

Abstract
INTRODUCTION:
We investigated associations of obesity with the expression of Alzheimer’s disease (AD)-related genes in a large community-based cohort.
METHODS:
The sample consisted of 5,619 participants from the Framingham Heart Study. Obesity metrics included Body mass index (BMI) and Waist-to-Hip Ratio (WHR). Gene expression was measured for a set of 74 AD-related genes, derived by integrating GWAS results with functional genomics data.
RESULTS:
Obesity metrics were associated with the expression of 21 AD-related genes. The strongest associations were observed with CLU, CD2AP, KLC3 and FCER1G. Unique associations were noted with TSPAN14, SLC24A4 for BMI and ZSCAN21, BCKDK for WHR. After adjustment for cardiovascular risk factors, 13 associations remained significant for BMI and 8 for WHR. Dichotomous obesity metrics exhibited unique associations with EPHX2 for BMI, and with TSPAN14 for WHR.
DISCUSSION:
Obesity was associated with AD-related gene expression; these findings shed light on the molecular pathways linking obesity to AD.
Keywords: Alzheimer’s disease, obesity, gene expression, dementia, GWAS, functional genomics
1. Introduction
The number of people that live with dementia is estimated to reach 131 million by 2050. This projection has momentous social and financial implications for patients, caregivers and healthcare systems.1 Alzheimer’s disease (AD) is the most common type of dementia.2 Ongoing research efforts, have expanded our comprehension of AD pathogenesis beyond the fundamental amyloid cascade and tau hyperphosphorylation hypotheses,3 to include biological pathways associated with immunity,4 inflammation,5 and lipid metabolism.6
Similarly, obesity is a growing public health concern. In the United States, up to 85% of adults are projected to be overweight or obese by 2030.7 Obesity has been linked to late-onset AD.8, 9 It has been hypothesized that obesity creates an environment of chronic low-grade systemic inflammation and increased oxidative stress that can contribute to neurodegeneration.10 Furthermore, neurobiological changes related to vascular pathology, often coexist with AD pathology and possibly have an additive or synergistic effect on cognitive decline.11 Therefore, since obesity is a known risk factor for type 2 diabetes, heart disease, and stroke,7 it might contribute to dementia risk by increasing the burden of vascular pathology in the brain. Nevertheless, the exact biological substrate of the relationship between obesity and AD has yet to be clarified.
One potential pathway linking obesity to AD may involve molecular changes in gene expression.12 Recent investigations have identified several genetic variants associated with AD through Genome-Wide Association Studies (GWAS).13 However, whether obesity is associated with differential expression of AD-related genes during the life course, remains unknown.
The present study aimed to shed light on the molecular links between obesity and AD by investigating associations of obesity metrics with expression profiles of AD-related genes in a large community-based cohort. Moreover, considering prior evidence that obesity during midlife is a stronger predictor of dementia,8 and that the underlying biology linking obesity to dementia might be affected by sex-related differences,14 potential effect modification by age and sex was explored.
2. Methods
2.1. Participants
The Framingham Heart Study (FHS) is a community-based, longitudinal cohort study initiated in 1948 to investigate the risk factors associated with cardiovascular disease. Since its inception, the study has followed three generations of participants who are invited for follow-up examinations every two to six years.15–17 The present cross-sectional analysis included 5,619 non-Hispanic White participants with whole-blood gene expression profiling from the Offspring16 and Third Generation15 cohorts of the FHS,17 who attended the 8th (2005–2008) and 2nd examination cycles (2008–2011), respectively, and had available data on obesity metrics. All participants have provided written informed consent. Study protocols and consent forms have been approved by the institutional review board of the Boston University Medical Center.
2.2. Selection of AD-related genes and expression profiling
To functionally characterize and prioritize individual AD-related genetic risk loci, we performed transcriptome-wide association studies (TWAS) using TWAS-Fusion.18 The descriptive statistics from the AD SNP-main effects of the most recent large-scale meta-analyses of AD GWAS,13 and precomputed SNP-expression weights from 22 gene expression reference panels from blood tissue Netherlands-Twin-Registry [NTR]; Young-Finns-Study, YFS; Genotype-Tissue-Expression [GTEx]) were used (Suppl. Methods 1 in supporting information). To rule out that potential associations might reflect the random overlap between expression quantitative trait loci (eQTLs) and non-causal AD risk variants, colocalization analysis (COLOC) was performed at each significant locus,19 to estimate the posterior probability of a shared causal variant (PP4≥0.75) between gene expression and trait association, using a prior probability of 1.1×10−5 for the AD association. Functional validation of the eGenes was performed by testing for positional overlap of the best eQTLs from TWAS with enhancer (H3K4me1) and/or promoter (H3K4me3) regions across a broad category of relevant tissue types (blood, brain, heart) using Haploreg v4.1 software.20 Transcription-wide significant eGene-eQTL pairs with either high colocalization probability (≥0.75), or evidence of regulatory epigenome overlap in at least one tissue, were retained for studying their association with obesity metrics. Moreover, accounting for the pairwise correlation between gene expression features, we conducted the multiple degree-of-freedom omnibus analysis to test for shared effects of eGenes across the gene reference panels; a significance threshold of p=5.3*10−6 was set to identify eGenes functional across multiple reference panels, after Bonferroni correction for the total number of genes (N=9,435) tested. Then, the final set of AD-related genes was derived by combining eGenes satisfying TWAS selection criteria with eGenes reaching the significance threshold in omnibus analysis. Finally, the APP, PSEN1, PSEN2, and APOE genes (that were either not included in the GWAS summary statistics [APOE, PSEN2],13 or not identified as high priority loci [APP, PSEN1]), were added in the final set, considering their association with AD risk.21, 22 The methodology followed for gene expression profiling has been described elsewhere.23
2.3. Obesity metrics
The Body Mass Index (BMI) was used as a measure of generalized obesity, and the Waist to Hip Ratio (WHR) as a measure of abdominal obesity. Although BMI is the most commonly used indicator of obesity, we also included WHR, based on prior evidence suggesting it might be a better predictor of health-related outcomes, including the risk for cardiovascular disease, stroke, diabetes, and overall mortality.24
BMI was calculated by dividing the participant’s weight by height squared (kg/m2); generalized obesity was defined as BMI≥30.0kg/m2.25 WHR was calculated by dividing the participant’s waist circumference (cm) by hip circumference (cm); abdominal obesity was defined as a WHR>0.90 for males and >0.85 for females, based on WHO guidelines.25
2.4. Cardiovascular risk factors
Blood pressure was calculated as the average of two measurements obtained 10 minutes apart with the participant resting in a sitting position. Laboratory values such as fasting blood glucose, total cholesterol (TC), high-density lipoprotein (HDL) cholesterol, and triglycerides were measured using standard assays.
Cardiovascular risk factors (systolic blood pressure, smoking, diabetes mellitus, and history of cardiovascular disease) were defined in accordance with the Framingham Stroke Risk Profile (Suppl. Methods 2 in supporting information).26
2.5. Lifestyle factors
Adherence to potentially neuroprotective dietary habits was assessed by calculating the 2015 Dietary Guidelines for Americans Adherence Index (DGAI) score,27 which reflects adherence to the Dietary Guidelines for Americans.28 Physical activity was self-reported using the physical activity index.29
2.6. Statistical analysis
Associations between different obesity metrics and AD-related gene expression were explored using linear mixed models with gene expression as the outcome. For each gene, two sets of models were constructed; one with BMI and the other with WHR as the main predictors. BMI and WHR were treated both as continuous as well as dichotomous measures (using the WHO-defined cutoffs for generalized obesity and abdominal obesity, respectively) to investigate for the potential presence of threshold effects. Familial relatedness was included as a random variance-covariance factor in the models. Confounder adjustment was conducted as follows: Model 1 was adjusted for age, sex, and differential cell counts; Model 2 was further adjusted for smoking, SBP, use of antihypertensive medications, diabetes, TC/HDL, and CVD.
Potential effect modification by age and sex was assessed by stratifying the participants into different groups based on age (<55 years of age, ≥55 years of age) and sex (males, females), and repeating the aforementioned analyses in the respective subgroups.
Exploratory analyses were performed by further adjusting Model 2 for dietary habits and physical activity (Model 3), to account for potential confounding effects of lifestyle factors on the observed relationships.
The significancy cutoff was set at 3.4*10−4 after Bonferroni correction for multiple hypotheses testing, accounting both for outcome multiplicity (the number of the different genes tested=74) as well as the number of different obesity metrics studied (BMI, WHR).
Statistical analyses were performed using SAS Software, version 9.4 (SAS Institute). Graphics were created using R (R Core Team, 2022). Statistical tests were two-tailed.
3. Results
3.1. Participant characteristics
Clinical and demographic characteristics of study participants, overall, as well as across different age and sex subgroups are presented in Table 1. Mean age was 55 years and 54% of study participants were women. Thirty-one percent of participants met the definition of obesity based on BMI, and 78% had excess WHR. The cohort had a relatively low prevalence of cardiovascular risk factors and CVD (<10%), whereas 33% of study participants reported use of antihypertensive medications. The vast majority of participants were cognitively healthy; only a small proportion had prevalent dementia (n=22, 0.4%).
Table 1:
Clinical and demographic characteristics of study participants.
| Total sample (N=5619) |
<55 years (n=2779) |
≥55 years (n=2840) |
Women (n=3041) |
Men (n=2578) |
|
|---|---|---|---|---|---|
| Age (years) | 55 ± 13 | 44 ± 7 | 66 ± 8 | 55 ± 14 | 55 ± 13 |
| Women | 3041 (54) | 1502 (54) | 1539 (54) | 3041 (100) | 0 (0) |
| BMI (Kg/m2) | 28.19 ± 5.68 | 27.88 ± 5.90 | 28.50 ± 5.44 | 27.46 ± 6.18 | 29.06 ± 4.89 |
| Obese (BMI ≥ 30 Kg/m2) | 1751 (31) | 818 (29) | 933 (33) | 839 (28) | 912 (35) |
| WHR | 0.94 ± 0.09 | 0.91 ± 0.08 | 0.96 ± 0.09 | 0.90 ± 0.09 | 0.98 ± 0.07 |
| Increased WHR | 4359 (78) | 1947 (70) | 2412 (85) | 2079 (68) | 2280 (88) |
| Current smoker (n%) | 530 (9) | 306 (11) | 224 (8) | 279 (9) | 251 (10) |
| Systolic blood pressure | 122 ± 17 | 115 ± 14 | 128 ± 17 | 119 ± 18 | 124 ± 15 |
| Anti-hypertensive medication | 1859 (33) | 357 (13) | 1502 (53) | 902 (30) | 957 (37) |
| Diabetes | 501 (9) | 102 (4) | 399 (14) | 210 (7) | 291 (11) |
| TC/HDL | 3.43 ± 1.12 | 3.41 ± 1.16 | 3.44 ± 1.09 | 3.10 ± 1.00 | 3.81 ± 1.14 |
| CVD | 431 (8) | 16 (0.6) | 415 (15) | 170 (6) | 261 (10) |
Values are presented as means ± SD or counts (relative frequencies) for continuous and categorical variables, respectively.
3.2. TWAS, COLOC, functional validation, and omnibus analysis
TWAS-Fusion identified 47 eGene-eQTL pairs with evidence for significant transcriptome-wide association with AD (Figure 1, Table S1 in supporting information). Colocalization was observed for 25 TWAS-significant eQTLs (53%). In functional validation, positional overlap with enhancer and/or promoter elements was noted for the majority (83%) of TWAS signals, including eQTLs with weaker colocalization probability (PP4<75%). A total of 32 genes satisfied TWAS selection criteria. An additional of 57 AD-related genes were identified by the omnibus analysis (Table S2 in supporting information). Overall, AD was associated with upregulated or downregulated expression of 89 genes (Table S3 in supporting information). After inclusion of the APP, PSEN1, PSEN2, and APOE genes, the final set consisted of 93 genes; expression profiles for 74 of those genes were available for analysis in the FHS.
Figure 1:
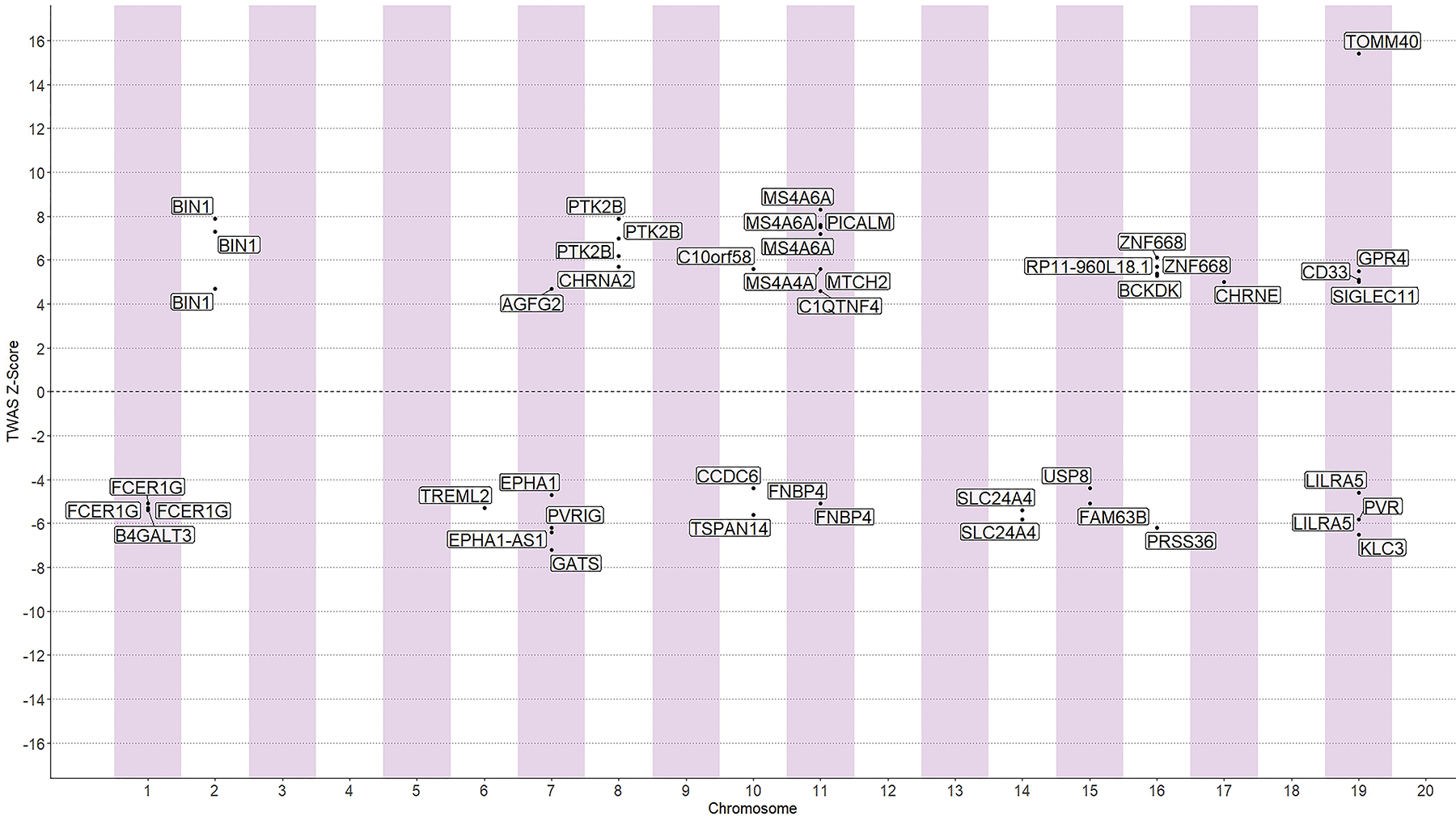
Summary of TWAS results. Each data point represents a gene grouped by chromosome (x-axis) and its respective z-score (y-axis), illustrating the direction of effect for expression of transcripts encoding AD-related genes. Only significant transcriptome-wide associations are presented.
3.3. Obesity metrics and expression profiles of AD-related genes
Overall, obesity metrics were associated with the expression profiles of 21 AD-related genes (Figure 2; Tables S4,S7 in supporting information). After adjustment for cardiovascular risk factors, 14 of those associations remained significant (Figure 3; Tables S5,S8 in supporting information).
Figure 2:
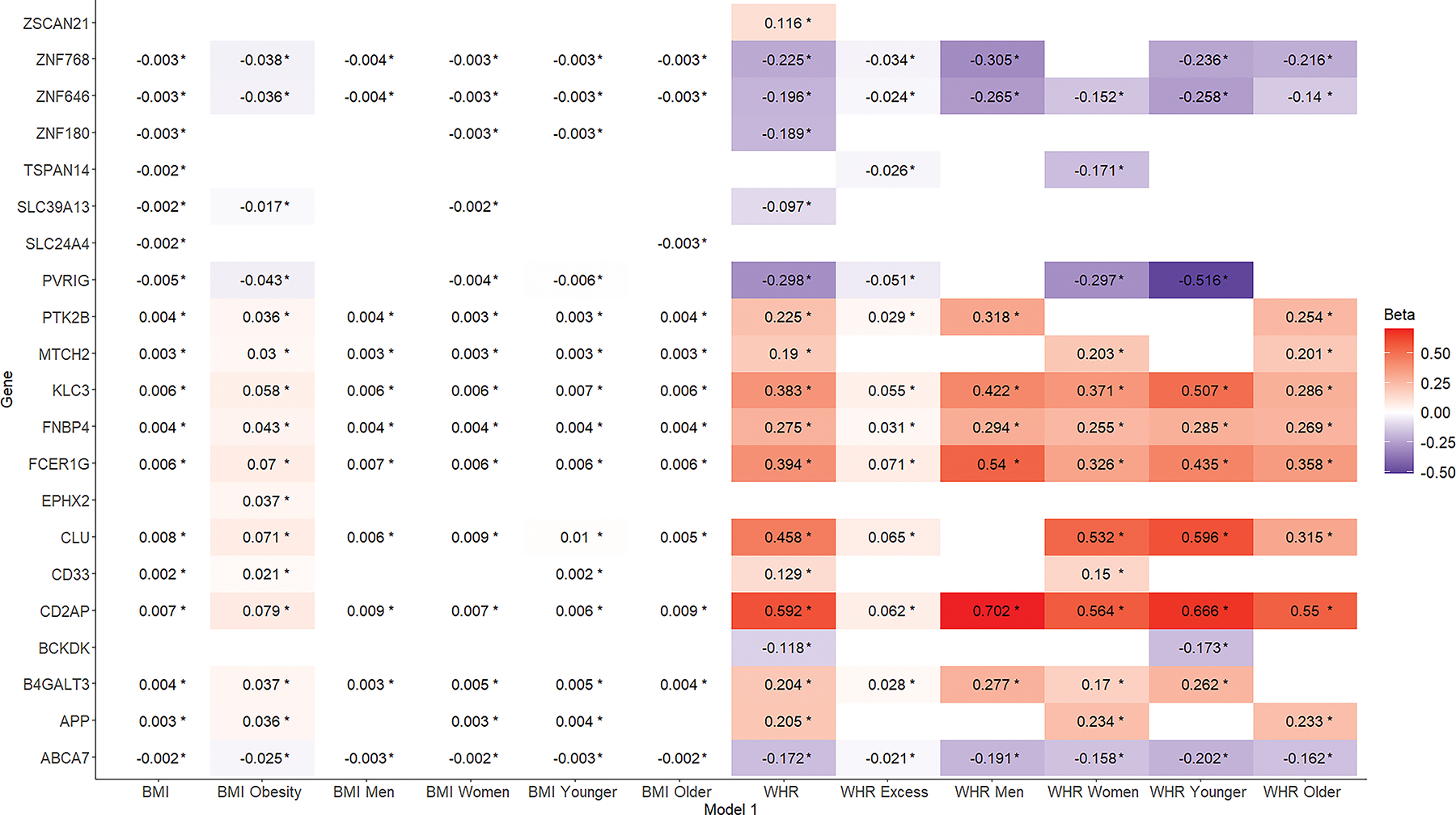
Associations of different obesity metrics (x-axis) with Alzheimer’s disease-related gene expression (y-axis). Results from linear mixed models with gene expression as the outcome and the different obesity metrics as the main predictors. Models are adjusted for age, sex, and differential cell counts. BMI, body mass index; WHR, waist-to-hip ratio.
Figure 3:
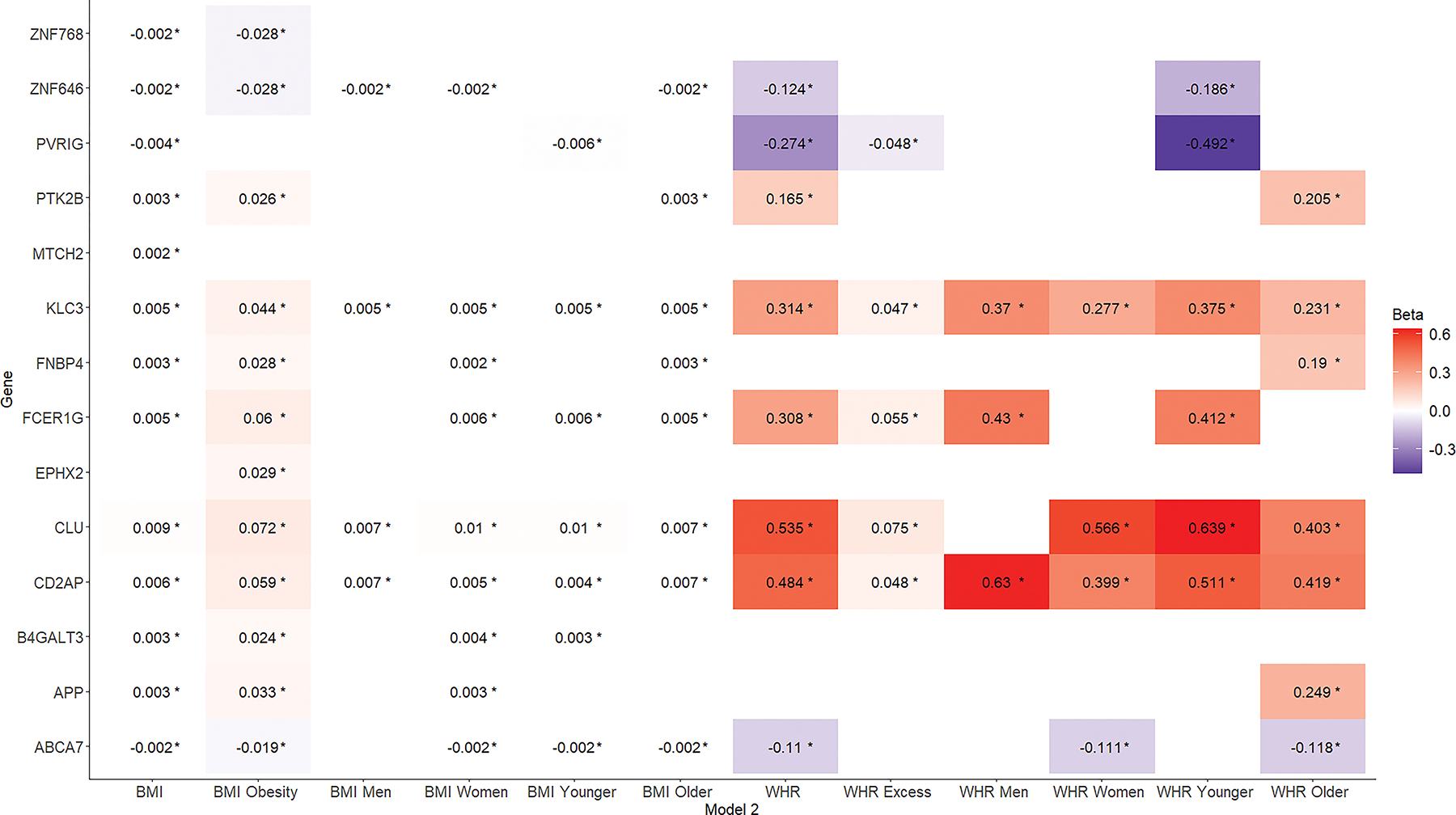
Associations of different obesity metrics (x-axis) with Alzheimer’s disease-related gene expression (y-axis). Results from linear mixed models with gene expression as the outcome and the different obesity metrics as the main predictors. Models are adjusted for age, sex, differential cell counts, and cardiovascular risk factors. BMI, body mass index; WHR, waist-to-hip ratio.
Linear mixed models with BMI and WHR as continuous measures, revealed significant associations with the expression of 20 AD-related genes (Figure S1 in supporting information). Associations with TSPAN14 and SLC24A4 were unique to the BMI set, and associations with ZSCAN21 and BCKDK were unique to the WHR set. Increased BMI and WHR were associated with increased expression of 10 and 11 genes, respectively, and with decreased expression of 8 and 7 genes, respectively. The strongest associations were observed with gene expression of CLU, CD2AP, FCER1G, and KLC3. No differences in the directionality of relationships between the BMI and WHR sets were observed.
Results from models with BMI and WHR as dichotomous measures (indicating generalized and abdominal obesity, respectively), are presented in Figure S2 in supporting information. Compared to models treating obesity as a continuous measure (Figure S3 in supporting information), one new association was identified for the BMI set (EPHX2), and one for the WHR set (TSPAN14). In contrast, 3 associations for the BMI set and 7 for the WHR set that were significant in the continuous models, became non-significant. Compared to models treating obesity as a continuous measure, the directionality of the overlapping relationships remained unchanged and the corresponding effect sizes were increased for the BMI set and decreased for the WHR set. Similar to the continuous models, the strongest associations were observed with gene expression of CLU, CD2AP, FCER1G, and KLC3.
After further adjustment for cardiovascular risk factors, 13 and 8 associations remained significant for the BMI and WHR sets, respectively (Figure S1). The directionality of the surviving relationships remained unchanged, with relatively small reductions in the corresponding effect estimates. Similar results were observed when obesity metrics were treated as dichotomous measures (Figure S2).
3.4. Effect modification by sex
Stratified analyses by sex, revealed that 4 out of the 18 identified associations between BMI and AD-related gene expression were only present in women (Figure S4A in supporting information). Furthermore, 3 associations, albeit present in the total sample, were not significant in the individual sex subgroups. In the models further adjusted for cardiovascular risk factors, 5 out of 13 associations were only present in women, and 4 associations were only present in the total sample but not in the individual sex subgroups; only 4 associations remained significant in men (Figure S4B). For the WHR set, 5 out of the 18 associations were only present in women, 2 were only present in men, one (TSPAN 14) was only present in women but not in the total sample or men, and 4 were only present in the total sample but not in the individual sex subgroups (Figure S5A in supporting information). Results from models further adjusted for cardiovascular risk factors are presented in Figure S5B.
3.5. Effect modification by age
In stratified analyses by age, 4 out of the 18 associations for the BMI set were present only in the younger subgroup, one was present only in the older subgroup, and 2 were present in the total sample but not in the individual age subgroups (Figure S6A in supporting information). Correspondingly, for the WHR set, 3 out of the 18 associations were present only in the younger subgroup, 3 only in the older subgroup, and 4 were only present in the total sample but not in the individual age subgroups (Figure S7A in supporting information). Further adjustment for cardiovascular risk factors, yielded similar findings, with the exception of the associations pertaining to the FNBP4 and APP genes in the WHR set, which albeit not present in the total sample, were significant in the older subgroup. (Figure S6B, S7B). Smaller effect sizes were noted in the older subgroup for the majority of overlapping relationships.
3.6. Exploratory analyses
Further adjustment for lifestyle factors led to almost identical findings (Figure S8; Tables S6,S9 in supporting information).
4. Discussion
In the present study, obesity was associated with the expression of 21 AD-related genes. These relationships were studied in a large sample of more than 5500 community-dwelling adults with a broad age range. After adjustment for cardiovascular risk factors, 14 associations remained significant, indicating that obesity might be modulating the expression of AD-related genes both directly and indirectly through vascular pathways. Further adjustment for lifestyle factors did not change the results. The fact that some associations became non-significant when obesity measures were treated as dichotomous, could indicate the linear nature of underlying relationships, leading to increased power for continuous models to capture potential effects.
4.1. Obesity and upregulated expression of AD-related genes
Obesity was associated with upregulated expression of 10 genes, after accounting for cardiovascular risk factors. The genes CLU, CD2AP, FCER1G, and KLC3 exhibited the most robust associations in terms of effect size as well as significance, and were present across all obesity metrics. The largest body of evidence regarding a potential role in AD pathogenesis exists for the CLU gene. CLU is the third most associated risk gene for late-onset AD, explaining around 9% of the attributable risk for the disease.30, 31 The encoded protein (clusterin), has been implicated in various molecular pathways of AD neurobiology, including amyloid aggregation and clearance, lipid metabolism, neuroinflammation, as well as neuronal cell cycle control and apoptosis.32 Clusterin levels have been found to be elevated in the brain and cerebrospinal fluid of individuals with AD,32 and plasma levels have been associated with brain atrophy, baseline disease severity, and rapid clinical progression in patients with AD.33, 34 Furthermore, CLU genotypes were related to β-amyloid (Aβ) deposition on amyloid PET imaging and hippocampal volumes on structural brain MRI in the Alzheimer’s Disease Neuroimaging Initiative (ADNI).35 Plasma clusterin levels have also been positively correlated with BMI and WHR.36 In fact, on array analysis, CLU was one of the top 15 extracellular matrix-related genes overexpressed in human obesity,37 further highlighting its potential role as a molecular intermediary in the relationship between obesity and AD.38 Interestingly, the CD2AP gene is also among the top 10 genetic predisposition factors for AD.31 Accruing evidence suggests that its encoding protein, a scaffolding molecule regulating cell-cell interactions, cytoskeletal remodeling, and membrane trafficking, might also be implicated in AD pathogenesis, although the exact mechanisms remain unknown.39 Similarly, FCER1G is a microglial-specific gene with higher expression in the adipose tissue of obese than that of non-obese individuals40; evidence for its potential role in AD pathogenesis comes from GWAS, DNA sequencing, and expression network analyses, supporting the involvement of microglial pathways in the development and progression of AD.41
4.2. Obesity and downregulated expression of AD-related genes
After adjustment for cardiovascular risk factors, obesity was associated with downregulated expression of 4 genes (PVRIG, ZNF646, ZNF768, ABCA7). Strong genetic evidence suggests that ABCA7 protects from AD pathogenesis.42 The gene encodes a protein expressed in neurons, astrocytes, and microglia, responsible for mediating the assembly of lipids and exchangeable apolipoproteins, such as apolipoprotein E, into HDL particles that are released into the extracellular space (lipid efflux). ABCA7 deletion in mouse models has been associated with significant increases in insoluble Aβ deposition in the brain, either by increased production, or by reduced clearance.43 Remarkably, prior studies in European ancestry populations have shown that AD-associated variants at ABCA7 locus raise the risk of late-onset AD by approximately 20%, whereas ABCA7 loss-of-function mutations raise the risk of early-onset AD by 100–400%. Moreover, a loss-of-function mutation present in African ancestry populations increases AD risk by approximately 80%.42
4.3. Obesity, AD-related gene expression, and the role of cardiovascular risk factors
Obesity contributes directly to incident cardiovascular risk factors and leads to the development of cardiovascular disease.44 In turn, cardiovascular risk factors have been associated with the risk of AD,45 therefore, they might act as mediators in the relationships between obesity and AD-related gene expression. Indeed, a number of those relationships were no longer significant after adjustment for cardiovascular risk factors. The fact that this phenomenon was more prominent for WHR, which is more closely related to cardiovascular disease risk than BMI24 (56% of the observed associations were affected vs 28% for BMI), further supports this hypothesis. In the brain microenvironment, vascular pathology can drastically affect gene expression by a protein called hypoxia-inducible factor-1 (HIF-1), which controls the expression of over 700 target genes46; through this pathway, it might drive gene expression profiles that promote amyloidogenesis and initiation/propagation of AD pathobiology, especially in regions particularly vulnerable to hypoxia such as the hippocampus.47 For instance, HIF-1 induction increases the expression of BACE1 by binding to its promoter region.48 BACE1 is the major protease for β-cleavage of APP and upregulation of this enzyme leads to increased generation of Aβ40 and Aβ42.48
4.4. Effect modification by sex
Two-thirds of AD cases affect women, and notable sex differences in disease risk, presentation, and progression have been observed.49 Those differences are likely multifactorial in etiology but might in part be related to differential susceptibility to disease risk factors such as obesity. Results from the English Longitudinal Study of Ageing (ELSA), revealed that women with abdominal obesity had a 39% increased risk for incident dementia compared to those with non-abdominal obesity; however, no such association was observed in men.50 This differential susceptibility might originate from the molecular level. The present results support this hypothesis, since subgroup analysis by sex revealed that many associations between obesity and AD-related gene expression were observed only in female participants.
4.5. Effect modification by age
Stratification of study participants by age, revealed that the corresponding effect sizes for the majority of the associations between obesity and AD-related gene expression were smaller for older participants. Prior studies and meta-analyses have consistently demonstrated that midlife rather than late-life obesity correlates with incident dementia.8, 9 The present results support, and also provide a putative biological explanation for, prior epidemiological data, by suggesting that the regulatory effects of obesity on the expression of AD-related genes might weaken with age. Nevertheless, the definitive presence, and the exact nature of, molecular variations that might lead to differential responses of gene expression to obesity based on age and/or sex, remain for future mechanistic studies to determine.
4.6. Limitations
GWAS findings are often difficult to interpret, since the causal variants driving the associations can be concealed by linkage disequilibrium, and the causal genes mediating variant effects on the trait of interest are rarely ascertainable from GWAS results alone.51 To overcome this issue, we used a series of cutting-edge integrative bioinformatics methods that combine GWAS results with functional genomics knowledge (TWAS, COLOC, and functional validation using histone regulatory mark information from blood, brain and heart tissues) to functionally characterize and prioritize the individual AD genomic risk loci studied. Moreover, the observed associations of obesity with AD-related gene expression identified genes that have been implicated in AD pathobiology by prior research, further increasing the biological plausibility of our findings. Nevertheless, the possibility that some of the observed relationships might have been with genes that are not part of AD-related pathways cannot be excluded. Additionally, the cross-sectional design of the present analysis does not allow for conclusions about causation or derivation of temporal relationships, potentially raising the issue of reverse causality. This is particularly pertinent for studies exploring associations between risk factors and AD, given the long latency period between the onset of neuropathological changes in the brain and AD-related clinical symptoms.52 However, the majority of study participants were cognitively healthy, and most of the observed associations were in fact stronger in younger participants (that are less likely to have accumulated AD-related pathology), decreasing the possibility that reverse causality accounts for the present findings. Finally, the study sample consisted of non-Hispanic White individuals; therefore, further studies are needed to evaluate the generalizability of the present findings to other racial/ethnic groups.
4.7. Conclusion
The present study identified associations of obesity with the expression of 21 AD-related genes in a large sample of individuals living in the community. These associations, among others, implicated a number of genes, known to be part of multiple pathways involved in AD neurobiology (e.g., CLU, CD2AP, FCER1G, ABCA7, APP, CD33, PTK2B). Therefore, one of the molecular mechanisms through which obesity contributes to AD risk might be its modulating effects on the expression profiles of genes regulating the underlying neurodegenerative cascade. The present findings provide important insights into the mechanistic intermediates of the pathway leading from obesity to dementia. Furthermore, they could assist public health and dementia prevention policies to target lifestyle interventions, such as weight loss, to population groups that might have the most benefit.
Supplementary Material
5. Acknowledgements
We thank the study participants, as well as the study team for their contributions to data collection without which the present work would not be feasible.
This study was supported by grants from the National Heart, Lung, and Blood Institute contract for the Framingham Heart Study (contract No. N01-HC-25195, No. HHSN268201500001I, and No. 75N92019D00031), the National Institute on Aging (R01 AG054076, R01 AG049607, U01 AG052409, R01 AG059421, RF1 AG063507, RF1 AG066524, U01 AG058589), and the National Institute of Neurological Disorders and Stroke (R01 NS017950 and UH2 NS100605). Gene expression profiling was supported by the Systems Approach to Biomarker Research (SABRe) Initiative through the Division of Intramural Research (Principal Investigator, Daniel Levy), National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD. C.L. Satizabal was supported by a New Investigator Research Grant to Promote Diversity from the Alzheimer’s Association (AARGD-16-443384), and also receives support from NIA (R01 AG059727), and TARCC (2020-58-81-CR). S. Seshadri and C.L. Satizabal report funding from P30 AG066546 and UF1 NS125513.
Footnotes
Conflicts of Interest
The authors report no disclosures relevant to the manuscript. Author disclosures are available in the supporting information.
References
- 1.Comas Herrera A, Prince M, Knapp M, et al. World Alzheimer Report 2016: Improving healthcare for people with dementia. Coverage, quality and costs now and in the future. 2016.
- 2.Rizzi L, Rosset I, Roriz-Cruz M. Global epidemiology of dementia: Alzheimer’s and vascular types. Biomed Res Int. 2014;2014:908915. doi: 10.1155/2014/908915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long JM, Holtzman DM. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell. Oct 3 2019;179(2):312–339. doi: 10.1016/j.cell.2019.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Britschgi M, Wyss‐Coray T. Systemic and Acquired Immune Responses in Alzheimer’s Disease. International Review of Neurobiology. Academic Press; 2007:205–233. [DOI] [PubMed] [Google Scholar]
- 5.Giunta B, Fernandez F, Nikolic WV, et al. Inflammaging as a prodrome to Alzheimer’s disease. Journal of neuroinflammation. Nov 11 2008;5:51. doi: 10.1186/1742-2094-5-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nature reviews Neuroscience. May 2011;12(5):284–96. doi: 10.1038/nrn3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hruby A, Hu FB. The Epidemiology of Obesity: A Big Picture. Pharmacoeconomics. Jul 2015;33(7):673–89. doi: 10.1007/s40273-014-0243-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kivipelto M, Ngandu T, Fratiglioni L, et al. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol. Oct 2005;62(10):1556–60. doi: 10.1001/archneur.62.10.1556 [DOI] [PubMed] [Google Scholar]
- 9.Lloret A, Monllor P, Esteve D, et al. Obesity as a Risk Factor for Alzheimer’s Disease: Implication of Leptin and Glutamate. Mini Review. 2019-May-22 2019;13doi: 10.3389/fnins.2019.00508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pugazhenthi S, Qin L, Reddy PH. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim Biophys Acta Mol Basis Dis. May 2017;1863(5):1037–1045. doi: 10.1016/j.bbadis.2016.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Attems J, Jellinger KA. The overlap between vascular disease and Alzheimer’s disease--lessons from pathology. BMC Med. Nov 11 2014;12:206. doi: 10.1186/s12916-014-0206-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shinohara M, Kikuchi M, Onishi-Takeya M, et al. Upregulated expression of a subset of genes in APP;ob/ob mice: Evidence of an interaction between diabetes-linked obesity and Alzheimer’s disease. FASEB Bioadv. May 2021;3(5):323–333. doi: 10.1096/fba.2020-00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schwartzentruber J, Cooper S, Liu JZ, et al. Genome-wide meta-analysis, fine-mapping and integrative prioritization implicate new Alzheimer’s disease risk genes. Nat Genet. Mar 2021;53(3):392–402. doi: 10.1038/s41588-020-00776-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moser VA, Pike CJ. Obesity and sex interact in the regulation of Alzheimer’s disease. Neurosci Biobehav Rev. Aug 2016;67:102–18. doi: 10.1016/j.neubiorev.2015.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Splansky GL, Corey D, Yang Q, et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol. Jun 1 2007;165(11):1328–35. doi: 10.1093/aje/kwm021 [DOI] [PubMed] [Google Scholar]
- 16.Kannel WB, Feinleib M, McNamara PM, et al. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol. Sep 1979;110(3):281–90. doi: 10.1093/oxfordjournals.aje.a112813 [DOI] [PubMed] [Google Scholar]
- 17.Dawber TR, Meadors GF, Moore FE Jr. Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health. Mar 1951;41(3):279–81. doi: 10.2105/ajph.41.3.279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gusev A, Ko A, Shi H, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. Mar 2016;48(3):245–52. doi: 10.1038/ng.3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. May 2014;10(5):e1004383. doi: 10.1371/journal.pgen.1004383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. Jan 2012;40(Database issue):D930–4. doi: 10.1093/nar/gkr917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cruchaga C, Haller G, Chakraverty S, et al. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PloS one. 2012;7(2):e31039. doi: 10.1371/journal.pone.0031039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu CC, Liu CC, Kanekiyo T, et al. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nature reviews Neurology. Feb 2013;9(2):106–18. doi: 10.1038/nrneurol.2012.263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joehanes R, Zhang X, Huan T, et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome biology. Jan 25 2017;18(1):16. doi: 10.1186/s13059-016-1142-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Czernichow S, Kengne AP, Stamatakis E, et al. Body mass index, waist circumference and waist-hip ratio: which is the better discriminator of cardiovascular disease mortality risk?: evidence from an individual-participant meta-analysis of 82 864 participants from nine cohort studies. Obes Rev. Sep 2011;12(9):680–7. doi: 10.1111/j.1467-789X.2011.00879.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.World Health O. Waist circumference and waist-hip ratio : report of a WHO expert consultation, Geneva, 8–11 December 2008. Geneva: World Health Organization; 2011. [Google Scholar]
- 26.Wolf PA, D’Agostino RB, Belanger AJ, et al. Probability of stroke: a risk profile from the Framingham Study. Stroke. Mar 1991;22(3):312–8. doi: 10.1161/01.str.22.3.312 [DOI] [PubMed] [Google Scholar]
- 27.Lin H, Rogers GT, Lunetta KL, et al. Healthy diet is associated with gene expression in blood: the Framingham Heart Study. Am J Clin Nutr. Sep 1 2019;110(3):742–749. doi: 10.1093/ajcn/nqz060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sauder KA, Proctor DN, Chow M, et al. Endothelial function, arterial stiffness and adherence to the 2010 Dietary Guidelines for Americans: a cross-sectional analysis. Br J Nutr. Jun 14 2015;113(11):1773–81. doi: 10.1017/S0007114515000859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kannel WB, Sorlie P. Some health benefits of physical activity. The Framingham Study. Arch Intern Med. Aug 1979;139(8):857–61. [PubMed] [Google Scholar]
- 30.Wu ZC, Yu JT, Li Y, et al. Clusterin in Alzheimer’s disease. Adv Clin Chem. 2012;56:155–73. doi: 10.1016/b978-0-12-394317-0.00011-x [DOI] [PubMed] [Google Scholar]
- 31.Bertram L, McQueen MB, Mullin K, et al. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. Jan 2007;39(1):17–23. doi: 10.1038/ng1934 [DOI] [PubMed] [Google Scholar]
- 32.Yu JT, Tan L. The role of clusterin in Alzheimer’s disease: pathways, pathogenesis, and therapy. Mol Neurobiol. Apr 2012;45(2):314–26. doi: 10.1007/s12035-012-8237-1 [DOI] [PubMed] [Google Scholar]
- 33.Thambisetty M, Simmons A, Velayudhan L, et al. Association of plasma clusterin concentration with severity, pathology, and progression in Alzheimer disease. Arch Gen Psychiatry. Jul 2010;67(7):739–48. doi: 10.1001/archgenpsychiatry.2010.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schrijvers EM, Koudstaal PJ, Hofman A, et al. Plasma clusterin and the risk of Alzheimer disease. JAMA. Apr 6 2011;305(13):1322–6. doi: 10.1001/jama.2011.381 [DOI] [PubMed] [Google Scholar]
- 35.Tan L, Wang H-F, Tan M-S, et al. Effect of CLU genetic variants on cerebrospinal fluid and neuroimaging markers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. Scientific Reports. 2016/May/27 2016;6(1):26027. doi: 10.1038/srep26027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Won JC, Park CY, Oh SW, et al. Plasma clusterin (ApoJ) levels are associated with adiposity and systemic inflammation. PloS one. 2014;9(7):e103351. doi: 10.1371/journal.pone.0103351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bradley D, Blaszczak A, Yin Z, et al. Clusterin Impairs Hepatic Insulin Sensitivity and Adipocyte Clusterin Associates With Cardiometabolic Risk. Diabetes care. Mar 2019;42(3):466–475. doi: 10.2337/dc18-0870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bradley D. Clusterin as a Potential Biomarker of Obesity-Related Alzheimer’s Disease Risk. Biomarker insights. 2020;15:1177271920964108. doi: 10.1177/1177271920964108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tao Q-Q, Chen Y-C, Wu Z-Y. The role of CD2AP in the Pathogenesis of Alzheimer’s Disease. Aging Dis. 2019;10(4):901–907. doi: 10.14336/AD.2018.1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu Z, Meng L, Sun Z, et al. Differentially Expressed Genes and Enriched Signaling Pathways in the Adipose Tissue of Obese People. Front Genet. 2021;12:620740. doi: 10.3389/fgene.2021.620740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. May 26 2017;12(1):43. doi: 10.1186/s13024-017-0184-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lyssenko NN, Pratico D. ABCA7 and the altered lipidostasis hypothesis of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. Feb 2021;17(2):164–174. doi: 10.1002/alz.12220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim WS, Li H, Ruberu K, et al. Deletion of Abca7 increases cerebral amyloid-beta accumulation in the J20 mouse model of Alzheimer’s disease. J Neurosci. Mar 6 2013;33(10):4387–94. doi: 10.1523/JNEUROSCI.4165-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Powell-Wiley TM, Poirier P, Burke LE, et al. Obesity and Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation. 2021/May/25 2021;143(21):e984–e1010. doi: 10.1161/CIR.0000000000000973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Viswanathan A, Rocca WA, Tzourio C. Vascular risk factors and dementia: how to move forward? Neurology. Jan 27 2009;72(4):368–74. doi: 10.1212/01.wnl.0000341271.90478.8e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitroshina EV, Savyuk MO, Ponimaskin E, et al. Hypoxia-Inducible Factor (HIF) in Ischemic Stroke and Neurodegenerative Disease. Review. Frontiers in Cell and Developmental Biology. 2021;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Helton R, Cui J, Scheel JR, et al. Brain-specific knock-out of hypoxia-inducible factor-1alpha reduces rather than increases hypoxic-ischemic damage. J Neurosci. Apr 20 2005;25(16):4099–107. doi: 10.1523/JNEUROSCI.4555-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang X, Zhou K, Wang R, et al. Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. The Journal of biological chemistry. Apr 13 2007;282(15):10873–80. doi: 10.1074/jbc.M608856200 [DOI] [PubMed] [Google Scholar]
- 49.Dumitrescu L, Barnes LL, Thambisetty M, et al. Sex differences in the genetic predictors of Alzheimer’s pathology. Brain. Sep 1 2019;142(9):2581–2589. doi: 10.1093/brain/awz206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma Y, Ajnakina O, Steptoe A, et al. Higher risk of dementia in English older individuals who are overweight or obese. International Journal of Epidemiology. 2020;49(4):1353–1365. doi: 10.1093/ije/dyaa099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wainberg M, Sinnott-Armstrong N, Mancuso N, et al. Opportunities and challenges for transcriptome-wide association studies. Nature Genetics. 2019/April/01 2019;51(4):592–599. doi: 10.1038/s41588-019-0385-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dubois B, Hampel H, Feldman HH, et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2016;12(3):292–323. doi: 10.1016/j.jalz.2016.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.