

Abstract
While apolipoprotein E (apoE) expression by myeloid cells is recognized to control inflammation, whether such benefits can be communicated via extracellular vesicles is not known. Through the study of extracellular vesicles produced by macrophages derived from the bone marrow of Wildtype (WT‐BMDM‐EV) and ApoE deficient (EKO‐BMDM‐EV) mice, we uncovered a critical role for apoE expression in regulating their cell signaling properties. WT‐BMDM‐EV communicated anti‐inflammatory properties to recipient myeloid cells by increasing cellular levels of apoE and miR‐146a‐5p, that reduced NF‐κB signalling. They also downregulated cellular levels of miR‐142a‐3p, resulting in increased levels of its target carnitine palmitoyl transferase 1A (CPT1A) which improved fatty acid oxidation (FAO) and oxidative phosphorylation (OxPHOS) in recipient cells. Such favorable metabolic polarization enhanced cell‐surface MerTK levels and the phagocytic uptake of apoptotic cells. In contrast, EKO‐BMDM‐EV exerted opposite effects by reducing cellular levels of apoE and miR‐146a‐5p, which increased NF‐κB−driven GLUT1‐mediated glucose uptake, aerobic glycolysis, and oxidative stress. Furthermore, EKO‐BMDM‐EV increased cellular miR‐142a‐3p levels, which reduced CPT1A levels and impaired FAO and OxPHOS in recipient myeloid cells. When cultured with naïve CD4+ T lymphocytes, EKO‐BMDM‐EV drove their activation and proliferation, and fostered their transition to a Th1 phenotype. While infusions of WT‐BMDM‐EV into hyperlipidemic mice resolved inflammation, infusions of EKO‐BMDM‐EV increased hematopoiesis and drove inflammatory responses in myeloid cells and T lymphocytes. ApoE‐dependent immunometabolic signaling by macrophage extracellular vesicles was dependent on transcriptional axes controlled by miR‐146a‐5p and miR‐142a‐3p that could be reproduced by infusing miR‐146a mimics & miR‐142a antagonists into hyperlipidemic apoE‐deficient mice. Together, our findings unveil a novel property for apoE expression in macrophages that modulates the immunometabolic regulatory properties of their secreted extracellular vesicles.
Keywords: ApoE, extracellular vesicles, immunometabolism, inflammation, macrophage, microRNA, oxidative stress
1. INTRODUCTION
Apolipoprotein (Apo) E, a 34 kDa protein expressed as three common human isoforms, was first identified for its central role in controlling levels of cholesterol‐rich lipoproteins in plasma, thereby protecting against atherosclerosis and cardiovascular disease (Mahley, 1988; Weisgraber, 1994). Soon thereafter, apoE became increasingly recognized for its ability to exert pleiotropic properties to maintain tissue homeostasis (Curtiss & Boisvert, 2000). Among these included a capacity to regulate immune cell activity and inflammation (Curtiss & Boisvert, 2000). While its expression is restricted to myeloid cells, apoE exerts a control over numerous types of leukocytes through paracrine mechanisms that remain incompletely understood (Bonacina et al., 2018; He et al., 2021; Igel et al., 2021; Riddell et al., 1997).
Macrophages, the second most important cellular source of apoE expression in mammals, are recognized to contribute up to 10% of apoE in plasma (Hasty et al., 1999; Linton et al., 1991). While this source of apoE expression has been shown to protect against atherosclerosis by enhancing plasma lipoprotein clearance (Boisvert et al., 1995; Linton et al., 1995), numerous other pathways have emerged. Macrophage apoE expression improves cellular cholesterol efflux (Rosenson et al., 2012) and the phagocytic uptake of apoptotic cells (Grainger et al., 2004), while also reducing the expression of inflammatory cytokines (Curtiss & Boisvert, 2000) and co‐stimulatory molecules on the cell surface (Bonacina et al., 2018; Tenger & Zhou, 2003) that together limit lesion inflammation. Studies also revealed that apoE expression in hematopoietic stem and progenitor cells (HSPC) in the bone marrow and spleen restricts myelopoiesis by limiting receptor complexes in cholesterol‐rich lipid rafts in the plasma membrane, which reduces proliferative signaling (Murphy et al., 2011). Furthermore, apoE expression in macrophages has recently been identified to regulate microRNA‐controlled NF‐κB signaling to limit inflammatory cytokine production and atherosclerosis in mice with hyperlipidemia (Li et al., 2015).
Although cytokines produced by macrophages are recognized to differentially modulate inflammation and its resolution in hyperlipidemia and atherosclerosis (Moore et al., 2013), extracellular vesicles (EVs) have recently emerged as a novel source of intercellular signaling (Bouchareychas et al., 2020, 2021; Nguyen et al., 2018). Indeed, studies have shown that EVs produced by macrophages can differentially control inflammatory properties in recipient cells, including in models of cancer (Zheng et al., 2018), atherosclerosis (Bouchareychas et al., 2020, 2021), and obesity (Phu et al., 2022). While macrophage apoE expression is recognized to play a central role in modulating cellular inflammatory and tissue‐reparative properties, whether it also controls the production and cell signaling properties of EVs is not known. Our study sought to address this question in the context of hyperlipidemia.
Our findings show that while a loss of apoE expression does not alter the rate or size of EV secretion by cultured macrophages, it substantially impacts their cellular signaling properties. Consistent with our recent observations (Bouchareychas et al., 2020), EVs produced by Wildtype macrophages (WT‐BMDM‐EV) communicated anti‐inflammatory properties by driving fatty acid oxidation (FAO) and oxidative phosphorylation (OxPHOS) in recipient myeloid cells. In contrast, EVs produced by Apoe −/− macrophages (EKO‐BMDM‐EV) communicated inflammatory signaling by increasing glycolysis and oxidative stress. Unlike WT‐BMDM‐EV which, as previously shown (Bouchareychas et al., 2020), resolved systemic inflammation when infused into hyperlipidemic mice, the infusion of EKO‐BMDM‐EV increased hematopoiesis and activated myeloid cells and T lymphocytes. Together, our findings reveal a novel property through which macrophage apoE expression serves to control immunity and limit inflammation in hyperlipidemia.
2. RESULTS
2.1. Characterization and in vitro assessment of cell signaling properties of extracellular vesicles produced by Apoe −/− and Wildtype macrophages
Fully differentiated Apoe −/− and Wildtype BMDM were cultured in EV‐depleted medium for 24 h. EVs secreted by these BMDM were purified using cushioned‐density gradient ultracentrifugation (C‐DGUC), a method that allows for a gentle concentration and purification of EVs from conditioned culture medium and biofluids as reported in our prior studies (Duong et al., 2019). Nanoparticle tracking analysis revealed similar particle concentration of 5.2 × 1010 and 5.5 × 1010 particles/mL and average mode size of 98 and 95 nm for EVs derived from EKO‐BMDM‐EV and WT‐BMDM‐EV, respectively (Figure 1a–c). Our data show that both Apoe −/− and Wildtype BMDM secreted the same quantity of EVs in a 24 h period, averaging 6.5 × 109 nano‐particles per million cells for both conditions (Figure S1A). Morphological assessment of EKO‐BMDM‐EV and WT‐BMDM‐EV using transmission electron microscopy revealed an expected cup‐shaped morphology and size averaging 100 nm (Figure 1d). Such isolates also showed similar average protein concentrations of 59 and 63 μg/mL for EKO‐BMDM‐EV and WT‐BMDM‐EV, respectively (Figure S1B). Western blot analysis showed the presence of EV proteins, including CD9, CD63, and CD81, and the absence of cell‐associated proteins Calnexin and GM130 in 1.5 × 109 particles of EKO‐BMDM‐EV or WT‐BMDM‐EV (Figure 1e). Despite sharing similar biophysical characteristics, only WT‐BMDM‐EV displayed apoE immunoreactivity (Figure 1e), a finding consistent with prior studies of EVs derived from macrophages (Zheng et al., 2018) and neutrophils (He et al., 2021). Importantly, using size‐exclusion chromatography, we demonstrate that apoE‐containing WT‐BMDM‐EV isolated by our C‐DGUC approach are free of apoE‐HDL (Figure 1f) that are likely also produced by the cultured BMDM and could otherwise serve as a confounding mediator of cellular signaling.
FIGURE 1.
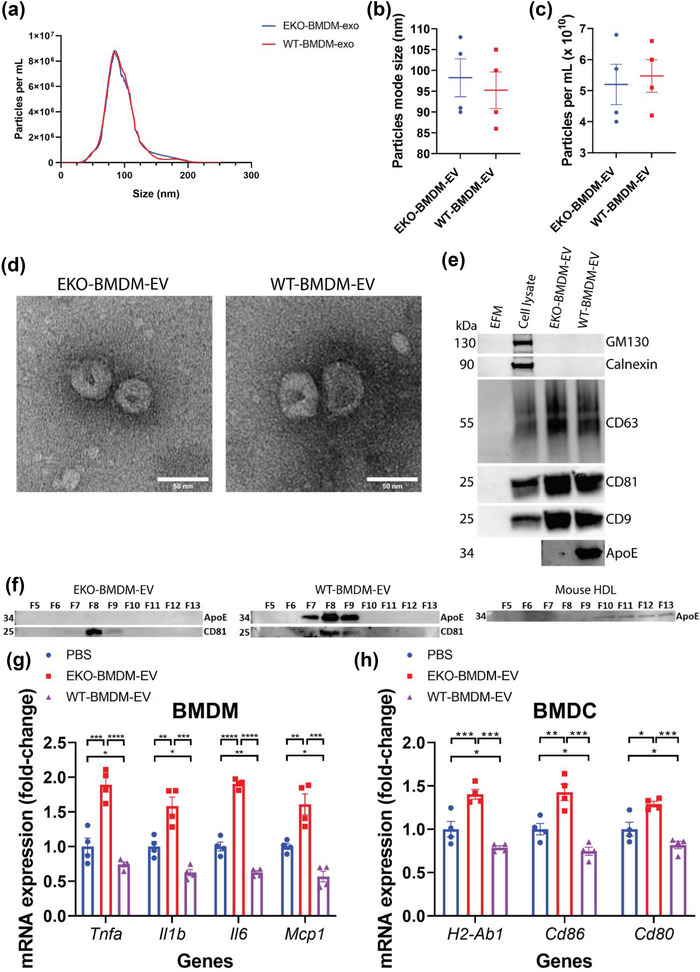
Biophysical parameters and immune‐modulation effects of BMDM‐derived extracellular vesicles. (a) Representative concentration and size distributions of EKO‐BMDM‐EV & WT‐BMDM‐EV purified from BMDM cell culture supernatants after a 24 h period of culture as determined using nanoparticle tracking analysis. (b and c) Average mode of particle diameter (b) and concentration of purified EVs in particles/mL (c) (n = 4 samples per group). (d) Electron micrograph of purified EVs from BMDM. Scale bar: 50 nm. (e) Western blot analysis of Calnexin, GM130, CD9, CD63, CD81, and apoE in EV‐free media (EFM), cell lysate, and 1.5 × 109 particles of BMDM‐derived EVs (representative of three independent experiments). (f) Western blot analysis of apoE and CD81 in EKO‐BMDM‐EV, WT‐BMDM‐EV, and mouse HDL fractionated by size‐exclusion chromatography. (g) qRT‐PCR analysis of Tnf, Il1b, Mcp1, and Il6 mRNA expression in wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h and stimulated with LPS (100 ng/mL) for 6 h. qRT‐PCR results were normalized to B2m or Gapdh, one representative experiment out of three independent replicates is shown; n = 4 per group. (h) qRT‐PCR analysis of H2‐Ab1, Cd86, and Cd80 mRNA expression in wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h and stimulated with LPS (100 ng/mL) for 6 h. qRT‐PCR results were normalized to B2m or Gapdh, one representative experiment out of three independent replicates is shown; n = 4 per group. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 as determined using one‐way ANOVA followed by Holm‐Sidak post‐test. Data are presented as mean ± SEM.
We next assessed whether EKO‐BMDM‐EV and WT‐BMDM‐EV displayed altered cell‐signaling properties. We did so by first testing their rate of cellular uptake by treating naïve BMDM with 2 × 109 particles/mL of PKH26‐labeled EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS as control and measured the fluorescent intensity in recipient cells after a 2‐h incubation period using fluorescent microscopy. Data in Figures S1C,D shows similar cellular internalization efficiencies for both forms of EVs.
Next, we incubated naïve BMDM & BM‐derived dendritic cells (BMDC) with EKO‐BMDM‐EV or WT‐BMDM‐EV at a concentration of 2 × 109 particles/mL for 18 h and subsequently stimulated the cells with 100 ng/mL lipopolysaccharides (LPS) for 6 h. Data in Figure 1g,h, show that BMDM/BMDC treated with EKO‐BMDM‐EV displayed increased expression of pro‐inflammatory cytokines and M1 macrophage marker genes (Tnf, Il6, Il1b, and Mcp1), as well as antigen‐presenting and co‐stimulatory molecules (H2‐Ab1, Cd86, and Cd80) following LPS stimulation. In contrast, WT‐BMDM‐EV treatments attenuated the expression of these genes in LPS‐stimulated BMDM and BMDC, even when compared to PBS treatments (Figure 1g,h). Taken together, our results demonstrate a previously unsuspected role for apoE expression by macrophages in modulating protective cell‐signaling properties of their secreted EVs.
2.2. Macrophage extracellular vesicles modulate cellular apoE levels and phagocytosis in recipient macrophages
We next tested the hypothesis that Apoe expression by macrophages could influence the ability for their EVs to modulate apoE levels in recipient macrophages, a process central to macrophage polarization and inflammatory activity (Baitsch et al., 2011). We did so by incubating naïve wildtype BMDM with EVs produced by both cell types at a concentration of 2 × 109 particles/mL for 18 h. While we did not observe a change in Apoe mRNA levels in recipient cells (Figure S1E), we noted that exposure to WT‐BMDM‐EV increased apoE protein levels by 2‐fold as compared to PBS exposure (Figure 2a,b). In contrast, exposure to EKO‐BMDM‐EV reduced apoE protein levels by 1.5‐fold as compared to PBS exposure (Figure 2a,b).
FIGURE 2.
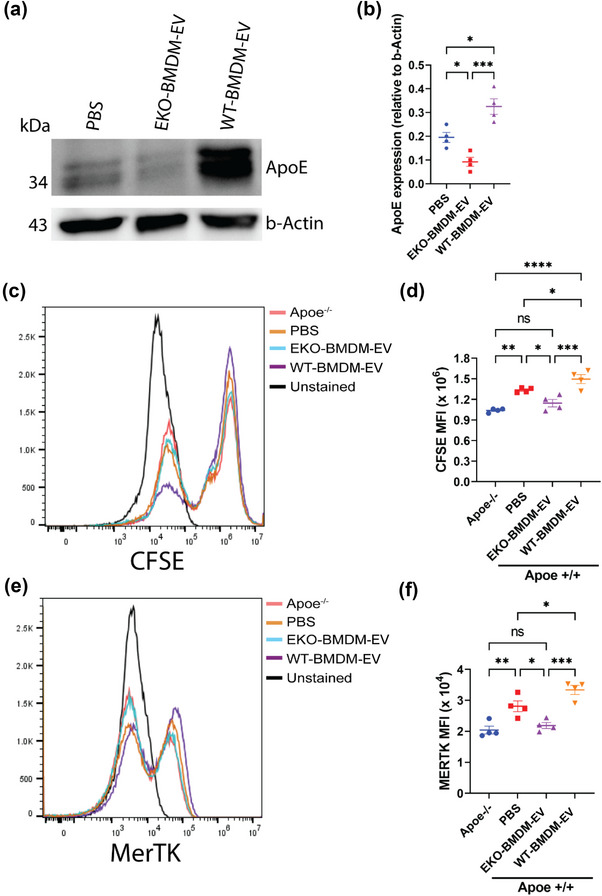
Macrophage EVs modulate cellular apoE protein levels and the phagocytic capacity of recipient macrophages. (a‐b) Western blot analysis (a) and quantification (b) of ApoE protein levels in cell lysates of wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h. (c,d) Representative histogram (c) and quantitative graph (d) showing MFI of CFSE‐labeled apoptotic Jurkat cells uptake in Apoe −/− BMDM or wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h measured by flow cytometry. (e,f) Representative histogram (e) and quantitative graph (f) showing MFI of MERTK surface expression in Apoe −/− BMDM or wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h measured by flow cytometry. One representative experiment out of two independent replicates is shown for all experiments; n = 4 per group. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 as determined using one‐way ANOVA followed by Holm‐Sidak post‐test. Data are presented as mean ± SEM.
We then wondered whether the modulation of cellular apoE levels by macrophage EVs extended beyond modulating inflammatory gene expression levels in response to LPS. Thus, we tested whether an enrichment or depletion of cellular apoE levels produced by an exposure to the two forms of BMDM EVs, respectively, could differentially impact the phagocytic properties of recipient macrophages, a process previously reported to be sensitive to cellular apoE levels (Grainger et al., 2004). Remarkably, while the exposure to WT‐BMDM‐EV enhanced the phagocytic uptake of apoptotic cells by naïve BMDM (Figure 2c,d), the exposure to EKO‐BMDM‐EV exerted an opposite effect. Specifically, EKO‐BMDM‐EV reduced the phagocytic properties of naïve wildtype BMDM to levels observed in naïve Apoe −/− BMDM (Figure 2c,d). Mechanistically, data shown in Figure 2e,f demonstrate that the exposure to WT‐BMDM‐EV caused an enrichment of MerTK on the macrophage cell surface, a receptor central to the clearance of apoptotic cells (Thorp et al., 2008). In contrast, an exposure to EKO‐BMDM‐EV reduced MerTK cell‐surface density on recipient macrophages that paralleled their reduced phagocytic capacity of apoptotic cells. Together, our findings show that ApoE expression by macrophages increases the capacity for their EVs to control inflammatory signaling and effector functions in recipient macrophages by increasing cellular apoE levels.
2.3. ApoE expression dictates the capacity for macrophage extracellular vesicles to suppress glucose uptake and glycolysis in recipient myeloid cells via a miR‐146a/NF‐κB axis
Building on our prior studies documenting a capacity for macrophage EVs to modulate bioenergetic metabolism in recipient naïve BMDM (Bouchareychas et al., 2020, 2021; Phu et al., 2022), we wondered whether such effects were sensitive to cellular apoE expression in the parental cell. We thus tested the capacity for EKO‐BMDM‐EV to communicate metabolic signaling to recipient myeloid cells. We did so by first examining the ability of EKO‐BMDM‐EV to modulate both basal and LPS‐stimulated aerobic glycolysis in recipient BMDM using a Seahorse Glycolytic Rate Assay to measure the glycolytic proton efflux rate (glycoPER). As shown in Figure 3a and b, naïve macrophages treated with EKO‐BMDM‐EV displayed increased levels of basal and compensatory glycolysis, as well as an increase in proton efflux rate produced from glycolytic activity as compared to macrophages treated WT‐BMDM‐EV and PBS both under basal condition and following LPS‐stimulation.
FIGURE 3.
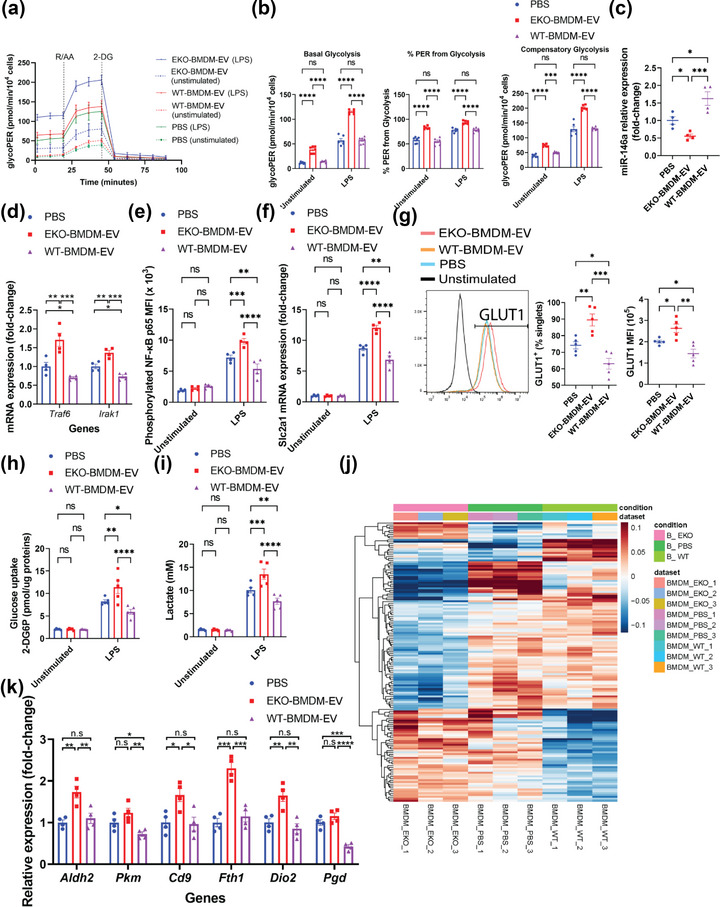
ApoE expression dictates the capacity for macrophage EVs to suppress glucose uptake and glycolysis in recipient macrophages via a miR‐146a/NF‐κB axis. (a) Graph showing representative Seahorse Glycolytic Rate Assay. R/AA, rotenone/antimycin A (0.5 μM) and 2‐DG, 2‐Deoxy‐D‐glucose (50 mM). (b) Graph showing quantified cell‐normalized glycolysis‐associated proton efflux rate (glycoPER) from the Seahorse Glycolytic Rate Assay. (c) qRT‐PCR analysis of miR‐146a‐5p expression in wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h. (d) qRT‐PCR analysis of Irak1 and Traf6 mRNA levels in wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h and subsequently stimulated with LPS (100 ng/mL) for 6 h. (e) MFI of nuclear NF‐κB phospho‐p65 subunit measured by flow cytometry in wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h and subsequently cultured in basal or LPS‐stimulated condition (100 ng/mL) for 6 h. (f) qRT‐PCR analysis of Slc2a1 mRNA expression in wildtype BMDM exposed to 2×109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h and subsequently cultured in basal or LPS‐stimulated condition (100 ng/mL) for 6 h. (g) Graphs showing percentage of GLUT1+ cells and mean fluorescent intensity (MFI) of GLUT1 in wildtype BMDM exposed to 2×109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h and subsequently stimulated with LPS (100 ng/mL) for 6 h. (h) 2‐DG uptake assay in wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h and subsequently cultured in basal or LPS‐stimulated condition (100 ng/mL) for 6 h. (i) Lactate production to the conditioned media by wildtype BMDM exposed to 2×109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h and subsequently cultured in basal or LPS‐stimulated condition (100 ng/mL) for 6 h as measured by the L‐Lactate Assay Kit. (j) Unannotated heatmap showing the distinct mRNA expression profiles between wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h (n = 3 per group, p <0.05). (k) qRT‐PCR analysis of Aldh2, Pkm, Cd9, Fth1, Dio2, and Pgd mRNA expression in wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h. qRT‐PCR results were normalized to B2m or Gapdh for mRNA analysis and U6 snRNA or miR‐16‐5p for microRNA analysis. One representative experiment out of three independent replicates is shown for all experiments; n = 3–5 per group. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 as determined using one‐way or two‐way ANOVA followed by Holm‐Sidak post‐test. Data are presented as mean ± SEM.
In seeking to uncover mechanisms to explain these observations, we next tested whether treatments of EKO‐BMDM‐EV altered miR‐146a‐5p levels in recipient myeloid cells, a microRNA that we previously observed as downregulated in apoE‐deficient monocytes and macrophages (Li et al., 2015). Interestingly, we noted that a quantity of 2 × 109 particles/mL of EKO‐BMDM‐EV suppressed miR‐146a‐5p levels by 50% in cultured naïve BMDM and BMDC as compared to a similar number of PBS‐treated cells (Figures 3c and S2A), a finding that paralleled our observed reduction in cellular apoE levels (Figure 2a,b). Next, we examined whether the downregulation of miR‐146a‐5p exerted by EKO‐BMDM‐EV could alter cellular inflammatory signaling via mRNA target genes. Findings shown in Figures 3d and S2B confirm an expected upregulation of canonical miR‐146a‐5p targets Traf6 & Irak1, two central mediators of NF‐κB signaling (Boldin et al., 2011; Li et al., 2015; Taganov et al., 2006), in naïve BMDM and BMDC treated with EKO‐BMDM‐EV and subsequently stimulated with LPS for 6 h. We also observed an enhanced nuclear accumulation of phosphorylated NF‐κB p65 subunit in LPS‐stimulated BMDM pre‐treated with EKO‐BMDM‐EV as compared to BMDM treated with WT‐BMDM‐EV or PBS (Figure 3e). As expected, we observed that an upregulation of apoE protein levels in naïve BMDM and BMDC exposed to WT‐BMDM‐EV (Figure 2a,b) exerted protective properties by increasing miR‐146a‐5p levels (Figures 3c and S2A) resulting in suppressed NF‐κB signaling (Figures 3d,e and S2B).
As the NF‐κB signaling pathway has been recognized as a major driver of GLUT1‐mediated glucose uptake and glycolysis (Obaid et al., 2021), we next tested whether the miR‐146a/NF‐κB axis controlled by EKO‐BMDM‐EV contributed to control cellular glucose uptake and glycolytic activity. Our findings show that both BMDM and BMDC treated with 2×109 particles/mL of EKO‐BMDM‐EV upregulated levels of GLUT1 mRNA (Slc2a1) (Figures 3f and S2C) and surface protein density (Figure 3g) upon stimulation with LPS. Increased cell‐surface GLUT1 in BMDM treated with EKO‐BMDM‐EV led to enhanced glucose uptake in these cells as measured by the cellular uptake of 2‐deoxyglucose (2‐DG) (Figure 3h). We also recorded increased levels of lactate, the end‐product of aerobic glycolysis caused by LPS‐stimulation (Kelly & O'Neill, 2015), in the conditioned media of LPS‐stimulated BMDM pre‐treated with EKO‐BMDM‐EV (Figure 3i). In contrast, we observed that WT‐BMDM‐EV treatments suppressed levels of GLUT1 mRNA (Slc2a1) and cell‐surface levels in naïve BMDM and BMDC (Figures 3f,g and S2C) that resulted in reduced 2‐DG uptake and lactate production upon LPS‐stimulation (Figure 3h,i).
Building on our observations linking EKO‐BMDM‐EV to miR‐146a‐5p control of NF‐κB‐driven glycolytic activity, we next sought to explore whether treatments of EKO‐BMDM‐EV could modulate the transcriptomic profile in recipient macrophages. We did so via unbiased sequencing of RNA isolated from cultured naïve BMDM treated with 2 × 109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h. Figures 3j and S2D revealed 150 genes that were differentially expressed between these three sets of BMDM. Among these included two genes involved in the glycolytic pathway (Aldh2 and Pkm) and six genes recognized to drive glycolytic activity (Cd9, Fth1, Dio2, and Pgd) (Daneshmandi et al., 2021; Sagliocchi et al., 2019; Scaramuzzino et al., 2021; Suárez et al., 2021; Sun et al., 2019) that were all found to be highly enriched in naïve BMDM treated with EKO‐BMDM‐EV as compared to cells treated with WT‐BMDM‐EV or PBS. This pattern of gene expression was further confirmed by qRT‐PCR in naïve BMDM and BMDC treated with 2 × 109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS (Figures 3k and S2E). Taken together, our data show that pro‐glycolytic signaling effects communicated by EKO‐BMDM‐EV are exerted due to a lack of apoE expression in the parental cells.
2.4. ApoE expression dictates the capacity for macrophage extracellular vesicles to communicate FAO and OxPHOS to recipient myeloid cells via a miR‐142a/CPT1A axis
In light of our findings documenting the capacity for EKO‐BMDM‐EV to drive glycolytic activity in recipient cells, we next examined whether they had the ability to modulate OxPHOS. Consistent with this idea, the RNA‐seq data shown in Figures 3j and S2D led us to identify the gene coding for carnitine palmitoyltransferase 1A (Cpt1a), an important driver of FAO and OxPHOS, to be suppressed in naïve BMDM treated with EKO‐BMDM‐EV. In contrast, this gene was enriched in BMDM treated with WT‐BMDM‐EV as compared to those treated with PBS treatment. qRT‐PCR confirmed this pattern of gene expression by showing a 30% increase in Cpt1a mRNA levels in naïve BMDM/BMDC treated with WT‐BMDM‐EV and a 40% decrease in Cpt1a mRNA levels in naïve BMDM/BMDC treated with EKO‐BMDM‐EV as compared to cells treated with PBS (Figures 4a and S3A). We also observed a 20% increase and 45% decrease in CPT1A protein levels in naïve BMDM treated with WT‐BMDM‐EV and EKO‐BMDM‐EV, respectively, as compared to cells treated with PBS (Figure 4b,c). We next sought to identify the mechanism responsible for the bidirectional control of CPT1A by EKO‐BMDM‐EV and WT‐BMDM‐EV treatments. Based on a recent study that highlighted the role of miR‐142a‐3p in downregulating OxPHOS and FAO by targeting CPT1A (Sun et al., 2019), we tested the expression of this microRNA in naïve BMDM/BMDC treated with the two forms of EVs or PBS as control. We uncovered that miR‐142a‐3p levels were enriched by 90% in BMDM/BMDC treated with EKO‐BMDM‐EV (Figures 4d and S3B), while they were reduced by 40% in cells treated with WT‐BMDM‐EV (Figures 4d and S3B).
FIGURE 4.
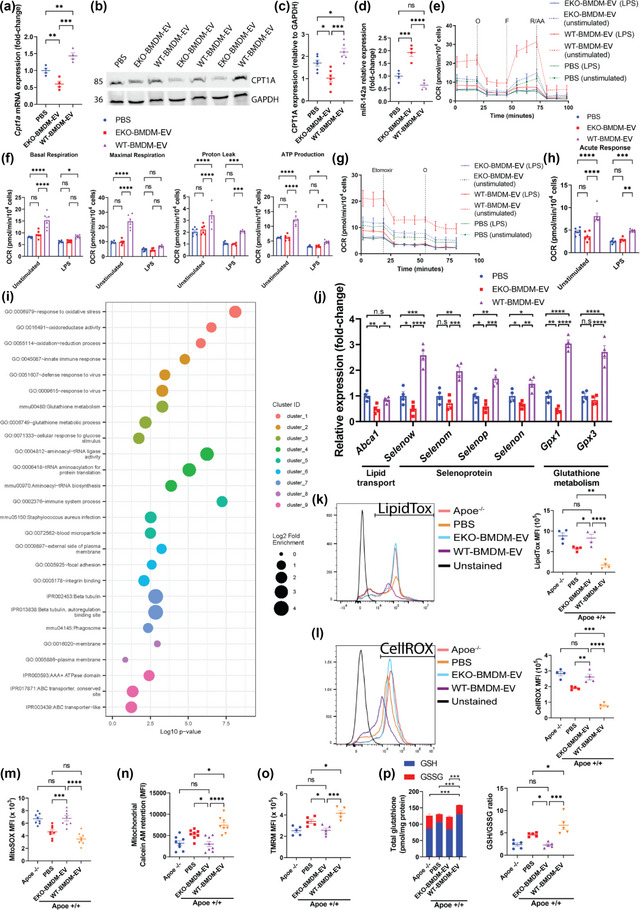
ApoE expression dictates the capacity for macrophage EVs to improve mitochondrial health & functions while suppressing neutral lipids accumulation & oxidative stress in recipient macrophages. (a) qRT‐PCR analysis of Cpt1a mRNA expression in wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h. (b‐c) Western blot analysis (b) and quantification (c) of CPT1A protein levels in cell lysates of wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h. (d) qRT‐PCR analysis of miR‐142a‐3p expression in wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h. (e) Graph showing representative Seahorse Mito Stress Assay. O, oligomycin (1 μM); F, FCCP (2 μM); and R/AA, rotenone/antimycin A (0.5 μM). (f) Graph showing quantified cell‐normalized mitochondrial OCR from Mito Stress test. (g) Graph showing representative OCR measurement in response to etomoxir treatment as measured by the Agilent Seahorse instrument. Etomoxir (4 μM) and O, oligomycin (1 μM). (h) Graph showing quantified cell‐normalized mitochondrial OCR drop upon CPT1a inhibition by etomoxir. (i) GO enrichment analysis (Biological process) of the genes differentially expressed between wildtype BMDM exposed to EKO‐BMDM‐EV or WT‐BMDM‐EV. The minimum count of genes considered for the analysis was >10 and p <0.05. (j) qRT‐PCR analysis of Abca1, Selenow, Selenom, Selenop, Selenon, Gpx1 and Gpx3 mRNA expression in wildtype BMDM exposed to 2×109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h. (k) Graph showing MFI of LipidTOX staining measured by flow cytometry in Apoe −/− BMDM or wildtype BMDM exposed to 2×109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h measured by flow cytometry. (l) Graph showing MFI of CellROX staining measured by flow cytometry in Apoe −/− BMDM or wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h measured by flow cytometry. (m‐o) Graphs showing MFI of MitoSOX (m), Calcein AM (n), and TMRM (o) signals in Apoe −/− BMDM or wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h measured by flow cytometry. (p) Graphs showing detection of total glutathione, including reduced glutathione (GSH) and oxidized glutathione (GSSG), in Apoe −/− BMDM or wildtype BMDM exposed to 2 × 109 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h measured by flow cytometry. qRT‐PCR results were normalized to B2m or Gapdh for mRNA analysis and U6 snRNA or miR‐16‐5p for microRNA analysis. One representative experiment out of three independent replicates is shown for all experiments; n = 3–5 per group. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 as determined using one‐way or two‐way ANOVA followed by Holm‐Sidak post‐test. Data are presented as mean ± SEM.
We next assessed whether a bidirectional control of the miR‐142a/CPT1A axis by EKO‐BMDM‐EV or WT‐BMDM‐EV treatments could serve to differentially modulate FAO and OxPHOS in recipient cells. To test this hypothesis, we measured the oxygen consumption rate (OCR) in BMDM treated with 2 × 109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS under basal and LPS‐stimulated conditions for 18 h using the Seahorse Mito Stress Assay. Our data show that unstimulated naïve BMDM treated with WT‐BMDM‐EV displayed enhanced OxPHOS as seen by elevated basal and maximal respiration associated with a higher proton leak and ATP production compared to PBS‐treated cells (Figure 4e and f). In contrast, naïve BMDM treated with EKO‐BMDM‐EV displayed similar levels of OxPHOS as compared to PBS‐treated cells in basal conditions (Figure 4e and f). We also noted a substantial reduction of OxPHOS in BMDM across all three treatments upon LPS stimulation, with cells treated with WT‐BMDM‐EV retaining more robust basal respiration and ATP production (Figure 4e and f).
Stemming from our data shown in Figure 4a–c that support a role for EKO‐BMDM‐EV in suppressing CPT1A expression, we next tested whether levels of OCR were associated with CPT1A‐mediated mitochondrial FAO by treating BMDM with the CPT1A inhibitor etomoxir following their exposure to EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS, respectively. We did so by using etomoxir at a working concentration of 4 μM, which allows to target CPT1A activity without causing cellular oxidative stress known to occur at concentrations greater than 5 μM (O'Connor et al., 2018). Data shown in Figure 4g,h reveal that, despite higher OCR at basal state, unstimulated BMDM treated with WT‐BMDM‐EV display a more substantial drop in OCR upon etomoxir treatment as compared to cells treated with EKO‐BMDM‐EV or PBS, indicating that the elevated OxPHOS in BMDM treated with WT‐BMDM‐EV is driven primarily by CPT1A‐dependent FAO. Interestingly, EKO‐BMDM‐EV treatments further lowered CPT1A‐dependent FAO as compared to PBS treatments as seen by a smaller drop in OCR upon etomoxir treatment (Figure 4g,h). Furthermore, we observed that LPS‐stimulated BMDM displayed minor drops in OCR upon etomoxir treatment as compared to unstimulated cells. In contrast, we noted that naïve BMDM treated with WT‐BMDM‐EV displayed a more pronounced drop in OCR as compared to BMDM treated with EKO‐BMDM‐EV or PBS (Figure 4g,h). Taken together, our data uncover a novel role for apoE expression in macrophages that is responsible for producing EVs capable of communicating OxPHOS and FAO to recipient myeloid cells.
2.5. ApoE expression dictates the capacity for macrophage extracellular vesicles to drive lipid mobilization, oxidative stress responses, and mitochondrial metabolism in recipient myeloid cells
We next performed a gene ontology (GO) enrichment assessment in naïve BMDM treated with WT‐BMDM‐EV vs. cells treated with EKO‐BMDM‐EV by subjecting the RNA‐seq data in Figures 3j and S2D to the Database for Annotation, Visualization and Integrated Discovery (DAVID) tool (Huang et al., 2007). This approach led us to uncover an enrichment in sets of genes involved in oxidative stress responses, glutathione metabolism, and ABC transporters in naïve BMDM treated with WT‐BMDM‐EV (Figure 4i), processes known to control mitochondrial oxidative stress (Kerksick & Willoughby, 2005) and cellular lipid efflux (Tall et al., 2008), respectively. Indeed, WT‐BMDM‐EV treatments enhanced the expression of the selenoproteins and glutathione peroxidase genes (Selenow, Selenom, Selenop, Selenon, Gpx1, and Gpx3) (Figures 3j, 4j, S2D and S3C). Interestingly, we also observed an enrichment in genes involved in the phagosome pathway (Figure 4i) in naïve BMDM treated with WT‐BMDM‐EV, which parallels our data in Figure 2c,d, supporting their increased capacity for the phagocytic uptake of apoptotic Jurkat cells. In sharp contrast, an assessment of the RNA‐seq data in Figures 3j and S2D, confirmed by qRT‐PCR data in BMDM & BMDC (Figures 4j and S3C), revealed a substantial reduction in the expression of genes involved in these biological processes in naïve BMDM treated with EKO‐BMDM‐EV. This includes the lipid transporter Abca1, the selenoproteins Selenow, Selenom, Selenop, and Selenon, as well as the glutathione peroxidases Gpx1 and Gpx3. Together, these findings support the notion that apoE expression is critical for macrophages to communicate OxPHOS and mitochondrial metabolism to recipient cells via EVs.
Next, we assessed whether changes in the transcriptomic profiles recorded in BMDM treated with the two forms of EVs could lead to functional changes in cellular metabolism. We first measured the accumulation of neutral lipids in naïve BMDM with LipidTOX staining followed by detection using flow cytometry upon treatment with EVs using a dose of 2 × 109 particles/mL. Data shown in Figure 4k revealed a 54% increase in LipidTOX mean fluorescent intensity (MFI) in naïve BMDM treated with EKO‐BMDM‐EV as compared to those treated with PBS. In contrast, WT‐BMDM‐EV treatments reduced LipidTOX MFI by 71% in naïve BMDM as compared to PBS treatments, and 81% as compared to EKO‐BMDM‐EV‐treatments and Apoe −/− BMDM alone (Figure 4k).
We next measured the accumulation of reactive oxygen species in naïve BMDM treated with both forms of EVs at a concentration of 2 × 109 particles/mL using CellROX staining detected by flow cytometry. Data in Figure 4l reveal a 39% increase in CellROX MFI in naïve BMDM treated with EKO‐BMDM‐EV as compared to PBS‐treated cells. In contrast, WT‐BMDM‐EV treatments reduced the CellROX MFI by 72% as compared to PBS‐treated cells and 81% as compared to naïve BMDM treated with EKO‐BMDM‐EV or Apoe −/− BMDM alone (Figure 4l). Consistent with our data shown in Figure 4l, we observed a 48% and 110% increase in mitochondrial superoxide (MitoSOX) accumulation in naïve BMDM treated with EKO‐BMDM‐EV as compared to naïve BMDM treated with PBS or WT‐BMDM‐EV, respectively (Figure 4m). Such increase in cellular and mitochondrial oxidative stress conferred by EKO‐BMDM‐EV treatments contributed to the prolonged opening of the mitochondrial transition pores as shown by Figure 4n, which resulted in a 41% and 58% reduction in mitochondrial Calcein AM retention as compared to naïve BMDM treated with PBS or WT‐BMDM‐EV, respectively (Figure 4n). Such adverse effects caused by EKO‐BMDM‐EV treatments on reducing mitochondrial health contributed to a lower mitochondrial membrane potential (ΔΨm) as detected by Tetramethylrhodamine (TMRM) staining. Figure 4o revealed a 29% and 40% drop in the TMRM MFI in naïve BMDM treated with EKO‐BMDM‐EV as compared to naïve BMDM treated with PBS or WT‐BMDM‐EV, respectively.
Finally, we measured the levels of glutathione (GSH) and glutathione disulfide (GSSG) in cells treated with 2 × 109 particles/mL of both forms of BMDM EVs. Results in Figure 4p show that naïve BMDM treated with WT‐BMDM‐EV displayed greater levels of GSH as well as the GSH:GSSG ratio, indicative of more robust protection against oxidative stress and improved cellular health (Owen & DA, 2010). In contrast, BMDM treated with EKO‐BMDM‐EV showed reduced levels of GSH and a GSH:GSSG ratio (Figure 4p), highlighting detrimental effects caused by these EVs in predisposing recipient cells to oxidative stress. Remarkably, treatments with EKO‐BMDM‐EV communicated a similar magnitude of detrimental effects to naïve BMDM as was detected when examining Apoe −/− BMDM alone across the different measurements in Figure 4k–p. Together, these data further support a critical role for apoE expression in macrophages that results in the production of extracellular vesicle capable of communicating mitochondrial metabolism and oxidative stress control in recipient myeloid cells.
2.6. Loss of apoE expression in macrophages results in extracellular vesicles that promote CD4+ T lymphocyte activation and proliferation
Next, we assessed whether macrophage EVs can control adaptive immune responses, and whether a loss of apoE expression can be detrimental. We did so by treating αCD3/αCD28‐stimulated CD4+ T lymphocytes with 2 × 109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h. We then performed unbiased RNA sequencing to assess changes in the transcriptomic profiles of these cells upon EV treatments and identified 37 differentially expressed genes (Figure 5a). Among these, we identified three genes that are key members of the T‐cell receptor complex (Cd3e, Cd247, and Cd4) and Il2rg, a key component of cytokine receptors on CD4+ T lymphocytes (Figure 5a). GO enrichment analysis using the DAVID tool (Huang et al., 2007) of the RNA‐seq data from CD4+ T lymphocytes treated with EKO‐BMDM‐EV or WT‐BMDM‐EV (Figure 5a), identified an enrichment of genes involved in the positive regulation of T cell activation, T cell receptor signaling pathway, and cell surface receptor pathway (Figure 5b). In light of these findings, we performed a T lymphocyte proliferation assay using carboxyfluorescein succinimidyl ester (CFSE)‐labeled CD4+ T lymphocytes stimulated with αCD3/αCD28 beads and treated every two days with 2×109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for a period of 4 days. We found that the mitotic index of CD4+ T lymphocytes treated with EKO‐BMDM‐EV was 36% higher than in cells treated with WT‐BMDM‐EV or PBS (Figure 5c). This finding was consistent with data shown in Figure 5d, demonstrating reduced levels of Annexin V, a marker of apoptosis, in αCD3/αCD28‐stimulated CD4+ T lymphocytes treated with 2 × 109 particles/mL of EKO‐BMDM‐EV, indicating that the lower number of apoptotic cells in this treatment group is associated with increased cellular proliferation. Next, we examined the cellular activation of naïve CD4+ T lymphocytes exposed to EKO‐BMDM‐EV or WT‐BMDM‐EV upon stimulation with αCD3/αCD28 beads. Our data in Figure 5e show that an exposure to EKO‐BMDM‐EV increased the cell surface density of the activation markers CD69 & CD25 in the recipient cells. Furthermore, this also led the cells to produce higher levels of the Th1 inflammatory cytokine IFN‐γ (Figure 5f). Together, our data uncover a critical new property for macrophage apoE expression in controlling T lymphocyte activation via communication by EVs.
FIGURE 5.
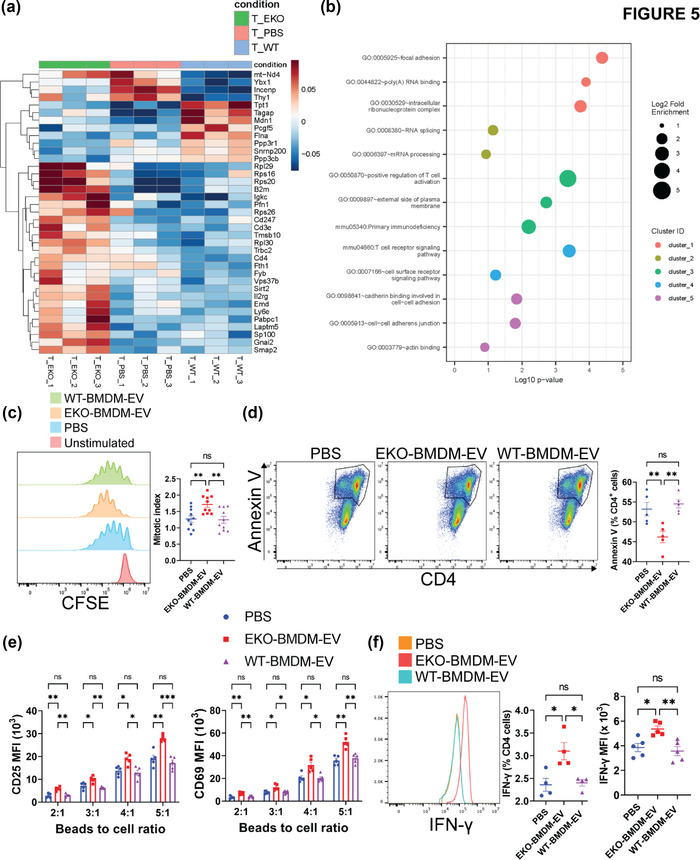
EKO‐BMDM‐EV drive the activation and proliferation of CD4+ T lymphocytes. (a) Heatmap showing the distinct mRNA expression profiles between wildtype CD4+ T lymphocytes exposed to 2 × 109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 24 h (n = 3 per group, p < 0.05) while stimulated with αCD3/αCD28 beads. (b) GO enrichment analysis (Biological process) of the genes differentially expressed between wildtype CD4+ T lymphocytes exposed to EKO‐BMDM‐EV or WT‐BMDM‐EV while stimulated with αCD3/αCD28 beads. The minimum count of genes considered for the analysis was >10 and p <0.05. (c) Graphs showing CD4+ T lymphocytes proliferation measured by CFSE labeling of CD4+ T lymphocytes stimulated with αCD3/αCD28 beads for 4 days. 2 × 109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS were added to the culture on day 1 and 3 of the experiment. (d) Graphs showing percentage of Annexin V+ CD4+ T lymphocytes upon stimulation with αCD3/αCD28 beads for 4 days. 2 × 109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS were added to the culture on day 1 and 3 of the experiment. (e) Graphs showing MFI of CD25 and CD69 in CD4+ T cells co‐cultured with αCD3/αCD28 beads, 5 ng/mL of murine IL‐2, and 2 × 109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 48 h. (f) Graphs showing IFN‐γ+ cells and IFN‐γ MFI in CD4+ T cells co‐cultured with αCD3/αCD28 beads, 5 ng/mL of murine IL‐2, and 2 × 109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 12 h. One representative experiment out of three independent replicates is shown for all experiments; n = 3–5 per group. *p < 0.05, ** p < 0.01, ***p < 0.001, and ****p < 0.0001 as determined using one‐way ANOVA followed by Holm‐Sidak post‐test. Data are presented as mean ± SEM.
2.7. ApoE expression dictates the capacity for macrophage extracellular vesicles to suppress systemic inflammation and activation of myeloid cells in hyperlipidemic mice
In light of our provocative in vitro findings supporting the capacity for EKO‐BMDM‐EV to shift macrophage cellular metabolism toward a glycolytic phenotype, we next assessed whether these EVs could exert such properties in vivo to drive inflammatory responses sensitive to elevated glycolysis (Koelwyn et al., 2008; Tabas & Bornfeldt, 2020). We did so by repeatedly infusing EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS intraperitoneally (i.p.) to hyperlipidemic mice. Prior to EV infusions, recipient 6‐week‐old C57BL/6J mice were first treated intravenously with a recombinant adeno‐associated virus vector serotype 8 encoding a gain‐of‐function mutant of proprotein convertase subtilisin/kexin type 9 (AAV8‐PCSK9) and fed a high‐fat Western Diet (WD) as previously reported (Bjørklund et al., 2014; Lu et al., 2016; Maxwell & Breslow, 2004; Roche‐Molina et al., 2015). These mice displayed similar levels of hyperlipidemia, with cholesterol and triglycerides levels of 660 and 215 mg/dL, respectively, after 6 weeks of high‐fat WD feeding (Figure S4A‐B). Furthermore, an assessment of the plasma lipoprotein profile in these mice showed an accumulation of remnant lipoproteins that is similar to the profile displayed by the Ldlr −/− mouse model fed a high‐fat diet (Figure S4C) as previously reported (Bjørklund et al., 2014; Lu et al., 2016; Maxwell & Breslow, 2004; Roche‐Molina et al., 2015).
Next, we assessed the biodistribution of EKO‐BMDM‐EV and WT‐BMDM‐EV by infusing i.p. 1 × 1010 particles of 1,1'‐Dioctadecyl‐3,3,3',3'‐Tetramethylindotricarbocyanine Iodide (DiR)‐labeled EVs or PBS as control into WD‐fed AAV8‐PCSK9 mice. Following 6 h post infusion, the presence of DiR‐positive EVs was detected in the circulation (Figures 6a & S5), as well as the epididymal white adipose tissues, lungs, livers, brains, kidneys, aortas, spleens, bones, and intestines (Figures 6b & S5). Our findings did not show any noticeable difference in the biodistribution of DiR‐labeled EKO‐BMDM‐EV and WT‐BMDM‐EV in hyperlipidemic mice.
FIGURE 6.
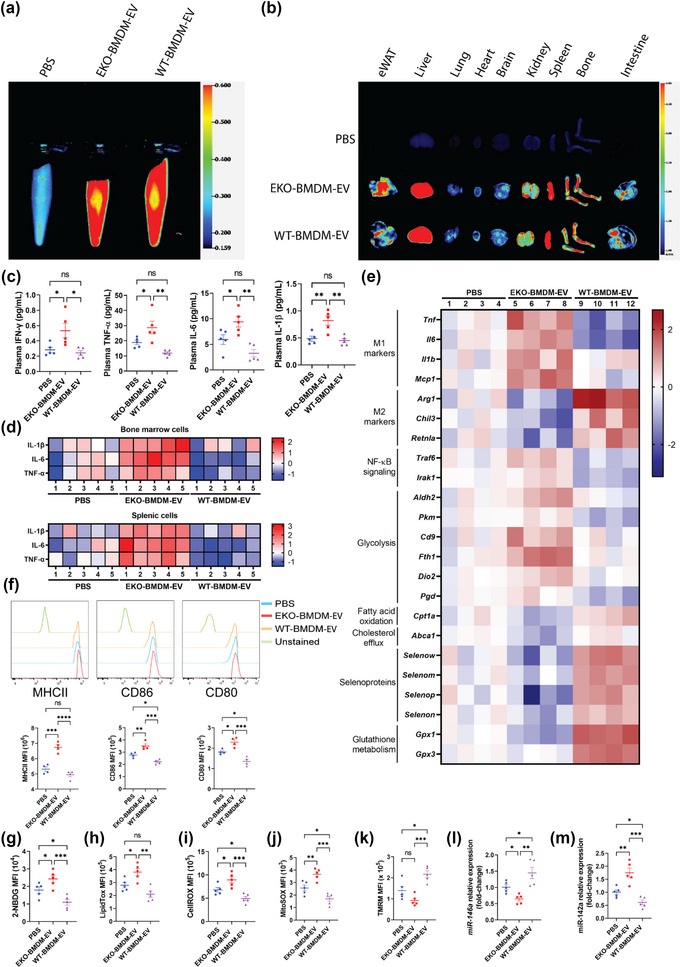
ApoE expression dictates the capacity for macrophage EVs to improve mitochondrial health & functions while suppressing glucose uptake, oxidative stress, activation of myeloid cells & systemic inflammation in hyperlipidemic mice. (a‐b) Images of DiR fluorescence in blood (a) and organs (b) 6 h post‐injection from 8‐week‐old Western diet‐fed AAV8‐PCSK9‐injected mice infused i.p. with PBS as control or 1 × 1010 particles of EKO‐BMDM‐EV or WT‐BMDM‐EV. (c) Multiplex immunoassay analysis of TNF‐α, IFN‐γ, IL‐6, and IL‐1β from plasma of Western diet‐fed AAV8‐PCSK9‐injected mice repeatedly infused with 1×1010 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS. (d) Heat maps representing multiplex immunoassay analysis of TNF‐α, IL‐6, and IL‐1β cytokines released by LPS‐stimulated splenic and bone marrow cells (100 ng/mL for 6 h) from Western diet‐fed AAV8‐PCSK9‐injected mice repeatedly infused with 1 × 1010 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS. Data are displayed as log2 fold‐change relative to PBS group. (e) Heat map representing qRT‐PCR analysis of Tnf, Il1b, Mcp1, Il6, Arg1, Retnla, Chil3, Traf6, Irak1, Aldh2, Pkm, Cd9, Fth1, Dio2, Pgd, Cpt1a, Abca1, Selenow, Selenom, Selenop, Selenon, Gpx1 and Gpx3 mRNA expression in peritoneal macrophages of Western diet‐fed AAV8‐PCSK9‐injected mice repeatedly infused with 1 × 1010 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS. Data are displayed as log2 fold‐change relative to PBS group. (f) MFI of MHC‐II, CD86, and CD80 expression in splenic Ly6C− MHCII+ CD11c+ cells measured by flow cytometry. (g‐k) Graphs showing MFI of 2‐NBDG (g), LipidTOX (h), CellROX (i), MitoSOX (j), and TMRM (k) signals in circulating Ly6Chi monocytes of Western diet‐fed AAV8‐PCSK9‐injected mice repeatedly infused with 1 × 1010 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS. (l‐m) qRT‐PCR analysis of miR‐146a‐5p (l) and miR‐142a‐3p (m) expression in peritoneal macrophages of Western diet‐fed AAV8‐PCSK9‐injected mice repeatedly infused with 1 × 1010 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS. qRT‐PCR results were normalized to B2m or Gapdh for mRNA analysis and U6 snRNA or miR‐16‐5p for microRNA analysis. One representative experiment out of two independent replicates is shown for all experiments; n = 4–5 per group. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 as determined using one‐way ANOVA followed by Holm‐Sidak post‐test. Data are presented as mean ± SEM.
We then proceeded to treat the mice with either 1 × 1010 particles of EKO‐BMDM‐EV or WT‐BMDM‐EV, a dose that represents approximately 2%–5% of total EVs in mouse plasma (Bouchareychas et al., 2021), or PBS, three times/week (every two days) for 4 weeks while maintaining the mice on WD. We first measured the impact of such treatments on the accumulation of plasma cytokines using a multiplex immunoassay. Our data show that EKO‐BMDM‐EV infusions raised levels of the inflammatory cytokines IFN‐γ, TNF‐α, IL‐6, and IL‐1β in the circulation (Figure 6c). Interestingly, WT‐BMDM‐EV infusions reduced levels of TNF‐α and IL‐6 in the plasma of these mice (Figure 6c). Next, we measured the accumulation of these cytokines in the conditioned media of LPS‐stimulated bone marrow and splenic cells derived from these mice. Data in Figure 6d indicates a substantial accumulation of TNF‐α, IL‐6, and IL‐1β in the conditioned media of these cells when derived from mice infused with EKO‐BMDM‐EV. Consistent with our plasma cytokine data in Figure 6c, we also noted a reduction in the production of TNF‐α and IL‐6 cytokines from these cells when derived from mice infused with WT‐BMDM‐EV. We also observed elevated gene expression in these cytokines and M1 macrophage markers (Tnf, Il6, Il1b, and Mcp1) and reduced gene expression in M2 macrophage markers (Arg1, Retnla, and Chil3) in peritoneal macrophages derived from mice that received EKO‐BMDM‐EV infusions as compared to those that received infusions of WT‐BMDM‐EV or PBS (Figure 6e). Moreover, WT‐BMDM‐EV infusions reduced the mRNA levels of the pro‐inflammatory cytokines Tnf and Il6 while increasing the M2 markers (Arg1, Retnla, and Chil3) in the peritoneal macrophages (Figure 6e). An assessment of dendritic cells (Ly6C− MHCII+ CD11c+) derived from the spleens of these mice also revealed an increase in the antigen‐presenting major histocompatibility complex class II (MHC‐II), as well as the co‐stimulatory molecules CD86 and CD80, on the surface of these cells when derived from mice infused with EKO‐BMDM‐EV (Figure 6f). Interestingly, WT‐BMDM‐EV infusions led to reduced expression of CD86 and CD80 on the surface of splenic dendritic cells even when compared to those derived from PBS‐treated mice (Figure 6f).
2.8. ApoE expression dictates the capacity for macrophage extracellular vesicles to improve mitochondrial health & function while suppressing glucose uptake & oxidative stress in myeloid cells of hyperlipidemic mice
Next, we assessed the capacity of the two forms of BMDM EVs to modulate the metabolic properties of primary myeloid cells derived from hyperlipidemic mice. Using flow cytometry, we found that circulating Ly6Chi monocytes derived from WD‐fed AAV8‐PCSK9 mice infused with EKO‐BMDM‐EV displayed increased glucose uptake as seen by the absorption of (2‐(N‐(7‐Nitrobenz‐2‐oxa‐1,3‐diazol‐4‐yl)Amino)‐2‐Deoxyglucose) (2‐NBDG) (Figure 6g) and neutral lipid accumulations as seen by LipidTOX staining (Figure 6h). We also observed increased levels of cellular reactive oxygen species (Figure 6I) and mitochondrial superoxides (Figure 6j) in circulating Ly6Chi monocytes examined from these mice. Such increased cellular levels of oxidative stress led to lower mitochondrial ΔΨm as revealed by TMRM staining (Figure 6k). In contrast, circulating Ly6Chi monocytes of WD‐fed AAV8‐PCSK9 mice infused with WT‐BMDM‐EV displayed decreased cellular glucose uptake, neutral lipid and reactive oxygen species accumulation, along with mitochondrial superoxides (Figure 6g–j). Furthermore, they displayed improved ΔΨm (Figure 6k).
Building on our in vivo data indicating a capacity of macrophage apoE to communicate immunometabolic signaling via EVs, we next assessed whether infusions of EKO‐BMDM‐EV into WD‐fed AAV8‐PCSK9 mice could modulate the expressions of target genes and microRNA revealed through our in vitro experiments with cultured primary BMDM and BMDC. Our findings revealed that peritoneal macrophages collected from mice infused with EKO‐BMDM‐EV also displayed reduced levels of miR‐146a‐5p while displaying increased levels of miR‐142‐3p (Figure 6l,m), consistent with our findings in cultured naïve BMDM treated with these EVs in Figures 3c and 4D. Furthermore, peritoneal macrophages derived from WD‐fed AAV8‐PCSK9 mice infused with EKO‐BMDM‐EV displayed increased expression of genes involved in the propagation of NF‐κB signaling, and those involved in glycolytic activity (Traf6, Irak1, Aldh2, Pkm, Cd9, Fth1, Dio2, and Pgd) (Boldin et al., 2011; Daneshmandi et al., 2021, Li et al., 2015; Sagliocchi et al., 2019; Scaramuzzino et al., 2021; Suárez et al., 2021; Sun et al., 2019; Taganov et al., 2006E) (Figure 6e). In contrast, these cells exhibited reduced expression of genes involved in FAO, cholesterol efflux, and oxidative stress protection (Cpt1a, Abca1, Selenow, Selenom, Selenop, Selenon, Gpx1, and Gpx3) (Figure 6e). Taken together, these data provide robust evidence supporting a role for macrophage apoE expression in regulating the immune and metabolic modulatory effects of their secreted EVs when tested in a hyperlipidemic in vivo environment.
2.9. Loss of apoE expression in macrophages results in extracellular vesicles that enhance hyperlipidemia‐driven hematopoiesis and myelopoiesis
As apoE deficiency in hematopoietic stem & progenitor cells (HSPC) has been reported to drive hematopoiesis and myelopoiesis in hyperlipidemic mice (Murphy et al., 2011), we next investigated whether infusions of EKO‐BMDM‐EV could communicate similar effects to drive excessive hyperlipidemia‐driven HSPC proliferation. Unlike our prior findings documenting a reduction of hematopoiesis by WT‐BMDM‐EV (Bouchareychas et al., 2020, 2021), we found that hyperlipidemic WD‐fed AAV8‐PCSK9 mice infused with EKO‐BMDM‐EV displayed increased cell numbers across all subsets of bone marrow HSPC (Figure 7a,b). This included elevated numbers of Lin− Sca‐1+ c‐Kit+ (LSK) cells, hematopoietic stem cells (HSC), multipotent progenitor cells (MPP), lymphomyeloid multipotent progenitor cells (LMPP), multipotent progenitor cells 1–4 (MPP1‐4), common myeloid progenitors (CMP), granulocyte‐macrophage progenitors (GMP), and megakaryocyte‐erythroid progenitors (MEP) (Figure 7a,b). We further detected a similar phenotype among progenitor cells examined from the spleens of mice infused with EKO‐BMDM‐EV as compared to those infused with WT‐BMDM‐EV or PBS (Figures S6A and 7c). Such acceleration of hyperlipidemia‐driven hematopoiesis in WD‐fed AAV8‐PCSK9 mice infused with EKO‐BMDM‐EV augmented myelopoiesis as revealed by increased numbers of circulating and splenic CD11b+ cells, Ly6Chi monocytes, and neutrophils (Figures S6B and 7d–f). Together, our data provide evidence supporting a role for macrophage apoE expression for the production of EVs capable of controlling hyperlipidemia‐driven hematopoiesis and myelopoiesis. Our findings thereby unveil a novel feedback property of myeloid cell apoE expression in modulating chronic leukocytosis & inflammation in hyperlipidemia.
FIGURE 7.

EKO‐BMDM‐EV enhance hyperlipidemia‐driven hematopoiesis and myelopoiesis. (a) Representative plots of flow cytometric analyses of hematopoietic stem and progenitor cells in the bone marrow. (b‐c) Graphs showing the percentages of hematopoietic stem and progenitor cell subsets (LSK, LMPP, MPP, MPP1‐4, HSC, CMP, GMP, and MEP) in the bone marrow (b) and spleen (c) of Western diet‐fed AAV8‐PCSK9‐injected mice repeatedly infused with 1 × 1010 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS. (d‐e) Representative flow cytometric analyses of circulating myeloid cells (d) and measurements of myeloid cell subsets (CD11b+ cells, neutrophils, Ly6Chi monocytes, and Ly6Clo monocytes) (e) in the circulation of Western diet‐fed AAV8‐PCSK9‐injected mice repeatedly infused with 1×1010 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS. (f) Flow cytometric analyses of splenic myeloid cell subsets (monocytes, neutrophils, Ly6Chi monocytes, and Ly6Clo monocytes) in the spleen of Western diet‐fed AAV8‐PCSK9‐injected mice repeatedly infused with 1 × 1010 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS. One representative experiment out of two independent replicates is shown for all experiments; n = 5 per group. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 as determined using one‐way ANOVA followed by Holm‐Sidak post‐test. Data are presented as mean ± SEM.
2.10. Systemic infusions of miR‐146a mimics or miR‐142a inhibitors substitute for apoE in suppressing hyperlipidemia‐driven hematopoiesis and myelopoiesis in hyperlipidemic mice
Next, we sought to explore whether the control of hyperlipidemia‐driven hematopoiesis and myelopoiesis by macrophage‐derived EVs was in part dependent on the modulation of miR‐146a‐5p and miR‐142a‐3p in recipient cells. We did so by performing repeated intravenous (i.v.) infusions of 1 nmol of miR‐146a mimics or miR‐142a inhibitors to WD‐fed Apoe −/− mice twice a week for a total of 4 weeks (Figure 8a) as previously reported (Li et al., 2015; Sun et al., 2014). While infusions of RNA mimics or antagonists did not alter the levels of plasma cholesterol in these mice (Figure S7A), delivery of miR‐146a mimics elevated the miR‐146a levels in splenic monocytes by 50% and had no impact on levels of miR‐142a‐3p (Figure S7B–C). Moreover, similar treatments using miR‐142a inhibitors reduced cellular miR‐142a‐3p levels by 95% in splenic monocytes (Figure S7C). Infusions of miR‐146a mimics or miR‐142a inhibitors suppressed the numbers of LSK, HSC, MPP, LMPP, MPP1‐4, CMP, GMP, and MEP in the bone marrows of WD‐fed Apoe −/− mice (Figure 8b,c). Interestingly, we noted a greater decrease in the number of HSPC in mice treated with miR‐142a inhibitors (Figure 8b,c). Such downregulation in hematopoiesis mirrored an observed decrease in CD11b+ cells, Ly6Chi monocytes, and neutrophils in the circulation of these mice, with miR‐142a inhibitors exerting a more profound effect (Figure 8d,e). Together, these data support a role for apoE expression in dictating the capacity of macrophage‐derived EVs to suppress hyperlipidemia‐driven hematopoiesis and myelopoiesis via the regulation of miR‐146a‐5p and miR‐142a‐3p in recipient cells.
FIGURE 8.
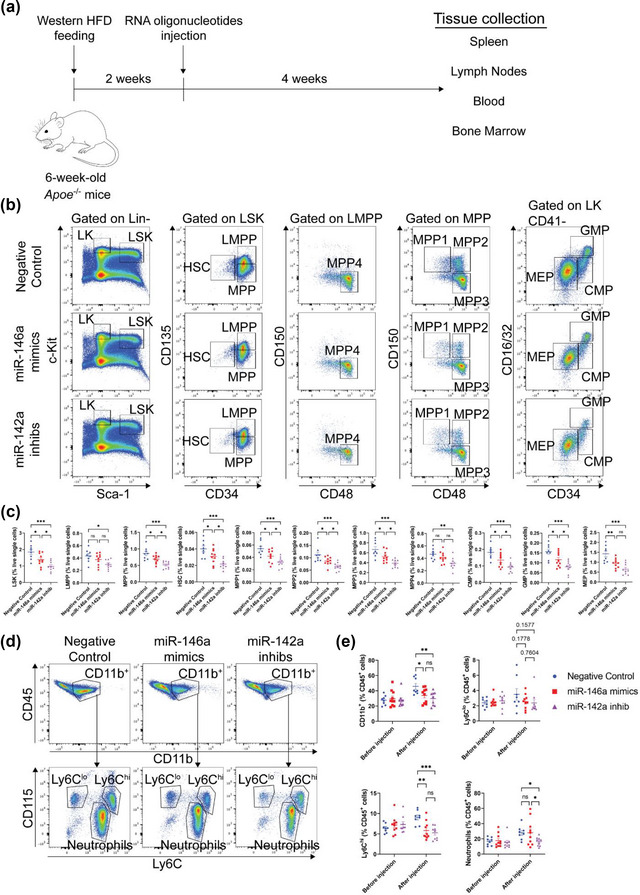
Systemic infusions of miR‐146a mimics or miR‐142a antagonists suppress hyperlipidemia‐driven hematopoiesis and monocytosis in Apoe −/− mice. (a) Schematic diagram depicting the injections of RNA oligonucleotides in Western diet‐fed Apoe −/− mice. (b) Representative plots of flow cytometric analyses of hematopoietic stem and progenitor cells in the bone marrow. (c) Graphs showing the percentages of hematopoietic stem and progenitor cell subsets (LSK, LMPP, MPP, MPP1‐4, HSC, CMP, GMP, and MEP) in the bone marrow of Western diet‐fed Apoe −/− mice repeatedly infused with 1 nmol of miR‐146a mimics, miR‐142a inhib, or negative control. (d‐e) Representative flow cytometric analyses of circulating myeloid cells (d) and measurements of myeloid cell subsets (CD11b+ cells, neutrophils, Ly6Chi monocytes, and Ly6Clo monocytes) (e) in the circulation of Western diet‐fed Apoe −/− mice repeatedly infused with 1 nmol of miR‐146a mimics, miR‐142a inhib, or negative control. Pooled data from two independent replicates is shown for all experiments; n = 8–10 per group. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 as determined using one‐way ANOVA followed by Holm‐Sidak post‐test. Data are presented as mean ± SEM.
2.11. Loss of apoE expression in macrophages results in extracellular vesicles that drive the proliferation, activation, and production of IFN‐γ in T lymphocytes
In light of our data shown in Figure 5a–f demonstrating a capacity for EKO‐BMDM‐EV to drive the activation and proliferation of CD4+ T cells in vitro, we next investigated whether infusions of these EVs into hyperlipidemic WD‐fed AAV8‐PCSK9 mice could adversely impact the activity of T lymphocytes in these mice. Interestingly, mice that received EKO‐BMDM‐EV infusions for 4 weeks displayed elevated levels of circulating T lymphocytes (CD45+ CD3e+) when compared to those that received WT‐BMDM‐EV or PBS (Figure 9a,b), while no differences were noted for circulating B lymphocytes (CD45+ B220+) (Figure 9a,b). Consistent with our findings in the circulation, we also observed an accumulation of CD4+ and CD8+ T lymphocytes in the spleens of mice infused with EKO‐BMDM‐EV (Figure 9c–d). Moreover, these cells exhibited increased cell‐surface levels of CD69 (Figure 9c and e), an established marker for recently activated T lymphocytes (Cibrián & Sánchez‐Madrid, 2017). These findings are consistent with our data in Figure 9c,f–g, which uncovered greater levels of effector memory cells T cells (TEM, CD44hi CD62Llo) and lower levels of naïve T cells (Tnaive, CD44lo CD62Lhi) in both splenic CD4+ and CD8+ populations derived from mice infused with EKO‐BMDM‐EV. Furthermore, these mice exhibited increased levels of splenic CD44+ CD4+ and CD44+ CD8+ T lymphocytes expressing the activation marker and chemokine receptor C‐X‐C Motif Chemokine Receptor 3 (CXCR3) (Figure 9c and h), which participates in modulating T lymphocyte activation and trafficking (Groom & Luster, 2011).
FIGURE 9.
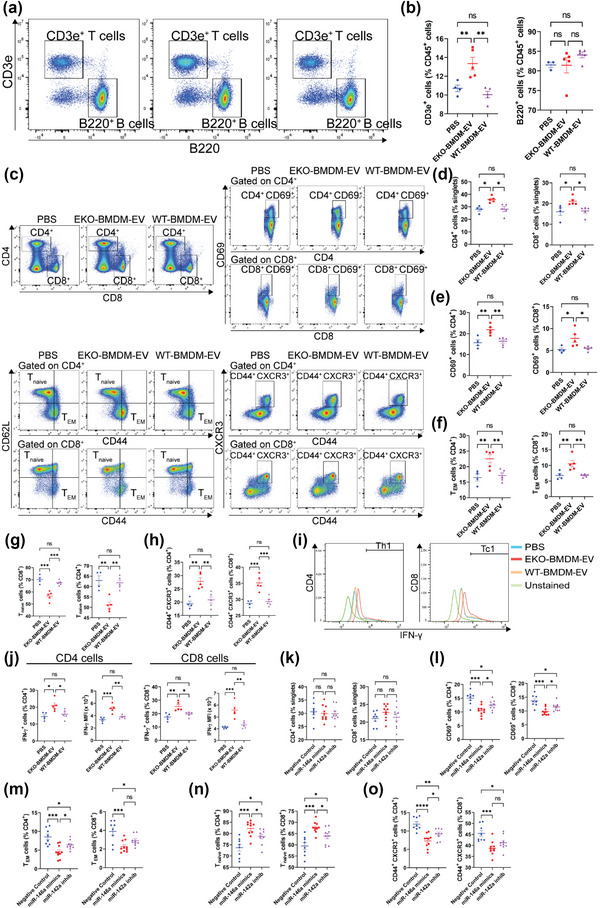
EKO‐BMDM‐EV drive the proliferation, activation, and IFN‐γ release from T lymphocytes of hyperlipidemic mice via the modulation of miR‐146a and miR‐142a levels. (a‐b) Representative flow cytometric analyses of circulating lymphocytes (a) and measurements of lymphocyte subsets (CD3e+ T lymphocytes and B220+ B lymphocytes) (b) in the circulation of Western diet‐fed AAV8‐PCSK9‐injected mice repeatedly infused with 1×1010 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS. (c‐h) Representative flow cytometric analyses of splenic T lymphocytes (c) and measurements of total CD4+ & CD8+ cells (d), CD4+ CD69+ & CD8+ CD69+ cells (e), CD4+ CD44+ CD62L− & CD8+ CD44+ CD62L− cells (TEM) (f), CD4+ CD44− CD62L+ & CD8+ CD44− CD62L+ cells (Tnaïve) (g), and CD4+ CD44+ CXCR3+ & CD8+ CD44+ CXCR3+ cells (h) in the spleens of Western diet‐fed AAV8‐PCSK9‐injected mice repeatedly infused with 1×1010 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS. (i‐j) Representative flow cytometric analyses of splenic Th1 and Tc1 lymphocytes (i) and measurements of IFN‐γ+ cells and IFN‐γ MFI (j) within the CD4+ and CD8+ T lymphocyte populations derived from Western diet‐fed AAV8‐PCSK9‐injected mice repeatedly infused with 1×1010 particles of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS. One representative experiment out of two independent replicates is shown for all experiments; n = 5 per group. (k‐o) Measurements of total CD4+ & CD8+ cells (k), CD4+ CD69+ & CD8+ CD69+ cells (l), CD4+ CD44+ CD62L− & CD8+ CD44+ CD62L− cells (m), CD4+ CD44− CD62L+ & CD8+ CD44− CD62L+ cells (n), and CD4+ CD44+ CXCR3+ & CD8+ CD44+ CXCR3+ cells (o) in the lymph nodes of Western diet‐fed Apoe −/− mice repeatedly infused with 1 nmol of miR‐146a mimics, miR‐142a inhib, or negative control. Pooled data from two independent replicates is shown for all experiments; n = 8–10 per group. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 as determined using one‐way ANOVA followed by Holm‐Sidak post‐test. Data are presented as mean ± SEM.
Next, we detected the impact of macrophage EVs on regulating the production of IFN‐γ, a pro‐inflammatory cytokine associated with helper T cell type I (Th1) and cytotoxic T cell type I (Tc1), in splenic CD4+ and CD8+ T lymphocytes that had been stimulated with 20 ng/mL of phorbol 12‐myristate 13‐acetate (Zhang et al., 2019) and 1 μg/mL of ionomycin for 4 h. Consistent with our in vitro data in Figure 5f, our ex vivo data in Figure 9i,j show that levels of IFN‐γ+ cells derived from the spleens of mice infused with EKO‐BMDM‐EV increased by 33% and 44% among CD4+ and CD8+ populations, respectively, as compared to T cells examined from WD‐fed AAV8‐PCSK9 mice infused with either WT‐BMDM‐EV or PBS. Furthermore, these cells displayed 41% and 45% greater MFI for IFN‐γ among CD4+ and CD8+ populations, respectively, as compared to cells derived from mice infused with WT‐BMDM‐EV and PBS (Figure 9i,j).
Lastly, we assessed whether systemic infusions of miR‐146a mimics or miR‐142a inhibitors could reverse T lymphocyte activation in hyperlipidemic WD‐fed Apoe −/− mice. While such treatments did not change the total numbers of lymph node‐derived CD4+ and CD8+ T lymphocytes (Figure 9k), they did suppress the expression of the activation marker CD69 in both CD4+ and CD8+ T lymphocyte populations (Figure 9l). Furthermore, both treatments successfully lowered levels of the TEM subset while elevating levels of the Tnaive subset in both CD4+ and CD8+ T lymphocytes (Figure 9m,n). Treatments with both forms of RNA oligonucleotides also suppressed the levels of CD44+ CD4+ and CD44+ CD8+ T lymphocytes expressing CXCR3 (Figure 9o). Interestingly, miR‐146a mimics exerted a more profound effect in reducing T lymphocyte activation and trafficking (Figure 9k–o).
Collectively, findings from this study bring to light a previously unappreciated role for macrophage apoE expression in controlling hyperlipidemia‐driven innate and adaptive immunity and inflammation via their secreted extracellular vesicles.
3. DISCUSSION
The expression of ApoE by myeloid cells has long been recognized to serve as a source of immune regulation (Curtiss & Boisvert, 2000; Linton et al., 1995; Mahley, 1988). Beyond contributing to the pool of apoE in plasma that benefits liver‐mediated remnant lipoprotein clearance (Mahley & Ji, 1999), apoE expression by myeloid cells has the ability to exert both cell‐intrinsic and cell‐extrinsic properties that together shape the immune repertoire and inflammatory status of leukocytes in hyperlipidemia (Davignon, 2005). Findings from this study uncover yet another immune‐regulatory property for macrophage apoE expression. Our data show that apoE expression by macrophages is critical for their ability to communicate immunometabolic signaling to recipient cells via extracellular vesicles.
While the expression of apoE did not impact the size or number of EVs produced by cultured primary macrophages, it substantially altered their capacity to communicate immunometabolic signaling to myeloid cells, T lymphocytes, and HSPCs. Indeed, the loss of apoE expression in macrophages led to the production of EKO‐BMDM‐EV that promoted the maturation of BMDC, primed BMDM to become more susceptible to inflammatory signaling, fostered pro‐survival and inflammatory signaling in T lymphocytes, and drove hematopoiesis in mice with hyperlipidemia. Furthermore, beyond impacting the production of inflammatory cytokines, EKO‐BMDM‐EV substantially impaired a key anti‐inflammatory process in recipient macrophages. By downregulating cell‐surface levels of MerTK in naïve cultured macrophages, EKO‐BMDM‐EV decreased their phagocytic uptake of apoptotic cells, hampering a process recognized for augmenting tissue repair and anti‐inflammatory properties (Thorp et al., 2008; Yurdagul et al., 2017; Zhang et al., 2019).
The role of apoE in serving as a checkpoint against inflammatory extracellular vesicle production by macrophages is supported by numerous lines of evidence. Exposure to WT‐BMDM‐EV attenuated the expression of inflammatory cytokines in BMDM and the maturation of BMDC in response to Toll‐like receptor (TLR) signaling following LPS stimulation. It also enhanced cell‐surface MerTK levels and improved the phagocytic capacity of recipient naïve BMDM. Our findings also identify apoE as molecular cargo in macrophage extracellular vesicles that could account for an observed enrichment of apoE in recipient macrophages, which has been shown to serve a functional role in improving their anti‐inflammatory activity (Curtiss & Boisvert, 2000). Conversely, the apoE‐depleting effect, caused by EKO‐BMDM‐EV exposure to recipient macrophages, could account for the observed heightened inflammatory properties. This effect is akin to that of IFN‐γ signaling that reduces apoE levels in macrophages polarizing the cells into an inflammatory phenotype (Brand et al., 1993). Interestingly, the modulation of cellular apoE levels by cytokine signaling was reported to occur through post‐translational mechanisms (Brand et al., 1993). Similar mechanisms could explain our observations of reduced apoE levels in BMDM stimulated with EKO‐BMDM‐EV. In contrast, increased apoE levels in BMDM stimulated with WT‐BMDM‐EV could have derived from direct delivery of apoE as EV cargo, as no changes in apoE mRNA were detected in the recipient cells. Future studies will thus be required to determine the mechanism associated with the modulation of cellular apoE levels in recipient myeloid cells exposed to EVs derived from macrophages.
It is interesting to speculate on the functional consequences conferred by the modulation of cellular apoE levels by the two forms of BMDM EVs. Robust changes in cellular apoE levels could have directly contributed to driving bioenergetic fuel utilization, lipid homeostasis, redox stress, and microRNA modulation in recipient macrophages. Support for this idea stems from our observations demonstrating the ability of endogenous apoE expresssion to upregulate levels of miR‐146a‐5p (Li et al., 2015) while simultaneously downregulating levels of miR‐142a‐3p (pending publication) in myeloid cells and their progenitors. The reciprocal control in levels of these two microRNAs by cellular apoE expression favors mitochondrial metabolism by fostering FAO and OxPHOS over glycolysis, resulting in suppressed myeloid cell activation, hyperlipidemia‐driven inflammation, and hematopoiesis (pending publication).
Together, our findings support the possibility that the modulation of apoE levels in macrophages serves as a rheostat for the production of EVs capable of communicating a spectrum of immunometabolic‐regulatory properties. A downregulation of cellular apoE levels caused by inflammatory cytokine signaling (Brand et al., 1993) likely polarizes cells to produce EVs that serve to drive glycolytic metabolism in recipient leukocytes to exacerbate inflammation. Conversely, an upregulation of apoE levels, such as in response to interleukin‐4−stimulated PPARγ signaling (Daniel et al., 2018; Kidani & Bensinger, 2012) that benefits cellular cholesterol efflux (Akiyama et al., 2002), also results in the production of EVs that communicate anti‐inflammatory and pro‐resolution signaling as reported in our studies of macrophages exposed to this cytokine (Bouchareychas et al., 2020; Phu et al., 2022). In line with such reasoning, ongoing studies are underway to determine whether apoE expression is required for the production of macrophage extracellular vesicles with potent anti‐inflammatory properties following interleukin‐4 treatment. Results of such studies could provide new insight to account for observations reporting impaired intercellular communication by EVs produced from apoE‐deficient M2‐like macrophages in a model of tumor cell metastasis (Zheng et al., 2018).
While our studies did not investigate bioactive cargo associated with macrophage EVs beyond identifying apoE in WT‐BMDM‐EV, they revealed mechanisms responsible for their differential signaling properties when produced by ApoE−/− or wildtype macrophages. Pro‐inflammatory properties displayed by EKO‐BMDM‐EV were found to derive from their ability to upregulate cellular aerobic glycolytic metabolism through the increase of GLUT1‐mediated glucose uptake upon LPS‐stimulation. EKO‐BMDM‐EV communicated this metabolic property by suppressing miR‐146a‐5p levels that we had previously demonstrated to be controlled by apoE expression (Li et al., 2015). These findings show that a paucity of miR‐146a‐5p levels seen in apoE‐deficient BMDM can be communicated to naïve wildtype macrophage via EVs to drive NF‐κB signaling, thereby increasing GLUT1‐driven immune cell activation (Obaid et al., 2021). Furthermore, beyond facilitating the uptake of glucose in naïve macrophages, that in itself is insufficient to drive inflammatory gene activation (Nishizawa et al., 2014), EKO‐BMDM‐EV robustly upregulated the expression of numerous glycolytic genes to accentuate aerobic glycolysis. Whether such complex bioenergetic polarization resulted from reduced cellular apoE levels alone, or if additional molecular cargo delivered by EKO‐BMDM‐EV contributed to the effects, is unclear and are topics of ongoing investigations.
In marked contrast, while WT‐BMDM‐EV exerted a modest increase in glycolytic function in recipient naïve wildtype macrophages, they exerted a profound increase in OxPHOS that was absent in cells treated with EKO‐BMDM‐EV. These observations are consistent with findings reported in our recent studies of macrophage EV function revealing their capacity to resolve inflammatory immune cell activation (Bouchareychas et al., 2020; Phu et al., 2022). Such reciprocal modulation of OxPHOS in recipient macrophages correlated with an upregulation of the mitochondrial fatty acid transporter CPT1A in cells treated with WT‐BMDM‐EV and its downregulation in those treated with EKO‐BMDM‐EV. A mechanism to explain this opposing mode of bioenergetic control is centered on the capacity for WT‐BMDM‐EV to reduce miR‐142a‐3p levels in recipient cells, a process we recently uncovered to be dependent on cellular apoE expression (pending publication). Further support for the involvement of these two microRNA‐signaling axes as central to apoE control of macrophage EV activity in the regulation of immunometabolism and inflammation derived from in vivo rescue experiments in which miR‐146a mimics or miR‐142a inhibitors were infused into hyperlipidemic Apoe −/− mice. Whether WT‐BMDM‐EV exert this form of cellular control solely by augmenting apoE levels in recipient cells is unclear, and ongoing studies exploring additional signaling cargo are underway.
Irrespective of the exact mode of cellular signaling conferred by the two forms of EVs, unbiased RNA‐seq, and gene ontology assessments revealed their distinct capacity to modulate the expression levels of genes associated with inflammation and its resolution. Among these include pathways responsible for oxidative stress control regulated by selenoproteins and glutathione peroxidases, ABC lipid transporters, and genes mediating phagocytosis. Consistent with these findings, a broad array of functional assays performed in this study revealed the capacity of EKO‐BMDM‐EV to drive neutral lipid accumulation and impair the phagocytic uptake of apoptotic cells in naïve macrophages. These assays also identified the capacity of EKO‐BMDM‐EV to increase both cellular and mitochondrial oxidant stress, which augmented the opening of the mitochondrial pore. Such effects likely contributed to impair mitochondrial activity, detected in recipient cells as decreases in membrane potential and levels of the reduced form of glutathione that mediates oxidative stress control. Together, such deleterious properties conferred by EKO‐BMDM‐EV unveil new insights that extend earlier observations of myeloid apoE expression in regulating cellular responses to redox stress (Igel et al., 2021). Furthermore, our study reports the first evidence for macrophage apoE expression acting in a paracrine manner through WT‐BMDM‐EV to profoundly oppose cellular and mitochondrial redox stress, while improving mitochondrial function by driving fatty acid utilization and OxPHOS in recipient myeloid cells.
Together, our findings reveal that cellular apoE expression in macrophages is critical for the production of EVs capable of communicating immunometabolic benefits to neighboring cells and those at a distance. Their functional targets included mature leukocytes in the circulation and lymphoid tissues, as well as their progenitor stem cells in the bone marrow and the spleen to control hematopoiesis. Based on our observations, it is not unreasonable to speculate that in the context of chronic inflammatory disorders including hyperlipidemia and diabetes, macrophage EVs could serve to reinforce deleterious leukocytosis and leukocyte activation to accelerate the pathogenesis of atherosclerosis. In contrast, in situations of plasma lipid control and PPARγ signaling, macrophage EVs could serve as a negative feedback loop to control hematopoiesis and actively stimulate the resolution of inflammation. This could also include functions such as driving the phagocytic clearance of apoptotic cells and restoring positive cellular lipid efflux in atheroma, which are both recognized as critical mechanisms responsible for driving lesion stabilization in human coronary artery disease (Doran et al., 2020). Furthermore, based on recent observations including from our group (Bouchareychas et al., 2020; Phu et al., 2022), macrophage EVs could also serve as a source of cardiometabolic protection by controlling metabolic signaling in other cell types, including adipocytes to regulate the production of adipokines, thereby controlling insulin sensitivity and obesity (Phu et al., 2022). Together, our findings support more in‐depth studies of macrophage EVs, and the contribution of isoform‐specific apoE expression, for their differential role as mediators of immunometabolic communication in cardiometabolic disease.
3.1. Limitations of the study
Our study focused on EVs isolated within the 1.07 – 1.10 g/mL density fractions using the C‐DGUC method as previously reported (Duong et al., 2019). While our studies of this population of vesicles support this class of EVs as potent mediators of intercellular communication by macrophages, other types of EVs produced by these cells may serve a role in controlling inflammation. Furthermore, while our study has identified the presence of apoE in macrophage‐derived EVs, whether all EVs contain apoE and whether it is localized inside the vesicles or on their outer membrane, as in EVs derived from the melanocytic cell line MNT‐1 (Van Niel et al., 2015), is not known. Studies will thus be required to further characterize the diversity of macrophage‐derived EVs and test whether apoE cargo facilitates their interaction with plasma lipoproteins and cell surface receptors.
4. MATERIALS AND METHODS
4.1. Animal studies
In vivo studies were conducted using C57BL/6J mice intravenously injected with AAV8‐PCSK9. Six‐week‐old male AAV8‐PCSK9‐injected C57BL/6 wildtype mice were fed a Western diet (Research Diets, USA) for 2 weeks before being randomly assigned to be infused with EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS as control for 4 weeks while remaining on the Western diet for the duration of the study (n = 4–5 for each treatment group).
For RNA oligonucleotide infusions, 6‐week‐old male Apoe −/− mice were fed a Western diet (Research Diets, USA) for 2 weeks before being randomly assigned to be infused i.v. with 1 nmol of miR‐146a mimics, miR‐142a inhibitors, or negative control RNA (scrambled) twice a week for a total of 4 weeks while remaining on Western diet (Research Diets, USA) as reported in prior studies (Li et al., 2015; Sun et al., 2014). Briefly, 1 μl of 1 nmol/ μl antisense oligonucleotides targeting miR‐142a (miR‐142a inhibitors), miR‐146a mimics, or negative control (Thermo Fisher, USA) was mixed with 100 μl PBS (Corning, USA). The transfection reagent was prepared by mixing 30 μl Lipofectamine RNAiMAX (Thermo Fisher, USA) with 70 μl PBS (Corning, USA). The 100 μl of oligonucleotides + PBS mixture was subsequently mixed with the 100 μl transfection reagent to create a 200 μl final mixture by pipetting 100x. This mixture was infused i.v. to WD‐fed Apoe −/− mice via retro‐orbital injection twice a week (1 nmol/mouse/injection).
For bone marrow derived macrophage (BMDM) EV collection, bone marrow cells were collected from age‐matched 6–12‐week‐old male Apoe −/− or wildtype mice on C57BL/6J background. These cells were then differentiated into macrophages prior to EV collection. Detailed elaboration of the cell culture and EV isolation methods are described in the sections below.
Data collection and analyses were conducted in a blinded‐fashion. All mice were housed and bred in specific pathogen–free conditions in the Animal Research Facility at the San Francisco Veterans Affairs Medical Center.
4.2. Cell culture
Murine BMDM were obtained as previously described (Bouchareychas et al., 2020, 2021, Phu et al., 2022). Briefly, bone marrow cells were flushed from the tibia and femurs of age‐matched 6–12‐week‐old male Apoe −/− or wildtype mice on C57BL/6J background. Cells were cultured in complete media containing DMEM (Corning, USA) supplemented with 10% fetal bovine serum (GIBCO, USA), 1% GlutaMax (GIBCO, USA), and 1% penicillin‐streptomycin (GIBCO, USA) and differentiated with 25 ng/ml mouse M‐CSF (Peprotech, USA) for 6 days in 37C and 5% CO2. Cells were then cultured in extracellular vesicle‐free media for the production and isolation of EVs.
For in vitro experiments, BMDM were seeded into 12‐well culture plates (Corning, USA) at a concentration of 3×105 cells/well and treated with 2×109 particles/mL of EVs or equal volume of PBS for 18 h. The cells were then collected for analysis or stimulated with lipopolysaccharides (LPS) (Sigma Aldrich, USA) for 6 h.
For bone marrow derived dendritic cells (BMDC), bone marrow cells were flushed from the tibia and femurs of 6‐ to 12‐week‐old male Apoe −/− or wildtype mice on C57BL/6J background. Cells were cultured in complete media and differentiated with 25 ng/ml mouse GM‐CSF (Peprotech, USA) for 6 days in 37C and 5% CO2. Cells were collected in media suspension as immature BMDC. Cells were then seeded into 12‐well culture plates (Corning, USA) at a concentration of 3×105 cells/well and treated with 2×109 particles/mL of EVs or equal volume of PBS for 18 h. The cells were then collected for analysis or stimulated with lipopolysaccharides (LPS) (Sigma Aldrich, USA) for 6 h.
4.3. BMDM extracellular vesicle isolation and nanoparticle tracking analysis
Our EV isolation and characterizations were performed in adherence to the MISEV2018 guidelines (Théry et al., 2018). BMDM were seeded into 15‐cm plates (Corning, USA) at a density of 5 × 106 cells/plate as described above. 25 ng/ml mouse M‐CSF (Peprotech, USA) was added every 2 days for 6 days. The cells were then washed twice with PBS (Corning, USA) and cultured in EV‐depleted media prepared by ultracentrifugation for 18 h at 100,000 × g (Type 45 Ti rotor, Beckman Coulter, USA) and filtration (0.2 um). After 24 h of incubation, the conditioned media was collected. EVs were isolated from conditioned media using Cushioned‐Density Gradient Ultracentrifugation (C‐DGUC) as previously described (Duong et al., 2019). Briefly, the conditioned media was centrifuged at 400 × g for 10 min at 4°C to pellet dead cells and debris followed by centrifugation at 2000 × g for 20 min at 4°C to eliminate debris and larger vesicles. The supernatant was then filtered (0.2 um) and centrifuged on a 60% iodixanol cushion (Sigma‐Aldrich, USA) at 100,000 × g for 3 h (Type 45 Ti, Beckman Coulter, USA). OptiPrep density gradient (5%, 10%, 20% w/v iodixanol) was employed to further purify the EVs at 100,000 × g for 18 h at 4C (SW 40 Ti rotor, Beckman Coulter, USA). Afterwards, twelve 1 mL fractions were collected starting from the top of the tube. Fraction 7 of the gradient was dialyzed in PBS with the Slide‐A‐Lyzer MINI Dialysis Device (Thermo Fisher Scientific, USA) and used for subsequent experiments and analyses.
Particles in Fraction 7 were subjected to size and concentration measurement by NanoSight LM14 (Malvern Instruments, Westborough, USA) at a 488‐nm detection wavelength. The analysis settings were optimized and kept identical for each sample. With a detection threshold set at 3, three videos of 1 min each were analyzed to give the mean, mode, median, and estimated concentration for each particle size. Samples were diluted in 1:100 or 1:200 PBS and measured in triplicates. Data were analyzed with the NTA 3.2 software. All EV samples were stored at 4C and used within 1 month after isolation. Details relevant to EV isolation and physical characterization data have been submitted to the EV‐TRACK knowledgebase (EV‐TRACK ID: EV220298).
4.4. Labeling and in vitro/in vivo tracking of BMDM extracellular vesicles
Fluorescently detectable BMDM EVs were generated using PKH26 (Sigma‐Aldrich, USA) or DiR (DiIC18(7) (1,1’‐Dioctadecyl‐3,3,3′,3′ Tetramethylindotricarbocyanine Iodide) (Invitrogen, USA). The dye was added to the 3 mL iodixanol cushion layer containing EV or to 3ml of PBS to achieve a final concentration of 3.5 mM for PKH26 or 1 μM for DiR and incubated for 20 min at room temperature. Labeled EVs and control were loaded below an iodixanol step gradient as described above in the EV isolation section. Free dye and non‐specific protein‐associated dye were eliminated from labeled EVs or from PBS control during this separation step. For in vitro experiments, naïve wildtype BMDM were exposed to 2×109 PKH26‐labeled EV for 2 h, washed three times with PBS and imaged using a Zeiss Observer microscope. Fluorescence intensity of the PKH26‐positive cells was measured by using ImageJ. For in vivo experiments, 10‐week‐old Western diet‐fed AAV8‐PCSK9‐injected mice were infused i.p. with PBS or 1×1010 DiR‐labeled EKO‐BMDM‐EV or WT‐BMDM‐EV for 6 h. The mice were then extensively perfused with PBS. Blood, aortas, hearts, livers, eWAT, bones, spleen, lungs, brains, intestines, and kidneys were collected, imaged, and quantified for DiR fluorescence signal using the Odyssey Infrared Imaging System and Image Studio software.
4.5. Transmission electron microscopy
EV morphology was assessed by Electron microscopy by loading 7×108 EVs onto a glow discharged 400 mesh Formvar‐coated copper grid (Electron Microscopy Sciences, USA). The nanoparticles were left to settle for 2 min, then the grids were washed four times with 1% Uranyl acetate. Excess Uranyl acetate was blotted off with filter paper. Grids were then allowed to dry and subsequently imaged at 120kV using a Tecnai 12 Transmission Electron Microscope (FEI, USA).
4.6. Protein extraction and immunoblotting
Each fraction of the C‐DGUC purified EVs (37.5 uL sample) was mixed with 12.5 mL of 4x Laemmli buffer (Bio‐Rad, USA). For cell lysates, cells were lysed in RIPA Buffer (Cell Signaling, USA) containing cOmplete, Mini, EDTA‐free Protease Inhibitor Cocktail (Roche, Switzerland) and 1 mM PMSF (Cell Signaling, USA). Protein concentrations were measured using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, USA). A total of 15 ug of proteins was diluted with PBS to 37.5 uL, then mixed with 12.5 uL 4x Laemmli buffer (Bio‐Rad, USA). Samples were subsequently heated at 95C for 5 min. Samples were then loaded on a 10% SDS‐PAGE gel and transferred onto a PVDF membrane (Bio‐Rad, USA). The membranes were blocked with 5% non‐fat milk dissolved in PBS for 1 h and then incubated with primary antibodies overnight at 4C. Primary antibodies for EV markers include anti‐CD9 (1:100, BD Biosciences, USA), anti‐CD63 (1:100, BD Biosciences, USA), and anti‐CD81 (1:100, Santa Cruz, USA), and anti‐apoE (1:1000) (Raffaï et al., 2001). Primary antibodies for cell lysate include anti‐Calnexin (1:500, Abcam, USA), anti‐GM130 (1:250, BD Biosciences, USA), anti‐CPT1A (1:200, Santa Cruz, USA), anti‐apoE (1:1000) (Raffaï et al., 2001), anti‐GAPDH (1:1000, Cell Signaling, USA), and anti‐b‐Actin (1:1000, Abcam, USA). After 4 washes in PBS containing 0.1% Tween (PBST), membranes were incubated with corresponding HRP‐conjugated secondary antibodies: anti‐Mouse IgG‐HRP (1:1000, Santa Cruz, USA) or anti‐Rabbit IgG‐HRP (1:1000, Thermo Fisher Scientific, USA) for 1 h and washed in PBST. Signals were visualized after incubation with Amersham ECL Prime substrate and imaged using an ImageQuant LAS 4000. Quantification was analyzed using ImageJ.
4.7. RNA extraction and gene expression analysis using qRT‐PCR
Total RNA isolated from cells was extracted using Qiazol Lysis Buffer (QIAGEN, Germany) and purified using the RNeasy Mini Kit (QIAGEN, Germany) according to the manufacturer's protocol. RNA was quantified using Nanodrop (Thermo Fisher Scientific, USA) and reverse transcribed using the iScript Reverse Transcription Supermix (Bio‐Rad, USA) for mRNA or the miRCURY LNA RT Kit (QIAGEN, Germany) for microRNA analysis. qPCR reactions were performed using the Fast SYBR Green Master Mix (Applied Biosystems, USA) for mRNA or the miRCURY LNA SYBR Green PCR Kit (QIAGEN, Germany) for microRNA and processed using a QuantStudio 7 Flex Real‐Time PCR System. Ct values were normalized to the housekeeping genes Gapdh and B2m. For microRNA expression, UniSp6 was used as a spike‐in control while U6 snRNA and miR‐16‐5p (QIAGEN, Germany) were used as reference genes. All reactions were done in triplicates.
4.8. Whole transcriptome library preparation, sequencing, and analysis
Total RNA isolated from cells was extracted using Qiazol Lysis Buffer (QIAGEN, Germany) and purified using the RNeasy Mini Kit (QIAGEN, Germany) according to the manufacturer's protocol. Isolated RNA sample was then DNase treated with TURBO DNA‐free (Thermo Fisher), then purified and concentrated with Zymo RNA Clean & Concentrator – 5 (Zymo Research). The RNA was measured for quantity with Quant‐iT Ribogreen RNA Assay (Thermo Fisher) and quality with Agilent High Sensitivity RNA Screen Tape and buffer (Agilent). For mouse RNA samples, an indexed, Illumina‐compatible, double‐stranded cDNA whole transcriptome library was synthesized from 10ng of total RNA with Takara Bio's SMARTer Stranded Total RNA‐Seq kit v2 Pico Input Mammalian (Takara Bio) and SMARTer RNA Unique Dual Index Kit (Takara Bio). Library preparation included RNA fragmentation (94°C for 4 min), cDNA synthesis, a 5‐cycle indexing PCR, ribosomal cDNA depletion, and a 12‐cycle enrichment PCR. Each library was measured for size with Agilent's High Sensitivity D1000 ScreenTape and reagents (Agilent) and concentration with KAPA SYBR FAST Universal qPCR Kit (Kapa Biosystems). Libraries were then combined into an equimolar pool which was also measured for size and concentration. The pool was clustered onto a flowcell (Illumina) with a 1% v/v PhiX Control v3 spike‐in (Illumina) and sequenced on Illumina's NovaSeq 6000 at a final flowcell concentration of 400pM. The first and second reads were each 100 bases.
For data processing, the SMARTer Total RNA pico v2 reads are quality filtered and trimmed as recommended by Takara Bio with the removal of the first 3 bases of read2. After trimming and filtering reads are genome and transcriptome mapped using STAR (v. 2.5.3a). Aligned BAM files are converted into gene counts matrices for further analysis using FeatureCounts (v.2.0.1), using read2 as the sense strand. For RNAseq analysis, differential expression was conducted using the DESeq2 package (version 1.20.0) in R (version 3.5.0) for all gene expression analyses. The raw read counts for the samples were normalized using the median ratio method (default in DESeq2). The significant differentially expressed genes (by Benjamini‐Hochberg adjusted p values) are reported in the paper. Heatmaps were created using the pheatmap (v.1.0.10) package in R.
GO analyses were performed using PANTHER GO‐slim Biological Process and DAVID with an FDR threshold at ≤ 0.05. RNA‐seq data have been deposited at GEO accession number: GSE237731 & GSE237730.
4.9. Phagocytotic uptake of CFSE‐labeled apoptotic cells
Naïve wildtype BMDM were seeded at a density of 1×106 cells/well in a 6‐well culture plate and treated with 2×109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h. The next day, BMDM were treated with 3×106 Jurkat cells that had undergone UV‐induced apoptosis for 50 min and labeled with CFSE. BMDM were then washed three times with PBS and dissociated from the cell culture plates. The cells were then assessed for the uptake of CFSE‐labeled apoptotic Jurkat cells using a Beckman CytoFLEX S cytometer (Beckman Coulter, USA).
4.10. I n vitro CD4+ T lymphocyte activation assay and IFN‐γ detection
Naive splenic CD4+ T lymphocytes were captured using negative selection magnetic beads (Miltenyi Biotec, Germany). These cells were stimulated with αCD3/αCD28 beads (Thermo Fisher, USA) and 5 ng/ml of recombinant murine IL‐2 (Peprotech, USA) for 48 h while also being co‐cultured with 2×109 particles/mL of EKO‐BMDM‐EV, or WT‐BMDM‐EV, or PBS. Measurements of T lymphocyte activation were assessed using flow cytometric detection of CD69 and CD25. For detection of IFN‐γ, naive splenic CD4+ T lymphocytes were stimulated with αCD3/αCD28 beads (Thermo Fisher, USA) and 5 ng/ml of recombinant murine IL‐2 (Peprotech, USA) for 12 h while also being co‐cultured with 2×109 particles/mL of EKO‐BMDM‐EV, or WT‐BMDM‐EV, or PBS. Cells were cultured in the presence of the Protein Transport Inhibitor cocktail (Invitrogen, USA). They were then permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience, USA) and stained with anti‐IFNγ (clone XMG1.2) at 1:100 concentration for 60 min in room temperature according to the manufacturer's protocol.
4.11. Glucose uptake assay in cultured macrophages and circulating monocytes
BMDM were seeded at a density of 150,000 cells/well in a 24‐well culture plate and treated with 2×109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS for 18 h. The next day, BMDM were stimulated with 100 ng/mL LPS for 6 h. The cells were then preincubated with KRPH buffer containing 2% bovine serum albumin, 20 mM HEPES, 5 mM KH2PO4, 1 mM MgSO4, 1 mM CaCl2, 136 mM NaCl, and 4.7 mM KCl, pH 7.4 (all from Sigma Aldrich, USA) for 40 min. Subsequently, 10 uL/well of 10 mM 2‐deoxyglucose (2‐DG) was added and incubated for 20 min. Next, cells were washed 3x with PBS to remove exogenous 2‐DG. BMDM were then lysed and 2‐DG uptake was processed using a Glucose Uptake Assay Kit (Abcam, USA) according to the manufacturer's protocol. Absorbance reading was measured at OD 412 nm on a microplate reader (Molecular Devices, USA). For 2‐NBDG uptake, pre‐stained circulating Ly6Chi monocytes were incubated with 2‐NBDG (Invitrogen, USA) for 30 min in 37C with 5% CO2. The cells were then washed with PBS and analyzed for 2‐NBDG uptake using a CytoFLEX S cytometer (Beckman, USA).
4.12. Assessments of glutathione levels, neutral lipids accumulation, oxidative stress, and mitochondrial health in cultured macrophages and circulating monocytes
Measurements of glutathione levels and GSH:GSSG ratio in BMDM were conducted using the GSH/GSSG Ratio Detection Assay Kit (Abcam, USA). BMDM were plated at a density of 1×106 cells/well in a 6‐well plate and treated with 2×109 particles/mL of EKO‐BMDM‐EV, WT‐BMDM‐EV, or PBS as control for 18 h. Cells were then washed twice with PBS (Corning, USA) and lysed with the kit's Mammalian Lysis Buffer (Abcam, USA). Measurements of total glutathione and GSH levels were conducted using the manufacturer's protocol.
For analysis of neutral lipids accumulation, BMDM or pre‐stained circulating Ly6Chi monocytes were stained with LipidTOX (Invitrogen, USA) (1:250) for 30 min in room temperature and analyzed using a CytoFLEX S cytometer (Beckman, USA). For analysis of cellular oxidative stress, BMDM or pre‐stained circulating Ly6Chi monocytes were stained with CellROX (Invitrogen, USA) (5 μM) for 30 min at 37°C. Cells were then analyzed using a CytoFLEX S cytometer (Beckman, USA).
For analysis of mitochondrial health and functions, BMDM or pre‐stained circulating Ly6Chi monocytes were stained with MitoSOX (Thermo Fisher, USA) or TMRM (Thermo Fisher, USA) at final concentrations of 5 μM and 0.1 μM, respectively. The cells were then incubated in 37°C for 30 min. Cells were then analyzed using a CytoFLEX S cytometer (Beckman, USA).
To measure mitochondrial transition pore opening, BMDM were analyzed using the MitoProbe Transition Pore Assay Kit (Invitrogen, USA) according to the manufacturer's protocol. Briefly, cell suspensions were mixed with 2 μM Calcein AM and 160 μM CoCl2. For negative control, cells were also mixed with 0.2 μM ionomycin. Cells were then analyzed for mitochondrial Calcein AM retention using the CytoFLEX S cytometer (Beckman, USA).
4.13. Multiplex immunoassay analysis of cytokines production
Total splenic cells or bone marrow cells were plated at 900,000 cells/well in a 6‐well plate and stimulated with 100 ng/mL LPS for 6 h. The conditioned media was then collected and spun at 400 x g for 10 min to remove the cells. TNF‐α, IL‐6, and IL‐1β cytokine levels in the conditioned media were measured using the V‐Plex Mouse Custom Cytokine Kit (Meso Scale Discovery, USA) according to the manufacturer's protocol. Plasma cytokines (TNF‐α, IFN‐γ, IL‐6, and IL‐1β) were also measured using the V‐Plex Mouse Custom Cytokine Kit (Meso Scale Discovery, USA) according to the manufacturer's protocol.
4.14. Measurements of lactate production by cells
BMDM were plated at 900,000 cells/well in a 6‐well plate and treated with EVs for 18 h. Cells were then either unstimulated or stimulated with 100 ng/mL LPS for 6 h. The conditioned media was then collected and spun at 400 x g for 10 min to remove the cells. The lactate levels in these conditioned media were measured using the L‐Lactate Colorimetric Assay Kit (Abcam, USA) according to the manufacturer's protocol. Absorbance reading was measured at OD 450 nm on a microplate reader (Molecular Devices, USA).
4.15. Assessments of leukocyte numbers and cellular markers using flow cytometry
Mice were anesthetized with isoflurane (Forane, Baxter, USA) and peripheral blood was collected by retro‐orbital bleeding with heparinized micro‐hematocrit capillary (Fisher Scientific, USA) in tubes containing 0.5M EDTA. Red blood cells were lysed in RBC lysis buffer (BioLegend, USA). Nonspecific binding was blocked with TruStain FcX Ab (BioLegend, USA) for 10 min at 4C in FACS buffer (Ca2+/Mg2+‐free PBS with 2% FBS and 0.5 mM EDTA) before staining with appropriate Abs: CD11b (clone M1/70), Ly‐6C (clone HK1.4), CD115 (clone AFS98), and CD45 (clone 30‐F11) (all BioLegend, USA) for 30 min at 4C. The antibody dilutions ranged from 1:200 to 1:100.
Splenocytes were isolated using mechanical dissociation. Briefly, spleens were mashed using the bottom of a 3 mL syringe (BD Biosciences). The cells were then passed through a 70 um cell strainer and incubated in RBC lysis buffer (BioLegend, USA). Nonspecific binding was blocked with TruStain FcX Ab (BioLegend, USA) for 10 min at 4C in FACS buffer before staining with appropriate Abs: CD11b (clone M1/70), Ly‐6C (clone HK1.4), Ly‐6G (clone 1A8), and CD11c (clone N418). Splenic dendritic cells were analyzed using the following Abs panel: CD11b (clone M1/70), Ly‐6C (clone HK1.4), CD11c (clone N418), I‐A/I‐E (clone M5/114.15.2). Splenic T lymphocytes were analyzed using the following Abs panel: CD4 (clone RM4‐5), CD8a (clone 53–6.7), CXCR3 (clone CXCR3‐173), CD69 (clone H1.2F3), CD62L (clone MEL‐14), and CD44 (clone IM7). The antibody dilutions ranged from 1:200 to 1:100.
Lymph node‐derived cells were collected from the inguinal, mesenteric, axillary, and mediastinal lymph nodes. The cells were then passed through a 70 um cell strainer and incubated in RBC lysis buffer (BioLegend, USA). T lymphocytes were analyzed using the following Abs panel: CD4 (clone RM4‐5), CD8a (clone 53–6.7), CXCR3 (clone CXCR3‐173), CD69 (clone H1.2F3), CD62L (clone MEL‐14), and CD44 (clone IM7). The antibody dilutions ranged from 1:200 to 1:100.
For detection of GLUT1 on cellular surface, BMDM were incubated with anti‐GLUT1 (clone SA0377) at 1:50 concentration in FACS buffer for 30 min in 4C. Cells were then washed once with PBS and incubated with APC‐conjugated Goat anti‐Rabbit (1:200 concentration) in FACS buffer for 30 min in 4C. For analysis of nuclear NF‐κB activity, the nuclei of BMDM were permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience, USA) and stained with anti‐phospho‐p65 subunit (clone 93H1) at 1:100 concentration for 60 min in room temperature according to the manufacturer's protocol.
For intracellular cytokine staining, 2 × 106 splenocytes were cultured in 1 ug/mL ionomycin (Sigma Aldrich, USA), 20 ng/mL phorbol 12‐myristate 13‐acetate (Zhang et al., 2019) (Fisher Scientific, USA), and Protein Transport Inhibitor cocktail (Invitrogen, USA) for 4 h. The cells were then collected and stained with anti‐CD4 (clone RM4‐5), anti‐CD8a (clone 53–6.7), anti‐CXCR3 (clone CXCR3‐173). Cells were then permeabilized using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience, USA) and stained with anti‐IFNγ (clone XMG1.2) at 1:100 concentration for 60 min in room temperature according to the manufacturer's protocol. All flow cytometry experiments were conducted using a CytoFLEX S cytometer (Beckman, USA).
4.16. Isolation and analysis of mature immune cells and HSPC subsets
Peritoneal cells were collected by lavage with 10 mL DPBS (Corning, USA) using a 16‐G needle. Cells were then incubated with red cell lysis buffer (BioLegend, USA) for 5 min and cultured in 6‐well cell culture plates (Corning, USA) in complete media. After 2 h, cells were washed once with DPBS (Corning, USA) and replenished with fresh complete media. Adherent cells were then collected for downstream analysis as peritoneal macrophages after 1 h.
For isolation and analysis of HSPC, cells were collected from the bone marrows or spleens and centrifuged at 300 × g, 5 min at 4°C, resuspended in red cell lysis buffer (BioLegend, USA) for 5 min and run through a 40 μm strainer. The cells were stained as previously described (Yamamoto et al., 2013) with a lineage‐marker cocktail of biotinylated anti‐CD4 (RM4‐5), ‐CD8 (53‐6.7), ‐B220/CD45RA (RA3‐6B2), ‐TER‐119 (TER‐119), ‐Gr‐1 (RB6‐8C5), and ‐CD127 (IL‐7Ra/A7R34) antibodies (all from BioLegend, USA). These cells were then stained with anti‐CD34 (RAM34, eBioscience, USA), anti‐CD150 (TC15‐12F12.2, BioLegend, USA), anti‐CD48 (Invitrogen, USA), anti‐Sca‐1 (D7, Invitrogen, USA) anti‐CD135 (A2F10, Invitrogen, USA) anti‐c‐Kit (2B8, Life Technologies, USA), anti‐CD16/32 (93, BioLegend, USA), anti‐CD41 (MWReg30, BioLegend, USA) and streptavidin‐BV786 (BD Biosciences, USA) to detect biotinylated antibodies. Cells were then analyzed using a CytoFLEX S cytometer (Beckman, USA).
4.17. Seahorse extracellular flux analysis
BMDM were plated at 60,000 cells/well into XFe24 cell culture microplates (Agilent, USA) and incubated overnight at 37°C and 5% CO2 while being treated with EVs or PBS for 18 h. The cells were then incubated with or without 100 ng/mL of LPS for 6 h. Cells were then washed with Seahorse XF DMEM assay buffer (Agilent, USA) supplemented with 10 mM glucose (Agilent, USA), 1 mM pyruvate (Agilent, USA), and 2 mM glutamine (Agilent, USA) and incubated for 1 h at 37°C without CO2. For measurements of oxidative phosphorylation, OCR and ECAR were measured using the mitochondrial stress test kit (Agilent, USA) in response to 1 μM Oligomycin, 2 μM Carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone (FCCP), and 0.5 μM Rotenone/Antimycin A (R/AA). For measurements of fatty acid oxidation, OCR and ECAR were measured in cells treated with 4 μM Etomoxir (Sigma Aldrich, USA) followed by 1 μM Oligomycin (Agilent, USA). For measurements of glycolytic activity, glycoPER was measured using the glycolytic rate assay (Agilent, USA) in response to 0.5 μM R/AA and 50 mM 2‐deoxy‐D‐glucose (2‐DG). All measurements were performed with the Seahorse XFe‐24 Bioanalyzer (Agilent, USA). After OCR measurements, cells were incubated in Hoechst (1:1000) diluted in Live Cell Imaging Solution (Invitrogen, USA) and imaged under a Zeiss Observer microscope. Total cell counts were measured using ImageJ.
4.18. Statistical Analysis
Statistical analysis was performed with GraphPad Prism v8, using the one‐way or two‐way analysis of variance (ANOVA) with post‐tests, Holm‐Sidak, as indicated in figure legends for multiple groups. *p < 0.05, ** p < 0.01, *** p < 0.001 **** p < 0.0001. Normality test was performed using the Shapiro‐Wilk test on GraphPad Prism v8, with p > 0.05 indicating normal distribution. All error bars represent the mean ± the standard error of the mean (SEM unless stated). All experiments were repeated at least twice or performed with independent samples.
4.19. Study approvals
All animal experiments were approved by the Institutional Animal Care and Use Committee at the San Francisco Veterans Affairs Medical Center.
AUTHOR CONTRIBUTIONS
Tuan Anh Phu: Conceptualization; data curation; formal analysis; investigation; methodology; resources; supervision; validation; visualization; writing—original draft; writing—review and editing. Martin Ng: Investigation; methodology; validation; writing—review and editing. Ngan K. Vu: Data curation; investigation; methodology; visualization; writing—review and editing. Alex S. Gao: Investigation; validation; writing—review and editing. Robert L. Raffai: Conceptualization; funding acquisition; investigation; methodology; project administration; resources; supervision; validation; writing—original draft; writing—review and editing.
CONFLICT OF INTEREST STATEMENT
There is no conflict of interest in writing this manuscript.
Supporting information
supplementary information
ACKNOWLEDGEMENTS
We thank the Diabetes Research Center at the University of California, San Francisco for a Pilot & Feasibility Award and assistance with the instruments at the Parnassus Flow Cytometry Core, which are supported by Grant P30 DK063720 from the National Institutes of Health. We also thank the UCSF Genomics CoLab facility for assistance with RNA‐seq experiments.
Phu, T. A. , Ng, M. , Vu, N. K. , Gao, A. S. , & Raffai, R. L. (2023). ApoE expression in macrophages communicates immunometabolic signaling that controls hyperlipidemia‐driven hematopoiesis & inflammation via extracellular vesicles. Journal of Extracellular Vesicles, 12, e12345. 10.1002/jev2.12345
[Correction added on 18‐October‐2023, after first online publication: GSE accession number in Section 4.8. is corrected from GSE237727 to GSE237730 in this version]
DATA AVAILABILITY STATEMENT
All data presented in this manuscript are available upon request.
REFERENCES
- Akiyama, T. E. , Sakai, S. , Lambert, G. , Nicol, C. J. , Matsusue, K. , Pimprale, S. , Lee, Y.‐H. , Ricote, M. , Glass, C. K. , Brewer, H. B , & Gonzalez, F. J., Jr (2002). Conditional disruption of the peroxisome proliferator‐activated receptor gamma gene in mice results in lowered expression of ABCA1, ABCG1, and apoE in macrophages and reduced cholesterol efflux. Molecular and Cellular Biology, 22(8), 2607–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baitsch, D. , Bock, H. H. , Engel, T. , Telgmann, R. , Müller‐Tidow, C. , Varga, G. , Bot, M. , Herz, J. , Robenek, H. , Von Eckardstein, A. , & Nofer, J.‐R. (2011). Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology, 31(5), 1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørklund, M. M. , Hollensen, A. K. , Hagensen, M. K. , Dagnæs‐Hansen, F. , Christoffersen, C. , Mikkelsen, J. G. , & Bentzon, J. F. (2014). Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circulation Research, 114(11), 1684–1689. [DOI] [PubMed] [Google Scholar]
- Boisvert, W. A. , Spangenberg, J. , & Curtiss, L. K. (1995). Treatment of severe hypercholesterolemia in apolipoprotein E‐deficient mice by bone marrow transplantation. Journal of Clinical Investigation, 96(2), 1118–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boldin, M. P. , Taganov, K. D. , Rao, D. S. , Yang, L. , Zhao, J. L. , Kalwani, M. , Garcia‐Flores, Y. , Luong, M. , Devrekanli, A. , Xu, J. , Sun, G. , Tay, J. , Linsley, P. S. , & Baltimore, D. (2011). miR‐146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. Journal of Experimental Medicine, 208(6), 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonacina, F. , Coe, D. , Wang, G. , Longhi, M. P. , Baragetti, A. , Moregola, A. , Garlaschelli, K. , Uboldi, P. , Pellegatta, F. , Grigore, L. , Da Dalt, L. , Annoni, A. , Gregori, S. , Xiao, Q. , Caruso, D. , Mitro, N. , Catapano, A. L. , Marelli‐Berg, F. M. , & Norata, G. D. (2018). Myeloid apolipoprotein E controls dendritic cell antigen presentation and T cell activation. Nature Communications, 9(1), 3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchareychas, L. , Duong, P. , Covarrubias, S. , Alsop, E. , Phu, T. A. , Chung, A. , Gomes, M. , Wong, D. , Meechoovet, B. , Capili, A. , Yamamoto, R. , Nakauchi, H. , Mcmanus, M. T. , Carpenter, S. , Van Keuren‐Jensen, K. , & Raffai, R. L. (2020). Macrophage Exosomes Resolve Atherosclerosis by Regulating Hematopoiesis and Inflammation via MicroRNA Cargo. Cell reports, 32(2), 107881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchareychas, L. , Duong, P. , Phu, T. A. , Alsop, E. , Meechoovet, B. , Reiman, R. , Ng, M. , Yamamoto, R. , Nakauchi, H. , Gasper, W. J. , Van Keuren‐Jensen, K. , & Raffai, R. L. (2021). High glucose macrophage exosomes enhance atherosclerosis by driving cellular proliferation & hematopoiesis. iScience, 24(8), 102847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand, K. , Mackman, N. , & Curtiss, L. K. (1993). Interferon‐gamma inhibits macrophage apolipoprotein E production by posttranslational mechanisms. Journal of Clinical Investigation, 91(5), 2031–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibrián, D. , & Sánchez‐Madrid, F. (2017). CD69: from activation marker to metabolic gatekeeper. European Journal of Immunology, 47(6), 946–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtiss, L. K. , & Boisvert, W. A. (2000). Apolipoprotein E and atherosclerosis. Current Opinion in Lipidology, 11(3), 243–251. [DOI] [PubMed] [Google Scholar]
- Daneshmandi, S. , Cassel, T. , Higashi, R. M. , Fan, T. W. , & Seth, P. (2021). 6‐Phosphogluconate dehydrogenase (6PGD), a key checkpoint in reprogramming of regulatory T cells metabolism and function. Elife, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel, B. , Nagy, G. , Horvath, A. , Czimmerer, Z. , Cuaranta‐Monroy, I. , Poliska, S. , Hays, T. T. , Sauer, S. , Francois‐Deleuze, J. , & Nagy, L. (2018). The IL‐4/STAT6/PPARgamma signaling axis is driving the expansion of the RXR heterodimer cistrome, providing complex ligand responsiveness in macrophages. Nucleic Acids Research, 46(9), 4425–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davignon, J. (2005). Apolipoprotein E and atherosclerosis: beyond lipid effect. Arteriosclerosis, Thrombosis, and Vascular Biology, 25(2), 267–269. [DOI] [PubMed] [Google Scholar]
- Donath, M. Y. , Meier, D. T. , & Böni‐Schnetzler, M. (2019). Inflammation in the Pathophysiology and Therapy of Cardiometabolic Disease. Endocrine Reviews, 40(4), 1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doran, A. C. , Yurdagul, A. , & Tabas, I. (2020). Efferocytosis in health and disease. Nature Reviews Immunology, 20(4), 254–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong, P. , Chung, A. , Bouchareychas, L. , & Raffai, R. L. (2019). Cushioned‐Density Gradient Ultracentrifugation (C‐DGUC) improves the isolation efficiency of extracellular vesicles. PLoS ONE, 14(4), e0215324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainger, D. J. , Reckless, J. , & Mckilligin, E. (2004). Apolipoprotein E modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein E‐deficient mice. Journal of Immunology, 173(10), 6366–6375. [DOI] [PubMed] [Google Scholar]
- Groom, J. R. , & Luster, A. D. (2011). CXCR3 in T cell function. Experimental Cell Research, 317(5), 620–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty, A. H. , Linton, M. F. , Swift, L. L. , & Fazio, S. (1999). Determination of the lower threshold of apolipoprotein E resulting in remnant lipoprotein clearance. Journal of Lipid Research, 40(8), 1529–1538. [PubMed] [Google Scholar]
- He, Y. , Rodrigues, R M. , Wang, X. , Seo, W. , Ma, J. , Hwang, S. , Fu, Y. , Trojnár, E. , Mátyás, C. , Zhao, S. , Ren, R. , Feng, D. , Pacher, P. , Kunos, G. , & Gao, B. (2021). Neutrophil‐to‐hepatocyte communication via LDLR‐dependent miR‐223‐enriched extracellular vesicle transfer ameliorates nonalcoholic steatohepatitis. Journal of Clinical Investigation, 131(3), e141513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Da. , Sherman, B. T. , Tan, Q. , Collins, J. R. , Alvord, W. G. , Roayaei, J. , Stephens, R. , Baseler, M. W. , Lane, H. C. , & Lempicki, R. A. (2007). The DAVID Gene Functional Classification Tool: a novel biological module‐centric algorithm to functionally analyze large gene lists. Genome biology, 8(9), R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igel, E. , Haller, A. , Wolfkiel, P R. , Orr‐Asman, M. , Jaeschke, A. , & Hui, D Y. (2021). Distinct pro‐inflammatory properties of myeloid cell‐derived apolipoprotein E2 and E4 in atherosclerosis promotion. Journal of Biological Chemistry, 297(3), 101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly, B. , & O'neill, L. A. (2015). Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Research, 25(7), 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerksick, C. , & Willoughby, D. (2005). The antioxidant role of glutathione and N‐acetyl‐cysteine supplements and exercise‐induced oxidative stress. Journal of the International Society of Sports Nutrition, 2, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidani, Y. , & Bensinger, S. J. (2012). Liver X receptor and peroxisome proliferator‐activated receptor as integrators of lipid homeostasis and immunity. Immunological Reviews, 249(1), 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelwyn, G. J. , Corr, E. M. , Erbay, E. , & Moore, K. J. (2008). Regulation of macrophage immunometabolism in atherosclerosis. Nature Immunology, 19(6), 526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, K. , Ching, D. , Luk, F. S. , & Raffai, R. L. (2015). Apolipoprotein E enhances microRNA‐146a in monocytes and macrophages to suppress nuclear factor‐kappaB‐driven inflammation and atherosclerosis. Circulation Research, 117(1), e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton, M. F. , Atkinson, J. B. , & Fazio, S. (1995). Prevention of atherosclerosis in apolipoprotein E‐deficient mice by bone marrow transplantation. Science, 267(5200), 1034–1037. [DOI] [PubMed] [Google Scholar]
- Linton, M. F. , Gish, R. , Hubl, S. T. , Bütler, E. , Esquivel, C. , Bry, W. I. , Boyles, J. K. , Wardell, M. R. , & Young, S. G. (1991). Phenotypes of apolipoprotein B and apolipoprotein E after liver transplantation. Journal of Clinical Investigation, 88(1), 270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, H. , Howatt, D. A. , Balakrishnan, A. , Graham, M. J. , Mullick, A. E. , & Daugherty, A. (2016). Hypercholesterolemia Induced by a PCSK9 Gain‐of‐Function Mutation Augments Angiotensin II‐Induced Abdominal Aortic Aneurysms in C57BL/6 Mice‐Brief Report. Arteriosclerosis, Thrombosis, and Vascular Biology, 36(9), 1753–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley, R. W. , & Ji, Z.‐S. (1999). Remnant lipoprotein metabolism: key pathways involving cell‐surface heparan sulfate proteoglycans and apolipoprotein E. Journal of Lipid Research, 40(1), 1–16. [PubMed] [Google Scholar]
- Mahley, R W. (1988). Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science, 240(4852), 622–630. [DOI] [PubMed] [Google Scholar]
- Maxwell, K. N. , & Breslow, J. L. (2004). Adenoviral‐mediated expression of Pcsk9 in mice results in a low‐density lipoprotein receptor knockout phenotype. PNAS, 101(18), 7100–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, K. J. , Sheedy, F. J. , & Fisher, E. A. (2013). Macrophages in atherosclerosis: a dynamic balance. Nature Reviews Immunology, 13(10), 709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, A. J. , Akhtari, M. , Tolani, S. , Pagler, T. , Bijl, N. , Kuo, C.‐L. , Wang, M. , Sanson, M. , Abramowicz, S. , Welch, C. , Bochem, A. E. , Kuivenhoven, J. A. , Yvan‐Charvet, L. , & Tall, A. R. (2011). ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. Journal of Clinical Investigation, 121(10), 4138–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, My. A. , Karunakaran, D. , Geoffrion, M. , Cheng, H. S. , Tandoc, K. , Perisic Matic, L. , Hedin, U. , Maegdefessel, L. , Fish, J. E. , & Rayner, K. J. (2018). Extracellular Vesicles Secreted by Atherogenic Macrophages Transfer MicroRNA to Inhibit Cell Migration. Arteriosclerosis, Thrombosis, and Vascular Biology, 38(1), 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa, T. , Kanter, J. E. , Kramer, F. , Barnhart, S. , Shen, X. , Vivekanandan‐Giri, A. , Wall, V. Z. , Kowitz, J. , Devaraj, S. , O'brien, K. D. , Pennathur, S. , Tang, J. , Miyaoka, R. S. , Raines, E. W. , & Bornfeldt, K. E. (2014). Testing the role of myeloid cell glucose flux in inflammation and atherosclerosis. Cell reports, 7(2), 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obaid, M. , Udden, S. M. N , Alluri, P. , & Mandal, S. S. (2021). LncRNA HOTAIR regulates glucose transporter Glut1 expression and glucose uptake in macrophages during inflammation. Scientific Reports, 11(1), 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'connor, R. S. , Guo, L. , Ghassemi, S. , Snyder, N. W. , Worth, A. J. , Weng, L. , Kam, Y. , Philipson, B. , Trefely, S. , Nunez‐Cruz, S. , Blair, I. A. , June, C. H. , & Milone, M. C. (2018). The CPT1a inhibitor, etomoxir induces severe oxidative stress at commonly used concentrations. Scientific Reports, 8(1), 6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen, J. B. , & DA, B. (2010). Measurement of oxidized/reduced glutathione ratio. Methods in Molecular Biology, 648, 269–77. [DOI] [PubMed] [Google Scholar]
- Phu, T. A. , Ng, M. , Vu, N. K. , Bouchareychas, L. , & Raffai, R. L. (2022). IL‐4 polarized human macrophage exosomes control cardiometabolic inflammation and diabetes in obesity. Molecular Therapy, 30(6), 2274–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaï, R. L. , Dong, Li.‐M. , Farese, R. V. , & Weisgraber, K. H. (2001). Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. PNAS, 98(20), 11587–11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell, D R. , Graham, A. , & Owen, J. S. (1997). Apolipoprotein E inhibits platelet aggregation through the L‐arginine:nitric oxide pathway. Implications for vascular disease. Journal of Biological Chemistry, 272(1), 89–95. [DOI] [PubMed] [Google Scholar]
- Roche‐Molina, M. , Sanz‐Rosa, D. , Cruz, F. M. , García‐Prieto, J. , López, S. , Abia, R. , Muriana, F. J. G. , Fuster, V. , Ibáñez, B. , & Bernal, J. A. (2015). Induction of sustained hypercholesterolemia by single adeno‐associated virus‐mediated gene transfer of mutant hPCSK9. Arteriosclerosis, Thrombosis, and Vascular Biology, 35(1), 50–59. [DOI] [PubMed] [Google Scholar]
- Rosenson, R. S. , Brewer, H. B , Davidson, W. S , Fayad, Z. A. , Fuster, V. , Goldstein, J. , Hellerstein, M. , Jiang, X. C. , Phillips, M. C. , Rader, D. J. , Remaley, A. T. , Rothblat, G. H. , Tall, A. R. , & Yvan‐Charvet, L. (2012). Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation, 125(15), 1905–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagliocchi, S. , Cicatiello, A. G. , Di Cicco, E. , Ambrosio, R. , Miro, C. , Di Girolamo, D. , Nappi, A. , Mancino, G. , De Stefano, M. A. , Luongo, C. , Raia, M. , Ogawa‐Wong, A. N. , Zavacki, A. M. , Paladino, S. , Salvatore, D. , & Dentice, M. (2019). The thyroid hormone activating enzyme, type 2 deiodinase, induces myogenic differentiation by regulating mitochondrial metabolism and reducing oxidative stress. Redox Biology, 24, 101228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaramuzzino, L. , Lucchino, V. , Scalise, S. , Lo Conte, M. , Zannino, C. , Sacco, A. , Biamonte, F. , Parrotta, E. I. , Costanzo, F. S. , & Cuda, G. (2021). Uncovering the metabolic and stress responses of human embryonic stem cells to FTH1 gene silencing. Cells, 10(9), 2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suárez, H. , Andreu, Z. , Mazzeo, C. , Toribio, V. , Pérez‐Rivera, A. E. , López‐Martín, S. , García‐Silva, S. , Hurtado, B. , Morato, E. , Peláez, L. , Arribas, E. A. , Tolentino‐Cortez, T. , Barreda‐Gómez, G. , Marina, A. I. , Peinado, H. , & Yáñez‐Mó, M. (2021). CD9 inhibition reveals a functional connection of extracellular vesicle secretion with mitophagy in melanoma cells. J Extracell Vesicles, 10(7), e12082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, X. , He, S. , Wara, A. K. M. , Icli, B. , Shvartz, E. , Tesmenitsky, Y. , Belkin, N. , Li, D. , Blackwell, T. S. , Sukhova, G. K. , Croce, K. , & Feinberg, M. W. (2014). Systemic delivery of microRNA‐181b inhibits nuclear factor‐kappaB activation, vascular inflammation, and atherosclerosis in apolipoprotein E‐deficient mice. Circulation Research, 114(1), 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Y. , Bandi, M. , Lofton, T. , Smith, M. , Bristow, C. A. , Carugo, A. , Rogers, N. , Leonard, P. , Chang, Q. , Mullinax, R. , Han, J. , Shi, X. , Seth, S. , Meyers, B. A. , Miller, M. , Miao, L. , Ma, X. , Feng, N. , Giuliani, V. , & Marszalek, J. R. (2019). Functional genomics reveals synthetic lethality between phosphogluconate dehydrogenase and oxidative phosphorylation. Cell reports, 26(2), 469–482.e5 e5. [DOI] [PubMed] [Google Scholar]
- Sun, Y. , Oravecz‐Wilson, K. , Bridges, S. , Mceachin, R. , Wu, J. , Kim, S. H. , Taylor, A. , Zajac, C. , Fujiwara, H. , Peltier, D. C. , Saunders, T. , & Reddy, P. (2019). miR‐142 controls metabolic reprogramming that regulates dendritic cell activation. Journal of Clinical Investigation, 129(5), 2029–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas, I. , & Bornfeldt, K. E. (2020). Intracellular and intercellular aspects of macrophage immunometabolism in atherosclerosis. Circulation Research, 126(9), 1209–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taganov, K. D. , Boldin, M. P. , Chang, K.‐J. , & Baltimore, D. (2006). NF‐kappaB‐dependent induction of microRNA miR‐146, an inhibitor targeted to signaling proteins of innate immune responses. PNAS, 03(33), 12481–12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall, A. R. , Yvan‐Charvet, L. , Terasaka, N. , Pagler, T. , & Wang, N. (2008). ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell metabolism, 7(5), 365–375. [DOI] [PubMed] [Google Scholar]
- Tenger, C. , & Zhou, X. (2003). Apolipoprotein E modulates immune activation by acting on the antigen‐presenting cell. Immunology, 109(3), 392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry, C. , Witwer, K. W. , Aikawa, E. , Alcaraz, M. J. , Anderson, J. D. , Andriantsitohaina, R. , Antoniou, A. , Arab, T. , Archer, F. , Atkin‐Smith, G. K. , Ayre, D. C. , Bach, J.‐M. , Bachurski, D. , Baharvand, H. , Balaj, L. , Baldacchino, S. , Bauer, N. N. , Baxter, A. A. , Bebawy, M. , & Zuba‐Surma, E. K. (2018). Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles, 7(1), 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorp, E. , Cui, D. , Schrijvers, D. M. , Kuriakose, G. , & Tabas, I. (2008). Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe‐/‐ mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 28(8), 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Niel, G. , Bergam, P. , Di Cicco, A. , Hurbain, I. , Lo Cicero, A. , Dingli, F. , Palmulli, R. , Fort, C. , Potier, M. C. , Schurgers, L. J. , Loew, D. , Levy, D. , & Raposo, G. (2015). Apolipoprotein E regulates amyloid formation within endosomes of pigment cells. Cell reports, 13(1), 43–51. [DOI] [PubMed] [Google Scholar]
- Weisgraber, K. H. (1994). Apolipoprotein E: structure‐function relationships. Advances in Protein Chemistry, 45, 249–302. [DOI] [PubMed] [Google Scholar]
- Yurdagul, A. , Doran, A. C. , Cai, B. , Fredman, G. , & Tabas, I. A. (2017). Mechanisms and consequences of defective efferocytosis in atherosclerosis. Frontiers in Cardiovascular Medicine, 4, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, S. , Weinberg, S. , Deberge, M. , Gainullina, A. , Schipma, M. , Kinchen, J M. , Ben‐Sahra, I. , Gius, D. R. , Yvan‐Charvet, L. , Chandel, N. S. , Schumacker, P. T. , & Thorp, E. B. (2019). Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell metabolism, 29(2), 443–456.e5 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, P. , Luo, Q. , Wang, W. , Li, J. , Wang, T. , Wang, P. , Chen, L. , Zhang, P. , Chen, H. , Liu, Y. , Dong, P. , Xie, G. , Ma, Y. , Jiang, L. , Yuan, X. , & Shen, L. (2018). Tumor‐associated macrophages‐derived exosomes promote the migration of gastric cancer cells by transfer of functional Apolipoprotein E. Cell death & disease, 9(4), 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary information
Data Availability Statement
All data presented in this manuscript are available upon request.