

Abstract
Respiratory syncytial virus (RSV) infection of airway epithelial cells results in persistent NF-κB activation and NF-κB-mediated interleukin-8 production. Previous studies in airway epithelial cells demonstrated that tumor necrosis factor alpha (TNF-α)-induced NF-κB activation is transient due to regulation by IκBα. However, during RSV infection, IκBα has only a partial inhibitory effect on NF-κB activation. Studies presented here demonstrate that neither increased IκBα production which occurs as a result of RSV-induced NF-κB activation nor inhibition of proteasome-mediated IκBα degradation results in a reversal of RSV-induced NF-κB activation. Thus, while manipulation of IκBα results in reversal of TNF-α-induced NF-κB activation, manipulation of IκBα does not result in a reversal of RSV-induced NF-κB activation.
Respiratory syncytial virus (RSV) infection in the airway is a major cause of morbidity and mortality in children (7). In vitro and in vivo studies have demonstrated that the pathophysiology of RSV infection involves airway inflammation (17). A key component to the inflammatory response is the production of proinflammatory mediators, such as interleukin-1 (IL-1), IL-6, IL-8, tumor necrosis factor alpha (TNF-α), and granulocye-macrophage colony-stimulating factor, by airway epithelial cells (18). All of these proinflammatory mediators are regulated at the level of gene transcription by the nuclear factor NF-κB (3). Studies in our and other labs have demonstrated that RSV replication in airway epithelial cells is associated with NF-κB activation (5, 8, 12). Therefore, amelioration of NF-κB activation offers a potential means of reversing RSV-induced inflammation.
In unstimulated cells, NF-κB is sequestered in the cytoplasm by inhibitors in the IκB family (3). Studies using A549 cells have demonstrated that with TNF-α stimulation, IκBα is targeted for degradation within 5 to 10 min (10). This is associated with NF-κB activation (10, 14). However, when the cells are treated with the proteasome inhibitor MG-132, IκBα degradation and NF-κB activation are reversed. The studies presented here were designed to determine whether augmentation of IκBα protein levels was associated with a reversal of RSV-induced NF-κB activation.
Studies in our lab and others have demonstrated that A549 cells respond to RSV infection similarly to primary airway cells in culture (2, 11). For all experiments, the A549 cells were between passages 80 and 95. The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with lipopolysaccharide-free 8% fetal calf serum and 2 mM l-glutamine. No antimicrobial agents were used at any time, and the cells were free of mycoplasma infection. The cells were infected with RSV by adding the virus at a multiplicity of infection of 1.0 in DMEM for 2 h. The RSV-containing DMEM was then removed, and the cells were washed several times with DMEM.
Previous studies have demonstrated that RSV infection of airway epithelial cells results in persistent NF-κB activation. Therefore, our focus in this study was on whether newly synthesized IκBα could inhibit RSV-induced NF-κB activation and whether inhibition of IκBα degradation would limit RSV-induced NF-κB activation.
Following RSV infection, NF-κB activation was observed for up to 72 h by electrophoretic mobility shift assays (EMSA) (9) of nuclear extracts (Fig. 1B). The NF-κB probe consisted of a 32P-labeled double-stranded DNA corresponding to the NF-κB binding site present in the IL-8 gene. The sequence of the probe is CAGCTACGCAGCGTGGAATTTCCT, which corresponds to a mutated NF-IL-6 site that does not bind NF-IL-6 (data not shown) and an intact NF-κB site from the IL-8 gene (15, 16). As illustrated in Fig. 1B (which is representative of five experiments), control cells had minimal NF-κB activation. In contrast, in RSV-infected cells, NF-κB activation was apparent 24 h after infection and remained elevated at 48 and 72 h after infection.
FIG. 1.

(A) Western blot analysis of cytosolic proteins. Fifty micrograms of cytosolic protein extract from control (C) or RSV-infected (R) cells was subjected to Western blot analysis using a polyclonal antibody to IκBα. Numbers at the top indicate hours after RSV infection. Protein detection was performed with a commercially available enhanced chemiluminescence detection kit. (B) EMSA analysis of nuclear extract proteins, performed with a DNA probe consisting of the IL-8 NF-κB binding region for control (C) and RSV-infected (R) cells. Numbers at the top indicate hours after RSV infection; 10 μg of total protein was used for each condition.
Western blot analysis was used to determine the effects of RSV infection on IκBα protein levels. A549 cells were infected with RSV, and cytoplasmic extracts were obtained 24, 48, and 72 h after infection. As illustrated in Fig. 1A (which is representative of five experiments), 24 h after infection, IκBα levels are lower in RSV-infected cells than in control cells. However, IκBα is still detectable in the RSV-infected cells. This is in contrast to TNF-α stimulation, which results in complete loss of IκBα, as previously reported (10, 14). Furthermore, in RSV-infected cells, IκBα levels approach those of control cells at 48 and 72 h. Thus, RSV infection is associated with an initial decline in IκBα levels, though near-control levels are detected later in the infection. However, NF-κB activation persists despite the near-control levels of IκBα, suggesting that IκBα cannot inhibit NF-κB nuclear translocation and DNA binding in the RSV-infected cells.
In other cell types, activation of NF-κB has been associated with increased IκBα gene expression (13). Three NF-κB binding sites are present in the 5′ flanking region of the IκBα gene (13). By increasing IκBα gene expression and protein production, NF-κB essentially down-regulates its own activation. To determine whether RSV-induced NF-κB activation was associated with IκBα gene expression, Northern blot analysis was performed. As illustrated in Fig. 2A, at 24, 48, and 72 h after infection IκBα mRNA expression is higher in RSV-infected cells than in controls. The increase in IκBα mRNA expression occurs during viral replication, as evidenced by expression of the RSV F gene (8). Thus, this result indicates that increased IκBα mRNA expression is associated with viral replication during RSV infection.
FIG. 2.

(A) Northern blot analysis of control (C) and RSV-infected (RSV) cellular RNA for IκBα, RSV F, and β-actin mRNA expression. For each condition, 10 μg of total RNA was used. Numbers at the top indicate hours after infection. (B) Effects of actinomycin D on IκBα mRNA expression. Control cells (C) or cells infected with RSV for 2 days (RSV) were treated with 10 μg of actinomycin D (Act-D) per ml for the indicated lengths of time. Ten micrograms of total RNA was subjected to Northern blot analysis using a cDNA probe for IκB or β-actin. (C) Degradation curve for IκBα mRNA expression for control and RSV-infected cells. Control cells (C) or cells infected with RSV for 2 days (RSV) were subjected to treatment with actinomycin D for the lengths of time given. Total RNA was subjected to Northern blot analysis using cDNA probes for IκBα and β-actin. Phosphorimaging analysis was used to detect signal intensity. The results are reported as IκBα mRNA expression normalized for β-actin mRNA expression.
The next experiment was designed to determine whether the increase in IκBα mRNA levels observed with RSV infection was due to prolongation of the IκBα mRNA half-life. Control and RSV-infected cells were treated with actinomycin D for 1, 2, or 4 h, and Northern blot analysis performed on cellular RNA. As illustrated in Fig. 2B, treatment with 10 μg of actinomycin D per ml results in decreased IκBα mRNA expression in both control and RSV-infected cells. Furthermore, as illustrated in Fig. 2C, the slope of the degradation curve for the IκBα mRNA signal in RSV-infected cells was identical to the slope of the curve in control cells. The curve for the RSV-infected cells was shifted upward due to a higher initial expression level of IκBα in RSV-infected cells. This result indicates that the increase in IκBα mRNA expression in RSV-infected cells is not due to stabilization of the message but likely is due to increased gene transcription.
The next experiment was designed to determine whether RSV infection resulted in the synthesis of IκBα protein. Control and RSV-infected cells were placed in methionine- and cysteine-free medium for 4 h, and then 35S-labeled methionine and cysteine were added. Cells lysates were obtained and subjected to protein precipitation using an antibody to IκBα. The resulting precipitate was subjected to electrophoresis on a 10% Tris-glycine gel. As illustrated in Fig. 3, minimal IκBα is detected in control cells (lane 1). However, in RSV-infected cells (lane 2), two bands are detected at the appropriate size for IκBα and phosphorylated IκBα. This result indicates that RSV infection is associated with increased IκBα protein synthesis in A549 cells.
FIG. 3.
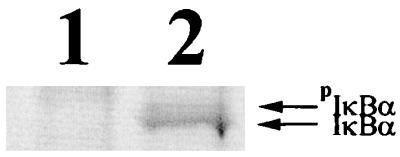
Production of IκBα protein during RSV infection. Control cells (lane 1) or cells infected with RSV for 2 days (lane 2) were labeled with [35S]cysteine and [35S]methionine for 4 h. The cell lysates were subjected to protein precipitation using an antibody to IκBα. IκBα, phosphorylated IκBα.
It was intriguing that IκBα protein levels were increasing after 24 h, but there was no apparent inhibition of NF-κB activation. This result indicates that NF-κB-regulated IκBα production did not result in effective inhibition of NF-κB activation as was previously reported for TNF-α stimulation (22). To determine whether IκBα production had any effect on RSV-induced NF-κB activation, the next set of experiments were performed to determine the effects of limiting IκBα gene expression and the effects of limiting IκBα protein production during RSV infection.
To determine whether limiting RSV-induced IκBα mRNA expression altered NF-κB activation, control and RSV-infected cells were treated with actinomycin D for 4 h. As illustrated in Fig. 2B, this results in decreased IκB∝ mRNA expression. Treatment with actinomycin D also resulted in decreased IκBα protein detection (data not shown). EMSA analysis of nuclear extracts for NF-κB activation is illustrated in Fig. 4A. Treatment of control cells with actinomycin D resulted in greater NF-κB activation than in untreated control cells (Fig. 4A; compare lanes 1 and 2). Furthermore, treatment of RSV infected cells with actinomycin D for 4 h resulted in a greater NF-κB response than in untreated RSV-infected cells (Fig. 4A; compare lanes 3 and 4). This result is consistent with the notion that inhibition of IκBα mRNA expression results in an exaggerated NF-κB response during RSV infection.
FIG. 4.

(A) EMSA analysis of control (C) and RSV-infected (RSV) cells treated with actinomycin D (ACT D; 10 μg for 4 h). Ten micrograms of nuclear extract for each condition was subjected to EMSA analysis using a DNA probe consisting of the IL-8 NF-κB binding region. OLIGO, oligonucleotide. (B) Western blot analysis of cells infected with the protein synthesis inhibitor cycloheximide. Control (C) cells and cells infected with RSV for 2 days (RSV) were treated with 10 μg of cyclohixamide (CYCLO) per ml for 2 h. Fifty micrograms of cytosolic extract was subjected to Western blot analysis using a polyclonal antibody to IκBα. (C) EMSA analysis of control cells (C) and cells infected with RSV for 2 days (RSV) treated with 10 μg of cycloheximide (CYCLO) per ml for 4 h. Ten micrograms of nuclear extract for each condition was subjected EMSA analysis using a DNA probe consisting of the IL-8 NF-κB binding region.
The next experiment focused on determining whether inhibition of IκBα protein synthesis would alter RSV-induced NF-κB activation. Control and infected cells were treated with 10 μg of cycloheximide per ml for 4 h. As illustrated in the Western blot in Fig. 4B, a small difference in IκBα protein detection is observed between control (lane 1) and RSV-infected (lane 2) cells. Treatment of control cells with cycloheximide had little effect on IκBα protein detection in control cells (lane 3) but had a marked effect on IκBα protein detection in RSV-infected cells (lane 4).
Analysis of nuclear extracts for NF-κB activation by EMSA analysis is illustrated in Fig. 4C. Treatment of control cells with cycloheximide resulted in no apparent difference in NF-κB activation (compare lanes 1 and 3). However, treatment of RSV-infected cells with cycloheximide resulted in an increase in NF-κB activation (compare lanes 2 and 4). Together, the data from Figs. 4B and C indicate that inhibition of IκBα protein synthesis during RSV infection is associated with an increase in NF-κB activation.
The combined results from the experiments using actinomycin and those using cycloheximide indicate that inhibition of IκBα mRNA expression and IκBα protein synthesis results in a exaggerated response to RSV infection with respect to NF-κB activation. The results of these experiments are consistent with (but not proof of) the notion that RSV-induced IκBα mRNA expression and protein synthesis are important in at least partially limiting NF-κB activation.
As illustrated in Fig. 1A, on days 1, 2, and 3, RSV-infected cells contained less IκBα than control cells, which indicates that IκBα degradation was occurring during RSV infection. The next set of experiments was designed to determine whether inhibition of RSV-induced IκBα degradation would result in altered NF-κB activation in response to RSV. For these studies, control cells or cells infected with RSV were treated with various concentrations of the proteasome inhibitor MG-132 for 2 h. Previous studies in our lab demonstrated that treatment of A549 cells with MG-132 reversed TNF-α-induced NF-κB activation by limiting IκBα degradation (10).
The results of the experiments using the proteasome inhibitor MG-132 are illustrated in Fig. 5. Western blot analysis (Fig. 5A) demonstrates that treatment of control and RSV-infected cells with increasing concentrations of MG-132 results in increased IκBα protein detection. The IκBα bands detected in the MG-132 treated cells migrated slightly more slowly than those not treated with MG-132. Previous studies in our lab using MG-132 indicated that the bands represented phosphorylated IκBα (10). Western blot analysis using an antibody to phosphorylated IκBα confirmed that the slower-migrating bands were phosphorylated IκBα (data not shown). Thus, treatment of RSV-infected cells with the proteasome inhibitor MG-132 resulted in reversal of IκBα protein loss but not IκBα phosphorylation.
FIG. 5.

(A) Western blot analysis of cytosolic proteins using a polyclonal antibody to IκBα. Control cells (C) or cells infected with RSV for 2 days (RSV) were treated with MG-132 for 2 h at the concentrations given at the top. Fifty micrograms of protein was subjected to analysis for each condition. (B) EMSA analysis of nuclear extracts from control cells (C) and cells infected with RSV (RSV) for 2 days. The cells were treated with the indicated concentration of MG-132 for 2 h. OLIGO, oligonucleotide.
To determine whether inhibition of proteasome-mediated IκBα degradation resulted in a reversal of RSV-induced NF-κB activation, EMSA analysis of nuclear extracts was performed. As illustrated in Fig. 5B, treatment with increasing concentrations of MG-132 had a partial effect on reversing RSV-induced NF-κB activation. Thus, despite the observation that treatment with MG-132 resulted in complete reversal of RSV-induced IκBα degradation, only a partial reversal of RSV-induced NF-κB activation was observed.
An interesting finding from the EMSA analysis in Fig. 5B was that treatment with MG-132 resulted in a slower-migrating NF-κB–DNA complex. The next experiment was designed to characterize this complex. As illustrated in Fig. 6, comparison of lane 1 (control cells) to lane 2 (RSV-infected cells) confirms that NF-κB activation is present during infection. Treatment of infected cells with 100 μM MG-132 results in a decrease in the NF-κB signal which is slower migrating (lane 3). When EMSA analysis of the same extracts from lane 3 is performed in the presence of a 100-fold excess of unlabeled NF-κB DNA probe, the signal is completely abolished (lane 4), indicating that the slower-migrating signal represents NF-κB–DNA binding.
FIG. 6.

EMSA analysis of RSV-infected cells, using the IL-8 NF-κB DNA probe. Lanes 3 to 10 represent cells treated with MG-132 for 2 h. Lane 1, control; lanes 2 to 10, RSV infected; lane 4, addition of 100-fold excess of unlabeled competitor. Lanes 5 to 10 represent addition of antibodies to p65, c-Rel, p50, p52, IκBα (C-15), and IκBα (C-21), respectively. Arrows at the right indicate bands that resulted from the supershift with either p65 or p50. OLIGO, oligonucleotide.
Lanes 5 to 10 in Fig. 6 represent attempts to supershift the NF-κB–DNA complex with polyclonal antibodies to individual potential components of NF-κB. The nuclear extracts used are from RSV-infected cells treated with 100 μM MG-132 for 2 h. The addition of antibodies to p65 (lane 5) and p50 (lane 7) resulted in supershifting of the NF-κB DNA complex. However, addition of antibodies specific for c-Rel (Lane 6) and p52 (Lane 8) did not result in any alterations in binding of NF-κB to the DNA probe, nor did they affect migration of the complex. Our results confirms an earlier finding by Bitko et al. (5). Furthermore, this result indicates that the slower-migrating complex observed in lane 3 is composed at least in part of p65 and p50 and does not represent heterodimers involving c-Rel or p52.
Since MG-132 inhibits the degradation of IκBα, we attempted to determine whether IκBα could be complexed to NFκB while bound to the DNA probe. Therefore, two different commercially available polyclonal antibodies to IκBα (C-15 [Fig. 6, lane 9] and C-21 [lane 10]; Santa Cruz Biotechnology) were added to the EMSA reactions. Neither of these antibodies resulted in supershifting of the NF-κB–DNA signal or competitive inhibition of binding of NF-κB to DNA probe. To further investigate whether IκBα remained bound to NF-κB during treatment with MG-132, whole-cell extracts were subjected to protein precipitation using a polyclonal antibody to p65. The precipitated protein was subjected to Western blot analysis using an antibody to IκBα (C-21). As illustrated in Fig. 7, the IκBα protein is easily detected in control cells (lane 1). However, in RSV-infected cells, less IκBα coprecipitated with the p65 antibody (lane 2). Furthermore, no increase in IκBα coprecipitation with the p65 antibody was observed with RSV-infected cells treated with 100 μM MG-132 (lane 3). Thus, treatment with MG-132 does not result in increased p65-IκBα binding. Combined, the results of the EMSA analysis and coprecipitation studies do not support the notion that the slower-migrating NF-κB–DNA signal observed in MG-132-treated cells infected with RSV is due to persistent IκBα binding. Furthermore, the coprecipitation study indicates that despite the observation that MG-132 results in decreased IκBα degradation, it does not increase binding of IκBα to p65.
FIG. 7.

Western blot of p65-IκBα coprecipitates. Control cells (lane 1), RSV-infected cells (lane 2), or RSV-infected cells treated with 100 μg of MG-132 per ml for 2 h (lane 3) were precipitated with a polyclonal antibody to p65 followed by Western blot analysis using a polyclonal antibody to IκBα.
The EMSA analysis in Fig. 5B indicates that treatment of RSV-infected cells with 100 μM MG-132 resulted in a small decrease in NF-κB activation. To determine whether this was of biological significance, the effects of RSV infection on IL-8 gene transcription and IL-8 protein production were determined. IL-8 gene transcription was assessed in a luciferase reporter assay previously described (9). In this assay, NF-κB-mediated IL-8 gene transcription is responsible for luciferase activity measured in cell lysates. Luciferase activity was measured in cells transfected with the reporter plasmid (controls) and cells transfected with the reporter plasmid and infected with RSV. For the control cells (n = 12), each of the individual values is divided by the mean in order to obtain a standard deviation. For the RSV-infected cells, each of the individual values (n = 12) is divided by the mean of the control cell values. Therefore, the values are reported as an increase in luciferase activity as a function of RSV infection. As illustrated in Fig. 8A, RSV infection of A549 cells results in a nearly sevenfold increase in luciferase activity (and therefore IL-8 gene transcription). Treatment with 100 μM MG-132 for 4 h resulted in a small but statistically significant (P < 0.05 by analysis of variance [ANOVA], n = 12) decrease in luciferase activity (Fig. 8; compare RSV to RSV+MG-132). However, similar to the observed changes in NF-κB activation, the luciferase activity in RSV-infected cells treated with 100 μM MG-132 was markedly greater than that in control cells.
FIG. 8.

Transcriptional activity of the IL-8 gene in response to RSV infection (RSV) and RSV infection with the addition of 100 μg of MG-132 per ml for 2 h (RSV+MG-132). Transcriptional activity is reported as an increase in luciferase activity over control cells; (C); error bars represent standard deviations. To control for transfection efficiency, cells were cotransfected with a plasmid containing the β-galactosidase cDNA under the transcriptional control of the cytomegalovirus early gene. Luciferase activity is controlled for measured β-galactosidase activity. The difference between C and RSV was significant (P < 0.001 by ANOVA); the difference between RSV and RSV+MG-132 was also significant (P < 0.05 by ANOVA). Error bars represent standard deviations. (B) Analysis of IL-8 protein released into the supernatant and intracellular IL-8 detected in cell lysates. Error bars represent standard deviations. (B) Analysis of IL-8 protein released into the supernatant and intracellular IL-8 detected in cell lysates. Error bars represent standard deviations. The difference in IL-8 released into the medium was statistically significant in comparisons of control to RSV (P < 0.001 by ANOVA) and control to RSV+MG-132 (P < 0.001 by ANOVA). Comparison of IL-8 released into the medium for RSV-infected cells compared to RSV-infected cells treated with MG-132 was not statistically significant. Error bars represent standard deviations; n = 12 for all conditions.
The next set of experiments focused on determining whether the small but statistically significant decrease in IL-8 gene transcription correlated with IL-8 protein production and release. For these experiments, the medium from control cells and RSV-infected cells was collected 4 h after the medium was changed. Cells treated with MG-132 received the agent at a concentration of 100 μM for 4 h. Cell supernatants, representative of the release of IL-8 over 4 h, were subjected to a previously described (9) enzyme-linked immunosorbent assay (ELISA) for IL-8. Cell lysates were also obtained after 4 h and subjected ELISA. As illustrated in Fig. 8B, RSV infection is associated with a marked increase in the amount of IL-8 released from the cells (error bars represent standard deviation for the supernatant, P < 0.001 by ANOVA, n = 12) and the amount of cell-associated IL-8 (lysates, P < 0.001 by ANOVA, n = 12). However, treatment of RSV-infected cells with 100 μM MG-132 for 4 h did not result in any statistically significant difference in IL-8 release into the supernatant or the cell-associated IL-8 (lysates) compared to untreated RSV-infected cells. Thus, although inhibition of proteasome-mediated IκBα degradation resulted in a small reversal of RSV-induced NF-κB activation and NF-κB-mediated IL-8 gene transcription, no significant change in IL-8 release or IL-8 production was observed.
The purpose of the study reported here was to determine whether augmentation of endogenous IκBα protein levels would reverse RSV-induced NF-κB activation and NF-κB-mediated inflammation. Our findings indicate that RSV-induced IκBα protein production partially limits virus-induced NF-κB activation in A549 cells. Data presented here indicate that RSV infection is associated with increased IκBα mRNA expression and protein production. Limiting either IκBα mRNA expression or protein production results in exaggerated NF-κB activation in response to RSV infection. Additionally, data presented here indicate that inhibition of endogenous IκBα degradation did not reverse RSV-induced NF-κB activation. Combined, these results indicate that endogenous IκBα plays a role in limiting RSV-induced NF-κB activation but cannot be recruited to completely reverse RSV-induced NF-κB activation.
Recently Thomas et al. reported that adenovirus-mediated expression of a nondegradable form of IκBα resulted in inhibition of RSV-induced NF-κB activation and NF-κB-mediated RANTES production in airway epithelial cells (20). The studies reported by Thomas et al. differ from those presented here in two major respects. First, the cells were transfected with the adenovirus-containing IκBα construct simultaneously with RSV infection. This enabled the cells to produce nondegradable IκBα during the entire course of RSV infection. In contrast, we did not treat the infected cells until RSV infection had progressed for 2 days. Thus, one reason for the differences in results observed maybe accounted for by experimental design. Our studies were focused on reversing NF-κB activation after it was initiated, whereas the studies of Thomas et al. were designed to inhibit NF-κB activation from the initial point of infection.
A second difference between the studies involves their use of a mutant, exogenous IκBα as opposed to our attempts to augment endogenous IκBα protein levels. The recombinant IκBα was rendered resistant to phosphorylation and subsequent degradation by mutating serines at positions 32 and 36. Thus, any recombinant protein made by the cells theoretically would not be degraded. In contrast, our studies focused on maintaining the endogenous IκBα protein at control levels, as this approach was successful with TNF-α stimulation (10). Inhibition of proteasome degradation does not alter IκBα phosphorylation. It is possible that nonphosphorylated IκBα maintains interactions with NF-κB to a greater degree than phosphorylated IκBα. Determining whether phosphorylation of IκBα alters its interactions with p65 was beyond the scope of this study.
Recently, Jamaluddin et al. reported that the major component of IκBα proteolysis occurs independent of the proteasome pathway in RSV-infected cells (14). In this study, the authors found that treatment with 25 μM Mg-132 for 4.5 h did not reverse IκBα degradation. The results of our study, in comparison, indicate that treatment with MG-132 in dose range of 1 to 100 μg/ml for 2 h (all nontoxic as assayed by neutral red uptake) resulted in a significant reversal of IκBα degradation. The 2-h treatment course was chosen based on preliminary studies comparing treatment courses with MG-132 of 20 min to 4 h. These studies demonstrated that peak inhibition of IκBα degradation occurred at 2 h (data not shown). The differences in our reported findings may be explained by the fact that MG-132 is a reversible inhibitor of the proteasome, and the optimal time course was chosen for our study.
Bitko and Barik recently reported on the persistent activation of the RelA component of NF-κB by RSV (4); they demonstrated that MG-132 (100 μM) reversed IκBα degradation and NF-κB activation at certain time points. The effects of MG-132 were reported as modest. The authors also presented data which suggest that IκBα may act as a chaperone for NF-κB, protecting it from IκBα inhibition. The results presented here are consistent with the finding of Bitko and Barik. The unsuccessful attempts to inhibit RSV-induced NF-κB activation by increasing IκBα protein levels may be due to binding of IκBβ to NF-κB. This would suggest that a pool of NF-κB (which can no longer be regulated by IκBα) is present in the RSV-infected cells. An analysis of the role of IκBβ was beyond the scope of our study.
In summary, the study presented here focused on testing the hypothesis that manipulation of IκBα levels would alter RSV-induced NF-κB activation and NF-κB-mediated IL-8 production; the results do not support this hypothesis. While RSV-induced IκBα synthesis does limit NF-κB activation, complete reversal does not occur. Furthermore, augmentation of IκBα levels by inhibition of its degration does not reverse RSV-induced NF-κB activation.
While our results are not in complete agreement with those of Thomas et al. (20) and Jamaluddin et al. (14), a recurrent finding is that RSV induces persistent NF-κB activation. The results presented here, along with those of Jamaluddin et al. and Bitko and Barik, may begin to explain why some children with RSV infection have persistent respiratory symptoms (1, 6, 21). It has been well documented that inflammation plays a key role in RSV-induced morbidity and mortality (6, 21). Persistent NF-κB-mediated inflammatory gene production in airway epithelial cells likely contributes to the morbidity associated with RSV infection. Furthermore, this persistent inflammatory response may help explain the morbidity associated with RSV infection in children with chronic inflammatory airway disorders such as cystic fibrosis (1). It will be critical in future studies to define the mechanisms by which persistent NF-κB activation occurs, in order to begin to reverse the inflammatory effects associated with its activation.
Acknowledgments
This study was supported by grants from the NIH (HL03451 to M.A.F.) and the Cystic Fibrosis Foundation (FIEDLEGO97).
REFERENCES
- 1.Abman S H, Ogle J W, Butler-Simon N, Rumack C M, Accurse F J. Role of respiratory syncytial virus in early hospitalizations for respiratory distress of young infants with cystic fibrosis. J Pediatr. 1988;113:826–830. doi: 10.1016/s0022-3476(88)80008-8. [DOI] [PubMed] [Google Scholar]
- 2.Arnold R, Humber B, Werchau H, Galiati H, Konig W. Interleukin-8, interleukin-6, and soluble tumor necrosis factor type II release from a human pulmonary epithelial cells line (A549) exposed to respiratory syncytial virus. Immunology. 1994;82:126–133. [PMC free article] [PubMed] [Google Scholar]
- 3.Baldwin A S. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–681. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 4.Bitko V, Barik S. Persistent activation of RelA by respiratory syncytial virus involves protein kinase C, underphosphorylated IκBα, and sequestration of protein phosphatase 2A by the viral phosphoprotein. J Virol. 1998;72:5610–5618. doi: 10.1128/jvi.72.7.5610-5618.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bitko V, Velayquey A, Yang L, Yang Y-C, Barik S. Transcriptional induction of multiple cytokines by human respiratory syncytial virus requires activation of NF-κB and is inhibited by sodium salicylate and aspirin. Virology. 1997;232:369–378. doi: 10.1006/viro.1997.8582. [DOI] [PubMed] [Google Scholar]
- 6.Breese Hall C W, Hall J, Gala C L, McGill F B, Leddy J P. Long term prospective study in children after respiratory syncytial virus infection. J Pediatr. 1984;105:358–364. doi: 10.1016/s0022-3476(84)80005-0. [DOI] [PubMed] [Google Scholar]
- 7.Centers for Disease Control and Prevention. Update: respiratory syncytial virus activity—United States, 1994–1995 season. Morbid Mortal Weekly Rep. 1994;43:920–922. [PubMed] [Google Scholar]
- 8.Fiedler M A, Wernke-Dollries K, Stark J M. Inhibition of viral replication reverses respiratory syncytial virus-induced NF-κB activation and interleukin-8 gene expression in A549 cells. J Virol. 1996;70:9079–9082. doi: 10.1128/jvi.70.12.9079-9082.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fiedler M A, Wernke-Dollries K, Stark J M. Mechanism of RSV-induced IL-8 gene expression in A549 cells before viral replication. Am J Physiol. 1996;271:L963–L971. doi: 10.1152/ajplung.1996.271.6.L963. [DOI] [PubMed] [Google Scholar]
- 10.Fiedler M A, Wernke-Dollries K, Stark J M. Inhibition of TNFα-induced NF-κB activation and IL-8 release in A549 cells using the proteasome inhibitor MG-132. Am J Respir Cell Mol Biol. 1998;19:259–268. doi: 10.1165/ajrcmb.19.2.3149. [DOI] [PubMed] [Google Scholar]
- 11.Fiedler M A, Wernke-Dollries K, Stark J M. Respiratory syncytial virus increases IL-8 gene expression and protein release in A549 cells. Am J Physiol. 1995;269:L872. doi: 10.1152/ajplung.1995.269.6.L865. [DOI] [PubMed] [Google Scholar]
- 12.Garafalo R, Sabry M, Jamaluddin M, Yu R K, Casola A, Orga P L, Brasier A R. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J Virol. 1996;70:8773–8781. doi: 10.1128/jvi.70.12.8773-8781.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ito C Y, Kazantsev A G, Baldwin A S. Three NF-κB sites in the IκBα promoter are required for induction for gene expression by TNFα. Nucleic Acids Res. 1994;22:3787–3792. doi: 10.1093/nar/22.18.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jamaluddin M, Casola A, Garofalo R P, Han Y, Elliot T, Ogra P L, Basier A R. A major component of IκBα proteolysis occurs independently of the proteasome pathway in respiratory syncytial virus-infected pulmonary epithelial cells. J Virol. 1998;72:4849–4857. doi: 10.1128/jvi.72.6.4849-4857.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukaida N, Okamoto S, Ishikawa Y, Matsushima K. Molecular mechanism of interleukin-8 expression. J Leukoc Biol. 1994;56:554–558. [PubMed] [Google Scholar]
- 16.Mukaida N S, Shiroo M, Matsushima K. Genomic structure of the human monocyte-derived neutrophil chemotactic factor IL-8. J Immunol. 1989;143:366–371. [PubMed] [Google Scholar]
- 17.Murphy E, Shibuya K, Hosken N, Openshaw P, Maino B, Davis K, Murphy K, O’Garra A. Reversibility of helper 1 and 2 populations is lost after long-term stimulation. J Exp Med. 1996;183:901–913. doi: 10.1084/jem.183.3.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noah T L, Henderson F W, Wortman I A, Devlin R B, Handy J, Koren H S, Becker S. Nasal cytokine production in viral acute upper respiratory infection of childhood. J Infect Dis. 1995;171:584–592. doi: 10.1093/infdis/171.3.584. [DOI] [PubMed] [Google Scholar]
- 19.Openshaw P, Murphy E E, Hosken N A, Maino B, Davis K, Murphy K, O’Garra A. Heterogeneity of intracellular cytokine synthesis at the single-cell level in polarized T helper 1 and T helper 2 populations. J Exp Med. 1995;182:1357–1367. doi: 10.1084/jem.182.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas L H, Friedland J S, Sharland M, Becker S. Respiratory syncytial virus induced RANTES production from human bronchial epithelial cells is dependent on nuclear factor-κB nuclear binding and is inhibited by adenovirus-mediated expression of inhibitor κBα. J Immunol. 1998;161:1007–1016. [PubMed] [Google Scholar]
- 21.Webb M S C, Henry R L, Milner A D, Stokes G M, Swarbuck A S. Continuing respiratory problems three and a half years after acute viral bronchiolitis. Arch Dis Child. 1985;60:1064–1067. doi: 10.1136/adc.60.11.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong H R, Ryan M, Wispe J R. Stress response decreases NF-kappa B nuclear translocation and increases I-kappa B alpha expression in A549 cells. J Clin Investig. 1997;99:2423–2428. doi: 10.1172/JCI119425. [DOI] [PMC free article] [PubMed] [Google Scholar]