

Abstract
Assessment of minimal residual disease in acute lymphoblastic leukemia by immune repertoire NGS requires spiking CDR3 sequences at known quantities into the patient's sample. Recently, the EuroClonality‐NGS group released one of the most comprehensive protocols for this purpose. ARResT/Interrogate is a closed‐source software for processing these NGS libraries, developed by this same group. Vidjil, an open‐source alternative, currently cannot handle libraries prepared using this protocol. Here, we present a Vidjil add‐on to solve this issue. EuroClonality‐NGS prepared samples analyzed with Vidjil and ARResT/Interrogate were highly concordant (r = 0.998) and presented low error (root‐mean‐square error, RMSE = 0.112).
Keywords: immune repertoire, immunoglobulin genes, leukemia, minimal residual disease, T‐cell receptor, Vidjil
1.
Minimal residual disease (MRD), the assessment of the remaining load of leukemia cells in the patient, is one of the most important prognosis and decision‐making factors in acute lymphoblastic leukemia (ALL) [1]. Real‐time PCR (qPCR) of immunoglobulin (IG) and T‐cell receptor (TR) gene rearrangements, with a special focus on their CDR3 region, is the current gold standard for MRD assessment [2]. Recently, next‐generation sequencing (NGS) of this same region has been used for the same purpose with many advantages over qPCR. Some of these include the possibility of better tracking clonal evolution and assessment of the patient's immune repertoire as a whole, making it possible to follow all of its potential leukemia clonotypes [3].
NGS results, however, are given in reads, while MRD is a cell frequency measure given by the number of cells of a leukemia clonotype divided by the number of cells present in the sample. One of the challenges for NGS‐based MRD assays, therefore, is converting the read count for a given leukemia clonotype into number of cells, so the clonotype's cell frequency can be determined. This is solved by spiking the sample DNA with plasmids or cell line's DNA containing a known CDR3 region at predetermined copy numbers. These control clonotypes are then used to establish a cell/read ratio for the sample, which in turn is used to find the number of cells for leukemia clonotypes. In recent years, protocols for this purpose have been established by several groups, with one of the most comprehensive and prominent examples being released by the EuroClonality‐NGS group [4, 5, 6, 7, 8].
Analysis of immune repertoire NGS assays requires specialized software capable of grouping the resulting reads into clonotypes. Furthermore, compatibility with these spike‐in‐based normalization methods needs to be specifically built into the software in order to obtain a final result expressed in cell frequency. The EuroClonality‐NGS method is integrated into ARResT/Interrogate, a closed‐source platform developed by this same group [9]. An open‐source alternative for ARResT/Interrogate is Vidjil, which also provides a web environment for immune repertoire NGS analysis [10, 11]. Being open‐source, it allows one to either use the official Vidjil server or perform a local installation and customization. Vidjil, however, is not currently compatible with the EuroClonality‐NGS normalization protocol. This work, therefore, aimed to create an add‐on to implement this method into the Vidjil platform.
Implementation was performed by adding two files to the Vidjil file structure, both available as Supplemental Data. The first, cIT‐QC.json, contains the definition and DNA sequences for all spike‐in controls used in the EuroClonality‐NGS protocol (n = 46), named central in‐tube quality/quantification control (cIT‐QC). A few sequences used for cIT‐QC identification were slightly adjusted to account for particularities of Vidjil's clonotype labeling method. The second file, cIT‐QC_normalization.py, is a Python script responsible for the normalization process, which outputs the frequency of each clonotype in clonotype cells/total cells instead of clonotype reads/total V(D)J reads. The cIT‐QC.json file must be placed in the docker directory, while cIT‐QC_normalization.py belongs in tools. Making this analysis available in the web application requires a new process configuration with the fuse command –pre ‘cIT‐QC_normalization.py’, created by an administrator account. Finally, the cell frequency result may be displayed in the result interface of Vidjil by selecting normalization from input data from the settings menu. This method will also be made available in the Vidjil official server in the future.
The process performed by the normalization script consists of four steps: (1) determine the primer set used for library preparation (IGH‐VJ, IGH‐DJ, IGK‐VJ‐Kde, intron‐Kde, TRB‐VJ, TRB‐DJ, TRG, TRD) based on the maximum read count of each of these IG/TR markers, from here on called the prevalent marker for the sample; (2) retrieve all cIT‐QC clonotypes in the sample related to the prevalent IG/TR marker; (3) calculate the total number of reads and cells for these cIT‐QC clonotypes; and (4) calculate the cell/read ratio for the sample and normalize the number of reads of all clonotypes into cell frequency. As per the EuroClonality‐NGS protocol, the script uses 15,000 as the initial number of input cells for each sample and 40 as the number of copies of each cIT‐QC sequence. To analyze libraries prepared with different specifications, these values must be changed in cIT‐QC_normalization.py. Further details regarding cIT‐QC_normalization.py, such as challenges in the identification of the prevalent marker in IGK‐VJ‐Kde samples, may be found as Supplemental Data.
In order to validate this add‐on, 32 NGS files from diagnosis (4 samples, 18 libraries) and aplastic posttreatment (4 samples, 14 libraries) B/T‐ALL bone marrow, which were analyzed using ARResT/Interrogate in the cIT‐QC original publication, were reanalyzed using Vidjil [6]. The cell frequency for the reported clonotypes of interest (n = 62) was compared (Figure 1A). These values, in log10 scale, were highly concordant between both software tools (r = 0.997) and presented low error (root‐mean‐square error, RMSE = 0.115). The Vidjil web application allows the user to manually cluster clonotypes, which were not automatically clustered due to small sequencing errors. Applying this technique to clonotypes differing in only one nucleotide had a low, but positive impact on the concordance (r = 0.998) and error (RMSE = 0.112) of the method (Figure 1B). It is important to note that this error was highly impacted by one clonotype where Vidjil and ARResT/Interrogate disagreed in their results by almost five times. This clonotype, however, belongs to a file (S17) containing only 500 rearrangement reads, which is considerably lower than the minimum expected to permit this type of analysis. As a frame of reference, the second lowest rearrangement read count (file S9) was 23205 in ARResT/Interrogate and 26537 in Vidjil. Without accounting for this outlier, the RMSE drops to 0.064 and 0.080, with and without manual clusterization, respectively (Figure S1). In addition, the Pearson's correlation also goes up to 0.999 in both cases.
FIGURE 1.
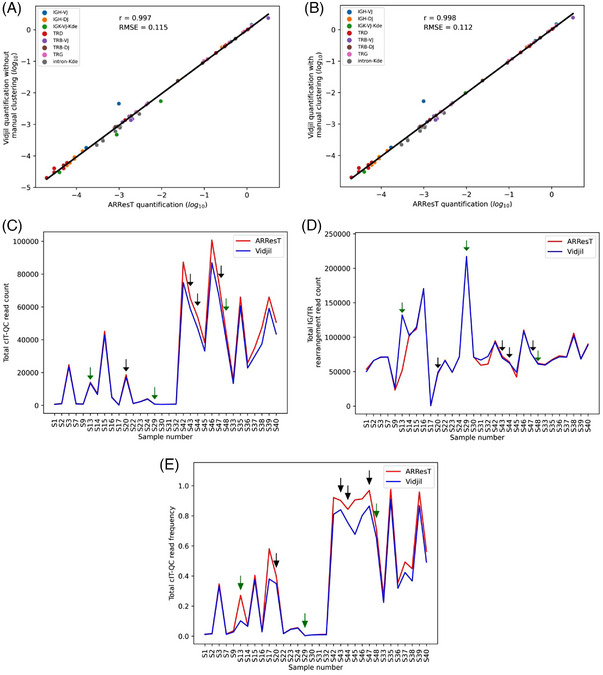
EuroClonality versus ARResT/Interrogate: IG/TR marker quantification and cIT‐QC identification. Analysis of bone marrow NGS files from the original cIT‐QC publication [6]. Samples S1‐S32 correspond to diagnostic B/T‐ALL, while S33‐S48 to aplastic posttreatment. (A and B) Linear regression of the log10 cell frequency for IG/TR clonotypes calculated by ARResT/Interrogate and Vidjil. Pearson's correlation coefficient represented by r, and root‐mean‐square error by RMSE. Vidjil analysis without (A) and with manual clonotype clustering (B). (C, D, and E) ARResT/Interrogate and Vidjil comparison per sample of total cIT‐QC read count (C), total IG/TR rearrangement read count (D), and total cIT‐QC read frequency (total cIT‐QC reads/total V(D)J reads) (E). Green and black arrows indicate samples where a cIT‐QC sequence was missing in both software tools and only in Vidjil, respectively.
Also of note, in sample S46, there are two clonotypes of interest with the same CDR3 region in ARResT/Interrogate. Since Vidjil uses a 50‐bp window centered around the CDR3 to cluster reads into clonotypes, it considered these two clonotypes as a single one and the same. Therefore, for this comparison, only one of the clonotypes reported by ARResT/Interrogate was taken into account, with its cell frequency being the sum of the frequencies observed for its two separate instances. This explains why the cIT‐QC publication reports 63 clonotypes of interest, while this work only reports 62.
The number of different cIT‐QC control molecules Vidjil identified for each sample was also analyzed. While the cIT‐QC system consists of 46 different control molecules in total, only a few of them (3–11) are expected to be found in each sample, since according to the EuroClonality‐NGS protocol each marker undergoes PCR for library preparation in different tubes with its own set of primers. In this context, Vidjil was able to identify all expected cIT‐QC sequences in 25/32 samples (78.1%), while ARResT/Interrogate did so in 29/32 samples (90.6%). In the seven cases where Vidjil failed to identify all cIT‐QC, no more than one sequence was missing. Importantly, while the total cIT‐QC reads identified by both software tools was always very similar, Vidjil always identify slightly less cIT‐QC sequences, which explains its tendency to generate marginally higher quantification values in comparison to ARResT/Interrogate (Figure 1C).
Total IG/TR read counts per sample were mostly very similar between both programs, with the exception of sample S13 where Vidjil was able to identify more than double the amount of reads in comparison to ARResT/Interrogate (Figure 1D). The cIT‐QC read frequencies (total cIT‐QC reads/total V(D)J reads) reported by the two programs showed some discrepancy (Figure 1E) in a few samples, but differences seem to be more associated with high cIT‐QC frequencies rather than with missing sequences. While samples S13 and S17 did present a considerable discrepancy even at low cIT‐QC frequencies, these were mostly due to their aforementioned anomalous behaviors. A summary for the main parameters yielded by each software tool can be found in Table 1.
TABLE 1.
Rearrangement and cIT‐QC read counts for 32 samples.
| ARResT/interrogate | Vidjil | |
|---|---|---|
| Samples analyzed by both software tools, n | 32 | |
| IGH‐DH | 3 | |
| IGH‐VJ‐FR1 | 4 | |
| IGK‐VJ‐Kde | 3 | |
| TRB‐DJ | 4 | |
| TRB‐VJ | 4 | |
| TRD | 6 | |
| TRG | 5 | |
| intron‐Kde | 3 | |
| Rearrangement reads per sample, mean (range) | 75,827.2 (525–217,101) | 78,568.2 (500–217,133) |
| IGH‐DH | 90,435.7 (66,049–110,460) | 89,015 (66,058–108,511) |
| IGH‐VJ‐FR1 | 29,849.5 (525–53,563) | 31,505.5 (500–50,018) |
| IGK‐VJ‐Kde | 73,352.3 (71,234–76716) | 72,475.3 (69,706–76,884) |
| TRB‐DJ | 86,919.5 (66,696–105,852) | 85,167 (65,977–102,104) |
| TRB‐VJ | 100,131.8 (51,795–217,101) | 119,950.5 (59,496–217,133) |
| TRD | 71,295.5 (48,781–111,744) | 72,782.7 (49,276–114,753) |
| TRG | 93,302.8 (60,888–170,711) | 94,822 (71,487–17,0351) |
| intron‐Kde | 57,739 (47,130–64,060) | 57,471 (49,008–62,465) |
| cIT‐QC reads per sample, mean (range) | 28,130 (305–100,754) | 24,858.4 (190–86,970) |
| IGH‐DH | 63,094 (1,174–100,754) | 54,281.3 (984–86,970) |
| IGH‐VJ‐FR1 | 9,995.5 (305–38,150) | 8,653.5 (190–33,136) |
| IGK‐VJ‐Kde | 54,711.7 (24,794–74,322) | 49,553 (23,566–66,503) |
| TRB‐DJ | 14,106 (607–47,478) | 11,433.3 (582–37,441) |
| TRB‐VJ | 16,420.8 (725–35,146) | 14,401.3 (682–30,053) |
| TRD | 30,209.8 (681–66,035) | 27,728.8 (646–60,598) |
| TRG | 17,299.6 (766–50,672) | 15,128 (702–43,496) |
| intron‐Kde | 38,965.7 (18,684–54,020) | 34,666.7 (17,109–47,102) |
In summary, this work produced an add‐on to Vidjil that makes it capable of properly processing immune repertoire NGS libraries prepared using the EuroClonality‐NGS protocol. The 63 clonotypes analyzed yielded highly concordant results (r = 0.998 and RMSE = 0.115) when compared to ARResT/Interrogate in the cIT‐QC original publication. Integration into a local Vidjil installation requires only the addition of two files to the software file structure, as well as the addition of a new process configuration on the web application. In addition, the add‐on will also be available in the official Vidjil server in a future release.
AUTHOR CONTRIBUTIONS
Guilherme N N Giusti, Andrés J Yunes, and João Meidanis conceived the study. Guilherme N N Giusti designed the study. Guilherme N N Giusti and Antônio V Ribeiro wrote the software. Guilherme N N Giusti, Antônio V Ribeiro, Patrícia Y Jotta, and Florian Thonier analyzed data. Guilherme N N Giusti wrote the manuscript, with input from all authors. Andrés J Yunes and João Meidanis supervised the study.
CONFLICT OF INTEREST STATEMENT
The institution Centro Infantil Boldrini is a part of the VidjilNet Consortium. Florian Thonier is a developer in the Vidjil team.
FUNDING INFORMATION
Guilherme N N Giusti was financed by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001 (PROEX 88887.342097/2019‐00). Antônio V Ribeiro was supported by the Programa de Educação em Oncologia Pediátrica (PEOp) from Centro Infantil Boldrini. José A Yunes received a productivity fellowship from the National Counsel of Technological and Scientific Development (CNPq, 308399/2021‐8). This work was supported by research funding from PRONON (Programa Nacional de Apoio à Atenção Oncológica, SIPAR 25000.057709/2015) together with Tetra Park Ltda, Raízen Combustiveis S/A, Globo S.A, Flextronics International Tecnologia Ltda, Águas Guariroba, Antibioticos do Brasil, Renovias Concessionária, Riguesa de Celulose, Empresa Catarinense de Transmissores de Energia, 3 M Manaus Ind. Prod. Químicos, STVD Holdings S.A, Bradesco Vida e Previdência, Prolagos S.A, Banco Haitongi, Astra S.A. Indústria e Comércio, Buckman Laboratórios, AEGEA Saneamento e Participações S.A, Riguesa do Nordeste, Rud Correntes, Rico Corretora, Finamax S/A Credito Financ. e Investimento, Japi Indústria e Comércio S.A, Manoel Fernandes Flores, LVE—Locadora de Veículos e Equipamentos Ltda and Danilo Rabetti.
ETHICS STATEMENT
The authors have confirmed ethical approval statement is not needed for this submission.
PATIENT CONSENT STATEMENT
The authors have confirmed patient consent statement is not needed for this submission.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We are thankful to Dr. Anton W Langerak for providing access to the files analyzed in the original cIT‐QC publication. We are also grateful to the Vidjil team for helping to ease our introduction to the software.
Giusti GNN, Ribeiro AV, Jotta PY, Thonier F, Yunes JA, Meidanis J. Vidjil add‐on for MRD quantification of samples processed using the EuroClonality‐NGS protocol. eJHaem. 2023;4:770–774. 10.1002/jha2.749
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in European Nucleotide Archive (ENA) at EMBL‐EBI, reference number under accession number PRJEB32195 (https://www.ebi.ac.uk/ena/browser/view/PRJEB32195) [12]. The files used are listed in the spreadsheet cIT‐QC_normalization.xlsx, available as supplementary material.
REFERENCES
- 1. van Dongen JJM, Seriu T, Panzer‐Grümayer ER, Biondi A, Pongers‐Willemse MJ, Corral L, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352(9142):1731–8. [DOI] [PubMed] [Google Scholar]
- 2. van der Velden VHJ, Hochhaus A, Cazzaniga G, Szczepanski T, Gabert J, van Dongen JJM. Detection of minimal residual diseasein hematologic malignancies by real‐time quantitative PCR: principles, approaches, and laboratory aspects. Leukemia. 2003;17(6):1013–34. [DOI] [PubMed] [Google Scholar]
- 3. Tran TH, Hunger SP. The genomic landscape of pediatric acute lymphoblastic leukemia and precision medicine opportunities. Semin Cancer Biol. 2020;84:144–52. [DOI] [PubMed] [Google Scholar]
- 4. Faham M, Zheng J, Moorhead M, Carlton VEH, Stow P, Coustan‐Smith E, et al. Deep‐sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120(26):5173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheng S, Inghirami G, Cheng S, Tam W. Simple deep sequencing‐based post‐remission MRD surveillance predicts clinical relapse in B‐ALL. J Hematol Oncol. 2018;11(1):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Knecth H, Reigl T, Kotrová M, Appelt F, Stewart P, Bystry V, et al. Quality control and quantification in IG/TR next‐generation sequencing marker identification: protocols and bioinformatic functionalities by EuroClonality‐NGS. Leukemia. 2019;33(9):2254–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brüggemann M, Kotrová M, Knecht H, Bartram J, Boudjogrha M, Bystry V, et al. Standardized next‐generation sequencing of immunoglobulin and T‐cell receptor gene recombinations for MRD marker identification in acute lymphoblastic leukaemia: a EuroClonality‐NGS validation study. Leukemia. 2019;33(9):2241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Giusti GNN, Jotta PY, Lopes CO, Ganazza MA, Azevedo AC, Brandalise SR, et al. Test trial of spike‐in immunoglobulin heavy‐chain (IGH) controls for next generation sequencing quantification of minimal residual disease in acute lymphoblastic leukaemia. Br J Haematol. 2020;189(4):e150–4. [DOI] [PubMed] [Google Scholar]
- 9. Darzentas N. ARResT/interrogate immunoprofiling platform: concepts, workflows, and insights. Methods Mol Biol. 2022;2453:571–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Giraud M, Salson M, Duez M, Villenet C, Quief S, Caillault A, et al. Fast multiclonal clusterization of V(D)J recombinations from high‐throughput sequencing. BMC Genomics. 2014;15(1):409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duez M, Giraud M, Herbert R, Rocher T, Salson M, Thornier F. Vidjil: a web platform for analysis of high‐throughput repertoire sequencing. PLoS One. 2016;11(11):e0166126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Toribio AL, Alako B, Amid C, Cerdeño‐Tarrága A, Clarke L, Cleland I, et al. European Nucleotide Archive in 2016. Nucleic Acids Res. 2017;45(D1):D32–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in European Nucleotide Archive (ENA) at EMBL‐EBI, reference number under accession number PRJEB32195 (https://www.ebi.ac.uk/ena/browser/view/PRJEB32195) [12]. The files used are listed in the spreadsheet cIT‐QC_normalization.xlsx, available as supplementary material.