter baumannii harbored blaVEB, blaVIM-2, aph(3’), strA/B, blaOXA-23, aph(3′) variants, and amrA, respectively. Regardless of the type of ARG, the MGEs linked with ARGs were also pathogen-specific. The sequence type of these pathogens was identified as high-risk international clones, with only a few lineages being predominant and region-specific. Whole-cell proteome analysis of extensively drug-resistant K. pneumoniae, A. baumannii, E. coli, and P. aeruginosa strains revealed differential abundances of resistance-associated proteins in the presence and absence of different classes of antibiotics. The pathogen-specific resistance signatures and differential abundance of AMR-associated proteins identified in this study should add value to AMR diagnostics and the choice of appropriate drug combinations for successful antimicrobial therapy.
Widespread antimicrobial resistance (AMR) in health care and even in community settings has drastically reduced the effectiveness of most antimicrobials routinely used in clinical practice. In 2019, an estimated 4.95 million patients died worldwide due to drug-resistant bacterial infections (1). Six pathogens—Escherichia coli, Staphylococcus aureus, Klebsiella pneumoniae, Streptococcus pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa—are the leading cause of death worldwide (1).
Rising AMR in bacterial pathogens is mostly due to the lateral acquisition of AMR genes or the accumulation of spontaneous mutations in the bacterial genome. Different mobile genetic elements (MGEs) genetically linked with AMR genes (ARGs) contribute significantly to the emergence and spread of drug-resistant bacterial pathogens (2, 3). Currently, the predominant AMR encountered globally is due to the widespread prevalence of mobile β-lactamases, aminoglycoside modifying enzymes (AMEs), 16S RMTases (16S ribosomal RNA methyltransferases), and plasmid-mediated colistin resistance (4). Further, the types of ARGs and their variants differ geographically at both regional and global levels. It is therefore imperative to investigate the prevailing resistance mechanisms, so as to be able to choose appropriate antimicrobials.
Molecular profiling of Gram-negative pathogens using whole-genome sequencing (WGS) is critical, primarily to understand the sequence heterogeneity in resistance alleles. Although a few studies have documented the prevalence of molecular AMR profiles in India (5, 6), in a multicentric study involving different regions of India, we sought to investigate resistance allele heterogeneity and compare Indian isolates to global lineages. The key objectives of the present study are to understand the genetic heterogeneity of ARGs, functional potency associated with ARGs, and the genetic linkage of ARGs with different MGEs in clinically important Gram-negative bacterial pathogens. Furthermore, global phylogeographic analyses of the drug-resistant isolates help us to understand the lineages currently circulating in India.
1. Results
1.1. Distribution of Gram-Negative Clinical Pathogens.
This study included 203 gram-negative bacterial strains isolated from 2016 to 2022 from five different sites covering the North (n = 2), South (n = 2), and East-Central (n = 1) regions of India. This predominantly comprises multidrug-resistant (MDR) Gram-negative extracellular pathogens, i.e., E. coli (n = 35), K. pneumoniae (n = 49), P. aeruginosa (n = 40), and A. baumannii (n = 36), and intracellular pathogen, i.e, Salmonella enterica Serovar Typhimurium (n = 28), and the rest are described in Fig. 1A. These isolates were obtained from diverse clinical specimens of invasive and noninvasive infections (Fig. 1B). The participating sites were tertiary care hospital settings in North and South, while the central India site (Chhattisgarh) was a secondary care hospital for rural tribal people (SI Appendix, Fig. S1 and Dataset S1).
Fig. 1.
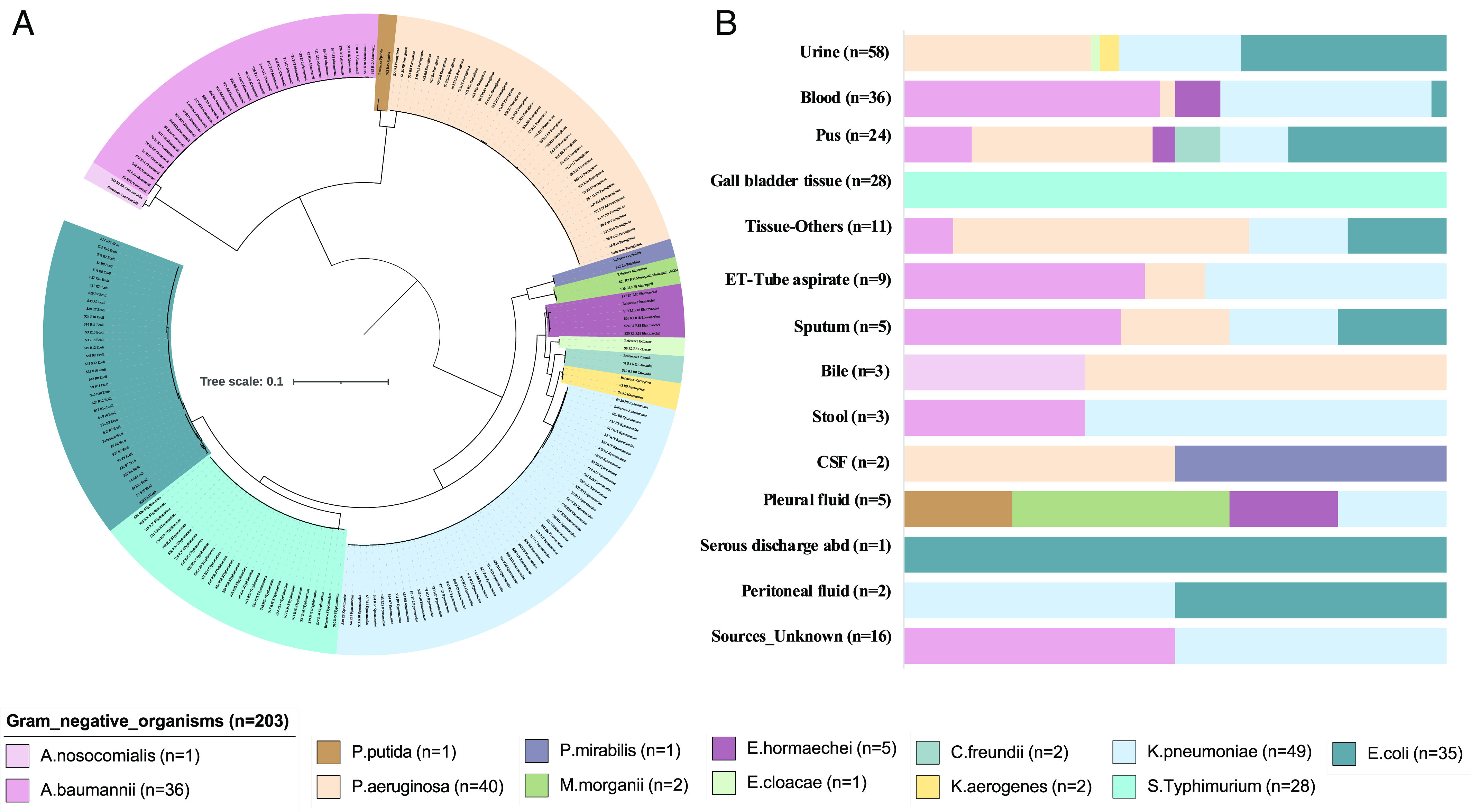
(A) Distribution of each pathogenic bacterial genome included in this study depicted by the maximum likelihood 16S rRNA sequence–based phylogenetic analysis. (B) Represents the diverse clinical specimen sources from where these gram-negative pathogens were isolated. The tree scale indicates the number of substitutions per genome per site.
1.2. Phenotypic Antimicrobial Susceptibility Testing (AST) Showed Distinct Profiles with Each Bacterial Pathogen.
All the 203 Gram-negative isolates were subjected to phenotypic AST against 17 antibiotics (Dataset S2). As illustrated in SI Appendix, Fig. S2, S. typhimurium was found to be the most susceptible pathogen against all antimicrobial classes, except aminoglycosides and fluoroquinolones. E. coli, P. aeruginosa, and A. baumannii appeared to show resistance to multiple classes of antimicrobials that are designated as MDR pathogens. Although differential resistance was observed in K. pneumoniae, carbapenem resistance was seen in a significant number of isolates (n = 34/49). Overall, 188/203 isolates were resistant to at least one of the antimicrobial agents. The maximum resistance noted was for 16 out of 17 agents tested in a proportion of isolates (n = 11/203) (Dataset S2).
1.3. Antimicrobial-Resistance Determinants Are Heterogeneous among Gram-Negative Pathogens.
The comparative genome sequence analysis of the study isolates indicates the presence of pathogen-specific ARGs, except for a few genes (Fig. 2). The presence of intrinsic ARGs blaOXA-51, blaPAO, and blaSHV in A. baumannii, P. aeruginosa, and K. pneumoniae, respectively, further confirms the specificity. In β-lactamases, very few genes such as blaTEM, blaOXA-9, and blaCTX-M-15 were observed among ESBLs (extended-spectrum beta-lactamases). Among clinically important carbapenemases, blaNDM was present among the four most common pathogens and predominantly in E. coli (n = 31/35) (Fig. 2). Unlike E. coli, very few isolates of K. pneumoniae harbored blaNDM-1 (n = 2) and blaOXA-232 (n = 5) variants. The overall prevalence of aminoglycoside modifying enzymes (AMEs) was higher as compared to the 16S rRNA methyl transferases (16S RMTases) (Fig. 2). We observed that aph(3′) and armA were prevalent in nonfermenters and aadA and rmtB in the Enterobacterales. Similarly, in macrolides, the nonfermenters harbored exclusively mphE and msrE, while most of the Enterobacterales harbored ermB and mphA, respectively. Other notable differences in the AMR genes were the dominance of catB, dfrB, and blaVIM-2 in P. aeruginosa; fosA in P. aeruginosa, K. pneumoniae, and Enterobacter spp; and oqxA and oqxB in K. pneumoniae and Enterobacter spp. Interestingly, only E. coli was found to harbor both dfrB and sul1/sul2 to confer resistance to trimethoprim/sulfamethoxazole, whereas all the other pathogens predominantly harbored either one or the other of these genes. AMR profiles in the other pathogens could not be compared due to the small number of isolates (n < 10).
Fig. 2.
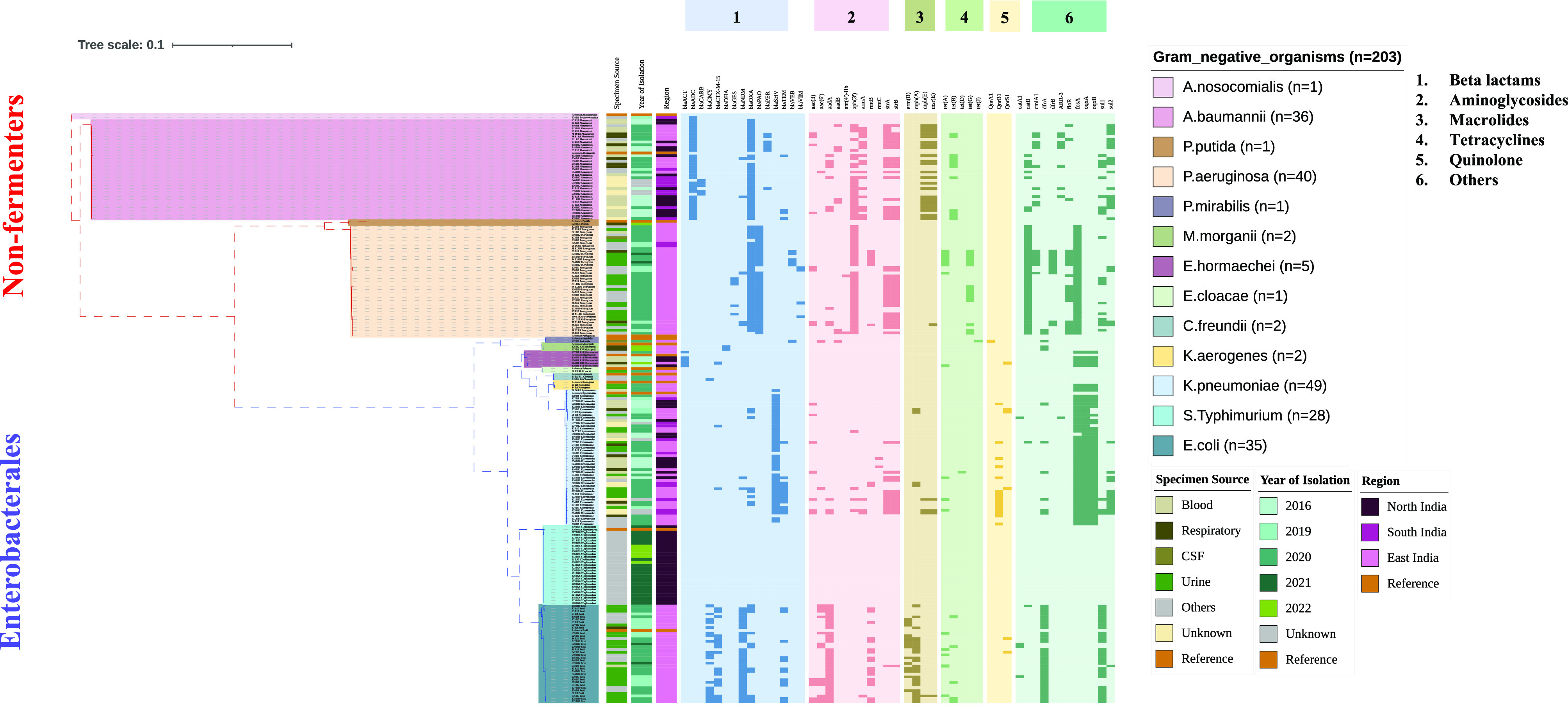
Molecular antimicrobial-resistant profile overlaid on the maximum likelihood phylogeny based on 16S, 23S, and 5S rRNA sequences for gram-negative pathogens (n = 203). The tree depicts the difference between the nonfermenters and Enterobacterales along with specimen type, year of isolation, and region. The heat map represents the classification of acquired AMR genes with five major antimicrobials (darker shade represents presence, and lighter shade represents absence). The tree scale indicates the number of substitutions per genome per site.
1.4. Pathogen-Specific Antimicrobial-Resistant Determinants Appear to Be Linked with Their Respective Lineages.
Phylogenetic trees have been generated based on the seven housekeeping gene sequences as per the pubMLST/Pasteur schemes. MLST-based E. coli tree discriminated the isolates based on their respective sequence type (ST) and their associated single/double loci variants (SI Appendix, Fig. S3). The isolates were clustered in four large STs (22/35); however, seven isolates could not be assigned ST due to single nucleotide polymorphisms (SNPs). Each of the isolates harbored multiple ARGs, with the exception of certain ARGs being more predominant and associated with specific STs. Among β-lactamases, blaCMY and blaCTX-M-15 were the common AmpC and extended spectrum beta-lactamses (ESBL) genes, while carbapenemase variants of blaNDM were present in 31/35 isolates across all STs and with two isolates coharbored blaOXA-181 and blaNDM-4 in the unknown STs. In K. pneumoniae, 19 different STs were identified, with two large clusters belonging to ST147 and ST14, and a few clusters such as ST395, ST22, and ST231 (SI Appendix, Fig. S4). Unlike E. coli, the burden of acquired ARGs was less in K. pneumoniae. Notably, ST147 appeared to harbor multiple ARGs, especially ESBLs (blaTEM), carbapenemases (blaOXA-232), AMEs, macrolides (mphE, msrE), fluoroquinolones (qnrB), sulfonamides (dfrA, sul1/sul2), and fosfomycin (fosA7) modifying functions. We observed that K. pneumoniae harbored either AMEs or 16S RMTases genes, but none of the isolates carried both genes. In addition, 16S RMTases in K. pneumoniae were found to be ST-specific. Although all 49 isolates harbored fosA genes, the variants were mostly specific to their respective STs.
In P. aeruginosa, eight different STs were identified with the four most common STs belonging to ST1047, ST357, ST316, and ST664 (SI Appendix, Fig. S5). Five unknown STs were found; however, three appeared to be a variant of known STs. ST357 displayed an MDR profile with multiple ARGs such as blaVEB, rmtB, tetA, and cmlA. Very few isolates displayed the presence of both dfrB and sul1/sul2 genes together to confer trimethoprim/sulfamethoxazole resistance. In A. baumannii, eight different known STs and four unknown STs were identified, predominantly ST1 and ST164 (SI Appendix, Fig. S6). All ST164 isolated exclusively harbored blaCARB, and blaOXA-91, but none of the other ARGs. Overall, ST2 displayed MDR profile as compared to the others. We observed that intrinsic resistance gene blaOXA-51 has at least nine variants, and they are specific to each ST. Further, the acquired resistance was typed as blaOXA-23 variant, and was present in all isolates, except the four unknown STs. Besides these, only one acquired blaOXA-235 carbapenemase was identified, which was isolated from a sepsis patient in Northern India.
1.5. Phylogeographical Analysis of the Indian Isolates in the Global Context Reveals the Dominance of Geo-Specific Lineage Diversity.
WGS-based epidemiological analysis was performed using Indian isolates and the global representative collection of E. coli, K. pneumoniae, P. aeruginosa, and A. baumannii, i.e, total of 2,720 Gram-negative bacterial genomes including 203 study isolates (Datasets S3–S6). This was done to look for the regional differences in the circulating ST/lineages in the global context and to understand the emergence and spread of any particular AMR-associated endemic lineages nationally and globally. The global phylogeographical analysis of 995 E. coli genomes showed that all these were typed into eight phylogroups (A, B1, B2, and C to G) (Fig. 3A). The present study’s isolates of uropathogenic E. coli (n = 22) were phylogroup as A and B1 predominantly, followed by B2, C, D, and F, while phylogroups A, B1, and D were also seen in E. coli isolated from other specimen types. The prevalence of the globally dominant B2 phylogroup was limited in our study collection (n = 2); however, phylogroups A and B1 were notable. The isolates of phylogroup D largely clustered with other Indian isolates collected from 2016 to 2019. In contrast, only a subcluster of phylogroup A isolates was closely related to a few other Indian isolates collected during 2016 to 2020, but the other isolates appeared distinct and were found adjacent to European isolates. This was mainly attributed to the highly diverse genomic nature of E. coli, and therefore, we could not identify any major lineages of high geographical prevalence.
Fig. 3.
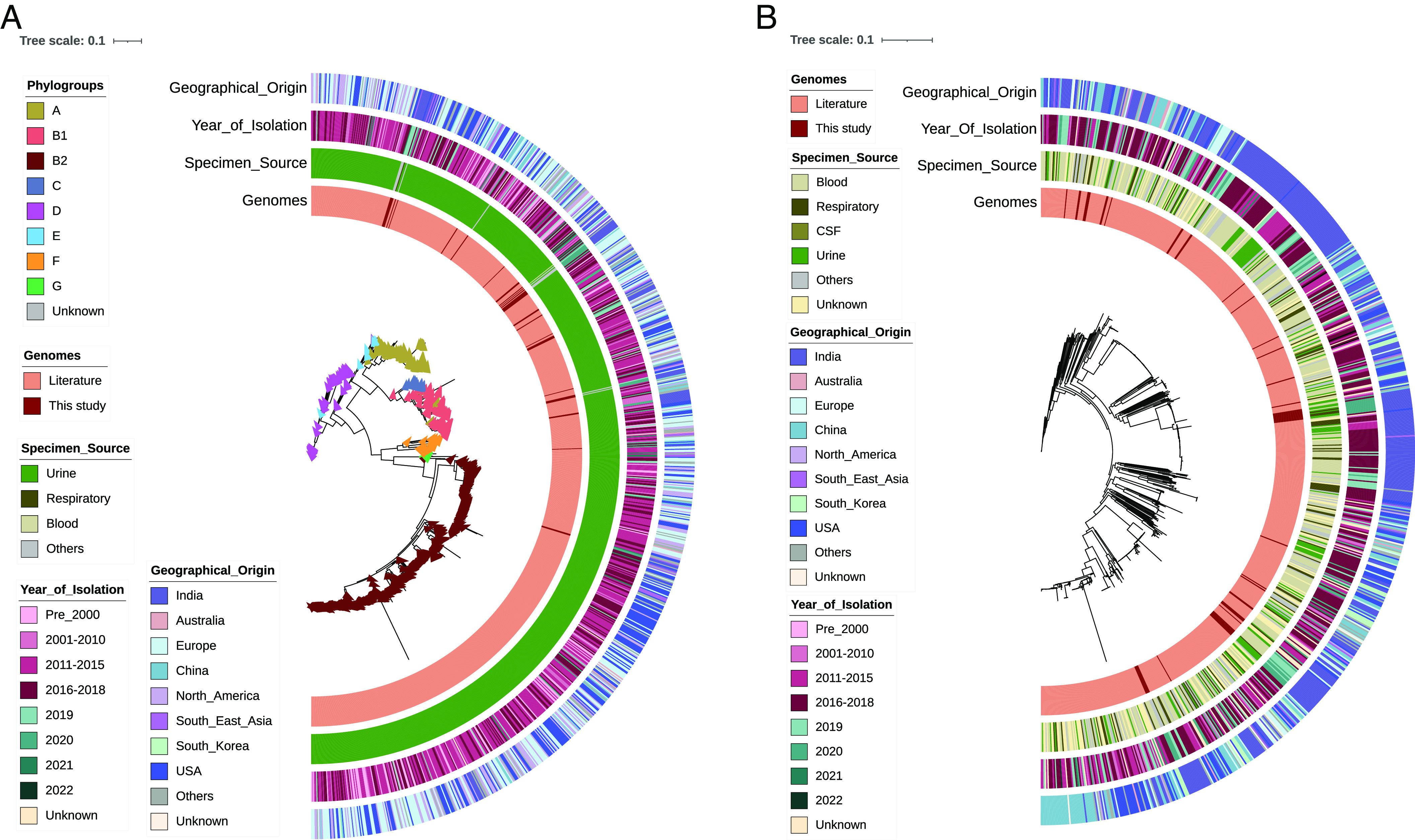
Global phylogeographical analysis of (A) E. coli (n = 995) and (B) K. pneumoniae (n = 776) along with their specimen source, geographical origin, and year of isolation. Phylogroups are mentioned for E. coli. The tree scale indicates the number of substitutions per genome per site.
Unlike E. coli, the global phylogeography of K. pneumoniae was highly clonal and geographic-specific. As shown in Fig. 3B, the majority of the Indian isolates were classified into five different STs (ST14, ST231, ST147, ST101, and ST395), and these were found to be predominant and endemic. Also, these STs were found to harbor multiple AMR genes including blaNDM and blaOXA-48 like carbapenemase. Most of the study isolates were placed adjacent to the Indian isolates collected during a similar time period of 2016 to 2022. These isolates were sourced mainly from bloodstream and respiratory tract infections. Such a close genetic relatedness of isolates in the present study with the other Indian isolates signifies the regional dominance, persistence, and circulation of certain AMR-associated STs/lineages. The population structure of P. aeruginosa with global representative genomes of all five phylogroups along with the study isolates showed distinct lineages (Fig. 4A). Groups 1 and 2 comprised >90% of all the isolates, followed by groups 3 to 5. In the phylogeny, all the study isolates were clustered in the major phylogroup 2 (n = 31), followed by 1 (n = 9), but none in the other distant groups (3 to 5). Although the phylogroup 1 isolate of ST664 (n = 7) and one each of ST245 and ST234 were placed adjacent to the Indian genomes, group 1 isolates were mostly from the United States and China, with very few Indian isolates. In phylogroup 2, three dominating STs such as ST357, ST1047, and ST316 displayed an MDR profile with ARGs against multiple agents. All the group 2 isolates were found to cluster with the Indian isolates that formed a separate branch within its respective STs, indicating their regional dominance. Although all these STs belonged to the international high-risk clones, it was evident that both the prevalence and AMR abundance were high in India for phylogroup 2 as compared to group 1 genomes.
Fig. 4.
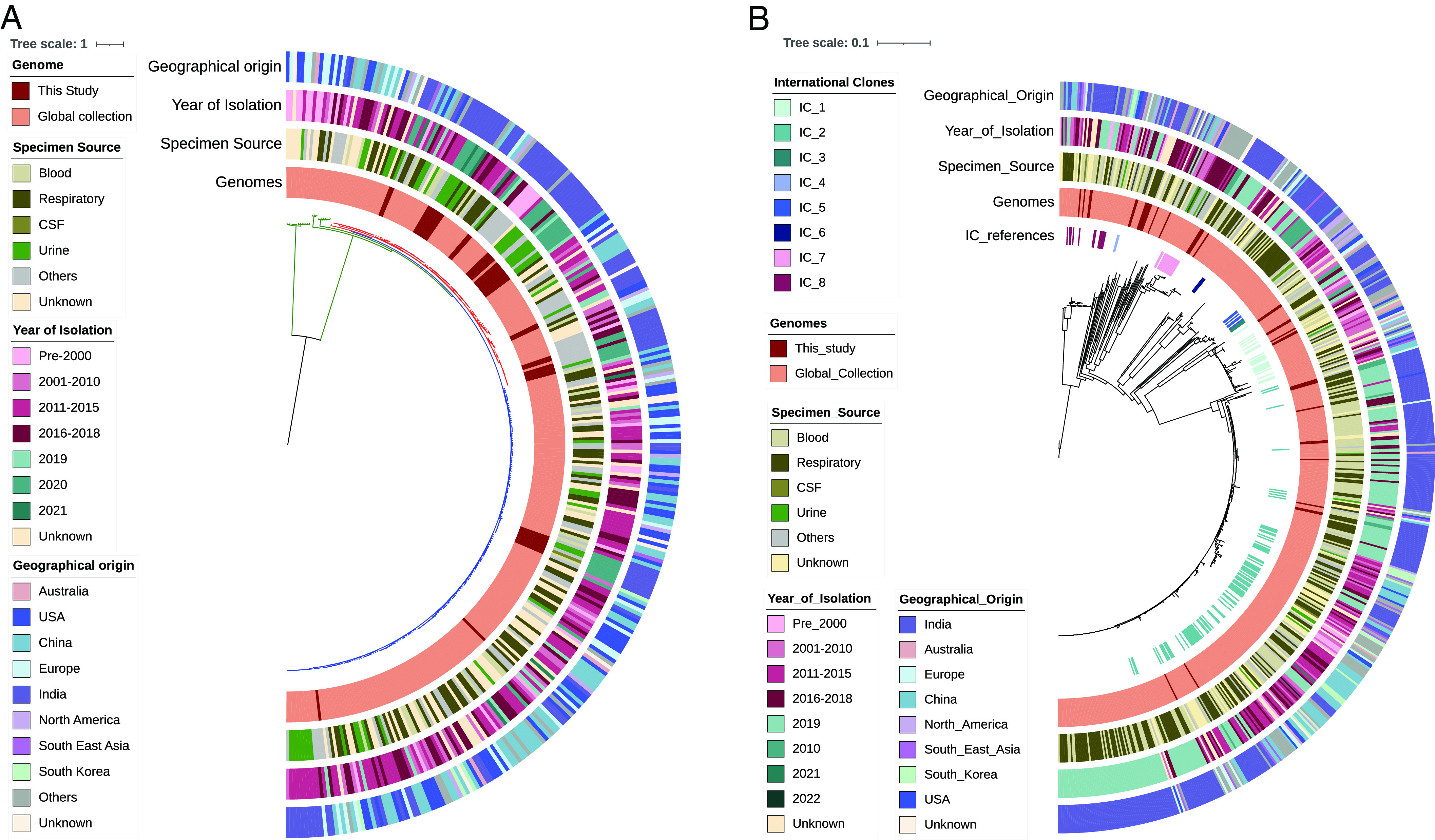
Global phylogeographical analysis of (A) P. aeruginosa (n = 323) and (B) A. baumannii (n = 582) along with their specimen source, geographical origin, and year of isolation. International clones are shown for A. baumannii. The tree scale indicates the number of substitutions per genome per site.
The global phylogeography of A. baumannii revealed the distribution of the present study isolates into four major international clones (ICs). This includes the most common and widespread IC-2 (n = 12, 33%) and IC-1 (n = 7, 19%), followed by four isolates each in IC-7 and IC-8 (Fig. 4B). It was evident from the phylogeny that all these study isolates were placed adjacent to the other Indian isolates collected during the same time period. It is to be noted that ST164 identified in this study has not been classified into any IC previously; however, this has formed a separate cluster with no significant geospatial signal from the other global genomes. Similarly, two unknown STs formed a distinct branch from the other genomes.
1.6. The Pan-Genome Analysis Highlights the Gene Flux Impacting the Differences in the Genome Composition.
After decoding the high-resolution phylogeny and geographical distribution of the Indian isolates in the global context, we analyzed the pan-genome of E. coli, K. pneumoniae, P. aeruginosa, and A. baumannii to determine the core genes in addition to the differential accessory genes, which could be either acquired or lost due to gene flux. Besides the traditional pangenome analysis by Panaroo (identity threshold of 98%), the accessory genes were further classified as per their distribution frequency by the “twilight” application. In the overall gene classification, each pathogen showed a unique profile with respect to each of the bacterial genome characteristics. On comparing the coding sequence annotated in the respective reference chromosomes, E. coli (2784/4298–64.77 %) and A. baumannii (2311/3582–64.51 %) showed a relatively lower number of genes being captured as core genomes as compared to K. pneumoniae (4077/5316–76.6 %) and P. aeruginosa (4598/5572–82.5 %) (SI Appendix, Figs. S7–S10). The number of core genes covered in P. aeruginosa was notable, despite its genome size (6.6 Mbp) and high GC content (66.6). However, the proportion of intermediate and rare genes was particularly high. In contrast, in accessory genes, lineage-specific rare genes were higher in E. coli, K. pneumoniae, and P. aeruginosa. Such a remarkable difference in these accessory genes indicates the genetic diversity of these pathogens. Further, a subset analysis was performed for each of the pathogen collections which were classified into three groups as sourced from global (countries other than India), India, and isolates in the present study. The modified sentence should be-In all the pathogens, the proportion of intermediate and rare genes was notable and was predominantly hypothetical and linked with MGEs. This gave us a clue to further investigate the role of MGEs linked with ARGs in the present study isolates.
1.7. Transmissibility of AMR Determinants Often Can Be Associated with Unique Pathogen-Specific MGEs.
The pan-genome analysis of the current isolates provided an important clue about the genome-specific distribution of MGEs linked with ARGs. All 203 assemblies were screened for the presence of acquired plasmids through the plasmid finder database and identified 38 different plasmid types in 106 isolates. This includes E. coli (n = 35/35), K. pneumoniae (n = 47/49), S. typhimurium (n = 14/28), Enterobacter hormaechei (n = 4/5), P. aeruginosa (n = 4/40), and A. baumannii (n = 2/36) (Dataset S7). On average E. coli harbored 3.5 plasmids (ranging from 2 to 9), followed by K. pneumoniae which was 3.2 (ranging from 1 to 7), and E. hormaechei (average, 1.75; range, 1 to 4). The nonfermenters had only one type of plasmid in each taxon, such as IncQ1 in A. baumannii (n = 2/36) and IncQ2 in P. aeruginosa (n = 4/40). A contrasting profile was seen in the other Enterobacterales, with a few plasmids that were pathogen-specific. Although the plasmids were abundantly seen, not all the plasmids carried ARGs. Rather, very few isolates showed a genetic linkage of the acquired plasmids with the presence of ARGs in them. This could possibly be explained by the draft genome nature of isolates, due to which the contigs could not be completely covered to infer the relatedness of MGEs carrying ARGs. Except for S. typhimurium, all the other isolates with plasmids were also detected positive to carry one or more ARGs.
Based on the MGE finder, 115/203 genomes were found to show a linkage of ARG with IS and a few transposons (Dataset S8). A. baumannii (89%) and P. aeruginosa (88%) harbored abundant IS elements as compared to E. coli (69%) and K. pneumoniae (49%). These MGEs were mostly species-specific, except a few types like ISVsa3 which was seen exclusively in the P. aeruginosa and A. baumannii that were associated with sul2, tetB, floR, and AMEs [aph(3″)-Ib, aph(6)-Id]. Similarly, IS6100 and Tn5403 were common in E. coli, K. pneumoniae, and P. aeruginosa; however, the type of ARGs present in both the MGEs were distinct. E. coli and K. pneumoniae carried mphA and rmtB, and P. aeruginosa carried aph(6)-Id and aph(3″)-Ib on a transposon Tn6082. A. baumannii harbored a species-specific ISAba24 (IS66 family). Further, armA was found to be genetically linked with ISEc29 (IS4 family). Despite the abundance and diversity of the IS elements typed in this study isolate, the species-specific types highlight the dissemination of these ARGs on a diverse MGE genetic background.
Besides associations, we further derived the genetic arrangements of ARGs and IS/Transposon elements within the plasmid sequence. To achieve this, larger contigs that covered these regions were mapped onto their respective references as identified by BLAST analysis. The abundance of blaNDM-5 variant in E. coli was looked for in the flanking regions and identified the association of bleMBL with all the strains, irrespective of the lineages (Fig. 5B). Contrastingly, blaNDM-1 variant in E. coli that was carried on an IncA/C plasmid (180 Kbp in size, S36_R7) showed the maximum coverage of the plasmid backbone; however, the hotspot regions that carried ARGs and IS could not be captured well (SI Appendix, Fig. S11). The other blaNDM-1 present in multiple organisms revealed diverse genetic backgrounds (Fig. 5A), unlike the blaNDM-5 variant. In case of blaCMY-42, the allele was present on IncI_Alpha plasmid in E. coli, revealing the presence of ARGs. However, its adjacent IS1 family transposases were missed may be due to less sequence coverage (SI Appendix, Fig. S12). In P. aeruginosa with a few plasmids, an integrative conjugative element (ICE) containing Tn3 family transposons carrying aph and dfr was found inserted in the chromosome between the genes encoding for guanosine monophosphate (GMP) synthase and inosine-5′-monophosphate dehydrogenase (SI Appendix, Fig. S13). The other regions could not be drawn due to small contig sizes. In A. baumannii, the presence of ISAba1 flanking blaOXA-23 was found in 28/36 isolates. Therefore, a few representatives of these positives and negatives for ISAba1 were considered to plot the genetic background as shown in SI Appendix, Fig. S14. Isolates that were negative for ISAba1 in the same contig further confirmed the absence of these elements adjacent to blaOXA-23.
Fig. 5.
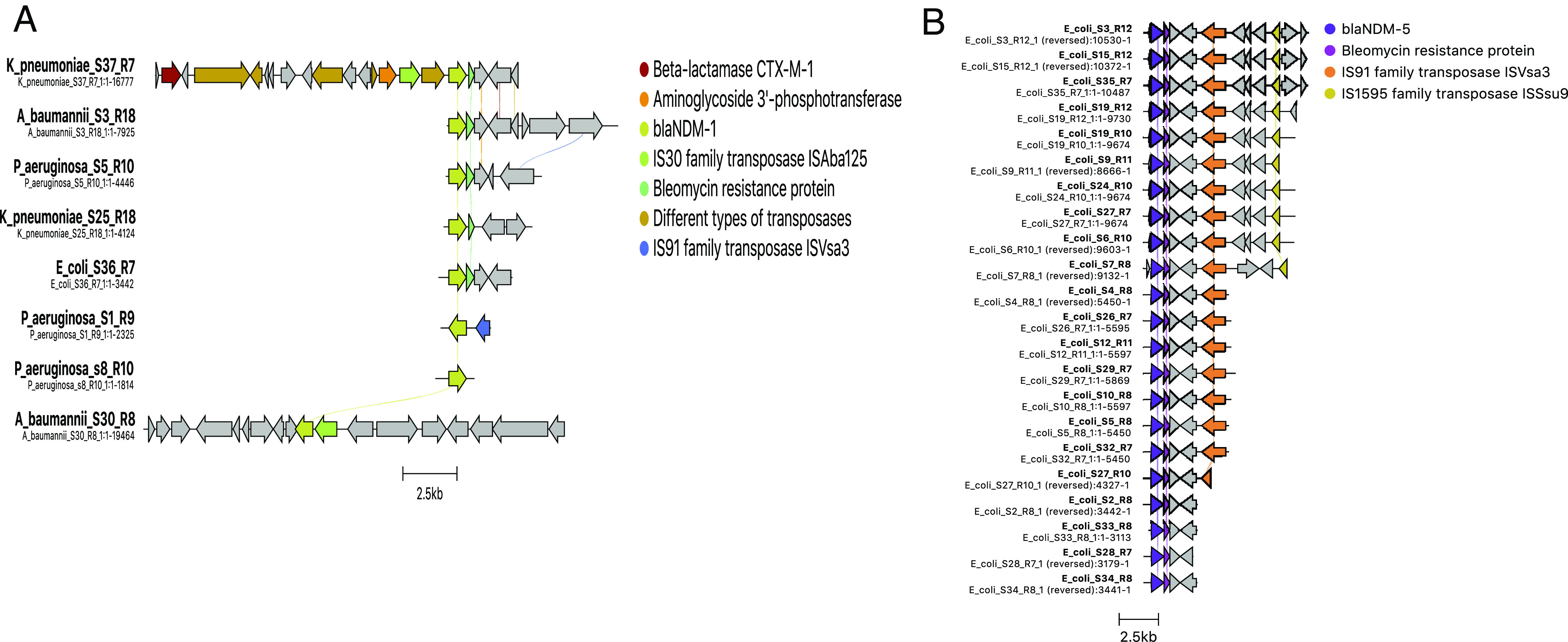
Comparison of genetic arrangements for (A) blaNDM-1 vs. (B) blaNDM-5 in different organisms to decode diversity in genetic background. Cluster alignments are drawn to scale based using the Clinker tool. Arrows indicate the direction of the open reading frame.
1.8. Monitoring Functionality of the ARGs by Exploring Whole-Cell Proteome of MDR K. pneumoniae, A. baumannii, E. coli, and P. aeruginosa.
Understanding the genomic potency of AMR is important, but such findings need additional support for their functionality. For a better understanding of the functionality of the ARGs, the whole-cell proteome of susceptible and MDR K. pneumoniae, A. baumannii, E. coli, and P. aeruginosa was explored in the presence and absence of different antibiotics. A total of 1,840, 1,554, 1,472, and 824 proteins were identified and quantified in K. pneumoniae, A. baumannii, E. coli, and P. aeruginosa, respectively, using SWATH-MS platform. Principal component analysis (PCA) was carried out to obtain the proteome profiles of different isolates of these MDR pathogens. A clear separation of intensities between control and drug-resistant isolates on both components of the X axis and Y axis was observed. Hierarchical clustering analysis further corroborated the differential results. The proteomic analysis detected the expression of the majority of the AMR proteins, including clinically relevant proteins such as 16S rRNA methyltransferase, O-phosphotransferase, N-acetyltransferase, macrolide phosphotransferase, nucleotidyltransferase, beta-lactamase, PBP1b, MFS and RND efflux pumps, sulfonamide resistant sul, and bifunctional polymyxin resistance protein (arnA) (SI Appendix, Figs. S15–S20). However, other than AMR-related proteins, we also found many proteins showing differential expression in K. pneumoniae, A. baumannii, E. coli, and P. aeruginosa strains in the presence and absence of antibiotics. These are predominantly related to metabolism, transcriptional and translational regulation, and stress response. These findings suggest the alteration of additional pathways, which may directly and/or indirectly correlate with AMR and thus could promote the functionality and dissemination of ARGs in a suitable environmental condition (http://www.pnas.org/lookup/doi/10.1073/pnas.2305465120#supplementary-materialsSI Appendix, Figs. S15–S20 and Dataset S9).
2. Discussion
Heterogeneity in ARGs in clinically significant bacterial pathogens and their functional potency to reduce the efficacy of different generations of antibiotics widely vary across geography and pose a major challenge in the molecular detection of AMR functions and the selection of appropriate antibiotics to treat drug-resistant infections. India has the highest AMR burden; however, very limited studies have characterized the ARGs in the clinical isolates collected from India. Also, a recently established national multicentric AMR surveillance network has documented the AMR rates at the phenotypic level and with a baseline molecular profile (https://iamrsn.icmr.org.in/index.php/resources/amr-icmr-data). Therefore, the present study characterized the clinical isolates for drug-resistant profiling indicating AMR diversity, predominantly from a community healthcare center in the East-Central Indian region and compared them with other tertiary care hospitals. The profile presented in the study signifies the burden of acquired resistance in extracellular pathogens such as E. coli, K. pneumoniae, P. aeruginosa, and A. baumannii, and the ARGs were found to be pathogen- and regional-specific. These data when added to the existing profile clearly distinguish the regional diversity in the AMR profile within the country. However, the disparity in the number of strains from each location is a limitation of the present study and may need further investigation for decoding the geo-specific resistance functions of each of the pathogens.
The study report was well compared with the previously documented resistance profile generated through the ICMR AMRSN report (7). The global dominance of blaCTX-M-15, in E. coli and K. pneumoniae was not seen in this study with K. pneumoniae isolates (8, 9). In contrast, the occurrence of AmpC β-lactamase blaCMY-42 was high in E. coli. In addition, the blaOXA-encoded β-lactamases found abundant in the previous Indian collection concur with this study (7). Significant differences were found with the abundance of blaOXA-48 in K. pneumoniae in India, which was seen as limited in this study. Moreover, the overall resistance in K. pneumoniae was lower as compared to the other studies with higher resistance reported, especially to carbapenems (7). In aminoglycoside resistance, the common aac and ant variants of AMEs in India are noted at lower rates in this study. However, the prevalence of amrA was found to be similar to other Indian studies (10). Cumulative analysis of the acquired AMR diversity in the 203 isolates highlights the distinct pattern of ARGs in each of the pathogens, despite their geographical origin. However, the differences within the pathogen profile were mainly associated with regional variations within India, which was interesting. The key factor responsible for the rapid dissemination of ARGs between bacterial species is linked with different MGEs like plasmids, IS, and transposons (2). The type of plasmids prevalent in E. coli and K. pneumoniae is in concordance with the other studies, especially those belonging to IncF and col groups (11). These plasmids carry multiple ARGs encoding ESBLs-blaCTX-M-15, AmpC-blaCMY, and carbapenemases-blaNDM. These plasmids also frequently carry aminoglycoside resistance genes, thus conferring resistance to multiple antimicrobials contributing to MDR and extensively drug-resistant (XDR) phenotypes. In contrast, plasmids are generally less in nonfermenter pathogens such as P. aeruginosa and A. baumannii (11). Further, the presence of ICE in P. aeruginosa carrying AMR on Tn3 transposons, ISAba1 with blaOXA23 in A. baumannii was previously reported in India (12, 13). Such commonalities among MGEs across Indian isolates suggest rapid exchange of ARGs between indigenous isolates through horizontal gene transfer (HGT) and highlight the basis of geo-specificity of some of the resistance functions.
Despite the diversity of MGEs, the success of any drug-resistant pathogen to dominate a niche depends on the type of lineage that harbors MGE linked ARGs. Genomic epidemiological studies have identified that the drug-resistant lineages carrying multiple ARGs in their genome are the fittest and most prevalent in clinical settings. However, these may vary geographically. Among MDR E. coli, ST131 predominates globally, while ST410 is emerging as the fastest-spreading high-risk clone in India (9). Similarly, in K. pneumoniae, the globally prevalent drug-resistant lineage is ST258 carrying blaKPC; however, it is rare in India. The other dominant STs are ST11, ST14, ST15, ST26, ST101, ST147, ST149, ST231, ST627, and ST977, whereas, ST231, ST14, and ST147 are endemic in Indian settings (14, 15). In P. aeruginosa, it was reported that 90% of the XDR isolates belong to three major high-risk international clones ST175, ST111, and ST235, whereas carbapenem-resistant P. aeruginosa in India belong to ST357, ST235, ST233, and ST244 (16, 17). In A. baumannii, the globally predominant STs are ST1, ST2, ST79, and ST25, with ST2 as the most dominant type. In India, ST146, ST110, ST69, ST103, ST194, ST108, and ST188 are the common STs in North India, and ST538, ST539, ST103, and ST576 are common in South India (18–20). However, ST2 is also seen in India, along with ST848, ST451, and ST195 (18, 21). These aforementioned global and Indian-specific lineages overall concur well with the present study lineages, except for a few STs. This clearly captures the regional differences in ST types for different pathogens within India, which could be linked with antimicrobial usage in the respective geographical location.
Although the WGS-based prediction of ARG is generally accurate, it needs functional validation. In addition, the expression of ARGs might be constitutive or inducible and may depend on the presence of relevant antibiotics or other xenobiotics in the growth medium. To get insights, the total proteome of drug-sensitive and resistant isolates of K. pneumoniae, A. baumannii, E. coli, and P. aeruginosa was explored. The protein profiles clearly distinguished the sensitive and resistant strains of the same bacterial isolate. Expression of several proteins associated with AMR functions, as well as proteins linked with metabolism, transcription, translational regulation, and stress response, was significantly different in the resistant isolates compared to the control group. Such an expression pattern of AMR-associated functions in the MDR strains may help them reduce energy expenses by limiting the expression of ARGs when they are not needed. Overall, high resistance rates in most of the clinical pathogens, including carbapenems and aminoglycosides resistance, both in the community-level healthcare setting as well as the hospital settings have been captured. This study has two clinical implications in terms of the diagnostics and treatment aspects. Diversity in the AMR determinants observed in this study would help to design and develop rapid molecular diagnostics tests with the target gene of interest being customized based on the AMR profile in a given geographical location. Further, the basis and prevalence of acquired resistance at the community level would help clinicians to choose appropriate antimicrobial therapy to improve clinical success rates.
3. Materials and Methods
3.1. Patient Recruitment and Sample Collection.
A total of 203 clinical gram-negative isolates from five different participating sites were included in this study (Dataset S1). Different biospecimen types included urine, blood, sputum, endotracheal tube aspirate, stool, pus, and tissue. Samples were collected from 2016 to 2022 (Dataset S1), irrespective of the phenotypic susceptibility profile.
3.2. AST and DNA Extraction.
AST was done for 203 samples using the commercial antibiotic discs (BD BBL, Sensi Discs) against 17 antibiotics. Results were interpreted using recent Clinical and Laboratory Standards Institute (CLSI) (M100-2021) (22) and EUCAST (2021) (23) guidelines. E. coli American Type Culture Collection (ATCC) 25922, Vibrio cholerae O395 (in-house), and S. typhimurium MV32691 (in-house) were used as control strains. The concentration of antibiotics and other relevant information are included in the SI Appendix, Materials and Methods.
3.3. WGS, Raw Reads Processing, Genome Assembly, Quality Assessment, and Annotation.
WGS was performed by a high-throughput Illumina MiSeq sequencing platform (in-house) using the Nextera XT DNA Library preparation kit (Illumina, Inc.). High-quality raw reads were used for assembly using a hybrid assembly pipeline unicycler version-v0.4.8 (24). Assembled genome quality was assessed using the CheckM program version v1.1.3 for any contamination at the genome level (25). Genomes were included for analysis only if the contamination level was <5% (25, 26). Annotation was done using Rapid Annotation using Subsystem Technology (RAST) version 2.0 (27).
3.4. Multilocus Sequence Typing.
The WGS was scanned to obtain the MLST profile for the different pathogens. MLST profiles for E. coli were analyzed using the Achtman scheme. For K. pneumoniae and P. aeruginosa, the PubMLST scheme has been followed. For A. baumannii, the Pasteur scheme was used (28).
3.5. Global Phylogenomic Framework.
For the global phylogeographical analysis, we have included genomes that were submitted from India (irrespective of draft/complete), complete genomes (irrespective of location) available in NCBI, isolation type selected as clinical, and host as Homo sapiens for the initial screening (as on 13.06.2022). The downloaded genomes were first screened for the quality check by the checkM tool, and only the genomes that had >90% completeness and <5% contamination were included for analysis (25). Phylogenetic trees were constructed using GTR-GAMMA-based maximum likelihood (ML) tree by RaxML (29). The generated phylogenies were annotated with the associated metadata using iTOL (30).
3.6. Pan-Genome Analysis—Panaroo, Twilight Zone of Analysis.
Pan-genome analysis was performed for the four pathogens using the panaroo pipeline (31). The obtained output was further analyzed with the twilight codes for further subclassification within the accessory genomes (32).
3.7. Screening of AMR Determinants, MGEs, IS Elements, and Virulence Factors.
All the genomes were screened through the Abricate tool for acquired AMR determinants and plasmids (https://github.com/tseemann/abricate) and IS elements through ISFinder (https://isfinder.biotoul.fr/). Genetic arrangements of AMR genes along with MGEs were generated using tools mobile element finder (https://cge.food.dtu.dk/services/MobileElementFinder/), clinker (33), and proksee (https://proksee.ca/projects/new).
3.8. LC-MS/MS (SWATH)-Based Proteome Profiling and Data Analysis.
Multidrug resistant K. pneumoniae, A. baumannii, E. coli, and P. aeruginosa carrying multiple ARGs were selected for proteome analysis. The organisms were grown in Luria broth (LB) and induced with different antibiotics viz. ampicillin (100 µg/mL), kanamycin (40 µg/mL), and nalidixic acid (20 µg/mL). The whole-cell peptide mixture was desalted and analyzed by Eksigent micro LC, connected with a TripleTOF 5600 mass spectrometer. Spectra were acquired using an ESI ion source with 250 ms accumulation time. SWATH-MS was performed using a set of 60 overlapping variable windows covering the mass range 400 to 1,250 Da. Identification of proteins was carried out via search against Uniprot protein databases (K. pneumoniae, A. baumannii, E. coli, and P. aeruginosa, released in April 2023) with ProteinPilot to obtain spectral library. The proteins and associated peptides were filtered by PeakView and MarkerView for relative quantitative analysis.
3.9. Nucleotide and Protein Sequence Submission.
Raw read fastq sequences of 203 bacterial strains are submitted to the NCBI SRA database with the Bio Project ID PRJNA925003 (Submission ID SUB12423051), and Indian Biological Data Center with the Inda accession INRP000050. Sequences will be released immediately after the article is published. The SWATH-MS data have been deposited to the ProteomeXchange consortium via the PRIDE partner repository (Project accession: PXD039154).
3.10. Biosafety and Ethical Clearance.
The institute’s Biosafety Committee (IBSC#293/2021) and Human Ethics Committee (Ref No: THS 1.8.1/(119) approved the study. Written informed consent was obtained from each patient.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Acknowledgments
We acknowledge Prof. Pramod Garg for his critical reading of the manuscript and valuable suggestions. We acknowledge Dr. Anamika Gambir and Dr. Neha Bansal for critical support during the study period. B.D. acknowledges Translational Health Science and Technology Institute (Intramural 2021 to 2023) and Department of Biotechnology (DBT), Govt. of India (GOI), for financial support (Grant No. BT/PR38173/MED/97/474/2020). T.M. and A.P. receive research fellowships from the University Grants Commission and Department of Science and Technology, GOI, respectively. R.K.B. acknowledges Council of Scientific and Industrial Research Emeritus Scheme (No. 21(1100)/20/EMR-II), GOI, for supporting his Lab. D.P. and L.N. receive MK Bhan fellowships from DBT, GOI.
Author contributions
D.K., G.B.N., and B.D. designed research; T.M., D.K., A.K.P., P.J., P.B., D.P., A.P., S.T., S.B., S.D., J.V., D.T., L.N., A.K., S.P.K., and B.D. performed research; D.K., S.C., S.P., K.J., C.V.S., M.J.S., K.A., R.A., R.G., M.B., N.K., R.K.B., T.R., G.B.N., and B.D. contributed new reagents/analytic tools; T.M., D.K., A.K.P., S.K., P.B., D.P., A.P., G.B.N., and B.D. analyzed data; and T.M., A.K.P., S.K., G.B.N., and B.D. wrote the paper.
Competing interests
B.D. and D.K.C. have published a commentary in 2019.
Footnotes
Reviewers: D.K.C., NIH; V.J.D., Michigan State University; A.U.K., Aligarh Muslim University; and C.P., CNRS-Institute of Integrative Biology of the Cell (I2BC) France.
Contributor Information
G. Balakrish Nair, Email: gbnair_2000@yahoo.com.
Bhabatosh Das, Email: bhabatosh@thsti.res.in.
Data, Materials, and Software Availability
All data generated and used in the present study are available in the SI Appendix, the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/), and the Functional Genomics Laboratory (THSTI). Submission IDs for all the WGSs are included in the Materials and Methods. Genome Sequences and Peptide sequences data have been deposited in NCBI SRA (https://dataview.ncbi.nlm.nih.gov/object/PRJNA925003?reviewer=r2ldqq4aqa50rp6mkfmmri5pi5) and PRIDE (https://www.ebi.ac.uk/pride/archive/projects/PXD039154), respectively. Details of genome sequences and Proteomes are available in Mehrotra et al. (2023a), and Mehrotra et al. (2023b), respectively (34, 35).
Supporting Information
References
- 1.Murray C. J., et al. , Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 399, 629–655 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Partridge S. R., Kwong S. M., Firth N., Jensen S. O., Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31, e00088–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu G., Thomsen L. E., Olsen J. E., Antimicrobial-induced horizontal transfer of antimicrobial resistance genes in bacteria: A mini-review. J. Antimicrob. Chemother. 77, 556–567 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Serio A. W., Keepers T., Andrews L., Krause K. M., Aminoglycoside revival: Review of a historically important class of antimicrobials undergoing rejuvenation. EcoSal Plus 8 (2018), 10.1128/ecosalplus.ESP-0002-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar P., et al. , Molecular insights into antimicrobial resistance traits of multidrug resistant enteric pathogens isolated from India. Sci. Rep. 7, 14468 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramamurthy T., et al. , Vibrio cholerae O139 genomes provide a clue to why it may have failed to usher in the eighth cholera pandemic. Nat. Commun. 13, 3864 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veeraraghavan B., Walia K., Antimicrobial susceptibility profile & resistance mechanisms of Global Antimicrobial Resistance Surveillance System (GLASS) priority pathogens from India. Indian J. Med. Res. 149, 87–96 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peirano G., Pitout J. D. D., Extended-Spectrum ptibility profile & resistance mechanisms of Global Antimicrobial Resistance Surveillance Sysons. Drugs 79, 1529 (2019).31407238 [Google Scholar]
- 9.Ragupathi N. K. D., et al. , First Indian report on genome-wide comparison of multidrug-resistant Escherichia coli from blood stream infections. PLOS One 15, e0220428 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pragasam A. K., et al. , Expected plazomicin susceptibility in India based on the prevailing aminoglycoside resistance mechanisms in Gram-negative organisms derived from whole-genome sequencing. Indian J. Med. Microbiol. 38, 313–318 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Ragupathi N. K. D., et al. , Plasmid profiles among some ESKAPE pathogens in a tertiary care centre in South India. Indian J. Med. Res. 149, 222–231 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan M., Willcox M. D. P., Rice S. A., Sharma S., Stapleton F., Development of antibiotic resistance in the ocular Pseudomonas aeruginosa clone ST308 over twenty years. Exp. Eye Res. 205, 108504 (2021). [DOI] [PubMed] [Google Scholar]
- 13.Vijayakumar S., et al. , Insertion sequences and sequence types profile of clinical isolates of carbapenem-resistant A. baumannii collected across India over four year period. J. Infect Public Health 13, 1022–1028 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Enany S., Zakeer S., Diab A. A., Bakry U., Sayed A. A., Whole genome sequencing of Klebsiella pneumoniae clinical isolates sequence type 627 isolated from Egyptian patients. PLOS One 17, e0265884 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sundaresan A. K., Vincent K., Mohan G. B. M., Ramakrishnan J., Association of sequence types, antimicrobial resistance and virulence genes in Indian isolates of Klebsiella pneumoniae: A comparative genomics study. J. Glob. Antimicrob. Resist 30, 431–441 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Horcajada J. P., et al. , Epidemiology and treatment of multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa infections. Clin. Microbiol. Rev. 32, e00031–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pragasam A. K., et al. , Dominance of international high-risk clones in carbapenemase-producing Pseudomonas aeruginosa: Multicentric molecular epidemiology report from India. Indian J. Med. Microbiol. 36, 344–351 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Vijayakumar S., et al. , Molecular characterization & epidemiology of carbapenem-resistant Acinetobacter baumannii collected across India. Indian J. Med. Res. 149, 240–246 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saranathan R., et al. , Emergence of carbapenem non-susceptible multidrug resistant Acinetobacter baumannii strains of clonal complexes 103B and 92B harboring OXA-type carbapenemases and metallo-β-lactamases in Southern India. Microbiol. Immunol. 59, 277–284 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Rynga D., Shariff M., Deb M., Multi-locus sequence types of Acinetobacter baumanii clinical isolates from India. J. Infect Dev. Ctries 7, 358ec60 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Hamidian M., Nigro S. J., Emergence, molecular mechanisms and global spread of carbapenem-resistant Acinetobacter baumannii. Microb. Genom. 5, e000306 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.CLSI M-100 guidelines-2021. Available at- https://clsi.org/standards/products/free-resources/access-our-free-resources [Accessed on -27th April 2022].
- 23.EUCAST guidelines -2021. Available at- https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v _ 11.0 _ Breakpoint_Tables.pdf [Accessed on -15th September 2022].
- 24.Wick R. R., Judd L. M., Gorrie C. L., Holt K. E., Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parks D. H., Imelfort M., Skennerton C. T., Hugenholtz P., Tyson G. W., CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pasolli E., et al. , Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649–662.e20 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aziz R. K., et al. , The RAST server: Rapid annotations using subsystems technology. BMC Genomics 9, 75 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jolley K. A., Bray J. E., Maiden M. C. J., Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3, 124 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stamatakis A., RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Letunic I., Bork P., Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–138 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Tonkin-Hill G., et al. , Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 21, 180 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horesh G., et al. , Different evolutionary trends form the twilight zone of the bacterial pan-genome. Microb. Genom. 7, 000670 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilchrist C. L. M., Chooi Y.-H., clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 37, 2473–2475 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Mehrotra T., et al. , Heterogeneity in antimicrobial resistance traits among multidrug resistant Gram-negative bacterial pathogens isolated from India: phenotypic, genotypic and proteomic analyses. National Center for Biotechnology Information. https://dataview.ncbi.nlm.nih.gov/object/PRJNA925003?reviewer=r2ldqq4aqa50rp6mkfmmri5pi5. Deposited 18 January 2023.
- 35.Mehrotra T., et al. , MDR K. penumoniae, A. baumannii, E. coli, and P. aeruginiosa SWATH. UniProt. https://www.ebi.ac.uk/pride/archive/projects/PXD039154. Deposited 2 January 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Data Availability Statement
All data generated and used in the present study are available in the SI Appendix, the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/), and the Functional Genomics Laboratory (THSTI). Submission IDs for all the WGSs are included in the Materials and Methods. Genome Sequences and Peptide sequences data have been deposited in NCBI SRA (https://dataview.ncbi.nlm.nih.gov/object/PRJNA925003?reviewer=r2ldqq4aqa50rp6mkfmmri5pi5) and PRIDE (https://www.ebi.ac.uk/pride/archive/projects/PXD039154), respectively. Details of genome sequences and Proteomes are available in Mehrotra et al. (2023a), and Mehrotra et al. (2023b), respectively (34, 35).