

Significance
STING is an essential adaptor protein required for the inflammatory response to cytosolic DNA. dsDNA activates cGAS to generate cGAMP, which binds and activates STING, triggering oligomerization, IRF3- and NFκB-dependent transcription of type I IFNs and inflammatory cytokines, as well as the activation of autophagy. Aberrant activation of STING leads to inflammatory diseases, such as AGS, SAVI, COPA syndrome, ALS, Parkinson’s disease, and SLE. Thus, there is an urgent need to develop specific small-molecule STING antagonists. Small-molecule inhibitors of STING oligomerization will advance our understanding of STING biology and create a paradigm in how we think about strategies for antagonizing STING function.
Keywords: stimulator of interferon genes, protein arginine deiminases, cytokines, small-molecule inhibitor, Trex-1
Abstract
Stimulator of interferon genes (STING) is an essential adaptor protein required for the inflammatory response to cytosolic DNA. dsDNA activates cGAS to generate cGAMP, which binds and activates STING triggering a conformational change, oligomerization, and the IRF3- and NFκB-dependent transcription of type I Interferons (IFNs) and inflammatory cytokines, as well as the activation of autophagy. Aberrant activation of STING is now linked to a growing number of both rare as well as common chronic inflammatory diseases. Here, we identify and characterize a potent small-molecule inhibitor of STING. This compound, BB-Cl-amidine inhibits STING signaling and production of type I IFNs, IFN-stimulated genes (ISGs) and NFκB-dependent cytokines, but not other pattern recognition receptors. In vivo, BB-Cl-amidine alleviated pathology resulting from accrual of cytosolic DNA in Trex-1 mutant mice. Mechanistically BB-Cl-amidine inhibited STING oligomerization through modification of Cys148. Collectively, our work uncovers an approach to inhibit STING activation and highlights the potential of this strategy for the treatment of STING-driven inflammatory diseases.
The cytosolic detection of pathogen-derived dsDNA is an essential component of antiviral immunity. This pathway has two major components: cGAMP synthase (cGAS) and Stimulator of interferon genes (STING) (1, 2). Upon binding to dsDNA in the cytosol, cGAS activity is allosterically increased and it catalyzes the conversion of GTP and ATP into cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) (3). cGAMP then binds STING, an endoplasmic reticulum (ER)-localized adaptor protein. cGAMP binding is associated with large-scale conformational changes that promote STING oligomerization, trafficking from the ER to the Golgi, palmitoylation, and subsequent lysosome-mediated degradation (4, 5). Once oligomerized, the C-terminal tail of STING recruits TBK1 and induces autoactivation and transphosphorylation of TBK1 followed by phosphorylation of STING on Ser365 (5, 6). TBK1 then promotes IRF3 and NFκB activation, thereby promoting their translocation into the nucleus where they bind DNA and activate the transcription of type I interferons (IFNs) and numerous cytokines (1, 4–7). Given the damaging consequences of DNA accrual in the cytosol, several regulatory mechanisms exist to restrict spontaneous cGAS-STING signaling. In the absence of stimulation, STING translocation is inhibited by the ER protein STIM1 and via its retrograde transport to the ER through COPI vesicles and the adaptor protein SURF4 (8, 9).
Protein citrullination is catalyzed by the Protein Arginine Deiminases (PADs), a family of enzymes whose activity controls a wide number of biological processes and contributes to numerous inflammatory diseases, including rheumatoid arthritis (RA). In RA, patients produce antibodies to citrullinated proteins (ACPA) whose levels correlate with disease severity. In addition to antigen generation, aberrant PAD activity is associated with neutrophil extracellular trap (NET) formation, a proinflammatory form of neutrophil cell death that has been implicated in the pathogenesis of SLE, ulcerative colitis, sepsis, and fibrosis (10). Notably, COPA, a major component of COPI vesicles, and SURF4 are citrullinated (11). Given the potential impact of protein citrullination on STING trafficking, we evaluated the ability of a pan-PAD inhibitor to modulate STING signaling. This compound, BB-Cl-amidine (Fig. 1A), has been demonstrated to possess potent anti-inflammatory properties in several experimental models (12–15). Interestingly, BB-Cl-amidine blocked STING-dependent signaling, but not other innate immune signaling pathways, in the subnanomolar range. Using a chemo proteomic strategy, we showed that BB-Cl-amidine covalently modifies STING in a PAD-independent manner to impair oligomerization and all proximal downstream pathways, such as activation of TBK1-IRF3 signaling leading to type I IFN production, NFκB activation, and the production of associated cytokines and autophagy. Furthermore, BB-Cl-amidine efficiently alleviated STING-dependent disease in the Trex1D18N/D18N mouse model of Aicardi–Goutières syndrome (AGS). In summary, our data identify a chemical entity that inhibits STING oligomerization, thereby providing a previously unknown scaffold for the development of therapeutics for treating STING-dependent inflammatory diseases.
Fig. 1.
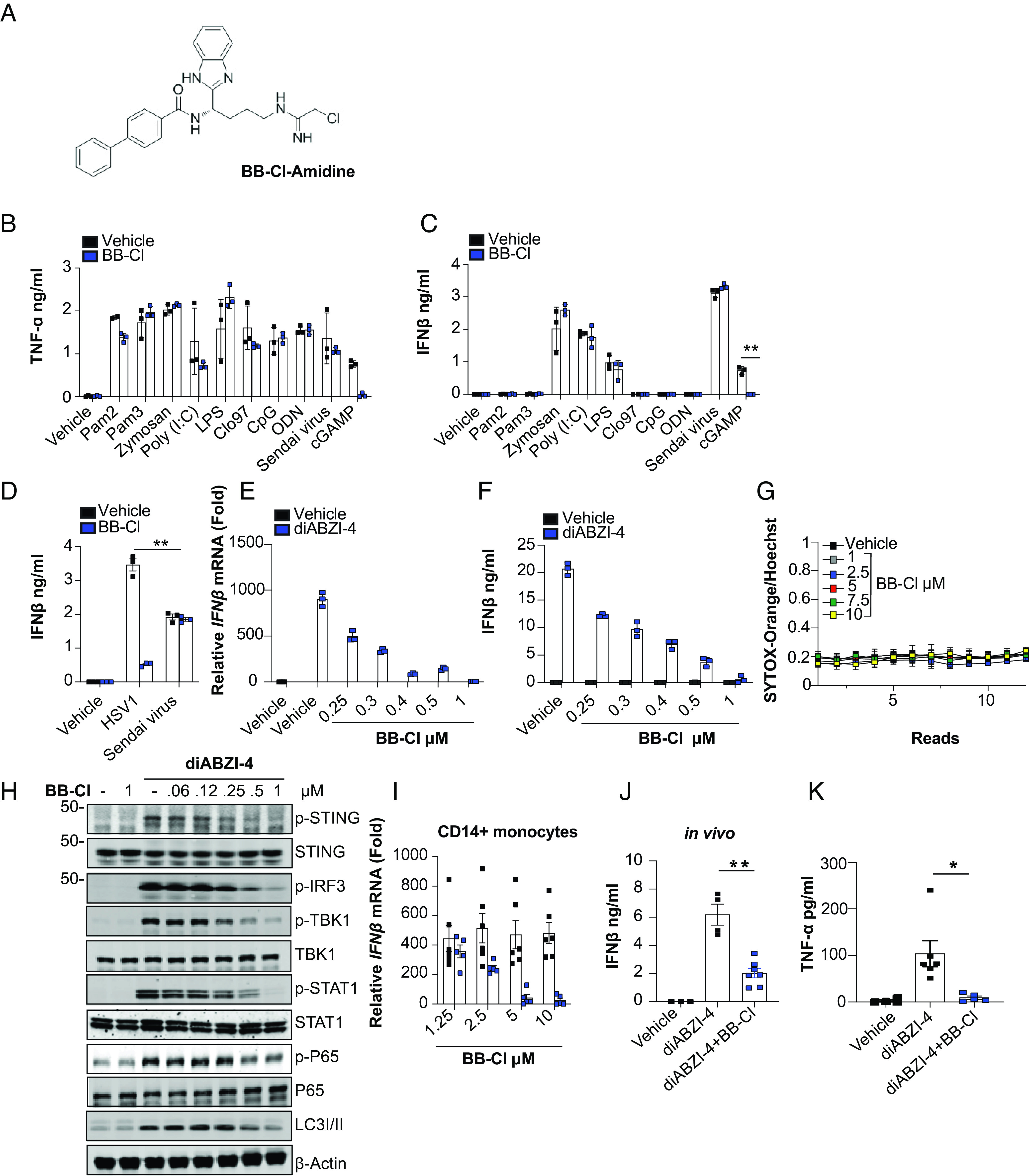
BB-Cl-amidine inhibits STING-dependent signaling. (A) Structure of BB-Cl-amidine. (B and C) ELISA analysis of TNF-α and IFN-β in conditioned medium from BMDMs pretreated with vehicle control (DMSO) or BB-Cl-amidine (1 μM) for 1 h followed by treatment with the indicated ligands for 24 h. (D) ELISA analysis of IFN-β in conditioned medium from BMDMs pretreated with vehicle control (DMSO) or BB-Cl-amidine (1 μM) for 1 h followed by infection with HSV1 (MOI 10) or Sendai virus 20 (20 Units) for 24 h. (E) qPCR analysis of Ifnβ expression in BMDMs pretreated with the indicated concentrations of BB-Cl-amidine followed by treatment with diABZI-4 for 2 h. (F) ELISA analysis of IFNβ from BMDMs pretreated with the indicated concentrations of BB-Cl-amidine followed by treatment with diABZI-4 (500 nM) for 24 h. (G) Kinetic cell death analysis of primary BMDMs treated with the indicated concentrations of BB-Cl and stained with Sytox Orange and Hoechst. Cells were imaged using the Cytation5 microscope. One read per hour for 12 h. (H) Immunoblot analysis of phosphorylated STING, IRF3, TBK1, STAT1, P65, and LC3 conversion in whole-cell lysates from BMDMs pretreated with the indicated concentrations of BB-Cl-amidine for 1 h followed by treatment with diABZI-4 for 1 h. (I) qPCR analysis of Ifnβ in human primary monocytes pretreated with BB-Cl-amidine (1 μM) followed by treatment with 500 nM diABZI-4 for 2 h. (J and K) Serum analysis of IFN-β and TNF-α in mice administered BB-Cl-amidine (10 mg/kg) for 1 h followed by diABZI (0.5 mg/kg) for 5 h. B, C, and G representative data. D–F, and H pooled data from three independent experiments. I and J vehicle (n = 4), diABZI-4 (n = 6), diABZI-4 + BB-Cl-amidine (n = 5). *P < 0.05; **P < 0.001. Error bars show mean ± SEM.
Results
Beyond their role in NET formation and their ability to generate TLR4 agonists (16, 17), little is known about how PADs contribute to innate immune signaling, especially with regard to their impact on the regulation of Toll-like receptors (TLRs) and nucleic acid–sensing pathways. As such, we sought to determine how the PADs broadly contribute to innate immune signaling. For these studies, bone marrow-derived macrophages (BMDMs) were pretreated with BB-Cl-amidine (Fig. 1A) and then stimulated with an array of innate immune receptor ligands. Changes in IFNβ and TNF-α levels were used as proximal readouts of activation. Vehicle-treated cells had comparable responses across all ligands. The one exception was cGAMP, where BB-Cl-amidine treatment led to a marked reduction in both TNF-α and IFNβ levels (Fig. 1 B and C). We next confirmed that BB-Cl-amidine blocks DNA sensing and not RNA sensing, by comparing its ability to inhibit IFNβ production induced by Herpes Simplex Virus (HSV1), a DNA virus, to that of Sendai virus, an RNA virus (Fig. 1D). Together, these data indicate that BB-Cl-amidine selectively inhibits signaling downstream of cGAS and STING.
Next, BMDMs were stimulated with the synthetic STING agonist diABZI in the presence of increasing concentrations of BB-Cl-amidine. Inhibition of Ifnβ transcription and secretion was used as readouts of STING activation (Fig. 1 E and F). Notably, BB-Cl-amidine inhibited diABZI-induced IFNβ production at an EC50 of ~0.5 μM. The inhibitory effects were independent of any toxicity issues as BB-Cl-amidine did not induce cell death (Fig. 1G). To confirm that BB-Cl-amidine directly inhibits STING signaling, we showed that BB-Cl-amidine also inhibited diABZI-induced phosphorylation of STING, IRF3, TBK1, p65, and STAT1, all of which are readouts of STING or STING-dependent responses, in a dose-dependent manner. BB-Cl-amidine also impaired diABZI-induced conversion of LC3, indicating that BB-Cl-amidine blocks the activation of autophagy, a key downstream effector response following STING activation (Fig. 1H). In addition to its ability to inhibit murine STING, BB-Cl-amidine blocked the STING-dependent induction of IFNβ in CD14+ monocytes isolated from peripheral blood (Fig. 1I). Thus, BB-Cl-amidine inhibits STING activation in both mouse and human cells. We next assessed the efficacy of BB-Cl-amidine in vivo. Mice administered diABZI i.p. induced robust production of IFNβ and TNF-α in serum. However, mice pretreated with BB-Cl-amidine showed a significant reduction in the production of diABZI-induced IFNβ and TNF-α in vivo (Fig. 1 J–K). Thus, BB-Cl-amidine blocks activation of the STING pathway in vivo.
To further assess the inhibitory effects of BB-Cl-amidine on STING-dependent signaling, we profiled the transcriptional response to diABZI by RNA-sequencing in the presence or absence of BB-Cl-amidine. diABZI treatment induced robust induction of the type I IFN and IFN-stimulated genes (ISGs) (Fig. 2 A–C). However, in the presence of BB-Cl-amidine, the transcriptional program induced downstream of STING was completely abolished (Fig. 2 A–C). Given that BB-Cl-amidine was initially characterized as a pan-PAD inhibitor, we next assessed whether BB-Cl-amidine mediated its effect via the inhibition of PADs. Surprisingly, WT and Padi4-deficient mice displayed comparable expression levels of diABZI-induced Ifnβ (Fig. 3A) and IRF3 phosphorylation (Fig. 3B). We next assessed if BB-Cl-amidine could also inhibit STING activation independently of PAD4. Indeed, BB-Cl-amidine impaired both diABZI-induced expression of Ifnβ and Cxcl10 (Fig. 3 C and D) and IRF3 phosphorylation (Fig. 3E) in WT and PAD4-deficient macrophages, comparably. In addition to PAD4, myeloid cells also express PAD2 at high levels. To rule out redundancy between PAD family members, we used CRISPR/Cas9 to generate PAD2- and PAD2/4-deficient mice and assessed STING responses in BMDM. Notably, diABZI induced the expression of Ifnβ and Cxcl10 to a similar degree in BMDM from WT, PAD2 knockout, and PAD2/4 knockout mice (Fig. 3 F and G).
Fig. 2.
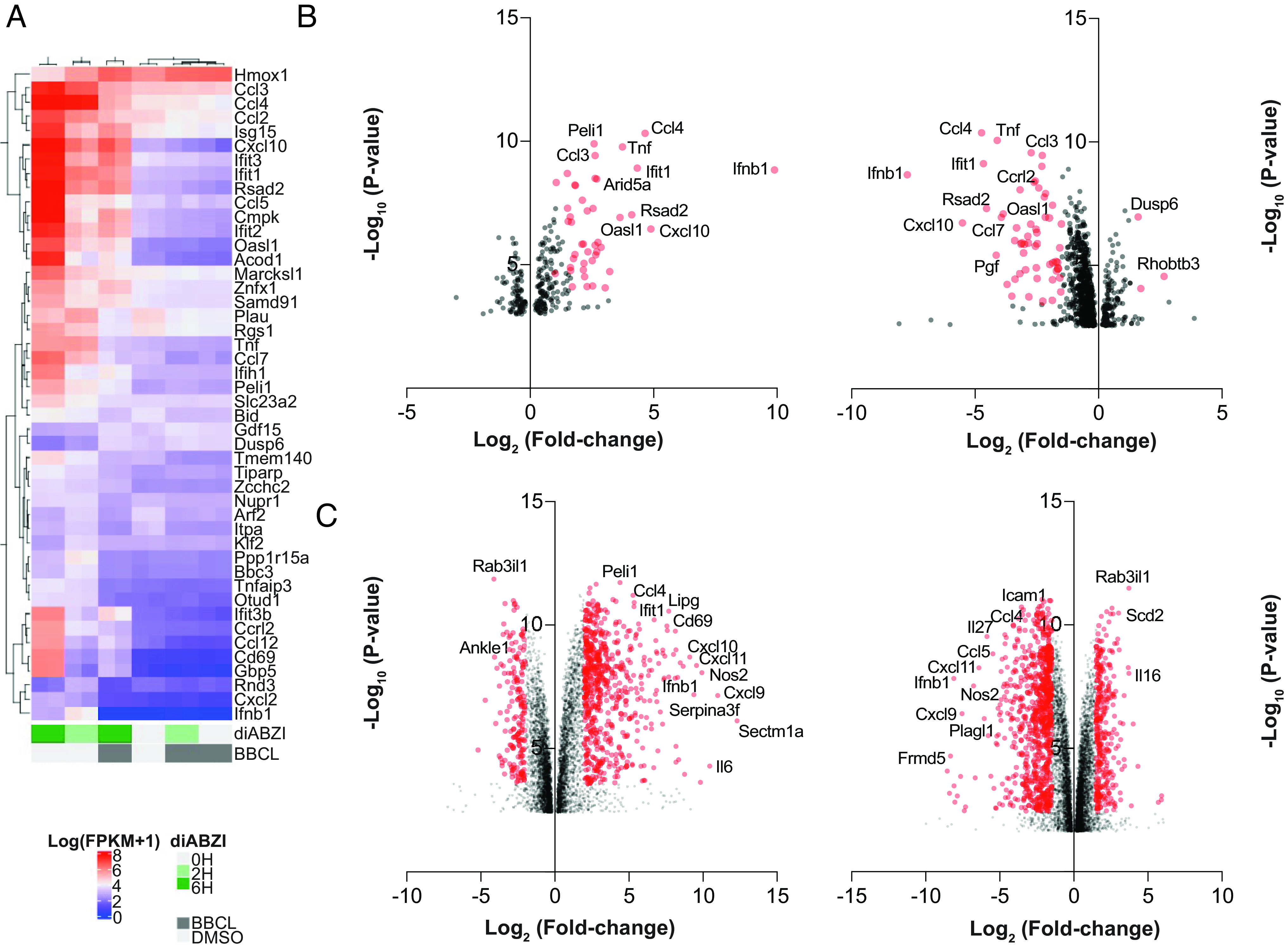
Transcriptome analysis of BB-Cl-amidine-induced STING inhibition. (A) Heat-map analysis of top 46 expression changes in genes calculated from log (FPKM+1) values from RNA sequencing analysis on RNA extracted from BMDMs pretreated with vehicle control (DMSO) or BB-Cl-amidine (1 μM) followed by treatment with diABZI-4 for 2 or 6 h. (B and C) Log2 fold change in vehicle control (DMSO) vs. diABZI-4-treated cells (Left) and BB-Cl-amidine-treated cells vs. diABZI-4-treated cells (Right) for 2 h (B) or 6 h (C). Data are the average of two replicates sequenced from each of the indicated samples.
Fig. 3.

BB-Cl-amidine inhibits STING signaling independent of PADs. (A) qPCR analysis of Ifnβ expression in WT and Padi4−/− BMDMs treated with diABZI-4 for 2 h. (B) Immunoblot analysis of phosphorylated IRF3, STING, and PAD4 in whole-cell lysates from WT and Padi4−/− BMDMs treated with diABZI-4 for the indicated times. (C and D) qPCR analysis of Ifnβ (C) and Cxcl0 (D) expression in BMDMs pretreated with 1 μM BB-Cl-amidine for 1 h followed by treatment with diABZI-4 for 2 h. (E) Immunoblot analysis of phosphorylated IRF3, PAD4, and β-actin in whole-cell lysates from WT and Padi4−/− BMDMs pretreated with 1 μM BB-Cl-amidine for 1 h followed by treatment with diABZI-4 for 1 h. (F and G) qPCR analysis of Ifnβ (F) and Cxcl0 (G) expression in WT, Padi2−/− and Padi2/4−/− BMDMs treated with diABZI-4 for 2 h. ***P < 0.0001. Error bars show mean ± SEM.
Given that BB-Cl-amidine retained its inhibitory effects on STING signaling in the absence of PAD2 and PAD4, we sought to account for these PAD-independent effects by identifying additional targets of BB-Cl-amidine. To that end, we used an alkyne-containing derivative of BB-Cl-amidine, BB-Cl-Yne (BB-Cl-Yne) (Fig. 4A), which can be rapidly coupled to an azide-containing reporter tag (e.g., biotin-azide) via “click chemistry” (18); the reporter tag enables the rapid visualization or enrichment of modified proteins. To ensure that the alkyne handle does not impact the inhibition of STING signaling, we first compared the inhibitory effects of BB-Cl-amidine and BB-Cl-Yne. As a control, we also tested BB-F-Yne, a more selective probe of PAD activity due to the relatively poorer leaving group ability of the fluoro group (Fig. 4B). Importantly, the potencies of BB-Cl-amidine and BB-Cl-Yne were comparable. By contrast, BB-F-Yne showed no inhibition of STING signaling (Fig. 4C). These data not only confirm that PAD inhibition does not impact STING signaling but also demonstrate that BB-Cl-Yne can be used to identify host factors bound by this drug.
Fig. 4.

BB-Cl-amidine directly targets STING. (A and B) Structure of BB-Cl-Yne and BB-F-Yne alkyne probes. (C) ELISA analysis of IFN-β in conditioned medium from BMDMs pretreated with vehicle control (DMSO), BB-Cl-amidine (1 μM), BB-Cl-Yne (1 μM), or BB-F-Yne (1 μM) for 1 h followed by treatment with 500 nM diABZI-4 for 24 h. (D) Log10 fold change enrichment of proteins from copper-clicked lysates from cells treated with BB-Cl-Yne (1 μM) for 1 h. (E) Representative mass spectrometry spectra of STING identified from streptavidin pull downs of clicked lysates from cells treated with BB-Cl-Yne or cells cotreated with BB-Cl-amidine and BB-Cl-Yne. (F) Peptide coverage analysis of peptides identified in streptavidin bead pull downs from clicked lysate cells treated with BB-Cl-Yne or cells cotreated with BB-Cl-amidine and BB-Cl-Yne. (G) Immunoblot of STING and strep-IR-dye in streptavidin bead pull downs and input from clicked lysates of cells treated with or without BB-Cl-Yne and BB-Cl-amidine. (C) pooled data from two independent experiments performed in triplicate. (D and E) performed in triplicate. (G) representative data of two independent experiments.
To identify such factors, BMDMs were treated with BB-Cl-Yne, BB-Cl-Yne plus BB-Cl-amidine, or vehicle control. Lysates were prepared, “clicked” to biotin-azide, and modified proteins enriched on streptavidin beads. Mass spectrometry revealed that STING itself was highly enriched by BB-Cl-Yne across all replicate samples but not in the control samples (Fig. 4 D and E and SI Appendix, Fig. S1A). Consistent with STING being a predominant target of BB-Cl-amidine, cotreatment of cells with BB-Cl-Yne and BB-Cl-amidine reduced STING enrichment as judged by relative STING coverage and by western blotting (Fig. 4F and SI Appendix, Fig. S1B). Further evaluation of BB-Cl-Yne binding of STING was confirmed by western blot (Fig. 4G).
To assess the efficacy of BB-Cl-amidine in a STING-dependent disease context, we used a mouse model of AGS. TREX1 is an abundant 3′-5′ exonuclease, which digests cytoplasmic DNA and prevents unwanted activation of cGAS (19). TREX1 mutations were first identified in AGS patients presenting with severe encephalitis, intracranial calcifications, and elevated type I IFN in the cerebrospinal fluid (20). Several studies have confirmed that Trex1 deficiency results in the abnormal accumulation of cytosolic DNA and the constitutive activation of cGAS/STING signaling (19–24). Trex1-deficient mice also exhibit systemic inflammation, production of autoantibodies to dsDNA, renal disease, reduced postnatal survival, and severe myocarditis (21, 22). To more easily study the in vivo role of Trex1, a nuclease-deficient knockin model was developed by generating a D18N mutation; this mutation compromises enzymatic activity (22). Trex1D18N/D18N mutant mice have shortened lifespans and develop severe myocarditis. To first demonstrate the dependency on STING and IFN in the Trex1D18N/D18N model, we intercrossed Trex1D18N/D18N mice to Irf3-deficient, STING knockout mice, or STINGR237A/R237A mutant mice; arginine 237 lies within the ligand-binding domain of STING and is critical for cGAMP binding. Thus, STINGR237A/R237A mice do not respond to cGAMP. Consistent with previous studies, Trex1D18N/D18N mice had reduced survival, displayed mild splenomegaly, accumulation of serum Cxcl10, and severe myocarditis (SI Appendix, Fig. S2 A–E). By contrast, Trex1D18N/D18N mice crossed to Irf3-deficient mice were completely rescued from disease (SI Appendix, Fig. S2 A–E). Thus, disease in the Trex1D18N/D18N model is dependent on IRF3 and the induction of type I IFNs. Trex1D18N/D18N mice were also protected by crossing them to either STING-deficient mice or STINGR237A/R237A mice (SI Appendix, Fig. S3 A–D). Thus, Trex1D18N/D18N myocarditis is dependent on cGAS/STING signaling. Having established that the development of myocarditis by Trex1D18N/D18N mice is STING-dependent, we next determined whether BB-Cl-amidine could alleviate disease. To that end, WT and Trex1D18N/D18N mice were administered BB-Cl-amidine or vehicle control daily in the chow for 8 wk starting at 2 mo of age. BB-Cl-amidine alleviated disease pathology in the Trex1D18N/D18N mice as evident from improved survival, reduced splenomegaly, and reduced myocarditis when compared to Trex1D18N/D18N mice receiving control chow (Fig. 5 A–E). BB-Cl-amidine also alleviated cardiac fibrosis in Trex1D18N/D18N mice (Fig. 5F).
Fig. 5.

BB-Cl-amidine alleviates STING-dependent experimental AGS. (A–C) Survival analysis (A), spleen weight (B) and heart:body weight ratio (C) of TrexD18N/D18N mice administered a control diet or a BB-Cl-amidine-embedded diet. (D) Representative H&E staining of tissue sections from hearts of WT and Trex1D18N/D18N mice administered a control diet or BB-Cl-amidine embedded diet. (E) Pathology scoring of heart sections from (D). (F) Trichrome staining of tissue sections from hearts of WT and Trex1D18N/D18N mice administered a control diet or BB-Cl-amidine embedded diet. (A) vehicle (n = 10) BB-Cl-amidine (n = 17), data representative of pooled mice from two independent experiments. (B and C) WT vehicle (n = 5), WT BB-Cl-amidine (n = 5), Trex1D18N/D18 vehicle (n = 6), Trex1D18N/D18N BB-Cl-amidine (n = 9). (D) representative images. (E) WT vehicle (n = 3), WT BB-Cl-amidine (n = 3), Trex1D18N/D18N vehicle (n = 4), Trex1D18N/D18N BB-Cl-amidine (n = 6). *P < 0.05, two-way ANOVA. Error bars show mean ± SEM.
BB-Cl-amidine is a cysteine-reactive molecule that derivatizes cysteine residues to generate a 423 Da posttranslational modification that alters protein function. Thus, we next assessed if BB-Cl-amidine could modify cysteine residues in STING. Using an in vitro reaction system, we incubated full-length recombinant STING with BB-Cl-amidine and performed LCMS/MS to determine if BB-Cl-amidine modified STING. Peptide mapping analysis revealed that treatment of recombinant human STING with BB-Cl-amidine led to a 423-Da modification on Cys148 (Fig. 6A). In addition to Cys148, BB-Cl-amidine also modified STING on Cys206, Cys257, and Cys309 (SI Appendix, Fig. S4 A–C). Interestingly, Cys148 is critical for STING oligomerization following activation (25). Previous studies have identified Cys148 in human STING and Cys147 in murine STING as an essential residue for forming disulfide bridges and stabilizing STING oligomers. High molecular weight oligomerization of STING creates a structural platform for recruitment and activation of TBK1. Upon oligomerization, the STING C-terminal tail recruits TBK1 and facilitates the phosphorylation of protruding C-terminal tails of proximal STING molecules in the oligomer followed by the recruitment of IRF3 (26–28). We evaluated the importance of Cys147 by generating mutations at this site and evaluating STING function. HEK293T cells expressing WT STING or the STINGCys147A mutant were stimulated with diABZI. diABZI induced robust oligomerization of WT STING. However, cells expressing STINGCys147A had a significant reduction in diABZI-induced STING oligomerization (Fig. 6B). Thus, we hypothesized that BB-Cl-amidine targets Cys148 to impair oligomerization. We next assessed the effect of BB-Cl-amidine on STING oligomerization in primary cells. WT BMDMs were pretreated with a titration of BB-Cl-amidine followed by treatment with diABZI to activate STING. BB-Cl-amidine inhibited diABZI-induced STING oligomerization in a dose-dependent manner (Fig. 6C). In addition, diABZI induced comparable levels of STING oligomerization in both WT and PAD4-deficient cells (Fig. 6D). Thus, BB-Cl-amidine inhibits STING oligomerization independently of PAD4.
Fig. 6.

BB-Cl-amidine impairs STING oligomerization via the modification of Cys148. (A) Representative mass spectrometry spectra of STING modified by BB-Cl-amidine identified in tryptic digests from recombinant STING (10 μg) incubated with BB-Cl-amidine (10 μM) for 1 h at 37 °C. (B) Immunoblot analysis of STING in native and reduced fractions from lysates of HEK293T cells expressing WT murine STING or a murine STING-C147S mutant and treated with diABZI-4 for 15 min. (C) Immunoblot analysis of STING in native and reduced fractions of lysates from BMDMs pretreated with the indicated concentrations of BB-Cl-amidine followed by treatment with diABZI-4. (D) Immunoblot analysis of STING in native and reduced fractions of lysates from WT and Padi4−/− BMDMs treated with 500 nM diABZI for the indicated times.
Discussion
STING activation directly leads to the robust IRF3-dependent production of type I IFNs as well as increased NFκB signaling (29). Aberrant STING activation has been linked to several Mendelian and non-Mendelian inflammatory diseases (30–32), and, as such, STING inhibitors have the potential to directly modulate inflammatory responses in these diseases. In this study, we showed that BB-Cl-amidine directly inhibits the STING-dependent activation of IRF3, NFκB, and autophagy in vitro and in vivo. Given that BB-Cl-amidine was initially developed as a pan-PAD inhibitor, we initially explored the role of protein citrullination in modulating STING. BB-Cl-amidine retained its inhibitory effects on STING in PAD-deficient cells. Mechanistically, BB-Cl-amidine modified the functionally important Cys148, conserved as Cys147 in mice, and blocked oligomerization. Given that STING oligomerization is a prerequisite for its activity, BB-Cl-amidine inhibited STING-dependent activation of IRF3, NFκB, and autophagy thus leading to impaired production of type I IFNs and inflammatory cytokines. Administration of BB-Cl-amidine also alleviated the inflammatory phenotype in an experimental model of AGS. Interestingly, BB-Cl-amidine targeted Cys148 and prevented oligomerization of STING. By contrast, H-151, a previously reported STING inhibitor with a comparable IC50 value, has been reported to block palmitoylation by modifying Cys91 (29). While BB-Cl-amidine derivatized three other functionally inert cysteine residues in STING, we were unable to detect any modification of Cys91. Thus, BB-Cl-amidine inhibits STING in a mechanistically distinct manner.
There are several implications of these studies. First, BB-Cl-amidine has been evaluated in several animal models where aberrant PAD activity has been implicated (12–15). These models include the collagen-induced arthritis (CIA) model of RA and the MRL/lpr model of lupus (12–15). STING knockout has either no effect or aggravates disease in these models, suggesting that the efficacy observed in these models is principally driven by PAD inhibition. For other models, care should be taken in interpreting the effects of BB-Cl-amidine as any observed efficacy may relate to STING inhibition. Given this issue, we recommend the use of BB-F-amidine, which shows excellent proteome-wide selectivity or combinations of isozyme-specific inhibitors, including the PAD1-selective inhibitor SM26 (33), the PAD2-selective inhibitor AFM30a (34, 35), and the PAD4-selective inhibitor GSK484 (34).
In summary, we report the identification of a small-molecule STING antagonist that inhibits STING activation by covalently modifying Cys148 and blocking oligomerization.
Materials and Methods
Mice.
B6.Cg-Padi4tm1.1Kmow/J (Padi4−/−) mice were obtained from the Jackson Laboratory, strain number 030315. Padi2−/− mice were generated using the sgRNA sequence: GCACGTACACCGCCTCCACG. Embryos were microinjected with the target sgRNA sequence (20 ng/μL) with Alt-R® S.p. Cas9 Nuclease V3 (50 ng/μL) and implanted into C57BL6 or PAD4−/− mice to generate Padi2−/− mice or Padi2−/− Padi4−/− double knockout (DKO), respectively. Resulting founder mice had a 25-nt or 29-nt deletion for the DKO, or 8-nt, 10-nt deletion for the single Padi2−/−. Trex1D18N/D18N STING−/− and STINGR237A/R237A were made and provided by GSK. IRF3−/− were from T. Taniguchi (U. Tokyo, Japan). All animal experiments were approved by the Institutional Animal Care Use Committees at the University of Massachusetts Chan Medical School. Animals were kept in a specific pathogen-free (SPF) environment. Animals were housed in groups and fed standard chow diets. Sample sizes used are in line with other similar published studies.
Reagents.
All chemicals were purchased from Cayman chemical or Sigma Aldrich, Inc. Compounds were dissolved in Dimethylsulfoxide (DMSO) and further diluted in media. Cells were transfected using gene juice according to the manufacturer’s instructions.
Plasmids.
Murine WT STING and Cysteine mutant constructs were kindly provided by Tomohiko Taguchi, Department of Health Chemistry, Graduate School of Pharmaceutical Sciences, University of Tokyo.
Cell Culture.
Human kidney cell line HEK293T was cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Human peripheral blood monocyte cell line, THP1 cells were cultured in RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. For isolation of BMDMs, tibias, and femurs were removed from wild-type and KO mice and bone marrow was flushed with complete DMEM-medium. Cells were plated in medium containing 20% (v/v) conditioned medium of L929 mouse fibroblasts cultured for 7 d at 37 °C in a humidified atmosphere of 5% CO2. Medium was replaced every 3 d.
ELISA.
Conditioned media or serum was collected as indicated and mouse IL-1β or TNF-α, were quantified by sandwich ELISA (R&D Systems).
Immunoblotting and Immunoprecipitation.
Primary BMDMs from WT or KO mice were cultured in 12-well plates (1 × 106 cells per mL; 1 mL). HEK293T cells (2.5 × 105 cells per mL) were cultured in 6-well plates and transfected with the relevant constructs where indicated. For cell lysate analysis, cells were lysed directly in 1X Laemmli sample buffer. For native gel analysis, cells were lysed in NP-40 lysis buffer (50 mM Tris-HCl, pH 7.4, containing 150 mM NaCl, 0.5% (w/v) IgePal, 50 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail). For immunoprecipitation of STING, cells were treated as indicated and then collected in 250 μL of lysis buffer, followed by incubation for 15 min on ice. Lysates were incubated with STING antibody and protein A–protein G-agarose was added to each sample, followed by incubation overnight at 4 °C. Immunoprecipitates were collected by centrifugation and washed four times with 1 mL of lysis buffer. Immunoprecipitates were eluted from beads using 1 × sample buffer. Samples were resolved by SDS-PAGE and transferred to nitrocellulose membranes and analyzed by immunoblot. Immunoreactivity was visualized by the Odyssey Imaging System (LICOR Biosciences). All signaling antibodies (antibodies to pIRF3, IRF3, STING, pSTING, TBK1, pTBK1, LC3, pSTAT1, STAT1, pp65, and p65) were from Cell signaling. Antibodies to PAD2 were from Proteintech and those to PAD4 were from Abcam. Anti-β-actin (AC-15; A1978) was from Sigma; anti-mouse IRDye® 680 (926-68070) and anti-rabbit IRDye® 800 (926-32211) were from LI-COR Biosciences
RNA Sequencing.
RNA sequencing and preliminary data analysis were performed by Beijing Genomics Institute (BGI), Shanghai, China. Heatmaps were generated using the R Complex Heatmap package (36) based on the union of the top 50 most differentially expressed genes in the diABZI vs. unstimulated (2H) and BBCLA vs. vehicle (2H) conditions using hierarchal clustering and Euclidean distance. Differential gene expression for volcano plots was performed using Salmon v1.6.0 (37) on the GRCm39 transcriptome and the R DESeq2 (38) package. Prefiltering was performed on low-expressed genes (counts < 10) and log-fold-change shrinkage based on the apeglm (39) algorithm was utilized. Genes were considered significant with a |Log2(Fold Change)| ≥ 1 and adjusted P-value < 0.05.
cDNA Synthesis and Real-Time PCR.
Total RNA was extracted from whole-lung tissue or cells. One microgram of RNA was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad). Then, 5 ng of cDNA was subjected to qPCR analysis using iQ SYBR Green supermix reagent (Bio-Rad). Gene expression levels were normalized to TATA-binding protein (TBP) or HPRT as indicated. Relative mRNA expression was calculated by a change in cycling threshold method as 2−ddC(t). Specificity of qRT-PCR amplification was assessed by melting curve analysis. The sequences of primers used in this study are listed in Table 1.
Table 1.
Primer sequences
| Primer name | Sequence |
|---|---|
| Murine IFN-β Forward | ATAAGCAGCTCCAGCTCCAA |
| Murine IFN-β Reverse | CTGTCTGCTGGTGGAGTTCA |
| Murine Cxcl10 Forward | CCAAGTGCTGCCGTCATTTTC |
| Murine Cxcl10 Reverse | GGCTCGCAGGGATGATTTCAA |
| Human IFN-β Forward | GTCTCCTCCAAATTGCTCTC |
| Human IFN-β Reverse | ACAGGAGCTTCTGACACTGA |
| Murine TBP Forward | GAAGCTGCGGTACAATTCCAG |
| Murine TBP Reverse | CCCTTGTACCCTTCACCAAT |
| Human HPRT Forward | ATCAGACTGAAGAGCTATTGTAATGA |
| Human HPRT Reverse | TGGCTTATATCCAACACTTCGTG |
Peptide Mapping by Nano LC-MS/MS.
The resulting peptides were lyophilized, resuspended in 5% acetonitrile, 0.1% (v/v) formic acid in water and injected onto a NanoAcquity UPLC (Waters) coupled to a Q Exactive (Thermo Scientific) hybrid quadrupole orbitrap mass spectrometer. Peptides were trapped on a 100 µm I.D. fused-silica precolumn packed with 2 cm of 5 µm (200 Å) Magic C18AQ (Bruker-Michrom) particles in 5% acetonitrile, 0.1% (v/v) formic acid in water at 4.0 µL/min for 4.0 min. Peptides were then separated over a 75-µm I.D. gravity-pulled 25-cm long analytical column packed with 3-µm (100 Å) Magic C18AQ particles, at a flow rate of 300 nL/min containing mobile phase A, 0.1% (v/v) formic acid in water and mobile phase B, 0.1% (v/v) formic acid in acetonitrile, using a biphasic gradient: 0 to 60 min (5 to 35% B), 60 to 90 min (35 to 60% B), 90 to 93 min (60% B), 93 to 94 min (60 to 90% B), 94 to 109 (90% B), followed by equilibration to 5% B. Nano-ESI source was operated at 1.4 kV via liquid junction. Mass spectra were acquired over m/z 300 to 1,750 at 70,000 resolution (m/z 200) with an Automatic Gain Control (AGC) target of 1e6. Data-dependent acquisition (DDA) selected the top 10 most abundant precursor ions for tandem mass spectrometry by HCD fragmentation using an isolation width of 1.6 Da, max fill time of 110 ms, and AGC target of 1e5. Peptides were fragmented by a normalized collisional energy of 27, and product ion spectra were acquired at a resolution of 17,500 (m/z 200). Raw data files were peak processed with Proteome Discoverer (version 2.1, Thermo Scientific) followed by identification using Mascot Server (Matrix Science) against the Mouse (Swissprot) FASTA file (downloaded 07/2019). Search parameters included full tryptic enzyme specificity, and variable modifications of N-terminal protein acetylation, oxidized methionine, glutamine conversion to glutamic acid, and a 423 Da cysteine mass shift corresponding to modification by BB-Cl-Amidine. Assignments were made using a 10 ppm mass tolerance for the precursor and 0.05 Da mass tolerance for the fragment ions. All nonfiltered search results were processed by Scaffold (version 4.8.4, Proteome Software) utilizing the Trans-Proteomic Pipeline (Institute for Systems Biology, Seattle, WA) at 1% false-discovery rate (FDR) for peptides and 99% threshold for proteins (two peptides minimum).
diABZI Treatment In Vivo.
Male and female mice 8 to 12-wk old were anesthetized with isoflurane and administered 0.5 mg/kg diABZI-4 by intraperitoneal injection for the indicated times. BB-Cl-Amidine was dissolved in 100% DMSO and diluted in 20% PEG/PBS for intraperitoneal injection at 10 mg/kg.
Histology.
Tissue blocks were sectioned at 5 µm thick. Tissues were fixed in 4% paraformaldehyde overnight. Tissue sections were stained with H&E for evaluation of inflammation. Pathology evaluation was performed by applied pathology systems. Heart tissue was assessed for myocarditis as follows.
BB-CL-Amidine Mouse Diet.
BB-Cl-amidine embedded mouse diet was formulated by Lab Diets®. BB-Cl-amidine was added to a base diet of laboratory lab diet 5001 at a dose of 20 mg/kg/day of consumption. Mice received the control base diet or the BB-Cl-amidine containing diet for the indicated times.
Copper Click Chemistry.
Cells treated with or without the BB-Cl-Yne probe were lysed and quantified by protein DC assay. Proteome samples (2 mg/mL) were incubated with TCEP, TBTA ligand, copper sulfate, and Biotin Azide for 1 h at room temperature with vortexing every 15 min. Precipitated proteins were centrifuged for 5 min at 4,600 g. Protein pellets were washed twice with ice-cold methanol and sonicated in 1.2% SDS. Samples were heated at 95 °C for 5 min and diluted to a final volume of 6 mL with PBS (0.2% SDS). An aliquot of the postclicked lysate was retained, and the remainder was incubated with streptavidin beads on a rotator overnight at 4 °C. Samples were rotated at room temperature for 2 h to resolubilize the SDS. Beads were washed five times with 0.2% SDS/PBS and placed on a rotator for 10 min in between washes. Beads were washed with ultrapure water a further three times. At this point, beads were eluted in 1× sample buffer or further processed for mass spec as described.
PBMC Isolation.
PBMCs were isolated from whole blood of consenting donors. Blood was diluted 1:1 in sterile PBS and layered over 15 mL of Lymphoprep. Blood was spun at 450 g with no break. The interphase was transferred to a fresh tube using a Pasteur pipette and washed twice in PBS. Red blood cells were lysed in red blood cell lysis buffer for 10 min at room temperature. Cells were washed once more in PBS and counted.
CD14+ Monocyte Isolation.
PBMC were isolated from Leukopacs purchased from New York Biologics. Blood was diluted 1:1 in sterile PBS and layered over 15 mL of Lymphoprep. Blood was spun at 450 g with no break. The interphase was transferred to a fresh tube using a Pasteur pipette and washed twice in PBS. Red blood cells were lysed in red blood cell lysis buffer for 10 min at room temperature. Cells were washed once more in PBS and counted. CD14+ monocytes were isolated using human CD14 magnetic microbeads (Miltenyi), washed twice in ice-cold macs buffer and plated in RPMI medium.
Ethics.
All animal studies were performed in compliance with the federal regulations set forth in the Animal Welfare Act, the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH, and the guidelines of the UMass Chan Medical School Institutional Animal Use and Care Committee. All protocols used in this study were approved by the Institutional Animal Care and Use Committee at the UMass Chan Medical School (protocol A-1633). Human PBMCs were sourced ethically from New York Biologics as leukopacs. No institutional review board protocol was required for use of these samples.
Statistical Analysis.
For comparisons of two groups two-tailed Students’ t test was performed. Multiple comparison analysis was performed using two-way ANOVA. Mantel–Cox analysis was used for survival analysis. Three-to-ten mice were used per experiment, sufficient to calculate statistical significance, and in line with similar studies published in the literature.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported by grants from the Li Weibo Institute for Rare Diseases (UMass Chan, to K.A.F.), the UMass OTCV technology fund (to K.A.F.), the UMass Chan BRIDGE fund (to K.A.F. and P.R.T.), and NIH T32 grants GM107000 and AI132152 (to J.Y.Z.).
Author contributions
F.H., L.S.-G., L.B., S.M., P.R.T., and K.A.F. designed research; F.H., L.S.-G., Z.J., J.Y.Z., R.W., N.S., and S.A.S. performed research; S.-L.N. and G.S.P. contributed new reagents/analytic tools; F.H., L.S.-G., Z.J., and J.Y.Z. analyzed data; L.B. and S.M. provided experimental advice; S.-L.N. and G.S.P. advice on experimental design; P.R.T. supervised the research; and F.H., L.S.-G., and K.A.F. wrote the paper.
Competing interests
K.A.F. serves on the scientific advisory board of NodThera Inc., Generation Bio, and Janssen and is a scientific founder of Danger Bio, LLC. P.R.T. is a scientific founder of Danger Bio. At the time of the study, S.-L.N. and G.S.P. were employed by GSK. All GSK authors have equity holdings in GSK. U.S. Patent Application No.: 63/147,387 Title: STING inhibitors filing Date: Feb 9, 2021 Inventor(s): F.H., L.S.-G., P.R.T., and K.A.F.
Footnotes
Reviewers: D.B.S., University of Washington; and I.Z., Boston Children’s Hospital.
Contributor Information
Fiachra Humphries, Email: Fiachra.Humphries@umassmed.edu.
Paul R. Thompson, Email: Paul.Thompson@umassmed.edu.
Katherine A. Fitzgerald, Email: Kate.Fitzgerald@umassmed.edu.
Data, Materials, and Software Availability
All source data is available from the authors upon request. All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Ishikawa H., Barber G. N., STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ishikawa H., Ma Z., Barber G. N., STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ablasser A., et al. , cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu D., et al. , STING directly activates autophagy to tune the innate immune response. Cell Death Differ. 26, 1735–1749 (2019), 10.1038/s41418-018-0251-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gui X., et al. , Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanaka Y., Chen Z. J., STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 5, ra20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitzgerald K. A., et al. , IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol. 4, 491–496 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Deng Z., et al. , A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. J. Exp. Med. 217, e20201045 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mukai K., et al. , Homeostatic regulation of STING by retrograde membrane traffic to the ER. Nat. Commun. 12, 61 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian Y., et al. , Peptidylarginine deiminase 2 has potential as both a biomarker and therapeutic target of sepsis. JCI Insight 5, e138873 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tilvawala R., et al. , The rheumatoid arthritis-associated citrullinome. Cell Chem. Biol. 25, 691–704.e696 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knight J. S., et al. , Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/<em>lpr</em> mice. Ann. Rheumatic Dis. 74, 2199–2206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghari F., et al. , Citrullination-acetylation interplay guides E2F–1 activity during the inflammatory response. Sci. Adv. 2, e1501257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kawalkowska J., et al. , Abrogation of collagen-induced arthritis by a peptidyl arginine deiminase inhibitor is associated with modulation of T cell-mediated immune responses. Sci. Rep. 6, 26430 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sodre F. M. C., et al. , Peptidylarginine deiminase inhibition prevents diabetes development in NOD mice. Diabetes 70, 516–528 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y., et al. , Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 184, 205–213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li F. J., et al. , Citrullinated vimentin mediates development and progression of lung fibrosis. Sci. Transl. Med. 13, eaba2927 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nemmara V. V., et al. , The development of benzimidazole-based clickable probes for the efficient labeling of cellular protein arginine deiminases (PADs). ACS Chem. Biol. 13, 712–722 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stetson D. B., Ko J. S., Heidmann T., Medzhitov R., Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crow Y. J., et al. , Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genetics 38, 917–920 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Morita M., et al. , Gene-Targeted Mice Lacking the Trex1 (DNase III) 3′→5′ DNA Exonuclease Develop Inflammatory Myocarditis. Mol. Cell Biol. 24, 6719–6727 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grieves J. L., et al. , Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease. Proc. Natl. Acad. Sci. U.S.A. 112, 5117–5122 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao D., et al. , Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. U.S.A. 112, E5699–E5705 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gray E. E., Treuting P. M., Woodward J. J., Stetson D. B., Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutières Syndrome. J. Immunol. 195, 1939–1943 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ergun S. L., Fernandez D., Weiss T. M., Li L., STING polymer structure reveals mechanisms for activation, hyperactivation, and inhibition. Cell 178, 290–301.e210 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Liu S., et al. , Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Zhao B., et al. , Structural basis for concerted recruitment and activation of IRF-3 by innate immune adaptor proteins. Proc. Natl. Acad. Sci. U.S.A. 113, E3403–E3412 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang C., et al. , Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398 (2019), 10.1038/s41586-019-1000-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haag S. M., et al. , Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Lepelley A., et al. , Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling. J. Exp. Med. 217, e20200600 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melki I., et al. , Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J. Allergy Clin. Immunol. 140, 543–552.e545 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Uggenti C., et al. , cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat. Genetics 52, 1364–1372 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Mondal S., et al. , Halogen bonding increases the potency and isozyme selectivity of protein arginine deiminase 1 inhibitors. Angew. Chem. Int. Ed. 58, 12476–12480 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis H. D., et al. , Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 11, 189–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muth A., et al. , Development of a selective inhibitor of protein arginine deiminase 2. J. Med. Chem. 60, 3198–3211 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gu Z., Eils R., Schlesner M., Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C., Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhu A., Ibrahim J. G., Love M. I., Heavy-tailed prior distributions for sequence count data: Removing the noise and preserving large differences. Bioinformatics 35, 2084–2092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All source data is available from the authors upon request. All study data are included in the article and/or SI Appendix.