

Abstract
The A3 adenosine receptor has been implicated in modulation of cell growth. As a first step to the characterization of the underlying mechanisms, we exposed Chinese hamster ovary (CHO) cells transfected with the human A3 receptor (A3R-CHO) to selective A3 receptor ligands. At micromolar concentrations, the A3 agonists N6-(3-iodobenzyl)-adenosine-5’-N-methyluronamide (IB-MECA) and its 2-chloro derivative Cl-IB-MECA reduced cell number, with no effects on either parental CHO cells (not expressing any adenosine receptor), or CHO cells transfected with the human A1 receptor. Cl-IB-MECA also reduced cell number in the human HEK293 cell line transfected with the human A3 receptor cDNA as opposed to the respective untransfected wild-type cells. In A3R-CHO, agonist-induced effects were antagonized by nanomolar concentrations of A3 antagonists, including the triazoloquinazoline derivative MRS1220, the dihydropyridine derivative MRS1191, and the triazolonaphthyridine derivative L-249,313. A3 agonist-induced effects were not due to modulation of cell adhesion, nor to necrosis or apoptosis. Growth curves revealed highly impeded growth, and flow-cytometric analysis showed markedly reduced bromodeoxyuridine incorporation into nuclei. The effect on cell cycle was completely antagonized by MRS1191. Hence, activation of the human A3 receptor in A3R-CHO results in markedly impaired cell cycle progression, suggesting an important role for this adenosine receptor subtype in cell cycle regulation and cell growth.
Keywords: Adenosine, Adenosine receptor, Human A3, CHO transfected cells, HEK293 transfected cells, Cell growth modulation, Cell cycle
Introduction
Adenosine acts through four G-protein-coupled membrane receptors, the A1, A2A, A2B and A3 receptors (Fredholm et al. 1994). Whereas the existence of the A1, A2A and A2B receptors was postulated before they were cloned, the A3 receptor was discovered by cloning, initially from rat and subsequently from human tissues (Linden 1994). The development of selective agonists, such as N6-(3-iodobenzyl)-adenosine-5’-N-methyluronamide (IB-MECA; Gallo-Rodriguez et al. 1994) and its 2-chloro-derivative (Cl-IB-MECA; Kim et al. 1994), and, more recently, antagonists, has played a crucial role in the characterization of this receptor. By modifying the structures of both flavonoids and dihydropyridines, a series of highly potent and competitive A3 receptor antagonists has been obtained (Jacobson et al. 1997), including the dihydropyridine derivative 3-ethyl-5-benzyl-2-methyl-6-phenyl-4-phenylethynyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate (MRS1191; 1,300-fold selective for human A3 vs. rat A1 receptors; Jiang et al. 1996) and the triazoloquinazoline derivative 9-chloro-2-(2-furyl)-5-phenyl-acetylamino-[1,2,4]-triazolo-[1,5-c]-quinazoline (MRS1220; Ki-value of 0.65 nM at the human A3 receptor), which is the most potent A3 antagonist yet reported (Kim et al. 1996). At the same time, the Merck group (Jacobson et al. 1996) independently identified two antagonists highly selective for A3 human receptors, the non-competitive triazolonaphthyridine derivative 6-carboxymethyl-5,9-dihydro-9-methyl-2-phenyl-[1,2,4]-triazolo-[5,1-a]-[2,7]-naphthyridine (L-249,313) and the competitive triazolopyrimidine derivative (L-268,605).
Use of these ligands has generated important information on the pathophysiological role of this receptor. The A3 receptor is involved in inflammation (Linden 1994), hypotension and mast cell degranulation (Hannon et al. 1995), ischemic heart pre-conditioning (Linden 1994), behavioral depression (Jacobson et al. 1993), and modulation of cerebral ischemic damage (von Lubitz et al. 1994). Use of selective A3 agonists also revealed that this receptor profoundly affects cell survival by promoting both cell protection and cell death, depending upon the cell type and/or the agonist concentrations. At low concentrations in the nanomolar range, A3 agonists reduce hypoxic heart damage (Stambaugh et al. 1997), protect HL-60 and U-937 cells from apoptosis (Yao et al. 1997), and, in mammalian astrocytes, promote reinforcement of the cytoskeleton and intracellular redistribution of the anti-apoptotic protein Bcl-xL (Abbracchio et al. 1997). At high concentrations in the micromolar range, these same agonists induce death of cerebellar granule neurons (Sei et al. 1997), HL-60 cells (Kohno et al. 1996) and human lymphocytes (Barbieri et al. 1997). In some instances, A3 receptor-mediated cell death is apoptotic (Kohno et al. 1996; Barbieri et al. 1997; Yao et al. 1997), whereas in other instances (Sei et al. 1997) it is necrotic. Hence, depending upon differential coupling to transduction mechanisms, expression of cell- or tissue-specific factors, and/or the degree of cellular interaction, adenosine may act as a positive or negative regulator of cell survival through the A3 receptor. This makes the A3 receptor a promising therapeutic target (Jacobson et al. 1995) in pathologies where modulation of cell viability may be crucial. In particular, agonists at this receptor may prove useful in anti-cancer therapy, whereas antagonists may be beneficial in counteracting ischemia- and aging-associated neurodegeneration.
However, a clear elucidation of A3 receptor-mediated effects on cell survival is hampered by several factors, such as significant pharmacological differences between rat (where most preclinical studies are performed) and human receptors (Linden 1994), its dual effects on cell viability (see above), and the possible complicating presence of other adenosine receptors. This raises the need for simple experimental models, in which the biochemical, pharmacological and functional correlates of this receptor could be easily studied and new potentially selective agonist/antagonists could be tested. Recombinant expression of receptors in heterologous cell systems has become a valuable method to provide such models (Kenakin 1996). Expression of single receptors allows for the investigation of their transductional and functional correlates in sub-type-selective models where conflicting effects via multiple receptors do not occur. Recently, the human A3 receptor has been successfully transfected into CHO cells (Klotz et al. 1998). We have taken advantage of this clone (A3R-CHO) to characterize the functional effects evoked in these cells by A3 receptor agonists and antagonists, with particular attention to the effects on cell survival and on the cell cycle.
Materials and methods
Cell culture
Chinese hamster ovary (CHO) cells, both wild-type (WT-CHO) and transfected with either the A1 or A3 human adenosine receptors (A1R-CHO and A3R-CHO; Klotz et al. 1998), were maintained at 37°C in a humidified atmosphere in DMEM NUT MIX F-12 (Gibco, Life Technologies, European Division) supplemented with 10% fetal calf serum (FCS; HyClone, Celbio, Milan, Italy), penicillin (100 I.U./ml; Gibco, Life Technologies, European Division) and streptomycin (100 μg/ml; Gibco, Life Technologies, European Division); 0.5 mg/ml G418 (Boehringer-Mannheim, Germany) was added to the culture medium utilized for both the A1- and the A3-transfected clone. Human embryonal kidney cells (HEK293 cells), both wild-type (WT-HEK293) and transfected with the human A3 receptor (A3R-HEK293), were maintained at 37°C in a humidified atmosphere in DMEM (Gibco, Life Technologies, European Division) supplemented with penicillin (100 I.U./ml) and streptomycin (100 μg/ml); 0.5 mg/ml G418 was added to the culture medium utilized for the A3-transfected clone.
Experiments were performed by plating cells on glass coverslips in 24-well dishes (103 cells/well). Cultures were exposed to the various pharmacological agents by direct addition to the culture medium (see also Drugs and treatments).
Membrane preparation, binding studies and adenylyl cyclase assay
Membranes for binding studies and adenylyl cyclase assays were prepared as described recently (Klotz et al. 1998). Binding studies at A3 receptors were carried out in a 96-well microplate formate with 10 nM [3H]NECA as a radioligand (for details see Klotz et al. 1998). A3 receptor-mediated inhibition of adenylyl cyclase in membranes prepared from A3R-CHO was measured similarly to the procedure described for A2B receptor-mediated cyclase stimulation (Klotz et al. 1998) with minor modifications as described recently (Klotz et al. 1999).
Hoechst 33258 staining of cell nuclei and microscope analysis
To quantify cell number and to assess the possible induction of apoptosis, cells grown on glass coverslips for the time periods indicated in the legends were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS; pH 7.4) for 20 min at room temperature. Cultures were then incubated for 20 min at room temperature with 1 μg/ml Hoechst 33258 dye in 0.1% Triton X-100, 0.1% NaN3, 0.01% bovine serum albumine (BSA; all purchased from Sigma–Aldrich, Milan, Italy) in PBS. After three washes with PBS, coverslips were mounted with a glycerol-PBS (2:1) solution. Cell number was evaluated by using a fluorescence microscope (Zeiss, Germany) equipped with a UV filter and by counting cells in an identical area for each coverslip. At least 50 optical fields were counted for each coverslip: for control cultures, this corresponded to approximately 500 cells.
Cell viability assay
To assess the possible induction of necrosis, a well-established colorimetric bioassay based on the mitochondrial conversion of a soluble precursor, 3-(4,5-dimethylthyazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT; Sigma–Aldrich, Milan, Italy), into an insoluble blue formazan product was used with minor modifications (Manthorpe et al. 1986). Briefly, A3R-CHO cells were plated in the absence (control) or presence of the maximally effective IB-MECA concentrations (60 μM). After 17 h the medium was changed, and a serum-free medium containing 0.5 mg/ml MTT was added to cells and incubated for 2.5 h. The medium was then replaced with 0.08% HCl in isopropanol, and after 10 min the optical density (OD) of samples was evaluated (at a test wavelength of 570 nm, reference wavelength of 630 nm). Healthy cells with functional mitochondria significantly metabolize the precursor molecule into a blue product, which can be quantified by subtracting OD at 630 nm from the corresponding OD-value at 570 nm; dying or damaged cells show a proportional reduction of their ability to metabolize MTT. To validate this procedure in our experimental system, in initial experiments cells were exposed to increasing concentrations of damaging agents (e.g., HCl) and a proportional loss of functional mitochondrial activity demonstrated. Results are expressed as percent of metabolizing activity in control healthy cells.
Flow cytometric studies
Evaluation of apoptosis.
After 48 h or 72 h in culture, adhering cells were detached from the culture substrate using 0.125% trypsin (37°C, 5 min). Cells were then incubated with the fluorescent dye propidium iodide (PI; 50 μg/ml in 0.1% sodium citrate containing 0.1% Triton X-100) for 30 min. DNA content was then quantified using a FACScan flow cytometer (Becton Dickinson, Mountain View, Calif., USA) equipped with a single 488-nm argon laser.
Cell cycle analysis.
Incorporation of bromodeoxyuridine (BrdU) into cellular DNA was used as a marker of the cell cycle. Cells in the active synthesis phase incorporate BrdU which is detected by fluorescein isothiocyanate (FITC)-conjugated anti-BrdU monoclonal antibody, according to Dolbeare et al. (1983) with some minor modifications (Barbieri et al. 1997). After 72 h in culture, cells were pulsed with 10 μM BrdU at 37°C for 30 min to allow incorporation of this precursor into the DNA of living cells, centrifuged, washed and resuspended in HCl 2 N. After a 30-min incubation at room temperature, cells were centrifuged, washed once in 0.1 M borax and resuspended in anti-BrdU antibody-containing solution, incubated for 60 min at 4°C, washed, and incubated with diluted FITC-conjugated secondary antibody. After several washes, cells were resuspended in PI working solution as described above. Cells were then analyzed by flow cytometry (see above).
Drugs and treatments
The A3-selective agonists IB-MECA and Cl-IB-MECA and the A3-selective antagonists MRS1220 and MRS1191 were synthesized as previously reported (Gallo-Rodriguez et al. 1994; Kim et al. 1994, 1996; Jiang et al. 1996). The nucleoside transporter inhibitor S-(4-nitrobenzyl)-6-thioinosine (NBTI) was purchased from Sigma–Aldrich (Milan, Italy). The adenosine antagonist 1,3-dipropyl-8-(4-acrylate)-phenylxanthine (BW-A1433) was a kind gift of Dr. Susan Daluge (Glaxo-Wellcome, Research Triangle Park, NC, USA); L-249,313 was a kind gift of Dr. Marlene Jacobson (Merck Research Laboratories, West Point, Pa, USA). The radiolabeled [3H]5’-N-ethylcarboxamidoadenosine ([3H]NECA) was purchased from NEN Life Science Products (Cologne, Germany).
Cultures were exposed to the various pharmacological agents by direct addition to the culture medium at the time of plating, except for HEK293 cells which were maintained in culture for 24 h before addition of the pharmacological agents. NBTI was dissolved in Hanks’ balanced salt solution (HBSS)/dimethylsulfoxide (DMSO) (2:1) to obtain a 1 mM stock solution. For all the other compounds 10 mM stock solutions were prepared in DMSO, and serial dilutions were obtained in HBSS without calcium and magnesium. For each condition a tenfold-concentrated solution was added to reach the final concentration in culture medium. For concentrations ≥30 μM, Cl-IB-MECA and IB-MECA were prepared by directly dissolving the drugs at their final concentration in culture medium.
Statistical analysis
Non-parametric one-way analysis of variance (ANOVA) was performed with the Scheffe’s or the Fisher’s test, using the software StatView for Macintosh. Statistical significance of the single concentration data was determined with the Student’s t-test. P<0.05 was considered significant. Cell growth curves were analyzed by computer-assisted analysis using the program Allfit (De Lean et al. 1978). All data are expressed as means ± SEM unless stated otherwise in the legends.
Results
Untransfected WT-CHO cells do not constitutively express any adenosine receptors, including the A3 receptor subtype, as shown by lack of specific binding to the non-selective adenosine receptor ligand [3H]NECA, which exhibits a remarkably high affinity for the human A3 adenosine receptor (Klotz et al. 1998). Transfection of the A3 receptor cDNA into these cells resulted in stable expression of this receptor, as shown by specific binding of [3H]NECA (KD=6 nM, Bmax=800 fmol/mg membrane protein; Klotz et al. 1998). Radioligand competition studies also showed that N6-benzyladenosine-5’-N-methyluronamides (e.g., IB-MECA) were more potent than other agonists tested at the transfected receptor in A3R-CHO cells (Klotz et al. 1998), as expected from previous pharmacological studies at the rat A3 receptor (Gallo-Rodriguez et al. 1994; Kim et al. 1994).
In a similar way, transfection of CHO cells with the cDNA for the human A1 receptor resulted in stable expression of this receptor subtype, as shown by radioligand binding studies for the A1 receptor (Klotz et al. 1998).
Binding and adenylyl cyclase studies
The ability of A3 agonists to bind the A3 receptor transfected in CHO cells and hence to induce a functional response was tested by evaluating Cl-IB-MECA inhibition of both [3H]NECA binding and forskolin-stimulated adenylyl cyclase activity. Cl-IB-MECA inhibited [3H]NECA binding with a mean Ki-value of 11.1 nM (95% confidence limits: 9.4–13.0), indicating a high affinity for the transfected A3 receptor (Fig. 1). The mean IC50 value for Cl-IB-MECA inhibition of adenylyl cyclase activity was in the micromolar range (1,170 nM; 95% confidence limits: 585–2320; Fig. 1), showing a discrepancy between Cl-IB-MECA affinity to the transfected A3 receptor and its potency in eliciting a functional response. A similar discrepancy has already been reported for other experimental models and appears to be a quite common feature of A1 and A3 adenosine receptors (Martens et al. 1988; Tawfik-Schlieper et al. 1989; Kim et al. 1994; Abbracchio et al. 1995; see also Discussion).
Fig. 1.
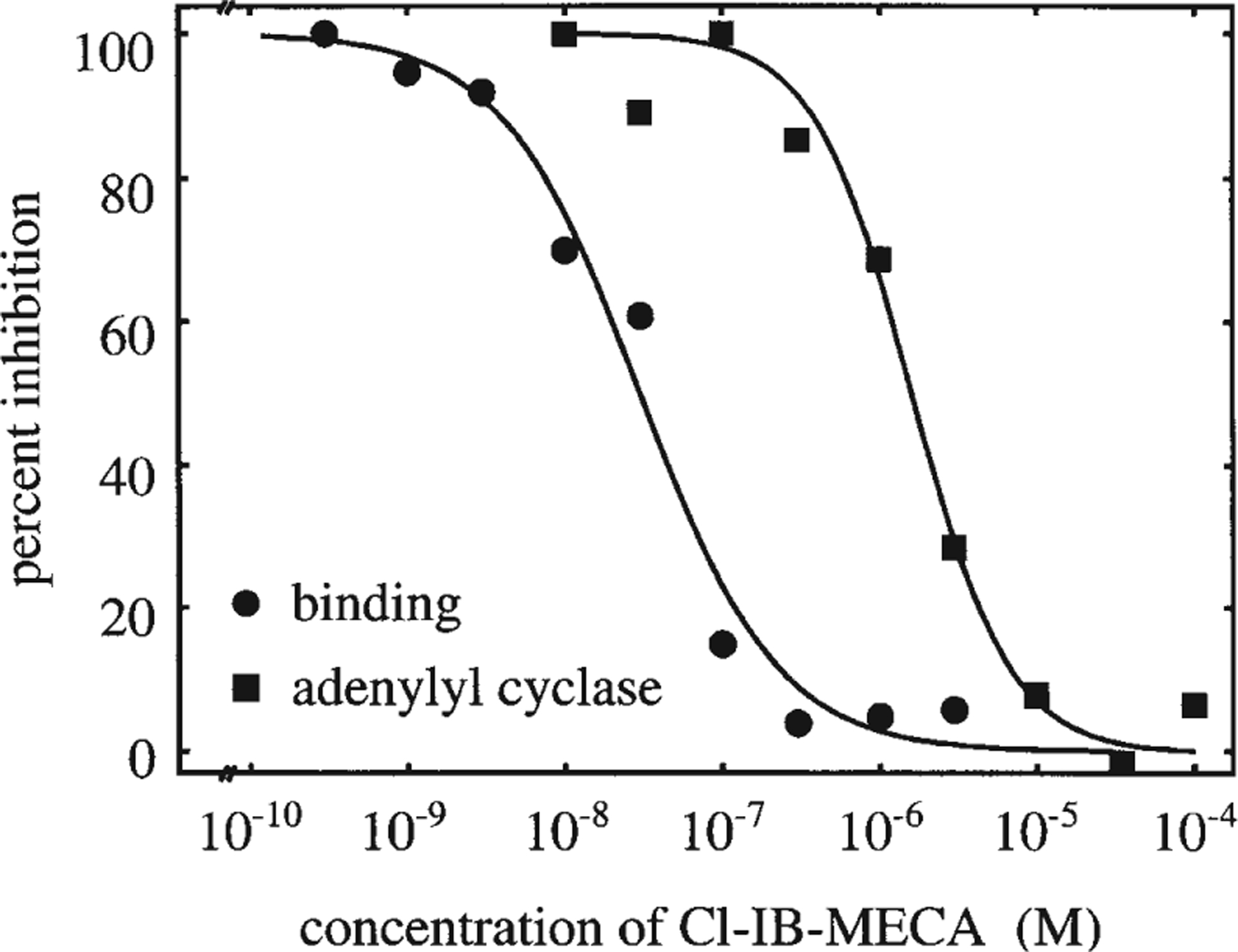
Inhibition of [3H]NECA binding (●) and inhibition of forskolin-stimulated adenylyl cyclase activity (■) by Cl-IB-MECA. The Ki-value for Cl-IB-MECA at human A3 adenosine receptors in CHO cells was determined in competition binding experiments with [3H]NECA as a radioligand. The curve (●) shows a representative experiment with a Ki-value of 10.0 nM. Specific binding amounted to 2.4 fmol/20 μg membrane protein (100% specific binding). The IC50-value for adenylyl cyclase inhibition (■) was determined by measurement of adenylyl cyclase activity in membrane preparations and was 1,620 nM in the experiment shown. Adenylyl cyclase activity stimulated by 30 μM forskolin amounted to 162 pmol/mg membrane protein per min (100% adenylyl cyclase activity) and was maximally inhibited to 118 pmol/mg membrane protein per min by Cl-IB-MECA. The mean Ki- and IC50-values from four independent experiments were 11.1 nM (9.4–13.0 nM) and 1,170 nM (585–2320 nM) for binding and cyclase experiments, respectively, with 95% confidence limits in parentheses
A3 adenosine receptor-mediated reduction of cell number
Exposure of A3R-CHO cells to the A3 adenosine receptor agonists IB-MECA and Cl-IB-MECA for 72 h (Fig. 2) resulted in a concentration-dependent reduction of cell number. No statistically significant reductions in cell number were induced by either agonist in the parental WT-CHO cell line, suggesting that the effect described above is related to the presence of the receptor. Very similar reductions of cell number were obtained with either 60 μM IB-MECA or 30 μM Cl-IB-MECA, and hence these agonist concentrations were indifferently utilized to activate the A3 receptor in subsequent experiments.
Fig. 2.
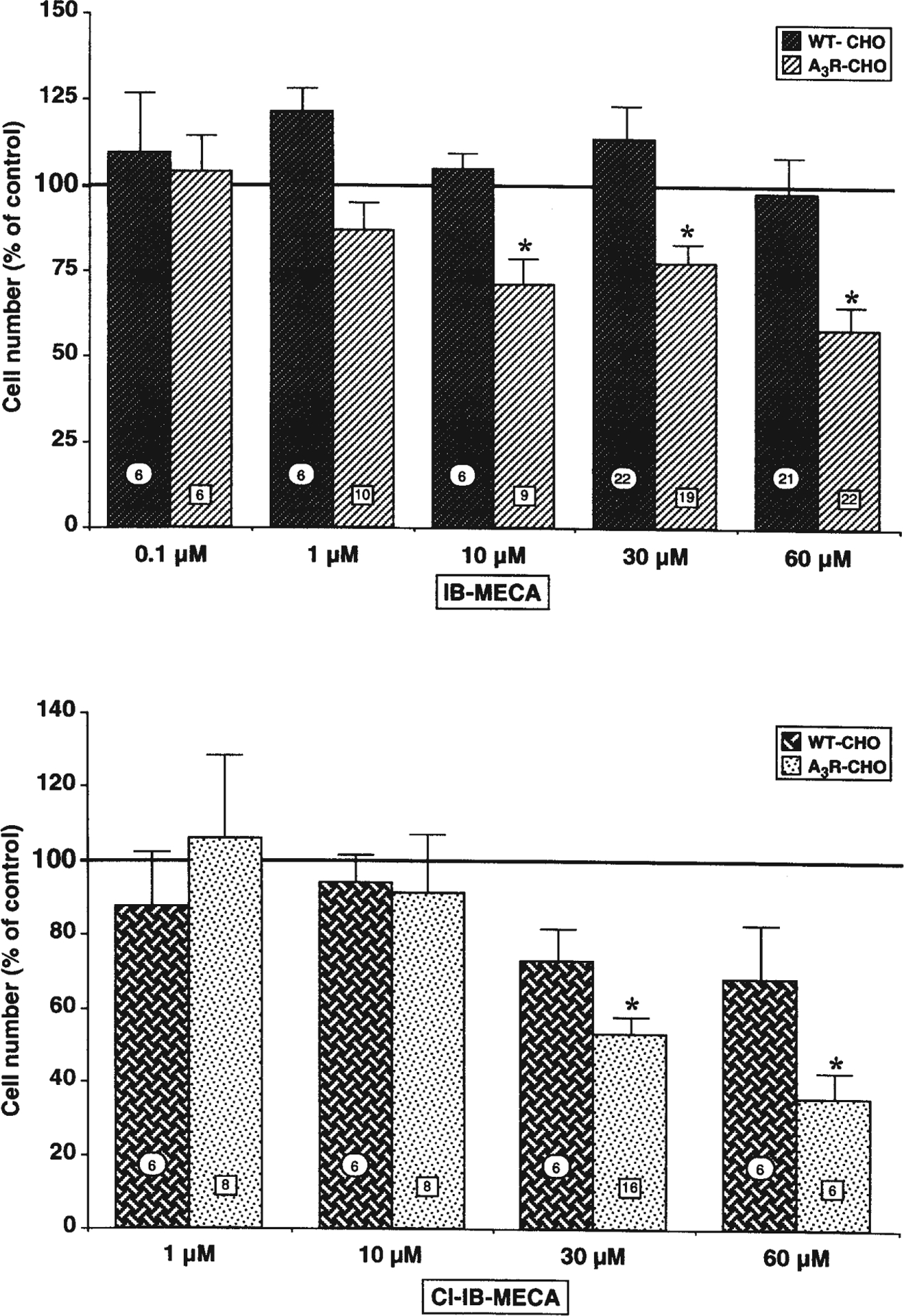
Both IB-MECA and Cl-IB-MECA concentration-dependently reduced A3R-CHO cell number, with no effects on corresponding WT-CHO cells. Cells were plated in the presence of the indicated concentrations of either IB-MECA (upper panel) or Cl-IB-MECA (lower panel). After 72 h in culture, cells were fixed and counted as described in Materials and methods. Results (means ± SEM) were calculated as percent of corresponding control values. Numbers on columns indicate the number of replications. *P<0.05 with respect to corresponding control; one-way ANOVA (Scheffe’s F-test)
The reduction of cell number induced by IB-MECA was not counteracted by the adenosine uptake inhibitor NBTI (Table 1), suggesting that these effects are likely due to the activation of extracellular receptors.
Table 1.
The adenosine uptake inhibitor NBTI does not prevent the reduction of A3R-CHO cell number induced by IB-MECA. Cells were plated in the absence (control) or presence of 60 μM IB-MECA with or without 1 μM NBTI and after 72 h fixed, stained and analyzed as described in Materials and methods. The number of replications/condition are shown in parentheses
| Condition | A3R-CHO cell number (% of control) |
|---|---|
| IB-MECA | 65.7± 5.6 (12)* |
| IB-MECA + NBTI | 45.6± 5.1 (12)* |
| NBTI | 118.8±21.7 (12) |
P<0.05 with respect to corresponding control; one-way ANOVA (Scheffe’s F-test)
Consistent with the previously demonstrated sensitivity of the human A3 receptor to 8-phenyl-xanthines with acidic para-substituents (Linden 1994), the effects induced by IB-MECA on the A3R-CHO cells were partially antagonized by BW-A1433, tested in the 50–500 nM range (Fig. 3A). Neither BW-A1433 nor any of the selective A3 receptor antagonists utilized had a statistically significant effect on cell number when tested alone. A variety of selective A3 receptor antagonists (for review, see Jacobson and Suzuki 1996) were much more potent than BW-A1433 in counteracting the effects induced by micromolar concentrations of A3 agonists on A3R-CHO cells. At a concentration as low as 10 nM, the triazoloquinazo-line derivative MRS1220 (Kim et al. 1996) completely antagonized the reduction of cell number induced by 60 μM IB-MECA (Fig. 3B). Similar effects were obtained with the dihydropyridine derivative MRS1191 (Jiang et al. 1996) which, when utilized at a 10-nM concentration against 30 μM Cl-IB-MECA, fully reversed the agonist-induced reduction of cell number. Indeed, cell number was 107.5±24.2 (% of control) in the agonist + antagonist group (not statistically different from control), compared to 62.0±13.5 (% of control) in the agonist-treated cells (P<0.04 with respect to corresponding control value; Student’s t-test). A statistically significant reversion of the effects induced by 30 μM Cl-IB-MECA was also demonstrated with the triazolonaphthyridine derivative L-249,313 (Jacobson et al. 1996) in the low nM range (data not shown).
Fig. 3.
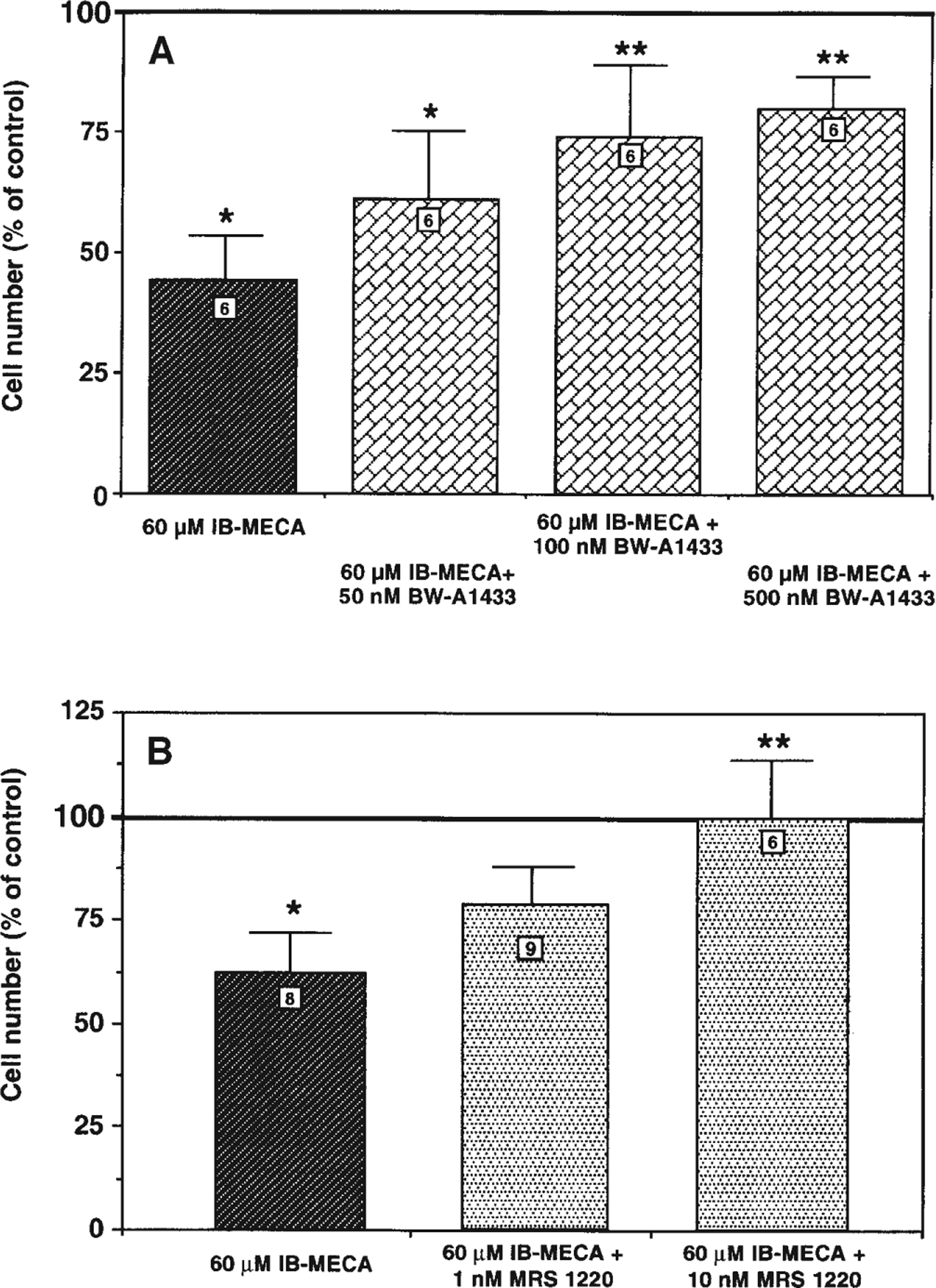
Effect of A BW-A1433 and B MRS1220 on IB-MECA-induced reduction of A3R-CHO cell number. Cells were plated in the presence of 60 μM IB-MECA with or w/o the indicated concentrations of A3 receptor antagonists. After 72 h in culture, cells were fixed and counted as described in Materials and methods. Results (means ± SEM) are calculated as percent of corresponding control values. Numbers on columns indicate the number of replications. *P<0.05 with respect to control, **P<0.05 with respect to 60 μM IB-MECA; one-way ANOVA (Fisher’s test). Neither BW-A1433 nor MRS1220 alone significantly affected the number of cells in either of the two clones (n=8; data not shown)
A specific role for the A3 receptor in the induction of the above described effects is confirmed by results obtained in independent control experiments with other cell clones. In addition to CHO cells, we also transfected HEK293 cells with the same A3 receptor cDNA, resulting in cell clones with A3 receptor expression similar to the A3R-CHO cells (not shown). Exposure of these human A3R-HEK cells to micromolar concentrations of Cl-IB-MECA resulted in a statistically significant decrease of cell number (Table 2). In contrast, the same concentration of Cl-IB-MECA had no effects on untransfected HEK293 cells. These results suggest that the effects observed on cell growth are indeed mediated by the transfected A3 adenosine receptor. Further support for this notion stems from experiments with CHO cells transfected with the human A1 receptor, where no effects on cell number were detected following exposure to micromolar concentrations of Cl-IB-MECA and IB-MECA that effectively reduced cell number in A3R-CHO (Table 3).
Table 2.
Cl-IB-MECA reduces number of A3R-HEK293 cells with no effects on the corresponding wild-type clone. Twenty-four hours after cell plating, 30 μM Cl-IB-MECA was added to cultures. After additional 48 h in culture, cells were fixed and counted as described in Materials and methods. Results (means ± SEM of six replicates from three independent experiments) were calculated as percent of corresponding control values
| Condition | WT-HEK293 cell number (% of control) | A3R-HEK293 cell number (% of control) |
|---|---|---|
| Control | 100 ±17.9 | 100 ± 6.3 |
| 30 μM Cl-IB-MECA | 103.2±20 | 62.1±13.7* |
P<0.05 with respect to corresponding control; one-way ANOVA (Fisher’s test)
Table 3.
Effects of selective A3 agonists on CHO cells transfected with the human A1 receptor (A1R-CHO). Cells were plated in the absence (control) or presence of the indicated adenosine agonists at the indicated concentrations. After 72 h cells were fixed, stained and analyzed as described in Materials and methods. Values refer to 4–12 replications/condition. No significant differences in any experimental conditions with respect to control cells
| A1R-CHO | Cell number (% of control) |
|---|---|
| 30 μM Cl-IB-MECA | 102.3±3.3 |
| 60 μM IB-MECA | 97.9±6.1 |
Characterization of the mechanisms responsible for A3 agonist-induced effects in A3R-CHO cells
Several putative mechanisms could be responsible for the selective reduction of cell number induced by A3 adenosine receptor agonists in A3R-CHO cells, namely: (1) effects on cell adhesion; (2) induction of cell death by either apoptosis or necrosis; or (3) modulation of cell cycle and hence of cell growth. We tested all these different possibilities via specific methodologies.
In our routine protocol, agonists were added to cells at the time of plating (see Materials and methods). In order to rule out the possibility that the detected reduction of cell number was due to effects on cell adhesion, A3 agonists were added 24 h after cell plating, when cells were already well attached to the culture dishes. A 72-h exposure to 30 μM Cl-IB-MECA yielded a reduction of cell number (57.7±4.8% of control, n=7, P<0.0001; Student’s t-test) that was identical to that obtained with the standard protocol of cell exposure (56.5±9.0% of control, n=7, P<0.0001; Student’s t-test). This suggests that the effects induced by A3 agonists on A3R-CHO cells cannot be attributed to an influence on cell adhesion.
Moreover, reduction of cell number by the activated A3 receptor is not due to non-specific toxicity, as demonstrated by the fact that A3 agonist-treated cells could metabolize the mitochondrial functional marker MTT to the same extent as control cells. In cultures exposed to 60 μM IB-MECA for 20 h, the mitochondrial transformation of MTT was 109.8±17.0% of control (mean of six experiments, not statistically different from control cultures; Student’s t-test); in parallel cultures, damaging agents (e.g., acidification of the culture medium with brief exposures to HCl) significantly reduced the mitochondrial metabolism of MTT (data not shown). The lack of non-specific toxicity is also confirmed by the lack of effect of A3 agonists on both non-transfected cells (Fig. 2) and A1R-CHO (Table 3).
To assess whether A3 agonist-induced reduction of cell number was due to apoptosis, a flow-cytometric analysis of PI-stained cell nuclei, in control and treated cells after different times in culture, was performed. Induction of apoptosis can be detected by reduced fluorescence of the nuclear dye PI, i.e., with the appearance of a hypodiploid DNA peak at lower fluorescence values (Ceruti et al. 1997). Analysis of adhering cells obtained from control and 60 μM IB-MECA-treated cultures revealed a typical diploid DNA peak. Only a very small percentage of cells with a hypodiploid DNA content was evident (indicated by percent values in Fig. 4). The fraction of apoptotic cells never exceeded 1% of total cell number in any of the experimental groups, after either 48 h (Fig. 4; upper panel) or 72 h in culture (Fig. 4; lower panel); no differences in the extent of apoptosis were found between control and IB-MECA-treated cells (Fig. 4) and after exposure to 30 μM Cl-IB-MECA (data not shown). Lack of induction of apoptosis by A3 receptor agonists was also confirmed by a morphological analysis of nuclei both from cells detached in the culture medium and cells still adhering to the culture dish after labelling with the chromatin dye Hoechst 33258 (data not shown). On the whole, these results suggest that spontaneous apoptosis is very low in A3R-CHO cells and, more importantly, that it is not increased by exposure to A3 receptor agonists.
Fig. 4.
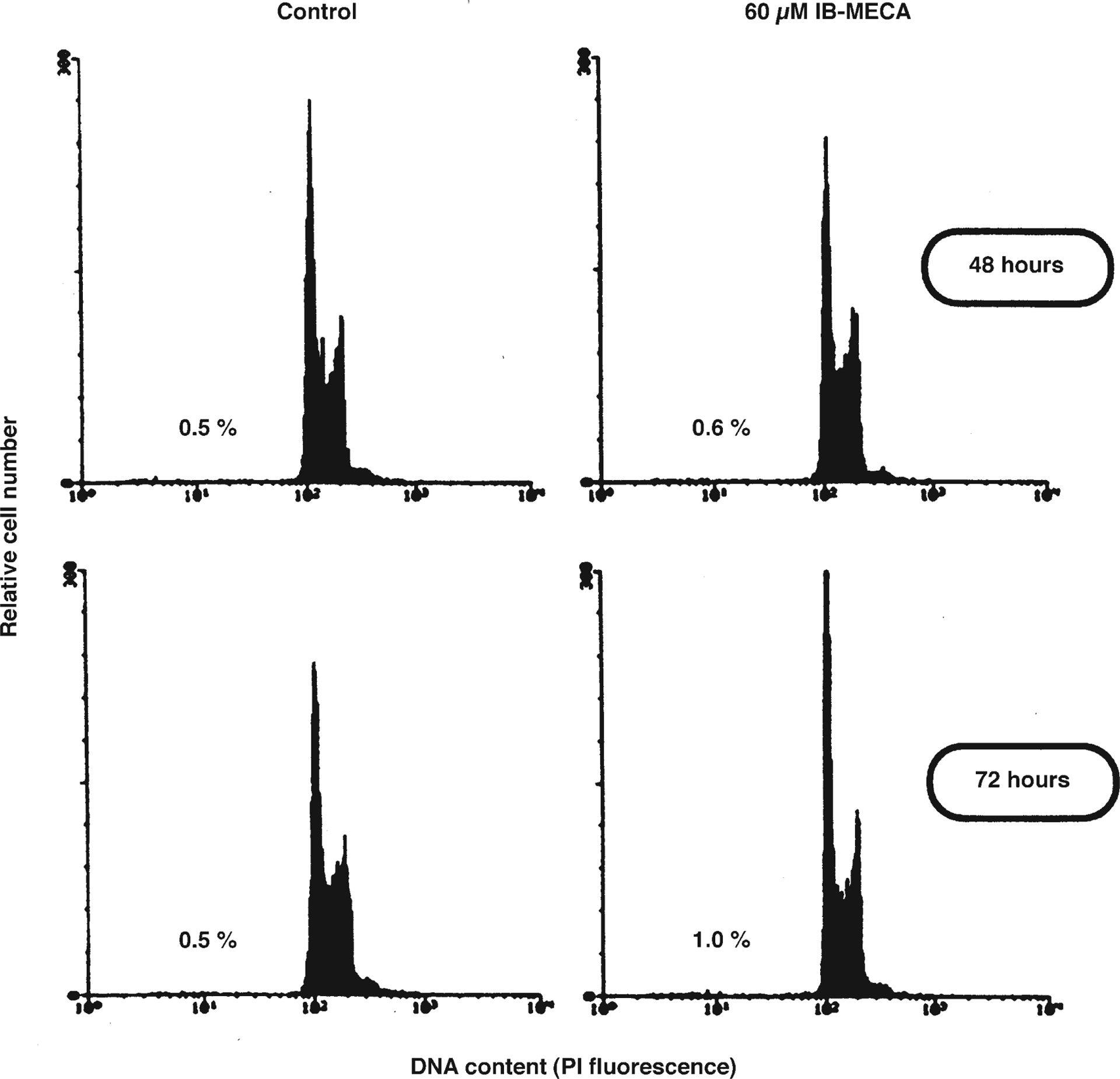
Lack of induction of apoptosis by IB-MECA in A3R-CHO cells. Cells were plated in the absence (control) or presence of 60 μM IB-MECA. After 48 h or 72 h (upper and lower panel, respectively) cells were detached from the culture dish, incubated with PI and apoptosis quantified by reduced fluorescence by flow cytometry, as described in Materials and methods. A typical diploid DNA peak was obtained for both control and treated cultures, and the extent of apoptosis (indicated by percent values in graphs) was very low and comparable in all of the experimental groups. Shown are the results of a typical analysis; similar data were obtained in three independent experiments
To understand whether A3 agonists could affect the growth rate of A3R-CHO cells, detailed growth curves were measured after initially plating cells at the same density utilized for all the other experiments shown in this study. Growth of A3R-CHO was delayed by about 40%–50% with respect to WT-CHO, even in the absence of adenosine agonists (data not shown), suggesting that there is a constitutive activation of the transfected receptor. Differently from the effects induced by addition of A3 agonists (Fig. 3), retardation of cell growth due to constitutive receptor activation could not be reversed by growing A3R-CHO in the presence of the A3 receptor antagonist MRS1191 (10 nM; data not shown). This is highly consistent with the proposal that full (“neutral”) antagonists (such as the MRS compounds) have no effects on constitutive receptor activation (Costa et al. 1991). Maintenance of A3R-CHO in the continuous presence of 60 μM IB-MECA resulted in highly retarded growth with respect to untreated A3R-CHO (Fig. 5), an effect that was already evident at day 2 in culture, maximal at day 4 (when cells in the agonist-treated group were 49±2.4% of control cell number, P<0.05; Scheffe’s F-test), maintained up to day 8 and then progressively reduced as cells approached confluency (Fig. 5). Statistical analysis revealed highly significant differences between the two curves (P<0.02, F-value in the sum of squares test; Allfit analysis) with both a reduction of the upper plateau for the IB-MECA-treated cells compared to controls (maximum number of cells ×105): 6.89±0.17, CV=2.4% in agonist-treated cultures (with respect to 7.44±0.16, CV=2.1% in control cultures) and a delay in the time necessary to achieve the half-maximum number of cells (4.5±0.1 days, CV=2.7% in agonist-treated cells compared to 5.0±0.1 days, CV=2.6% in control cultures).
Fig. 5.
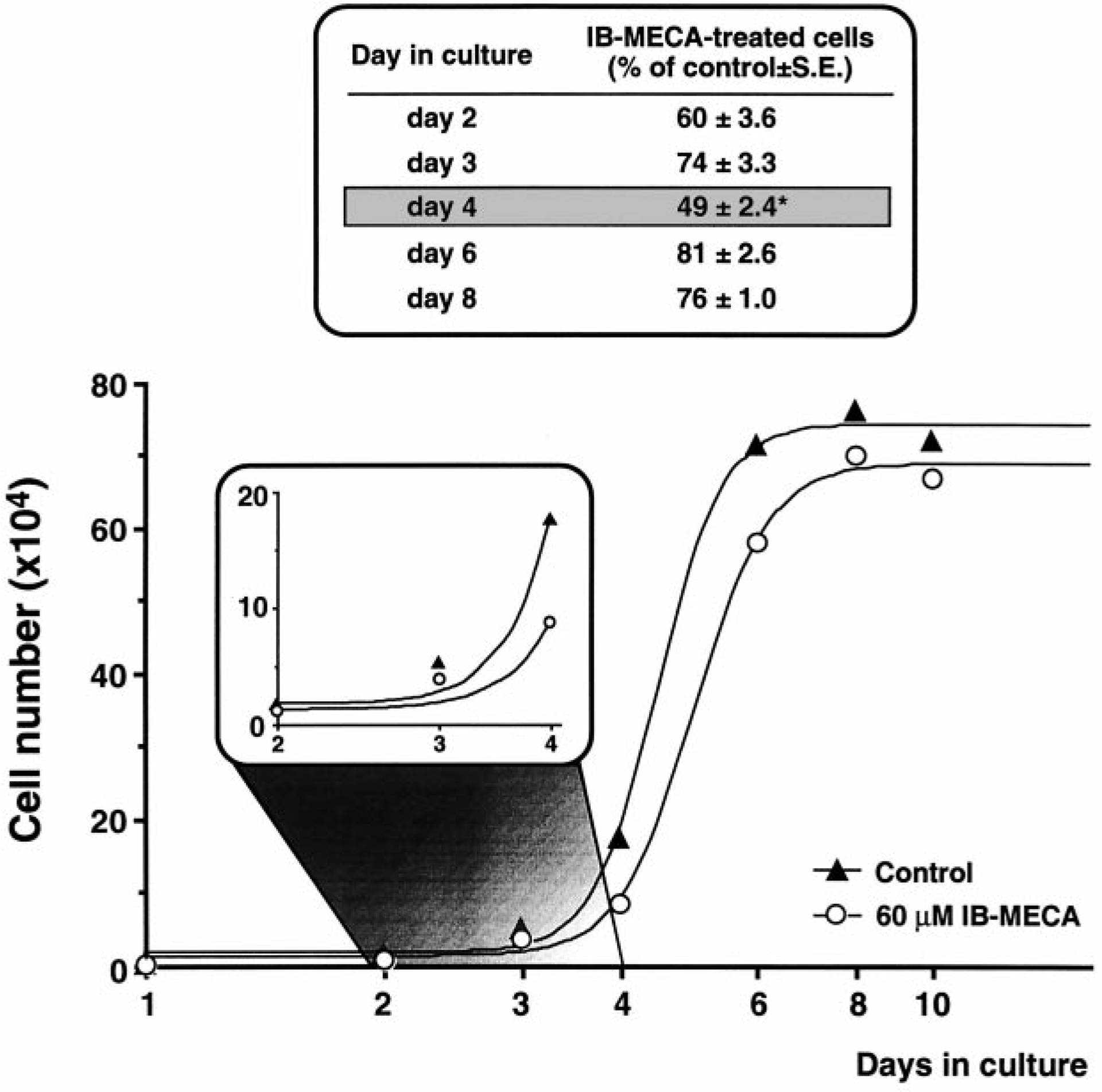
Reduction of the rate of cell growth by IB-MECA in A3R-CHO cells. Cells were plated in the absence (control) or presence of 60 μM IB-MECA and cell number evaluated as described in Materials and methods at the time points indicated on the x-axis. Raw data were fitted with Allfit to obtain the reported growth curves. The two curves were highly different from each other (P<0.02, F-value, sum of squares test). *P<0.05 with respect to control; one-way ANOVA (Scheffe’s F-test)
To assess whether retardation of cell growth by A3 agonists was due to effects on the cell cycle, we performed a flow-cytometric analysis of PI- and BrdU-double-labelled cells. This analysis allows the exact quantification of the percentage of cells at the G0/G1, S and G2/M phases of the cell cycle that are represented, respectively, by regions R2, R3 and R4 in Fig. 6. After growing cells for 72 h in the presence of IB-MECA, it is still possible to detect cells with a DNA content typical of the S phase in the area between the R2 and R4 regions (cells falling between 200 and 400 arbitrary fluorescence units in the abscissa), but the percentage of these cells that actively incorporated BrdU during the 30-min in vitro pulse was dramatically reduced compared to controls (in treated cultures, the R3-value was equal to 5.9% vs. 70.5% in control cells; panels B and A, respectively, of Fig. 6). This suggests that the A3 agonist markedly impairs the ability of cells to neosynthe-size DNA. A marked increase in the number of cells in the G2/M phase was also found in A3 agonist-treated cells. Hence, the highly impeded growth shown in Fig. 5 is due to an alteration of cell cycle progression. Both the reduced BrdU incorporation and the associated increase of cells in the G2/M phase were specifically mediated by the activation of the A3 receptor, being almost completely antagonized by the concomitant exposure to 10 nM MRS1191 (R3-value equal to 54.0%; panel C of Fig. 6).
Fig. 6A–C.
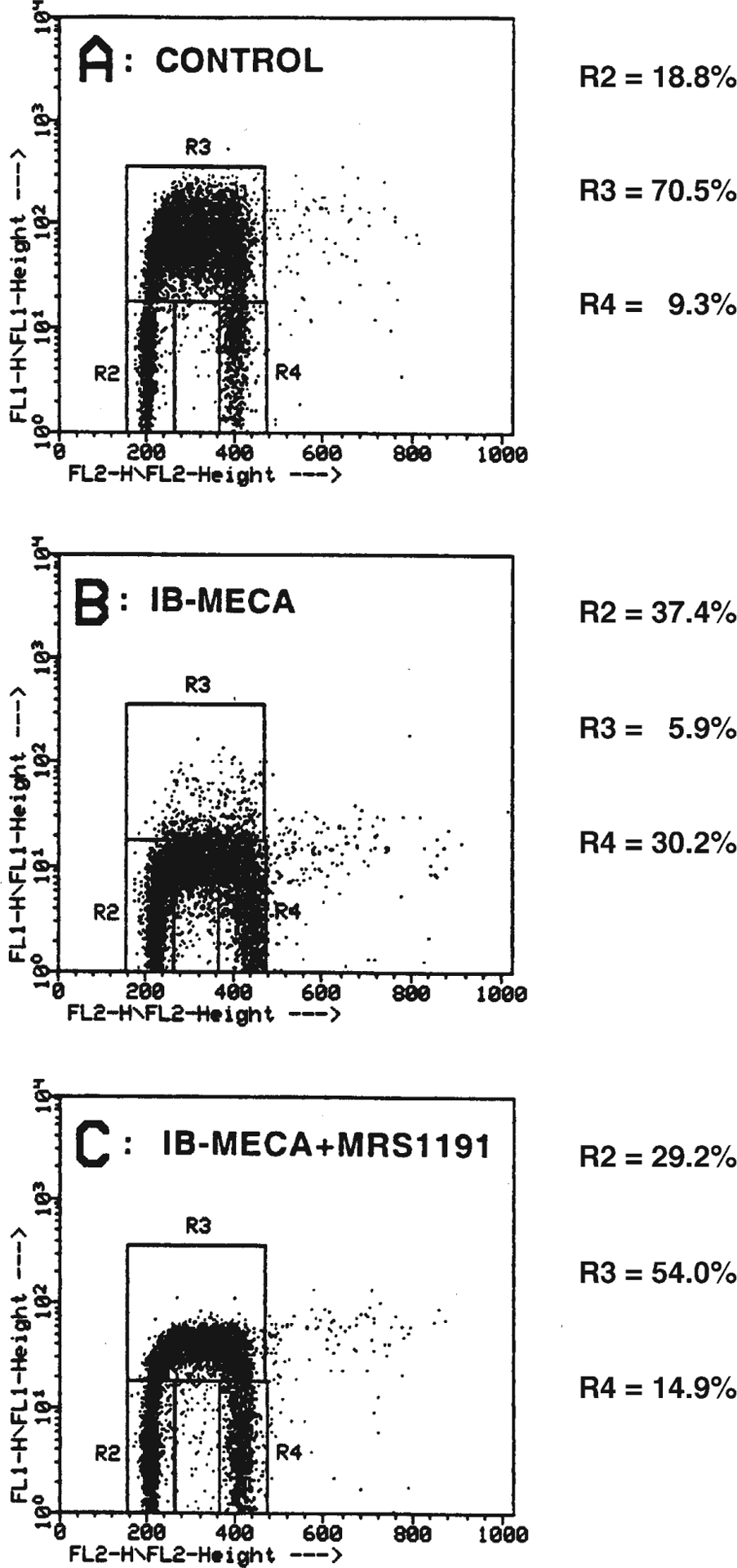
IB-MECA-induced alteration of cell cycle progression in A3R-CHO cells and reversion by MRS1191. Cells were plated in the absence (control; A) or presence of 60 μM IB-MECA (B) with or w/o 10 nM MRS1191 (C). After 72 h, cells were pulsed with BrdU for 30 min and then incubated with PI as described in detail in Materials and methods. Cells were analyzed by flow cytometry for their DNA content (reported on the x-axis) as well as for BrdU incorporation (y-axis) to allow calculations of regions corresponding to cells at the G0/G1 phases (R2), at the S phases (R3) and at the G2/M phases (R4). For the different experimental conditions, these values are reported in the right part of each panel. The results of a typical analysis are shown; similar data were obtained in three independent experiments
Discussion
Transfection of CHO cells with the human A3 receptor cDNA resulted in stable and efficient expression of this receptor subtype (Klotz et al. 1998), providing a valuable experimental model to allow a detailed characterization of the functional effects evoked in these cells by the activation of this receptor. Exposure of A3R-CHO cells to micromolar A3 receptor agonists (Kim et al. 1994) resulted in a marked and concentration-dependent reduction of the number of cells in culture. This effect was not due to any influence on cell adhesion, nor to induction of cell death by necrosis or apoptosis, but it was rather caused by an alteration of cell cycle progression. In particular, a decreased ability to neo-synthesize DNA was detected in A3 agonist-treated cells and this was associated with an increase of the number of cells in the G2/M phase, suggesting that A3 agonists may affect cell cycle progression at multiple levels.
Several lines of evidence support a specific involvement of the A3 receptor in these effects: (1) in WT-CHO cells, which do not express the A3 adenosine receptor (Klotz et al. 1998), no effect on the number of cells was detected following exposure to A3 receptor agonists (IB-MECA and Cl-IB-MECA), which effectively reduced this parameter in the transfected clone; (2) in A3R-CHO, both the reduction of cell number and the cell cycle alteration induced by A3 receptor agonists were counteracted by selective A3 receptor antagonists, such as MRS1220, MRS1191 and L-249,313, when utilized at concentrations in the nanomolar range. These data are also consistent with previous studies demonstrating antagonism of the human A3 receptor with MRS1220 and MRS1191. A partial reversal of agonist-induced effects was obtained with the weak xanthine A3 antagonist BW-A1433, in agreement with previous results (Linden 1994). (3) Transfection of the same A3 receptor cDNA in another cell line (human HEK293 cells) also resulted in the appearance of sensitivity to A3 agonists. Conversely, IB-MECA and Cl-IB-MECA did not induce any effect in CHO cells transfected with the human A1 receptor. Hence, the effects observed on the A3R-CHO are specifically related to the transfection of the A3 adenosine receptor subtype.
Despite the nanomolar affinity demonstrated by Cl-IB-MECA at the human A3 receptor both in the present (Fig. 1) and in previous radio-receptor binding studies (Gallo-Rodriguez et al. 1994; Kim et al. 1994; Klotz et al. 1998), the effective concentration in inhibiting forskolin-stimulated adenylyl cyclase activity and in influencing cell cycle progression resulted in the micromolar range. A discrepancy between affinities of A3 agonists in receptor binding compared to potencies in functional assays was previously noted (Kim et al. 1994; Abbracchio et al. 1995; Klotz et al. 1999) and seems to be a relatively common characteristic of the A3 adenosine receptor. Micromolar concentrations of agonists are required for A3 receptor-mediated necrosis of cerebellar granule cells (Sei et al. 1997), and for elevation of [Ca2+]i as well as apoptosis of human eosinophils and HL-60 cells (Kohno et al. 1996; Yao et al. 1997). The reason for this discrepancy is not clear at the moment, but may be related to similar observations for A1 adenosine receptors in the rat and guinea pig heart where functional potency typically corresponds to low affinity agonist binding (e.g., Martens et al. 1988; Tawfik-Schlieper et al. 1989). In addition, currently unknown complex protein interactions downstream of receptor activation that modulate the final effect may be involved. The reasons at the basis of such discrepancy are currently under examination in our laboratory.
As mentioned above, activation of the A3 adenosine receptor by micromolar agonist concentrations was shown to reduce the number of cells also in other experimental systems (Sei et al. 1997; Yao et al. 1997; Barbieri et al. 1998). However, in all these other cases, reduction of cell number was shown to be due to induction of cell death, by either necrosis (Sei et al. 1997) or apoptosis (Kohno et al. 1996; Yao et al. 1997; Barbieri et al. 1998). No signs of either non-specific toxicity or apoptosis were detected upon activation of this receptor in A3R-CHO cells. Such a different outcome (impairment of cell cycle progression in the present study vs. cell death in the above examples) may be due to differences in the intracellular targets expressed downstream of receptor activation in the different cellular systems (e.g., proteins involved in regulation of cell survival and in progression of cells through the cell cycle). This intriguing issue is actually under study in our laboratories by evaluation of several members of the Bcl-2 family of proteins (e.g., bak; Yao et al. 1997) and of key cyclins involved in the transition between the different phases of the cell cycle. These studies will also help to clarify the exact nature of the cell cycle alteration reported in the present study.
In conclusion, our data suggest a specific role for the A3 adenosine receptor in modulation of the cell cycle and modulation of cell growth. This is particularly important in view of the potential of selective A3 receptor ligands in diseases characterized by either abnormally increased cell growth (e.g., cancer) or by massive cell death (e.g., neurodegeneration), where the availability of novel and selective pharmacological agents able to modulate cell growth and cell survival would be highly desirable. The A3R-CHO cells provide a suitable model to characterize such compounds and to further investigate the molecular mechanisms of the A3 receptor-mediated control of cell growth.
Acknowledgements
Partially supported by the European Commission (Concerted Action ADEURO, BIOMED 1), by the Italian Research National Council CNR 96.03340.CT04, by grants from the Italian Ministero per la Ricerca Scientifica e Tecnologica (MURST 60% 1995 and 1996 to M.P.A. and Cofinanziamento di ricerche di interesse nazionale 1999 to F.C. on “Recettori Purinergici e Neuroprotezione”) and by Deutsche Forschungsgemeinschaft to M.J.L. Authors are grateful to Dr. Marlene Jacobson, Merck Research Laboratories, West Point, Pa, USA; Dr. Mauro Corsi, Glaxo-Wellcome, Verona, Italy and Dr. G.E. Rovati, University of Milan, Italy, for useful discussion. Cl-IB-MECA was kindly provided by Research Biomedical International as part of the Chemical Synthesis Program of the National Institute of Mental Health, contract no. 01MH30003.
Contributor Information
Roberta Brambilla, Institute of Pharmacological Sciences, School of Pharmacy, University of Milan, Via Balzaretti 9, I-20133 Milan, Italy.
Flaminio Cattabeni, Institute of Pharmacological Sciences, School of Pharmacy, University of Milan, Via Balzaretti 9, I-20133 Milan, Italy.
Stefania Ceruti, Institute of Pharmacological Sciences, School of Pharmacy, University of Milan, Via Balzaretti 9, I-20133 Milan, Italy.
Daniela Barbieri, Department of Biomedical Sciences, Section of General Pathology, University of Modena, Via Campi 187, Modena, Italy.
Claudio Franceschi, Department of Biomedical Sciences, Section of General Pathology, University of Modena, Via Campi 187, Modena, Italy.
Yong-Chul Kim, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Kenneth A. Jacobson, Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, USA
Karl-Norbert Klotz, Institut für Pharmakologie und Toxikologie, Universität Würzburg, Versbacher Strasse 9, D-97078 Würzburg, Germany.
Martin J. Lohse, Institut für Pharmakologie und Toxikologie, Universität Würzburg, Versbacher Strasse 9, D-97078 Würzburg, Germany
Maria P. Abbracchio, Institute of Pharmacological Sciences, School of Pharmacy, University of Milan, Via Balzaretti 9, I-20133 Milan, Italy
References
- Abbracchio MP, Brambilla R, Ceruti S, Kim HO, Lubitz DKJE von, Jacobson KA, Cattabeni F (1995) G-protein-dependent activation of phospholipase C by adenosine A3 receptors in rat brain. Mol Pharmacol 48:1038–1045 [PubMed] [Google Scholar]
- Abbracchio MP, Rainaldi G, Giammarioli AM, Ceruti S, Brambilla R, Cattabeni F, Barbieri D, Franceschi C, Jacobson KA, Malorni W (1997) The A3 adenosine receptor mediates cell spreading, reorganization of actin cytoskeleton, and distribution of Bcl-xL. Studies in human astroglioma cells. Biochem Biophys Res Commun 241:297–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri D, Abbracchio MP, Salvioli S, Monti D, Cossarizza A, Ceruti S, Brambilla R, Cattabeni F, Jacobson KA, Franceschi C (1998) Apoptosis by 2-chloro-2’-deoxy-adenosine and 2-chloro-adenosine in human peripheral blood mononuclear cells. Neurochem Int 32:493–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceruti S, Barbieri D, Veronese E, Cattabeni F, Cossarizza A, Giammarioli AM, Malorni W, Franceschi C, Abbracchio MP (1997) Different pathways of apoptosis revealed by 2-chloro-adenosine and deoxy-D-ribose in mammalian astroglial cells. J Neurosci Res 47:372–383 [DOI] [PubMed] [Google Scholar]
- Costa T, Ogino Y, Munson PJ, Onaran HO, Rodbard D (1991) Drug efficacy at guanine nucleotide-binding regulatory protein-linked receptors: thermodynamic interpretation of negative antagonism and of receptor activity in the absence of ligand. Mol Pharmacol 41:549–560 [PubMed] [Google Scholar]
- De Lean A, Munson PJ, Rodbard D (1978) Simultaneous analysis of sigmoidal curves: application to bioassay, radioligand assay, and physiological dose-response curves. Am J Physiol 235: E97–E102 [DOI] [PubMed] [Google Scholar]
- Dolbeare F, Gratzner H, Pallavicini M, Gray JW (1983) Flow cytometric measurement of total DNA content and incorporated bromodeoxyuridine. Proc Natl Acad Sci USA 80:5573–5577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M (1994) Nomenclature and classification of purinoceptors. Pharmacol Rev 46:143–156 [PMC free article] [PubMed] [Google Scholar]
- Gallo-Rodriguez C, Ji X-D, Melman N, Siegman BD, Sanders LH, Orlina J, Pu Q, Olah ME, Galen PJM van, Stiles GL, Jacobson KA (1994) Structure-activity-relationships of N6-benzyladenosine-5’-uronamides as A3-selective adenosine agonists. J Med Chem 37:636–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon JP, Pfannkuche HJ, Fozard JR (1995) A role for mast cells in adenosine A3 receptor-mediated hypotension in the rat. Br J Pharmacol 115:945–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Suzuki F (1996) Recent developments in selective agonists and antagonists acting at purine and pyrimidine receptors. Drug Dev Res 39:289–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Nikodijevic O, Shi D, Gallo-Rodriguez C, Olah ME, Stiles GL, Daly JW (1993) A role for central A3-adenosine receptors: mediation of behavioral depressant effects. FEBS Lett 336:57–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Kim HO, Siddiqi SM, Olah ME, Stiles G, Lubitz DKJE von (1995) A3 adenosine receptors: design of selective ligands and therapeutic prospects. Drugs Future 20:689–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Park KS, Jiang JI, Kim YC, Olah ME, Stiles GL, Ji XD (1997) Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology 36:1157–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M, Chakravarty PK, Johnson RG, Norton R (1996) Novel selective non-xanthine A3 adenosine receptor antagonists. Drug Dev Res 37:131 [Google Scholar]
- Jiang JL, Rhee AM van, Melman N, Ji XD, Jacobson KA (1996) 6-Phenyl-1,4-dihydropyridine derivatives as potent and selective A3 adenosine receptor antagonist. J Med Chem 39:4667–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin TP (1996) The classification of seven transmembrane receptors in recombinant expression systems. Physiol Rev 48:413–463 [PubMed] [Google Scholar]
- Kim HO, Ji XD, Siddiqi SM, Olah ME, Stiles GL, Jacobson KA (1994) 2-Substitution of N6-benzyladenosine-5’-uronamides enhances selectivity for A3-adenosine receptors. J Med Chem 37:3614–3621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Ji XD, Jacobson KA (1996) Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. J Med Chem 39:4142–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ (1998) Comparative pharmacology of human adenosine receptor subtypes. Characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’s Arch Pharmacol 357:1–9 [DOI] [PubMed] [Google Scholar]
- Klotz KN, Camaioni E, Volpini R, Kachler S, Vittori S, Cristalli G (1999) 2-Substituted N-ethylcarboxamidoadenosine derivatives as high-affinity agonists at human A3 adenosine receptors. Naunyn-Schmiedeberg’s Arch Pharmacol 360:103–108 [DOI] [PubMed] [Google Scholar]
- Kohno Y, Sei Y, Koshiba M, Kim HO, Jacobson KA (1996) Induction of apoptosis in HL-60 human promyelocytic leukemia cells by selective adenosine A3 receptor agonists. Biochem Biophys Res Commun 219:904–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden J (1994) Cloned adenosine A3 receptors: pharmacological properties, species-differences and receptor functions. Trends Pharmacol Sci 15:298–306 [DOI] [PubMed] [Google Scholar]
- Lubitz DKJE von, Lin RCS, Popik P, Carter MF, Jacobson KA (1994) Adenosine A3 receptor stimulation and cerebral ischemia. Eur J Pharmacol 263:59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manthorpe M, Fagnani R, Skaper SD, Varon S (1986) An automated colorimetric micro assay for neurotrophic factors. Dev Brain Res 25:191–198 [DOI] [PubMed] [Google Scholar]
- Martens D, Lohse MJ, Schwabe U (1988) [3H]-8-Cyclopentyl-1,3-dipropylxanthine binding to A1 adenosine receptors of intact rat ventricular myocytes. Circ Res 63:613–620 [DOI] [PubMed] [Google Scholar]
- Sei Y, Lubitz DKJE von, Abbracchio MP, Ji XD, Jacobson KA (1997) Adenosine A3 receptor agonist-induced neurotoxicity in rat cerebellar granule neurons. Drug Dev Res 40:267–273 [Google Scholar]
- Stambaugh C, Jiang JL, Jacobson KA, Liang BT (1997) Novel cardioprotective function of adenosine A3 receptor during prolonged simulated ischemia. Am J Physiol 273:H501–H505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik-Schlieper H, Klotz KN, Kreye VAW, Schwabe U (1989) Characterization of the K+-channel-coupled adenosine receptor in guinea pig atria. Naunyn-Schmiedeberg’s Arch Pharmacol 340:684–688 [DOI] [PubMed] [Google Scholar]
- Yao Y, Sei Y, Abbracchio MP, Jiang JL, Kim YC, Jacobson KA (1997) Adenosine A3 receptor agonists protect HL-60 cells and U-937 cells from apoptosis induced by A3 antagonists. Biochem Biophys Res Commun 232:317–322 [DOI] [PMC free article] [PubMed] [Google Scholar]