

Abstract
Protein phosphorylation, which regulates many critical aspects of cell biology, is dynamically governed by kinases and phosphatases. Many diseases are associated with dysregulated hyperphosphorylation of critical proteins, such as retinoblastoma protein in cancer. Although kinase inhibitors have been widely applied in the clinic, growing evidence of off-target effects and increasing drug resistance prompts the need to develop a new generation of drugs. Here, we propose a proof-of-concept study of Phosphorylation TArgeting Chimeras (PhosTACs). Similar to PROTACs in their ability to induce ternary complexes, PhosTACs focus on recruiting a Ser/Thr phosphatase to a phosphosubstrate to mediate its dephosphorylation. However, distinct from PROTACs, PhosTACs can uniquely provide target gain-of-function opportunities to manipulate protein activity. In this study, we applied a chemical biology approach to evaluate the feasibility of PhosTACs by recruiting the scaffold and catalytic subunits of the PP2A holoenzyme to protein substrates such as PDCD4 and FOXO3a for targeted protein dephosphorylation. For FOXO3a, this dephosphorylation resulted in transcriptional activation of a FOXO3a-responsive reporter gene.
Graphical Abstract
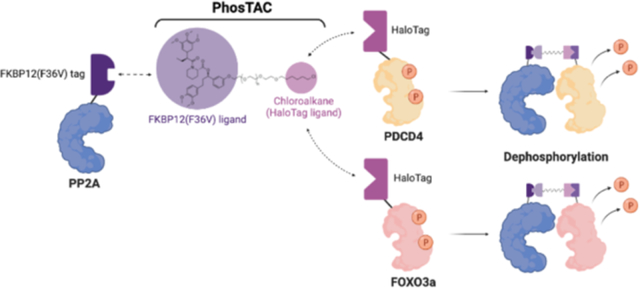
INTRODUCTION
Dynamically and reversibly mediated by kinases1 and phosphatases2, phosphorylation can occur on proteins, sugars, lipids or metabolites and is one of the most critical posttranslational modifications (PTMs). While the human genome encodes more than 500 protein kinases (Ser/Thr/Tyr kinase), only ~137 protein phosphatases have been identified. It has been estimated that there are more than 10,000 experimentally identified phosphorylated proteins and 86,000 protein phospho-sites in the human proteome.3 Protein phosphorylation not only directly regulates many important biological processes but can also crosstalk and/or compete with other PTMs on the same protein to fine-tune biological function.4
Protein dephosphorylation is governed by 107 protein tyrosine phosphatases (PTPs) and ~30 protein serine/threonine phosphatases (PSPs). Given their relatively low number in the human genome and the dominant ratio of pSer/pThr (pSer/pThr/pTyr, ~86%/12%/2%) in mammalian cell lines,5 PSPs were once considered to be promiscuous in substrate selectivity. However, it is now clear that some PSPs use accessory regulatory subunits to determine their substrate specificity.6, 7 One of the well-known examples is PP2A phosphatase, which acts as a holoenzyme with one catalytic subunit (PP2A C); one of 26 regulatory subunits (PP2A B); and one scaffold subunit (PP2A A).8 It is known that the regulatory subunit determines the substrate specificity by directly interacting with substrates and recruiting the other subunits of the holoenzyme. In this way, PP2A-mediated dephosphorylation is controlled in a spatial and temporal manner by the expression of the regulatory subunits. Similar modes of regulation are also observed for other phosphoprotein phosphatase (PPP) family members.6
Many diseases are associated with dysregulated protein phosphorylation. One well-known example is Alzheimer’s disease, which is characterized by Tau hyperphosphorylation. Aberrant Tau phosphorylation correlates with filament formation, microtubule dysfunction, and neuronal death9,. Indeed, several phosphorylated Tau peptides were used to generate therapeutic vaccines for preclinical studies.10 Many kinases and phosphatases have been reported to regulate Tau phosphorylation. For example, conditional knockout of murine B56δ, the PP2A regulatory subunit in neural tissue, increased Tau phosphorylation at Ser202/Thr205 and Thr231.11 Another classic example of pathogenic dysregulation of protein phosphorylation is the inactivation of the tumor suppressor protein retinoblastoma (Rb) by hyperphosphorylation. Upon Rb phosphorylation by cyclin-dependent kinases (CDK), transcription factor E2F is released from Rb and is free to initiate transcription programs for the synthesis phase. However, this mechanism is often hijacked by tumor cells. Amplification mutations of CDK4/6 as well as inactivating mutations of CDKN2A, a negative regulator of CDK4/6, often lead to hyperphosphorylation and inactivation of Rb in many cancers.12, 13 Currently, kinase inhibitors are widely used in the clinic to decrease phosphorylation of particular kinase substrate proteins. As an alternative approach, phosphatase activation is promising complementary method to down regulate phosphorylation.14, 15 However, given that many phosphatases govern multiple signaling pathways simultaneously, global activation of any particular phosphatase may lead to unfavorable outcomes in various signaling pathways.
In this study, we developed a new approach different than non-discriminatorily activating phosphatase activity to effect widespread substrate dephosphorylation. This strategy, called “PhosTACs”: Phosphorylation TArgeting Chimera, uses heterobifunctional molecules to mediate complex formation between a phosphatase and a targeted phosphoprotein to affect focused, targeted dephosphorylation of the latter. This is an adaptation of our PROTAC technology, wherein bifunctional small molecules alter target protein ubiquitination to induce degradation. PROTACs have demonstrated advantages over traditional inhibitors, such as a catalytic mechanism16, high selectivity17, and an extended period of effect.18 By recruiting a phosphatase, rather than an E3 ligase, to a desired substrate protein we hypothesize that induced proximity will lead to substrate protein dephosphorylation with similar mechanistic advantages.
Herein, we provide proof-of-concept evidence for our targeted protein dephosphorylation strategy. Using chemical biology approaches, we provide evidence that by recruiting the 65 kDa scaffold subunit A of serine/thereonine-protein phosphatase 2A (PP2A A), we can dephosphorylate phosphorylated POIs in cells.
RESULTS AND DISCUSSION
Proof-of-Concept of FKBP12(F36V) ligand-mediated Phosphorylation Targeting Chimera.
To test the feasibility of proximity-induced dephosphorylation, we employed the FKBP12(F36V) and HaloTag7 (abbreviated as halo) fusion protein systems described in our previous E3 ligase exploration study of the PROTAC technology.19 Given that PP2A is the most abundant protein phosphatase in mammalian cells, we generated a PP2A phosphatase construct that expresses the scaffold subunit (PP2A A) fused with FKBP12(F36V). Additionally, we generated Halotag7 fusions with PDCD4 (Programmed Cell Death 4) and FOXO3a (Forkhead-box O3a transcription factor) as two phosphorylated substrate candidates since both are key tumor suppressors of well-characterized oncogenic pathways. Phosphorylation of PDCD4 by Akt and RSK (p90 ribosomal S6 kinase) negatively correlates with its ability to inhibit translation20–22. However, the native phosphatase responsible for PDCD4 dephosphorylation has not yet been reported. On the other hand, FOXO3a phosphorylation by Akt or casein kinase 1a inhibits its transactivation activity23, 24, and PP2A has been reported to dephosphorylate FOXO3a.25 Given that PP2A holoenzymes regulate the majority of Ser/Thr phosphorylation events, we proposed that deliberate recruitment of PP2A A to PDCD4 or FOXO3a may induce their dephosphorylation. Stable cell lines expressing both FKBP12(F36V)-PP2A A and Halo-PDCD4 or Halo-FOXO3a were generated using the FlpIn system and lentiviral transduction in HeLa cells (Figure 1A). Meanwhile, we synthesized a series of heterobifunctional molecules with a FKBP12(F36V) ligand and a HaloTag7 ligand (chloroalkane) connected by linkers of varying numbers of polyethylene glycol (PEGs) groups. For example, PhosTAC2 indicates a PhosTAC molecule composed of a linker with 2 PEG units. In addition, the non-HaloTag-binding fluoroalkane PhosTAC7F was used as an inactive PhosTAC control (Figure 1B).26
Figure 1.
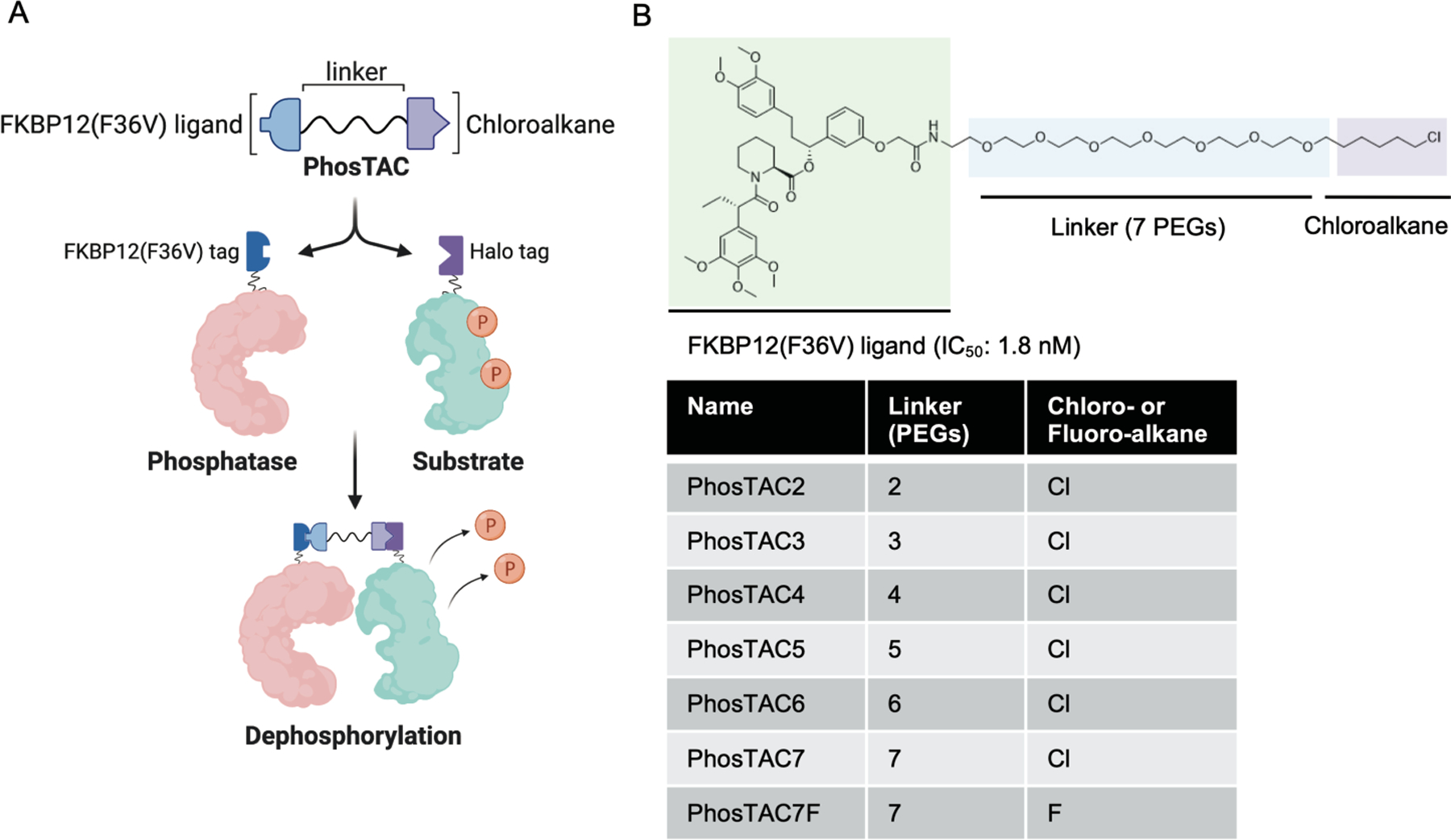
Proof-of-Concept of phosphorylation targeting chimeras (PhosTACs). (a) Design of PhosTACs and mechanism of action. (b) Chemical structure of PhosTACs and controls.
PhosTAC mediates ternary complex formation.
To test whether PhosTACs can mediate ternary complex formation between phosphatases and protein substrates, we performed a Halotrap pulldown assay after treatment of PP2A A/PDCD4 stably-expressing cells with a panel of PhosTACs varying in linker length (PEG 2–7) (Figure 1B). While we did not observe PDCD4 and PP2A A interaction in the vehicle control group, PhosTAC-mediated PP2A A and PDCD4 interaction was strongly favored by longer linker PhosTACs, PhosTAC6 and 7 (Figure 2A). As a control experiment, we used a mutant (P179R) form of the scaffold subunit PP2AA that can no longer interact with the catalytic and regulatory subunits, thereby abrogating PP2A holoenzyme formation.27 We verified that both wild type and P179R mutant FKBP12(F36V)-PP2A A stably interact with Halo-PDCD4 in the presence of PhosTAC7 but not with DMSO, PhosTAC2, or non-HaloTag7 binding PhosTAC7F (fluoroalkane). These data suggest that PhosTAC7 induces specific and stable ternary protein complex (Figure 2B). Moreover, we also observed that by recruiting the scaffold subunit (PP2A A), PhosTAC7 can recruit endogenous catalytic subunit (PP2A C) of PP2A as well (Figure 2C). Overall, we have established a tractable system for PhosTAC-induced ternary complex for phosphatases and protein substrates, where PhosTACs with longer linkers, up to 7 PEGs, strongly favor ternary complex formation.
Figure 2.
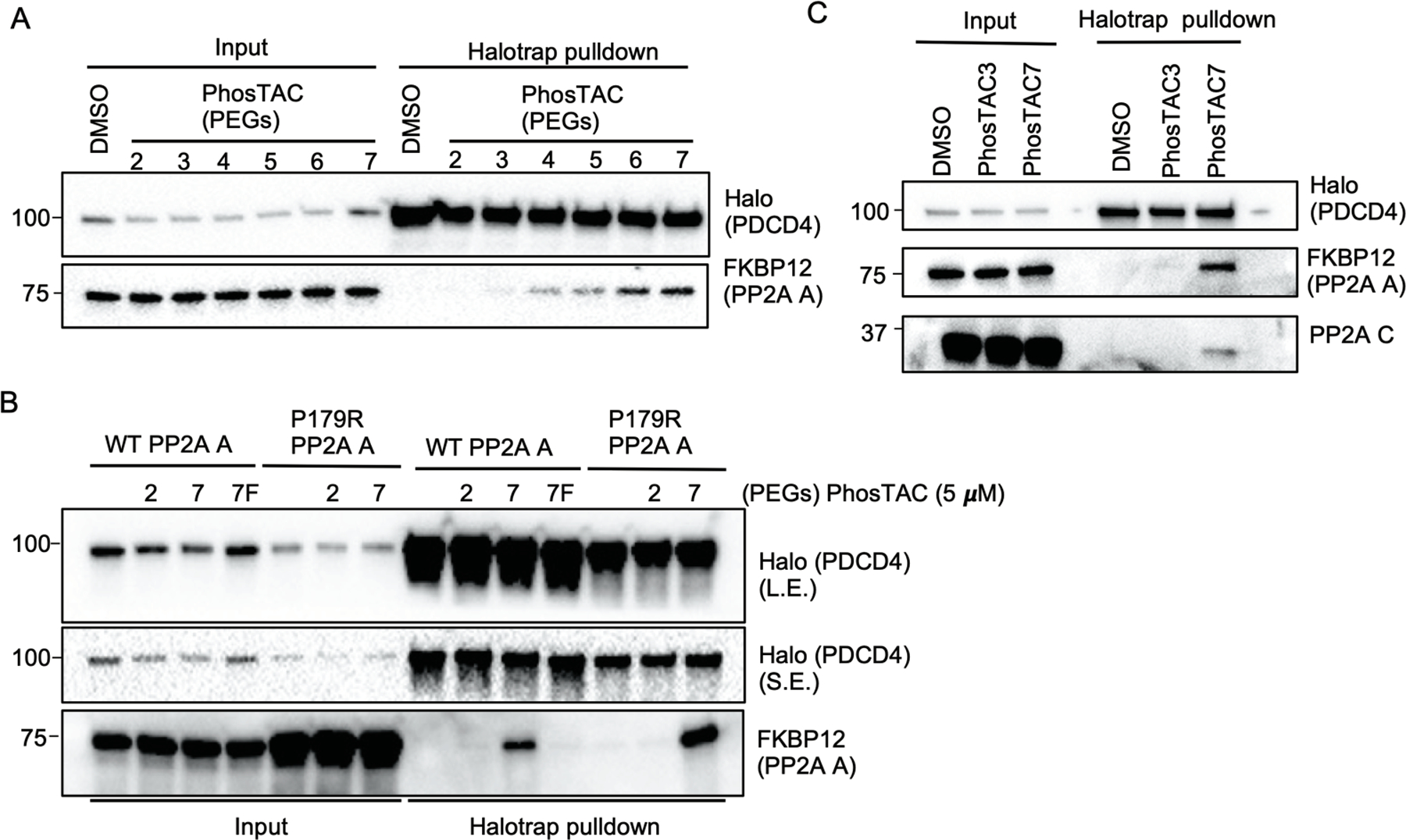
PhosTACs mediate ternary complex formation between Halo-PDCD4 and FKBP12(F36V)-PP2A A. (a) PhosTACs form a stable ternary complex with Halo-PDCD4 and FKBP12(F36V)-PP2A A in a linker length dependent manner. Halo-PDCD4/FKBP12(F36V)-PP2A A wild type HeLa cells were treated with indicated PhosTACs at a concentration of 5 μM for 24h, lysed for Halotrap pulldown, and analyzed by Western blot using indicated antibodies. (b) Inactive PhosTAC7F does not form ternary complex with Halo-PDCD4 and Halo-PP2A A. Halo-PDCD4/FKBP12(F36V)-PP2A A wild type or P179R mutant HeLa cells treated with indicated PhosTACs at a concentration of 5 μM for 24h, lysed for Halotrap pulldown, and analyzed by Western blot using indicated antibodies. (L.E.: long exposure; S.E.: short exposure) (c) PhosTAC7 efficiently recruits PP2A A and C subunits to Halo-PDCD4. Halo-PDCD4/FKBP12(F36V)-PP2A A wild type HeLa cells treated with indicated PhosTACs at a concentration of 5 μM for 24h, lysed for Halotrap pulldown, and analyzed by Western blot using indicated antibodies.
Recruitment of PP2A A subunit dephosphorylates PDCD4.
Given that PhosTAC7 provides optimal induced proximity between PP2A phosphatase and PDCD4, we evaluated substrate dephosphorylation efficiency after incubating cells with PhosTAC7. PhosTAC7-treated cells induced PDCD4 dephosphorylation at serine 67 in a dose-dependent manner, reaching 50% dephosphorylation at 10 uM (DePhos50 value) after 12 hours of incubation (Figure 3A). On the other hand, PhosTAC7F, which cannot facilitate ternary complex formation (Figure 2B) failed to induce PDCD4 dephosphorylation (Figure 3A). Additionally, the P179R mutant form of PP2A A, while able to form a ternary complex with PDCD4, could not induce PDCD4 dephosphorylation due to defective protein-protein-interactions between P179R PP2A A and the catalytic and regulatory subunits27 (Figure 3B). In testing the kinetics of PhosTAC7-mediated dephosphorylation, we noticed a significant reduction of PDCD4 phosphorylation at serine 67 and 457 after 8 hours of PhosTAC7 incubation that reached maximum dephosphorylation at 90% (DePhosMax value) at ~16 hours of incubation (Figure 3C). Collectively, these data demonstrated that PhosTAC provides on-target and efficient PDCD4 dephosphorylation. It is worth noting that a biologically relevant phosphatase or particular PP2A-B subunit for PDCD4 has not yet been identified, although future proteomic studies may provide further insights regarding the native PDCD4 phosphatase.
Figure 3.
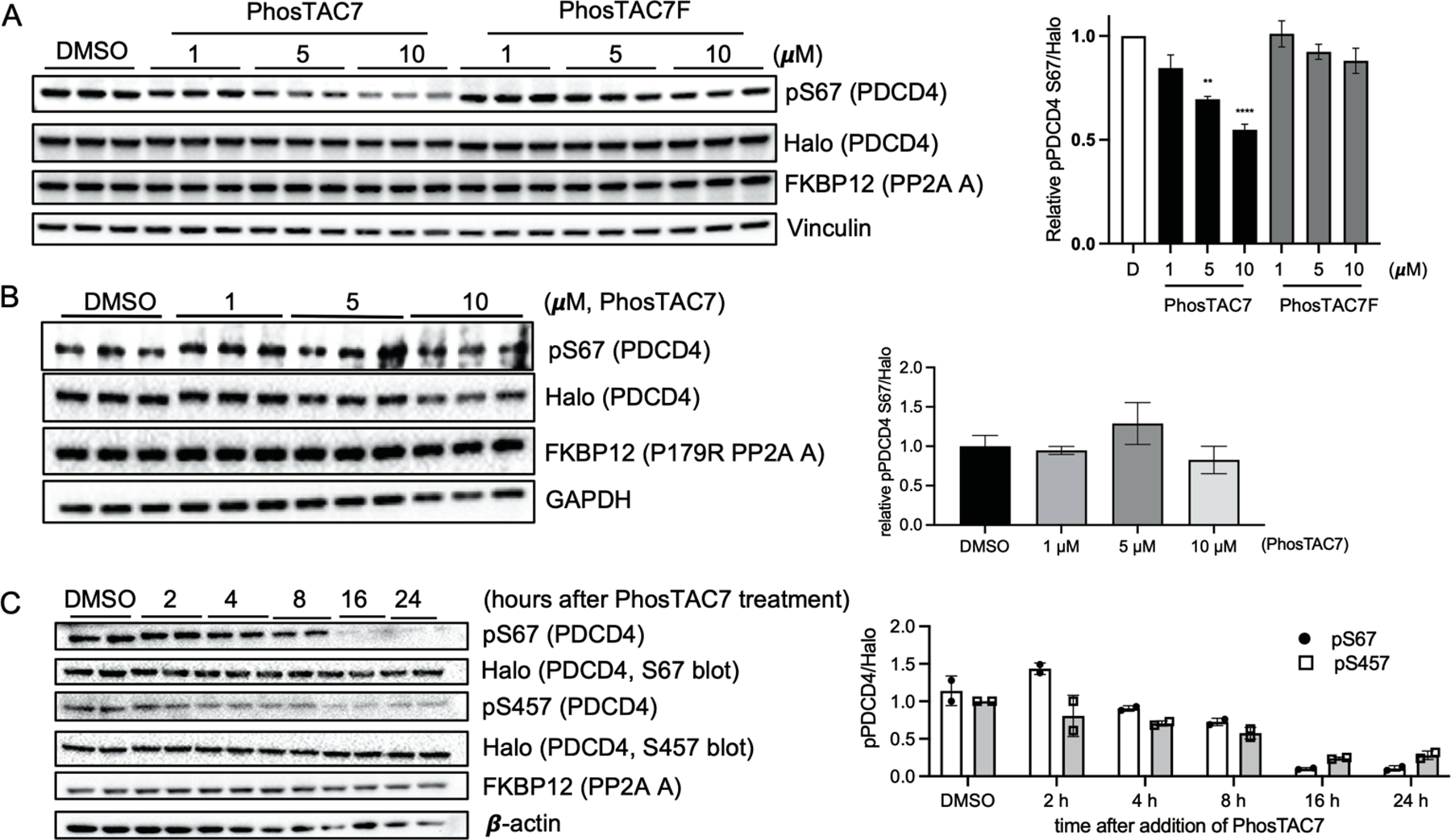
Recruitment of PP2A A subunit induces dephosphorylation of PDCD4. (a) PhosTAC7 but not inactive PhosTAC7F induces PDCD4 dephosphorylation at serine 67. Halo-PDCD4/FKBP12(F36V)-PP2A A wild type HeLa cells were treated with indicated concentrations of PhosTAC7, PhosTAC7F, or DMSO. Cell lysates were collected 12 h after treatment and analyzed by Western blot using indicated antibodies. Data were quantified from three biological samples and summarized as mean and standard error of the mean. (**p= 0.0016, ****p<0.0001, one-way ANOVA) (b) PP2AA(P179R) mutant cannot induce PDCD4 dephosphorylation at serine 67. Halo-PDCD4/FKBP12(F36V)-PP2A A P179R mutant HeLa cells were treated with indicated concentrations of PhosTAC7 or DMSO. Cell lysates were collected 12 h after treatment and analyzed by Western blot using indicated antibodies. Data were quantified from three biological replicates and summarized by their means and standard deviations. (c) Dephosphorylation kinetics of PDCD4 by PhosTAC7. Halo-PDCD4/FKBP12(F36V)-PP2A A wild type HeLa cells were treated with DMSO or PhosTAC7 (5 μM). Cell lysates were collected at indicated time points after treatment and analyzed by Western blot using indicated antibodies. Data were quantified from two biological replicates and summarized as means and standard deviations.
Recruitment of PP2A A subunit alters phosphorylation kinetics of FOXO3a.
Based on information from our PP2A/PDCD4 experiments, we next confirmed that PhosTAC7 can also induce ternary complexes of the PP2A A fusion protein with other another fusion protein substrate, FOXO3a (Figure S1A). We also observed ternary complex formation in the presence of PhosTAC3 to a lesser extent and no complex formation upon treatment with DMSO or PhosTAC7F. With PhosTAC7, we observed about a 30% of FOXO3a dephosphorylation at serine 318/321 when normalized to its total protein level (Figure S1B). It was observed that the total protein level of FOXO3a fluctuates with phosphorylation status. In particular, serum stimulation drastically increases FOXO3a phosphorylation at serine 318/321 and serine 253 and stabilizes the protein but the serum-induced FOXO3a phosphorylation was reversed by the treatment of PhosTAC7. Indeed, FOXO3a phosphorylation status at specific residues has been shown to affect overall FOXO3a protein stability.28, 29 Importantly, PhosTAC7-mediated FOXO3a dephosphorylation can be reverted by okadaic acid treatment, a potent PP2A inhibitor (Figure S1C). We next turned to examine FOXO3a phosphorylation kinetics in the presence of PhosTACs. We found that upon EGF challenge, FOXO3a phosphorylation at serine 318/321 increased by 50% in both DMSO-(Figure S1D) or PhosTAC7F (Figure 4A) pre-treatment, while PhosTAC7 pre-treated samples maintained a steady phosphorylation level throughout the time course of EGF challenge. These data suggest that FOXO3a phosphorylation kinetics at serine 318/321 sites were regulated through PhosTAC recruitment of the PP2A phosphatase fusion protein. Since FOXO3a serine 318/321 phosphorylation inhibits its transcriptional activity,24, 30 we sought to determine whether PhosTAC-mediated FOXO3a dephosphorylation has any biological significance. We co-transfected a FHRE (forkhead response element) firefly luciferase reporter and a renilla luciferase coreporter as an internal control into cells expressing FKBP12(F36V)-PP2A A and Halo-FOXO3a. The FHRE firefly luciferase reporter contains three copies of FHRE cloned from the endogenous Fas ligand promoter.23 The transactivation activity of FOXO3a will induce FHRE luciferase expression. Consistent with our hypothesis, treatment with PhosTAC7 significantly increased FOXO3a transactivation activity when compared with DMSO or PhosTAC7F (Figure 4B). Treatment with wortmannin, a PI3K inhibitor that causes suppression of Akt-mediated FOXO3a phosphorylation also demonstrated increased transactivation of FOXO3a (Figure 4C). Interestingly, inhibition of PP2A phosphatase activity using okadaic acid completely suppressed PhosTAC7-induced transactivation of FOXO3a (Figure 4D). That okadaic acid by itself reduced the FHRE-driven luciferase expression is consistent with previous reports that PP2A serves as a native phosphatase for FOXO3a.25 In summary, our results demonstrated that through PhosTAC7-induced proximity, recruited PP2A can efficiently alter FOXO3a phosphorylation kinetics at serine 318/321 and is associated with its upregulation of transactivation activity.
Figure 4.
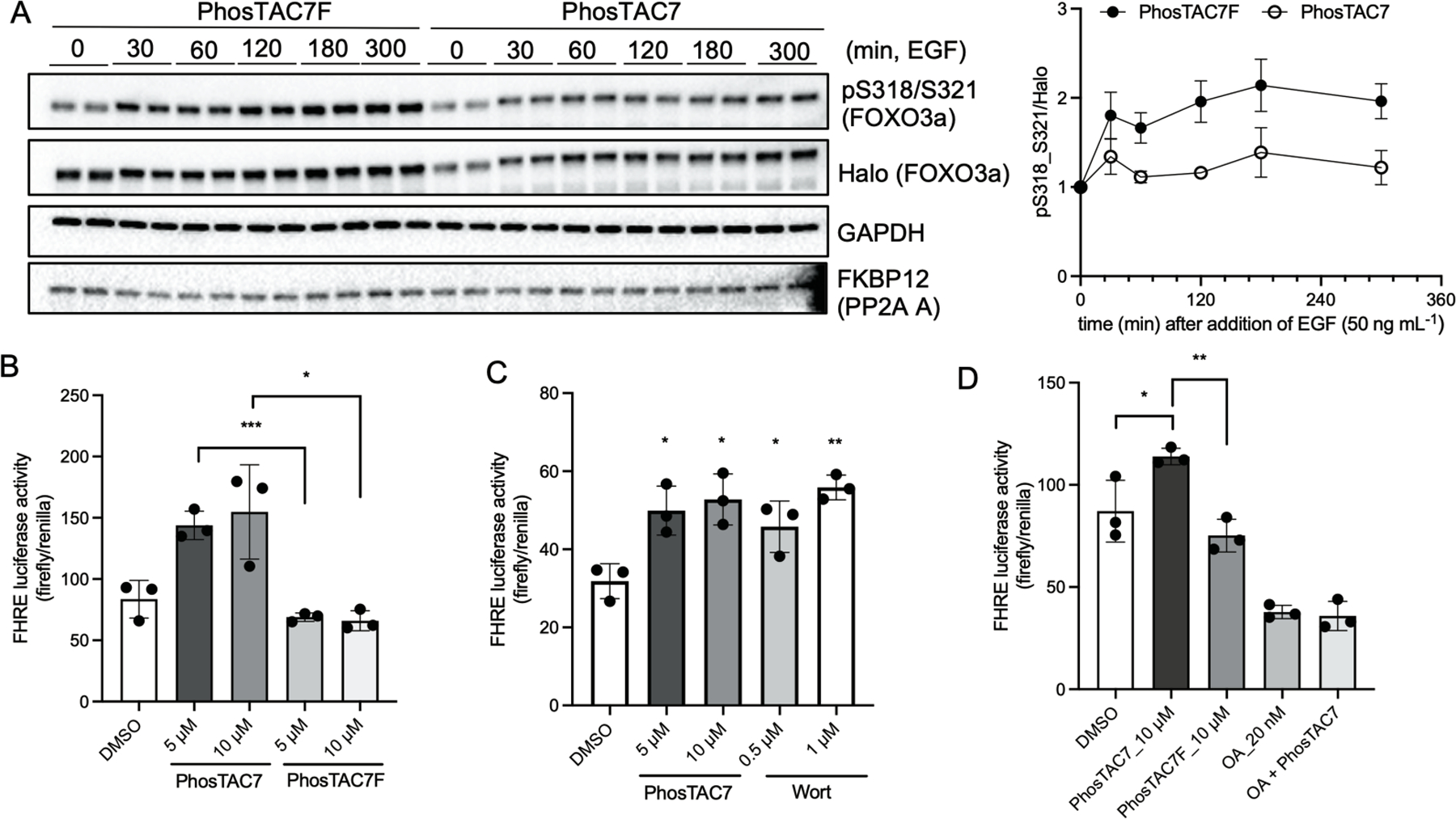
PhosTAC7 alters phosphorylation kinetics and transactivation activity of FOXO3a. (a) PhosTAC7 but not inactive PhosTAC7 alters the phosphorylation kinetics of FOXO3a serine 318/321. Halo-FOXO3a/FKBP12(F36V)-PP2A A wild type HeLa cells were serum starved for 17 hours and pre-treated with PhosTAC7 (5 μM) or PhosTAC7F (5 μM) in serum-free media for additional 3 hours before stimulating the cells with of 50 ng mL−1 EGF (epidermal growth factor). Cell lysates were collected after EGF treatment and analyzed by Western blot using indicated antibodies. Right panel: Data were quantified from four biological replicates and summarized as means and standard deviations. (b-d) PhosTAC7 increases FOXO3a transactivation activity. FHRE luciferase activity was normalized by renilla in each group. Data were presented as means and standard deviations. (*p<0.05, **p<0.01, ***p<0.001, unpaired, two-tailed, Student’s t-test)(Wort: wortmannin; OA: okadaic acid)
CONCLUSIONS
Due to aberrant kinase hyperactivation that can lead to numerous diseases, kinase inhibitors are a common therapy in the clinic for treatment of cancer and other diseases. Although kinase inhibitors are initially effective, acquired resistance to those inhibitors often occurred in many clinical cases. For example, gefitinib, which targets the ATP-binding site of EGFR, was initially effective for NSCLC patients with EGFR L858R mutation, but the majority of patients acquired resistance due to a secondary mutation (T790M) within EGFR, or in other signaling pathways.31, 32 Similarly, more than 16% of chronic myeloid leukemia patients acquired T315I mutation in BCR-ABL after treatment with imatinib33, thereby conferring resistance to the drug. Furthermore, acquired resistance to Ser/Thr kinase CDK4/6 inhibitors such as ribociclib has also been reported34. Also, because most kinase inhibitors target the ATP-binding sites of kinases, they often show off-target toxicity via their interaction with other kinases. Thus, developing an alternative strategy to target dysregulated kinase activity in clinical application is urgently needed.
A recent study by Yamazoe and colleagues has demonstrated the catalytic subunit of protein phosphatase 1 (PPP1CA) can be harnessed for induced dephosphorylation of Akt and EGFR.35 In another study, PP2A Bα was recruited to dephosphorylate Tau through heterobifunctional peptides.36 However, many questions related to targeted protein dephosphorylation remain poorly understood. Here, we used a chemical biology approach that complements previous studies and further provides a foundation for a tractable targeted protein dephosphorylation platform.
Small-molecule kinase inhibitors negate POI function via occupying pockets on target proteins. Since their effect is lost upon dissociation from the POI, this is referred to as the “occupancy-driven model”. The protein inhibition is stoichiometric: one inhibitor molecule can inhibit only one target protein molecule. On the other hand, we envision that PhosTACs work via the “event-driven model” similar to PROTACs: one PhosTAC molecule can cycle through many rounds of induced target dephosphorylation, which leads to the removal of phosphorylation on protein substrates in super-stoichiometric quantities.16 In summary, we have demonstrated that by directly recruiting the scaffolding subunit A of PP2A and subsequently indirectly recruiting the catalytic subunit C of the PP2A holoenzyme, a PhosTAC-mediated ternary complex formation can induce the dephosphorylation of PDCD4 and FOXO3a, with the latter having measurable biological consequences. Our study suggests that unlike loss-of-function degradation by PROTACs, targeted protein dephosphorylation may provide either a loss or a gain-of-function window to manipulate specific protein functions in a more precise manner. The unique model of action provides PROTACs with enhanced selectivity17, extended period of effect18, and the ability to overcome resistance.37 PhosTACs works through a similar mechanism as PROTACs, and could be of great potential as both a chemical biology tool and a clinical therapeutic approach.
METHODS
Cell culture, reagents, and DNA constructs.
HeLa Flp-In TREx cells (Invitrogen) were cultured and maintained in DMEM, supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) penicillin and streptomycin at 37°C with 5% CO2. Halo-PDCD4 and Halo-FOXO3a lentiviral constructs (pReceiver vector) were purchased from GeneCopoeia. PPP2R1A cDNA (encoding PP2A Aα scaffolding subunit, referred as PP2A A in our study) were fused with Flag-FKBP12(F36V) at its N-terminal end and cloned into pcDNA5/FRT vector (ThermoFisher) by Gibson assembly (NEB, #E5510S). pcDNA5/FRT/Flag-FKBP12(F36V)-PP2A A construct was then co-transfected with pOG44 into HeLa Flp-In TREx cells and selected with hygromycin (200 μg mL−1) to generate FKBP12(F36V)-PP2A A stable cell lines. To generate lentiviral particle, the lentiviral constructs were co-transfected with pMD2.G and psPAX2 by lipofectamine 2000 (ThermoFisher) into 293T cells. The lentiviral particles were then filtered and transduced into HeLa Flp-In TREx cells expressing Flag-FKBP12(F36V)-PP2A A and selected with puromycin (2 μg mL−1) to generate FKBP12(F36V)-PP2A A/Halo-PDCD4 or Halo-FOXO3a cell lines. The same method was applied to generate FKBP12(F36V)-PP2A A P179R/Halo-PDCD4 HeLa stable cells. All cell lines were regularly tested for mycoplasma contamination.
Western blotting and Antibody.
Whole cell lysates were collected and lysed in lysis buffer (150 mM NaCl, 1 mM EDTA, 25 mM Tris-HCl, 1% (v/v) NP40, and 1% (w/v) SDS) supplied with protease (Roche, cOmplete) and phosphatase inhibitor cocktails (Roche, PhosStop). All lysates were then passed through 22G needle 15 times and spun down at 15000 rpm for 20 min before BCA assay. In general, 10–20 μg of protein samples were loaded for SDS-PAGE and blotted by indicated antibodies. Anti-HaloTag (Promega, #G9211), anti-PDCD4 Ser67 (Abcam, #AB73343), anti-PDCD4 Ser457 (ThermoFisher, #200–301-964), anti-FOXO3a (Cell Signaling, #2497), anti-FOXO3a Ser318/321 (Cell Signaling, #9465), anti-FOXO3a Ser253 (Cell Signaling, #9466), anti-GAPDH (Cell Signaling, #2118), anti-FKBP12 (Santa Cruz, #SC-133067), anti-β-actin (Cell Signaling, #4970), anti-a-Tubulin (Cell Signaling, #2144) and anti-PP2AC (Cell Signaling, #2259) antibodies.
Halotrap pulldown.
HeLa stable cells expressing Halo-PDCD4 (or Halo-FOXO3a) and FKBP12(F36V)-PP2A A were treated with DMSO, 5 μM PhosTAC7 or other related compounds in 10 cm dishes. After 24 hours of incubation, cells were washed with cold PBS, and collected in lysis buffer (150 mM NaCl, 1 mM EDTA, 25 mM Tris-HCl, and 1% (v/v) NP40) supplied with protease and phosphatase inhibitor cocktails and passed through 22G needles 7 times. The soluble supernatants were then collected for BCA assay after being spun down at 15000 rpm for 20 min at 4 °C. To perform halotrap pulldown, halotrap agarose (25 μl, Chromotek, #ota20) was prewashed with lysis buffer and incubated with gently rotation with 1 mg HeLa lysates in 500 μl total volume at 4 °C. After 16 hours of incubation, halotrap agarose was spun down at 2500 x g for 5 min at 4°C and washed with 500 μL wash buffer (75 mM NaCl, 0.5 mM EDTA, 12.5 mM Tris-HCl, 0.5% (v/v) NP40) three times. After the last wash, protein samples were eluted by adding 2x sample buffer to the halotrap agarose and boiled at 95 °C for 5 min. The samples were analyzed by Western blot with indicated antibodies.
Luciferase assay.
To test the transactivation activity of FOXO3a in the presence of PhosTAC, we transfected a forkhead responsive element with luciferase reporter (FHRE-Luc, Addgene #1789)23 along with renilla luciferase into HeLa cells expressing FKBP12(F36V)-PP2A A/Halo-FOXO3a. HeLa cells were transfected with FHRE-Luc and renilla at a 20 to 1 ratio by TransIT-LT1 (Mirus, #MIR2300) for 24 hours, and then split into 96 wells for overnight. Cells were seeded and treated in triplicate with DMSO, PhosTAC7 (5 or 10 μM), PhosTAC7F (5 or 10 μM), Wortmannin (1 μM), or okadaic acid (20 nM) as indicated for 24 hours. The luciferase assay was performed with the Dual-Glo luciferase kit (Promega, #E2920) according to manufacturer’s protocol and measured by plate reader (PerkinElmer EnVision 2101 Multilabel Reader).
General protocol for the use of PhosTAC molecules.
PhosTAC molecules were dissolved in dimethyl sulfoxide (DMSO) at 10 mM as stock solution and stored in −20 °C. For the treatment of PhosTACs, serial diluted PhosTACs or control molecules were first added in serum-free or full serum media (supplemented with antibiotics) and incubated with cells for indicated time point in each experiment.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge critical discussion from members of the Crews Lab (especially M. Bond, J. Swartzel, D. Nalawansha, and M. Krone) and Halda Therapeutics. We appreciated J. Hines’ comments on the manuscript. The graphic abstract was created with BioRender.com.
FUNDING
This study is supported by NIH R35 CA197589 and by the Defense Advanced Research Project Agency (HR0011-20-2-0051). This study does not reflect the sponsor’s position or the policy of the Government and no official endorsement was inferred. S. Z. is supported by the NIH Medical Scientist Training Program Training Grant (T32GM136651).
Footnotes
Supporting Information
The Supporting Information is available free of charge at website.
Additional data related to FOXO3a dephosphorylation and chemical synthesis of all PhosTACs are listed in supporting information.
Notes
The authors declare the following conflict of interests: C.M.C. is a shareholder and consultant to Arvinas, Inc., which supports research in his lab. C.M.C is the founder of Halda Therapeutics, LLC, which provides research supports for this project.
REFERENCES
- (1).Graves LM; Duncan JS; Whittle MC; Johnson GL The dynamic nature of the kinome. Biochem. J 2013, 450 (1), 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bertolotti A The split protein phosphatase system. Biochem. J 2018, 475 (23), 3707–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vlastaridis P; Kyriakidou P; Chaliotis A; Van de Peer Y; Oliver SG; Amoutzias GD Estimating the total number of phosphoproteins and phosphorylation sites in eukaryotic proteomes. Gigascience 2017, 6 (2), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hart GW; Slawson C; Ramirez-Correa G; Lagerlof O Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem 2011, 80, 825–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Olsen JV; Blagoev B; Gnad F; Macek B; Kumar C; Mortensen P; Mann M Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127 (3), 635–48. [DOI] [PubMed] [Google Scholar]
- (6).Casamayor A; Arino J Controlling Ser/Thr protein phosphatase PP1 activity and function through interaction with regulatory subunits. Adv. Protein Chem. Struct. Biol 2020, 122, 231–288. [DOI] [PubMed] [Google Scholar]
- (7).Seshacharyulu P; Pandey P; Datta K; Batra SK Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett 2013, 335 (1), 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lambrecht C; Haesen D; Sents W; Ivanova E; Janssens V Structure, regulation, and pharmacological modulation of PP2A phosphatases. Methods Mol. Biol 2013, 1053, 283–305. [DOI] [PubMed] [Google Scholar]
- (9).Johnson GV; Stoothoff WH Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci 2004, 117 (Pt 24), 5721–9. [DOI] [PubMed] [Google Scholar]
- (10).Jadhav S; Avila J; Scholl M; Kovacs GG; Kovari E; Skrabana R; Evans LD; Kontsekova E; Malawska B; de Silva R, et al. A walk through tau therapeutic strategies. Acta Neuropathol. commun 2019, 7 (1), 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Louis JV; Martens E; Borghgraef P; Lambrecht C; Sents W; Longin S; Zwaenepoel K; Pijnenborg R; Landrieu I; Lippens G, et al. Mice lacking phosphatase PP2A subunit PR61/B’delta (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3beta. Proc. Natl. Acad. Sci. U.S.A 2011, 108 (17), 6957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Knudsen ES; Pruitt SC; Hershberger PA; Witkiewicz AK; Goodrich DW Cell Cycle and Beyond: Exploiting New RB1 Controlled Mechanisms for Cancer Therapy. Trends Cancer 2019, 5 (5), 308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Dick FA; Goodrich DW; Sage J; Dyson NJ Non-canonical functions of the RB protein in cancer. Nat. Rev. Cancer 2018, 18 (7), 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Leonard D; Huang W; Izadmehr S; O’Connor CM; Wiredja DD; Wang Z; Zaware N; Chen Y; Schlatzer DM; Kiselar J, et al. Selective PP2A Enhancement through Biased Heterotrimer Stabilization. Cell 2020, 181 (3), 688–701 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Morita K; He S; Nowak RP; Wang J; Zimmerman MW; Fu C; Durbin AD; Martel MW; Prutsch N; Gray NS, et al. Allosteric Activators of Protein Phosphatase 2A Display Broad Antitumor Activity Mediated by Dephosphorylation of MYBL2. Cell 2020, 181 (3), 702–715 e20. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (16).Lai AC; Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discov 2017, 16 (2), 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Testa A; Hughes SJ; Lucas X; Wright JE; Ciulli A Structure-Based Design of a Macrocyclic PROTAC. Angew. Chem. Int. Ed 2020, 59 (4), 1727–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bond MJ; Chu L; Nalawansha DA; Li K; Crews CM Targeted Degradation of Oncogenic KRAS(G12C) by VHL-Recruiting PROTACs. ACS Cent. Sci 2020, 6 (8), 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ottis P; Toure M; Cromm PM; Ko E; Gustafson JL; Crews CM Assessing Different E3 Ligases for Small Molecule Induced Protein Ubiquitination and Degradation. ACS Chem. Biol 2017, 12 (10), 2570–2578. [DOI] [PubMed] [Google Scholar]
- (20).Dorrello NV; Peschiaroli A; Guardavaccaro D; Colburn NH; Sherman NE; Pagano M S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314 (5798), 467–71. [DOI] [PubMed] [Google Scholar]
- (21).Galan JA; Geraghty KM; Lavoie G; Kanshin E; Tcherkezian J; Calabrese V; Jeschke GR; Turk BE; Ballif BA; Blenis J, et al. Phosphoproteomic analysis identifies the tumor suppressor PDCD4 as a RSK substrate negatively regulated by 14–3-3. Proc. Natl. Acad. Sci. U.S.A 2014, 111 (29), E2918–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Palamarchuk A; Efanov A; Maximov V; Aqeilan RI; Croce CM; Pekarsky Y Akt phosphorylates and regulates Pdcd4 tumor suppressor protein. Cancer Res 2005, 65 (24), 11282–6. [DOI] [PubMed] [Google Scholar]
- (23).Brunet A; Bonni A; Zigmond MJ; Lin MZ; Juo P; Hu LS; Anderson MJ; Arden KC; Blenis J; Greenberg ME Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96 (6), 857–68. [DOI] [PubMed] [Google Scholar]
- (24).Cheong JK; Zhang F; Chua PJ; Bay BH; Thorburn A; Virshup DM Casein kinase 1alpha-dependent feedback loop controls autophagy in RAS-driven cancers. J. Clin. Invest 2015, 125 (4), 1401–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Singh A; Ye M; Bucur O; Zhu S; Tanya Santos M; Rabinovitz I; Wei W; Gao D; Hahn WC; Khosravi-Far R Protein phosphatase 2A reactivates FOXO3a through a dynamic interplay with 14–3-3 and AKT. Mol. Biol. Cell 2010, 21 (6), 1140–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Los GV; Encell LP; McDougall MG; Hartzell DD; Karassina N; Zimprich C; Wood MG; Learish R; Ohana RF; Urh M, et al. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol 2008, 3 (6), 373–82. [DOI] [PubMed] [Google Scholar]
- (27).Taylor SE; O’Connor CM; Wang Z; Shen G; Song H; Leonard D; Sangodkar J; LaVasseur C; Avril S; Waggoner S, et al. The Highly Recurrent PP2A Aalpha-Subunit Mutation P179R Alters Protein Structure and Impairs PP2A Enzyme Function to Promote Endometrial Tumorigenesis. Cancer Res 2019, 79 (16), 4242–4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wu C; Ba Q; Lu D; Li W; Salovska B; Hou P; Mueller T; Rosenberger G; Gao E; Di Y, et al. Global and Site-Specific Effect of Phosphorylation on Protein Turnover. Dev. Cell 2021, 56 (1), 111–124 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Huang H; Tindall DJ Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim. Biophys. Acta 2011, 1813 (11), 1961–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Rena G; Woods YL; Prescott AR; Peggie M; Unterman TG; Williams MR; Cohen P Two novel phosphorylation sites on FKHR that are critical for its nuclear exclusion. EMBO J 2002, 21 (9), 2263–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Paez JG; Janne PA; Lee JC; Tracy S; Greulich H; Gabriel S; Herman P; Kaye FJ; Lindeman N; Boggon TJ, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004, 304 (5676), 1497–500. [DOI] [PubMed] [Google Scholar]
- (32).Pao W; Miller VA; Politi KA; Riely GJ; Somwar R; Zakowski MF; Kris MG; Varmus H Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med 2005, 2 (3), e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).O’Hare T; Eide CA; Deininger MW Bcr-Abl kinase domain mutations, drug resistance, and the road to a cure for chronic myeloid leukemia. Blood 2007, 110 (7), 2242–9. [DOI] [PubMed] [Google Scholar]
- (34).Formisano L; Lu Y; Servetto A; Hanker AB; Jansen VM; Bauer JA; Sudhan DR; Guerrero-Zotano AL; Croessmann S; Guo Y, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat. Commun 2019, 10 (1), 1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Yamazoe S; Tom J; Fu Y; Wu W; Zeng L; Sun C; Liu Q; Lin J; Lin K; Fairbrother WJ, et al. Heterobifunctional Molecules Induce Dephosphorylation of Kinases-A Proof of Concept Study. J. Med. Chem 2020, 63 (6), 2807–2813. [DOI] [PubMed] [Google Scholar]
- (36).Zheng J; Tian N; Liu F; Zhang Y; Su J; Gao Y; Deng M; Wei L; Ye J; Li H, et al. A novel dephosphorylation targeting chimera selectively promoting tau removal in tauopathies. Signal Transduct. Target Ther 2021, 6 (1), 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Buhimschi AD; Armstrong HA; Toure M; Jaime-Figueroa S; Chen TL; Lehman AM; Woyach JA; Johnson AJ; Byrd JC; Crews CM Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry 2018, 57 (26), 3564–3575. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.