

Summary
Cystatin C (CyC), a secreted cysteine protease inhibitor, has unclear biological functions. Many patients exhibit elevated plasma CyC levels, particularly during glucocorticoid (GC) treatment. This study links GCs with CyC’s systemic regulation by utilizing genome-wide association and structural equation modeling to determine CyC production genetics in the UK Biobank. Both CyC production and a polygenic score (PGS) capturing predisposition to CyC production were associated with increased all-cause and cancer-specific mortality. We found that the GC receptor directly targets CyC, leading to GC-responsive CyC secretion in macrophages and cancer cells. CyC-knockout tumors displayed significantly reduced growth and diminished recruitment of TREM2+ macrophages, which have been connected to cancer immunotherapy failure. Furthermore, the CyC-production PGS predicted checkpoint immunotherapy failure in 685 patients with metastatic cancer from combined clinical trial cohorts. In conclusion, CyC may act as a GC effector pathway via TREM2+ macrophage recruitment and may be a potential target for combination cancer immunotherapy.
Keywords: cystatin C, renal function, glucocorticoids, GWAS, PGS, immunotherapy, macrophages
Graphical abstract
Highlights
-
•
Glucocorticoids are linked to the systemic regulation of cystatin C (CyC)
-
•
Elevated CyC predicts higher mortality rates
-
•
CyC impacts tumor growth and Trem2+ macrophage recruitment
-
•
CyC production polygenic score predicts immunotherapy failure
Kleeman et al. reveal a connection between cystatin C, a blood marker often used to measure kidney function, and glucocorticoid signaling. The authors further demonstrate that cystatin C plays a role in immunosuppression and failure of cancer immunotherapy. This discovery may open new avenues for combination cancer immunotherapy targeting.
Introduction
Multilevel phenotyping paired with quantitative models can direct discovery of the molecular determinants of complex biomedically relevant phenotypes such as organ function. Previously, we developed a metabolite-based model for the accurate estimation of kidney filtration function, defined as the estimated glomerular filtration rate (eGFR), in patients with cancer.1,2 Like others before, we used creatinine,3 a breakdown and metabolic end product of muscle creatine metabolism that is renally excreted,4 as a predictor variable. In non-cancer patients, creatinine use has been compared with use of cystatin C (CyC; gene name CST3),5 a secreted, paracrine cysteine protease inhibitor.1,6 Serum levels of both molecules depend on latent (unmeasured) components, including their production (synthesis and secretion) and the GFR. While creatinine production is well characterized and relates to muscle mass and diet,7 CyC production is poorly defined.8
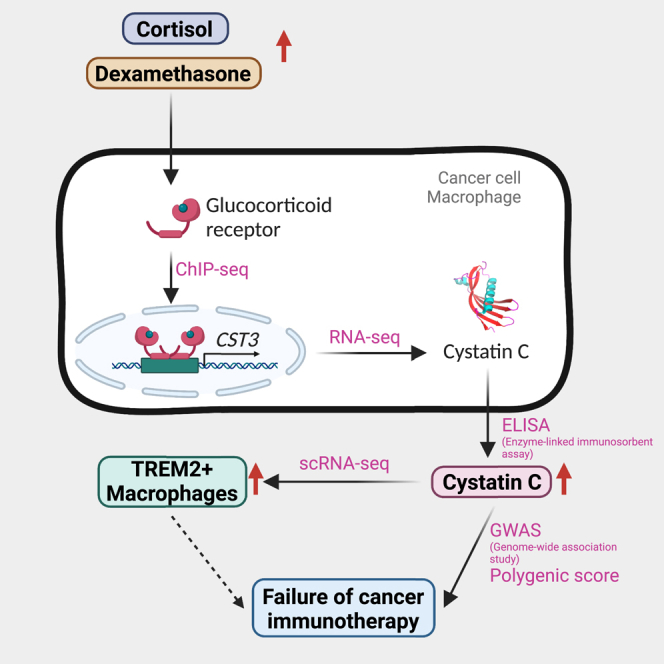
There are multiple indications that CyC production is likely systemically regulated. Patients who have had an organ transplant tend to have higher serum CyC levels for a given measured GFR.9 These patients are routinely prescribed exogenous glucocorticoids (GCs), such as prednisolone or dexamethasone, as part of their immunosuppressive regimen.10 Paired analyses demonstrated that exogenous GCs increase CyC production,11 an effect also observed in patients with excess endogenous GC production (Cushing’s syndrome).12 Moreover, CyC production is increased in diseases that induce GC elevations, including viral infection,6 inflammatory disease,8 and cancer.13,14,15,16,17 This positive association between GC exposure and CyC production has been recapitulated experimentally in vitro,18 in vivo,19 and in patients.11,20,21
Cortisol, the endogenous GC in humans, is produced by the adrenal gland22 in a circadian rhythm peaking in the early morning.23 Through action on the cytosolic GC receptor (GR), GCs profoundly modulate the cellular transcriptional landscape,24 affecting up to 20% of all genes25 and driving systemic reprogramming of metabolism and immunity.26 GCs are therapeutically employed as immunosuppressors across a wide range of autoimmune and inflammatory diseases27 and to mitigate immune-mediated damage to normal organ systems, a common and potentially severe side effect of T cell activation by checkpoint immunotherapy (CPI) in cancer.28 However, in vivo models of cancer also suggests that even low doses of GCs can impair the efficacy of CPI29,30 and suppress anti-tumor immunity,30 leading to enhanced metastasis and reduced survival.31 This has remained difficult to investigate in patients with cancer due to confounding by performance status and comorbidities,32 inconsistent CPI trial inclusion criteria,29 and the difficulties in performing well-controlled trials in this context.
We hypothesized that, rather than being a passive marker of renal function, CyC is directly associated with disease states and that this association might be mediated by GC signaling. Here, to empirically investigate this question, we leverage the UK Biobank (UKB), a prospective population-based cohort comprising approximately 480,000 subjects who provided germline genetics, serum CyC, and serum creatinine. Using conventional genome-wide association studies (GWASs) for eGFR-CyC/eGFR-creatinine followed by structural equation modeling (SEM), we estimate single-nucleotide polymorphism (SNP)-level associations with the latent trait of CyC production. We characterize patient-level predisposition to CyC production via construction of a polygenic score (PGS), which is validated in a held-out cohort. Through multimodal genomics, in vitro, in vivo, and experimental medicine approaches, we link CyC to GC signaling, recruitment of Trem2+ macrophages, and failure of cancer immunotherapy.
Results
Genomic architecture of CyC production
To investigate the genomic architecture of CyC production (Figure 1A), we first performed a discovery GWAS for eGFR-CyC and eGFR-creatinine (eGFR-Cr) in 381,764 European subjects in the UKB, using linear mixed models to account for population stratification and cryptic relatedness. We randomly selected 50,000 unrelated subjects from the overall UKB European population and excluded their data from the GWAS to enable later validation analyses (Figure 1B). Using linkage disequilibrium (LD) score regression, we identified a strong genetic correlation (r2 = 0.61) between eGFR-CyC and eGFR-Cr, consistent with both traits sharing a common factor that reflects renal filtration function. We reasoned that the genetic variance in eGFR-CyC that was not explained by this common factor represented the latent trait of CyC-production given that the CyC plasma level is a function of both CyC excretion in the kidney and its cellular production. Thus, we estimated the SNP-level effects on CyC production and renal function by constructing a SEM (Figures 1C, S1A, and S1B) implemented in Genomic-SEM,33 assuming no covariance between CyC production and renal function. Concoradantly, loci known to directly regulate renal function such as SHROOM334 and UMOD35 were predominantly associated with the renal function latent trait, while the locus coding for CyC (CST3) was predominantly associated with the CyC-production latent trait (Figure 1D). Other loci associated with CyC production, such as SH2B336 and FLT3,37 identify components of immune cell signaling cascades and are strongly associated with autoimmune disease. The index SNP at the SH2B3 locus is a missense variant (R262W) and exhibits a markedly larger effect size than would be expected for its allele frequency (minor allele fraction = 0.48; Figure S1C), consistent with evidence that this variant is under active positive selection.38 The CPS1 locus, coding for carbamoyl-phosphate synthase 1, notably had divergent effects on renal function and CyC-production, probably reflecting its independent roles in creatine metabolism39 and immune signaling.40 We next performed tissue-specific partitioned heritability analysis using gene expression and chromatin accessibility datasets (including Genotype-Tissue Expression [GTEx]41 and Roadmap Epigenomics Project42). This confirmed enrichment of heritability of the renal function rather than the CyC-production component of CyC levels in kidney tissues (Figures S1D and S1E). This analysis also demonstrated enriched heritability for the renal function trait in liver tissues, in keeping with coupled hepatic and kidney function, observed clinically as hepatorenal syndrome.43
Figure 1.

Genomic architecture of cystatin C production
(A) Schematic of study plan. The analysis of CyC-production latent trait in UK Biobank (UKB) is leveraged to determine the biological and clinical relevance of CyC.
(B) Consort diagram and summary of UKB genome-wide association analysis strategy in the European ancestry population. The software packages utilized for each step are displayed in red.
(C) Structural equation model to estimate latent traits of CyC production and renal function. The model schematic, heritability (h2) of eGFR-creatinine and eGFR-CyC, and their genetic correlations derived from LD score regression are shown. Circular arrows refer to variance of each component, and dashed lines refer to covariance between components. RF, renal function.
(D) Latent trait effect sizes (CyC production and renal function) for single-nucleotide polymorphisms (SNPs) corresponding to each clumped locus in eGFR-CyC summary. Gene names are annotated per OpenTargets V2G pipeline.
(E) Linear model of eGFR-CyC as a function of eGFR-creatinine across all paired blood samples in UKB, including sex as a covariate. The deviation of the eGFR-CyC from the linear fit as indicated by the red arrow is defined as the CyC residual, a surrogate for CyC production. p value refers to Pearson correlation test.
(F) Correlation of CyC residual with CyC-production polygenic score (PGS). The continuous PGS has been converted into deciles (1 = lowest, 10 = highest PGS).
Only data from the independent validation set (see B) were used. Boxplots show median (central line) with interquartile range (IQR; box) and extrema (whiskers at 1.5× IQR). p value refers to Pearson correlation test.
Using the discovery dataset, we captured the polygenic architecture of CyC production by deriving a PGS, implemented in LDpred244 using HapMap3 variants, that could be reliably imputed in all UKB, The Cancer Genome Atlas (TCGA), and GTEx cohorts (Figure S1F; supplemental information). To maximize portability to clinical sequencing cohorts where only exome sequencing is available, we derived a second PGS from HapMap3 variants that could be reliably imputed from exome sequencing data (Figure S1G). To validate both PGSs with the data from the 50,000 unrelated European patients (Figure 1B), it was necessary to define an independent, measurable patient-level estimate for CyC production. This is possible because the discordance between eGFR-CyC and eGFR-Cr approximates CyC production. Therefore, we modeled eGFR-CyC as a function of eGFR-Cr and sex and computed the residual (termed CyC residual; Figure 1E), a proxy of CyC production. As CyC residual is estimated from blood-protein levels, it is more likely to be confounded (for example by exogenous steroid treatment) than the germline-derived CyC production PGS. However, it can be conveniently scored using routinely available blood tests in the absence of germline genotypes. Using CyC residual, we confirmed that the genome-wide CyC-production PGS had predictive power in the validation cohort (r2 = 0.08, p < 1e−300, Pearson correlation test; Figure 1F). As expected, predictive performance was reduced for the exome-wide PGS in the validation cohort (r2 = 0.04, p < 1e−300, Pearson correlation test).
To investigate the trans-ancestral portability of the genome-wide CyC-production PGS, we measured performance versus CyC residual in African (AFR; n = 8,152) and Central and South Asian (CSA, n = 9,845) genetic ancestry groups in the UKB. We observed poor trans-ancestral portability of this PGS in these ancestry groups (Figures S2A and S2B). In order to derive a PGS in each non-European (EUR) population, we performed GWAS and SEM as described above (Figure 1B) in these two ancestry groups, but these analyses were underpowered (Figures S2C and S2D). While the genetic correlation between eGFR-CyC and eGFR-Cr in CSA subjects (r2 = 0.65) was comparable to EUR subjects (r2 = 0.61), genetic correlation was substantially diminished in AFR subjects (r2 = 0.18). This indicates that eGFR-Cr and/or eGFR-Cy correlate weakly with true GFR in the AFR population, thus providing empirical genetic evidence to the observation that eGFR models have reduced performance in individuals self-identifying as Black or African American.45
CyC production is associated with accelerated onset of disease
We hypothesized that these quantitative measures of CyC production (CyC-residual and CyC-production PGSs) could be used to investigate its prognostic potential. Therefore, we used multivariate Cox regression to estimate the effect of the blood test-derived CyC residual on all-cause mortality, adjusted for relevant patient covariates known to predict mortality.46,47,48 We found that CyC residual was associated with significantly increased all-cause mortality (hazard ratio [HR] = 1.56, p < 1e−16; Figure 2A). We considered that CyC residual has the potential to be confounded by a multitude of environmental factors, including but not limited to inflammation and exogenous GC treatment. To mitigate this, we investigated whether the germline predisposition to CyC production, estimated as the CyC-production PGS, could predict lifespan in our UKB EUR validation set (Figure 1B). Using multi-variate Cox regression adjusted for sex, year of birth, and principal components capturing genetic ancestry, we found that the CyC-production PGS was associated with significantly reduced lifespan of UKB subjects (p = 0.00013), as well as their two parents (p < 1e−16; Figure 2B).
Figure 2.

CyC production is associated with multiple disease states and is prognostic in patients with cancer
(A) Multivariate Cox regression to measure effect size for CyC residual on overall survival in UKB. Covariates included age, sex, body mass index (BMI), hemoglobin, C-reactive protein, eGFR-creatinine, and operation status (for cancer-specific subanalysis). Error bars indicate 95% confidence interval.
(B) Multivariate Cox regression to measure effect size for CyC-production PGS on subject and parental lifespan in UKB validation cohort. Covariates included PC1-4, recruitment center, genotyping array, year of birth of subject, and sex of subject (if applicable).
(C) Phenome-wide association (Cox regression) between CyC-production PGS and 694 time-to-event phenotypes in UKB validation cohort. Covariates included principal components 1–4 (PC1–4), year of birth, and sex.
(D and E) Multivariate Cox regression to measure effect size for CyC-production PGS on disease-specific survival for specific cancers in (D) UKB validation cohort (cancers diagnosed since 2000, n = 3,954) and (E) TCGA cohort (n = 4,368). Covariates included PC1–4, age, sex, and a term reflecting whether the patient had curative surgery. Error bars indicate 95% confidence interval; gray squares indicate sample size.
We considered that increased all-cause mortality might be explained by either earlier onset of specific disease states or reduced prognosis following disease diagnosis. To investigate the former, we performed a phenome-wide association analysis (PheWAS) in the UKB validation set to identify time-to-event phenotypes (n = 694) that were significantly associated with CyC production using multivariate Cox regression (Figure 2C). We identified positive associations meeting phenome-wide significance (p < 1e−5) between the CyC-production PGS and multiple diseases linked to metabolic syndrome, including type 2 diabetes, obesity, hypertension, and ischemic heart disease. To investigate how CyC production could modulate disease prognosis, and as elevated plasma CyC is associated with cancer,13 we examined whether the blood test-derived CyC-residual and CyC-production PGSs were independent predictors of adverse outcomes in patients with cancer. Using UKB patients diagnosed with cancer since 2000 and with cancer-specific mortality, we found that CyC residual is an independent predictor of increased cancer-specific mortality in UKB (HR = 1.22, p < 1e−16, Cox regression; Figure 2A), consistent with the findings of others.17 For orthogonal validation, we performed multivariate Cox regression of cancer-specific mortality against CyC-production PGSs across 13 tumor groups in 2 independent cohorts (UKB validation set, TCGA EUR subjects). Both fixed and random effect meta-analyses in each independent cohort confirmed a significant positive association between CyC-production PGSs and cancer-specific mortality (Figures 2D and 2E). We noted that while there was variation in a single-cancer level, the overall effect size was concordant between the UKB and TCGA. Consistent with this, we have found that the CyC-production PGS is associated with increased odds of COVID-19 critical illness in four cohorts spanning EUR and AFR ancestry populations.49 In summary, the association between CyC-production PGSs and reduced lifespan likely reflects a combination of earlier disease onset and reduced disease-specific survival and is consistent with evidence for elevated plasma CyC in patients with cancer.14
CyC is a GC response gene in vitro
To better understand the mechanism by which CyC production could regulate disease incidence and prognosis, we reviewed the genetic loci most associated with CyC production in our GWAS summary statistics. The SERPINA1/6 locus on chromosome 14 had one of the largest effect sizes for CyC production (Figures 1D and 3A) and is known to be associated with plasma cortisol,50 implying the possibility of a link between cortisol and CyC. In a recent cortisol genome-wide meta-analysis, this signal was thought to be mediated by altered hepatic expression of SERPINA6,50 which encodes cortisol-binding globulin (CBG). To determine if there was a shared common variant, we performed co-localization analysis.51 We did not detect a shared causal variant (posterior probability = 1.45e−15), but trans-expression quantitative trait locus (trans-eQTL) analysis in the Stockholm Tartu Atherosclerosis Reverse Networks Engineering Task (STARNET)52 cohort identified a single SNP (rs2749527) at the SERPINA1/6 locus that was associated with significantly reduced plasma cortisol (p = 1.75e−13, fixed effect meta-analysis) and significantly reduced CST3 gene expression in visceral adipose fat (p = 0.0024 in additive model, p = 9.21e−6 in recessive model, Bonferroni-adjusted alpha level of 0.0025; Figure 3B). Visceral adipose fat is known to predominantly comprise adipocytes, endothelial cells, and macrophages.53 We investigated the potential cellular mediators of this association in detail and discuss the results below. In addition to acting as a trans-eQTL in adipose tissue, rs2749527 is independently associated with significantly reduced liver SERPINA6 expression in STARNET (p = 4.73e−9, additive model; Figure 3C) and GTEx (p = 0.004, additive model; Figures S3A–S3C) cohorts. As such, a single genetic instrument connects CBG, plasma cortisol, and CyC, thus providing genetic evidence for a direct link between GCs and CyC.
Figure 3.

CyC is a glucocorticoid response gene in vitro
(A) Co-localization of summary statistics for CyC-production from UKB and plasma cortisol from CORNET Consortium at SERPINA1/6 locus. rs2749527 variant is highlighted in red.
(B) Trans-eQTL analysis examining association between genetic instrument rs2749527 and CST3 gene expression in visceral adipose fat (VAF) in STARNET cohort.
(C) Cis-eQTL association between rs2749527 and SERPINA6 (encodes cortisol-binding globulin) in liver in STARNET cohort. p values for additive and recessive models are shown. See Figure S3 for replication analysis in GTEx.
(D) Gene set enrichment analysis (MAGMA) across CyC-production summary statistics (UKB) for steroid signaling-related gene sets.
(E) Functional genomics in A549 cell line (ENCODE project) treated with 100 nM dexamethasone for 0 min to 12 h. ChIP-seq (for glucocorticoid receptor/NR3C1) and ATAC-seq (at 0 h) at CST3 locus identifies a glucocorticoid-responsive and accessible distal enhancer element.
(F and G) Time course of (F) GR recruitment (at distal enhancer) and (G) CST3 gene expression (log-CPM) following dexamethasone (DEX) treatment in A549 cells (ENCODE project). Trendline and shaded 95% confidence interval correspond to regression of gene expression as a function of log-time.
(H and I) Extracellular CyC concentration in (H) A549 cells and (I) HeLa cells normalized to cellular protein content after 18-h treatment with 100 nM DEX or vehicle (VEH) control. Each condition comprises at least 5 biological replicates; horizontal bars indicate mean extracellular CyC. p values correspond to two-sided t tests. See Figure S3 for timecourse.
(B and C) Boxplots show median (central line) with IQR (box) and extrema (whiskers at 1.5× IQR). Outliers beyond 1.5× IQR are shown as dots. SH, steroid hormone; SP, signaling pathway; VEH, vehicle; DEX, dexamethasone.
To further examine the link between GCs and CyC, we mapped each SNP meeting genome-wide significance to overlapping genes (defined by transcriptional start and end sites) and performed gene set enrichment analysis for gene sets relating to GC signaling. This analysis identified significant enrichment of 7/15 GC signaling gene sets from the Gene Ontology Resource (Figure 3D); thus, we hypothesized that CST3 might be a direct transcriptional target of GR (gene name NR3C1). Using functional genomics data derived from the ENCODE project, including chromatin immunoprecipitation sequencing (ChIP-seq) for GR and assay for transposase-accessible chromatin using sequencing (ATAC-seq) data in the A549 cell line treated with dexamethasone, we identified dexamethasone-induced recruitment of GR to an accessible downstream enhancer element at the CST3 locus (Figures 3E and 3F). In the same experiment, dexamethasone significantly increased CST3 gene expression over time (p < 0.0001, linear model; Figure 3G). We next investigated whether, and on what timescale, the transcriptional induction of CST3 by dexamethasone results in increased cellular secretion of CyC, which would cause increased tissue and circulating CyC levels. We first repeated the ENCODE experimental protocol using A549 cells and found that extracellular CyC concentration was significantly increased after 18 h of dexamethasone treatment compared with 0 h (Figure S3D). We also detected increased extracellular CyC concentration 18 h after treatment with dexamethasone compared with vehicle control in A549 cells (Figure 3H) and HeLa cells (Figure 3I).
CyC is secreted in healthy individuals by monocytes in a GC-independent manner
CyC has been validated as a marker of renal function in multiple large clinical cohorts54 comprising patients without acute disease. Considering that it is dynamically regulated in disease states such as cancer,13 we hypothesized that GC-inducible expression of CyC would operate in a context-dependent manner. To investigate this hypothesis, it was first necessary to characterize the dominant source of secreted CyC in health. At first glance, CST3 gene expression was relatively consistent across all tissues examined as part of the GTEx project (GTEx Portal), but we reasoned that tissues that predominantly secrete CyC would exhibit a significant positive correlation between CyC-production PGSs and CST3 gene expression. Using expression quantitative trait score (eQTS) analysis, we detected a significant positive correlation in spleen tissues (n = 171; Figure 4A). In support of this, we identified circadian rhythmicity from cosinor regression of spleen CST3 gene expression against time of death, which was attenuated compared with the canonical, circadian-rhythm-dependent GC target FKBP5 (amplitude = 0.060 versus 0.24; Figure S4A). To understand which cell types might be driving this signal, we examined available single-cell RNA sequencing (scRNA-seq) data from human spleen.55 This showed that only myeloid-derived cell populations (dendritic cells, macrophages, and monocytes) expressed CST3 (Figure 4B). We confirmed myeloid-specific CST3 expression in peripheral blood mononuclear cells (PBMCs) with scRNA-seq56 (Figure 4C) and across multiple scRNA-seq datasets harmonized as part of the Human Protein Atlas57 (Figure 4D). As additional validation supporting the role of myeloid-derived cells, and specifically monocytes, as a dominant contributor to plasma CyC levels, we found a significant positive correlation between blood monocyte counts and CyC residual in the UKB cohort (Figure S4B; multivariate regression), and two-sample Mendelian randomization using blood-derived CST3 eQTLs (eQTLGen58) as exposure identified a highly significant positive association with CyC production (p = 6.13e−77; Figure 4E). With the limited circadian variability in CST3 gene expression in the spleen (Figure S4A), we hypothesized that monocytes would constitutively express CyC without GC inducibility. We confirmed this hypothesis in monocyte-like THP-1 cells by RNA (Figure 4F) and protein level (Figure 4G). As orthogonal verification, we detected constitutive expression of CyC unaffected by GC agonism in primary human monocytes (p = 0.39, two-sided t test; Figure S4C). Consistent with these findings, high-dose dexamethasone treatment did not elevate plasma CyC levels in healthy BALB/c (Figure 4H) and C57BL/6J (Figure 4I) mice, nor did near-physiological hydrocortisone treatment affect Cr-normalized CyC levels (ratio of eGFR-Cr to eGFR-CyC, termed C2 ratio) in patients with primary adrenal insufficiency59 (Figure 4J). Altogether, these findings indicate that CyC production is relatively constant in health and, in this context, does not significantly increase in response to GC agonism, helping to explain the validated utility of CyC as a marker of renal function in patients without acute illness.54
Figure 4.

CyC is predominantly produced by myeloid cells in health
(A) Tissue-specific expression quantitative trait score (eQTS) analysis to identify tissues with significant correlation (Spearman coefficient) between CyC-production PGS and tissue-specific CST3 gene expression in GTEx cohort. p values are uncorrected, as each correlation test is performed in a non-overlapping set of tissue-specific samples.
(B and C) Distribution of normalized single-cell CST3 expression (log-transcripts per million [TPM]) in cell clusters isolated from (B) spleen and (C) peripheral blood mononuclear cells (PBMCs). Clusters defined by correlation to reference PBMC data.60
(D) Mean CST3 gene expression (log-TPM) in each cell cluster from multiple tissue-specific single-cell RNA sequencing projects, harmonized by Human Protein Atlas. The top cell cluster and tissue-specific macrophage cell type (if not top cluster) by tissue is annotated.
(E) Two-sample Mendelian randomization using blood-specific cis-eQTLs for CST3 (eQTLGen) as exposure and CyC-production latent trait GWAS as outcome. Error bars correspond to standard errors, and point color refers to linkage with top cis-eQTL.
(F and G) Non-significant (p > 0.05) changes in (F) CST3 gene expression (reverse transcription PCR) during 0- to 18-h DEX (100 nM) treatment and (G) extracellular CyC concentration in human THP-1 cells (monocyte-like) normalized to cellular protein content after 18-h treatment with DEX (100 nM) or VEH control.
(H and I) Each condition comprises 10 biological replicates. Plasma CyC concentration in healthy (H) BALB/cJ and (I) C57BL/6J mice treated with VEH or 20 mg/kg DEX.
(J) Creatinine-normalized plasma CyC (C2 ratio) in patients with primary adrenal insufficiency treated with placebo (VEH) or hydrocortisone (CORT) in a crossover experimental medicine study. The administered intranveous (i.v.) CORT dose was 0.03 mg/kg/h between 12 and 7 a.m. (the time point of sampling), achieving near-physiological GC exposure.
(G–J) p values refer to two-sided t tests. VEH, vehicle; DEX, dexamethasone.
CyC secretion is dynamically and GC-dependently regulated in disease states
Inflammation is characterized by the recruitment of monocytes to diseased tissues, where they differentiate into macrophages.61 As GR is expressed in macrophages but not in monocytes,62 we hypothesized that while monocytes have constitutive basal CyC production (Figures 4F and 4G), macrophages would secrete CyC in response to GC agonism. To investigate this question, we treated monocytic human THP-1 cells with the protein kinase C activator PMA (phorbol 12-myristate 13-acetate) to induce macrophage-like differentiation and measured CST3 gene expression, total protein content, and extracellular CyC in each sample. Dexamethasone treatment of PMA-activated THP-1 cells significantly increased CST3 gene expression at 6 h (Figure 5A), while extracellular CyC protein concentration did not increase until 18 h (Figure 5B), mirroring the results found in A549 and HeLa cells (Figures 3H and 3I). In addition, we detected that 18-h dexamethasone exposure induced extracellular CyC elevations in an independent experiment using macrophage-like cells (Figure 5C). We also verified GC-inducible secretion of CyC in primary human macrophages. Here, we experimentally differentiated monocytes into M1 and M2 macrophages and then treated cells with dexamethasone for 18 h. We measured significantly increased extracellular CyC protein in M2 macrophages (p = 0.05, two-sided t test) and increased CyC in M1 macrophages (p = 0.07, two-sided t test; Figures S4D and S4E). In contrast, GC treatment did not induce CyC in monocyte-derived immature and mature dendritic cells (p > 0.39, two-sided t test; Figures S4F–S4G). To investigate the GC-CyC connection in these cell types on a regulatory level, we analyzed established estimates of enhancer-gene pair activity63 and found increased activation of the downstream enhancer element at the CST3 locus (Figure S4H) in macrophage-like versus monocyte-like THP-1 cells (Figure S4I).
Figure 5.

CyC production is dynamically regulated in disease states
(A and B) Significant (p < 0.05) changes in paired (A) CST3 gene expression (reverse transcription PCR) and (B) extracellular CyC concentration in PMA-treated human THP-1 cells (macrophage-like) normalized to cellular protein content during 0- to 18-h DEX (100 nM) treatment. There are 6 biological replicates per group.
(C) Change in normalized extracellular CyC concentration in macrophage-like THP-1 cells after 18-h treatment with DEX (100 nM) or VEH control.
(D and E) Creatinine-normalized plasma CyC (C2 ratio) at specific time points with sufficient data in hospitalized COVID-19 patients treated with DEX or standard of care (control [CTRL]) as part of cohorts based in (D) Calgary, Canada, and (E) Charité Hospital, Germany. Day 1 in the Calgary, Canada, cohort refers to a time window of 72 h after admission to the ICU. Error bars indicate standard error of the mean.
(F) Single-cell CST3 gene expression in each cell cluster in melanoma tumors (n = 12) from Jerby-Anon et al.64 Clusters defined by correlation to reference PBMC data,60 with unclassified cells that exhibit detectable clonal copy-number variation classified as tumors.
(G) Plasma CyC concentration in BALBc mice after inoculation with colon-26 (C26) tumor cells. Cachexia is defined by >15% body weight loss, and pre-cachexia refers to 14 days after tumor inoculation; tumor bearing refers to day 7 after tumor inoculation.
(H) Significant positive correlation between plasma corticosterone and plasma CyC during tumor progression in C26 model.
(I) Extracellular CyC concentrations in C26 cells normalized to cellular protein content after 0-, 6-, 12-, 18-, or 24-h treatment with 100 nM DEX. Each time point comprises at least 4 biological replicates. p values refer to two-sided t tests. VEH, vehicle; DEX, dexamethasone.
Severe COVID-19 infection is characterized by persistent lung inflammation associated with concomitant recruitment of monocyte-derived macrophages.65 Until the release of the RECOVERY trial,66 patients with severe COVID-19 were not routinely treated with GC agonists such as dexamethasone. As such, COVID-19 presents a unique opportunity to investigate the effect of dexamethasone on Cr-normalized CyC levels (C2 ratio). We collated plasma Cr and CyC measurements in two independent cohorts of patients (from Calgary, Canada,67 and Berlin, Germany68). In each cohort, a subset of patients received standard of care (pre-RECOVERY trial) and a subset received standard of care plus dexamethasone from admission (post-RECOVERY trial). We identified significantly increased C2 ratios in dexamethasone-treated patients at early time points (day 1 or 3; Figures 5D and 5E) that normalized by day 7 after admission.
The findings that CyC is constitutively expressed by myeloid cells and that GC-responsive CyC secretion occurs in macrophages, but not monocytes, have the potential to explain our finding that rs2749527 is a trans-eQTL for CST3 measured in visceral adipose fat (VAF) in STARNET but not GTEx (p = 0.77, additive model; Figure S3B). The STARNET study recruited patients with established coronary artery disease,52 while the GTEx study is a relatively unselected cohort of deceased donors.41 As metabolic syndrome is associated with significant macrophage accumulation in adipose tissue,69 we hypothesized that STARNET patients would have significantly increased macrophage gene signatures in VAF compared with GTEx donors. Using CIBERSORTx70 (absolute mode) analysis of RNA-seq data in each cohort, we identified highly significant enrichment of M2-like macrophages (demarcated by high expression of CCL18, TREM2, and CLEC4A) in STARNET versus GTEx (p = 3.03e−289, two-sided t test; Figure S5A). M2-like macrophages were by far the most abundant myeloid component in the STARNET VAF samples, suggesting that they are the cell type underlying the trans-eQTL signal. This finding both provides orthogonal validation for the role of macrophages in GC-responsive CyC secretion and illustrates the limitations of eQTL analysis using bulk RNA-seq data, as has been described previously.71
While we did not identify significant CST3 gene expression in epithelial tissues in the GTEx and Human Protein Atlas datasets, we detected high and GC-inducible CyC expression in cancer cell lines (Figures 3H and 3I). This raises the possibility that cancer cells co-opt a phenotype normally exhibited by macrophages and ectopically express CST3. We reanalyzed melanoma scRNA-seq data from 12 patients and confirmed high CST3 expression in the myeloid compartment and identified comparable ectopic CST3 expression in the tumor compartment (Figure 5F). Consistent with elevated intratumoral GC levels,72 expression of the canonical GC target FKBP5 could be identified in all cell populations profiled in the tumor (Figure S5B), demonstrating that GC signaling is necessary but not sufficient for CST3 expression. The murine colon-26 (C26) model of cancer progression is characterized by marked elevations in endogenous GC production during disease progression.30 As has been demonstrated in human patients with cancer,14 we hypothesized and subsequently confirmed that CyC levels would significantly increase during disease progression (Figure 5G) and that these increases would positively correlate with levels of the endogenous murine GC corticosterone (Figure 5H). Dexamethasone treatment of C26 cells in vitro was associated with significantly increased CyC secretion at 24 h (Figure 5I), suggesting that elevations in CyC during C26 cancer progression are at least in part mediated by GC-induced cancer cell-intrinsic CyC secretion. Altogether, these findings demonstrate that the capacity of GCs to induce CyC secretion is highly context dependent, and can be co-opted by cancer cells, suggesting a possible immunomodulatory selective advantage for cancer cells.
CyC directs recruitment of Trem2+ macrophages and failure of cancer immunotherapy
To investigate how CyC expression would provide a selective advantage to cancer cells, we used transient transfection with Cas9 and CST3-specific guide RNAs (gRNAs) to generate a CST3-knockout (CST3−/−) clone of the Mm1 cell line, which is derived from a liver metastasis of the autochthonous KPC model of pancreatic cancer,73 which recapitulates the low immunogenicity and immunotherapy responsiveness of the human pancreatic cancer. Knockout was confirmed for extracellular CyC protein levels (Figure S6A) and Sanger sequencing of the predicted edit site (Figure S6B), which confirmed 97% editing efficiency. Isogenic sgScrambled and CST3−/− Mm1 clones had equivalent doubling times in vitro (sgScrambled: 23.3 h, 95% confidence interval [CI] 21.6–25.4; CST3−/−: 24 h, 95% CI 22.4–25.8; Figure S6C). In contrast, CST3−/− tumors had markedly attenuated growth kinetics in vivo (Figure 6A, independent replication; Figure S6D) and significantly lower endpoint tumor weights (Figure S6E, independent replication; Figure S6F). Three findings linked together led us to hypothesize that CyC might have an immunosuppressive function: the growth defect of CST3−/− tumors was only detectable in vivo, CyC is a known potent inhibitor of cysteine proteases,74 such as those involved in antigen presentation,75 and observation that CyC is a GC response gene. To minimize the effect of mouse-specific factors and to maximize the immune selective pressure on tumors, we inoculated mice with a sgScrambled tumor on the left flank and a CST3−/− tumor on the right flank (termed biflank model), and we treated mice with 2–3 doses of anti-PD-L1 antibody. This experiment confirmed the suppressed growth of CST3−/− versus paired sgScrambled tumors (Figures 6B and S6G–S6I). Consistent with this, the proportion of Ki67+ cells was significantly lower in CST3−/− versus paired sgScrambled tumors (Figure 6C). In contrast, growth kinetics were similar between tumors formed from sgScrambled and CST3−/− Mm1 clones inoculated in immunodeficient (Rag1-null) mice (Figure S6J), consistent with an immune-dependent growth detect in CST3−/− tumors.
Figure 6.

CyC directs recruitment of TREM2+ macrophages and promotes failure of cancer immunotherapy
(A) Tumor growth curves (mean and standard error of the mean) for single-flank sgScrambled (n = 8) and CST3−/− (CST3 knockout [KO], n = 8) tumors; 100,000 cells were inoculated in right flank (cohort A).
(B) Tumor growth curves (mean and standard error of the mean) for biflank paired sgScrambled (n = 5) and CST3−/− (n = 5) tumors; 50,000 cells were inoculated in both flanks (cohort C). Mice received three doses of anti-PD-L1 antibody. p values refer to paired two-sided t tests.
(C and D) Proliferation index (proportion of Ki67+ cells/total cells) (C) and proportion (D) of non-epithelial cells in histological sections from paired biflank sgScrambled and CST3−/− tumors (pooled cohorts C and D). p values refer to paired two-sided t tests.
(E) Uniform manifold approximation and projection (UMAP) of 14,416 cells, annotated with cell type, from 4 tumor samples (2 sgScrambled, 2 CST3−/−).
(F) Proportion of Trem2+ macrophages in sgScrambled and CST3−/− tumors; p value is adjusted p value from linear model of logit-transformed proportions.
(G) Number of Trem2+ cells per mm2 from digital image analysis of Trem2 immunohistochemistry in paired biflank sections from sgScrambled and CST3−/− tumors. p value refers to paired two-sided t test.
(H) Multivariate (Cox and logistic) regression of Z scored CyC-production PGS against immuno-oncology biomarkers (progression-free survival [PFS], overall survival [OS], durable clinical benefit [DCB]) in meta-analysis of European patients (n = 685) treated with checkpoint immunotherapy (anti-CTLA4 or anti-PD1/PD-L1). Sample sizes for each clinical endpoint were n = 342, 685, and 670, respectively. In each model, covariates included PC1–4, sex, and primary cancer. Error bars reflect 95% confidence interval. Lower hazard ratios (survival, Cox regression) or higher odds ratios (durable clinical benefit, logistic regression) reflect better therapeutic outcomes (annotated with purple arrow).
(I) Sensitivity analysis indicating odds ratio and 95% confidence interval for DCB in each cancer type. p values refer to two-sided t tests unless otherwise stated. ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001.
Boxplots show median (central line) with interquartile range (IQR; box) and extrema (whiskers at 1.5× IQR).
To investigate whether altered growth kinetics reflected remodeling of the tumor microenvironment, we performed pan-cytokeratin immunohistochemistry and automated image segmentation to score the epithelial and non-epithelial areas in each tumor section (Figure S6K). The fraction of non-epithelial cells was markedly reduced in paired CST3−/− versus sgScrambled tumors (Figure 6D). In order to identify whether the depletion of specific non-epithelial cell types could explain this observation, we performed scRNA-seq on 2 sgScrambled and 2 CST3−/− uni-flank tumors, with 14,416 cells spanning 14 cell types passing quality control criteria (Figures 6E and S7A; Table S7). scRNA-seq profiles of cancer cells confirmed CST3 knockout in this compartment (Figure S7B; p = 9.16e−30, pseudobulk likelihood ratio test) but not in other compartments (p > 0.05). To identify enriched or depleted cell types, we implemented the propeller method,76 which models the logit-transformed cell type proportions as a function of the CST3 genotype. At 5% false discovery rate (FDR), we identified a single-cell population, annotated as Trem2+ macrophages, that was significantly depleted in CST3−/− tumors (adjusted p = 0.004, moderated ANOVA test, ratio = 0.098; Figure 6F). We validated depletion of Trem2+ cells by digital image analysis of Trem2 immunohistochemistry (IHC) in a non-overlapping cohort of biflank sgScrambled and CST3−/− tumor sections (Figure 6G). We identified a highly non-random distribution of Trem2+ cells in both sgScrambled and CST3−/− sections, with a marked enrichment of Trem2+ cells in the outer rim of the tumor (Figure S7C). These findings suggest that CyC can influence migration or expansion of Trem2+ macrophages and that Trem2+ macrophages might regulate trafficking of immune cells into the tumor.
As Trem2+ monocytes can be detected in blood samples,77 we hypothesized that GC treatment in critically ill patients would be associated with expansion of Trem2+ monocytes in blood. To investigate this, we reanalyzed CD14+ monocyte scRNA-seq profiles from patients admitted to the intensive care unit (ICU) with COVID-1967 and recovered dropped-out features to impute cluster gene expression,78 thereby identifying a cluster of Trem2+ monocytes (termed cluster 0; Figure S7D). We identified significant expansion of cluster 0 at day 7 versus day 1 in patients treated with dexamethasone (adjusted p = 0.004, moderated ANOVA test, ratio = 16.1; Figure S7E) but not in dexamethasone-naive patients (adjusted p = 0.749, ratio = 2.12; Figure S7F). This would support a stepwise model in which GC agonists increase extracellular CyC levels (Figure 5C), which in turn promotes recruitment or expansion of Trem2+ myeloid cells.
Others have shown that TREM2+ macrophages play a highly immunosuppressive role in the tumor microenvironment79 and are known to be associated with failure of CPI targeting the PD-1/PD-L1 axis.80,81 We hypothesized that increased CyC, by either inducing recruitment or expansion of Trem2+ macrophages or both, would be associated with reduced efficacy of CPI. Consistent with this, analyzing CST3 gene expression in TCGA tumors showed significantly elevated CST3 in the “immunologically quiet” immune subtype (C5 TCGA, p < 1e−10, Tukey’s test against all other subtypes; Figure S8A), which is characterized by the highest macrophage and lowest lymphocyte abundance. To investigate whether dynamically increased CST3 gene expression would be associated with resistance to CPI, we reviewed paired pre- and post-treatment tumor biopsy scRNA-seq from patients (n = 8) with metastatic basal cell carcinoma (BCC) treated with anti-PD-1.82 Patients were split into responders (n = 3) and non-responders (n = 5; Figure S8B) according to radiological response. Pre-treatment CST3 expression in macrophages, dendritic cells (DCs), cancer-associated fibroblasts (CAFs), and tumor clusters did not predict CPI responsiveness (p > 0.05, paired t test). However, we observed evidence for significant dynamic CST3 upregulation in CAFs and DCs in non-responder patients (p < 0.05, paired t test; Figures S8C–S8F).
We considered that the CyC-production PGS could reasonably capture the capacity to dynamically regulate secretion of CyC and thus predict failure of CPI. To estimate the CyC-production PGS in patients treated with CPI, we collated 8 published cohorts of patients with cancer treated with anti-PD-1, anti-PD-L1, or anti-CTLA-4 therapies with available germline exome sequencing (termed panIO cohort; Figure S6B; Table S2A). 685 patients with EUR ancestry passed quality control for inclusion (cohort characteristics summarized in Table S2B). Following imputation of common variants, the exome-wide CyC-production PGS was scored in each patient. Using multivariate Cox regression adjusted for sex, genetic ancestry, and tumor type, we demonstrated that the CyC-production PGS was associated with significantly worse progression-free survival (HR = 1.29, p = 0.0005) and worse overall survival (HR = 1.09, p = 0.10; Figure 6H). Using logistic regression with the same covariates, we further demonstrated that the PGS was associated with significantly reduced odds of durable clinical benefit (OR = 0.78, p = 0.003; Figure 6H). This latter effect was broadly consistent in each tumor type (Figure 6I). Altogether, these findings suggest that increased intratumoral CyC production may make a substantial contribution to failure of cancer immunotherapy and that this effect may be mediated by recruitment of TREM2+ macrophages.
Discussion
This work proposes a mechanistic link between GC signaling, CyC, and Trem2+ macrophages. We investigated CyC’s biological and clinical relevance using a combination of genetic analyses and in vitro and in vivo experimental medicine approaches, as well as clinically relevant prognostic and predictive studies and make contributions to two knowledge gaps.
Firstly, estimation of renal function is central to clinical practice by defining disease states, capturing acute systemic illness, and informing optimal medication dosing. Therefore, the relative strengths and weaknesses of CyC as a marker of renal function have substantial clinical relevance. CyC performs well as a renal function marker in healthy individuals, but its performance deteriorates in patients with acute disease and patients who receive GC agonists such as prednisolone, for example patients who have had a renal transplant.54 Our findings provide additional context for seemingly contradictory studies11,83 measuring the effect of GC treatment on CyC levels by demonstrating that GC-inducible CyC secretion is context dependent and not detected systemically in healthy mice and humans. An inflammatory stimulus could drive differentiation of monocytes to macrophages, in turn upregulating GR and enabling GC-dependent gene programs,62 such as GC-inducible expression of CST3. Such a regulatory system would function to precisely tune the GC response program to minimize the off-target effects of GCs, which are well recognized in clinical practice.84 However, we recognize that GCs have pleiotropic effects and that other mechanisms may contribute to our observations. Furthermore, our data demonstrate that GCs are necessary (Figures 5A and 5B) but not sufficient (Figures 4H–4J) for induction of CyC in all biological contexts. In the context of inflammatory diseases, such as COVID-19, however, we show that dexamethasone treatment induces detectable increases in systemic CyC levels and have recently reported that CyC levels are dynamically regulated in COVID-19 patients and correlate with in-hospital mortality.49 Altogether, this adds to the discourse of whether CyC is a robust marker of renal function in patients with significant inflammatory disease,54 for whom correct estimation of kidney function is important.
Secondly, despite the widespread adoption of exogenous GCs as a treatment for inflammatory conditions and for treating autoimmune adverse effects of CPI, the exact mechanisms by which GCs cause immunosuppression remain elusive.84 Our findings, together with work from others, suggest that CyC may be an effector of GC-induced immunosuppression: CyC is biologically active as a potent cysteine protease inhibitor,74 CyC secretion can be induced by GC agonists in inflammatory macrophages, and this process is co-opted by cancer cells, for which the immune system is a dominant selection pressure.85 Furthermore, CyC levels are highest in cerebrospinal and seminal fluid,74 suggesting a role in immune privilege. We recognize that the GC effect is not exclusive to CyC and that CyC may function as one of many mechanisms of GC-induced immunosuppression. In support of this model, cancer cell-intrinsic CyC knockout attenuates tumor growth kinetics and proliferation in immunocompetent mice, consistent with evidence that germline CST3 knockout abrogates metastasis in vivo.86 Using single-cell omics and population genetics, we propose a model in which CyC is associated with recruitment of immunosuppressive Trem2+ macrophages,80,81 which in turn promote failure of cancer immunotherapy. Alternatively, CyC may be necessary for the survival or maintenance of Trem2 expression in tumor-resident Trem2+ macrophages rather than promoting their recruitment. Future work has to determine the precise molecular mechanism by which this sequence occurs, as well as the origin of Trem2+ macrophages in the tumor microenvironment. One potential link is apolipoprotein E (ApoE), which is known to be secreted by cancer cells87 and is a high-affinity ligand for the Trem2 receptor (Kd = 6 nM).88 Ligation of the Trem2 receptor by ApoE is sufficient to promote phagocytosis in TREM2-expressing microglial (brain-resident macrophage) cells and, in turn, activates Apoe RNA expression,89 suggesting an autocrine positive feedback loop. Given that ApoE can be proteolytically processed90 and that CyC is a potent protease inhibitor, CyC may act to regulate ApoE availability in the tumor microenvironment, thereby regulating recruitment and proliferation of Trem2+ macrophages. Furthermore, evidence that M2-like macrophages appear to not only modulate the trans-eQTL association at rs2749527 between cortisol and CST3 expression but also express TREM280,91 suggests the existence of a single autocrine loop driving GC-induced expansion of TREM2+ macrophages. In support of connectivity between CyC, ApoE, and TREM2, Trem2-knockout mice have accelerated amyloid burden in mouse models of Alzheimer’s disease (AD),92 TREM2 R47H mutations impair ApoE binding88 and increase the risk of human AD,93 while CyC knockout is associated with reduced amyloid burden.94 Consistent with a direct immunosuppressive function of CyC, we demonstrate that germline predisposition to CyC production is significantly associated with substantial remodeling of the intertumoral immune landscape and failure of cancer immunotherapy. The evidence that the CyC-production PGS predicts failure of immunotherapy requires experimental confirmation in future work. If confirmed, a combination of PD-1/PD-L1 blockade and CyC inhibition may offer a therapeutic approach in patients who do not respond to CPI.
Limitations of the study
While the focus on human datasets allowed us to investigate clinically relevant questions, we acknowledge that many of the analyses presented are limited by their associative nature. We have necessarily adopted several surrogate measures of CyC production, including CST3 mRNA expression and CyC-residual (from blood tests) and CyC-production PGSs (from germline genetics). The time-dependent positive correlation between CST3 mRNA expression and extracellular CyC protein in vitro (Figures 5A and 5B), as well as the association between the CyC-production PGS and CST3 mRNA expression in the spleen (Figure 4A) indicate that these measures are linked and can probably be used interchangeably. Although associations between measured plasma CyC levels and clinical outcomes have the potential to be confounded in multiple directions, we and others argue that associations between patient-level PGSs and outcomes are more robust, potentially capturing causal associations.95 Also, we performed all PGS analyses in either a held-out validation cohort (for the UKB) or an independent non-overlapping cohort (TCGA, panIO) to mitigate against the risk of overfitting.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-TREM2 antibody | Proteintech | 13483-1-AP |
| Anti-Ki67 antibody | Thermo Fisher | 14-5698-82 |
| Anti-pan-cyclokeratin antibody (HRP-conjugated) | Novus Bio | NBP1-48348H |
| Anti-F4/80 antibody | Thermo Fisher | 14-4801-82 |
| Critical commercial assays | ||
| Human Cystatin C ELISA Kit | R&D | DSCTC0 |
| Mouse Cystatin C ELISA Kit | Abcam | ab119590 |
| Mouse Cystatin C ELISA Kit (SimpleStep) | Abcam | ab201280 |
| Corticosterone ELISA | IBL International | RE52211 |
| Deposited data | ||
| UK Biobank | https://www.ukbiobank.ac.uk/ | |
| Genotype-Tissue Expression (GTEx | https://gtexportal.org/home/ | phs000424 |
| The Cancer Genome Atlas | https://portal.gdc.cancer.gov/ | phs000178 |
| STARNET | N/A | |
| Pan-UK Biobank project | https://pan.ukbb.broadinstitute.org/ | N/A |
| ENCODE | https://www.encodeproject.org/ | N/A |
| panIO patient cohort | Accession codes detailed in Table S2A | N/A |
| Single-cell RNA sequencing of monocytes from COVID-19 patients – this paper | Figshare: https://doi.org/10.6084/m9.figshare.14330795.v13, ‘covid.combined_final.CD14.Mono.Robj’ | N/A |
| GWAS summary statistics (CyC-production) – this paper | GWAS Catalog: GCP000606 | N/A |
| Polygenic score (CyC-production) – this paper | Deposited on PGS Catalog: PGP000463, https://doi.org/10.1101/2021.08.17.21261668 | N/A |
| Raw data for single-cell RNA sequencing of mouse tumors – this paper | Deposited on Sequence Read Archive: PRJNA961746 | N/A |
| Processed data for single-cell RNA sequencing of mouse tumors – this paper | Figshare: https://doi.org/10.6084/m9.figshare.20063402 | N/A |
| Single-cell RNA sequencing from Yost et al.82 | NCBI GEO: GSE123813 | N/A |
| Single-cell RNA sequencing from Jerby-Anon et al.96 | Single cell portal: SCP109 | N/A |
| Single-cell RNA sequencing from Madissoon et al.55 | Human Cell Atlas: https://data.humancellatlas.org/explore/projects/c4077b3c-5c98-4d26-a614-246d12c2e5d7 | N/A |
| Single-cell RNA sequencing from Wilk et al.56 | Single cell portal: SCP345 | N/A |
| Digital pathology (scanned slides from Mm1 model) – this paper | Mendeley Data: https://doi.org/10.17632/kcwn7bpdf9.1 | N/A |
| Experimental models: Cell lines | ||
| Mm1 cell line | Gift from Tuveson Laboratory (Cold Spring Harbor Laboratory) | N/A |
| A549 cell line | ATCC | N/A |
| C26 cell line | Maintained in Janowitz Laboratory (Cold Spring Harbor Laboratory) | N/A |
| HeLa cell line | Gift from Cold Spring Harbor Laboratory | N/A |
| THP-1 cell line | ATCC | N/A |
| Primary human monocytes | STEMCELL Technologies | 70034 |
| Experimental models: Organisms/strains | ||
| C57BL/6J | Jax | 000664 |
| C57BL/6J; Rag1-KO | Jax | 002216 |
| BALB/c | Charles River | N/A |
| BALB/cJ | Jax | 000651 |
| Oligonucleotides | ||
| Primer sequences | Detailed in Table S5 | N/A |
| Guide RNA sequences | Detailed in Table S6 | N/A |
| Software and algorithms | ||
| Code to reproduce core analyses – this paper | Zenodo: https://doi.org/10.5281/zenodo.7921111 | N/A |
| PLINK | https://www.cog-genomics.org/plink/ | N/A |
| Hail | https://hail.is/ | N/A |
| R | https://www.r-project.org/ | N/A |
| TOPMED | https://imputation.biodatacatalyst.nhlbi.nih.gov/#! | N/A |
| Genomic-SEM | https://github.com/GenomicSEM/GenomicSEM | N/A |
| LDSC | https://github.com/bulik/ldsc | N/A |
| LDpred2 | https://privefl.github.io/bigsnpr | N/A |
| Seurat | https://github.com/satijalab/seurat | N/A |
Resource availability
Lead contact
Further information and requests for resources should be directed to the Lead Contact, Tobias Janowitz (janowitz@cshl.edu).
Materials availability
CyC−/− Mm1 cell line that was generated as part of this study is available from the lead contact with a completed material transfers agreement.
Experimental model and subject details
Cell line models
Human lung carcinoma cell line A549 was purchased from ATCC (CCL-185). Human cervical cancer cell line HeLa was obtained from Cold Spring Harbor Laboratory. Human acute monocytic leukemic cell line THP-1 was purchased from ATCC (TIB-202). Mm1 cells were a gift from D. Tuveson (Cold Spring Harbor Laboratory, NY), and are derived from a liver metastasis in the KPC model of pancreatic ductal adenocarcinoma.73 A549, HeLa and Mm1 cell lines were cultured in DMEM (Gibco 11965092, 4mM glutamine) supplemented with 10% FBS and 1% penicillin-streptomycin. THP-1 and C26 cells were cultured in RPMI (Gibco 11875093, 2mM glutamine) supplemented with 10% FBS and 1% penicillin-streptomycin. Macrophage-like differentiation in THP-1 cells was induced by treatment with 50nM PMA (Sigma) for 48 h, before replacement with PMA-free media and recovery for 24 h prior to treatment. Cell viability was checked by trypan blue method and was consistently above 95% prior to seeding. All cell lines were cultured at 37°C in 5% CO2. Dexamethasone and PMA (phorbol 12-myristate 13-acetate) were purchased from Sigma-Aldrich. DMEM and RPMI cell culture media, fetal bovine serum (FBS), penicillin/streptomycin (P/S) and Dulbecco’s phosphate-buffered saline (DPBS) were purchased from Gibco.
Mouse models
Wild-type BALB/c mice obtained from Charles River Laboratories (for C26 model of cancer progression) and Jax (for dexamethasone treatment); and wild-type and Rag1-KO C57BL/6J mice were obtained from Jax. All mice examined as part of this study were male as C26 and Mm1 lines were isolated from male mice. Mice were allowed to acclimatize for 7 days from arrival in the Cold Spring Harbor Laboratory animal facility. All animal experiments and care were performed in accordance with the Cold Spring Harbor Laboratory (CSHL) Institutional Animal Care and Use Committee (IACUC) and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice were kept in specific pathogen-free conditions on a 24 h 12:12 light-dark cycle. Tumor samples were obtained by dissection of mice after euthanasia by cervical dislocation and tumor weights were routinely recorded. Plasma samples were obtained from tail bleeds and terminal cardiac bleeds. Tail bleeds were performed using a scalpel via tail venesection, and terminal bleeds were obtained at endpoint (cachexia) through exsanguination via cardiac puncture under isoflurane anesthesia. Samples were kept on ice at all times. Plasma samples were collected into heparin-coated capillary tubes to avoid coagulation and were processed as follows: centrifuge spin at 14,000 rpm for 5 min at 4°C, snap frozen in liquid nitrogen, and stored at −80°C.
Human studies
This study incorporates human subjects from three independent studies. All human subjects gave informed consent, and all studies were approved by the respective institutional review boards.
-
1.
Cohort 1. This cohort has been reported previously59 and refers to a prospective, single-center, single-blind randomized crossover clinical trial that recruited 10 subjects (men and women) with primary adrenal insufficiency (Addison’s disease). The study was approved by the Ethics Review Board of the University of Gothenburg, Sweden (permit no. 374-13, 8 August 2013) and conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all subjects before participation. The study was registered at ClinicalTrials.gov with identifier NCT02152553.
-
2.
Cohort 2. This cohort has been reported previously67 and refers to a prospective study that recruited 14 patients with COVID-19 necessitating admission to ICU, of which 6 received dexamethasone treatment as part of their clinical course. All patients or their surrogate decision-makers gave informed consent for participation. This study was approved by the Conjoint Health Research Ethics Board at the University of Calgary (Ethics ID: REB20-0481) and is consistent with the Declaration of Helsinki.
-
3.
Cohort 3. This cohort (Pa-COVID-19 study) has been reported previously68 and refers to a prospective observational cohort study at Charité Universitätsmedizin Berlin. Patients with a PCR-confirmed diagnosis of SARS-CoV-2 infection were eligible for inclusion in the study. The Pa-COVID-19 study is carried out according to the Declaration of Helsinki and the principles of Good Clinical Practice (ICH 1996) where applicable and was approved by the ethics committee of Charité- Universitätsmedizin Berlin (EA2/066/20).
Method details
Differentiation of human monocytes to macrophages and dendritic cells
Human Peripheral Blood Monocytes were obtained from STEMCELL Technologies (70034). For monocyte-to-macrophage differentiation, monocytes were seeded in 96-well tissue-culture treated plates at 1x10 5 cells/well in ImmunoCult-SF Macrophage Medium (10961) containing 50 ng/mL macrophage colony stimulating factor (M-CSF, 78057.1) for 4 days. M0-like macrophages were further differentiated into M1-like macrophages by addition of 50 ng/mL interferon-gamma (IFNγ, 78020.1) and 10 ng/mL Lipopolysaccharides (LPS, Thermo Fisher) and M2-like macrophages by addition of 10 ng/mL interleukin-4 (IL-4,78045.1) for 2 days. For monocyte-to-dendritic cell differentiation, 1x105 cells/well were seeded in 96-well tissue-culture treated plates and grown in ImmunoCult Dendritic Cell Differentiation Medium (10985) for 5 days. On day 5, ImmunoCult Dendritic Cell Maturation Supplements (10989) were added into the immature dendritic cells for 2 days.
In vitro glucocorticoid treatment
For cancer cell line (A549, C26, THP-1) experiments, cells were plated in 6 well plates, at a density of approximately 500,000 cells/well. For primary cell experiments, cells were plated in 96-well plates as described above. For suspension cells (monocyte-like THP-1 cells, primary monocytes, primary mature dendritic cells), cell-repellent plates were used to seed cells prior to dexamethasone treatment (Nunc or CellStar). Cells reached confluence on day one or day two after being seeded. For time course experiments, cells were seeded and harvested at the same time, with the only variable being the duration of treatment with 100nM dexamethasone (varied between 0 and 18 h), with 0-h treatment acting as the control. For single-timepoint experiments, cells were treated with either 100nM dexamethasone (Sigma) or 0.01% ethanol for 18 h prior to harvesting. For mature dendritic cell experiments, 10nM dexamethasone was used to minimize cytotoxicity.97 For each experiment all samples were harvested concurrently.
For quantification of extracellular CyC, cell supernatant was collected at harvesting, spun at 10000 x g for 5 min to remove debris, and analyzed by ELISA (Human Cystatin C ELISA Kit, R&D Systems; Mouse Cystatin C ELISA Kit, Abcam/ab119590), with each sample profiled in duplicate. For quantification of cellular protein content, cells were washed with DPBS and ice-cold RIPA buffer with protease and phosphatase inhibitors (Thermo Fisher) or buffer APL (Qiagen) was added to each well. The cell lysate was passed through a 25G syringe for homogenization and spun for 10000 x g for 15 min, at 4°C. For RIPA lysates, protein content was determined by BCA assay (Thermo Fisher), with each sample profiled in duplicate. For APL lysates, samples were processed according to the AllPrep RNA/Protein Kit (Qiagen) manufacturer’s protocol, with protein content determined by Nanodrop spectrophotometry. Normalized extracellular CyC concentrations were determined by dividing the ELISA-derived CyC concentration (ng/mL) by the cellular protein content (mg).
For quantitative real-time PCR (RT-PCR), RNA was extracted using the RNeasy Mini Kit (Qiagen) or the AllPrep RNA/Protein Kit (Qiagen) and reverse transcribed using SuperScript IV VILO Master Mix (Thermo Fisher) according to the manufacturer’s protocol. Four housekeeping genes (GUSB, PPIA, RPL15, RPL19) with minimal variation on GC treatment were selected on the basis of a literature review98 and differential expression analysis in ENCODE RNA-seq data (accession ENCSR897XFT), implemented in edgeR. Primers were designed using NCBI Primer-BLAST, with exon-spanning primers designed where possible (primer sequences detailed in Table S5). PCR was performed using the PowerTrack SYBR Green Master Mix (Thermo Fisher) using the QuantStudio 6 Flex (Thermo Fisher) instrument, using a 10μL reaction volume in technical triplicate according to the manufacturer’s protocol. The threshold cycle was determined by the Second Derivative Maximum method and the expression of each target was normalized relative to the geometric mean of endogenous controls.
In vivo glucocorticoid treatment
Wild-type BALB/c and C57BL/6J were treated with a single high dose (20 mg/kg) of dexamethasone given intraperitoneally (IP) at 9a.m. Dexamethasone 21-phosphate disodium salt (Sigma) was dissolved in PBS and filter sterilized prior to injection. Tail vein samples were taken 24- and 48-h following IP dosing, and plasma levels of CyC were determined with Mouse Cystatin C ELISA Kit (ab119590), Abcam.
Glucocorticoid treatment in human subjects
Glucocorticoid treatment in human cohort 1 has been reported previously.59 Briefly, subjects were randomized to a 22-h treatment (commencing at 9a.m.) with placebo (intravenous 0.9% saline) or near-physiological glucocorticoid treatment with intravenous hydrocortisone. During the GC exposure, hydrocortisone was administered at a varying dose of 0.024 mg/kg/h between 9 a.m. and 12 p.m. (first day), 0.012 mg/kg/h between 12 p.m. and 8 p.m. (first day), 0.008 mg/kg/h between 8 p.m. and 12 a.m. (first day), and 0.030 mg/kg/h between 12 a.m. and 7 a.m. (second day). After 2 weeks, subjects were given whichever treatment they had no yet received, as part of a crossover study design. Blood samples were collected in the morning of the second intervention day (6 a.m.) and plasma was isolated. Plasma CyC and creatinine were measured used validated clinical assays (creatinine: Alinity c Creatinine (Enzymatic) Reagent Kit; CyC: Gentian Cystatin C Immunoassay) at the laboratory of Sahlgrenska University Hospital in Gothenburg, Sweden.
CyC quantification in patients with COVID-19
For human cohort 2, serum samples were collected as specified timepoints (timepoint 1: within 72 h of admission/referred to as day 1, and timepoint 2: 7 days after timepoint 1).67 ELISA-based serum cystatin C measurement was performed by Eve Technologies (Custom Human Kidney Injury Panel – Cystatin C). For human cohort 3, plasma sampling for plasma proteomics by mass spectrometry was performed three times per week subsequent to inclusion. Sample processing, mass spectrometry and data analysis were performed as described previously,2 allowing for quantification of plasma CyC levels in 309 patients. Out of these patients, 131 had available paired serum creatinine for at least one timepoint, as well as clinical outcome data (COVID-specific mortality). For patients with at least one creatinine measurement, missing data were imputed with the most recent value. Plasma CyC levels were scaled by a factor of 300, so that the cohort mean was comparable to the mean serum CyC recorded in the UKB cohort (field 30720, units mg/L). For each patient, a creatinine-CyC (C2) ratio was calculated at each timepoint, using CKD-EPI eGFR equations with the race term set to 0.
In vivo model of cancer progression
Experiments with the C26 model were performed using 8-weeks old wild-type BALB/c male mice. Mice were inoculated subcutaneously in their right flank with the syngeneic C26 colorectal cancer cell line (2x106 viable cells in 100μL RPMI vehicle) that induces cachexia. Prior to inoculation, C26 cells were dissociated with trypsin, followed by resuspension in FBS-free RPMI and counting of the viable cell concentration (trypan blue). C26-tumor bearing mice were termed pre-cachectic from 18 days post-inoculation and were defined as cachectic when their weight loss exceeded 15% from peak body weight. Plasma levels of CyC were determined with Mouse Cystatin C ELISA Kit (ab119590), Abcam. Corticosterone levels were quantified using Corticosterone ELISA (RE52211) from IBL International (TECAN).
Establishment of isogenic CyC−/− cell line
We prioritized experimentation with the Mm1 (KPC-derived) cell line as we have found this line to behave highly reproducibly across different experimenters over time, allowing us to place greater confidence on the findings from each experiment. Mm1 were transiently transfected with CRISPR plasmids (PX459, GenScript) encoding either a guide RNA (gRNA) specific to a coding region in mouse Cst3 or a non-targeting (scrambled) gRNA. We tested two Cst3-specific gRNAs and one scrambled gRNA from a pre-validated database.99 Guide RNA sequences are summarized in Table S6. Mm1 cells were seeded into 24-well plates with 50,000 cells per well and after 24 h, they were transfected with 500ng plasmid using Lipofectamine 3000 (Thermo Fisher) according to the manufacturer’s protocol. We included a GFP-expressing plasmid to assess transfection efficiency. After 48 h, the media was changed and replaced with DMEM media supplemented with 5 μg/ml puromycin. After 72 h, the media was replaced with DMEM media for 24 h, followed by isolation of monoclonal populations by serial dilutions in a 96-well plate. To identify clones with CyC knockout, we measured CyC in the cell supernatant for each clone using the Mouse Cystatin C ELISA Kit (ab201280), Abcam. To verify the presence of truncating mutations in the Cst3 coding region, we extracted genomic DNA from each clone (Qiagen DNeasy Blood and Tissue Kit) and performed targeted polymerase chain reaction (PCR) amplification and Sanger sequencing of the predicted gRNA binding sites. The editing efficiency was assessed using the Synthego ICE Analysis tool (https://ice.synthego.com/).
Characterization of isogenic CyC−/− cell line
To compare the in vitro growth kinetics of isogenic sgScrambled and CyC−/− cell lines, cells were seeded into 6-well plates with 200,000 cell per well, with three biological replicates per clonal cell line. Each well was scanned every 2 h using an IncuCyte S3 Live Cell Analysis Instrument using the phase channel according to the manufacturer’s protocol. Cell confluence was estimated using the Incucyte Cell-By-Cell analysis module, and was normalized to the first timepoint. The doubling time was estimated by fitting a model of log(time) as a function of confluence. To compare the in vivo growth kinetics of isogenic sgScrambled and CyC−/− cell lines, mice were inoculated subcutaneously with 50,000–200,000 cells in the flank. For uni-flank experiments, mice were inoculated in the right flank; for biflank experiments, mice were inoculated in both left and right flanks, with the sgScrambled tumors on the left flank and the CyC−/− tumor on the right flank. For tumor inoculation, Mm1 cells were dissociated with trypsin followed by resuspension in FBS-containing DMEM, counting of the viable cell concentration (trypan blue) and resuspension in sterile PBS. 10-20μL of PBS-suspended cell mixture was combined with an equal volume of Cultrex Reduced Growth Factor Basement Membrane Extract (3433-010-01, R&D Systems) on ice. Immediately prior to inoculation, the suspended cell mixture is thawed to room temperature and loaded into insulin syringes (328440, BD). Mice were monitored regularly until palpable tumors formed, after which point the longest and shortest dimensions of each tumor was measured every 3–4 days using calipers. For anti-PD-L1 treatment, mice were treated with 200μg of anti-PD-L1 monoclonal antibody (BioXCell, BP010) every 3 days, given intraperitoneally (IP). Unless otherwise stated, mice were sacrificed by cervical dislocation once tumors exceeded 20mm on one axis.
Single-cell RNA sequencing of mouse tumors
Tumors were finely minced at 4°C and transferred into tumor digestion medium containing collagenase/hyaluronidase and DNase I in RPMI 1640 with glutamine, then incubated on a shaker for 45 min at 37C and 300rpm. Freed cells were collected by passing through the dissociated tumor and media into a 70um cell strainer and quenching with FACS buffer (2% fetal bovine serum in sterile PBS) at 4°C. Cells were spun down at 300g for 5 min at 4°C, the pellet resuspended in ice-cold ammonium chloride solution for 5 min and quenched with FACS buffer. Cells were spun down again and resuspended in FACS buffer. Viable cells were quantified by trypan blue method and samples were then subject to dead cell removal (EasySep Dead Cell Removal Kit, STEMCELL). Prior to library preparation, viability and cell number were re-assessed with a Countess II FL using AOPI (PN- CS2-0106-5mL, Nexcelom Bioscience). Single-cell RNA-seq libraries targeting 8,000 cells per sample were generated using the Chromium Next GEM Single Cell 3ʹ Reagent Kits v3.1 (PN-1000121, 10x Genomics) according to the manufacturer’s instructions. Final libraries were sequenced to at least 25,000 reads per cell with the Illumina NextSeq 2000 and aligned with Cell Ranger (version 6.0.0, 10x Genomics) to the mm10 reference genome (refdata-gex-mm10-2020-A, 10x Genomics).
Tumor immunohistochemistry
Tumors were harvested and embedded in tissue molds containing OCT (Sakura) and frozen on dry ice prior to storage at −80°C. IHC staining were performed at CSHL Tissue Imaging Shared Facility. OCT embedded fresh tissue blocks were sectioned with Thermo #NX50 cryostat. 10μm thick sections were collected and mounted on positive charged glass slides (VWR superfrost plus micro slide) IHC slides were stained on DISCOVERY ULTRA IHC/ISH research platform (Roche) following standard protocols. Briefly, after fixation, slides were incubated with primary antibody at 37°C for 1h and Discovery multimer detection system (Discovery OmniMap HRP, Discovery DAB, Roche) was used to detect and amplify immuno-signals. Primary antibodies: Ki67 (Thermo Fisher 14-5698-82), 1:500 dilution; Pan-CK (Novus Bio NBP1-48348H), 1:100 dilution; TREM2 (Proteintech 13483-1-AP), 1:150 dilution.
Tumor in situ hybridization
Staining was performed using the RNAscope platform (ACD), according to the manufacturer’s protocol for the RNAscope 2.5 HD Detection Reagent (red, 322360) and technical note for fresh-frozen tissue (320536). Tissue sections were fixed with 4% PFA for 15 min at 4°C, and dehydrated with a series of ethanol washes (50%, 70%, 100%, 100%) for 5 min each. The sections were pretreated with hydrogen peroxide for 10 min, washed once with distilled water, pretreated with Protease IV for 30 min at RT and washed with 1X PBS. Sections were then individually hybridized for 2 h at 40°C, with probes targeting either TREM2 (404111, ACD), DapB (negative control; 310043, ACD) or PPIB (positive control; 313911, ACD). After hybridization, sections were washed twice with 1X PBS for 2 min and subject to 6 amplification steps (30 min at 40°C, 15 min at 40°C, 30 min at 40°C, 15 min at 40°C, 30 min RT, 15 min RT) prior to detection. Signal was detected using Fast Red reagent (322360, ACD) for 10 min at RT, and briefly washing with tap water prior to counterstaining with hematoxylin. Slides were mounted using xylene and EcoMount. Images were scanned using a Leica-Aperio Versa slide scanner.
Quantification and statistical analysis
Cohort genomic data quality control
UK Biobank
UK Biobank (UKB)-provided measured genotype, imputed genotype (GRCh37, imputed data release 3) and phenotype data100 was accessed as part of application 58510. We selected subjects with available imputed genomic data (field 22028) and at least one paired creatinine (field 30700) and CyC measurement (field 30720), and excluded subjects with sex chromosome aneuploidy (field 22019), discordant genetic sex (fields 31 and 22001), excess heterozygosity and missing rate (field 22027). To classify genetic ancestry, we lifted over directly genotyped and linkage disequilibrium (LD)-pruned high-quality variants (biallelic SNPs, MAF >0.1%, call rate >99%) to GRCh38 and merged with variants available from an integrated callset (call rate >95%) derived from 1000 Genomes and Human Genome Diversity Project (HGDP, gnomAD). LD pruning was implemented using PLINK1.9 with parameters ‘--indep-pairwise 50 5 0.2’. Principal components (1–10) were computed using the unrelated reference subjects (PC-relate kinship coefficient <0.05) then projected onto all reference and UKB subjects. Next, a random forest classifier was trained using ancestry data from the reference cohort, implemented in the gnomAD package for Hail. This classifier was applied to the UKB subjects, and genetic ancestry was assigned with a minimum probability of 70% (Table S1). Relatedness data was extracted from the UKB-provided kinship matrix, generated using KING software. For the EUR ancestry group, subjects were split into a discovery cohort (n = 381,764 subjects) and validation cohort (n = 50,000 subjects), with the validation cohort comprising a random selection from unrelated UKB subjects (KING kinship coefficient <0.0442). For all other ancestry groups, all subjects were used as discovery cohort. For all analyses using imputed data, we filtered to variants with INFO score >0.8 and MAF >1% across whole cohort.
GTEx project
Whole-genome sequencing data (GRCh38) and controlled-access metadata (including time of death) was accessed through dbGaP (phs000424.v8.p2) as part of application 26811. The provided imputed data had already undergone extensive quality control, however, we removed an additional 9 subjects with a PC-relate kinship coefficient >0.05. We identified EUR ancestry subjects (n = 678) as above using 1000G/HGDP reference data to train a random forest classifier that was applied to genotype-tissue expression (GTEX) subjects, using high-quality LD-pruned common variants (biallelic SNPs, MAF >0.1%, call rate >99%, r2 < 0.1), LD pruning was implemented using the ‘ld_prune’ function in Hail, subsequent to removal of high-LD regions.101 In this smaller cohort, ancestry was defined using a minimum probability of 50% followed by removal of PCA outliers with a PCA Z score >5.
The Cancer Genome Atlas
Germline array data (Birdseed format, GRCh37) was downloaded from the GDC Legacy archive as part of dbGaP application 26811, before conversion to VCF format. For sample QC, we started with a sample list defined by Sayaman et al.,102 which selected one germline sample per subject, prioritizing blood-derived or high call-rate samples, while removing samples with excess heterozygosity or hematological malignancies. For additional sample QC, we removed samples with discordant sex (using the impute_sex function in Hail), excess hetero- or homo-zygosity (Z score >3, using agg.inbreeding function in Hail), related subjects (PC-relate kinship coefficient >0.05) and called genetic ancestry as described for the GTEX cohort (n = 7260 EUR patients). For imputation in the unrelated EUR population, we selected variants with call rate >95% and MAF >0.1%. Imputation was performed using the TOPMED server, which automatically lifts over variants to GRCh38. For the final cleaned dataset, we selected autosomal variants imputed with r2 > 0.6 and MAF >0.1%.
STARNET
Stockholm Tartu Atherosclerosis Reverse Networks Engineering Task (STARNET) is a cohort of 600 Caucasian patents of Eastern European origin, with a confirmed diagnosis of coronary artery disease. Genomic data quality control has been described previously.52 Briefly, array-based genotyping was performed on germline DNA from blood, followed by imputation against the 1000 Genomes phase 1 SNPs. Comparison of population structure with 1000 Genomes cohort confirmed that all STARNET subjects had European genetic ancestry.
Immunotherapy meta-cohort
We requested access to 8 cohorts of patients treated with CPI (anti-PD-1, anti-PD-L1 and/or anti-CTLA4) with available germline exome sequencing and clinical outcome (Tables S2A and S2B). Clinical annotations were downloaded from the supplemental data from associated manuscripts or requested directly from principal investigators. Samples were excluded if there was insufficient data to report at least one outcome measure (overall survival, progression-free survival, durable clinical benefit). Durable clinical benefit (binary) was defined by patients with no radiological progression >6 months or overall survival >1 year. Harmonized germline short variant calling was implemented using nf-core/sarek pipeline, with Strelka mutation caller103 and GRCh38 reference genome. gVCFs were merged using Illumina gvcfgenotyper tool and imported into Hail for processing. Samples with discordant sex (n = 13) were identified by comparison of sex reported in clinical metadata and genetic sex determined from integration of X chromosome heterozygosity and Y chromosome genotype counts (via PLINK 1.9 impute-sex function). For the small minority of patients without supplied sex (n = 4), sex was genetically imputed. For variant QC, calls filtered by Strelka were removed, SNPs calls required a minimum depth of 7 while indel calls required a minimum depth of 10. Each variant required a call rate >90% and at least one ‘high-quality’ call defined as one homozygous ALT call or one heterozygous ALT call (with allele balance >15% for SNP or >20% for indel). Samples with a call rate <90% or excess hetero- or homozygosity (Z score >3) were removed. No subjects had >third degree relatedness, which also excludes the possibility of duplicates samples in the cohort. EUR ancestry subjects were identified as for GTEX cohort. Imputation of EUR population was performed using the TOPMED server. For the final cleaned dataset, we selected autosomal variants imputed with r2 > 0.6 and MAF >0.1%.
Computation of principal components
We computed 20 principal components (PCs) on all subjects (including related) and all genotyped variants, as per the BOLT-LMM manual, implemented in PLINK2 (--pca function). Due to computational complexity, the PLINK2 PCA approximation (--approx) was used for the EUR population. To account for genetic ancestry in downstream analyses, PCs (1–4) were computed on high-quality linkage disequilibrium (LD)-pruned variants (biallelic SNPs, MAF >0.1%, call rate >99%, r2 < 0.1), with SNPs in known high-LD regions removed.101 For UKB, high-quality SNPs were derived from the ‘in_PCA’ field from the UKB-provided SNP QC file. In the UKB cohort, PCs were computed with related subjects removed (approach described above), and then projected onto all remaining samples, using the ‘run_pca_with_relateds’ function in the gnomAD package for Hail. In other cohorts (where related subjects were removed), PCs were computed using the ‘hwe_normalized_pca’ function in Hail.
Genome-wide association analysis
eGFR-CyC and eGFR-Cr were calculated using CKD-EPI equations54 implemented in the nephro package for R, with race term set to 0 for all subjects. For subjects we more than 1 paired creatinine and CyC measurement, we selected the earliest complete datapoint. Genome-wide association analyses (GWAS) in the discovery cohorts (for eGFR-CyC and eGFR-Cr were performed in each ancestry group, including related subjects, using BOLT-LMM9 with covariates including age (field 21003), age,2 sex (field 31), genotyping array (binarized from field 22000), recruitment center (field 54), and genetic PCs 1–20 (described above). LD score matrices for each ancestry group were downloaded from the Pan-UK Biobank project (https://pan.ukbb.broadinstitute.org/). To assess for confounding we determined the attenuation ratio of each trait via LD score regression, which was within the expected range for polygenic traits (Table S3).104
Structural equation modeling
Structural equation modelling of eGFR-Cr and eGFR-Cy summary statistics was implemented in the Genomic-SEM package for R33 and performed as per the GWAS-by-subtraction tutorial (https://rpubs.com/MichelNivard/565885). Briefly, for EUR, AFR and CSA populations, we performed LD score regression using LD matrices from the Pan-UK Biobank Project (https://pan.ukbb.broadinstitute.org/). We designed a structural equation model (summarized in Figure 1C), with latent traits estimated using the userGWAS function parallelized across each chromosome. Summary statistics for each latent trait (renal function, CyC-production) were extracted and effective sample sizes were estimated using the script provided by the Genomic-SEM authors (https://github.com/GenomicSEM/GenomicSEM/wiki/5.-User-Specified-Models-with-SNP-Effects). CyC-production summary statistics were standardized by setting A1 as the GRCh37 ALT allele and A2 as the GRCh37 REF allele, and multiplying the effect size of CyC-production by −1 so a higher effect size reflects increased CyC production.
Processing of summary statistics
Clumping was performed in the EUR eGFR-CyC summary statistics, implemented in PLINK 1.9 with parameters clump-r2 0.001, clump-p1 5e-8, clump-p2 5e-8 and clump-kb 10000 using 1000 Genomes reference data (derived from European subjects). For each clump, the index SNP (SNP with lowest p value) was annotated using the OpenTargets Genetics (https://genetics.opentargets.org/) variant-to-gene pipeline,105 which integrates both proximity and functional genomics data. For the small minority of variants (n = 2) not represented in the OpenTargets database, the index SNP was annotated to the nearest coding gene. Partitioned heritability analysis was performed using the LDSC package for R106 using the provided datasets, as per the tutorial by the package authors (https://github.com/bulik/ldsc/wiki/Cell-type-specific-analyses). For each trait and tissue-sample pair, we extracted the t-statistic as the ratio of the coefficient and standard error. To compare cell type-specific enrichment between renal function and CyC-production latent traits, we computed the absolute difference in t-statistic between eGFR-CyC and each latent trait, for each tissue sample. Colocalization analysis was performed using the coloc package for R,51 using the single-variant assumption. Gene set enrichment analysis of CyC-production latent trait was performed using MAGMA,107 implemented in the FUMA web server (https://fuma.ctglab.nl/) with a 0kb gene window. Mendelian randomization analysis, using cis-eQTLs probes for CST3, was implemented in GCTA-SMR108 using SMR-formatted eQTL data from the eQTLGen Consortium.58
Derivation and application of polygenic scores
CyC-production polygenic scores (PGS) were derived using LDpred244 (automatic model) according to the package vignette (https://privefl.github.io/bigsnpr/articles/LDpred2.html). For the genome-wide score, HapMap3 variants were intersected with high-quality genomic variants available for all of UKB (array), TCGA (array) and GTEX (WGS) cohorts (n = 1,031,527). For the exome-wide score, HapMap3 variants were intersected with high-quality exonic variants from the panIO cohort (n = 352,549). The provided UKB LD reference was used for PGS derivation. Model fitting was confirmed by visual inspection of chain convergence for each PGS. The PLINK2 linear scoring function (--score) was used to apply the PGS to each cohort and to avoid exclusion of duplicate dbSNP IDs, the source data was filtered to the PGS variants according to position and alleles. The sample-level PGS was normalized by Z-scoring in each cohort. To generate a patient-level surrogate for CyC production, we modeled eGFR-CyC as a function of eGFR-Cr and sex, with intercept set as 0. We computed the residual of this model, termed CyC-residual, which is multiplied by −1 so that increasing CyC-residual reflects increased serum CyC relative to creatinine.
Functional genomics
ChIP-seq (for GR/NR3C1, timeseries accession: ENCSR210PYP) and ATAC-seq (timeseries accession: ENCSR385LRX) data for A549 cells treated with dexamethasone was downloaded from the ENCODE data portal (https://www.encodeproject.org/). Data was processed using the ENCODE data analysis pipeline, generating a p value for each signal peak that reflects enrichment of DNA sequences. Data at the CST3 locus was plotted using the karyoploteR package109 for R. Enhancer activity scores (‘ABC scores’) derived from the validated activity-by-contact model,63 applied to 131 biosamples, was downloaded from ftp://ftp.broadinstitute.org/outgoing/lincRNA/ABC/Nasser2021-Full-ABC-Output/. Scores for the distal enhancer element at the CST3 locus reflecting analysis of data derived THP-1 cells were extracted, and data from THP-1 cells treated with PMA was compared to data from naive THP-1 cells.
Gene expression profiling
For GTEX and ENCODE gene expression profiling, gene-level counts derived from STAR-aligned RNA sequencing (RNA-seq) reads were downloaded from the GTEX (https://GTExportal.org/home/datasets) and ENCODE (timeseries accession: ENCSR897XFT) data portals respectively. TMM and library size normalization were applied using the edgeR package110 for R, generating TMM-normalized log-counts per million (CPM) expression values that can be compared between samples. For TCGA gene expression profiling, batch- and expression quantile-normalized data (RNA-seq) was downloaded from the PanCancer Atlas repository (https://gdc.cancer.gov/about-data/publications/pancanatlas). For STARNET gene expression profiling was performed as previously described109 – briefly, gene counts were adjusted for GC content, library size and quantile-normalized implemented in EDAseq,111 prior to log-transformation. For digital cytometry analysis implemented in CIBERSORTx70 (for STARNET and GTEX cohorts), gene expression was normalized to gene length to generate transcripts per million (TPM) expression values. CIBERSORTx was run in absolute mode with LM22 reference set, 100 permutations and B-mode batch correction.
eQTL analysis
Expression quantitative loci (eQTLs) were identified in the STARNET52 cohort using the Kruskal Wallis test statistic (additive model), as implemented by the tool kruX,112 using individual-level genotype and gene expression data (data processing described above). To identify associations between CST3 and SNPs present at the SERPINA6/SERPINA1 loci, we carried out this association analysis using 72 SNPs previously shown to be significantly associated with plasma cortisol.50 This approach was applied to all non-vascular tissues (n = 5) in the STARNET cohort (subcutaneous fat, visceral abdominal fat, skeletal muscle, liver, blood). As the 72 SNPs reflecting 4 independent LD blocks, we modeled this analysis as 20 (4 LD blocks, 5 tissues) independent hypotheses and so the Bonferroni-corrected significance threshold was 0.0025. For independent validation of the significant eQTL associations in GTEX, we performed Kruskal Wallis tests in two tissues (visceral adipose fat, liver) using the ‘kruskal.test’ function for R, using individual-level genotype and gene expression data (data processing described above). For further characterization of significant eQTLs, we constructed a recessive linear model of CST3 gene expression as a function of genotype (binarized to 0/1 versus 2), using the ‘lm’ function for R.
scRNA-seq analysis
For analysis of single-cell RNA sequencing (scRNA-seq) profiles of human skin tumors, scRNA-seq expression matrices and metadata for Jerby-Anon et al.96 and Yost et al.82 were downloaded from Single Cell Portal (accession SCP109) and GEO (accession GSE123813), respectively. For analysis of scRNA-seq profiles of spleen and PBMCs, scRNA-seq expression matrices were downloaded from For Yost et al. the peritumoral T cell-specific samples were excluded from the analysis. Count or normalized expression data was imported into Seurat113 (version 4.0), filtered (according to number of features, <10,000, and mitochondrial content, <7.5%, per cell), log-normalized (if applicable) and scaled. Highly variable features (n = 2000) were used for principal component analysis followed by clustering (Louvain algorithm). Immune clusters were annotated by comparison to reference PBMC data, implemented in clustifyR package for R.60 Unannotated clusters (presumed to reflect one of tumor, cancer-associated fibroblast or endothelial cells) were manually annotated via established marker gene expression98 and clonal copy number variation profiles, examined using the inferCNV114 package for R. Patient-level pseudobulk cluster-specific expression data was extracted using the ‘AverageExpression’ function in Seurat.
For analysis of scRNA-seq profiles of murine Mm1 tumors, cellranger-processed data was imported into Seurat113 (version 4.0). Quality control steps included removal of putative doublet cells (implemented in DoubletFinder115) and removal of cells with >20% mitochondrial genome-aligned reads or fewer than 200 features (UMIs). Data processing in Seurat included normalization, scaling (regressing out the effect of cell cycle genes), integration, Louvain clustering, dimensionality reduction and visualization. Each cluster was annotated as one of 14 cellular populations according to expression of validated marker genes (summarized in Table S7, and Figure S7A) Differential gene expression was identified from pseudobulk data, implemented in the Libra package for R.116 Differentially enriched/depleted cell populations were identified by modeling logit-transformed cell proportions as a function of tumor genotype, implemented in the speckle package for R.76
To assess changes in the monocyte population following dexamethasone, we reanalyzed whole blood scRNA-Seq datasets from ICU-admitted COVID-19 patients treated with or without dexamethasone.67 Initial data pre-processing and cell identity annotations were performed as described previously. Briefly, cell identity annotations were generated by mapping single-cell transcriptomes to the PBMC scRNA/CITE-seq multi-omic reference (Azimuth).113 To identify a high-confidence TREM2+ monocyte population, we extracted cells annotated as CD14+ monocytes and repeated batch effect correction (implemented using the ‘FindIntegrationAnchors’ function in Seurat), dimensionality reduction, and clustering. High-confidence Trem2+ monocytes were identified by expression of known TREM2+ monocyte markers (TREM2, APOE, CSF1R81,89), identified by gene-weighted density estimation implemented in the Nebulosa package for R.78 Differentially enriched/depleted cell populations were identified as above.
Cosinor regression
Cosinor regression for gene expression (FKBP5, CST3) as a function of time was performed using the cosinor package for R. A cosinor model has 4 parameters – MESOR (intercept), period (assumed as 24 h), amplitude and acrophase (timing of activity peak). The aim of this analysis is to estimate the amplitude of variation in gene expression of a 24-h cycle. Gene expression was derived from TMM-normalized TPM data (implemented in edgeR), to facilitate intra- and inter-sample comparisons. For UKB, time referred to time of sampling and for GTEX, time referred to time of death; both rounded to the nearest hour in 24-h clock. Amplitude coefficients were extracted from the transformed coefficients table.
UKB cancer cohort
To identify patients who were treated with non-topical exogenous GCs, we reviewed field 20003 for coded medications bio-equivalent to dexamethasone or prednisolone. Subject lifespan was extracted from analysis of fields 40007 and 34. Parental lifespan was extracted from analysis of fields 2946, 1845, 1797, 1835, 1807 and 3526. Using cancer registry data (fields 40005, 40012, 40008, 40011), ICD10-coded cancer diagnoses were extracted and mapped to Phecodes (https://phewascatalog.org/). Using a curated list of operation codes (OPCS-4) reflecting curative procedures for 13 main tumor groups (Table S4), we mapped each cancer diagnosis to matched surgeries that occurred no more than 90 days prior to the coding entry. To account for variation in operation data availability prior to 2000, we filtered the data to cancers that were diagnosed after the year 2000. In cases where a patient was coded with a cancer of the same primary type more than once, the entries were merged. Patients with more than one discrete cancer diagnosis were excluded (n = 2435 subjects) due to the difficulty in defining the time since diagnosis. For recruited patients who had died, we manually reviewed details from the death certificate (field 40010) to identify descriptions that were consistent with cancer-specific mortality.
Survival analyses
For Cox regression of overall and cancer-specific survival against CyC-residual, the time variable used was time from blood sampling to death or last follow-up date (nominally June 2020). For subjects with multiple CyC-residual datapoints over time, each datapoint was annotated with survival time relative to blood sampling and treated independently. Model covariates included age (at blood sampling), sex, body mass index (BMI), hemoglobin, eGFR-Cr, C-reactive protein. For Cox regression of lifespan (for subject and parents) against CyC-production PGS in the UKB validation cohort, we used age at death or age at most recent follow-up as the time variable. Model covariates included year of birth (of subject, as parental birth years are not recorded) to account for historical increases in mean lifespan.
For Cox regression of cancer-specific survival against CyC-production PGS, it was necessary to consider bias from left truncation, where patients who died between diagnosis and the recruitment period would not be recruited. To account for this, the time interval used for Cox regression of overall and cancer-specific survival against CyC-production PGS in UKB referred to time from recruitment to death or data cut-off (June 2020). In contrast, TCGA patients were generally recruited close to the time of cancer diagnosis, prior to surgical resection of tumor and so time from diagnosis to death or last-follow-up date was used. Cancer-specific survival was extracted from the ‘DSS’ and ‘DSS.time’ fields in the TCGA clinical data resource available as part of the PanCancer Atlas (https://gdc.cancer.gov/about-data/publications/pancanatlas). Cancer-specific survival analyses with respect to CyC-production PGS were adjusted for age of diagnosis, genetic ancestry (PC1-4), sex (except sex-specific cancers), and a term reflecting whether curative surgery was performed. For UKB this term was derived from matching with curative operation codes as described above, for TCGA this term was derived from the field ‘residual_tumor’ in the clinical data resource. UKB-specific PGS-cancer survival analyses were additionally adjusted for recruitment center (to account for regional heterogeneity in cancer outcomes) and genotyping array. Pan-cancer inverse variance-weighted meta-analysis in each cohort (UKB, TCGA) was implemented in the meta package for R64 using both fixed and random effects models.
For phenome-wide time-to-event analysis in UKB, all UKB ‘first occurrence’ fields and cancer registry data (fields 40005 and 40006) were extracted, with ICD10 codes mapped to Phecodes. If multiple ICD10 codes mapped to a single Phecode, the earliest date of diagnosis was selected. For each time-to-event Phecode, the time variable was defined as time from birth to first occurrence of diagnosis or most recent follow-up date. To account for region-specific variability in health record linkage, this date was determined by either the most recent coded diagnosis or most recent UKB center visit. Each phenotype-specific Cox regression was adjusted for sex, genetic ancestry (PC1-4) and year of birth (to account for historical variation in disease risk).
Digital pathology analyses
Digital image analysis of H + E stains as well as Trem2, panCK and Ki67 IHC were performed using HALO digital image analysis software version v3.4.2986.151 (Indica Labs, Corrales, NM, USA). All H&E scans were reviewed by two pathologists (DL and VHK), and the regions of viable respectively necrotic tissue was determined and computed. Necrotic tissue was excluded for further analysis. In order to analyze the tumor/stroma interaction, a deep neural network algorithm was trained on the panCK scans to recognize epithelial and non-epithelial compartments. Graphical overlays for both compartments simplified the quality control of the tissue classifier. The total area of the two compartments was then calculated automatically.
The ‘Multiplex IHC v3.1.4’ algorithm of HALO was implemented for analysis of Ki67 and Trem2 IHC. The nuclei and the chromogens were detected by color deconvolution with thresholds determined by internal controls. For Ki67 IHC, a proliferation index (Ki67+ cells/total cells) was calculated. For Trem2 IHC, the proportion of Trem2+ per unit tissue area was calculated, and the distance of each Trem2+ cell from the tumor border was measured using the proximity analysis tool of HALO.
Statistical analysis
Significance testing refers to two-tailed unpaired t-tests with the assumption of unequal variance unless stated otherwise. For biflank tumor experiments, differences were assessed using two-tailed paired t-tests. For statistical and computational analyses, we used R (version 4.0.2) and Python (version 3.7.4) implemented as a Jupyter Notebook.
Acknowledgments
This work was conducted using the UKB (application 58510). All data access relating to this manuscript was approved by the institutional IRB at Cold Spring Harbor Laboratory (CSHL), which determined that it does not meet the definition of human subject research under the purview of the IRB according to federal regulations. We thank all patients and their families who have volunteered to participate in clinical research, without whom this study would not have been possible. T.J. acknowledges funding from CSHL and grants from the Simons Foundation, the Mark Foundation (20-028-EDV), and the Nancy Gay Foundation (58391101). H.V.M. receives funding from the Simons Center for Quantitative Biology at CSHL. S.O.K. was supported by the Starr Centennial Scholarship at the CSHL of Biological Sciences. M.F. is supported by the “la Caixa” Foundation (ID 100010434) in the framework of the “La Caixa” Fellowship Program under agreement LCF/BQ/AA18/11680037. B.R.W. is supported by a Wellcome Investigator Award (107049/Z/15/Z). S.B. was supported by a Medical Research Council PhD studentship. This work was further supported by the Ministry of Education and Research (BMBF), as part of the National Research Node “Mass spectrometry in Systems Medicine” (MSCoreSys), under grant agreements 031L0220A (to M.R.) and 161L0221 (to V.D.). Computational analyses were performed with assistance from the US National Institutes of Health grant S10OD028632-01. L.G.N.d.A. received Alberta Innovates and Killam doctoral scholarships. S.S. received CIHR Vanier, Alberta Innovates, and Killam doctoral scholarships. G.J. acknowledges funding from Swedish Research Council (project nos. 2015-02561 and 2019-01112). Histological processing was performed by the CSHL Tissue Imaging Shared Facility with assistance from the Cancer Center Support grant 5P30CA045508.
Author contributions
S.O.K., H.V.M., and T.J. conceived and designed the study. S.O.K. performed statistical and computational analyses with support from T.H. S.B. performed eQTL analyses in the STARNET cohort. H.L. contributed to data curation of clinical cohorts. T.M.T. and B.D. performed in vitro experiments, while S.O.K., N.M., B.D., and M.F. performed in vivo experiments. C.R. performed single-cell library preparation. S.O.K. performed single-cell analysis with support from Y.J.R.A.R.-K. and guidance from J.P. D.C. performed analyses of human clinical trial specimens under the supervision of G.J. S.S., N.R., B.Y., J.B., A.D., L.G.N.d.A., P.T.-L., M.R., F.K., and V.D. performed proteomic analyses of COVID-19 patient specimens. Q.G. optimized and executed histological staining. Scanned slides were computationally analyzed by D.L. under the supervision of V.H.K. B.R.W. provided input for interpretation of results and direction for further analyses. H.V.M. and T.J. supervised the project and guided all data analyses. S.O.K., H.V.M., and T.J. wrote the manuscript with input from all authors. All co-authors approved the final version of the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: June 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2023.100347.
Contributor Information
Hannah V. Meyer, Email: hmeyer@cshl.edu.
Tobias Janowitz, Email: janowitz@cshl.edu.
Supplemental information
Data and code availability
-
•
Due to the data use agreements for the datasets analyzed in this manuscript, we are unable to directly share or distribute any patient-level data except for COVID-19 patient scRNA-seq (reposited at https://doi.org/10.6084/m9.figshare.14330795.v13, filename ‘covid.combined_final.CD14.Mono.Robj’). GWAS summary statistics are published alongside the study (, and polygenic scores are deposited in PGS Catalog (https://doi.org/10.1101/2021.08.17.21261668). To facilitate dataset requests from applicable data use committees, we provide all accession codes for all datasets relating to this manuscript in the key resources table and in Table S2A. UK Biobank data can be requested through the application process detailed at https://www.ukbiobank.ac.uk/.
-
•
Single-cell RNA sequencing raw sequencing data (FASTQ) is reposited in the Sequence Read Archive (SRA) under BioProject PRJNA961746, while the processed Seurat matrix is available from Figshare (https://doi.org/10.6084/m9.figshare.20063402).
-
•
Digital pathology derived from histological and immunohistochemical analyses of mouse tumor sections (Mm1 model) has been deposited at https://data.mendeley.com/datasets/kcwn7bpdf9/1.
-
•
Code to reproduce core computational and statistical analyses has been reposited on Github at https://github.com/Janowitz-Lab/cystatinc (https://doi.org/10.5281/zenodo.7921111).
-
•
Where data use agreements allow, additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Janowitz T., Williams E.H., Marshall A., Ainsworth N., Thomas P.B., Sammut S.J., Shepherd S., White J., Mark P.B., Lynch A.G., et al. New model for estimating glomerular filtration rate in patients with cancer. J. Clin. Oncol. 2017;35:2798–2805. doi: 10.1200/JCO.2017.72.7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams E.H., Flint T.R., Connell C.M., Giglio D., Lee H., Ha T., Gablenz E., Bird N.J., Weaver J.M.J., Potts H., et al. CamGFR v2: a new model for estimating the glomerular filtration rate from standardized or non-standardized creatinine in patients with cancer. Clin. Cancer Res. 2021;27:1381–1390. doi: 10.1158/1078-0432.CCR-20-3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rehberg P.B. Studies on kidney function. Biochem. J. 1926;20:447–460. doi: 10.1042/bj0200447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevens L.A., Coresh J., Greene T., Levey A.S. Assessing kidney function — measured and estimated glomerular filtration rate. N. Engl. J. Med. 2006;354:2473–2483. doi: 10.1056/nejmra054415. [DOI] [PubMed] [Google Scholar]
- 5.BUTLER E.A., FLYNN F.V. The occurrence of post-gamma protein in urine: a new protein abnormality. J. Clin. Pathol. 1961;14:172–178. doi: 10.1136/jcp.14.2.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Odden M.C., Scherzer R., Bacchetti P., Szczech L.A., Sidney S., Grunfeld C., Shlipak M.G. Cystatin C level as a marker of kidney function in human immunodeficiency virus infection: the FRAM study. Arch. Intern. Med. 2007;167:2213–2219. doi: 10.1001/archinte.167.20.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levey A.S. Measurement of renal function in chronic renal disease. Kidney Int. 1990;38:167–184. doi: 10.1038/ki.1990.182. [DOI] [PubMed] [Google Scholar]
- 8.Stevens L.A., Schmid C.H., Greene T., Li L., Beck G.J., Joffe M.M., Froissart M., Kusek J.W., Zhang Y.L., Coresh J., Levey A.S. Factors other than glomerular filtration rate affect serum cystatin C levels. Kidney Int. 2009;75:652–660. doi: 10.1038/ki.2008.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bökenkamp A., Domanetzki M., Zinck R., Schumann G., Byrd D., Brodehl J. Cystatin C serum concentrations underestimate glomerular filtration rate in renal transplant recipients. Clin. Chem. 1999 doi: 10.1093/clinchem/45.10.1866. [DOI] [PubMed] [Google Scholar]
- 10.STARZL T.E., MARCHIORO T.L., WADDELL W.R. The reversal of rejection in human renal homografts with subsequent development of homograft tolerance. Surg. Gynecol. Obstet. 1963;117:385–395. [PMC free article] [PubMed] [Google Scholar]
- 11.Risch L., Herklotz R., Blumberg A., Huber A.R. Effects of glucocorticoid immunosuppression on serum cystatin C concentrations in renal transplant patients. Clin. Chem. 2001;47:2055–2059. doi: 10.1093/clinchem/47.11.2055. [DOI] [PubMed] [Google Scholar]
- 12.Naka M., Kadoya M., Kosaka-Hamamoto K., Morimoto A., Miyoshi A., Kakutani M., Shoji T., Koyama H. Overestimation of glomerular filtration rate calculated from serum creatinine as compared with cystatin c in patients with subclinical hypercortisolism: hyogo adrenal metabolic registry. Endocr. J. 2020;67:469–476. doi: 10.1507/endocrj.EJ19-0478. [DOI] [PubMed] [Google Scholar]
- 13.Jones M., Denieffe S., Griffin C., Tinago W., Fitzgibbon M.C. Evaluation of cystatin C in malignancy and comparability of estimates of GFR in oncology patients. Pract. Lab. Med. 2017;8:95–104. doi: 10.1016/j.plabm.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwon W.S., Kim T.S., Nahm C.H., Moon Y., Kim J.J. Aberrant cystatin-C expression in blood from patients with breast cancer is a suitable marker for monitoring tumor burden. Oncol. Lett. 2018;16:5583–5590. doi: 10.3892/ol.2018.9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kos J., Krašovec M., Cimerman N., Nielsen H.J., Christensen I.J., Brünner N. Cysteine proteinase inhibitors stefin A, stefin B, and cystatin C in sera from patients with colorectal cancer: relation to Prognosis1. Clin. Cancer Res. 2000;6:505–511. [PubMed] [Google Scholar]
- 16.Jung C.-Y., Kim H.W., Han S.H., Yoo T.-H., Kang S.-W., Park J.T. Creatinine–cystatin C ratio and mortality in cancer patients: a retrospective cohort study. J. Cachexia Sarcopenia Muscle. 2022;13:2064–2072. doi: 10.1002/jcsm.13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lees J.S., Ho F., Parra-Soto S., Celis-Morales C., Welsh P., Sullivan M.K., Jani B.D., Sattar N., Lang N.N., Pell J.P., et al. Kidney function and cancer risk: an analysis using creatinine and cystatin C in a cohort study. eClinicalMedicine. 2021;38:101030. doi: 10.1016/j.eclinm.2021.101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bjarnadóttir M., Grubb A., Ólafsson Í. Promoter-mediated, dexamethasone-induced increase in cystatin c production by hela cells. Scand. J. Clin. Lab. Invest. 1995 doi: 10.3109/00365519509110261. [DOI] [PubMed] [Google Scholar]
- 19.Zhu X.R., Ge N., Wang Y., Zhai J.L., Liu C. Corticosteroids significantly increase cystatin C levels in the plasma by promoting cystatin C production in rats. Ren. Fail. 2019;41:698–703. doi: 10.1080/0886022X.2019.1638798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhai J.-L., Ge N., Zhen Y., Zhao Q., Liu C. Corticosteroids significantly increase serum cystatin C concentration without affecting renal function in symptomatic heart failure. Clin. Lab. 2016;62:203–207. doi: 10.7754/clin.lab.2015.150701. [DOI] [PubMed] [Google Scholar]
- 21.Pöge U., Gerhardt T., Bökenkamp A., Stoffel-Wagner B., Klehr H.-U., Sauerbruch T., Woitas R.P. Time course of low molecular weight proteins in the early kidney transplantation period—influence of corticosteroids. Nephrol. Dial. Transplant. 2004;19:2858–2863. doi: 10.1093/ndt/gfh341. [DOI] [PubMed] [Google Scholar]
- 22.Mason H.L., Hoehn W.M., Kendall E.C. Chemical studies of supra-renal cortex iv structure of compounds C-D-E-F and G. J. Biol. Chem. 1938;124:459–474. [Google Scholar]
- 23.Weitzman E.D., Fukushima D., Nogeire C., Roffwarg H., Gallagher T.F., Hellman L. Twenty-four hour pattern of the episodic secretion of cortisol in normal subjects. J. Clin. Endocrinol. Metab. 1971;33:14–22. doi: 10.1210/jcem-33-1-14. [DOI] [PubMed] [Google Scholar]
- 24.Reddy T.E., Pauli F., Sprouse R.O., Neff N.F., Newberry K.M., Garabedian M.J., Myers R.M. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009;19:2163–2171. doi: 10.1101/gr.097022.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galon J., Franchimont D., Hiroi N., Frey G., Boettner A., Ehrhart-Bornstein M., O’shea J.J., Chrousos G.P., Bornstein S.R. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. Faseb. J. 2002;16:61–71. doi: 10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- 26.Oelkers W. Current concepts: adrenal insufficiency. N. Engl. J. Med. 1996;335:1206–1212. doi: 10.1056/NEJM199610173351607. [DOI] [PubMed] [Google Scholar]
- 27.Hench P.S., Kendall E.C. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone; compound E) and of pituitary adrenocorticotropic hormone on rheumatoid arthritis. Proc. Staff Meet. Mayo Clin. 1949;24:181–197. [PubMed] [Google Scholar]
- 28.Brahmer J.R., Lacchetti C., Schneider B.J., Atkins M.B., Brassil K.J., Caterino J.M., Chau I., Ernstoff M.S., Gardner J.M., Ginex P., et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American society of clinical oncology clinical practice guideline. J. Clin. Oncol. 2018;36:1714–1768. doi: 10.1200/JCO.2017.77.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connell C.M., Raby S., Beh I., Flint T.R., Williams E.H., Fearon D.T., Jodrell D.I., Janowitz T. Cancer immunotherapy trial registrations increase exponentially but chronic immunosuppressive glucocorticoid therapy may compromise outcomes. Ann. Oncol. 2017;28:1678–1679. doi: 10.1093/annonc/mdx181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flint T.R., Janowitz T., Connell C.M., Roberts E.W., Denton A.E., Coll A.P., Jodrell D.I., Fearon D.T. Tumor-induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metabol. 2016;24:672–684. doi: 10.1016/j.cmet.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obradović M.M.S., Hamelin B., Manevski N., Couto J.P., Sethi A., Coissieux M.M., Münst S., Okamoto R., Kohler H., Schmidt A., et al. Glucocorticoids promote breast cancer metastasis. Nature. 2019 doi: 10.1038/s41586-019-1019-4. [DOI] [PubMed] [Google Scholar]
- 32.Janowitz T., Kleeman S., Vonderheide R.H. Reconsidering dexamethasone for antiemesis when combining chemotherapy and immunotherapy. Oncol. 2021;26:269–273. doi: 10.1002/onco.13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grotzinger A.D., Rhemtulla M., de Vlaming R., Ritchie S.J., Mallard T.T., Hill W.D., Ip H.F., Marioni R.E., McIntosh A.M., Deary I.J., et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat. Human Behav. 2019;3:513–525. doi: 10.1038/s41562-019-0566-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeo N.C., O’Meara C.C., Bonomo J.A., Veth K.N., Tomar R., Flister M.J., Drummond I.A., Bowden D.W., Freedman B.I., Lazar J., et al. Shroom3 contributes to the maintenance of the glomerular filtration barrier integrity. Genome Res. 2015;25:57–65. doi: 10.1101/gr.182881.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hart T.C., Gorry M.C., Hart P.S., Woodard A.S., Shihabi Z., Sandhu J., Shirts B., Xu L., Zhu H., Barmada M.M., Bleyer A.J. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J. Med. Genet. 2002;39:882–892. doi: 10.1136/jmg.39.12.882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smyth D.J., Plagnol V., Walker N.M., Cooper J.D., Downes K., Yang J.H.M., Howson J.M.M., Stevens H., McManus R., Wijmenga C., et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N. Engl. J. Med. 2008;359:2767–2777. doi: 10.1056/nejmoa0807917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saevarsdottir S., Olafsdottir T.A., Ivarsdottir E.V., Halldorsson G.H., Gunnarsdottir K., Sigurdsson A., Johannesson A., Sigurdsson J.K., Juliusdottir T., Lund S.H., et al. FLT3 stop mutation increases FLT3 ligand level and risk of autoimmune thyroid disease. Nature. 2020;584:619–623. doi: 10.1038/s41586-020-2436-0. [DOI] [PubMed] [Google Scholar]
- 38.Zhernakova A., Elbers C.C., Ferwerda B., Romanos J., Trynka G., Dubois P.C., de Kovel C.G.F., Franke L., Oosting M., Barisani D., et al. Evolutionary and functional analysis of celiac risk loci reveals SH2B3 as a protective factor against bacterial infection. Am. J. Hum. Genet. 2010;86:970–977. doi: 10.1016/j.ajhg.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shin S.Y., Fauman E.B., Petersen A.K., Krumsiek J., Santos R., Huang J., Arnold M., Erte I., Forgetta V., Yang T.P., et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014;46:543–550. doi: 10.1038/ng.2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park M.J., D’Alecy L.G., Anderson M.A., Basrur V., Feng Y., Brady G.F., Kim D.I., Lahann J., et al. Constitutive release of CPS1 in bile and its role as a protective cytokine during acute liver injury. Proc. Natl. Acad. Sci. USA. 2019;116:9125–9134. doi: 10.1073/pnas.1822173116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carithers L.J., Moore H.M. The genotype-tissue expression (GTEx) project. Biopreserv. Biobanking. 2015;13:307–308. doi: 10.1089/bio.2015.29031.hmm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roadmap Epigenomics Consortium. Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ginès P., Solà E., Angeli P., Wong F., Nadim M.K., Kamath P.S. Hepatorenal syndrome. Nat. Rev. Dis. Prim. 2018;4:23. doi: 10.1038/s41572-018-0022-7. [DOI] [PubMed] [Google Scholar]
- 44.Privé F., Arbel J., Vilhjálmsson B.J. LDpred2: better, faster, stronger. Bioinformatics. 2020 doi: 10.1093/bioinformatics/btaa1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Diao J.A., Inker L.A., Levey A.S., Tighiouart H., Powe N.R., Manrai A.K. In search of a better equation — performance and equity in estimates of kidney function. N. Engl. J. Med. 2021;384:396–399. doi: 10.1056/nejmp2028243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emerging Risk Factors Collaboration. Kaptoge S., Di Angelantonio E., Lowe G., Pepys M.B., Thompson S.G., Collins R., Danesh J., Ford C.E., Pressel S.L., et al. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375:132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ganna A., Ingelsson E. 5 year mortality predictors in 498 103 UK Biobank participants: a prospective population-based study. Lancet. 2015;386:533–540. doi: 10.1016/S0140-6736(15)60175-1. [DOI] [PubMed] [Google Scholar]
- 48.Lees J.S., Welsh C.E., Celis-Morales C.A., Mackay D., Lewsey J., Gray S.R., Lyall D.M., Cleland J.G., Gill J.M.R., Jhund P.S., et al. Glomerular filtration rate by differing measures, albuminuria and prediction of cardiovascular disease, mortality and end-stage kidney disease. Nat. Med. 2019;25:1753–1760. doi: 10.1038/s41591-019-0627-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleeman S.O., Cordioli M., Timmers P.R.H.J., Khan A., Tober-Lau P., Kurth F., Demichev V., Meyer H.V., Wilson J.F., Ralser M., et al. Cystatin C is associated with adverse COVID-19 outcomes in diverse populations. iScience. 2022;25:105040. doi: 10.1016/j.isci.2022.105040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crawford A.A., Bankier S., Altmaier E., Barnes C.L.K., Clark D.W., Ermel R., Friedrich N., van der Harst P., Joshi P.K., Karhunen V., et al. Variation in the SERPINA6/SERPINA1 locus alters morning plasma cortisol, hepatic corticosteroid binding globulin expression, gene expression in peripheral tissues, and risk of cardiovascular disease. J. Hum. Genet. 2021;66:625–636. doi: 10.1038/s10038-020-00895-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giambartolomei C., Vukcevic D., Schadt E.E., Franke L., Hingorani A.D., Wallace C., Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franzén O., Ermel R., Cohain A., Akers N.K., Di Narzo A., Talukdar H.A., Foroughi-Asl H., Giambartolomei C., Fullard J.F., Sukhavasi K., et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science. 2016:eaad6970. doi: 10.1126/science.aad6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Glastonbury C.A., Couto Alves A., El-Sayed Moustafa J.S., Small K.S. Cell-type heterogeneity in adipose tissue is associated with complex traits and reveals disease-relevant cell-specific eQTLs. Am. J. Hum. Genet. 2019;104:1013–1024. doi: 10.1016/j.ajhg.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inker L.A., Schmid C.H., Tighiouart H., Eckfeldt J.H., Feldman H.I., Greene T., Kusek J.W., Manzi J., Van Lente F., Zhang Y.L., et al. Estimating glomerular filtration rate from serum creatinine and cystatin C. N. Engl. J. Med. 2012;367:20–29. doi: 10.1056/NEJMoa1114248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Madissoon E., Wilbrey-Clark A., Miragaia R.J., Saeb-Parsy K., Mahbubani K.T., Georgakopoulos N., Harding P., Polanski K., Huang N., Nowicki-Osuch K., et al. ScRNA-seq assessment of the human lung, spleen, and esophagus tissue stability after cold preservation. Genome Biol. 2019;21:1. doi: 10.1186/s13059-019-1906-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilk A.J., Rustagi A., Zhao N.Q., Roque J., Martínez-Colón G.J., McKechnie J.L., Ivison G.T., Ranganath T., Vergara R., Hollis T., et al. A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 2020;26:1070–1076. doi: 10.1038/s41591-020-0944-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., et al. Tissue-based map of the human proteome. Science. 2015;80 doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 58.Võsa U., Claringbould A., Westra H.J., Bonder M.J., Deelen P., Zeng B., Kirsten H., Saha A., Kreuzhuber R., Kasela S., et al. Unraveling the polygenic architecture of complex traits using blood eQTL meta-analysis. bioRxiv. 2018 doi: 10.1101/447367. Preprint at. [DOI] [Google Scholar]
- 59.Chantzichristos D., Svensson P.-A., Garner T., Glad C.A., Walker B.R., Bergthorsdottir R., Ragnarsson O., Trimpou P., Stimson R.H., Borresen S.W., et al. Identification of human glucocorticoid response markers using integrated multi-omic analysis from a randomized crossover trial. Elife. 2021;10:e62236. doi: 10.7554/eLife.62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Riemondy K.A., Fu R., Gillen A.E., Sheridan R.M., Tian C., Daya M., Hao Y., Hesselberth J.R. clustifyr: an R package for automated single-cell RNA sequencing cluster classification. F1000Research. 2020 doi: 10.12688/f1000research.22969.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ley K., Laudanna C., Cybulsky M.I., Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 62.Diaz-Jimenez D., Petrillo M.G., Busada J.T., Hermoso M.A., Cidlowski J.A. Glucocorticoids mobilize macrophages by transcriptionally up-regulating the exopeptidase DPP4. J. Biol. Chem. 2020;295:3213–3227. doi: 10.1074/jbc.RA119.010894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nasser J., Bergman D.T., Fulco C.P., Guckelberger P., Doughty B.R., Patwardhan T.A., Jones T.R., Nguyen T.H., Ulirsch J.C., Lekschas F., et al. Genome-wide enhancer maps link risk variants to disease genes. Nature. 2021;593:238–243. doi: 10.1038/s41586-021-03446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoyer A. Springer; 2017. Metaanalysis with R. G.Schwarzer, J. R.Carpenter, G.Rücker (2015) Biom. J. [DOI] [Google Scholar]
- 65.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 2020;26:842–844. doi: 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 66.The RECOVERY Collaborative Group Dexamethasone in hospitalized patients with Covid-19. N. Engl. J. Med. 2021;384:693–704. doi: 10.1056/NEJMoa2021436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sinha S., Rosin N.L., Arora R., Labit E., Jaffer A., Cao L., Farias R., Nguyen A.P., de Almeida L.G.N., Dufour A., et al. Dexamethasone modulates immature neutrophils and interferon programming in severe COVID-19. Nat. Med. 2022;28:201–211. doi: 10.1038/s41591-021-01576-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Messner C.B., Demichev V., Wendisch D., Michalick L., White M., Freiwald A., Textoris-Taube K., Vernardis S.I., Egger A.-S., Kreidl M., et al. Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst. 2020;11:11–24.e4. doi: 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weisberg S.P., McCann D., Desai M., Rosenbaum M., Leibel R.L., Ferrante A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003;112:1796–1808. doi: 10.1172/jci19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., Khodadoust M.S., Esfahani M.S., Luca B.A., Steiner D., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37:773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van der Wijst M.G.P., de Vries D.H., Groot H.E., Trynka G., Hon C.C., Bonder M.J., Stegle O., Nawijn M.C., Idaghdour Y., van der Harst P., et al. The single-cell eQTLGen consortium. Elife. 2020;9:e52155. doi: 10.7554/eLife.52155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Acharya N., Madi A., Zhang H., Klapholz M., Escobar G., Dulberg S., Christian E., Ferreira M., Dixon K.O., Fell G., et al. Endogenous glucocorticoid signaling regulates CD8+ T cell differentiation and development of dysfunction in the tumor microenvironment. Immunity. 2020;53:658–671.e6. doi: 10.1016/j.immuni.2020.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hingorani S.R., Wang L., Multani A.S., Combs C., Deramaudt T.B., Hruban R.H., Rustgi A.K., Chang S., Tuveson D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 74.Barrett A.J., Davies M.E., Grubb A. The place of human γ-trace (cystatin C) amongst the cysteine proteinase inhibitors. Biochem. Biophys. Res. Commun. 1984;120:631–636. doi: 10.1016/0006-291X(84)91302-0. [DOI] [PubMed] [Google Scholar]
- 75.Kitamura H., Kamon H., Sawa S.-I., Park S.-J., Katunuma N., Ishihara K., Murakami M., Hirano T. IL-6-STAT3 controls intracellular MHC class II alphabeta dimer level through cathepsin S activity in dendritic cells. Immunity. 2005;23:491–502. doi: 10.1016/j.immuni.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 76.Phipson B., Sim C.B., Porrello E.R., Hewitt A.W., Powell J., Oshlack A. propeller: testing for differences in cell type proportions in single cell data. bioRxiv. 2021 doi: 10.1101/2021.11.28.470236. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chan G., White C.C., Winn P.A., Cimpean M., Replogle J.M., Glick L.R., Cuerdon N.E., Ryan K.J., Johnson K.A., Schneider J.A., et al. CD33 modulates TREM2: convergence of Alzheimer loci. Nat. Neurosci. 2015;18:1556–1558. doi: 10.1038/nn.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Alquicira-Hernandez J., Powell J.E. Nebulosa recovers single-cell gene expression signals by kernel density estimation. Bioinformatics. 2021;37:2485–2487. doi: 10.1093/bioinformatics/btab003. [DOI] [PubMed] [Google Scholar]
- 79.Katzenelenbogen Y., Sheban F., Yalin A., Yofe I., Svetlichnyy D., Jaitin D.A., Bornstein C., Moshe A., Keren-Shaul H., Cohen M., et al. Coupled scRNA-Seq and intracellular protein activity reveal an immunosuppressive role of TREM2 in cancer. Cell. 2020;182:872–885.e19. doi: 10.1016/j.cell.2020.06.032. [DOI] [PubMed] [Google Scholar]
- 80.Binnewies M., Pollack J.L., Rudolph J., Dash S., Abushawish M., Lee T., Jahchan N.S., Canaday P., Lu E., Norng M., et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 2021;37 doi: 10.1016/j.celrep.2021.109844. [DOI] [PubMed] [Google Scholar]
- 81.Molgora M., Esaulova E., Vermi W., Hou J., Chen Y., Luo J., Brioschi S., Bugatti M., Omodei A.S., Ricci B., et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell. 2020;182:886–900.e17. doi: 10.1016/j.cell.2020.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yost K.E., Satpathy A.T., Wells D.K., Qi Y., Wang C., Kageyama R., McNamara K.L., Granja J.M., Sarin K.Y., Brown R.A., et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019;25:1251–1259. doi: 10.1038/s41591-019-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bökenkamp A., van Wijk J.A., Lentze M.J., Stoffel-Wagner B. Effect of corticosteroid therapy on serum cystatin C and β2-microglobulin concentrations. Clin. Chem. 2002;48:1123–1126. doi: 10.1093/clinchem/48.7.1123. [DOI] [PubMed] [Google Scholar]
- 84.Walker B.R. Cortisol - cause and cure for metabolic syndrome? Diabet. Med. 2006;23:1281–1288. doi: 10.1111/j.1464-5491.2006.01998.x. [DOI] [PubMed] [Google Scholar]
- 85.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 86.Huh C.G., Håkansson K., Nathanson C.M., Thorgeirsson U.P., Jonsson N., Grubb A., Abrahamson M., Karlsson S. Decreased metastatic spread in mice homozygous for a null allele of the cystatin C protease inhibitor gene. Mol. Pathol. 1999;52:332–340. doi: 10.1136/mp.52.6.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pencheva N., Tran H., Buss C., Huh D., Drobnjak M., Busam K., Tavazoie S.F. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell. 2012;151:1068–1082. doi: 10.1016/j.cell.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Atagi Y., Liu C.-C., Painter M.M., Chen X.-F., Verbeeck C., Zheng H., Li X., Rademakers R., Kang S.S., Xu H., et al. Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2) J. Biol. Chem. 2015;290:26043–26050. doi: 10.1074/jbc.M115.679043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Krasemann S., Madore C., Cialic R., Baufeld C., Calcagno N., El Fatimy R., Beckers L., O’Loughlin E., Xu Y., Fanek Z., et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–581.e9. doi: 10.1016/j.immuni.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tudorache I.F., Trusca V.G., Gafencu A.V. Apolipoprotein E - a multifunctional protein with implications in various pathologies as a result of its structural features. Comput. Struct. Biotechnol. J. 2017;15:359–365. doi: 10.1016/j.csbj.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Byers D.E., Wu K., Dang-Vu G., Jin X., Agapov E., Zhang X., Battaile J.T., Schechtman K., Yusen R., Pierce R.A., Holtzman M.J. Triggering receptor expressed on myeloid cells-2 expression tracks with M2-like macrophage activity and disease severity in COPD. Chest. 2018;153:77–86. doi: 10.1016/j.chest.2017.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang Y., Cella M., Mallinson K., Ulrich J.D., Young K.L., Robinette M.L., Gilfillan S., Krishnan G.M., Sudhakar S., Zinselmeyer B.H., et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J.S.K., Younkin S., et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sun B., Zhou Y., Halabisky B., Lo I., Cho S.-H., Mueller-Steiner S., Devidze N., Wang X., Grubb A., Gan L. Cystatin C-cathepsin B Axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron. 2008;60:247–257. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Richardson T.G., Harrison S., Hemani G., Davey Smith G. An atlas of polygenic risk score associations to highlight putative causal relationships across the human phenome. Elife. 2019;8 doi: 10.7554/eLife.43657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jerby-Arnon L., Shah P., Cuoco M.S., Rodman C., Su M.J., Melms J.C., Leeson R., Kanodia A., Mei S., Lin J.R., et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell. 2018;175:984–997.e24. doi: 10.1016/j.cell.2018.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cao Y., Bender I.K., Konstantinidis A.K., Shin S.C., Jewell C.M., Cidlowski J.A., Schleimer R.P., Lu N.Z. Glucocorticoid receptor translational isoforms underlie maturational stage-specific glucocorticoid sensitivities of dendritic cells in mice and humans. Blood. 2013;121:1553–1562. doi: 10.1182/blood-2012-05-432336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Franzén O., Gan L.M., Björkegren J.L.M. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database. 2019 doi: 10.1093/database/baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Anderson C.A., Pettersson F.H., Clarke G.M., Cardon L.R., Morris A.P., Zondervan K.T. Data quality control in genetic case-control association studies. Nat. Protoc. 2010;5:1564–1573. doi: 10.1038/nprot.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sayaman R.W., Saad M., Thorsson V., Hu D., Hendrickx W., Roelands J., Porta-Pardo E., Mokrab Y., Farshidfar F., Kirchhoff T., et al. Germline genetic contribution to the immune landscape of cancer. Immunity. 2021;54:367–386.e8. doi: 10.1016/j.immuni.2021.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim S., Scheffler K., Halpern A.L., Bekritsky M.A., Noh E., Källberg M., Chen X., Kim Y., Beyter D., Krusche P., Saunders C.T. Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods. 2018;15:591–594. doi: 10.1038/s41592-018-0051-x. [DOI] [PubMed] [Google Scholar]
- 104.Loh P.R., Kichaev G., Gazal S., Schoech A.P., Price A.L. Mixed-model association for biobank-scale datasets. Nat. Genet. 2018;50:906–908. doi: 10.1038/s41588-018-0144-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ghoussaini M., Mountjoy E., Carmona M., Peat G., Schmidt E.M., Hercules A., Fumis L., Miranda A., Carvalho-Silva D., Buniello A., et al. Open Targets Genetics: systematic identification of trait-associated genes using large-scale genetics and functional genomics. Nucleic Acids Res. 2021;49:D1311–D1320. doi: 10.1093/nar/gkaa840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Finucane H.K., Bulik-Sullivan B., Gusev A., Trynka G., Reshef Y., Loh P.R., Anttila V., Xu H., Zang C., Farh K., et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015;47:1228–1235. doi: 10.1038/ng.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.de Leeuw C.A., Mooij J.M., Heskes T., Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhu Z., Zhang F., Hu H., Bakshi A., Robinson M.R., Powell J.E., Montgomery G.W., Goddard M.E., Wray N.R., Visscher P.M., Yang J. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat. Genet. 2016;48:481–487. doi: 10.1038/ng.3538. [DOI] [PubMed] [Google Scholar]
- 109.Gel B., Serra E. KaryoploteR: an R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics. 2017;33:3088–3090. doi: 10.1093/bioinformatics/btx346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Risso D., Schwartz K., Sherlock G., Dudoit S. GC-content normalization for RNA-seq data. BMC Bioinf. 2011;12:480. doi: 10.1186/1471-2105-12-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Qi J., Asl H.F., Björkegren J., Michoel T. KruX: matrix-based non-parametric eQTL discovery. BMC Bioinf. 2014;15:11. doi: 10.1186/1471-2105-15-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Tickle T., Tirosh I., Georgescu C., Brown M.,H.B. Broad Inst. MIT Harvard; 2019. inferCNV of the trinity CTAT project. Klarman Cell Obs. [Google Scholar]
- 115.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Squair J.W., Gautier M., Kathe C., Anderson M.A., James N.D., Hutson T.H., Hudelle R., Qaiser T., Matson K.J.E., Barraud Q., et al. Confronting false discoveries in single-cell differential expression. Nat. Commun. 2021;12:5692. doi: 10.1038/s41467-021-25960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Due to the data use agreements for the datasets analyzed in this manuscript, we are unable to directly share or distribute any patient-level data except for COVID-19 patient scRNA-seq (reposited at https://doi.org/10.6084/m9.figshare.14330795.v13, filename ‘covid.combined_final.CD14.Mono.Robj’). GWAS summary statistics are published alongside the study (, and polygenic scores are deposited in PGS Catalog (https://doi.org/10.1101/2021.08.17.21261668). To facilitate dataset requests from applicable data use committees, we provide all accession codes for all datasets relating to this manuscript in the key resources table and in Table S2A. UK Biobank data can be requested through the application process detailed at https://www.ukbiobank.ac.uk/.
-
•
Single-cell RNA sequencing raw sequencing data (FASTQ) is reposited in the Sequence Read Archive (SRA) under BioProject PRJNA961746, while the processed Seurat matrix is available from Figshare (https://doi.org/10.6084/m9.figshare.20063402).
-
•
Digital pathology derived from histological and immunohistochemical analyses of mouse tumor sections (Mm1 model) has been deposited at https://data.mendeley.com/datasets/kcwn7bpdf9/1.
-
•
Code to reproduce core computational and statistical analyses has been reposited on Github at https://github.com/Janowitz-Lab/cystatinc (https://doi.org/10.5281/zenodo.7921111).
-
•
Where data use agreements allow, additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.