

Abstract
Background: DNA hypermethylation and instability due to inactivation mutations in Ten–eleven translocation 2 (TET2) is a key biomarker of hematological malignancies. This study aims at characterizing two intronic noncanonical splice‐site variants, c.3954+5_3954+8delGTTT and c.3954+5G>A.
Methods: We used in silico prediction tools, reverse transcription (RT)‐PCR, and Sanger sequencing on blood/bone marrow‐derived RNA specimens to determine the aberrant splicing.
Results: In silico prediction of both variants exhibited reduced splicing strength at the TET2 intron 7 splicing donor site. RT‐PCR and Sanger sequencing identified a 62‐bp deletion at the exon 7, producing a frameshift mutation, p.Cys1298*.
Conclusion: This study provides functional evidence for two intronic TET2 variants that cause alternative splicing and frameshift mutation.
Keywords: in silico prediction, myeloid neoplasm, next‐generation sequencing (NGS), noncanonical splicing site, ten‐eleven translocation 2, TET2, tumor suppressor
1. INTRODUCTION
Ten‐eleven translocation (TET) is a three‐member family of genes, located on chromosome 4q24, that encodes for enzymes Tet methylcytosine deoxygenate. TET2 are key epigenetic modifiers and tumor suppressor genes, crucial in hematological malignancies as well as normal hematopoiesis, immune cell activation, and clonal expansion [1, 2, 3]. TET2 loss of function and deletion leading to the hypermethylated state of the genome is frequently detected in myeloid and lymphoid malignancies and clonal hematopoiesis of indeterminate potential. In high throughput genome‐wide studies, somatic deletion and loss of function TET2 mutations are detected in 7–23% of acute myeloid leukemia (AML), 10%–20% of myelodysplastic syndrome (MDS)/myeloproliferative neoplasms (MPN), and 40%–50% chronic myelogenous leukemia patients [2, 4–6]. TET2 regulates gene expression primarily through the demethylation of DNA 5‐methylcytosine (5mC) to 5‐hydroxymethyl cytosine (5hmC), requiring Fe2+ and α‐ketoglutarate (α‐KG) for their activity. Subsequently, TET2 converts 5hmC to 5‐formylcytosine (5fC) and 5‐carboxy‐cytosine (5caC), facilitating the removal of 5fC and 5caC through the base‐excision repair pathway and playing a key role in DNA demethylation and transcription regulation [7]. TET2 deoxygenase domain resides in its exons 4 through 11, which consists of a cysteine‐rich domain, a double‐stranded β‐helix (DSBH) domain, and a low‐complexity linker region [4, 8]. The DSBH domain harbors binding sites for Fe(II) and α‐ketoglutarate that help TET2 binding to carry out the 5mC oxidation process.
The majority of inactivating or loss‐of‐function TET2 variants are exonic and located throughout the coding exons, including 76.7% truncating, 17.4% missense and 4.8% splice sites as reported in the cBioportal database (on 02/05/2023). However, the lack of study in TET2 noncanonical splicing site intronic variants has made it very challenging to interpret these variants for clinical usage. In fact, most clinical laboratories will either choose not to report these variants or classify them as unknown clinical significance. In a combination of in silico prediction, RT‐PCR, and Sanger sequencing analysis here we show the impact of two noncanonical splicing site intronic variants of TET2, a deletion c.3954+5_3954+8delGTTT and a recurrent substitution c.3954+5G>A, in producing frameshift mutations that expected to yield truncated, dysfunctional TET2 gene products.
2. MATERIALS AND METHODS
2.1. Patient samples, DNA and RNA extraction, next‐generation sequencing
Genomic DNA and total RNA were extracted from either bone marrow or peripheral blood of the same specimens. A nonleukemic patient was used as the negative control. Two TET2 intronic variants c.3954+5_3954+8delGTTT (present in one patient) and c.3954+5G>A (present in two patients) were identified from a 63‐gene hematologic neoplasm next‐generation sequencing (NGS) panel. For a given sample, the NGS minimum coverage requirement of targeted regions was >100X. Variants with variant allele fractions (VAFs) as low as 4% were identified. The study was conducted according to the approved protocols of Cleveland Clinic's Institutional Review Board (17–177 and 19–329).
2.2. In silico splicing prediction, RT‐PCR and Sanger sequencing
In silico splice prediction tools, including SpliceSiteFinder‐like, MaxEntScan, NNSplice, and GeneSplicer, integrated into the Alamut Visual Plus (Version v1.3, SOPHiA GENETICS), were used in this study [5]. PCR was performed using a forward primer specific to TET2 Exon 6, 5′‐CCGAGACGCTGAGGAAATAC−3′ and a reverse primer specific to TET2 Exon 8, 5′‐TGGACAGGTTTTGCAAATGA−3′. Sanger sequencing was conducted using BigDye Terminator 1.1 kits.
3. RESULTS
3.1. Identification of two intronic TET2 variants in hematoneoplasm patients
Two novel TET2 (NM_001127208.2) intron 7 variants with high VAFs were identified in three patients (Table 1). Patient 1 was diagnosed with acute myeloid leukemia, carried 4‐bp deletion, c.3954+5_3954+8delGTTT, with 42.5% VAF. Patient 2 and patient 3 with chronic myelomonocytic leukemia carry the substitution mutation, c.3954+5G>A, with 77.31% and 97.68% VAFs, respectively. As these variants are located very near to the intron 7 canonical splice donor sequence (GT) and within the 5′ splice site (5′ss) consensus sequence [6], we conducted an in silico splicing analysis to predict the impact of splicing. Our analyses showed a drastic decline of splicing strength at the TET2 intron 7 splicing donor site in both variants, c.3954+5_3954+8delGTTT and c.3954+5G>A, in compared with the TET2 wild‐type transcript (Figure 1A,B).
TABLE 1.
Clinical characteristics and variant information.
| Characteristics | Patient 1 | Patient 2 | Patient 3 |
|---|---|---|---|
| Tumor type | Acute myeloid leukemia | Chronic myelomonocytic leukemia | Acute myeloid leukemia (with a history of chronic myelomonocytic leukemia) |
| Specimen source | Peripheral blood | Bone marrow | Peripheral Blood |
| TET2 intronic variants | c.3954+5_3954+8delGTTT | c.3954+5G>A | c.3954+5G>A |
| VAF = 42.5% | VAF = 77.31% | VAF = 97.68% | |
| Depth = 647 | Depth = 335 | Depth = 689 |
FIGURE 1.

(A and B) In silico splicing prediction of TET2 intronic variants of (A) c.3954+5_3954+8del, and (B) c.3954+5G>A. Variants are indicated by red lines. Four splicing predictors were used; the impact on gene splicing is indicated with vertical blue bars. The predicted strength of canonical splice donor signals at the splicing junctions is reduced in both variants compared to the wild‐type reference transcript. The gains of splicing signals were observed at an internal cryptic splice donor site.
3.2. Confirmation of impact in splicing in two novel TET2 variants
The two intronic variants’ impact on TET2 gene splicing was further characterized by RT‐PCR and Sanger sequencing at the RNA level. As shown in Figure 2A,B, a band at approximately 246 bp region corresponding to the wild‐type sequence was seen in the negative (WT) control. However, for the patient with deletion mutation (patient 1), two bands were seen; one was at 246 bp, and the other appeared just below 200 bp. This ∼200 bp RT‐PCR product was also obtained for patients 2 and 3 (data not shown) along with 246 bp WT product. Sanger sequencing of the extracted 246 bp band from the WT control consists of all expected exonic sequences, including partial exon 6, full exon 7, and partial exon 8. Interestingly, sequencing of the ∼200 bp products from both cases of c.3954+5G>A and c.3954+5_3954+8delGTTT variants, as shown in Figure 3, revealed a 62‐bp deletion at the 3′ portion of the exon 7. This deletion used an exon 7 internal cryptic splicing site c.3892 (g.106180864) to produce an alternative splicing product that is predicted to cause a protein reading frameshift mutation, p.Cys1298*. These results demonstrate that both variants lead to the same premature termination codon that is expected to result in a truncating protein.
FIGURE 2.
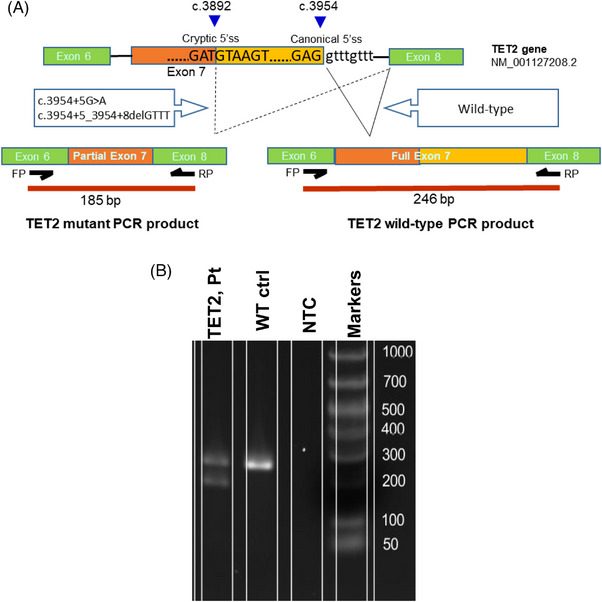
(A) Graphic representation showing TET2 splicing products with or without TET2 intronic variants (c.3954+5_3954+8delGTTT, c.3954+5G>A). Breakpoints at the cryptic and original 5′ splice site junctions are shown with blue triangles. The primer binding sites and subsequent PCR products in WT and mutant TET2 spliced mRNA are also shown. (B) RT‐PCR analysis on RNA from a negative control patient and a patient with TET2 c.3954+5_3954+8del variant. Gel electrophoresis image showing PCR products amplified using specific primers binding to Ex6 (forward) and Ex8 (reverse). Fragment size of 246 bp indicates TET2 wild‐type sequences (ex6+ex7+ex8) and fragment of 185 bp size (ex6+ partial ex7+ex8) indicates the TET2 splicing variant. Only wild‐type product was identified in the negative (WT) control. No visible product is shown in no template control (NTC).
FIGURE 3.

Sanger sequencing of the 185 bp size PCR products obtained from a polyacrylamide gel. Nucleotide sequences of wild‐type TET2 transcript are shown at the bottom of each chromatogram. The coding exon sequences were highlighted in blue. The splicing junctions connecting exon 7 and exon 8, leading to partial depletion of exon 7, are indicated with black arrows. (A) Chromatogram of patient 1 fragment of TET2 c.3954+5_3954+8del. (B) Chromatogram of patient 2 fragment of c.3954+5G>A substitution.
4. DISCUSSION
TET2 is an epigenetic modifier; its mutations commonly occur alongside other driver mutations across AML, MPN, and MDS malignancies. Mutations of TET2 are usually observed before the acquisition of other driver mutations contributing to genome instability and clonal evolution. Therefore, to understand the clonal structure, proliferative, and differentiation capacity of neoplastic cells in the hematologic neoplasm, and identification of loss‐of‐function variants in TET2 is crucial. We have shown two intronic variants, c.3954+5_3954+8delGTTT and c.3954+5G>A, allow the splicing machinery to bypass the canonical 5′ splice donor site and activate a cryptic splicing sequence within exon 7, leading to exclusion of a portion of exon 7. This results in a frameshift mutation expected to produce a Tet2 truncating protein (p.Cys1298*) devoid of the majority of Tet2 oxygenase domain and the downstream amino acids. In addition, this study warrants the adoption of follow‐up confirmatory steps when TET2 intronic variants are detected for accurate variant interpretation and classification.
The activation of 5′ cryptic or generation of de novo splice sites is mostly due to alteration in the first intron nucleotide (+1) or +5 position [9]. Position +5 in the intron is required for base pairing with U1 and U6 snRNA components of spliceosomes during the splicing process [10]. Since both variants, c.3954+5_3954+8delGTTT and c.3954+5G>A, in our study encompass the +5G intron position, our finding of the splicing product with a 62‐bp deletion of exon 7 corroborates with our understanding of pre‐mRNA splicing mechanism and demonstrates the usefulness of in silico prediction tools.
Sequence changes at the TET2 intron 7 canonical splice donor site have been widely documented in the COSMIC database, including c.3954+1G>A (COSV54396751) [11, 12], c.3954+1G>T (COSV54397716) [13], c.3954+2T>G (COSV54429204), and c.3954+2T>A (COSV105020527) [14, 15]. Because TET2 is a tumor suppressor gene, splicing site variants are predicted to cause gene inactivation and oncogenic events [13, 16]. In fact, shown in Figure 4, in silico predictions indicate that all the above‐mentioned COSMIC variants (positioned at c.3954+1 and c.3954+2) activate the identical exon 7 cryptic internal splicing site at c.3892 (g.106180864) shown in this study and are expected to produce the same truncating protein product, p.Cys1298*. The fact that the presented two TET2 intronic variants and the exon 7 canonical splice donor site variants, all result in the same truncating gene product, highlights the importance of classifying both c.3954+5G>A and c.3954+5_3954+8delGTTT variants as potential clinical significance. It is worth mentioning that the c.3954+5G>A variant (rs575928986, dbSNP151) has a relatively rare minor allele frequency of 0.0032% (gnomAD v2.1) in the general population. However, in the disease cohort of our study, we have identified three such cases in a total of 2164 myeloid neoplasm patients, equating to a 0.139% instance rate. This increase of two orders of magnitude in mutation rate in the disease cohort and the evidence of producing a truncating protein strongly suggest that these two intronic variants are highly associated with the pathogenicity of myeloid neoplasms.
FIGURE 4.

(A and B) Examples of in silico splicing prediction of TET2 canonical splice donor variants of (A) c.3954+1G>A, and (B) c.3954+2T>A substitutions. Four splicing predictors were used; the impact on gene splicing is indicated with vertical blue bars. The predicted strength of canonical splice donor signals at the splicing junctions is reduced in both variants compared to the wild‐type reference transcript. The gains of splicing signals were observed at the same internal cryptic splice donor site as shown in Figure 1.
Due to a lack of functional evidence TET2 intronic variants were not reported for the diagnosis and management of the patients’ disease. However, it is very likely that the TET2 intronic variants contributed to the myeloid clonal evolution in these patients and provided a persistent mutational burden. These variants were detected at very high variant allele frequency (Table 1). Additionally, follow‐up sequencing for patients 2 and 3 detected the TET2 intronic variants (c.3954+5G>A) at increased variant allele fractions with persistent disease phenotype. In addition to the TET2 variants, all patients carried other known driver mutations in either FLT3, KRAS, JAK2, or CBL genes. Since the sequencing test was not performed at the early stages of disease manifestation, one cannot say for certain that TET2 intronic variants alone acted as a founder mutation.
Altogether, this study identifies two TET2 intronic variants that are potentially pathogenic. We present a cost‐effective approach, by combining the in silico prediction, RT‐PCR, and Sanger sequencing to determine the significance of potential splicing site variants. A clinical laboratory may adopt this approach to the standard operating procedure to improve the variant triage process.
AUTHOR CONTRIBUTIONS
YWC designed the study. ZJT and DB collected data. DB provided clinical consultation and regulatory support for the cases. RD, ZJT, and YWC performed analyses. YWC supervised the study. RD and YWC drafted the manuscript. All authors approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
CLINICAL TRIAL REGISTRATION (INCLUDING TRIAL NUMBER)
This study has not been registered for any clinical trial.
PATIENT CONSENT STATEMENT
n/a
ETHICS STATEMENT
The study was conducted according to the approved protocols of Cleveland Clinic's Institutional Review Board (IRB; 17‐177 and 19‐329).
ACKNOWLEDGMENTS
This study was in part supported by the research fund of the Laboratory Medicine Department of the Robert J. Tomsich Pathology and Laboratory Medicine Institute, Cleveland Clinic.
Das R, Tu ZJ, Bosler DS, Cheng Y‐W. Identification and interpretation of TET2 noncanonical splicing site intronic variants in myeloid neoplasm patients. eJHaem. 2023;4:738–744. 10.1002/jha2.744
DATA AVAILABILITY STATEMENT
Data are available upon request due to privacy/ethical restrictions.
REFERENCES
- 1. Jiang S. Tet2 at the interface between cancer and immunity. Commun Biol. 2020;3(1):667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009;360(22):2289–301. [DOI] [PubMed] [Google Scholar]
- 3. Kunimoto H, Nakajima H. TET2: a cornerstone in normal and malignant hematopoiesis. Cancer Sci. 2021;112(1):31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bussaglia E, Anton R, Nomdedeu JF, Fuentes‐prior P. TET2 missense variants in human neoplasia. A proposal of structural and functional classification. Mol Genet Genomic Med. 2019;7(7):e00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Das R, Jakubowski MA, Spildener J, Cheng YW. Identification of novel MET exon 14 skipping variants in non‐small cell lung cancer patients: a prototype workflow involving in silico prediction and RT‐PCR. Cancers. 2022;14(19):4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roca X, Sachidanandam R, Krainer AR. Intrinsic differences between authentic and cryptic 5' splice sites. Nucleic Acids Res. 2003;31(21):6321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, et al. Tet proteins can convert 5‐methylcytosine to 5‐formylcytosine and 5‐carboxylcytosine. Science. 2011;333(6047):1300–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, et al. Crystal structure of TET2‐DNA complex: insight into TET‐mediated 5mC oxidation. Cell. 2013;155(7):1545–55. [DOI] [PubMed] [Google Scholar]
- 9. Buratti E, Chivers M, Kralovicova J, Romano M, Baralle M, Krainer AR, et al. Aberrant 5' splice sites in human disease genes: mutation pattern, nucleotide structure and comparison of computational tools that predict their utilization. Nucleic Acids Res. 2007;35(13):4250–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crispino JD, Sharp PA. A U6 snRNA:pre‐mRNA interaction can be rate‐limiting for U1‐independent splicing. Genes Dev. 1995;9(18):2314–23. [DOI] [PubMed] [Google Scholar]
- 11. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–74. [DOI] [PubMed] [Google Scholar]
- 12. Tefferi A, Lim KH, Abdel‐Wahab O, Lasho TL, Patel J, Patnaik MM, et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia. 2009;23(7):1343–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saint‐Martin C, Leroy G, Delhommeau F, Panelatti G, Dupont S, James C, et al. Analysis of the ten‐eleven translocation 2 (TET2) gene in familial myeloproliferative neoplasms. Blood. 2009;114(8):1628–32. [DOI] [PubMed] [Google Scholar]
- 15. Swierczek SI, Yoon D, Bellanne‐Chantelot C, Kim SJ, Saint‐Martin C, Delhommeau F, et al. Extent of hematopoietic involvement by TET2 mutations in JAK2V(6)(1)(7)F polycythemia vera. Haematologica. 2011;96(5):775–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bernard V, Gebauer N, Dinh T, Stegemann J, Feller AC, Merz H. Applicability of next‐generation sequencing to decalcified formalin‐fixed and paraffin‐embedded chronic myelomonocytic leukaemia samples. Int J Clin Exp Pathol. 2014;7(4):1667–76. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available upon request due to privacy/ethical restrictions.