

Summary
Studying endogenous proteins in mice has provided numerous insights into the physiological and pathological roles of these proteins. However, the availability and specificity of protein-specific antibodies often limit such studies. Here we present a protocol for generating epitope tag knock-in mice with CRISPR-Cas9-mediated gene editing. We discuss key considerations for tag selection and knock-in location and provide insights into CRISPR design. Subsequently, we outline the sequential steps involved in knock-in mouse generation, genotyping, and validation and explore potential applications.
For complete details on the use and execution of this protocol, please refer to Zhang et al. (2022).1
Subject areas: Genetics, CRISPR
Graphical abstract
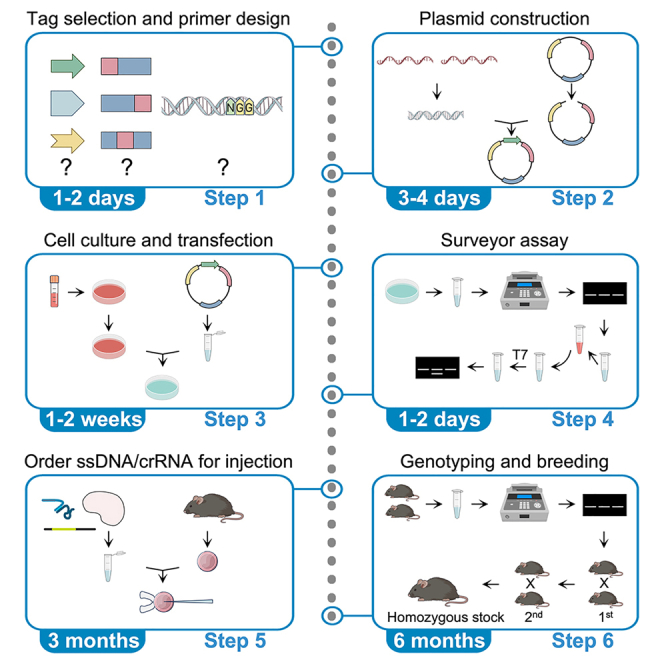
Highlights
-
•
Protocol for efficiently generating epitope tag knock-in mice using CRISPR-Cas9
-
•
Detailed discussion of tag selection, primer design, and plasmid construction
-
•
Testing the efficiency of CRISPR-Cas9 in cell lines with a surveyor assay
-
•
Step-by-step guide for epitope tag knock-in mouse generation and validation
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Studying endogenous proteins in mice has provided numerous insights into the physiological and pathological roles of these proteins. However, the availability and specificity of protein-specific antibodies often limit such studies. Here we present a protocol for generating epitope tag knock-in mice with CRISPR-Cas9-mediated gene editing. We discuss key considerations for tag selection and knock-in location and provide insights into CRISPR design. Subsequently, we outline the sequential steps involved in knock-in mouse generation, genotyping, and validation and explore potential applications.
Before you begin
One major limitation in mouse research is the lack of reliable and high-quality endogenous antibodies, particularly when the protein of interest is poorly studied. To overcome this challenge, we have developed a robust and standardized approach for generating epitope tag knock-in mice, which proves especially valuable when antibodies for the proteins of interest are either unavailable or unsatisfactory. The protocol outlined in this manuscript provides detailed instructions for generating a genetically modified mouse model with a 3×Flag tag knock-in at the N-terminus of Predicted gene 4951 (Gm4951). In principle, this protocol can be adapted to incorporate various tags for tagging any protein of interest in mice. However, careful consideration must be given to the selection of different tags and insertion sites (N-terminus, C-terminus, or within the protein) to ensure that the inserted tag does not interfere with the expression and functionality of the endogenous protein.
Institutional permissions
All experiments described in this protocol were conducted with the approval of the Institutional Animal Care and Use Committee (IACUC) at the University of Texas Southwestern Medical Center. As this protocol involves the use of live vertebrates, individuals intending to perform these procedures should ensure they obtain the necessary permissions from the appropriate institutions.
Tag, insertion location selection, primer design, and other preparations
Timing: 1–2 days
-
1.Choose a suitable tag to tag your protein of interest.
-
a.There are many options for tags with commercially available antibodies, including HA, Flag, Myc, V5, GFP, His, GST, etc. Depending on the size of the tagged protein, for in vivo studies, we prefer to use tags that are relatively small with reliable and specific antibodies.
-
b.The selection of the tag also depends on your major applications and the tissues of interest. If live cell/tissue imaging will be used, GFP or other fluorescence tags are preferred. While most tag antibodies can be used in different applications such as western blot, immunoprecipitation, and immunohistology, the non-specific signals might vary. Different cells/tissues may also exhibit different non-specific signals, which can be determined using wild-type cells/tissues in different applications. These non-specific signals might be problematic when studying proteins with low abundance.
-
c.The selected tag should not interfere with the expression and function of your protein. This can be tested ahead of your knock-in project by constructing tagged proteins and checking their expression in mammalian cells. The function of the tagged protein can also be tested in vitro if a functional assay system is available. The choice of functional assays depends on specific proteins and should be determined based on previous experience or published studies.
-
d.We prefer using 3×Flag as a versatile tag for generating knock-in mice. The 3×Flag tag improves the sensitivity and affinity of the original Flag tag by fusing three tandem Flag tags. 3×Flag tagged proteins are especially suitable for immunoprecipitation, as they can be eluted with 3×Flag peptides under neutral pH conditions.
-
a.
-
2.Decide where to insert the tag in your target protein.
-
a.The tag could be inserted at either the N-terminus or the C-terminus of your target protein. The key consideration here is to ensure that the tag does not interfere with the expression and function of your protein. Similarly, this can be tested by constructing N-terminal and C-terminal tagged proteins and checking their expression in mammalian cells or performing in vitro functional assays.
-
b.Check if your target protein has different isoforms that may have different start codons or stop codons. The location of the tag depends on which isoform (s) you would like to track.
-
c.Most membrane proteins and secreted proteins have a signal peptide at the N-terminus, which is usually cleaved off co-translationally.2 N-terminal tagging should be avoided for proteins with potential N-terminal signal peptides.
-
d.There might be some transcriptionally regulatory sites upstream of the coding sequence. N-terminal tagging has the potential to affect mRNA transcription efficiency. Refer to the potential solution to problem 4 in the troubleshooting section for more details.
-
a.
-
3.Choose single guide RNA (sgRNA) target loci and design primers.
-
a.Once a suitable terminus is identified, download the sequence of approximately 100 base pairs (bp) around the desired insertion site from NCBI.
-
b.Search for potential sgRNA targets with high efficiency and low off-targets. We routinely use the CRISPOR tool (http://crispor.tefor.net).3
-
c.Ideally, the cut site [3 bp upstream of the protospacer adjacent motif (PAM) in the case of Cas9] should be as close to the desired insertion site as possible. We achieved good tag knock-in efficiency with cut sites that are within 10 bp of the desired insertion sites. However, successful tag knock-ins can still be achieved with cut sites up to 20 bp away from the desired insertion sites, albeit with decreased efficiency.
-
d.Select three sgRNA sites and add linkers to the 20 bp guide sequence for cloning into vectors. Order two primers for each sgRNA site: forward primer sequence: 5′-CACCG [forward sequence] -3′, reverse primer sequence: 5′-AAAC [reverse complement sequence] C-3′.
-
e.Design a set of primers that will be used to amplify a short fragment (200–500 bp) of DNA surrounding the sgRNA target sites for the surveyor assay. Ideally, these target sites should not be in the middle of the amplification region to make it easier to distinguish two different lengths of cleaved DNA on the agarose gel.
-
a.
-
4.Prepare other required items.
-
a.An sgRNA expressing vector is required for the surveyor assay to test the efficiency of CRISPR-Cas9 editing. We normally use the PX458 vector, which contains an EGFP marker, or the PX459 vector, which contains a puromycin marker for plasmid construction.4 Both plasmids can be obtained from Addgene.
-
b.Choose a mouse cell line that is easy to grow and has a high transfection efficiency. We prefer to use Neuro-2a cells, which are mouse neuroblast. This cell can be obtained from ATCC.
-
c.Seek available spaces in animal resource facilities and consult mouse microinjection facilities for the project.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-FLAG M2 antibody, 1:5000 dilution | Sigma-Aldrich | F3165 |
| Rabbit anti-GAPDH (D16H11) antibody, 1:1000 dilution | Cell Signaling Technology | 5174 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG, Light Chain Specific, 1:5000 dilution | Jackson ImmunoResearch | 115-035-174 |
| Peroxidase AffiniPure Goat Anti-Rabbit IgG (H + L), 1:5000 dilution | Jackson ImmunoResearch | 111-035-144 |
| Bacterial and virus strains | ||
| Stellar Competent Cells | Clontech | 636763 |
| Chemicals, peptides, and recombinant proteins | ||
| EMEM | ATCC | 30–2003 |
| FBS | ATCC | 30–2020 |
| Penicillin-Streptomycin 100× | Gibco | 15-140-122 |
| 1 M Tris-Cl pH 8.3 | Teknova | T1083 |
| KCl | Sigma-Aldrich | P9541 |
| MgCl2.6H2O | Sigma-Aldrich | M2393 |
| IGEPAL | Sigma-Aldrich | I8896 |
| Tween 20 | Sigma-Aldrich | P9416 |
| Gelatin | Sigma-Aldrich | G9391 |
| 1 M Tris-Cl pH 7.6 | Teknova | T1076 |
| NaCl | Sigma-Aldrich | S3014 |
| Sodium deoxycholate | Sigma-Aldrich | 30970 |
| SDS | Sigma-Aldrich | L3771 |
| 10× CutSmart Buffer | NEB | B7204 |
| BbsI-HF | NEB | R3539S |
| 10× T4 Ligase Buffer | NEB | B0202S |
| Lipofectamine 2000 | Invitrogen | 11668019 |
| Opti-MEM | Gibco | 31985070 |
| Proteinase K | Roche | 3115879001 |
| JumpStart Taq ReadyMix | Sigma-Aldrich | P0982 |
| 10× NEBuffer 2 | NEB | B7002S |
| T7 endonuclease | NEB | M0302S |
| Protease inhibitor | Roche | 11873580001 |
| 4%–12% NuPAGE Bris-Tris gel | Invitrogen | NP0336BOX |
| 20× MOPS buffer | Invitrogen | NP001 |
| Nitrocellulose Transfer Kit | Bio-Rad | 1704270 |
| 10× TBST | Teknova | T9511 |
| Critical commercial assays | ||
| QIAquick Gel Extraction Kit | QIAGEN | 28706 |
| QIAprep Spin Miniprep Kit | QIAGEN | 27106 |
| Experimental models: Cell lines | ||
| Neuro-2a cells | ATCC | CCL-131 |
| Experimental models: Organisms/strains | ||
| C57BL/6J mice (4–6 weeks old, male and female) | The Jackson Laboratory | 000664 |
| Oligonucleotides | ||
| Primers | IDT | N/A |
| ssDNA oligos | IDT | N/A |
| crRNA | IDT | N/A |
| Recombinant DNA | ||
| PX458 | Addgene | 48138 |
| PX459 | Addgene | 62988 |
Materials and equipment
Neuro-2a cell culture medium
| Reagent | Final concentration | Amount |
|---|---|---|
| EMEM | N/A | 445 mL |
| FBS | 10% v/v | 50 mL |
| Penicillin-Streptomycin 100× | 1× | 5 mL |
| Total | N/A | 500 mL |
Note: After preparation, the medium is stable for up to 12 weeks at 4° C.
PCR buffer with nonionic detergents (PBND) buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 M Tris-Cl pH 8.3 | 10 mM | 445 mL |
| KCl | 50 mM | 1.87 g |
| MgCl2.6H2O | 2.5 mM | 0.255 g |
| IGEPAL | 0.45% v/v | 2.25 mL |
| Tween 20 | 0.45% v/v | 2.25 mL |
| Gelatin | 0.1 mg/mL | 0.05 g |
| ddH2O | N/A | To 500 mL |
| Total | N/A | 500 mL |
Note: The prepared medium needs to be autoclaved for sterilization. Gelation will dissolve only after autoclaving. The PBND buffer should be aliquoted and frozen at −20° C. When stored at 4° C, the PBND solution is stable for up to 4 weeks. Add 10 mg/mL Proteinase K stock solution at a 1:100 dilution into PBND buffer immediately before use to prepare the working solution. It is recommended to freshly prepare an adequate amount of PBND working solution each time.
RIPA lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 M Tris-Cl pH 7.6 | 25 mM | 12.5 mL |
| 5 M NaCl | 150 mM | 15 mL |
| IGEPAL | 1% v/v | 5 mL |
| Sodium deoxycholate | 1% w/v | 5 g |
| 10% SDS | 0.1% w/v | 5 mL |
| Total | N/A | 500 mL |
Note: Store the RIPA buffer at 4° C and add a proteasome inhibitor before use. The RIPA buffer remains stable for up to 6 months when stored at 4° C.
Step-by-step method details
Plasmid construction
This section describes how to generate plasmids that express selected sgRNAs for the surveyor assay to test the efficiency of CRISPR-Cas9 editing.
-
1.Preparation of digested vector.
-
a.Digest 1 μg of the vector with BbsI for 1 h at 37° C.Enzyme digestion mix
Reagent Amount Vector 1 μg 10× CutSmart Buffer (NEB) 2 μL BbsI-HF (NEB) 0.5 μL ddH2O to 20 μL -
b.Run the digested vector on a 1% agarose gel. You should observe ∼9 kb band corresponding to the linearized vector.
-
c.Excise the linearized vector band and perform gel purification using a QIAquick Gel Extraction Kit following the manufacturer’s protocol or other commercially available gel extraction kits.
-
a.
-
2.Annealing of oligos and ligation.
-
a.Prepare the DNA annealing mix using the ordered oligos as follows:DNA annealing mix
Reagent Amount Forward oligo (100 μM) 1 μL Reverse oligo (100 μM) 1 μL 10× T4 Ligase Buffer (NEB) 1 μL ddH2O to 10 μL -
b.Anneal the oligos using a thermocycler with the following program:DNA annealing program
Temperature Time 95° C 5 min 95° C (−1° C/cycle) 1 min × 70 cycles 4° C Hold -
c.Prepare the following DNA ligation mix and incubate at room temperature for 10 min or at 16° C overnight.DNA ligation mix
Reagent Amount Digested vector (50 ng/μL) 1 μL Annealed oilgos (1:200 dilution in ddH2O) 1 μL 10× T4 Ligase Buffer (NEB) 1 μL T4 DNA Ligase 0.5 μL ddH2O To 10 μL  CRITICAL: The annealed oligo duplex needs to be diluted at least 1:200 or further. A high concertation of the oligo duplex significantly reduces the ligation efficiency.
CRITICAL: The annealed oligo duplex needs to be diluted at least 1:200 or further. A high concertation of the oligo duplex significantly reduces the ligation efficiency.
-
a.
-
3.
Transform the ligation product using Stellar Competent Cells (Clontech) following the manufacturer’s protocol. Other recombination-deficient strains with high transformation efficiency can also be used.
-
4.
Pick single clones to culture them in 2 mL TB medium at 37° C overnight.
-
5.
Prepare plasmids using the QIAprep Spin Miniprep Kit (QIAGEN) following the manufacturer’s protocol.
-
6.
Send the purified plasmids for DNA Sanger sequencing to verify the correct insertion of sgRNA targets. We use the hU6-F (5′-GAGGGCCTATTTCCCATGATT-3′) primer to sequence the sgRNA insertion region.
-
7.
To improve the quality of the plasmids for transfection, re-amplify the sequencing-verified clone using QIAGEN Plasmid Plus Midi Kit following the manufacturer’s protocol.
Optional: Alkaline phosphatase treatment of the vector can be performed during the enzyme digestion. If the vector is dephosphorylated, use T4 polynucleotide kinase during the annealing step to add 5′ phosphorylation to the oligos. Since the sticky ends formed from two BbsI digestion sites are not compatible, the self-ligation rate is low for these CRISPR vectors.
Note: When analyzing the sequencing data, carefully check both the sequence of the sgRNA target and the adjacent vector sequence. Mutations can be induced during oligo production, and the adjacent vector sequence might be altered during enzyme digestion or DNA ligation.
Cell culture and transfection
This major step describes the culture and transfection of Neuro-2a cells with CRISPR plasmids to test the efficiency of different sgRNA targets.
-
8.
Thaw the frozen vial of Neuro-2a cells obtained from ATCC (CCL-131) by placing it in a 37° C water bath with gentle agitation. Transfer the cells to a centrifuge tube containing 9 mL of cell culture medium and centrifuge at 125 × g for 5 min.
-
9.
Discard the supernatant and resuspend the cell pellet with 10 mL cell culture medium. Transfer the cells into a 10 cm culture dish and incubate them at 37° C in an incubator with 5% CO2.
-
10.
Subculture the cells every 2–3 days at a subcultivation ratio of 1:3 to 1:6. Cells should be passaged for at least 1–2 generations before transfection to ensure they have fully recovered from the frozen stock.
-
11.
One day before transfection, plate 2–5 × 105 cells in 2 mL of cell culture medium in a well of a 6-well plate so that cells reach 70%–90% confluent at the time of transfection.
-
12.Transfect Neuro-2a cells using Lipofectamine 2000.
-
a.Dilute 10 μL of Lipofectamine 2000 in 125 μL of Opti-MEM medium for each well of the 6-well plate. Incubate for 5 min at room temperature.
-
b.Dilute 4 μg of plasmid DNA in 125 μL of Opti-MEM.
-
c.Add the diluted DNA to the diluted Lipofectamine 2000 (total volume = 250 μL), mix gently, and incubate for 20 min at room temperature.
-
d.Add the 250 μL DNA-lipid complex to each well and mix gently.
-
a.
Genomic DNA isolation and surveyor assay
This major step describes the preparation of genomic DNA from Neuro-2a cells and surveyor assay using the genomic DNA to test the efficiency of different sgRNA targets.
-
13.
48 h after transfection, remove the medium from cells and then harvest cells with 200 μL of PBND buffer (with added protease K).
-
14.
Transfer the cell lysate to a PCR tube and put it in a thermocycler with the following program:
Genomic DNA preparation program
| Temperature | Time |
|---|---|
| 56° C | 60 min |
| 95° C | 5 min |
| 4° C | Hold |
-
15.Surveyor PCR.
-
a.Prepare the PCR reaction mix with the following surveyor PCR oligos:Surveyor PCR mix
Reagent Amount Forward oligo (10 μM) 2 μL Reverse oligo (10 μM) 2 μL 2× JumpStart Taq ReadyMix 25 μL Crude genomic DNA 1 μL ddH2O to 50 μL -
b.Run the PCR reaction in a thermocycler with the following program:Surveyor PCR program
Temperature Time 95° C 2 min 95° C 30 s 35 cycles 55° C–68° C (Average Tm of primers - 5° C) 30 s 72° C 1 min 72° C 5 min 4° C Hold
-
a.
-
16.
Analyze the PCR product using an agarose gel. Cut out the band corresponding to the desired size and perform gel purification using QIAquick Gel Extraction Kit (QIAGEN) following the manufacturer’s protocol.
Note: Ideally, there should be only a single band that corresponds to the desired size. Exercise caution to only cut the target band if there are multiple bands present on the gel. Since the quality of the PCR product is critical for the subsequent T7 endonuclease assay, it is advisable to design a new set of primers to obtain more specific PCR products.
-
17.Perform the surveyor nuclease assay with T7 endonuclease.
-
a.Prepare the following T7 annealing mix:T7 annealing mix
Reagent Amount Purified PCR products 200 ng 10× NEBuffer 2 (NEB) 2 μL ddH2O to 19 μL -
b.Anneal the PCR products in a thermocycler using the same DNA anneal program as described in step 2b.
-
c.Add 1 μL of T7 endonuclease I (NEB) to the annealed PCR products and incubate the mixture at 37° C for 15 min.
-
d.Stop the reaction by adding 1.5 μL of 0.25 M EDTA.
-
e.Add DNA loading buffer and run the mixture on an agarose gel to assess the T7 cleavage efficiency.Note: For each sgRNA target, it is necessary to set up a non-T7 endonuclease cleaved control and run it alongside the T7 endonuclease cleaved sample. Half of the gel-purified PCR product can be used for the T7 reaction, while the remaining purified DNA can serve as the control. Compared to the non-cut controls, the T7 endonuclease cleavage should result in two fragments that are locates close to the sgRNA target site: one upstream and one downstream. Refer to Figure 1 for an example of a successful assay. Three targets were chosen for each Genes. The length of the surveyor assay PCR products and the predicted sizes after T7 endonuclease cleavage are listed in Figure 1A. In the actual result shown in Figure 1B, T1, T3, T4, and T5 exhibited higher efficiency compared to T2 and T6.
-
a.
Figure 1.

Example of surveyor assay results
(A) Predicted size of PCR products in the surveyor assay before and after T7 cleavage.
(B) Surveyor assay results of two different genes.
Order oligo template and CRISPR RNA (crRNA) and perform microinjection
This major step describes how to design and order oligo template and crRNA for microinjection. Both single-stranded DNA (ssDNA) oligonucleotide and donor plasmid have been used as templates for homology-directed repair (HDR). In general, the ssDNA template offers a higher insertion efficiency and requires shorter homologous arms compared to the dsDNA template.5,6 On the other hand, both short ssDNA (<200 bp) and long ssDNA (>200 bp) can be synthesized directly by major vendors within a reasonable time and cost. Compared to sgRNA, we prefer a two-part guide RNA system, which includes crRNA and trans-activating CRISPR RNA (tracrRNA). The tracrRNA pairs with complementary sequences within the crRNA to form a functional gRNA duplex (crRNA:tracrRNA) (Figure 2A). Using this system, only crRNA needs to be ordered for each CRISPR project, and tracrRNA is the same for different targets, providing high activity at an affordable price. For the form of Cas9 nuclease, recombinant S. pyogenes Cas9 protein is a better option compared to Cas9 mRNA due to increased stability and the absence of the need for protein translation. All these reagents were ordered from Integrated DNA Technologies, Inc (IDT).
-
18.Compare the genomic editing efficiency of different sgRNA targets from the surveyor assay and select one target site with the highest efficiency and the least distance to the tag insertion site. The HDR template will be designed according to the selected site.
-
a.When designing the HDR template, the length of the homolog arm and the selection of the target vs. non-target strand will affect the efficiency of insertion. In our hands, we achieved the highest HDR rate with asymmetric donor DNA and the target strand, as reported previously.7
-
b.As shown in Figure 2A, the crRNA:tracrRNA:Cas9 complex targets the DNA strand complementary to the gRNA and generates a double-strand break 3 bp upstream of the NGG PAM sequence.
-
c.Figure 2B shows an example of a ssDNA oligo template design when a 3×Flag tag is desired to be inserted at the N-terminus of a protein. The template was designed as the target strand with 36 bp before the 3×Flag tag and 91 bp after the 3×Flag tag. Keep in mind that the left homologous arm of the 3×Flag tag should not be less than 36 bp.
-
d.If the cut site is not exactly the site of insertion, the homologous arm on one side needs to be longer to take the extra DNA sequence between the cut site and insertion site into consideration.
-
a.
-
19.
Order the crRNA and ssDNA oligo from IDT. When ordering the crRNA, enter the 20 bp DNA sequence upstream of the PAM site in IDT’s online ordering tool, which automatically converts the DNA sequence to RNA during the ordering process. We normally order 2 nmol of crRNA and 4 nmol of Ultramer DNA oligo with standard desalting purification.
-
20.
Deliver the crRNA and ssDNA oligo to the UT Southwestern Transgenic Core for microinjection. Many institutions offer mouse microinjection services in core facilities. If microinjection is not available at the reader’s institution, there are other commercial alternatives, such as Charles River.
-
21.
Cas9 protein and tracrRNA were provided by the UT Southwestern Transgenic Core and mixed with crRNA and ssDNA oligo for the microinjection.
Note: C57BL/6 mice are the most commonly used inbred mice in biochemical research and are generally used to produce CRISPR mice. There are several different substrains of C57BL/6 mice, such as C57BL/6J and C57BL/6N, which carry different mutations.8 Readers need to check carefully and choose a strain that does not affect their research.
-
22.
At the UT Southwestern Transgenic Core, zygotes were prepared, injected with crRNA-tracrRNA/Cas9/ssDNA, and then transferred into pseudopregnant foster mothers. Pups were born and delivered to the requester around the weaning age (3–4 weeks).
Figure 2.

Design of HDR template
(A) Illustration depicting the binding of Cas9-crRNA-tracrRNA complex with the target DNA.
(B) Example of asymmetric donor DNA design with the target strand, resulting in a high HDR rate.
Genotyping and breeding of mice
This major step describes how to genotype and breed CRSIRP-modified mice. For genotyping, we typically use PCR genotyping as the initial step to check for the presence of the tag insertion. Subsequently, only the PCR-positive mice are sequenced to determine the exact nucleotide sequence of the tag and flanking regions. Genotyping-verified founders are then bred to establish individual knock-in mouse lines.
-
23.Design primers for PCR and sequencing (Figure 3).
-
a.Design a primer set (FT and RT in Figure 3A) to detect the 3×Flag tag in front of ATG. This primer set can only detect the tag insertion but cannot distinguish between one copy (hemizygous) and two copies (homozygous) of the tag insertion.
-
b.Design a primer set (FI and RI in Figure 3A) to detect the insertions on two chromosomes. This primer set can distinguish between hemizygous and homozygous insertions but cannot determine if the inserted DNA is the 3×Flag tag.
-
a.
Note: Keep in mind that these tags are very small (66 bp for 3×Flag tag), so the amplification regions should be designed to be small enough to differentiate a tag insertion.
-
24.During weaning, tag mice and perform tail snipping for genotyping.
-
a.Lysis tails with 200 μL of PBND buffer (with added protease K) and incubate on a 56° C shaker for 2 h to overnight until the tissue is completely lysed.
-
b.Heat the samples at 95° C for 5 min to inactivate the proteinase K.
-
c.Prepare the following genotyping PCR reaction mix:Genotyping PCR mix
Reagent Amount Forward oligo (10 μM) 1 μL Reverse oligo (10 μM) 1 μL 2× JumpStart Taq ReadyMix 12.5 μL Crude genomic DNA 0.5 μL ddH2O to 25 μL -
d.Run the PCR reaction in a thermocycler with the following program:Genotyping PCR program
Temperature Time 95° C 2 min 95° C 30 s 35 cycles 55° C–68° C (Average Tm of primers - 5° C) 30 s 72° C 30 s 72° C 5 min 4° C Hold -
e.Mice with positive results from both FT + RT and FI + RI PCR gels should be kept for sequencing to verify the correct sequence of the tag and flanking regions.
-
f.From the FI + RI PCR gel, cut the larger tag insertion band in hemizygous and homozygous mice and perform gel purification with QIAquick Gel Extraction Kit (QIAGEN) following the manufacturer’s protocol. Send the purified PCR product and use either FI or RI primer for Sanger sequencing.
-
a.
-
25.
For genotyping verified founders, set up breeding cages for each individual founder (G0) to breed with wild-type mice (C57BL/6J).
-
26.
Genotype the progeny (G1) and set up breeding cages for intercrossing these G1 mice for each founder line. Keep the homozygous progeny (G2) and establish breeding cages for a homozygous stock line.
Figure 3.

Design of primers for genotyping tag knock-in mice
(A) Illustration showing the design of two primer sets (FI + RI, FT + RT) for genotyping.
(B) Anticipated agarose gel results showing different genotypes.
Testing the expression of the tag in different mouse tissues
This major step describes how to test the expression of the tag in different mouse tissues. Although homozygous tag knock-in mice are more desirable for experiments, hemizygous tag knock-in mice might be sufficient to assess the tag expression of proteins with reasonable abundance. Age- and sex-matched wild-type mice should be dissected in the same manner as the tag knock-in mice to serve as controls. Western blot is typically used to test the tag expression.
-
27.Dissect different mouse tissues and prepare samples for western blot.
-
a.Dissect mouse tissues and place them in 2 mL beating tubes filled with ceramic beads. Snap-freeze these tubes in liquid nitrogen.CRITICAL: The weight of different tissues needs to be optimized to obtain sufficient protein for western blot while also ensuring complete lysis. As a rule of thumb, we use 100 mg of tissue per 1 mL of lysis buffer for most tissues, except for adipose tissues, which require 250 mg of tissue per 1 mL of lysis buffer.
-
b.Once the dissection is complete, fill the tube with 1 mL of cold RIPA lysis buffer (with freshly added protease inhibitor) and use bead mill homogenizer to lyse the tissues.CRITICAL: The tissue lysates should be kept at 4° C to slow down protein degradation. Bead mill homogenizers generate a significant amount of heat during high-speed or prolonged homogenization. For tough tissues, it is necessary to break down the homogenization process into separate sections and cool down samples on ice in between these sections.
-
c.Sonicate tissue lysates on ice to break down genomic DNA into small pieces to reduce viscosity.
-
d.Centrifuge at 12,000 rpm for 30 min at 4° C. Transfer the supernatant into a new tube and measure the protein concertation using the Pierce BCA Protein Assay Kit, following the manufacturer’s protocol.
-
e.Adjust the protein concentration with RIPA lysis buffer and 4× LDS loading buffer. The final protein concertation of all mouse tissue lysates should be the same in 1× LDS loading buffer (around 2–10 μg/μL).
-
f.Load 20 μg of samples into each well of 4%–12% NuPAGE Bris-Tris gels and run the gels with 1× MOPS buffer at 120V for 1–2 h.
-
g.Transfer the proteins from the gels to NC membranes at 15 V for 30 min, then incubate and wash the members for signal detection.
-
i.Block the membranes with 5% non-fat milk in 1×TBST at room temperature for 1 h.
-
ii.Incubate the membranes with the primary antibody diluted in 1% BSA 1×TBST at 4° C overnight.
-
iii.Wash the membranes with 1×TBST for 15 min three times.
-
iv.Incubate the membranes with the secondary antibody diluted in 1% BSA 1×TBST at room temperature for 1 h.
-
v.Wash the membranes with 1×TBST for 15 min three times.
-
vi.Visualize the membranes using a chemiluminescent substrate.
-
i.
-
a.
Expected outcomes
Using the method described above, we have generated seven different tag knock-in mouse models, incorporating either a 3×Flag tag or a GFP tag. The efficiency of the CRISPR-mediated tag knock-in approach has been remarkably high, and we have successfully detected tag expression in six of the knock-in mice lines. Figure 4 illustrates an example of the expected outcomes from Gm4951 knock-in mice with a 3×Flag tag. These established tag knock-in mouse models offer valuable tools for various applications. They enable the examination of tagged protein expression in different tissues and primary cells using western blotting, as well as the investigation of the subcellular localization of tagged proteins in tissues or cells through immunostaining. Moreover, the tag knock-in mice can be employed for protein pull-down experiments to analyze interacting proteins using mass spectrometry or identify interacting RNAs/DNAs through sequencing approaches. Furthermore, this protocol can be extended to a wide range of other applications. For instance, it can be utilized for introducing fluorescence tags to label and trace cell lineages or for inserting Cre or Cre/ERT2 to conditionally knock out specific genes.
Figure 4.

Expected outcomes of a 3×Flag tag knock-in project
(A) Expression of the GM4951 protein with a 3×Flag tag in several tissues from two different founders. The highest expression of the GM4951 protein was observed in the liver, which is consistent with the expression profile of the Gm4951 mRNA.
(B) Genotyping results of 3×Flag tag Gm4951 knock-in mice after breeding for two generations. Two sets of genotyping primers (FI + RI, FT + RT) were used.
Limitations
The protocol outlined above has certain limitations that need to be considered. One major limitation is the potential impact of knock-in tags on the function of tagged proteins. To address this, it is important to perform functional assays in vitro, if available, to assess the effects of different types of tags and tag insertion locations. For novel proteins without established in vitro assays, it is advisable to check conserved domains and AlphaFold predicted structures to avoid adding tags near crucial domains. The most reliable approach to assess the impact of a knock-in tag on protein function is to generate homozygous knock-in mice and compare them with homozygous knockout mice.
Additionally, it should be noted that not all tags are suitable for all applications. For instance, if a tag is not exposed on the surface of a protein, it may only be detectable by Western blot and not by immunoprecipitation or immunostaining. Different tags may also exhibit varying background signals in different applications. Therefore, it is essential to include age and sex-matched wild-type control mice during the initial characterization of tag knock-in mice to evaluate background signals. Considering the cost and time involved in generating tag knock-in mice, it is recommended to test different tags for overexpressed proteins in cells using major applications such as Western blot, immunoprecipitation, and immunostaining.
Regarding the specific insertion sites, this protocol typically adds a tag immediately before the start codon (N-terminus) or after the stop codon (C-terminus) of the protein of interest. However, the limitation arises from the requirement of a PAM sequence in the vicinity of the insertion site for CRISPR-Cas9 targeting. To overcome this limitation, alternative CRISPR systems such as Cas12 or Cas13, or even PAM-free nucleases, can be employed to generate knock-in animals.9,10
Furthermore, it is important to consider the potential off-target effects associated with CRISPR targeting, which may result in genetic mutations beyond the intended target loci. To minimize off-target effects, careful selection of sgRNA targets with minimal predicted off-target sites is recommended. Additionally, breeding different knock-in mouse lines and comparing the tag signal, as well as assessing any noticeable phenotypes, can provide valuable insights. Homozygous tag knock-in mice are more likely to exhibit phenotypic effects due to complete disruption of off-target sites. If hemizygous tag knock-in mice yield satisfactory results in tag detection, it is advisable to use hemizygous mice instead of homozygous mice.
Troubleshooting
Problem 1
No correct construct with designed target insertion or all constructs are empty vectors. Related to step 2.
Potential solution
A major cause of this problem is insufficient digestion of the vector. To solve this, the digestion time could be extended from 1 h to overnight. The quality of the vector plasmid is important for efficient enzyme digestion. During the lysis step of the alkaline lysis-based plasmid isolation protocol, prolonged exposure to the alkaline condition may cause the plasmid to denature and become resistant to enzyme digestion. If this is the reason for insufficient digestion, researchers are advised to prepare a new batch of the vector. Additionally, common errors such as adding adapter linker sequences in the wrong orientations to the 20 bp guide sequence or failure to dilute the annealed oligo duplex can also lead to failed construction. It is important to check for these errors and take necessary precautions to avoid them.
Problem 2
The transfection of sgRNA plasmids seems not working. Related to step 3.
Potential solution
Ensure that your method of transfection is working by checking the GFP expression (using the PX458 vector) or by using a control GFP-expressing plasmid (such as the PX459 vector). If the transfection efficiency is very low as indicated by GFP-positive cells, it is recommended to check the quality and integrity of the plasmids and, if necessary, re-prepare them. The status of the cells also plays a role in transfection efficiency. It is advisable to freeze multiple tubes of cells at the first one or two passages and to replace the culture with a new one for high-passaging cells. While it is not necessary in most cases, transfected cells can be selected using a GFP marker (PX458) or a puromycin marker (PX459) to enrich CRISPR-Cas9 targeting cells.
Problem 3
The surveyor assay is not working, no cleavage of genomic DNA for all sgRNA targets tested. Related to step 4.
Potential solution
If the problem is due to poor surveyor PCR, the surveyor assay primers should be redesigned to generate a clear single band on the agarose gel. PCR conditions can also be optimized to increase the specificity of the primers. Adding a positive control sgRNA target that has been proven to work for a different target is advised to ensure the experiment is performed correctly. If the positive control works well, it suggests that the sgRNA targets may need to be redesigned.
Problem 4
There are no positive knock-in mice detected after genotyping, and all founders are wild-type mice. Related to step 5.
Potential solution
There might be several reasons for the negative genotyping result. First, readers should check the PCR/genotyping step to ensure the correct functioning of the genotyping PCR. For instance, incomplete proteinase K digestion of tails or insufficient inactivation of proteinase K in the final samples could be potential issues. Setting up different controls would be helpful in ensuring the accurate execution of the genotyping PCR. If the genotyping is accurate, the problem might arise from the microinjection process. Readers should send the PCR products amplified by the FI + RI primers for Sanger sequencing to check for small insertions and deletions (indels). The presence of indels suggests the effectiveness of the sgRNA and the microinjection, and the absence of knock-in mice may be caused by issues with the ssDNA template. If no indels are detected, the problem is likely due to a failed microinjection, which should be improved and repeated.
Problem 5
Genotyping of knock-in mice is right, but tag expression could not be detected by a specific assay. Related to step 6.
Potential solution
The insertion of the tag may potentially decrease the transcription of the tagged gene, particularly when the tag is added at the N-terminus of a protein. To determine if this is the cause, researchers can design tag-specific primers to quantify the mRNA level of the "tagged-protein" using quantitative PCR. While tag knock-in mice can be utilized for various assays, we recommend starting with Western blotting as an initial method to assess tag expression in major mouse tissues. If the tag insertion leads to reduced protein expression or if the endogenous protein level is already low, researchers can employ immunoprecipitation to enrich the tagged protein for Western blot analysis. The tag signal can also be enhanced by testing different combinations of primary and secondary antibodies. For example, in our experiments, we employed various approaches to detect Flag tag expression by Western blot. One approach involved using a mouse anti-Flag M2 antibody coupled with a goat anti-mouse IgG light chain HRP antibody (to avoid the 50 kD IgG heavy chain band), while the other approach involved using a rabbit anti-Flag antibody coupled with a goat anti-mouse whole IgG HRP antibody. We found that the first approach worked better in improving the Flag tag signal.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Zhao Zhang (Zhao.Zhang@UTSouthwestern.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate/analyze any dataset or code.
Acknowledgments
Research reported in this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R00DK115766 and R01DK130959. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
Z.Z. designed and supervised the work, performed experiments, and wrote the manuscripts.
Declaration of interests
The author declares no competing interests.
References
- 1.Zhang Z., Xun Y., Rong S., Yan L., SoRelle J.A., Li X., Tang M., Keller K., Ludwig S., Moresco E.M.Y., Beutler B. Loss of immunity-related GTPase GM4951 leads to nonalcoholic fatty liver disease without obesity. Nat. Commun. 2022;13:4136. doi: 10.1038/s41467-022-31812-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Owji H., Nezafat N., Negahdaripour M., Hajiebrahimi A., Ghasemi Y. A comprehensive review of signal peptides: Structure, roles, and applications. Eur. J. Cell Biol. 2018;97:422–441. doi: 10.1016/j.ejcb.2018.06.003. [DOI] [PubMed] [Google Scholar]
- 3.Concordet J.P., Haeussler M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018;46:W242–W245. doi: 10.1093/nar/gky354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miura H., Quadros R.M., Gurumurthy C.B., Ohtsuka M. Easi-CRISPR for creating knock-in and conditional knockout mouse models using long ssDNA donors. Nat. Protoc. 2018;13:195–215. doi: 10.1038/nprot.2017.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang H., Wang H., Shivalila C.S., Cheng A.W., Shi L., Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richardson C.D., Ray G.J., DeWitt M.A., Curie G.L., Corn J.E. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol. 2016;34:339–344. doi: 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- 8.Simon M.M., Greenaway S., White J.K., Fuchs H., Gailus-Durner V., Wells S., Sorg T., Wong K., Bedu E., Cartwright E.J., et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013;14:R82. doi: 10.1186/gb-2013-14-7-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collias D., Beisel C.L. CRISPR technologies and the search for the PAM-free nuclease. Nat. Commun. 2021;12:555. doi: 10.1038/s41467-020-20633-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickar-Oliver A., Gersbach C.A. The next generation of CRISPR-Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019;20:490–507. doi: 10.1038/s41580-019-0131-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze any dataset or code.