

Abstract
The chromobox-containing protein CBX4 is an important regulator of epithelial cell proliferation and differentiation, and has been implicated in several cancer types. The cancer stem cell (CSC) population is a key driver of metastasis and recurrence. The undifferentiated, plastic state characteristic of CSCs relies on cues from the microenvironment. Cancer-associated fibroblasts (CAFs) are a major component of the microenvironment that can influence the CSC population through the secretion of extracellular matrix and a variety of growth factors. Here we show CBX4 is a critical regulator of the CSC phenotype in squamous cell carcinomas of the skin and hypopharynx. Moreover, CAFs can promote the expression of CBX4 in the CSC population through the secretion of interleukin-6 (IL-6). IL-6 activates JAK/STAT3 signaling to increase ∆Np63α—a key transcription factor that is essential for epithelial stem cell function and the maintenance of proliferative potential that is capable of regulating CBX4. Targeting the JAK/STAT3 axis or CBX4 directly suppresses the aggressive phenotype of CSCs and represents a novel opportunity for therapeutic intervention.
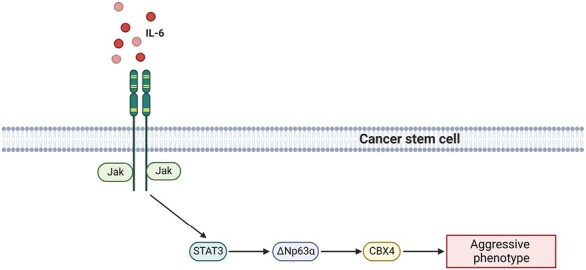
This study identifies a signaling cascade activated by cancer-associated fibroblast-secreted IL-6 that leads to enhanced ∆Np63α and CBX4 expression that drives aggressive cancer stem cell phenotype in squamous cell carcinoma.
Graphical Abstract
Graphical Abstract.
Introduction
Squamous cell carcinomas (SCC) are solid tumors that occur in epithelial tissue that can be classified as stratified, such as the skin, esophagus and oral cavity, or non-squamous epithelia such as airway epithelium (1). SCC occurs frequently in response to mutagens, with the most common locations being the skin, oral cavity, esophagus, lung and cervix (1). Treatment can include surgical resection of the primary tumor, radiation and chemotherapy (2). However, recurrence and metastasis are frequent events following initial treatment (3). The cancer stem cell (CSC) population is a highly lethal subpopulation of cells largely regarded as the primary drivers of recurrence, drug resistance and metastasis in multiple cancer types (4). Therefore, an important goal is to identify pathways responsible for the maintenance of the CSC population that can be targeted therapeutically. Chromatin-modifying proteins have been shown to be essential regulators of SCC CSCs, and have generated intense interest as therapeutic targets (5–7).
The chromobox family of proteins is a subgroup within the polycomb group (PcG) family of proteins that contains five members; CBX2, CBX4, CBX6, CBX7 and CBX8 (1). CBX4 belongs to the Polycomb Repressive Complex 1 (PRC1) family and has an H3K27me3 histone mark-interacting chromodomain which mediates transcriptional repression in coordination with the Polycomb Repressive Complex 2 (PRC2) (1). In normal human skin, CBX4 protects epithelial stem cells from cell cycle withdrawal, and senescence and controls differentiation (8). CBX4 has been implicated in the progression of several cancers, but its role in SCC, and CSCs in particular, has yet to be investigated (9–12).
The epithelial cells that comprise SCC coexist with several distinct stromal cell types that together, create the tumor microenvironment (TME) (13). The TME plays an important role in regulating multiple aspects of tumorigenesis through its crosstalk with cancer cells (14). Cancer-associated fibroblasts (CAFs) are a key part of the TME that influence tumor progression (15). One of the ways CAFs do this is through the secretion of chemokines (16). Chemokines exert their multifunctional roles in several physiologic and pathologic processes through interaction with their specific receptors. One such chemokine, interleukin-6 (IL-6), plays a critical role in modulating stem cell proliferation and survival through its canonical receptor (17–19). While the function and importance of IL-6 signaling has been clearly demonstrated, the mechanisms mediating IL-6-driven phenotypes are not fully characterized. In this study, we demonstrate that CAFs regulate the CSC phenotype through the secretion of IL-6 to activate JAK/STAT signaling in CSCs. JAK/STAT activation leads to the transcriptional activation of the transcription factor ∆Np63α, which in turn transactivates CBX4, a factor critical to epithelial stem cells. We propose that targeting this network of CSC survival proteins activated by CAFs offers a novel therapeutic strategy for SCC patients.
Materials and methods
Antibodies and reagents
DMEM (10-013-CV), Trypsin (25-054-CI) and 6-well ultralow attachment plates (3471) were purchased from Corning. DMEM F-12 (11320-033) was purchased from Thermo Fisher. Cell lysis buffer (9803), and antibodies specific for p63 (39692), JAK2 (3230), JAK2-P (3771), STAT3 (9139) and STAT3-P (9145) were purchased from Cell Signaling Technology. Anti-CBX4 (SC-517216) and CBX7 (SC-376274) were purchased from Santa Cruz. Peroxidase-conjugated anti-mouse IgG and antirabbit IgG were obtained from GE Healthcare. Matrigel (354234) and BD Biocoat cell inserts (353097) were from BD Biosciences. DAPI (D9542) and Anti-b-actin (A5441) were purchased from Sigma. Tocilizumab (A2012) and AZD1480 (S2162) were purchased from Selleckchem. STATTIC (2798) was purchased from Tocris and UNC3866 (19237) was purchased from Cayman Chemical. STAT3C.Ubc.GFP was a gift from Linzhao Cheng (Addgene plasmid # 24983; http://n2t.net/addgene:24983; RRID:Addgene_24983 (20)). ΔNp63α-FLAG was a gift from David Sidransky (Addgene plasmid # 26979; http://n2t.net/addgene:26979; RRID: Addgene_26979 (21)).
Cell culture
Human HSC-5 and HSC-1 cells were obtained from Sekisui Xenotech, LLC and Human FaDu cell lines were a generous gift from Leif Ellison in 2017. HSC-5, HSC-1 and FaDu cells were cultured in DMEM supplemented with 10% FBS. Cells lines were most recently authenticated using short tandem repeat (STR) in February 2023. All cell lines tested negative for Mycoplasma contamination using MycoAlert (LT07-218) from Lonza.
Isolation of fibroblasts
Tumor samples were washed with cold PBS and minced with a scalpel. The minced tissue was transferred to a 50 ml conical tube, suspended in PBS and centrifuged for 5 min. Red blood cell lysis buffer (ThermoFisher #00-4333-57) was added and tissue was gently mixed to remove red blood cells. The tissue was then centrifuged for 5 min, supernatant removed and the tissue resuspended in RPMI with 10% FBS. Cells were transferred to a 10 cm dish and incubated at 37°C for 2 h before the media was changed to remove non-adherent cells. The following day, cells were briefly trypsinized to remove fibroblasts, while keeping any cancer cells that adhered attached.
Fibroblast conditioned media
Normal adjacent and CAFs were maintained in RPMI with 10% FBS. To generate conditioned media, fibroblasts were trypsinized, and three million cells replated. Upon attachment, plates were washed three times with PBS, and cancer cell media was added. Conditioned media was collected 24 h after plating and sterile filtered (0.22 µm pores) prior to being utilized on the cancer cell lines.
Lentivirus production
Lentivirus was produced using 293T cells maintained in DMEM with 1 mmol/L l-glutamine, 1 mmol/L sodium pyruvate and 10% FBS. 293T cells were harvested and plated in 100 mm dishes at 5% confluence for 24 h prior to transfection. Media were removed and plates were washed with Hank balanced Salt Solution before serum-free media were added containing 7.5 µg pPax2, 5 µg VSVG and 5 µg single-guide RNA (sgRNA) and polyethyleneimine (PEI) for co-transfection. After 8 h, 10% FCS was added, and 48 h after transfection, the medium was collected, centrifuged for 15 min at 1500 rpm, sterile filtered (22 µm) and stored at 80°C in aliquots.
Creation of CRISPR-depleted cells
Wild-type cancer cells were first plated at 50% confluency in 6-well plates. Cells were then transduced with a Cas9-puro vector packaged in lentivirus with 1 ml of lentivirus-containing serum-free media with PEI for 5 h, after which media was replaced with serum-containing media. After 24 h, fresh media was added containing puromycin at a concentration of 1 µg/ml. Cells were selected in puromycin for 2 weeks, and Cas9 expression was confirmed with western blot. The Cas9-expressing cells were then transduced with sgRNAs cloned into neomycin-resistant vectors. Cas9-expressing cells were then selected with 300 µg/ml G418. CRISPR-mediated depletion was assessed via western blot after 2 weeks of selection with G418.
Creation of overexpression cells
∆Np63α and STAT3C were cloned into LentiV expression vectors from the original backbones listed in ‘Antibodies and Reagents’. Expression vectors were then packaged into lentivirus and used to transduce target cells. Hygromycin was used to select cells at a concentration of 100 µg/ml for 2 weeks. Overexpression of each target was confirmed by western blotting.
Spheroid formation
Monolayer cultures were harvested with trypsin and gently pipetted to form single-cell suspensions. Trypsin was inactivated by addition of serum-containing medium and cells were collected by centrifugation at 2000 rpm for 5 min. Cell pellets were resuspended in a spheroid medium consisting of DMEM/F12 (1:1) containing 2% B27 serum-free supplement (17504-044, Invitrogen), 20 ng/ml EGF (AF-100-15, PeproTech) and plated at 4 × 104 cells per 9.5 cm2 well in 6-well ultralow attachment plates (Corning; #3471).
Immunoblotting
For immunoblot analyses, equivalent amounts of protein were electrophoresed on denaturing and reducing 10% polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked in 5% non-fat dry milk for 30 min and then incubated with the appropriate primary antibody (1:1000) overnight at 4°C, followed by incubation with the corresponding secondary antibodies (1:5000) for 2 h at room temperature. Secondary antibody binding was visualized using SuperSignal West Atto (ThermoFisher #A38556) chemiluminescence detection technology.
Invasion assays
Matrigel was diluted in 0.01 Tris–HCL/0.7% NaCl, filter sterilized and 0.1 ml was used to coat individual BD BioCoat inserts (MillicellPCF, 0.4 mm, 12 mm, PIHP01250). Cells (2.5 × 104) were plated in 100 µl spheroid medium supplemented with 1% FBS into the upper chambers of the inserts. The lower chambers contained spheroid medium supplemented with 10% FBS. After invasion, membranes were harvested and the surface of the upper membranes were scraped and rinsed with PBS to remove unattached cells. Membranes were fixed in 4% paraformaldehyde, stained with 1 mg/ml DAPI and the underside of the membrane was photographed using an inverted fluorescent microscope and cells were quantitated.
Migration assays
Cancer cells (2 × 106) were plated in 10 cm dishes and grown as monolayer cultures in spheroid medium until confluent. A 10 µl pipette tip was used to prepare areas void of cells and the dishes were washed three times with PBS to remove the dislodged cells. Images were collected at 0–24 h after the scratches were made using a 10× objective, and the width of the openings was measured as a function of time as an index of cell migration potential.
qRT-PCR
Total RNA was isolated with the RNeasy Mini Kit (Qiagen) and reverse transcribed using the Superscript III First-Strand Synthesis System (Invitrogen). RNA (1 µg) was used for cDNA preparation. The Power SYBRTM GREEN PCR Master Mix (Thermo Fisher) was used to quantitate expression levels according to manufacturer’s protocol. Signals were normalized to the levels of cyclophilin A mRNA to determine expression.
Tumor xenografts
All animal experiments were approved by the Institutional Animal Care and Use Committee. Spheroid-derived cancer cells were dispersed with trypsin to produce single-cell suspensions, and cells were resuspended in 100 ml of spheroid medium and then mixed with Matrigel at a 1:1 ratio. The mixture, containing 1 × 106 cells, was injected subcutaneously into the flanks of nude mice using 26.5 gauge needles attached to 1 cc syringes. Cells and syringes were kept on ice throughout the procedure to prevent Matrigel from solidifying. Four mice were used per data point with two tumors per mouse. For drug treatments, drugs were administered Monday/Wednesday/Friday via intraperitoneal injection. Mice were randomized into different groups and no animals were excluded from analysis.
Statistical analyses
All data are presented as mean values SEM of at least three biological replicates unless otherwise indicated. Two-tailed Student t-test or ANOVA (one-way or two-way) were used to determine the effects of treatments. P < 0.05 was considered significant.
Results
CAFs induce CSC marker expression and an aggressive phenotype in SCC
CAFs have been implicated as drivers of a number of pathways linked to various aspects of tumorigenesis (22–24). Here, we set out to identify if CAFs isolated from SCC patients were capable of inducing a CSC phenotype in SCC cells. We have shown previously that SCC CSCs display enhanced spheroid formation, invasion and migration compared with their more differentiated counterparts (6). Therefore, we first examined whether conditioned media from CAFs or normal fibroblasts (NFs) could stimulate these CSC traits. To do this, we used SCC cell lines from the skin (HSC-1, HSC-5), pharynx (FaDu) and tongue (CAL-33), which we previously utilized to characterize the CSC phenotype. We found that cells grown under spheroid conditions in the presence of CAF-conditioned media (CAF-CM) produced a greater number of spheroids compared with those in NF-conditioned media (NF-CM) (Figure 1A, Supplementary Figure 1A, available at Carcinogenesis Online). In line with this result, CAF-CM led to an increase in the rate of invasion through Matrigel (Figure 1B, Supplementary Figure 1B, available at Carcinogenesis Online) and migration into a scratch wound (Figure 1C). Furthermore, we found this CSC phenotype stimulated by CAF-CM was associated with increases in known CSC markers SOX2 (25) and ∆Np63α (26) (Figure 1D, Supplementary Figure 1C, available at Carcinogenesis Online). Our previous work identified a number of chromatin-modifying proteins are essential for regulating the CSC phenotype (5–7). Therefore, we screened for chromatin-modifying proteins specifically altered in response to CAF-CM and identified CBX4 as increased in response (Figure 1D, Supplementary Figure 1C, available at Carcinogenesis Online).
Figure 1.
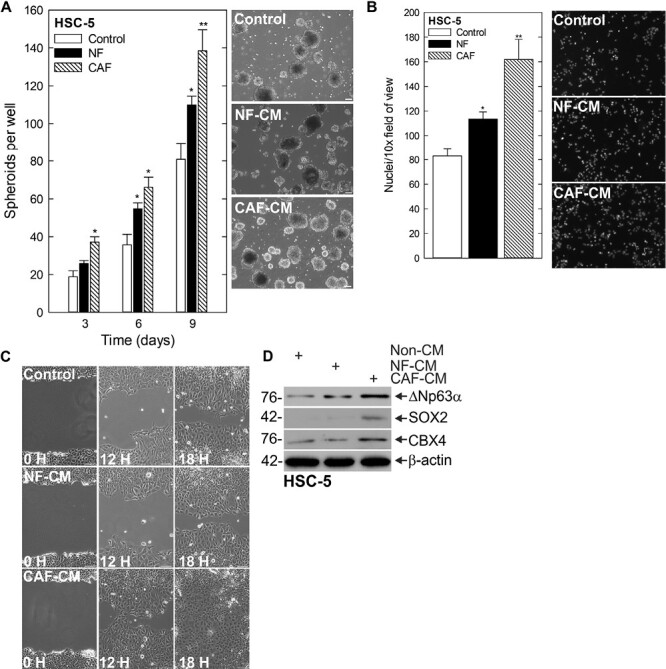
CAFs stimulate CSC phenotype and CBX4 expression. (A) HSC-5 monolayer cultures maintained in a growth medium were harvested and plated at 4 × 104 cells per well in spheroid growth conditions with normal or CAF-CM and spheroid number monitored for 9 days (left). Representative images on day 9 of growth are shown (right). Scale bars, 200 µm. (B) Spheroid cells were trypsinized and single-cell suspensions were seeded onto Matrigel-coated membranes in Millicell chambers for invasion assays with normal or CAF-CM in the bottom chamber (C) or replated as monolayer cultures and allowed to reach confluence, at which time they were scratched with a 10 ml pipette tip to create a wound; wound closure was monitored over time. (D) After 10 days of spheroid growth in normal or CAF-CM, lysates were electrophoresed for detection of the indicated epitopes.
CBX4 drives an aggressive phenotype
The above findings suggest that CAFs are capable of driving a CSC phenotype in SCC. CBX4 is involved in epithelial stem cell regulation and has been linked to a number of cancers (8–12). We found that CBX4 expression was elevated in cells cultured in CAF-CM (Figure 1D). To understand the necessity of CBX4 in this context, we studied how CBX4 deficiency impacted the CSC phenotype using cells with CRISPR-mediated depletion of CBX4 (Figure 2A) in spheroid formation, invasion and migration assays. As shown in Figure 2B and Supplementary Figure 2A, available at Carcinogenesis Online, CBX4-depleted cells formed fewer spheroids than their control counterparts. Furthermore, invasion through Matrigel (Figure 2C, Supplementary Figure 2B, available at Carcinogenesis Online) and migration into a scratch wound were also impaired following CBX4 depletion (Figure 2D). Using the small molecule inhibitor UNC3866, which binds to the chromodomain of CBX4 to inhibit the methyl-lysine reading function, we found that treatment of cells at the initiation of spheroid growth led to fewer spheroids forming (Figure 2E, Supplementary Figure 2C, available at Carcinogenesis Online). We further assessed the effect of UNC3866 on invasion and migration of spheroid-derived cells, revealing that UNC3866 slowed both of these critical endpoints (Figure 2F and G, Supplementary Figure 2D, available at Carcinogenesis Online). Because UNC3866 is capable of inhibiting CBX7, we treated CBX4-depleted cells with UNC3866 to determine if the phenotypic changes we observed could be due CBX7 inhibition. We found no changes in spheroid number or rate of invasion when CBX4-depleted cells were treated with UNC3866, indicating that this inhibitor function works through CBX4 (Supplementary Figure 3A and B, available at Carcinogenesis Online). An important goal was to determine if CBX4 was a valid target in vivo. To determine this, we injected 2.5 × 106 spheroid-derived cells subcutaneously into the flanks of nude mice and monitored tumor growth in Control- and UNC3866-treated mice. Tumors of UNC3866-treated mice were markedly smaller than controls (Figure 2H). These findings indicate CBX4 is a key driver of the CSC phenotype in SCC.
Figure 2.
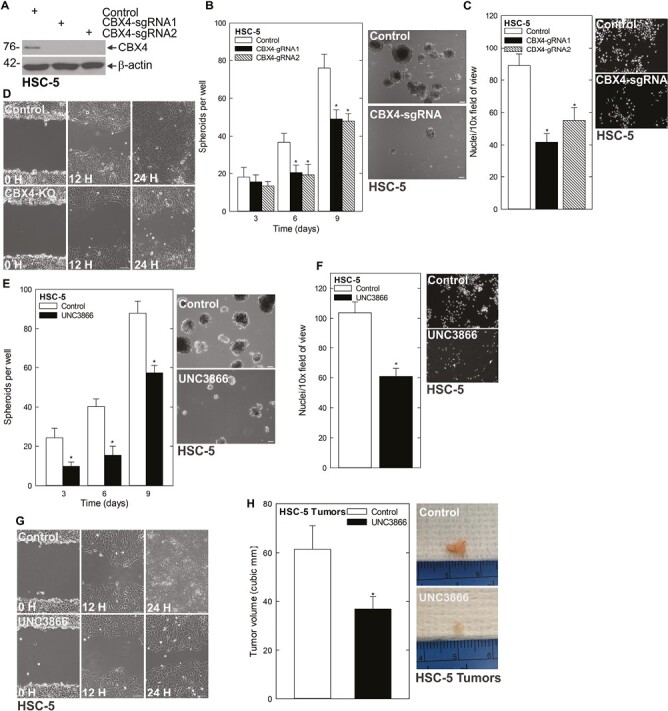
CBX4 regulates the CSC phenotype. (A) HSC-5 control empty vector (EV) and CBX4 CRISPR-depleted cells were grown in spheroid culture, and lysates were collected for immunoblotting. (B) HSC-5 control and CBX4 CRISPR-depleted cells were seeded in spheroid growth conditions and the spheroid number was monitored for 9 days (left). Representative images on day 9 of growth are shown (right). (C) HSC-5 control and CBX4 CRISPR-depleted cells were seeded in Matrigel-coated membranes in Millicell chambers for invasion assays. (D) HSC-5 control and CBX4 CRISPR-depleted cells were seeded as monolayer cultures and allowed to reach confluence before being scratched with a 10 µl pipette tip to monitor the rate of scratch-wound closure. H, hours. (E) HSC-5 cells (4 × 104) were plated in non-adherent 6-well plates, grown for 9 days in spheroid medium, with or without 20 µM UNC3866 and spheroid numbers were counted over 9 days. The values are mean ± SEM. The asterisks indicate significant differences compared with the control. Representative spheroid images following a 9 day treatment with 0 or 20 mmol/L UNC3866 are shown. (F and G) HSC-5 spheroids were trypsinized to form single-cell suspensions and reseeded for invasion and migration assays ± UNC3866.
CAF-secreted IL-6 regulates CBX4 expression
Because of the demonstrated importance of CBX4 to the CSC phenotype, we next sought to elucidate the mechanism behind the CAF activation of CBX4. We screened for secretome components that were enriched in CAF-CM compared with NF-CM and identified IL-6 levels as significantly elevated (Figure 3A). While the expression of multiple cytokines was altered in the CAF population compared with normal adjacent fibroblasts, we decided to focus on IL-6 due to fact that it was one of the more highly enriched factors in our screen, and its ability to activate STAT3, which we have previously linked to regulation of ∆Np63α and the CSC phenotype (6). To determine if IL-6 was the key component of the CAF secretome stimulating expression of CBX4, we treated with recombinant IL-6 (R-IL6) and found increased protein expression of CBX4 in the presence of R-IL6 (Figure 3B, Supplementary Figure 4A, available at Carcinogenesis Online). To assess whether R-IL6 induction of CBX4 was sufficient to stimulate the CSC phenotype, we monitored spheroid formation, invasion and migration following treatment with R-IL6 and found that each of these key endpoints was elevated in response to R-IL6 (Figure 3C–E). To further validate the role of IL-6 in the activation of CBX4 and determine if inhibiting upstream regulators of CBX4 was a potential therapeutic strategy, we utilized the monoclonal antibody Tocilizumab (which competitively inhibits the binding of (IL-6) to its receptor (27)) in the context of R-IL6 treatment. We found that whereas R-IL6 stimulated CBX4 expression, Tocilizumab was able to suppress the R-IL6-induced increase in CBX4 (Figure 3F). Furthermore, this is associated with a decrease in spheroid number, as well as compromised invasion through Matrigel (Figure 3G and H). Finally, to confirm that IL-6 was the key component responsible for driving CAF-induced CBX4 expression, we utilized Tocilizumab in the presence of CAF-CM, and again saw CBX4 expression was reduced (Figure 3I). This evidence indicates that CAF-secreted IL-6 induces the expression of CBX4 and stimulates the CSC phenotype, and that blocking IL-6 signaling with Tocilizumab is a means for impairing CAF activation of CBX4 and the CSC phenotype.
Figure 3.
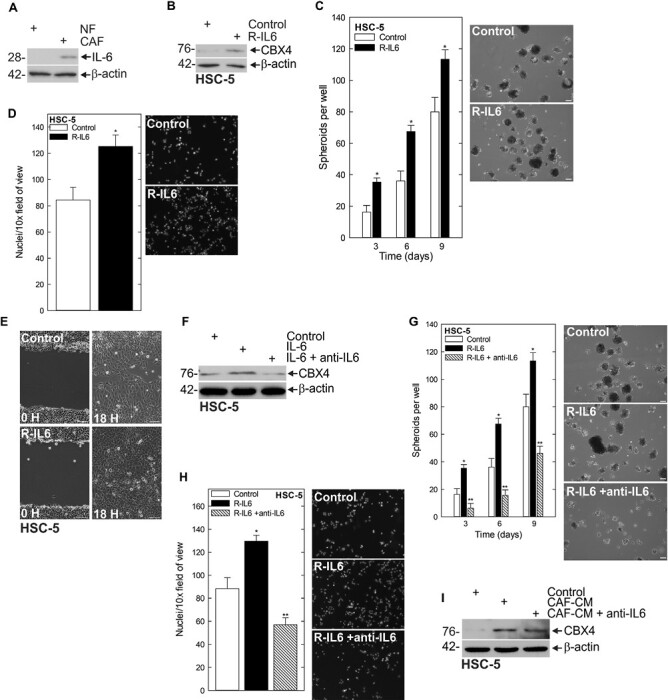
IL-6 is responsible for the CAF-stimulated upregulation of CBX4. (A) Chemokine dot blot array of normal and CAF. (B) Spheroids were grown for 8 days then IL-6 was added to the media for 48 h and lysates were collected for immunoblot. (C) HSC-5 cells (4 × 104) were plated in spheroid growth conditions, and at the time of seeding, 0 or 10 nmol/L R-IL-6 was added. Incubation continued for 9 days, and the spheroid number was counted at each time point. (D and E) HSC-5 spheroid-derived cells were seeded for invasion and migration assays ± R-IL-6 and monitored over time. (F) Spheroids were grown for 8 days with R-IL-6 with or without Tocilizumab, and lysates were collected for immunoblots to detect the indicated epitopes. (G) HSC-5 cells (4 × 104) were plated in spheroid growth conditions, and at the time of seeding, 0 or 10 nmol/L R-IL6 was added with or without 5 µg/ml Tocilizumab. Incubation continued for 9 days, and the spheroid number was counted at each time point. (H) HSC-5 spheroid-derived cells were seeded for invasion assays ± R-IL.26 with or without 5 µ/ml Tocilizumab and monitored over time. (I) HSC-5 cells (4 × 104) were seeded in spheroid growth conditions with or without CAF-CM and Tocilizumab, and the spheroid number was monitored over time. (J and K) HSC-5 spheroid-derived cells were seeded for invasion and migration assays in control or CAF-CM with or without Tocilizumab and monitored over time.
IL-6-mediated JAK/STAT activation is necessary for IL-6-induced CBX4
Canonical IL-6 signaling leads to the activation of JAK/STAT (28,29). Our finding that IL-6 was capable of inducing CBX4 expression prompted us to ask whether JAK/STAT downstream of IL-6 was involved in CBX4 regulation. We assessed the impact of CAF-CM media on JAK/STAT signaling and observed increased JAK2 and STAT3 activation in CAF-CM (Figure 4A). To confirm IL-6 was responsible for the increase in JAK2 and STAT3, we monitored the ability of Tocilizumab to suppress JAK2 and STAT3 phosphorylation in the presence of CAF-CM (Figure 4B). To further assess the importance of JAK2 signaling in the regulation of CBX4, we utilized the JAK inhibitor AZD1480 (30). We found that JAK inhibition effectively reduced CBX4 expression (Figure 4C, Supplementary Figure 4B, available at Carcinogenesis Online). Additionally, JAK inhibition targeted the CSC phenotype; it reduced spheroid formation, invasion and migration (Figure 4D–F). Furthermore, these changes in the CSC phenotype appeared to be dependent on the ability of STAT3 to be activated by JAK2, as expression of constitutively active STAT3, STAT3C, is able to rescue the compromised spheroid formation and invasion seen with JAK inhibition (Figure 4G and H). Additionally, STAT3C is able to rescue the expression of CBX4, further suggesting that CBX4 expression is a key regulator of the phenotype (Figure 4I). We showed previously that STAT3 is able to drive the CSC phenotype, and this can be suppressed with the STAT3 inhibitor STATTIC, an inhibitor of STAT3 activation, dimerization and nuclear translocation. Here we show that treatment of STATTIC results in reduced CBX4 expression, further implicating STAT3 in the CAF-induced signaling changes responsible for increased CBX4 expression (Figure 4J, Supplementary Figure 4C, available at Carcinogenesis Online). These findings indicate CAFs stimulate CBX4 expression through an IL-6/JAK/STAT-dependent signaling network.
Figure 4.
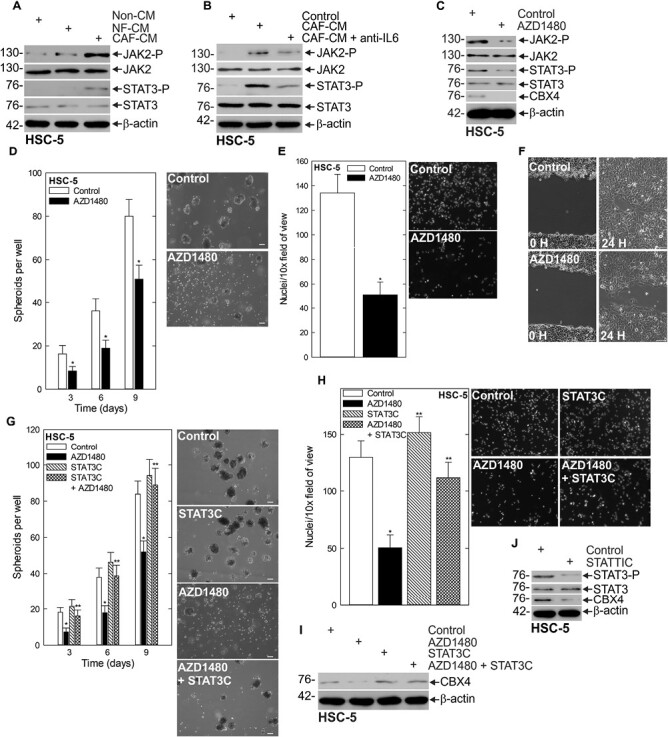
JAK/STAT is responsible for CAF-stimulated upregulation of CBX4. (A) HSC-5 spheroids were grown in Control, NF or CAF-CM, and lysates were collected for immunoblots of the indicated epitopes. (B) Spheroids cultured in Control or CAF-CM were treated with anti-IL6 Tocilizumab and lysates were collected for immunoblots. (C) Spheroids were grown in CAF-CM and treated with 1 µM of the JAK inhibitor AZD1480 and lysates were collected for protein detection of the indicated epitopes. (D) HSC-5 monolayer cultures maintained in a growth medium were harvested and plated at 4 × 104 cells per well in spheroid growth conditions in CAF-CM with or without AZD1480 and the spheroid number was monitored for 9 days. (E/F) HSC-5 spheroids were trypsinized to form single-cell suspensions and reseeded for invasion and migration assays ± AZD1480. (G) HSC-5 cells infected with lentivirus expressing STAT3C or empty vector were seeded in spheroid growth conditions and treated with AZD1480 or DMSO at the time of seeding. Spheroid number was monitored for 9 days. Representative spheroid images are shown from day 9 of growth. (H) Cells as described in (G) were seeded in Matrigel invasion assays ± AZD14800. (I) Immunoblots of the indicated epitopes following AZD1480 or DMSO treatment of empty vector- and STAT3C-expressing cells. (J) Immunoblots of the indicated epitopes following treatment of spheroids cultured in CAF-CM with DMSO or 5 µM STATTIC. Scale bars, 200 μm. *, **, P < 0.05 (the double asterisk indicates significance compared with the single asterisk group).
∆Np63α regulates CBX4 expression
∆Np63α is a transcription factor in the p53 protein family with important functions in stem cell status, fate and differentiation (26,31,32). ∆Np63α has been linked to the SCC CSC phenotype (33–35) and we have shown previously that ∆Np63α is a critical regulator of CSC survival in SCC that is regulated by STAT3 (6). Additionally, ∆Np63α has been shown to regulate the expression of CBX4 in thymic epithelial cells and keratinocytes (8,36). This prompted us to examine if ∆Np63α was involved in CBX4 regulation in this context. In that regard, we created pan-p63 CRISPR-depleted cells and observed a reduction in CBX4 protein level that was rescued with the reintroduction of ∆Np63α into the pan-p63-depleted cells (Figure 5A, Supplementary Figure 4D, available at Carcinogenesis Online). Following this, we sought to identify whether ∆Np63α was required for STAT3 activation of CBX4. To do this, we expressed STAT3C in the presence of p63-sgRNA and monitored CBX4 expression. As shown in Figure 5B and Supplementary Figure 4E, available at Carcinogenesis Online, STAT3C is unable to induce CBX4 expression in the context of p63 depletion. Furthermore, STAT3C expression is not capable of rescuing spheroid formation and invasion when p63 is depleted (Figure 5C and D). To further demonstrate ∆Np63α is downstream of STAT3 and necessary for STAT3-mediated activation of CBX4, we expressed ∆Np63α in the context of STAT3 inhibition with STATTIC (37). Western blotting showed that ∆Np63α expression can maintain CBX4 expression despite inhibition of STAT3 (Figure 5E). Having shown previously that ∆Np63α is critical to the CSC phenotype, we next sought to determine if ∆Np63α was driving the CSC phenotype through regulation of CBX4. To do this, we expressed CBX4 in p63-depleted cells (Figure 5F) and monitored the aggressive phenotype. While spheroid formation and invasion are significantly impaired following p63 depletion, expression of CBX4 provided a partial rescue of the phenotype, indicating CBX4 is a downstream mediator of the ∆Np63α driven CSC phenotype.
Figure 5.
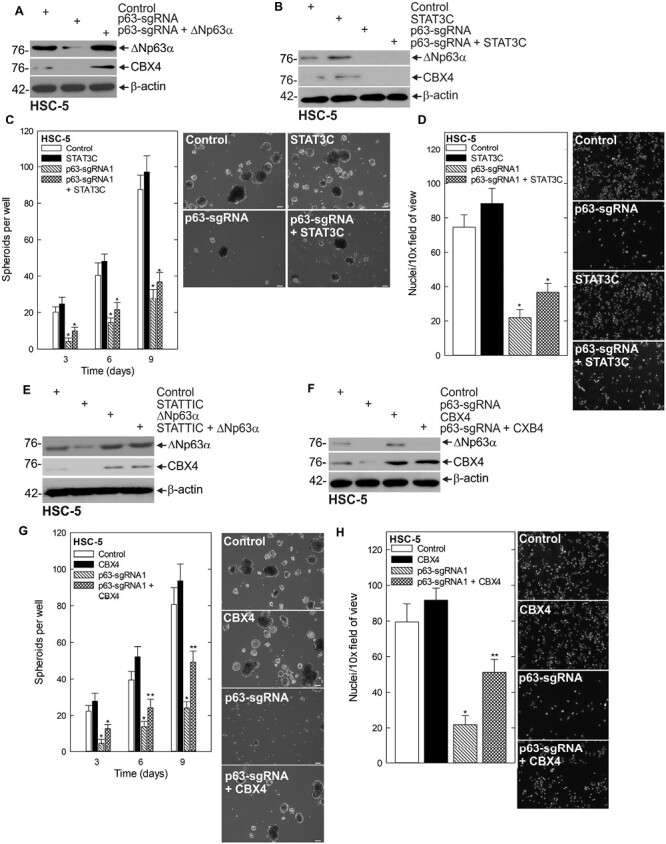
STAT3 regulates CBX4 via the regulation of ∆Np63α. (A) Control, p63-sgRNA and p63-sgRNA + ∆Np63α-expressing cells were harvested for protein for western blot. (B) Control, STAT3C, p63-sgRNA and STAT3C + p63-sgRNA-expressing cells were harvested for protein for western blotting of the indicated proteins. (C) Control, STAT3C, p63-sgRNA and STAT3C + p63-sgRNA-expressing cells were seeded in spheroid growth conditions and the spheroid number was monitored for 9 days. (D) Control, STAT3C, p63-sgRNA and STAT3C + p63-sgRNA-expressing cells were seeded on Matrigel-coated membranes for an invasion assay, and the number of invading cells was counted after 24 h. (E) HSC-5 cells infected with lentivirus expressing ∆Np63α or empty vector were treated with STATTIC or DMSO and lysates were collected for immunoblot. (F) Control- and p63-sgRNA ± CBX4-expressing cells were harvested for protein and seeded in spheroid conditions (G), or invasion assays (H) and spheroid number and rate of invasion were monitored, respectively, *, **, P < 0.05 (the double asterisk indicates significance compared with the single asterisk group).
Discussion
In the CSC model, a fraction of cancer cells in the tumor are capable of initiating and sustaining tumor growth (38). CSCs possess many of the same features as normal stem cells, including their slow-cycling nature and the ability to produce daughter cells with varying degrees of differentiation that contribute to tumor heterogeneity (38). Additionally, CSCs possess enhanced resistance to apoptosis and have metastatic potential (4). The CSC population represents a major therapeutic obstacle due to the ability of CSCs to resist therapy, metastasize and recur. Identification of signaling cascades critical to the maintenance of this cell population can lead to the development of therapeutic strategies that prevent metastasis, drug resistance and recurrence, thus improving patient survival.
The TME is composed of extracellular matrix (ECM), cytokines and stromal cells (39). The TME varies depending on the tissue of origin and regulates various aspects of cancer progression. An important aspect of the TME is the stem cell niche, which provides regulatory cues to CSCs (40). CAFs have emerged as a key factor of the CSC niche that regulates CSCs (41). CAFs are capable of crosstalk with CSCs through the secretion of ECM and growth factors that regulate the CSC phenotype (40). Therefore, it is essential to identify how CAFs alter CSC signaling to contribute to their aggressive phenotype. In this study, we reveal that CAFs derived from SCC are capable of driving an aggressive CSC phenotype in SCC cells. We show that the culture of SCC cells with CAF-conditioned media enhances the rate of spheroid formation, invasion and migration. Additionally, this is associated with increased expression of the canonical stem cell marker SOX2, which is overexpressed in several cancer types (42,43), as well as ΔNp63α, which is an essential regulator of epithelial stem cells and frequently overexpressed in cancer as well (32). We had shown previously a number of chromatin-modifying proteins are critical to regulating the CSC population of SCC (5,6). Therefore, we screened for chromatin-modifying proteins that have altered expression in response to CAF-conditioned media, identifying CBX4 as being enriched.
CBX4 is a chromobox family member within the PcG family of proteins (10). In normal human skin, CBX4 protects epithelial stem cells from senescence and controls differentiation, and has been implicated in cancer progression (8,9,11,12,36,44). Our gene editing screens identify CBX4 as critical to the aggressive phenotype, with CBX4 depletion impairing spheroid formation, invasion and migration. To further explore CBX4 as a potential therapeutic target in SCC CSCs, we utilized UNC3866, a small molecule inhibitor of CBX4. Treatment with UNC3866 results in reduced spheroid number and impaired invasion and migration. Importantly, tumor growth in vivo is impaired by UNC3866 as well. This data have important translational implications, as it demonstrates that therapeutic inhibition of CBX4 can target SCC CSCs. Because of the role of CBX4 in driving the aggressive phenotype, understanding how CAFs are regulating CBX4 expression was an important goal. This prompted us to look at the secretome of CAFs, and here we show that IL-6 is highly expressed. IL-6 is capable of driving an aggressive CSC phenotype measured by migration, invasion and spheroid formation in multiple SCC cell lines. Importantly, the addition of IL-6 to the media-induced CBX4 expression, shows that IL-6 can regulate this important chromatin-modifying protein. Moreover, we show that inhibition of IL-6 with the anti-IL6 antibody Tocilizumab prevents IL-6-mediated induction of CBX4 and impairs the CSC phenotype. Thus, this data create a case for the use of Tocilizumab in SCC patients.
Further mechanistic studies indicate that these changes in the CSC phenotype are mediated by IL-6 activating JAK2 signaling in a canonical fashion. Treatment with the JAK2 inhibitor AZD1480 prevents IL-6-mediated induction of CBX4, demonstrating IL-6 induces CBX4 through canonical JAK2 signaling. Furthermore, JAK2 inhibition impairs the CSC phenotype that is driven by CAF-conditioned media, providing rationale for yet another therapeutic target involved in CAF/CSC crosstalk. The observation that JAK2 is required for IL-6-driven CBX4 prompted us to look at known mediators of JAK2 activity. We focused on STAT3, as it is downstream of IL-6 and JAK2, and we have shown previously that it is critical for maintaining the CSC phenotype (6). Here we show that STAT3 is downstream from IL-6/JAK2 signaling, and required for CAF induction of CBX4. Overexpression of STAT3 in the presence of JAK2 inhibition is able to sustain CBX4 expression, demonstrating it is a key mediator of IL-6/JAK2-driven CBX4 expression. Furthermore, direct targeting of STAT3 with the small molecule inhibitor STATTIC leads to a reduction in CBX4 protein level. Interestingly, our data suggest that inhibition of JAK2 or STAT3 is more effective at reducing CBX4 levels than inhibition of IL-6 in CAF-CM, suggesting other CAF secretome components that also lead to JAK2 activation may regulate CBX4 expression as well. Therefore, drugs targeting JAK2 and STAT3 may prove more effective, either alone or in combination, than IL-6-targeted therapies, as they can not only impair essential downstream components of IL-6 signaling, but prevent compensatory activation of these pathways by other CAF-secreted cytokines.
To further investigate how STAT3 was regulating CBX4, we looked at the transcription factor p63, a key regulator of CSC survival (32). We have shown previously that STAT3 transcriptionally regulates ΔNp63α and that ΔNp63α is required for the maintenance of CSC spheroid formation, invasion and migration (6). This led us to believe that p63 may mediate STAT3-driven CBX4 expression, an idea consistent with previous work in the context of both thymic epithelial cells and keratinocytes (8,36). Our studies reveal an important link between STAT3, ΔNp63α and CBX4. Pan-p63 depletion using CRISPR results in the loss of CBX4 expression, which can be rescued with expression of the ΔNp63α isoform alone. However, expression of constitutively active STAT3 in p63-depleted cells is incapable of rescuing CBX4 expression. This implicates ΔNp63α as the key mediator between STAT3 and CBX4, and further demonstrates its essential role in the CSC phenotype. The observation that CBX4 drives the CSC phenotype and is downstream of ΔNp63α prompted us to examine whether CBX4 is essential to ΔNp63α-mediated regulation of the CSC phenotype. We show that depletion of ΔNp63α with CRISPR severely impairs the CSC phenotype, and CBX4 provides a partial rescue of spheroid formation and invasion. This supports our previous findings demonstrating ΔNp63α is a master regulator of the CSC phenotype through control of multiple downstream targets, CBX4 being one of them.
Targeting the dynamic interactions between tumor cells and the microenvironment is an important therapeutic strategy. Our data support a model in which CAFs regulate the CSC phenotype through activation of the ΔNp63α/CBX4 axis. Furthermore, the ΔNp63α/CBX4 axis is stimulated by CAF secretion of IL-6, activating an IL-6/JAK2/STAT3 signaling pathway upstream of ΔNp63α and CBX4. These findings highlight a novel CAF-stimulated signaling axis as a potential therapeutic target in SCC to effectively reduce CBX4 and the CSC phenotype. This work has important therapeutic implications in efforts to target the CSC population by impairing proteins essential to their aggressive phenotype.
While there are currently no therapeutic options being tested in the clinic that target ∆Np63α or CBX4 directly, there are several therapeutic strategies targeting upstream IL-6/JAK2/STAT3 signaling that may prove valuable in treating SCC. The anti-IL-6 therapy Tocilizumab is FDA approved for treatment of rheumatoid arthritis and COVID-19, and is currently being investigated in the context of lung (NCT04496674) and head and neck (NCT03708224) SCC. The JAK inhibitor Ruxolitinib is currently used to treat myelofibrosis, and a phase 2 trial is currently underway in head and neck SCC (NCT03153982). Napabucasin is a small molecule inhibitor of STAT3 that is FDA approved for gastric cancers, and is currently in phase 1 clinical trials for use in solid tumors in combination with immune checkpoint inhibitors (NCT02467361). An additional STAT3 inhibitor, TTI-101 is currently in phase 1 trials for advanced solid tumors (NCT03195699), as well as in combination with Pembrolizumab for head and neck SCC (NCT05668949). While additional preclinical studies are needed to fully appreciate all the potential TME components that can regulate the ∆Np63α/CBX4 axis, and the full range of effects this signaling has on the cancer cell population, we believe these clinical trials could provide important insights into the therapeutic potential of targeting this newly identified pathway.
Supplementary Material
Glossary
Abbreviations
- CAFS
cancer-associated fibroblasts
- CSC
cancer stem cell
- IL-6
interleukin-6
- SCC
squamous cell carcinomas
- TME
tumor microenvironment
- JAK
Janus Kinase
- STAT
Signal Transducer and Activator of Transcription
- IgG
Immunoglobulin G
- DMEM
Dulbecco's Modified Eagle Medium
- FBS
Fetal Bovine Serum
- RPMI
Roswell Park Memorial Institute
- PBS
Phosphate Buffered Saline
- CRISPR
clustered regularly interspaced short palindromic repeats
- DAPI
4',6-diamidino-2-phenylindole
- qRT-PCR
Quantitative Reverse Transcription Polymerase Chain Reaction
- cDNA
complementary DNA
- mRNA
messenger RNA
- SEM
Standard Error of the Mean
- ANNOVA
Analysis of Variance
Contributor Information
Matthew L Fisher, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Seamus Balinth, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA; Molecular and Cellular Biology Program, Stony Brook University, Stony Brook, NY 11794, USA.
Yon Hwangbo, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Caizhi Wu, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Carlos Ballon, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Gary L Goldberg, Zucker School of Medicine, Hofstra University/Northwell Health, Hempstead, NY 11549, USA.
Alea A Mills, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Funding
This work was supported by the Office of the Director, National Institutes of Health [5P30CA045508 Cancer Center Support Grant], [CA225134 to M.L.F.], [CA247400 to S.B.], as well as [R01CA190997 and R21OD018332 to A.A.M.]. This project was also supported through the Cold Spring Harbor Laboratory and Northwell Health Affiliation.
Conflict of Interest Statement: None declared.
Data availability
All experimental data necessary to evaluate the conclusions of this research are included in the published article and its supplementary files. The datasets and materials in this study are available on reasonable request from corresponding authors.
References
- 1. Sánchez-Danés, A., et al. (2018) Deciphering the cells of origin of squamous cell carcinomas. Nat. Rev. Cancer, 18, 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Marur, S., et al. (2016) Head and neck squamous cell carcinoma: update on epidemiology, diagnosis, and treatment. Mayo Clin. Proc., 91, 386–396. [DOI] [PubMed] [Google Scholar]
- 3. Specenier, P., et al. (2018) Optimizing treatments for recurrent or metastatic head and neck squamous cell carcinoma. Expert Rev. Anticancer Ther., 18, 901–915. [DOI] [PubMed] [Google Scholar]
- 4. Batlle, E., et al. (2017) Cancer stem cells revisited. Nat. Med., 23, 1124–1134. [DOI] [PubMed] [Google Scholar]
- 5. Balinth, S., et al. (2022) EZH2 regulates a SETDB1/ΔNp63α axis via RUNX3 to drive a cancer stem cell phenotype in squamous cell carcinoma. Oncogene, 41, 4130–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fisher, M.L., et al. (2021) BRD4 regulates transcription factor ΔNp63α to drive a cancer stem cell phenotype in squamous cell carcinomas. Cancer Res., 81, 6246–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Keyes, W.M., et al. (2011) ΔNp63α is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell, 8, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mardaryev, A.N., et al. (2016) Cbx4 maintains the epithelial lineage identity and cell proliferation in the developing stratified epithelium. J. Cell Biol., 212, 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu, C., et al. (2020) CBX4 promotes the proliferation and metastasis via regulating BMI-1 in lung cancer. J. Cell. Mol. Med., 24, 618–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van Wijnen, A.J., et al. (2021) Biological functions of chromobox (CBX) proteins in stem cell self-renewal, lineage-commitment, cancer and development. Bone, 143, 115659. [DOI] [PubMed] [Google Scholar]
- 11. Wang, Z., et al. (2021) Chromobox 4 facilitates tumorigenesis of lung adenocarcinoma through the Wnt/β-catenin pathway. Neoplasia, 23, 222–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zeng, J.S., et al. (2018) CBX4 exhibits oncogenic activities in breast cancer via Notch1 signaling. Int. J. Biochem. Cell Biol., 95, 1–8. [DOI] [PubMed] [Google Scholar]
- 13. Hinshaw, D.C., et al. (2019) The tumor microenvironment innately modulates cancer progression. Cancer Res., 79, 4557–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Denton, A.E., et al. (2018) Stromal cells in the tumor microenvironment. Adv. Exp. Med. Biol., 1060, 99–114. [DOI] [PubMed] [Google Scholar]
- 15. Sahai, E., et al. (2020) A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer, 20, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Biffi, G., et al. (2021) Diversity and biology of cancer-associated fibroblasts. Physiol. Rev., 101, 147–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson, D.E., et al. (2018) Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol., 15, 234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kumari, N., et al. (2016) Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol., 37, 11553–11572. [DOI] [PubMed] [Google Scholar]
- 19. Tanaka, T., et al. (2014) IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol., 6, a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hillion, J., et al. (2008) The high-mobility group A1a/signal transducer and activator of transcription-3 axis: an achilles heel for hematopoietic malignancies? Cancer Res., 68, 10121–10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chatterjee, A., et al. (2008) U-box-type ubiquitin E4 ligase, UFD2a attenuates cisplatin mediated degradation of ΔNp63α. Cell Cycle, 7, 1231–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cruz-Bermúdez, A., et al. (2019) Cancer-associated fibroblasts modify lung cancer metabolism involving ROS and TGF-β signaling. Free Radic. Biol. Med., 130, 163–173. [DOI] [PubMed] [Google Scholar]
- 23. Wen, S., et al. (2019) Cancer-associated fibroblast (CAF)-derived IL32 promotes breast cancer cell invasion and metastasis via integrin β3-p38 MAPK signalling. Cancer Lett., 442, 320–332. [DOI] [PubMed] [Google Scholar]
- 24. Zhai, J., et al. (2019) Cancer-associated fibroblasts-derived IL-8 mediates resistance to cisplatin in human gastric cancer. Cancer Lett., 454, 37–43. [DOI] [PubMed] [Google Scholar]
- 25. Rizzino, A., et al. (2016) Sox2/Oct4: a delicately balanced partnership in pluripotent stem cells and embryogenesis. Biochim. Biophys. Acta, 1859, 780–791. [DOI] [PubMed] [Google Scholar]
- 26. Blanpain, C., et al. (2007) p63: revving up epithelial stem-cell potential. Nat. Cell Biol., 9, 731–733. [DOI] [PubMed] [Google Scholar]
- 27. Sebba, A.T. (2008) the first interleukin-6-receptor inhibitor. Am. J. Health Syst. Pharm., 65, 1413–1418. [DOI] [PubMed] [Google Scholar]
- 28. Garbers, C., et al. (2015) The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr. Opin Immunol., 34, 75–82. [DOI] [PubMed] [Google Scholar]
- 29. Heinrich, P.C., et al. (1998) Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J., 334, 297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hedvat, M., et al. (2009) The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell, 16, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Candi, E., et al. (2008) p63 in epithelial development. Cell. Mol. Life Sci., 65, 3126–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fisher, M.L., et al. (2020) p63-related signaling at a glance. J. Cell Sci., 133, 228015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fisher, M.L., et al. (2017) Sulforaphane reduces YAP/ΔNp63α signaling to reduce cancer stem cell survival and tumor formation. Oncotarget, 8, 73407–73418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fisher, M.L., et al. (2016) Transglutaminase interaction with α6/β4-integrin stimulates YAP1-dependent ΔNp63α stabilization and leads to enhanced cancer stem cell survival and tumor formation. Cancer Res., 76, 7265–7276. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 35. Grun, D., et al. (2018) NRP-1 interacts with GIPC1 and α6/β4-integrins to increase YAP1/ΔNp63α-dependent epidermal cancer stem cell survival. Oncogene, 37, 4711–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu, B., et al. (2013) Cbx4 regulates the proliferation of thymic epithelial cells and thymus function. Development, 140, 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schust, J., et al. (2006) A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol., 13, 1235–1242. [DOI] [PubMed] [Google Scholar]
- 38. Chang, J.C. (2016) Cancer stem cells: role in tumor growth, recurrence, metastasis, and treatment resistance. Medicine (Baltimore), 95, S20–S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weber, C.E., et al. (2012) The tumor microenvironment. Surg. Oncol., 21, 172–177. [DOI] [PubMed] [Google Scholar]
- 40. Plaks, V., et al. (2015) The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell, 16, 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lacina, L., et al. (2015) Cancer microenvironment: what can we learn from the stem cell niche. Int. J. Mol. Sci., 16, 24094–24110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Novak, D., et al. (2020) SOX2 in development and cancer biology. Semin. Cancer Biol., 67, 74–82. [DOI] [PubMed] [Google Scholar]
- 43. Chaudhary, S., et al. (2019) Sox2: a regulatory factor in tumorigenesis and metastasis. Curr. Protein Pept. Sci., 20, 495–504. [DOI] [PubMed] [Google Scholar]
- 44. Luis, N.M., et al. (2011) Regulation of human epidermal stem cell proliferation and senescence requires polycomb-dependent and -independent functions of Cbx4. Cell Stem Cell, 9, 233–246. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental data necessary to evaluate the conclusions of this research are included in the published article and its supplementary files. The datasets and materials in this study are available on reasonable request from corresponding authors.