

Abstract
Background:
The placenta is critical for the overall development and life-long health of the fetus. Abnormal placental development and function occur in pregnancies with fetal congenital heart disease. However, studies that utilize standardized diagnostic criteria and incorporate control populations are lacking. This limits generalizability of current research and the ability to determine the specific placental abnormalities associated with congenital heart disease.
Objectives:
Apply consensus statement guidelines (known as the Amsterdam Criteria) for placental pathology interpretation to compare the frequency and pattern of abnormalities in pregnancies with fetal congenital heart disease to demographically matched control pregnancies and evaluate for differences in placental abnormalities by cardiac physiology.
Study Design:
A single-center retrospective cohort study was conducted from January 2013-June 2019. Infants with a prenatal diagnosis of moderate-severe congenital heart disease who were born at ≥37 weeks’ gestation were included. A control group born at ≥37 weeks’ but without fetal congenital heart disease or other major pregnancy complications was matched to the congenital heart disease group on maternal race and ethnicity and infant sex. Using the Amsterdam Criteria, placental pathology findings were categorized as delayed villous maturation, maternal vascular malperfusion, fetal vascular malperfusion, and inflammatory lesions. Frequency of placental abnormalities were compared between groups and logistic regression was performed to evaluate the association of clinical and sociodemographic factors with delayed villous maturation, maternal vascular malperfusion, and fetal vascular malperfusion.
Results:
There were 194 pregnancies with fetal congenital heart disease and 105 controls included, of whom 83% in the congenital heart disease group and 82% in the control group were of non-Hispanic Caucasian race and ethnicity. Compared to controls, pregnancies with fetal congenital heart disease had higher rates of delayed villous maturation (6% vs 19%, p < 0.001) and maternal vascular malperfusion (19% vs 34%, p = 0.007), but not fetal vascular malperfusion (6% vs 10%, p = 0.23). Congenital heart disease infants with two-ventricle anatomy displayed the highest odds of delayed villous maturation compared to controls (odds ratio 5.5, 95% confidence interval 2.2 – 15.7, p < 0.01]. Maternal vascular malperfusion was 2.2 times higher (p = 0.02) for infants with two ventricle anatomy and 2.9 times higher (p = 0.02) for infants with single ventricle physiology with pulmonary obstruction. Within the congenital heart disease group, delayed villous maturation was associated with higher maternal body mass index, polyhydramnios, larger infant birth head circumference, and infant respiratory support in the delivery room, whereas maternal vascular malperfusion was associated with oligohydramnios. In multivariable models adjusting for cardiac diagnosis, associations of delayed villous maturation persisted for infant birth head circumference (odds ratio 1.2, 95% confidence interval 1.0 – 1.5, p = 0.02) and infant respiratory support in the delivery room (odds ratio 3.0, 95% confidence interval 1.3 – 6.5, p = 0.007).
Conclusion:
Pregnancies with fetal congenital heart disease display higher rates of delayed villous maturation and maternal vascular malperfusion compared to controls, suggesting that placental maldevelopment may relate to maternal factors. Future investigations are needed to determine the association of these abnormalities with postnatal infant outcomes.
Keywords: Placenta, maternal vascular malperfusion, fetal vascular malperfusion, delayed villous maturation, congenital heart disease, fetal
Condensation:
Placental delayed villous maturation and maternal vascular malperfusion are more common in pregnancies with fetal congenital heart disease than demographically matched controls.
Introduction
The placenta is essential for oxygen and nutrient exchange,1 immune modulation,2 hormonal regulation,3, 4 and long-term health of the fetus.5 Both placental and fetal cardiac structures begin to develop during the third week of gestation through a complex cascade of shared molecular and genetic pathways, known as the placenta-heart axis.6–10 Increasing knowledge of these developmental processes have highlighted a potential link between congenital heart disease (CHD) and placental maldevelopment.
Smaller placental weight,11 placental histopathologic abnormalities,12, 13 and altered placental perfusion14 have all been reported in CHD. Genetic variants affecting both cardiac and placental development may contribute.6 Maternal conditions that are more common with fetal CHD, such as preeclampsia or diabetes, may also affect placental development.15–17 Finally, fetal hemodynamic disturbances, a hallmark of CHD, may lead to epigenetic changes in placental vascular development.6 Oxygen saturation and blood flow are reduced in the descending aorta and umbilical vein for some, but not all forms of CHD, suggesting placental maldevelopment could vary by diagnosis.18 Regardless of etiology, placental abnormalities are common in CHD. Yet, the exact patterns of abnormalities and the fetuses at highest risk remain unclear.
Two primary challenges of current literature are the use of heterogeneous methods for evaluating the placenta and a lack of control populations for comparison.7 A standardized approach to placental pathology assessment in studies of high-risk and healthy pregnancies is necessary to understand the magnitude of placental involvement, gain insight to potential pathophysiologic mechanisms, and link placental abnormalities with fetal and postnatal outcomes.
In 2016, a consensus statement was published to standardize placental sampling and evaluation (known as the Amsterdam Criteria).19 Terminology was updated to include the following: delayed villous maturation (DVM), an abnormality in the development of vasculosyncytial membrane; maternal vascular malperfusion (MVM), abnormalities that reflect under-perfusion and high-velocity malperfusion; and fetal vascular malperfusion (FVM), abnormalities that lead to obstruction in fetal blood flow.17, 19 This terminology has been increasingly applied to placental studies across a variety of populations, although data in fetal CHD have been limited by small sample size and a lack of controls.20–23 Recently, Leon and colleagues have begun to address these limitations with a large, retrospective cohort study that identified higher rates of MVM and FVM in pregnancies with fetal CHD compared to controls.13 However, DVM remains understudied in CHD and data are still limited by a lack of demographically matched controls.
The current study aimed to address these gaps in the literature by applying the Amsterdam Criteria to compare placental abnormalities between CHD and demographically matched controls. We hypothesized that DVM, MVM, and FVM would be more common in CHD and that the frequency of these abnormalities would vary by cardiac physiology.
Materials and Methods
Study Design
A single center, retrospective cohort study was conducted of mother/infant dyads with a prenatal diagnosis of fetal CHD who delivered their infant between January 1, 2013 through June 30, 2019. Inclusion criteria were birth at ≥37 weeks’ gestation and fetal moderate-severe CHD requiring cardiac intervention within the first year of life. Moderate-severe CHD was defined based on published data of CHD severity.24 Exclusion criteria were multiple gestation pregnancy, unknown gestational age at birth, and limited/no placental pathology data. A control group of pregnant women without fetal CHD who also delivered at ≥37 weeks’ gestation between January 1, 2013 and June 30, 2019 were selected from an existing placental pathology database to match the CHD group on maternal race and ethnicity and infant sex. Exclusion criteria for the controls mirrored that of the CHD group, with the following additions: hypertensive disorders (chronic hypertension, pregnancy-induced hypertension, preeclampsia, HELLP syndrome), pre-gestational or gestational diabetes, limited/no prenatal care, fetal growth restriction, known fetal genetic syndrome or congenital anomaly(ies), preterm labor, clinical chorioamnionitis, known placental (e.g., placenta previa) or uterine (e.g., bicornate uterus) structural abnormalities, and acute events at delivery (e.g., placental abruption, cord prolapse). Institutional Review Board approval was obtained prior to the initiation of the study.
Placental Pathology Data
Gross and histopathologic placental data were obtained through maternal chart review and included cord insertion (given previously reported abnormalities associated with CHD25), placental trimmed weight, presence/absence of DVM, MVM, and FVM (Table 1, Figure 1), and presence/absence of chorangiosis, inflammation, villous edema, thrombohematoma, and markers of meconium exposure. All findings were reviewed and categorized by a single rater (CBO). A random selection of 42 preserved histology slides from the CHD group (20% of the entire cohort) were independently categorized by a pediatric pathologist (MH) to ensure accuracy in interpretation of findings. Discrepancies between raters were reviewed and a consensus was reached for classification of pathology report terminology based on the Amsterdam Criteria (as some samples in this study were collected before these guidelines were published). This consensus was then applied across all CHD and control placental pathology data.
Table 1.
Definition and Categorization of Placental Pathology findings for Primary Outcomes.
| Delayed Villous Maturation |
|
| Maternal Vascular Malperfusion |
|
| Fetal Vascular Malperfusion |
|
Categorization is based on published data providing guidelines for placental pathology preparation and interpretation.19 These criteria were applied across all placental samples.
Diagnosis of delayed villous maturation was made based on hematoxylin and eosin staining and did not use CD-15 immunohistochemistry.
Figure 1.
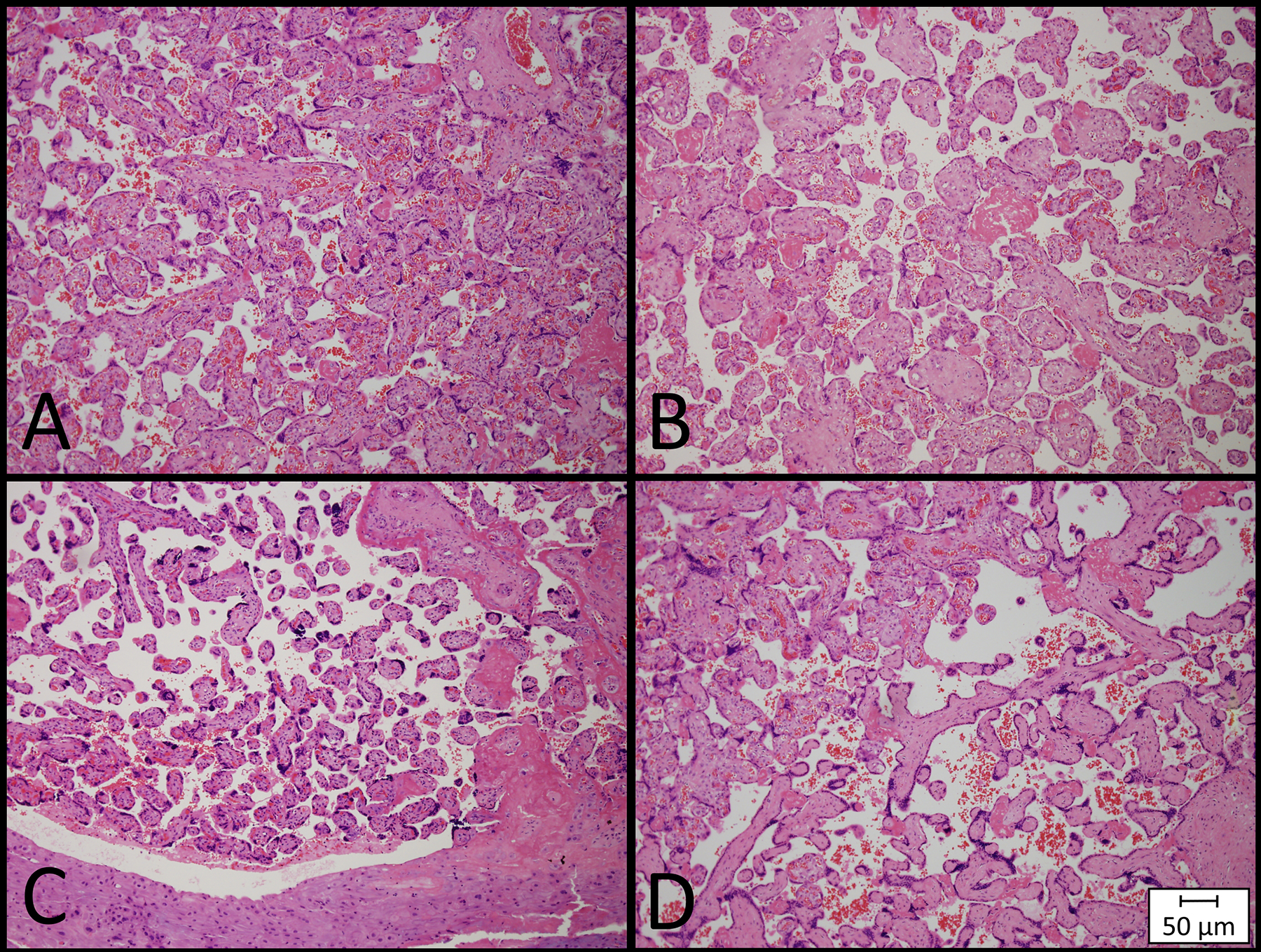
Examples of Placental Histopathologic Abnormalities. Histology slides are shown at a magnification of 100X and demonstrate representative examples of (A) normal placental histology, (B) delayed villous maturation, (C) distal villous hypoplasia, a feature of maternal vascular malperfusion, and (D) avascular villi, a feature of fetal vascular malperfusion.
Demographic and Clinical Data
Maternal and infant demographic and clinical factors were obtained from pre-existing clinical databases and chart review. Maternal factors included maternal age, race, and ethnicity (as self-identified within the medical record), earliest documented body mass index (BMI) as an indicator of pre-pregnancy BMI, pre-existing or ongoing hypertensive disorders (defined as chronic hypertension, pregnancy-induced hypertension, or preeclampsia), diabetic disorders (defined as pregestational, Type 1 or Type 2 diabetes mellitus, or gestational diabetes mellitus), oligohydramnios, and polyhydramnios. Self-reported alcohol, tobacco, and recreational drug use prior to and during pregnancy were also collected from the medical record.
Infant clinical factors included mode of delivery, infant sex, gestational age at birth, birth anthropometrics, intrauterine growth restriction (IUGR, defined as estimated fetal weight <10th percentile at any point during the pregnancy), small for gestational age (SGA, defined as <10th percentile at birth), Apgar scores, and infant respiratory support in the delivery room (defined as supplemental oxygen, positive pressure ventilation, or intubation). For the CHD group, cardiac diagnosis and presence of a genetic diagnosis/syndrome (based on clinical genetic testing) were also collected. To evaluate differences in placental abnormalities by cardiac diagnosis, infants were categorized based on their prenatal cardiac physiology into one of four groups: 1) single ventricle physiology with aortic obstruction (SV Ao), 2) single ventricle physiology with pulmonic obstruction (SV Po), 3) dextro-transposition of the great arteries (d-TGA, including those with d-TGA physiology), or 4) two ventricle (2V) anatomy.12 To ensure accurate categorization, the first postnatal echocardiogram was reviewed to confirm the prenatal diagnosis. This categorization approach was chosen to reflect distinct fetal cardiac physiologies that also vary with respect to blood flow and saturation to the placenta. For example, the SV Ao group primarily includes fetuses with hypoplastic left heart syndrome, a diagnosis with reduced descending aorta and umbilical vein saturation, whereas d-TGA has normal saturations within these vessels.18
Statistical Analyses
Statistical analyses were undertaken using SAS® (SAS Institute Inc., Cary, NC, USA) 9.4 version and IBM SPSS® Statistics (IBM Corporation) Version 27. DVM, MVM, and FVM were selected as the primary outcome variables, whereas all other placental pathology findings were secondary outcomes.
A Pearson’s Chi-square test, Fisher’s exact test, independent samples t-test, or analysis of variance was used to compare demographic and clinical variables between groups, as appropriate. Univariate logistic regression was used to determine the association of DVM, MVM, and FVM with CHD. Secondary analyses were conducted for a “Low Risk” CHD group in an effort to remove clinical factors that may affect placental development. This CHD group excluded patients with hypertensive disorders, diabetic disorders, or a fetal genetic diagnosis/syndrome. Finally, because the controls were selected to be a “healthy” population without significant pregnancy complications that may contribute to altered placental development, and because CHD was the primary group of interest, analyses to define predictors of placental abnormalities were only conducted within the CHD group. Univariate logistic regression was first performed to detect the effect of demographic and clinical factors on DVM, MVM, and FVM. Variables with p<0.10 in the univariate models were then treated as candidates in the stepwise logistic regression (p-value threshold of 0.1 to remain in the model) to determine risk factors associated with the outcome while adjusting for other variables. After the stepwise selection, cardiac group was added in the final multivariable logistic models as a covariate to determine the associations of pregnancy factors while adjusting for cardiac group.
Results
Cohort Characteristic
A total of 194 mother/infant dyads with a prenatal diagnosis of fetal moderate-severe CHD and 105 control dyads met inclusion criteria. Indications for placental evaluation in controls included meconium-stained amniotic fluid (n = 18, 17%), non-reassuring fetal status without hypoxic-ischemic encephalopathy or neonatal intensive care unit admission (n = 12, 11%), bilateral tubal ligation (n = 8, 7%), research (n = 1, 1%), or other maternal diagnoses such as hyperthyroidism, recurrent urinary tract infections, or failure to progress (n = 13, 12%). Eighteen (17%) of patients had more than one of the prior indications and 35 (33%) did not have an identifiable indication in the medical record.
Cohort characteristics are shown in Table 2. The CHD group had a younger maternal age (but no difference for advanced maternal age), higher maternal BMI, greater alcohol and tobacco use, earlier gestational age at birth, smaller infant birth weight, length, and head circumference, higher rate of intrauterine growth restriction, lower 5-minute Apgar score, and higher rate of respiratory support in the delivery room. The most common cardiac physiology was 2V anatomy, which occurred in 43% of the CHD group (Table 3). Cardiac groups differed on frequency of advanced maternal age, maternal BMI, infant respiratory support in the delivery room, and infant genetic diagnosis/syndrome. Advanced maternal age occurred in 14% of infants with SV Ao, 4% of infants with SV Po, 7% of infants with d-TGA, and 24% of infants with 2V anatomy (p = 0.04). Mean maternal BMI was 27.8 for SV Ao, 30.3 for SV Po, 25.6 for d-TGA, and 29.6 for 2V anatomy (p = 0.03). Respiratory support was provided in the delivery room for 31% of infants with SV Ao, 33% with SV Po, 63% with d-TGA, and 43% with 2V anatomy (p = 0.04). Genetic diagnosis/syndrome was present in 29% of infants with SV Ao, 30% with SV Po, 11% with d-TGA, and 45% with 2V anatomy (p = 0.007). There were no differences by cardiac group for the remainder of maternal and infant characteristics (p > 0.1 for all variables).
Table 2.
Cohort Characteristics
| Demographic and Clinical Variables | Control (n=105) |
CHD (n=194) |
P-value |
|---|---|---|---|
| Maternal Characteristics | |||
| Age, years | 29.7 (5.2) | 28.2 (6.1) | 0.03 |
| Advanced maternal age (≥35 years) | 16 (15) | 31 (16) | 0.87 |
| Race and Ethnicity | 0.69 | ||
| Non-Hispanic White | 86 (82) | 162 (83) | |
| Non-Hispanic Black or African | 14 (13) | 24 (12) | |
| American | |||
| Non-Hispanic Asian | 2 (2) | 1 (1) | |
| Hispanic | 3 (3) | 7 (4) | |
| BMIa | 26.6 (6.9) | 28.6 (6.4) | 0.04 |
| Hypertensive disorder | 0 (0) | 36 (19) | - |
| Diabetic disorder | 0 (0) | 31 (16) | - |
| Oligohydramnios | 0 (0) | 7 (4) | - |
| Polyhydramnios | 0 (0) | 11 (6) | - |
| Alcohol use | 5 (5) | 27 (14) | 0.02 |
| Tobacco use | 9 (9) | 51 (26) | <0.001 |
| Drug use | 6 (6) | 16 (8) | 0.45 |
| Infant Characteristics | |||
| Mode of delivery, vaginal | 62 (59) | 128 (66) | 0.26 |
| Infant sex, male | 58 (55) | 116 (60) | 0.46 |
| Gestational age at birth, weeks | 39.7 (1.0) | 38.7 (0.8) | <0.001 |
| Birth weight, kilograms | 3.4 (0.5) | 3.2 (0.5) | <0.001 |
| Birth length, centimeters | 51.2 (2.3) | 49.1 (2.9) | <0.001 |
| Birth head circumference, centimeters | 34.7 (1.4) | 33.7 (2.0) | <0.001 |
| Intrauterine growth restriction | 2 (2) | 61 (31) | <0.001 |
| Small for gestational age | 7 (7) | 11 (6) | 0.71 |
| Apgar Scores, 1 min | 8 (8–8) | 8 (7–8) | 0.08 |
| Apgar Scores, 5 min | 9 (9–9) | 8 (8–9) | <0.001 |
| Respiratory support in delivery room | 22 (21) | 79 (41) | <0.001 |
| Genetic abnormality/syndrome | 0 (0) | 65 (34) | - |
Data are reported as mean (standard deviation) or number (percentage). Statistical comparison was not performed for hypertensive disorder, gestational diabetes, oligohydramnios, polyhydramnios, or genetic abnormality/syndrome as these were exclusion criteria for the control group. Intrauterine growth restriction was also an exclusion criterion for the control group; two infants are listed as having this diagnosis as it was present but resolved during the pregnancy.
BMI was missing for 4 CHD and 44 controls secondary to incomplete medical record documentation.
Table 3.
Classification of Cardiac Diagnoses
| CHD Classification | Diagnoses | N (%) |
|---|---|---|
| SV, Ao | DILV, l-TGA | 56 (29) |
| DORV, interrupted aortic arch | ||
| DORV, mitral atresia | ||
| HLHS | ||
| Tricuspid atresia, d-TGA | ||
| Tricuspid atresia, l-TGA | ||
| Unbalanced AVC, right dominant | ||
| SV, Po | DILV | 27 (14) |
| DORV, pulmonary stenosis | ||
| Pulmonary atresia, intact ventricular septum or hemodynamically insignificant VSD | ||
| Tricuspid atresia, normally related great arteries | ||
| Unbalanced AVC, left dominant | ||
| d-TGA | d- TGA, hemodynamically insignificant or no VSD, no outflow obstruction | 27 (14) |
| DORV, sub-pulmonary VSD | ||
| 2V Anatomy | ASD | 84 (43) |
| Aortopulmonary window | ||
| Coarctation of the aorta, hypoplastic aortic arch | ||
| Complete AVC, balanced | ||
| DORV, no obstruction | ||
| Hemitruncus arteriosus | ||
| Incomplete AVC | ||
| Interrupted aortic arch | ||
| l-TGA | ||
| Partial AVC | ||
| TOF, pulmonary stenosis or pulmonary atresia | ||
| TAPVR | ||
| VSD |
CHD classification was determined based on prenatal echocardiography and confirmed on the first postnatal echocardiogram. Abbreviations: 2V, two ventricle anatomy; ASD, atrial septal defect; AVC, atrioventricular canal; d-TGA, dextro-transposition of the great arteries; DILV, double inlet left ventricle; DORV, double outlet right ventricle; HLHS, hypoplastic left heart syndrome; l-TGA, levo-transposition of the great arteries; TAPVR, total anomalous pulmonary venous return; SV Ao, single ventricle physiology with aortic obstruction; SV Po, single ventricle physiology with pulmonic obstruction; TOF, Tetralogy of Fallot; VSD, ventricular septal defect.
Placental Gross and Histopathology Findings
The CHD group demonstrated a higher frequency of DVM and MVM compared to controls (Table 4, Figure 2), whereas the control group was more likely to have markers of meconium exposure (Table 4). There were no group differences for the remainder of the placental pathology outcomes, including FVM. Findings persisted when comparing controls to the Low-Risk CHD group (Table 4), which also had a lower birth weight to placental weight ratio.
Table 4.
Group Differences for Placental Pathology Findings
| Placental Pathology | Control (n=105) |
CHD (n=194) |
P-value | Low-Risk CHD (n=88) |
P-value |
|---|---|---|---|---|---|
| Gross Findings | |||||
| Placental weight, grams | 471 (131) | 450 (115) | 0.16 | 455 (105) | 0.36 |
| BW:PW ratio | 7.7 (1.5) | 7.4 (1.8) | 0.13 | 7.2 (1.4) | 0.03 |
| Cord insertion | |||||
| Eccentric (referent) | 79 (75) | 156 (80) | -- | 70 (80) | -- |
| Central | 11 (11) | 18 (9) | 0.85 | 10 (11) | 0.96 |
| Other | 13 (12) | 15 (8) | 0.30 | 7 (8) | 0.32 |
| Not recorded | 2 (2) | 5 (3) | 1 (1) | ||
| Histopathologic Findings | |||||
| DVM | 6 (6) | 37 (19) | <0.001 | 14 (16) | 0.02 |
| MVM | 20 (19) | 66 (34) | 0.007 | 30 (34) | 0.02 |
| FVM | 6 (6) | 19 (10) | 0.23 | 9 (10) | 0.24 |
| Chorangiosis | 3 (3) | 11 (6) | 0.25 | 6 (7) | 0.19 |
| Inflammation | 33 (31) | 50 (26) | 0.30 | 20 (23) | 0.18 |
| Villous edema | 4 (4) | 16 (8) | 0.15 | 7 (8) | 0.22 |
| Thrombohematoma | 16 (15) | 37 (19) | 0.40 | 16 (18) | 0.58 |
| Meconium exposure | 66 (63) | 70 (36) | <0.0001 | 39 (44) | 0.01 |
Data are reported as mean (standard deviation) or number (percentage). The p-values represent the comparison of all CHD to control and the comparison of Low-Risk CHD to control. Abbreviations: BW:PW ratio, birth weight to placental weight ratio; DVM, delayed villous maturation; FVM, fetal vascular malperfusion; MVM, maternal vascular malperfusion.
Figure 2.
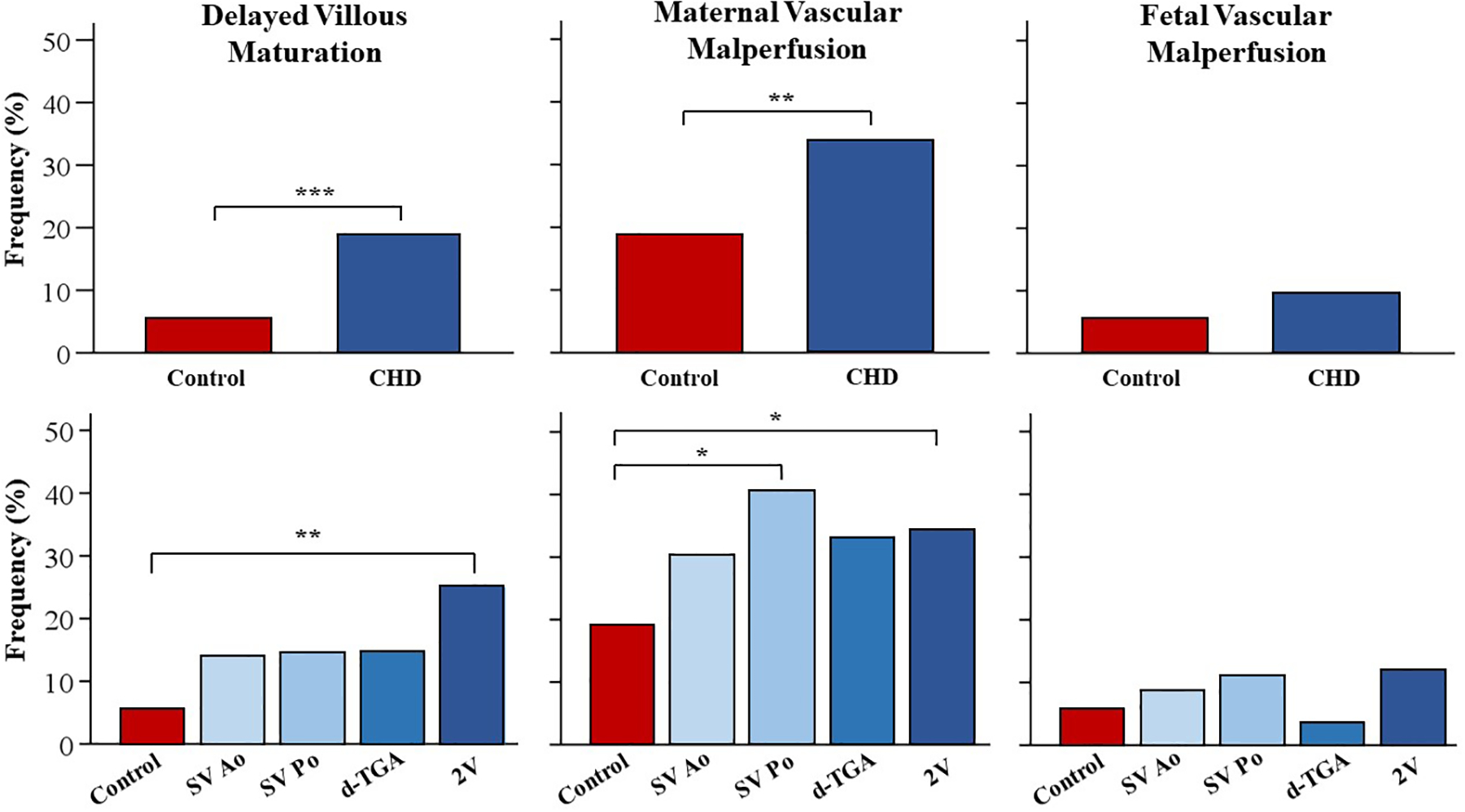
Frequency of histopathological findings in CHD compared to controls. Each graph represents the univariate logistic regression results for placental pathology outcomes. The top row displays the frequency of each pathology finding in pregnancies with fetal CHD compared to controls. The bottom row shows the subgroup analyses for each CHD group compared to controls. Abbreviations: 2V, two ventricle anatomy; d-TGA, dextro-transposition of the great arteries; SV Ao, single ventricle physiology with aortic obstruction; SV Po, single ventricle physiology with pulmonic obstruction.
*p < 0.05
**p < 0.01
***p < 0.001
To account for the possibility that meconium exposure reflected more subtle perinatal factors that may affect placental development beyond known pregnancy complications, and that inclusion of such patients might have skewed findings, analyses were repeated after removing all patients with meconium exposure. The CHD group continued to display higher rates of DVM (CHD: 25/124, 20%; control: 1/39, 3%; p = 0.003) and MVM (CHD: 50/124, 40%; control: 4/39, 10%; p = 0.002). There was no difference for FVM (CHD: 10/124, 8%; control: 11/39, 33%; p = 0.26).
Frequency of Histopathologic Abnormalities by Cardiac Group
Infants with 2V anatomy had an increased odds of DVM (odds ratio [OR] 5.5, 95% confidence interval [CI] 2.2 – 15.7, p<0.01) and MVM (OR 2.2, 95% CI 1.2 – 4.3, p = 0.02) compared to controls (Figure 2). Infants with SV Po had higher odds of MVM (OR 2.9, 95% CI 1.2 – 7.3, p = 0.02). There were no differences in DVM for SV Ao (OR 2.8, 95% CI 0.9 – 8.8, p = 0.90), SV Po (OR 2.9, 95% CI 0.7 – 10.9, p = 0.84), or d-TGA (OR 2.9, 95% CI 0.7 – 10.9, p = 0.85) or in MVM for SV Ao (OR 1.9, 95% CI 0.9 – 3.9, p = 0.11) or d-TGA (OR 2.1 95% CI 0.8 – 5.4, p = 0.12). There were no differences by cardiac group for FVM.
Factors Associated with Placental Abnormalities in CHD
Within the CHD cohort, DVM was associated with higher pre-pregnancy BMI (OR 1.1, 95% CI 1.0 – 1.1, p = 0.04), presence of polyhydramnios (OR 3.9, 95% CI 1.1 – 13.8, p = 0.04), larger head circumference at birth (OR 1.2, 95% CI 1.0 – 1.5, p = 0.03), and infant respiratory support in the delivery room (OR 3.1, 95% CI 1.4 – 6.6, p = 0.004). MVM was associated with oligohydramnios (OR 5.2, 95% CI 1.1 – 36.7, p = 0.05) and FVM was associated with IUGR (OR 2.7, 95% CI 1.0 – 7.0, p = 0.04). In multivariable models, birth head circumference and infant respiratory support remained significant for DVM and IUGR remained significant for FVM, whereas other factors including cardiac group became non-significant (Table 5).
Table 5.
Logistic Regression Models for Factors associated with Placental Abnormalities in Congenital Heart Disease Group.
| OR (95% CI) | |
|---|---|
| Delayed Villous Maturation | |
| Maternal BMI | 1.1 (1.0 – 1.1) |
| Birth Head Circumference, centimeters | 1.2 (1.0 – 1.5)* |
| Respiratory support in delivery room | 3.0 (1.3 – 6.5)* |
| Cardiac Group, SV Ao (referent) | -- |
| SV Po | 0.9 (0.2 – 3.5) |
| d-TGA | 0.9 (0.2 – 3.4) |
| 2V Anatomy | 1.9 (0.7 – 4.9) |
| Maternal Vascular Malperfusion | |
| Diabetic Disorder | 2.1 (0.9 – 4.6) |
| Oligohydramnios | 5.3 (1.0 – 29.6) |
| Cardiac Group, SV Ao (referent) | -- |
| SV Po | 1.8 (0.7 – 4.7) |
| d-TGA | 1.2 (0.5 – 3.4) |
| 2V Anatomy | 1.4 (0.7 – 3.0) |
| Fetal Vascular Malperfusion | |
| Intrauterine growth restriction | 2.6 (1.0 – 7.0)* |
| Cardiac Group, SV Ao (referent) | -- |
| SV Po | 1.0 (0.2 – 4.7) |
| d-TGA | 0.4 (0.1 – 3.5) |
| 2V anatomy | 1.3 (0.4 – 4.1) |
Variables with p-value < 0.1 on univariate analysis were included in the multivariable model, with stay threshold of 0.1. Polyhydramnios met this inclusion threshold for delayed villous maturation, but did not meet stay criteria for the final model.
Denotes p ≤ 0.05.
Abbreviations: 2V, two ventricle anatomy; BMI, body mass index; d-TGA, dextro-transposition of the great arteries; SV Ao, single ventricle physiology with aortic obstruction; SV Po, single ventricle physiology with pulmonic obstruction.
Comment
Principal Findings
Applying the Amsterdam consensus statement recommendations,19 we identified that DVM and MVM, but not FVM, are more common in pregnancies with a fetal diagnosis of moderate-severe CHD compared to demographically matched controls, even when excluding pregnancies with high risk factors for placental abnormalities. The odds of DVM were 5.5 times higher in fetuses with 2V anatomy compared to controls. For MVM, odds ratios were 2.2 for 2V anatomy and 2.9 for SV Po compared to controls. Maternal BMI, polyhydramnios, larger birth head circumference, and infant respiratory support in the delivery room were associated with DVM, whereas oligohydramnios was associated with MVM. However, associations of DVM and MVM with maternal factors and cardiac group did not persist in multivariable models, only fetal/infant factors remained significant.
Results in the Context of What is Known
There has been increasing recognition that uniform placental pathology interpretation across studies, and comparison to control groups, are necessary to understand the association of fetal CHD with placental abnormalities.7 Leon and colleagues have recently published a large, well-designed retrospective study that addressed these challenges by applying the Amsterdam Criteria to placental samples from 305 CHD and 40 control pregnancies.13 That work showed higher rates of MVM and FVM for the CHD group compared to controls, even in the absence of known genetic diagnoses. Our study adds to this work, and prior studies, in two ways. First, we have included a focus on DVM, and second, we have incorporated a larger, demographically matched control group.
DVM is an abnormality in vasculosyncytial membrane development,19 a region of the placenta essential for oxygen/nutrient exchange between maternal and fetal circulations.26, 27 DVM is typically identified beyond 36 weeks’ gestation and generally not reported before 34 weeks’.19 Within our cohort, DVM was present in 19% of the CHD group, compared to only 6% of controls. Rychik and colleagues previously reported that 15% of infants with CHD have hypomature villi, which was defined similarly to DVM,12 Additionally, infants with hypoplastic left heart syndrome and d-TGA have been shown to display decreased vasculosyncytial membranes (a key feature of DVM) compared to controls.28, 29 These data support our findings that DVM is a common placental abnormality with fetal CHD.
DVM has a well-established association with maternal pre-gestational and gestational diabetes mellitus.30–33 Hyperglycemia from diabetes mellitus can lead to significant macro- and micro-vascular effects across multiple organ systems.34 It has been proposed that angiogenic dysregulation, via a complex interplay between glucose, insulin, and oxygen, contributes to abnormal placental vascular development in diabetic disorders during pregnancy.35 Because CHD is associated with maternal diabetes mellitus, this is one possible explanation for our findings. While our CHD group included patients with diabetic disorders, our regression analyses did not identify this as a risk factor for DVM. Further, the Low-Risk CHD group (which excluded diabetes) still had higher rates of DVM than controls. However, we did identify an association with maternal BMI, which may indicate a similar but more subtle metabolic derangement contributing to DVM.
Higher rates of DVM may also result from hemodynamic alterations due to the cardiac defect. Fetuses with CHD display lower descending aorta and umbilical vein saturations and altered placental perfusion, the presence and severity of which vary by diagnosis.14, 18 For example, fetuses with hypoplastic left heart syndrome and tetralogy of Fallot have significant reductions in descending aorta and umbilical vein saturation, suggesting reduced oxygenation of the placenta.18 Smaller placental weight z-scores have been reported for tetralogy of Fallot, double outlet right ventricle, and large ventricular septal defects.11 Finally, placental abnormalities have been reported more commonly with d-TGA and left-sided obstructive lesions.12, 36 Within our cohort, fetuses with 2V anatomy (i.e., tetralogy of Fallot, double outlet right ventricle, ventricular septal defect) displayed the highest rates of DVM. The commonality of tetralogy of Fallot as an affected diagnosis in our cohort, and in existing data, is worthy of further investigation.
The link between CHD and DVM may also be genetic in origin, particularly given the number of shared molecular and genetic pathways of the placenta-heart axis and the frequency of genetic disorders with CHD.6–10, 37 Pathogenic variants in pro-angiogenic genes have been associated with abnormal umbilical artery Doppler in CHD, suggesting placental insufficiency is linked with genetic disturbances.38 We did not identify an association between genetic abnormalities and DVM, although those with 2V anatomy displayed the highest frequency of DVM and genetic diagnoses. Of interest, polyhydramnios, which often occurs with genetic syndromes, was also associated with DVM on univariate analyses. Routine clinical genetic testing at our institution includes chromosomal microarray analysis for all CHD infants, as well targeted testing based on clinical presentation (e.g., fluorescence in situ hybridization for 22q11 microdeletion). It is possible that genetic variants contributing to both CHD and DVM are only detectable on whole exome or genome sequencing and could explain the lack of association between genetics and DVM for our cohort.
Alterations in placental gene expression may also explain the association of DVM with fetal CHD. The placental transcriptome is a dynamic entity that varies throughout gestation for genetic syndromes and pregnancy disorders such as preeclampsia and gestational diabetes.39, 40 While current data have begun to identify specific variants that directly affect the placental transcriptome, further investigation is warranted to understand the role of placental mosaicism for histopathologic abnormalities in CHD.
The current study also showed higher rates of MVM with CHD. The pathophysiology of MVM is thought to relate to spiral artery remodeling resulting in abnormal intervillous oxygenation and oxidative stress.17 Preeclampsia, other hypertensive disorders, and IUGR are classically associated with MVM, but complications such as oligohydramnios are also reported.17 Consistent with this, we identified an association of oligohydramnios with MVM in the CHD group, although not for hypertensive disorders, which may relate to the small number of patients in the cohort with hypertension.
In contrast to prior work,13, 22 we did not identify group differences for FVM. One possible explanation for our lack of group differences may lie within the control population, which was intentionally selected to match the CHD group on demographic characteristics and to exclude factors that may alter placental development. Control groups from other studies have demonstrated similar rates of MVM20 and FVM23 as our data, suggesting our control cohort reflects the general population. Nonetheless, there may still be confounding variables not accounted for that led to our lack of group difference. Alternatively, it is possible that the pattern of placental abnormalities in CHD are most consistently developmental in origin, manifesting as DVM or MVM, and are less likely due to fetal blood flow obstruction.
Clinical Implications
DVM has been associated with late intrauterine fetal demise, preterm birth, and poorer neurological outcomes in preterm infants and infants with neonatal encephalopathy.23, 41–43 Similar adverse outcomes are also seen for MVM.20, 44 We speculate that these associations will also occur with CHD. Existing studies have reported that an adverse maternal-fetal environment (e.g., preeclampsia, growth restriction, preterm birth) increases postoperative mortality in CHD infants,45, 46 supporting the likelihood that placental abnormalities affect postnatal outcomes in CHD. Indeed, we identified that infants with DVM had a higher need for respiratory support in the delivery room, even after adjusting for cardiac physiology.
The potential relationship of DVM and MVM with altered brain development and neurodevelopmental risk is an important consideration for infant outcomes, as neurodevelopmental impairment is the most common adverse outcome for CHD.47 Our data demonstrated an association between DVM and larger head circumference at birth. This finding was contradictory to what we expected. However, head circumference is not a direct surrogate for brain development and alterations in head size may occur for a variety of reasons that could not be delineated in this retrospective study. Future investigations incorporating placental pathology with brain imaging and neurodevelopmental evaluation are needed to capture the relationships between DVM and neurological/neurodevelopmental outcomes in CHD.
Research Implications
The findings of this retrospective study suggest potential research pursuits across several arenas. First, an important next step will be to quantify placental development and function. This could be through histopathologic or novel in vivo imaging studies, some of which have already begun.14, 18, 28, 29 Advances in fetal magnetic resonance imaging may permit real-time assessment of the placenta that could inform underlying mechanisms of specific abnormalities and/or could identify infants at highest risk for adverse outcomes before birth. Studies that incorporate genetic and epigenetic approaches will also be vital for untangling the pathways contributing to placental maldevelopment. It is also worth noting that elevated amniotic fluid erythropoietin has been reported with DVM, outside of CHD.48 Altered expression of vascular endothelial growth factor, placental growth factor, and soluble fms-like tyrosine kinase-1 have been identified in maternal serum and/or fetal tissues with fetal CHD, supporting a complex interplay between hypoxic-induced dysregulation, vascular development, and placental histopathologic abnormalities.49, 50 These data underscore the importance of future studies that integrate biomarkers of fetal hypoxia with placental pathology. Finally, future investigations that incorporate infant outcome measures are of utmost importance for defining the long-term effects of placental abnormalities in CHD.
Strengths and Limitations
The greatest strength of the current study is standardized placental categorization in a large cohort of pregnancies with and without CHD. This provided adequate power to compare abnormalities across cardiac physiologies and to evaluate numerous demographic and clinical factors in association with placental findings. However, this placental categorization still has limitations due to its somewhat qualitative nature. Additionally, some of the placental pathology samples in our cohort were acquired before the Amsterdam Criteria were published, which may have influenced our ability to identify DVM, MVM, and FVM. Both CHD and control groups were included across the same time frame to account for this potential limitation. Further, the histopathologic features of DVM, MVM and FVM were not novel discoveries; rather they were new terminology to unify a constellation of findings, which makes retrospective application of these definitions feasible.
Another limitation is the retrospective study design, which relied on placental reports and preserved histology slides. A prospective study would facilitate more clear delineation of focal versus diffuse findings. The retrospective nature of this study also contributed to limitations with our control population because placental pathology evaluation is not standard of care for all pregnancies. We intentionally selected a relatively “healthy” control population so as not to over-represent any particular pregnancy complication. However, prospective studies are needed to address this limitation.
Conclusions
DVM is more common in pregnancies with CHD and likely reflects multifactorial physiologic and genetic pathways that disrupt typical placental development. The higher frequency of MVM, but not FVM, underscores the importance of maternal factors for placental maldevelopment in CHD. Future investigations that incorporate placental histopathology with detailed maternal factors, genetic/epigenetic evaluation, and measures of fetal hemodynamics will be important for identifying the mechanisms that underlie placental abnormalities. Longitudinal studies linking placental pathology with postnatal outcomes such as neurodevelopment are also vital for understanding the life-long impact of the placenta in CHD.
Supplementary Material
AJOG at a Glance:
- Why was this study conducted?
- To compare placental abnormalities in pregnancies with and without fetal congenital heart disease using consensus statement recommendations.
- What are the key findings?
- Delayed villous maturation and maternal vascular malperfusion were more common with fetal congenital heart disease and demonstrated diagnosis-specific variation.
- Within congenital heart disease, delayed villous maturation was associated with higher maternal body mass index, polyhydramnios, larger infant birth head circumference, and higher infant respiratory support, whereas maternal vascular malperfusion was associated with oligohydramnios.
- What does this study add to what is already known?
- Higher rates of delayed villous maturation and maternal vascular malperfusion in congenital heart disease underscore the importance of maternal factors for placental maldevelopment and may inform investigations of mechanism and subsequent infant outcomes.
Funding:
This work was supported by the National Institutes of Health/National Heart, Lung, and Blood Institute (K23HL141602) and the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The authors report no conflicts of interest.
Presented at 70th Annual Scientific Session of the American College of Cardiology, May 15–18, 2021 and 10th Annual Scientific Sessions of the Cardiac Neurodevelopmental Outcome Collaborative, November 17–19, 2021.
References:
- 1.Gude NM, Roberts CT, Kalionis B, King RG. Growth and function of the normal human placenta. Thromb Res 2004;114:397–407. [DOI] [PubMed] [Google Scholar]
- 2.Heerema-McKenney A Defense and infection of the human placenta. APMIS 2018;126:570–88. [DOI] [PubMed] [Google Scholar]
- 3.Toriola AT, Vaarasmaki M, Lehtinen M, et al. Determinants of maternal sex steroids during the first half of pregnancy. Obstet Gynecol 2011;118:1029–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Randhawa R, Cohen P. The role of the insulin-like growth factor system in prenatal growth. Mol Genet Metab 2005;86:84–90. [DOI] [PubMed] [Google Scholar]
- 5.Bronson SL, Bale TL. The Placenta as a Mediator of Stress Effects on Neurodevelopmental Reprogramming. Neuropsychopharmacology 2016;41:207–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Courtney JA, Cnota JF, Jones HN. The Role of Abnormal Placentation in Congenital Heart Disease; Cause, Correlate, or Consequence? Front Physiol 2018;9:1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leon RL, Mir IN, Herrera CL, et al. Neuroplacentology in congenital heart disease: placental connections to neurodevelopmental outcomes. Pediatr Res 2022;91:787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schleich JM. Images in cardiology. Development of the human heart: days 15–21. Heart 2002;87:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaufmann P, Mayhew TM, Charnock-Jones DS. Aspects of human fetoplacental vasculogenesis and angiogenesis. II. Changes during normal pregnancy. Placenta 2004;25:114–26. [DOI] [PubMed] [Google Scholar]
- 10.Andescavage NN, Limperopoulos C. Placental abnormalities in congenital heart disease. Transl Pediatr 2021;10:2148–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matthiesen NB, Henriksen TB, Agergaard P, et al. Congenital Heart Defects and Indices of Placental and Fetal Growth in a Nationwide Study of 924 422 Liveborn Infants. Circulation 2016;134:1546–56. [DOI] [PubMed] [Google Scholar]
- 12.Rychik J, Goff D, McKay E, et al. Characterization of the Placenta in the Newborn with Congenital Heart Disease: Distinctions Based on Type of Cardiac Malformation. Pediatric cardiology 2018;39:1165–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leon RL, Sharma K, Mir IN, et al. Placental vascular malperfusion lesions in fetal congenital heart disease. Am J Obstet Gynecol 2022. [DOI] [PubMed] [Google Scholar]
- 14.Zun Z, Zaharchuk G, Andescavage NN, Donofrio MT, Limperopoulos C. Non-Invasive Placental Perfusion Imaging in Pregnancies Complicated by Fetal Heart Disease Using Velocity-Selective Arterial Spin Labeled MRI. Sci Rep 2017;7:16126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auger N, Fraser WD, Healy-Profitos J, Arbour L. Association Between Preeclampsia and Congenital Heart Defects. JAMA 2015;314:1588–98. [DOI] [PubMed] [Google Scholar]
- 16.Oyen N, Diaz LJ, Leirgul E, et al. Prepregnancy Diabetes and Offspring Risk of Congenital Heart Disease: A Nationwide Cohort Study. Circulation 2016;133:2243–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ernst LM. Maternal vascular malperfusion of the placental bed. APMIS 2018;126:551–60. [DOI] [PubMed] [Google Scholar]
- 18.Sun L, van Amerom JFP, Marini D, et al. MRI characterization of hemodynamic patterns of human fetuses with cyanotic congenital heart disease. Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and Gynecology 2021;58:824–36. [DOI] [PubMed] [Google Scholar]
- 19.Khong TY, Mooney EE, Ariel I, et al. Sampling and Definitions of Placental Lesions: Amsterdam Placental Workshop Group Consensus Statement. Arch Pathol Lab Med 2016;140:698–713. [DOI] [PubMed] [Google Scholar]
- 20.Catov JM, Scifres CM, Caritis SN, Bertolet M, Larkin J, Parks WT. Neonatal outcomes following preterm birth classified according to placental features. Am J Obstet Gynecol 2017;216:411 e1–11 e14. [DOI] [PubMed] [Google Scholar]
- 21.Miremberg H, Gindes L, Schreiber L, Raucher Sternfeld A, Bar J, Kovo M. The association between severe fetal congenital heart defects and placental vascular malperfusion lesions. Prenat Diagn 2019;39:962–67. [DOI] [PubMed] [Google Scholar]
- 22.Ozcan T, Kikano S, Plummer S, Strainic J, Ravishankar S. The Association of Fetal Congenital Cardiac Defects and Placental Vascular Malperfusion. Pediatr Dev Pathol 2021;24:187–92. [DOI] [PubMed] [Google Scholar]
- 23.Vik T, Redline R, Nelson KB, et al. The Placenta in Neonatal Encephalopathy: A Case-Control Study. The Journal of pediatrics 2018;202:77–85 e3. [DOI] [PubMed] [Google Scholar]
- 24.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol 2002;39:1890–900. [DOI] [PubMed] [Google Scholar]
- 25.Albalawi A, Brancusi F, Askin F, et al. Placental Characteristics of Fetuses With Congenital Heart Disease. J Ultrasound Med 2017;36:965–72. [DOI] [PubMed] [Google Scholar]
- 26.Critchley GR, Burton GJ. Intralobular variations in barrier thickness in the mature human placenta. Placenta 1987;8:185–94. [DOI] [PubMed] [Google Scholar]
- 27.Huppertz B The anatomy of the normal placenta. J Clin Pathol 2008;61:1296–302. [DOI] [PubMed] [Google Scholar]
- 28.Jones HN, Olbrych SK, Smith KL, et al. Hypoplastic left heart syndrome is associated with structural and vascular placental abnormalities and leptin dysregulation. Placenta 2015;36:1078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Courtney J, Troja W, Owens KJ, et al. Abnormalities of placental development and function are associated with the different fetal growth patterns of hypoplastic left heart syndrome and transposition of the great arteries. Placenta 2020;101:57–65. [DOI] [PubMed] [Google Scholar]
- 30.Higgins M, McAuliffe FM, Mooney EE. Clinical associations with a placental diagnosis of delayed villous maturation: a retrospective study. Pediatr Dev Pathol 2011;14:273–9. [DOI] [PubMed] [Google Scholar]
- 31.Starikov R, Inman K, Chen K, et al. Comparison of placental findings in type 1 and type 2 diabetic pregnancies. Placenta 2014;35:1001–6. [DOI] [PubMed] [Google Scholar]
- 32.Evers IM, Nikkels PG, Sikkema JM, Visser GH. Placental pathology in women with type 1 diabetes and in a control group with normal and large-for-gestational-age infants. Placenta 2003;24:819–25. [DOI] [PubMed] [Google Scholar]
- 33.Huynh J, Dawson D, Roberts D, Bentley-Lewis R. A systematic review of placental pathology in maternal diabetes mellitus. Placenta 2015;36:101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fowler MJ. Microvascular and Macrovascular Complications of Diabetes. Clinical Diabetes 2011;29:116–22. [Google Scholar]
- 35.Cvitic S, Desoye G, Hiden U. Glucose, insulin, and oxygen interplay in placental hypervascularisation in diabetes mellitus. Biomed Res Int 2014;2014:145846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schlatterer SD, Murnick J, Jacobs M, White L, Donofrio MT, Limperopoulos C. Placental Pathology and Neuroimaging Correlates in Neonates with Congenital Heart Disease. Sci Rep 2019;9:4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morrish AM, Smith J, Enriquez A, et al. A new era of genetic testing in congenital heart disease: A review. Trends Cardiovasc Med 2022;32:311–19. [DOI] [PubMed] [Google Scholar]
- 38.Russell MW, Moldenhauer JS, Rychik J, et al. Damaging Variants in Proangiogenic Genes Impair Growth in Fetuses with Cardiac Defects. The Journal of pediatrics 2019;213:103–09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inno R, Kikas T, Lillepea K, Laan M. Coordinated Expressional Landscape of the Human Placental miRNome and Transcriptome. Front Cell Dev Biol 2021;9:697947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kikas T, Laan M, Kasak L. Current knowledge on genetic variants shaping placental transcriptome and their link to gestational and postnatal health. Placenta 2021;116:2–11. [DOI] [PubMed] [Google Scholar]
- 41.Jaiman S, Romero R, Pacora P, et al. Disorders of placental villous maturation in fetal death. Journal of perinatal medicine 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harteman JC, Nikkels PG, Benders MJ, Kwee A, Groenendaal F, de Vries LS. Placental pathology in full-term infants with hypoxic-ischemic neonatal encephalopathy and association with magnetic resonance imaging pattern of brain injury. The Journal of pediatrics 2013;163:968–95 e2. [DOI] [PubMed] [Google Scholar]
- 43.Redline RW. Placental pathology: a systematic approach with clinical correlations. Placenta 2008;29 Suppl A:S86–91. [DOI] [PubMed] [Google Scholar]
- 44.Kulkarni VG, Sunilkumar KB, Nagaraj TS, et al. Maternal and fetal vascular lesions of malperfusion in the placentas associated with fetal and neonatal death: results of a prospective observational study. Am J Obstet Gynecol 2021;225:660 e1–60 e12. [DOI] [PubMed] [Google Scholar]
- 45.Gaynor JW, Parry S, Moldenhauer JS, et al. The impact of the maternal-foetal environment on outcomes of surgery for congenital heart disease in neonates. Eur J Cardiothorac Surg 2018;54:348–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steurer MA, Peyvandi S, Baer RJ, et al. Impaired Fetal Environment and Gestational Age: What Is Driving Mortality in Neonates With Critical Congenital Heart Disease? J Am Heart Assoc 2019;8:e013194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marino BS, Lipkin PH, Newburger JW, et al. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation 2012;126:1143–72. [DOI] [PubMed] [Google Scholar]
- 48.Jaiman S, Romero R, Pacora P, et al. Placental delayed villous maturation is associated with evidence of chronic fetal hypoxia. Journal of perinatal medicine 2020;48:516–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Llurba E, Sanchez O, Ferrer Q, et al. Maternal and foetal angiogenic imbalance in congenital heart defects. Eur Heart J 2014;35:701–7. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez O, Ruiz-Romero A, Dominguez C, et al. Brain angiogenic gene expression in fetuses with congenital heart disease. Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and Gynecology 2018;52:734–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.